

Abstract
Simple Summary
Metabolic reprogramming is one of tumor and immune cells’ most significant metabolic alterations. Moreover, metabolic-related signaling pathways, such as phosphoinositide 3-kinases (PI3Ks), the mammalian target of rapamycin (mTOR), can induce growth, proliferation, and angiogenesis of tumor cells. Therefore, inhibiting these metabolic pathways can be considered a potential therapeutic strategy in human malignancies. On the other hand, according to previous studies, pharmacological inhibiting of metabolic pathways using dual-pathway inhibitors can considerably inhibit tumor growth and progression, more than suppressing each pathway separately. This review aims to summarize the latest metabolic interventions by dual pathway inhibitors and discuss the achievements and limitations of this therapeutic tactic.
Abstract
The metabolism of tumors and immune cells in the tumor microenvironment (TME) can affect the fate of cancer and immune responses. Metabolic reprogramming can occur following the activation of metabolic-related signaling pathways, such as phosphoinositide 3-kinases (PI3Ks) and the mammalian target of rapamycin (mTOR). Moreover, various tumor-derived immunosuppressive metabolites following metabolic reprogramming also affect antitumor immune responses. Evidence shows that intervention in the metabolic pathways of tumors or immune cells can be an attractive and novel treatment option for cancer. For instance, administrating inhibitors of various signaling pathways, such as phosphoinositide 3-kinases (PI3Ks), can improve T cell-mediated antitumor immune responses. However, dual pathway inhibitors can significantly suppress tumor growth more than they inhibit each pathway separately. This review discusses the latest metabolic interventions by dual pathway inhibitors as well as the advantages and disadvantages of this therapeutic approach.
Keywords: metabolic intervention, dual inhibitor, metabolic reprogramming, cancer therapy
1. Introduction
Metabolic processes turn nutrients into molecules termed metabolites through a complex network of biochemical reactions, generating energy, redox equivalents, and macromolecules, such as RNA, DNA, proteins, and lipids essential for cell functions and survival [1,2]. Cytosolic glycolysis under anaerobic conditions and mitochondrial oxidative phosphorylation under aerobic conditions are energy sources for normal cells, respectively [3]. In contrast, according to the “Warburg effect”, cancerous cells desire to obtain energy via cytosolic glycolysis than oxidative phosphorylation, even under aerobic conditions [4,5]. Following activation of glycolysis, glycolytic tumor cells produce lactate, which is considered an energetic fuel for oxidative tumor cells. Monocarboxylate transporters (MCTs) catalyze the proton-linked transport of lactate and other monocarboxylates across cell membranes [6] (Figure 1). The justification for this tendency of tumor cells is their uncontrollable proliferation and the need for a fast ATP supply that is only accessible via glycolysis [7,8]. On the other hand, various principal metabolism pathways can be dysregulated in tumor cells [1]. According to available knowledge, immune responses are associated with significant changes in tissue metabolism, such as nutrient depletion, consumption of oxygen, and the generation of reactive oxygen and nitrogen intermediates [9,10,11].
Figure 1.

The Warburg effect. Most tumor cells produce energy, principally through glycolysis in the cytosol, producing lactic acid even in the presence of oxygen. MCTs catalyze the proton-linked transport of produced lactate across cell membranes. On the other hand, normal cells use oxidative phosphorylation in the mitochondria to produce energy under aerobic conditions.
Moreover, in the TME, numerous metabolites can affect immune cells’ differentiation and effector function [12]. However, in the TME, there is always fierce competition between immune and tumor cells to consume nutrients, and tumor cells usually win this competition due to their proliferative power and aggressive characteristics [13]. Correspondingly, metabolic interventions may be a potential therapeutic approach for treating malignancies.
It has been revealed that various signaling pathways, such as mitogen-activated protein kinase (MAPK), AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR), hypoxia-inducible factor 1-alpha (HIF-1α), PI3K/AKT, Ras, and insulin receptor are involved in cell metabolism. Interestingly, these pathways and cross-regulation could affect tumor growth and T cell-mediated immunity [14,15]. In this regard, several studies showed that pharmacological intervention using various inhibitors of these pathways could determine T cells’ metabolic fitness and persistence of these immune cells [16]. For instance, sirolimus analogs such as mTOR inhibitors are now being studied in phase II and III clinical trials because mTOR signaling dysfunction induces cellular proliferation and has been associated with various human malignancies [17]. However, despite the benefits of this therapeutic method, using these inhibitors could have adverse reactions such as nephrotoxicity and increased risk of infections requiring conscientious monitoring of the treatment [18]. The PI3K is an essential mediator of tumor cell growth, proliferation, and survival because overactivated PI3K alpha (PI3KA) following tumor mutations is critical for downstream signals of receptor tyrosine. These data indicate that administering selective PI3KA inhibitors might be attractive therapeutic agents in cancer treatment. The mTOR is a PI3K downstream kinase pivotal in cell growth and metabolism. Therefore, inhibition of the mTOR is beneficial in clinical settings for several types of cancers [19].
Furthermore, dual pathway inhibitors could be more efficient than controlling the metabolic pathways separately. Simultaneous inhibition of glycolysis and oxidative phosphorylation, as well as PI3K/AKT/mTOR and other pathways and involved molecules with dual inhibitors, showed that this strategy is effective in most cases and helps to prevent the growth and development of the tumor [20,21,22,23]. However, this response to treatment can be different in different cancers.
This review summarized the metabolism of tumor and immune cells and their effect on each other. Furthermore, critical signaling pathways involved in tumor and immune cell metabolism, related therapeutic interventions with dual inhibitors but not dual inhibition of metabolic pathways with combination regimens, and the advantages and disadvantages of these dual inhibitors are discussed.
2. Metabolism of Tumor and Immune Cells
2.1. Tumor Cells
Due to the high proliferation rate of tumor cells, regardless of whether the condition is aerobic or anaerobic, cytosolic glycolysis is the preferred method of providing ATP to their growth [24]. Researchers have demonstrated that tumor cells generate pyruvate under hypoxic conditions via the glycolysis pathway, producing lactic acid by pyruvate kinase type M2 instead of entering the mitochondrial oxidative phosphorylation and acetyl-CoA formation [25]. The tumor cells also generate biological macromolecules to replicate themselves using serine metabolism and the pentose phosphate pathway (PPP) [26,27]. The environmental conditions and concentration of nutrients for tumor cells determine which path and which macromolecules they use to find the optimal conditions for their growth and development. Therefore, in addition to decomposing glucose, tumor cells can use other macromolecules, such as amino acids, lipids, and fatty acids, to produce energy and grow [28,29,30].
Interestingly, when the concentration of glucose or glutamine is low (nutrients deprivation), tumor cells induce c-Myc to promote their survival via regulation of metabolic enzyme expression in the serine synthesis pathway, including phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), phosphoserine phosphatase (PSPH), activating the de novo serine synthesis and preserving redox homeostasis [31]. Moreover, under nutrient-deficient conditions, tumor cells are able to use acetoacetate to produce acetyl-CoA and fatty acids, which guarantee their survival [32,33,34]. Ketone body decomposition by tumor cells also generates metabolites that can enter the tricarboxylic acid cycle (TCA), providing ATP for their survival [30]. Cell cycle arrest, autophagy, anoikis, and entosis are four forms of anchorage-independent survival [35]. Recently an investigation reported that tumor cells prioritize glutamine-derived TCA energy metabolism over glycolysis to support ATP and suppress augmented oxidative stress by interacting with cysteine, preserving an anchorage-independent survival [36]. These findings indicate that depending on the different conditions governing the TME, tumor cells can intelligently provide their required energy through metabolic reprogramming and using different pathways to prolong their survival.
2.2. Immune Cells
In general, energy consumption in immune cells is different in active and inactive states. Moreover, like cancer cells, immune cells also use the metabolic pathways mentioned in the previous section [37]. Different metabolic patterns can affect immune cell differentiation. Previous studies demonstrated that M1 macrophages, activated neutrophils, and inducible nitric oxide synthase (iNOS)-expressed dendritic cells (DCs) mainly use glycolysis for their energy supply [38]. In the resting state, DCs prefer to use oxidative phosphorylation for energy supply, but activation of these cells is associated with increased glycolysis and lipid metabolism changes, affecting their function [39,40]. Furthermore, neutrophils use pentose phosphate and aerobic glycolysis pathways, and glycolysis is involved in regulating several neutrophil functions, such as chemotaxis and respiratory burst [41].
T cells play a unique role in anti-tumor defense among immune cells, and according to the various microenvironment signals, their phenotypes are metabolically different from other immune cells. Evidence demonstrated that the metabolic pattern of naïve and memory T cells is in a basic nutrient intake mode, glycolysis rate is decreased, proliferation is in a minimum state, and ATP supply mainly depends on oxidative phosphorylation [42]. While in pathologic conditions such as cancer, naïve T cells have to differentiate into effector T cells to defend against tumor cells, which require metabolic changes and increased proliferation. These metabolic alterations intensify the absorption of nutrients and glycolysis rate and increase the synthesis of essential macromolecules, such as nucleotides, proteins, and lipids. Simultaneous with these metabolic changes, mitochondrial oxygen consumption is condensed, inducing an effector T cell proliferation [2].
In contrast, regulatory T cells (Tregs) and M2 macrophages principally use oxidative phosphorylation from fatty acid oxidation (FAO) to provide the energy they need [43]. B cells are other immune cells that are involved in humeral immunity. It has been reported that activated B cells prefer to use glycolysis. However, following B cell activation by lipopolysaccharide (LPS) or other antigens, mitochondrial metabolism and glycolysis are boosted in these cells [44,45]. Recently, it has been revealed that the upregulation of oncogene c-Myc and increased glycolysis are critical for generating functional regulatory B cells (Bregs) [46].
2.3. Nutritional Competition between Tumor Cells and Immune System Cells
A significant challenge for antitumor immune responses is the competition between tumor cells and immune cells to take up glucose, amino acids, fatty acids, growth factors, and other metabolites in the TME. The expression of related transporters on the surface of these cells can also influence the fate of tumors and the immune system’s response [13]. The most critical nutrient consumed and absorbed by tumor cells is glucose, which also serves as an essential energy substance for the differentiation, activation, and function of infiltrated immune cells in the TME, such as tumor-infiltrating lymphocytes (TILs) [47,48,49]. Competitive uptake of glucose by tumor cells to suppress the function of TILs is one of the tumor escape and immunosuppressive mechanisms of cancer [50]. Moreover, increased glycolytic activities of tumor cells, and generated metabolites, such as lactate, can suppress glucose consumption by TILs, their exhaustion, and damage to their functions [51,52]. Additionally, tumor heterogenicity, high acidity, hypoxia, and high concentrations of lactate and ROS in the TME stimulate immune escape and cancer development [52]. Consequently, targeting various involved metabolic pathways affecting T cell-mediated antitumor responses could be a potential approach to overcome the destructive effects of metabolic competition between immune and tumor cells [53] (Figure 2).
Figure 2.

Metabolic competition between cancer cells and immune cells in the TME. There is a competition between tumor cells and immune cells to take up glucose, amino acids, fatty acids, growth factors, and other metabolites in the TME. The most critical nutrient consumed and absorbed by tumor cells is glucose, which also serves as an essential energy substance for the differentiation, activation, and function of infiltrated immune cells in the TME, such as TILs. Competitive uptake of glucose by tumor cells to suppress the function of TILs. Increased glycolytic activities of tumor cells, and generated metabolites, such as lactate, can suppress glucose consumption by TILs, and their exhaustion.
3. The Most Important Metabolic Pathways in Cancer and Therapeutic Interventions
3.1. PI3K/AKT/mTOR Pathway
PI3K is known as a group of plasma membrane-related lipid kinases. These kinases comprise p55 (regulatory), p110 (catalytic), and p85 (regulatory) subunits [54]. PI3K is categorized into PI3KI, PI3KII, and PI3KIII classes based on various structures and substrates [55]. The p85 regulatory subunit can bind and integrate signals from protein kinase C (PKC), tyrosine kinase-linked receptors, hormonal receptors, Src homology 2 domain-containing protein tyrosine phosphatase 1 (SHP1), Src, mutated Ras, Rac, and Rho, activating the p110 catalytic subunit and other downstream molecules [56]. Stabilizing the p110 subunit depends on its dimerization with the p85 subunit. As extracellular stimuli, hormones, cytokines, and growth factors activate PI3K in normal and physiologic conditions [57]. Activated PI3K induces the phosphorylation of phosphatidylinositol 4,5-bisphosphate to produce phosphatidylinositol 3,4,5-trisphosphate (PIP3), stimulating downstream kinases, such as AKT and 3-phosphoinositide-dependent protein kinase-1 (PDK1), and inducing cell growth and cell survival pathways [58,59]. It has been revealed that phosphatase and tensin homolog (PTEN) regulates the PI3K pathway via dephosphorylation of PIP3 to PIP2, inhibiting downstream kinase activation [56].
One of the leading downstream PI3K signaling effectors is mTOR, a serine/threonine protein kinase that regulates cell growth, proliferation, and metabolism [60,61]. Based on available knowledge, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) are two structures of mTOR. These complexes have different functions; for instance, mTORC1 induces cell anabolism by promoting the synthesis of nucleic acid and protein while preventing cell catabolism-mediated processes such as autophagy. On the other hand, mTORC2 induces glutamine uptake via activating AGC kinases, resulting in the regulation of glutamine cell surface transporters [60]. Furthermore, mTORC1 induces glutamine synthesis by positively regulating glutamate dehydrogenase (GDH) and suppressing sirtuin 4 (SIRT4), which is responsible for GDH inhibition [62,63]. Since aerobic glycolysis is a hallmark of tumor cells, nitrogen and carbon are supplied by glutamine to facilitate anabolic processes and cell growth [64]. In tumor cells, it has been demonstrated that the mTOR pathway is responsible for stimulating tumorigenesis, inducing the expression of inhibitory molecules, such as programmed cell death ligand-1 (PDL-1), and suppressing anticancer immune responses [65].
In some human malignancies, mTOR gene mutations are reported because these malignancies can activate mTOR constitutively. According to the tumor genome sequencing datasets, thirty-three mTOR mutations involved in cancer have been identified. The discovered mutations are categorized into six distinct regions in the C-terminal half of mTOR. They are responsible for hindering the interactions between mTOR and DEP domain-containing mTOR-interacting protein (DEPTOR) (endogenous mTOR inhibitor), hyperactivating the mTOR pathway [66]. Other mutations are also related to mTORC1 and mTORC2-specific components and upstream elements, including oncogenes and tumor suppressors [67,68]. Moreover, several cancer-mediated mutations are reported in the PI3K pathway, the upstream of mTORC1 and mTORC2 [69]. For instance, mutations in PIK3CA, which encodes the p110α PI3K catalytic subunit, have been reported in several human malignancies, such as prostate, breast, endometrium, colon, and upper aerodigestive tract cancers [70].
As discussed, cancer cells require metabolic reprogramming to facilitate their proliferation, growth, biological functions, and survival. In this context, mTOR plays a regulatory role in cellular metabolism via upregulating the expression of ribosomal protein S6 kinase beta-1 (S6K1) and eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) [71]. In addition, the proliferation and growth of tumor cells are supported by mTOR-enhancing glucose metabolism by upregulating the transporter 1 (GlUT1), HIF1-α, and c-MYC, resulting in the enhancement of glycolytic enzymes, such as enolase (ENO), phosphofructokinase (PFK), and phosphoglucoisomerase (PGI) [72,73,74]. The signaling of mTORC1 and mTORC2 induces fatty acid uptake and lipogenesis to support tumor cell proliferation [74]. These complexes induce sterol regulatory element-binding protein 1 (SREBP-1) and the peroxisome proliferator-activated receptor γ (PPARγ), which are involved in promoting the expression of lipid and cholesterol homeostasis-associated enzymes, such as fatty acid transporter CD36, acetyl-CoA carboxylase 1 (ACC1), ATP citrate lyase (ACLY), and fatty acid synthase (FASN) [75,76,77]. It has been revealed that inhibiting the rapamycin-insensitive companion of the mammalian target of rapamycin (RICTOR) as a mTORC2 component, as well as inhibition of mTORC1, mTORC2, and PI3K, could remarkably interrupt the progression of pancreatic cancer and prolong survival in end-stage of the tumor [78]. Furthermore, overexpression of RICTOR is associated with lymph node metastasis, tumor progression, and poor prognosis [79]. Employing kinase inhibitors or using RICTOR knockdown are other therapeutic approaches in mTORC2-targeted cancer therapy, leading to suppression of tumor cell growth, migration, and metastasis [80,81]. In colorectal cancer (CRC), RICTOR deficiency could significantly decrease the pAktSer473 level and reduce the proliferation and growth of CRC cells [82]. AKT hyperactivation is another consequence of RICTOR upregulation, progressing tumor cells and decreasing overall survival. In human epidermal growth factor receptor 2 (EGFR2) positive breast cancer, the effectiveness of HER2/EGFR tyrosine kinase inhibitors such as lapatinib is increased following the knockdown of RICTOR or using kinase inhibitors [68].
According to the available evidence, it regulates the immune system components, including immune cell metabolism, differentiation, activation, effector function, and homeostasis in innate and adaptive immunity [83]. Moreover, the activation of PI3K/AKT/mTORC1 is essential for developing metabolic reprogramming effector CD4+ and CD8+ T cells [84,85]. Following the interaction of the T cell receptor (TCR) and the presented antigens, downstream signals sent by TCR, co-stimulatory molecules in immunologic synapses, as well as cytokine-mediated signals received by mTORC1 and mTORC2 and their complexes regulate the immune receptor pathways, transcription factors, migration, and metabolic reprogramming. In addition, mTOR signals are involved in determining the fate of T cells and which phenotype will be formed in them and go towards memory, regulatory, or effector T cells [85]. In this regard, an investigation demonstrated that T cells with Rheb deficiency could not differentiate into T helper 1 (Th1) and Th17 and generate related immune responses. In contrast, these T cells tend to differentiate into Th2 [86].
Interestingly, targeting mTORC2 signals through the knockdown of RICTOR in T cells prevents their differentiation into Th2 and enhances differentiation into Th1 and Th17 cells. Furthermore, the generation of Tregs depends on the selective deletion of mTORC1 and mTORC2 signals regardless of the existence of the exogenous transforming growth factor-beta (TGF-β) [86]. Therefore, rapamycin, as an mTOR inhibitor, can repress the activation and proliferation of T cells [87]. An experimental study showed that metabolic manipulation of naïve T cells and TILs during their expansion in vitro using Akt inhibitor VIII could induce the differentiation of T cells into memory T cells with suitable antitumor activity following reinfusion of these T cells to immune-deficient mice with multiple myeloma [88].
The metabolic interventions using pharmaceutic agents can affect metabolic fitness and T cell persistence [16]. An investigation on CD33-specific chimeric antigen receptor (CAR)-T cells showed that treating these engineered cells with LY294002, a PI3K inhibitor, in vitro led to less differentiation of these cells into shorter-lived effector forms with enhanced antitumor activity and persistence in mice. Inhibition of PI3K/AKT/mTOR was also associated with rising glycolytic flux following the activation of CAR-T cells [89]. In these CAR-T cells, using various co-stimulatory domains such as CD28 or 4-1BB could affect T cell metabolism and persistence. For instance, 4-1BB could induce mitochondrial biogenesis, oxidative phosphorylation, and differentiation into memory T cells, along with more in vivo persistence of T cells, while employing CD28 was associated with increasing glycolysis and effector differentiation of T cells [90]. These findings demonstrate that metabolic interventions could be related to improving the effectiveness of cell therapy in cancer; however, due to the metabolic alteration of T cells, it is possible to change the function and phenotype, and this type of intervention needs more studies.
3.2. AMPK Pathway
AMPK is considered a crucial molecule in regulating cell energy homeostasis by monitoring AMP, ADP, and ATP levels. AMPK comprises three subunits: α subunit (catalytic) and β and γ (regulatory) subunits and several tissues/organisms-specific isoforms, including α1, α2, β1, β2, γ1, γ2, γ3 [91]. Intracellular calcium ions through calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) and adenine nucleotides are able to activate the AMPK pathway [92]. In stress conditions, including hypoxia, low glucose concentrations, and ischemia associated with ATP depletion, the AMPK pathway is also activated. This activation is regulated by cellular AMP/ADP/ATP that competitively binds to the γ subunit. These occurrences can stimulate Thr172 phosphorylation on the α subunit via the tumor suppressor liver kinase B1 (LKB1) or suppress Thr172 phosphorylation through dephosphorylating α subunit by phosphatases [93,94]. AMPK can also be suppressed by fructose 1,6-bisphosphate (FBP), a glucose metabolite [91]. Activating the AMPK can induce autophagy and fatty acid oxidation to supply and reload the intracellular ATP [95]. Since gluconeogenesis, protein, and lipid synthesis are ATP-consuming, AMPK negatively regulates biosynthetic processes to preserve ATP and control energy metabolism, activating immune cells [96]. These findings indicate that the AMPK pathway controls the balance between immune responses and energy metabolism [2]. On the other hand, AMPK activation inhibits various immune signaling pathways involved in the proliferation and activation of immunosuppressive immune cells, such as myeloid-derived suppressor cells (MDSCs) [96]. Accordingly, the AMPK pathway, as a metabolic regulator, may play an antitumoral role in cancer. In contrast, other studies showed that AMPK activation could be associated with the suppression of pro-inflammatory pathways, such as NFκB, and the differentiation of macrophages from the M1 into the M2 phenotype, enhancing the expression of anti-inflammatory cytokines, such as IL-10 [97,98]. Activation of the AMPK pathway via controlling energy metabolism is involved in the differentiation of T cells, affecting the function of these immune cells [2].
3.3. Adenosine Pathway
Following tissue injury or hypoxic TME, nucleoside adenosine levels are significantly amplified and bind to adenosine 2A receptor (A2AR) on the cell surfaces, inhibiting cytotoxic T cell/natural killer cells (NK) cell-mediated antitumor immune responses. CD73 and CD39 regulate the production of adenosine via the catabolism of ATP. CD39 converts ATP to AMP, and CD73 converts AMP to adenosine [99]. Immunosuppressive cells such as Tregs can express CD39, and activation of the A2AR pathway in these immune cells leads to the downregulation of inflammatory mediators and upregulation of anti-inflammatory mediators, such as IL-10, resulting in dephosphorylation of signal transducer and activator of transcription 5 (STAT5), inhibiting the NFκB pathway, and reducing IL-2R-mediated signals in T cells. Tregs generate adenosine through the co-expression of CD39/CD73, activating the adenosine pathway and overexpressing prostaglandin E2 (PGE2) receptor, EP2 receptors (EP2R) on the surface of responder T cells. In addition, adenylate cyclase activity increased following the adenosine pathway’s activation, leading to increased cAMP and promoting immunosuppressive responses [100].
4. Dual Pathway Inhibitors
So far, numerous studies have been performed on metabolic pathway inhibitors in cancer therapy, and relatively satisfactory outcomes have been achieved. However, there is also a theory that utilizing dual pathway inhibitors increases the effectiveness of cancer therapy. This section discusses the properties of these dual inhibitors and the consequences of their use in cancer treatment (Table 1). The chemical structure and molecular formula of dual inhibitors are also shown in Table 2.
Table 1.
List of the most important dual pathway inhibitors.
| Inhibitor | Target | Mechanism and Outcomes | Adverse Effects | Ref |
|---|---|---|---|---|
| Dactolisib (BEZ235) |
PI3K/mTOR |
|
|
[101,102,103,104,105] |
| Gedatolisib (PKI-587) |
PI3K/mTOR |
|
|
[106,107,108,109] |
| Voxtalisib (SAR245409) | Class-I PI3Ks, mTORC1/ mTORC2 |
|
|
[110,111] |
| Bimiralisib (PQR309) | Pan-class I PI3K/mTOR |
|
|
[112,113,114] |
| Paxalisib (GDC-0084) |
PI3K/mTOR |
|
|
[115,116,117,118,119,120] |
| Omipalisib (GSK2126458) | PI3K/mTOR |
|
- | [121,122,123] |
| SF1126 | PI3K/mTOR BRD4 |
|
- | [124,125,126] |
| PF-04691502 | PI3K/mTOR |
|
- | [127,128,129,130] |
| Samotolisib (LY3023414) | PI3K/mTOR |
|
|
[131,132,133,134] |
| PWT33597 | PI3K/mTORC1 and mTORC2 |
|
- | [135] |
| Apitolisib (GDC-0980) | PI3K/mTOR |
|
|
[136,137] |
| AptCCN | Glycolysis & oxidative phosphorylation |
|
- | [138] |
| KPT-9274 | NAMPT/p21 PAK4 |
|
- | [139] |
| 20e and 20k | PDK/LDHA |
|
- | [140] |
| 3,5-diamino-1,2,4-triazole analogs | KDM1A/SMOX |
|
- | [141,142] |
PI3K, phosphatidylinositol 3-kinase; mTOR, mammalian target of rapamycin; STAT3, signal transducer and activator of transcription 3; PFS, progression-free survival; BBB, blood–brain barrier; DNA-PKcs, DNA-dependent protein kinase catalytic subunit; HIF, hypoxia-inducible factor; 4E-BP1, eukaryotic initiation factor 4E-binding protein 1; PERK, protein kinase R (PKR)-like endoplasmic reticulum kinase; NO, nitric oxide; IFN, interferon; DNMT1, DNA-methyltransferase 1; ROS, reactive oxygen species.
Table 2.
Chemical structure of dual pathway inhibitors.
| Name | Molecular Formula | 2D Structure |
|---|---|---|
| Dactolisib | C30H23N5O |
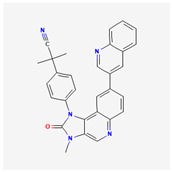
|
| Gedatolisib | C32H41N9O4 |
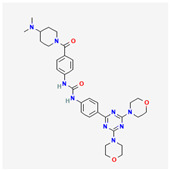
|
| Voxtalisib | C13H14N6O |
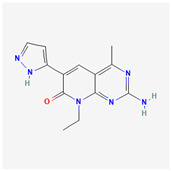
|
| Bimiralisib | C17H20F3N7O2 |
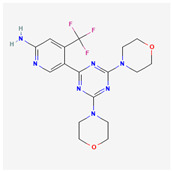
|
| Paxalisib | C18H22N8O2 |
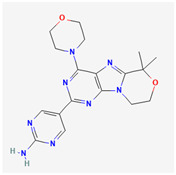
|
| Omipalisib | C25H17F2N5O3S |
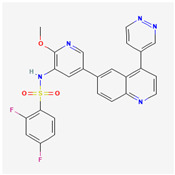
|
| SF1126 | C39H48N8O14 |
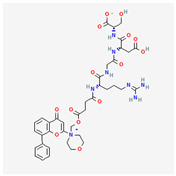
|
| PF-04691502 | C22H27N5O4 |
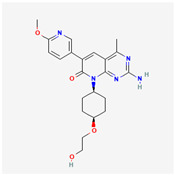
|
| Samotolisib | C22H27N5O4 |
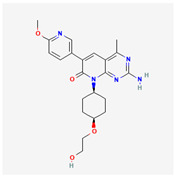
|
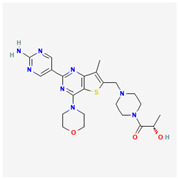
| Apitolisib | C23H30N8O3S |
|
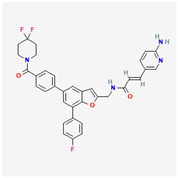
| KPT-9274 | C35H29F3N43 |
|
Figure source: PubChem®.
4.1. Dual PI3K/AKT/mTOR Inhibitors
PI3K and mTOR belong to the family of phosphatidylinositol 3-kinase-related kinases (PIKKs). According to the structural and functional similarities of PI3K and mTOR, as well as studies on mTOR inhibitors, researchers synthesized inhibitors with dual functions, suppressing both PI3K and mTOR [143].
4.1.1. Dactolisib
Dactolisib (BEZ235) is an imidazoquinoline targeting the PI3K and the mTOR, with robust antitumor activity. Dactolisib suppresses PI3K kinase and mTOR kinase in the PI3K/AKT/mTOR kinase pathway, inducing tumor cell apoptosis and inhibiting growth in PI3K/mTOR highly expressing cancer cells. In addition to causing tumor cell growth, proliferation, and survival, the PI3K/mTOR pathway also plays a crucial role in making the tumor resistant to conventional therapies, such as radiotherapy and chemotherapy [101].
It was investigated in non-small-cell lung cancer (NSCLC) cells with various EGFR statuses whether co-inhibiting PI3K and mTOR would improve the therapeutic outcomes. This study reported that BEZ235 repressed tumor growth in vitro and in vivo via promoting cell-cycle arrest at the G1 phase and reducing the cyclin D1/D3 expression. Additionally, BEZ235 synergistically promoted cisplatin-mediated apoptosis in NSCLC cells by boosting or persisting DNA damage. These data indicate that dual PI3K/mTOR inhibition by BEZ235 can be a potential anticancer agent that induces the efficacy of targeted therapy or chemotherapy [102].
An investigation on mantle cell lymphoma (MCL) cells showed that compared with everolimus (an mTOR inhibitor) or NVP-BKM120 (a PI3K inhibitor), BEZ235 could be more potent in suppressing the PI3K/Akt/mTOR pathway. Furthermore, BEZ235 can inhibit angiogenesis, migration, and invasion of tumor cells. Besides, it has been revealed that interleukin-4 (IL-4) and IL-6/signal transducer and activator of transcription 3 (STAT3) pathway are involved in chemoresistance. Regarding the role of IL-6 in inducing chemoresistance, it has been revealed that IL-6-mediated stem cell expansion and epithelial-mesenchymal transition (EMT) could be involved in this obstacle. Mechanistically, IL-6 induces upregulation of multidrug-resistant associated mediators, such as MDR1 and glutathione S transferase pi (GSTpi). Moreover, IL-6 protects tumor cells from the paclitaxel- and cisplatin-associated cytotoxic effects by downregulating caspase3 (Cas3) and upregulating antiapoptotic proteins, such as X-linked inhibitor of apoptosis (XIAP), B-cell lymphoma 2 (Bcl-2), and B-cell lymphoma-extra large (Bcl-xL) in resistant cancer cells. Further, IL-6 can induce activation of the PI3K/AKT pathway in resistant tumor cells [144]. There is no clear indication of the exact mechanism by which IL-4 contributes to chemoresistance in tumors; however, the evidence demonstrates that, similar to IL-6, IL-4 can regulate key antiapoptotic factors that may have functional effects on the chemoresistance [145].
Unlike everolimus and NVP-BKM120, BEZ235 can inhibit signals of these cytokines, improving the effectiveness of chemotherapy [103]. These findings indicate that dual pathway inhibitors can be more effective than single pathway inhibition, inhibiting the PI3K/Akt/mTOR pathway at multiple levels. Combining BEZ235 with dexamethasone in acute lymphoblastic leukemia (ALL) showed that along with inhibiting the PI3K/AKT/mTOR pathway, antileukemic effects of dexamethasone were improved in vitro and in vivo. AKT1 is responsible for repressing dexamethasone-induced tumor cell apoptosis. Therefore, BEZ235, by inhibiting AKT and downregulating myeloid cell leukemia-1 (MCL-1), can induce dexamethasone-mediated apoptotic pathways in malignant cells [104]. A phase Ib dose-escalation clinical trial demonstrated that combining everolimus and BEZ235 (orally in escalating doses of 200, 400, and 800 mg/day plus everolimus at 2.5 mg/day in 28-day cycles) and this therapeutic regimen was associated with poor efficacy and tolerance. The remarkable feature of BEZ235 administration was that its oral administration could not be a suitable option for treatment due to low bioavailability and gastrointestinal toxicity.
In contrast, the systemic administration of this inhibitor can have better effectiveness in a dose-dependent manner [146]. Another phase I/Ib, multicenter, open-label by administrating different doses of BEZ235 to patients with HER2+ breast cancer showed that the effect of this drug was partially observed in only 13% of patients. Side effects, including nausea, diarrhea, and vomiting, were reported in patients. Moreover, BEZ235 showed more variability and effect in doses higher than 100 mg, although high doses were associated with gastrointestinal toxicity [105].
On the other hand, patients with advanced pancreatic neuroendocrine tumors (pNET) were treated with oral everolimus 10 mg once daily or oral BEZ235 400 mg twice daily on a continuous dosing schedule. Findings showed that the median progression-free survival (PFS) in BEZ235 treated group was 8.2 months versus 10.8 months in patients treated with everolimus. The most frequent adverse effects in patients with BEZ235 were diarrhea, stomatitis, and nausea. These results show that BEZ235 cannot be more effective than everolimus, at least in terms of PFS. On the other hand, this dual inhibitor’s side effects are more than everolimus. However, this response to treatment may change in cancers and patients with different conditions [147].
4.1.2. Gedatolisib
Gedatolisib (PKI-587) is a dual inhibitor targeting the PI3K and mTOR kinases in the PI3K/mTOR signaling pathway, with potential antitumor activity. Evidence demonstrated that following intravenous administration of gedatolisib, it inhibits both mTOR and PI3K kinases, inducing apoptosis and suppressing the growth of tumor cells overexpressing PI3K/mTOR. Moreover, gedatolisib can enhance radio and chemosensitivity by inhibiting the PI3K/AKT/mTOR pathways to decrease DNA damage repair mechanisms [106]. Recently, an investigation reported that combining PKI-587 with Cofetuzumab Pelidotin, a protein tyrosine kinase 7 (PTK7)-targeted, auristatin-based antibody-drug conjugate in patients with metastatic triple-negative breast cancer (TNBC) was associated with promising clinical activity, two months median PFS, and moderate toxicity (anorexia nausea, mucositis, and fatigue) [107]. PKI-587 can increase radiosensitization. A study showed that DNA damage was increased in SK-Hep1 xenograft hepatocellular carcinoma (HCC) models, combining ionizing radiation with PKI-587, and G0/G1 cell-cycle arrest, as well as apoptosis, were induced in tumor cells. Accordingly, repressing the PI3K/AKT/mTOR and DNA damage repair pathways by PKI-587 can stimulate the radiosensitizing of HCC cells [108]. The prognosis in T-cell ALL patients (T-ALL) is poor. Changes in the PI3K/mTOR signaling pathway are responsible for relapse and treatment failure because the PI3K/mTOR pathway is overactivated in relapsed T-ALL patients. This study demonstrated that PKI-587 inhibited T-ALL cell line proliferation and colony formation via selective suppression of the PI3K/mTOR pathway without disturbing the mitogen-activated protein kinase (MAPK) pathway in vitro and in vivo. Furthermore, PKI-587 reduces tumor load and progression, lengthening survival rates in immune-deficient mice xenograft models without causing weight loss in the mice treated with the inhibitor [109]. It seems that PKI-587 can be a suitable option for treating human malignancies. However, combination therapy using PKI-587 can increase the effectiveness of the treatment by creating synergistic responses.
4.1.3. Voxtalisib
Voxtalisib (SAR245409) is a powerful class-I PI3Ks, mTORC1, and mTORC2 inhibitor [148]. It has been reported that voxtalisib could suppress the phosphorylation of PI3K and control the incorporation of mTOR effector in cancer cells [149]. In a phase Ib clinical trial on patients with advanced malignant tumors, 90 mg pimasertib (a MEK1/2 inhibitor) and 70 mg voxtalisib was administrated, and findings showed that this combination regimen was not well tolerated and did not have a significant effect on the survival of patients with advanced solid tumor. The most commonly observed adverse events in this study were diarrhea, nausea, and fatigue [110]. It appears that patient drug tolerance depends on voxtalisib dose and schedule. A phase I clinical trial administrated a combination of voxtalisib with temozolomide, with or without radiation therapy, to patients with high-grade glioma. Outcomes showed that the maximum tolerated doses (MTDs) for voxtalisib in combination with temozolomide were 90 mg once a day and 40 mg twice daily. The most frequently experienced adverse events in this study were nausea, fatigue, thrombocytopenia, diarrhea, and lymphopenia. This study showed that voxtalisib, combined with temozolomide with or without radiation therapy, could effectively treat high-grade gliomas with acceptable safety [111].
4.1.4. Bimiralisib
Bimiralisib (PQR309) is known as a pan-class I PI3K/mTOR antagonist that vigorously represses PI3Kα and mTOR. According to the biochemical experiments, bimiralisib has less influence on PI3Kβ and cannot remarkably inhibit other protein kinases [150]. It has been revealed that the PI3K/mTOR pathway is involved in several lymphoma types. Therefore, pharmacologic inhibition of this pathway may benefit patients with lymphoma. A preclinical lymphoma model demonstrated that bimiralisib showed anti-lymphoma activity in vitro alone or combined with other anticancer drugs, such as panobinostat, venetoclax, lenalidomide, ibrutinib, ARV-825, rituximab, and marizomib. This study demonstrated that bimiralisib could induce the expression of HRK, YPEL3, and TP63, while the gene expression of HSPA8 and HSPA1B, CCDC86, PAK1IP1, and MIR155HG was downregulated following treatment [112]. A dose-escalation, open-label phase I trial evaluated the anticancer effects and safety of bimiralisib (dose 10 to 150 mg) in patients with advanced solid tumors. Results showed that partial response was detectable following bimiralisib therapy in a patient with metastatic thymus malignancy.
Moreover, disease volume was reduced to one-quarter in a patient with sinonasal cancer, and a patient with clear cell Bartholin’s gland cancer experienced stable disease for more than sixteen weeks. The MTD and recommended phase 2 dose of bimiralisib was considered to be 80 mg orally once per day. Analysis of tumor biopsies revealed that bimiralisib exerts its antitumor effects by downregulating the PI3K pathway phosphoprotein. Additionally, common adverse events, including hyperglycemia, fatigue, nausea, constipation, diarrhea, rash, vomiting, and anorexia, were detected in about 30% of patients [113]. Interestingly, bimiralisib can effectively cross the brain–blood barrier (BBB) compared with BEZ235 and voxtalisib [112,114]. This feature of bimiralisib can facilitate its delivery to the tumor tissue in brain tumors and improve the effectiveness of the treatment.
4.1.5. Paxalisib
Paxalisib (GDC-0084) is known as a selective and potent oral brain-penetrant dual inhibitor of PI3K and mTOR kinase. Paxalisib was exclusively designed for treating brain tumors, such as progressive or recurrent glioma, because it can efficiently cross the BBB to improve drug delivery to the brain. Experimental studies have shown that paxalisib could inhibit the growth of tumor cells in a dose-dependent manner [115,116,117]. Based on available knowledge, the PI3K/Akt/mTOR pathway is overactivated due to the PIK3CA mutations in up to 70% of brain metastases in patients with breast cancer. A preclinical study showed that paxalisib significantly reduced cell viability and phosphorylation of AKT and p70 S6 kinase.
Moreover, apoptosis of PIK3CA-mutant breast cancer brain metastatic cells was increased following the treatment in lines in a dose-dependent fashion [118]. Therefore, using paxalisib may be effective in brain cancers and brain metastatic cancers. However, this dual inhibitor can be effective in other malignancies, such as cutaneous squamous cell carcinoma (cSCC). In this context, an investigation reported that paxalisib treatment at nanomole doses potently repressed the proliferation and survival of SCC-13, SCL-1, and A431 cell lines as well as primary human cSCC cells via induction of apoptosis and cell cycle arrest in the cSCC cells. Interestingly, in addition to its more lethal effect on tumor cells than other PI3K-Akt-mTOR pathway inhibitors, paxalisib was non-toxic to normal skin cells, including keratinocytes and fibroblasts [119]. The mechanism of action of paxalisib is inhibiting the phosphorylation of fundamental components of the PI3K-Akt-mTOR pathway, such as Akt, S6, p85, and S6K1. Furthermore, paxalisib hinders the activation of DNA-PKcs in cSCC cells [119].
4.1.6. Omipalisib
Omipalisib (GSK2126458) is an oral dual PI3K/mTOR inhibitor that suppresses the growth and progression of cancer cells [151]. It has been revealed that omipalisib treatment could prevent the colony formation of cancer stem cells and induce autophagic cell death because clonogenicity depends on basic fibroblast growth factor (bFGF) and Insulin-like growth factor 1 (IGF-1) signaling via AKT and ERK pathways and omipalisib in combination with an ERK inhibitor, such as MEK162 can suppress colony formation [121]. Anti-proliferative effect of omipalisib on AML cell lines was explored and revealed that omipalisib could considerably induce G0/G1 cell cycle arrest in OCI-AML3 HL60 and THP1 cell lines. As discussed, omipalisib downregulates the phosphorylation of mTOR, AKT, 4E-BP1, and S6K. Moreover, metabolic pathway enrichment analysis showed that metabolites related to amino acid metabolisms were remarkably decreased upon treatment with omipalisib.
Additionally, following treatment of the OCI-AML3 cells with omipalisib, the expression of several essential genes, including PHGDH, PSPH, PSAT1, MTHFD1/2, and SHMT1/2, in the glycine and serine synthesis pathway, were significantly downregulated in these cells. Due to reducing the energy levels, the biosynthesis and functions of the mitochondria could probably be affected by omipalisib [122]. Additionally, studies on mice models showed that 0.2 or 1 mg/kg oral administration of omipalisib could notably reduce tumor growth without apparent alteration in the body weight of treated animals [123].
4.1.7. SF1126
SF1126 is an RGD-conjugated LY294002 pro-drug with high solubility and antiangiogenic properties that can bind to specific integrins in the TME [152]. Therefore, the administration of SF1126 enhances delivery to the TME and tumor vasculature. Recent studies have shown that this compound can inhibit PI3K/AKT/mTOR and bromodomain-containing protein 4 (BRD4) pathways in cancer cells [124,125]. A study treated CRC cell lines as well as primary human colon cancer cells isolated from human tumors with SF1126, and findings showed that this drug could inhibit tumor cell growth and induce apoptosis. SF1126 also could lead to cell cycle arrest in cancer cells [124]. Another study reported that SF1126 treatment revokes the HIF-2α stabilization in VHL-mutated RCC cell lines under normoxic and hypoxic conditions. In addition, SF1126 subcutaneously administration to RCC-xenografted mice remarkably inhibited angiogenesis, tumor growth, and progression. SF1126 also could suppress integrin-mediated tumor cell migration and block the integrin-induced guanosine diphosphate (GDP)-Rac family small GTPase 1 (Rac1) conversion to its active state [126].
4.1.8. PF-04691502
PF-04691502 is another dual PI3K/mTOR inhibitor that can repress tumor growth and progression through the induction of apoptosis. PF-04691502 also improves the radiosensitivity of several human malignancies [127]. It has been reported that PF-04691502 could inhibit the growth, proliferation, migration, and invasion of bladder cancer cells. Additionally, it can enhance the apoptosis of these tumor cells through the intrinsic pathway. PF-04691502 reduces the expression of the PI3K/Akt/mTOR pathway and myeloid leukemia 1 (MCL-1) in bladder cancer cells. As with several of the dual inhibitors discussed, PF-04691502 can also increase the effectiveness of chemotherapy and increase tumor cells’ sensitivity to radiotherapy [128].
Advanced-stage gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are associated with poor prognosis despite radiotherapy and chemotherapy. Treatment of NET cell lines (QGP-1 and BON) with PF-04691502 downregulated the expression of pAKT for up to 72 h than in the control group. Surprisingly, concurrent treatment with PF-04691502 and radiotherapy did not enhance apoptosis in NET cells, while adding PF-04691502 48 h upon radiotherapy considerably induced apoptosis compared to radiotherapy or PF-04691502 therapy alone [129]. These outcomes indicate that combining radiation and PF-04691502 could be a novel and potential therapeutic approach for treating NETs [153].
In patients with T-cell lymphomas (CTCLs) and Sézary syndrome (SS), overactivation of the PI3K/AKT/mTOR pathway is demonstrable. Therefore, blocking this pathway signifies a potential therapeutic option against the cutaneous CTCLs [130]. Treatment with PF-04691502 suppressed the growth of the CTCL cell lines and derived tumor cells from SS patients. The PF-04691502 induced the apoptotic cascades and G1 cell arrest in the cell cycle of CTCL cell lines, whereas, in SS patients, its action was primarily due to the induction of strong apoptosis. Notably, PF-04691502 only mildly affected healthy donors obtained T cells.
Moreover, PF-04691502 suppressed CXCL12-related cell recruitment and migration in all studied groups. Following the treatment, along with increased survival, it was revealed that tumor volume reduced from 936 mm3 in the control group to 400 mm3 in treated mice. Additionally, tumor weight was decreased from 0.56 g in controls to 0.2 g in treated mice [153].
4.1.9. Samotolisib
Samotolisib (LY3023414) is an orally available dual kinase inhibitor of class I PI3K and mTOR [131]. Preclinical studies showed that combining samotolisib with prexasertib, a checkpoint kinase 1 inhibitor (samotolisib 200 mg orally twice daily plus prexasertib 105 mg/m2 intravenously every 14 days), could have anticancer activity in preclinical models and preliminary value in seriously pretreated patients; however, the clinical combination was accompanied by toxicity, which should be considered in future trials [131]. A double-blind, placebo-controlled phase Ib/II trial combined samotolisib with enzalutamide (a nonsteroidal antiandrogen medication used to treat prostate cancer) in patients with metastatic castration-resistant prostate cancer. This study showed that the combination of samotolisib with enzalutamide was well tolerated and pointedly improved PFS in studied patients [132]. Evidence demonstrated that fatigue, nausea, vomiting, and diarrhea were the most frequent adverse events following treatment with samotolisib [133]. In anal dysplasia and anal cancer, inhibition of the PI3K/AKT/mTOR pathway is a practical approach. In K14E6/E7 mice treated with topical samotolisib, squamous cell carcinoma was inhibited after 15 weeks of treatment initiation in a sex-dependent manner (only male mice) [134].
4.1.10. PWT33597
PWT33597 is another dual kinase inhibitor that, based on biochemical assays, represses PI3K alpha and mTOR. PWT33597 profiling showed little or no cross-reactivity with protein kinases, including tyrosine kinases or serine/threonine [19]. Treatment of mutationally activated PI3K alpha in HCT116 and NCI-H460 tumor cells with PWT33597 showed that this drug could inhibit mTOR pathway proteins and PI3K. Moreover, PWT33597 exhibited promising pharmacokinetic properties in multiple tumor xenograft models via an enduring reserve of PI3K and mTOR pathway signaling [19]. Several drugs that inhibit mTORC1 (rapalogs) are approved for the treatment of advanced renal cell carcinoma (RCC) [154]. However, the effectiveness of these drugs is limited to a specific subset of patients and is not enduring. It is proposed to administer PWT33597 to renal xenograft models in which both mTORC1 and mTORC2 inhibitions and PI3K inhibition may increase the effectiveness of the treatment by directly targeting multiple signaling nodes, including the vascular endothelial growth factor receptors (VEGFRs). PWT33597 was tested in VHL−/−, PTEN−/− xenografts compared to rapamycin as a mTORC1 inhibitor and sorafenib, a VEGFR/RAF inhibitor. The results showed that despite the tumor growth-inhibitory properties of sorafenib and rapamycin (64%), PWT33597 had a much higher growth-inhibitory effect (93%). PWT33597 was more efficient than paxalisib (a pan-PI3K inhibitor) in inhibiting tumor growth, significantly reducing tumor weight and size. Furthermore, PWT33597 increases cleaved caspase 3 (an apoptotic indicator) [135].
4.1.11. Apitolisib
Apitolisib (GDC-0980) is a novel dual PI3K/mTOR inhibitor. Apitolisib treatment strongly reduced the phosphorylation of AKT and mTOR and decreased growth in two cholangiocarcinoma (CCA) cell lines, SNU1196 and SNU478. Apitolisib also improved the effects of chemotherapeutic agents, such as cisplatin or gemcitabine, in vitro and boosted the cleavage of PARP. Additionally, combining apitolisib with chemotherapy in a mouse xenograft model of CCA decreased colony formation by SNU1196 and SNU478 cells and inhibited tumor cell growth [136]. Dysregulated PI3K/AKT/mTOR signals are responsible for tumorigenesis via inducing tumor growth, metastasis, and resistance to antitumor therapies in glioblastoma. Therefore, this axis could be an attractive therapeutic target for pharmacological manipulation. Glioblastoma multiforme (GBM) cell lines (A-172 and U-118-MG) were treated with apitolisib, and the treatment was associated with time- and dose-dependent cytotoxicity and apoptosis. The action mechanism of apitolisib is probably the downregulation of protein kinase RNA-like endoplasmic reticulum kinase (PERK) expression, blocking its inhibitory effect on protein synthesis, intensifying translation, and inducing apoptosis [137]. In contrast, a randomized open-label phase II trial reported that due to adverse events, such as hyperglycemia and rash, apitolisib could not effectively treat metastatic RCC, compared with everolimus [155]. Probably, the effect of this inhibitor can be different in different cancers.
4.2. Other Potential Dual Inhibitors
A cancer therapeutic approach is the dual inhibition of critical metabolic pathways, such as glycolysis and oxidative phosphorylation, that breaks cancer cells’ metabolic plasticity and limits the provided energy supply [156,157]. In this regard, an aptamer-based artificial enzyme was designed and constructed by arginine aptamer-modified carbon-dots-doped graphitic carbon nitride (AptCCN) to inhibit glycolysis and oxidative phosphorylation concurrently. The dAptCCN is able to capture intracellular arginine and convert arginine to nitric oxide (NO) via oxidation under red light irradiation. Evidence showed that the depletion of arginine and NO stress suppress glycolysis and oxidative phosphorylation, blocking energy supply and inducing tumor cell apoptosis [138]. Numerous tumor cells have been shown to increase the expression of nicotinamide phosphoribosyltransferase (NAMPT), which is essential for NAD+ salvage.
Consequently, employing NAMPT inhibitors could be an attractive option for cancer therapy [158]. KPT-9274 is a dual NAMPT/p21-activated kinase 4 (PAK4)/inhibitor that decreases the NAD+/NADH ratio in cancer cells, inhibiting tumor growth in sarcoma mice models and RCC [139,159]. KPT-9274 also induces antitumor immune responses via improving tumor antigen presentation and rising interferons (IFN)-α and IFN-γ responses [139]. GMX1778 is another NAMPT inhibitor that was used in murine GMB by microparticles. A study on GBM models reported that combining immune checkpoint inhibitors with GMX1778 increased the survival of treated animals [160]. GMX1778 increases the expression of programmed cell death ligan-1 (PD-L1) via NAD+ depletion and induces recruiting effector immune cells, such as CD4+ and CD8+ T cells. The frequency of M2-macrophages as immunosuppressive cells also decreased following treatment with GMX1778.
As discussed, tumor cells are capable of glucose metabolic alteration from oxidative phosphorylation to cytoplasmic glycolysis; pyruvate dehydrogenase kinases (PDKs) and lactate dehydrogenase A (LDHA) are crucial enzymes in this occurrence. Therefore, inhibiting these enzymes might be a promising approach in cancer therapy. An investigation designed two PDK/LDHA inhibitors (20e and 20k) that could decrease lactate formation and enhance oxygen consumption in A549 cells. These data indicate that these inhibitors can regulate the glucose metabolic pathways in cancer cells [140].
Type II topoisomerases are responsible for changing DNA topology via generating transient DNA double-strand breaks and are crucial for eukaryotic cells [161]. It has been revealed that dual inhibitors of kinases and topoisomerases II could be a potential therapeutic approach in cancer therapy. Designing dual inhibitors may also be a valuable and exciting strategy to overcome resistance to topoisomerase-targeted drugs due to the structural similarities between the topoisomerase II and other proteins, such as heat shock protein 90 (Hsp90), which is involved in DNA repair mechanisms [162].
Lysine (K)-specific demethylase 1A (KDM1A) is a flavin-dependent amine oxidase that is involved in the demethylation of lysine 3 and 4 in histone 3 tails (H3K4 and H3K9) [163]. Evidence showed that the upregulation of KDM1A is associated with multiple human disorders, such as cancer, through reduced methylation at the H3K4 and H3K9. Moreover, the demethylation of H3K4 and H3K9 leads to the condensation of chromatin, suppressing the transcription of several anticancer gene regions, such as DNA methyltransferase-1 (DNMT-1), p53, p21, GATA-binding factor (GATA)-1 and GATA-2. Accordingly, KDM1A inhibition can be beneficial in suppressing tumors [141]. On the other hand, spermine oxidase (SMOX) is an amine oxidase that can convert spermine and spermidine to spermidine and putrescine via deaminating aminopropyl [164]. Spermine and spermidine are involved in cellular functions, such as gene expression control, scavenging reactive oxygen species (ROS), cell cycle regulation, DNA structure maintenance, and protein synthesis [165]. Interestingly, SMOX has considerable sequence homology to KDM1A, which facilitates the design of dual inhibitors for cancer therapy [142]. In this context, an investigation reported that 3,5-diamino-1,2,4-triazole analogs could be used for dual inhibition of KDM1A and SMOX to treat pancreatic cancer [141].
5. Advantages and Disadvantages of Dual Pathway Inhibitors in Cancer Therapy
Evidence demonstrated that multitarget inhibitors are a promising tool for treating complicated disorders due to the inherent redundancy and robustness of numerous biological networks and pathways. In parallel, designing multitarget inhibitors is challenging for medicinal chemists [166] (Figure 3). One of the critical metabolic pathways that have been studied more is the PI3K/AKT/mTOR pathway, and significant dual inhibitors have been designed to inhibit the kinases of this pathway. There is a high prevalence of dysregulation of the PI3K/AKT/mTOR signaling pathway among cancerous cells [167,168,169]. There are different classes of PI3K/AKT/mTOR inhibitors, including mTOR inhibitors, PI3K/AKT inhibitors, and dual PI3K/AKT/mTOR inhibitors. The rationale of PI3K/AKT/mTOR inhibitor development is the existence of a negative feedback loop of S6K1 because durable inhibition of mTOR promotes the activation of PI3K/AKT [170].
Figure 3.

Advantages and disadvantages of using dual pathway inhibitors in cancer therapy.
Consequently, to overcome this challenge, employing dual PI3K/AKT/mTOR inhibitors could be a potential therapeutic approach. Lack of appropriate biomarkers that predict the activity of PI3K/AKT/mTOR inhibitors is another limitation in the clinical development of these dual inhibitors. However, in some human cancers, such as breast cancer, PIK3CA mutation is considered a biomarker for predicting PI3K/AKT/mTOR pathway activity [171]. Furthermore, WNT/β-catenin pathway-mediated PIK3CA mutations may reduce the sensitivity of tumor cells to the dual PI3K/mTOR inhibitor [172].
Clinical trials reported that common toxicities of administered PI3K/AKT/mTOR inhibitors were rash, gastrointestinal adverse events, fatigue, and asthenia. Furthermore, due to the impact of PI3K signaling on glucose metabolism, hyperglycemia has also been variable [173]. However, other adverse events may also be reported following the administration of dual pathway inhibitors. The induction of RICTOR acetylation by glucose is another challenge in targeting the PI3K/AKT/mTOR pathway because it leads to the activation of mTORC2 and therapeutic resistance to PI3K/AKT inhibitors. In glioblastoma cells, overactivation of mTORC2 following glucose-mediated RICTOR acetylation promotes the epidermal growth factor receptor vIII (EGFRvIII) signaling [174]. Besides, it has been demonstrated that monotherapy with mTOR inhibitors, such as rapamycin, suppresses antitumor immune responses via inhibiting effector CD8+ T cells, increasing Tregs frequency, and modulating dendritic cells and antigen presentation [175]. Therefore, determining the exact role of the mTOR pathway in the microenvironment of different tumors plays an essential role in the success of treatment using PI3K/AKT/mTOR inhibitors. For instance, it has been recently stated that inhibiting the mTOR pathway significantly stimulates antitumor immune response via increasing the frequency of long-lived CD8+ memory T cells and improving tumor cell eradication [16]. Moreover, inhibition of the PI3K/AKT/mTOR pathway could be associated with reducing tumor cell growth, proliferation, migration, invasion, and survival. On the other hand, PI3K/AKT/mTOR inhibitors can improve tumor immunosurveillance efficacy by downregulating the immunosuppressive pathways and activating antitumor immune responses in the TME.
ATP-binding cassette (ABC) drug transporters, including ABCB1 and ABCG2, are involved in multidrug resistance [176]. It has been revealed that overexpression of these transporters reduced the efficacy of dual PI3K/AKT/mTOR inhibitors, such as LY3023414, in tumor cells. Since LY3023414 is a substrate for ABCB1 and ABCG2, these transporters, by their drug efflux function, significantly reduce intracellular levels of LY3023414 in tumor cells [177]. Moreover, pharmacokinetic alterations in PI3K/AKT/mTOR inhibitors should be noted in pharmacological interventions when the drugs are prescribed together. For instance, drug–drug interactions between these inhibitors, such as everolimus and BEZ235, can affect their steady-state pharmacokinetic parameters [146]. It is realized that everolimus is a substrate of the CYP3A4 enzyme as well as the P-glycoprotein (a drug transporter) enzymes. This drug is highly susceptible to any alterations in the level of the enzyme CYP3A [178]. Available metabolic-related findings demonstrate that BEZ235 may modulate the expression and activation of CYP3A4. It was hypothesized that everolimus and BEZ235 could interact due to their absorption, metabolism (pharmacokinetic properties), and pharmacodynamic pathways [179].
How the inhibitors are metabolized is also a critical issue in the effectiveness of the treatment. Some PI3K/AKT/mTOR dual inhibitors, such as PWT33597, are metabolized more slowly in vivo and interact less with cytochrome P450 enzyme, resulting in an enduring inhibition of the PI3K/AKT/mTOR pathway in xenograft tumors. However, PWT33597 administration in mice could be accompanied by transient increases in insulin plasma concentrations [19]. Therefore, considering a drug’s positive and negative aspects is critical in managing and increasing the success of cancer treatment with metabolic intervention.
6. Concluding Remarks
Pharmacological intervention in different metabolic pathways can lead to fundamental alterations in tumor cell metabolism and pathological function, affecting immune responses in the TME. The dual inhibitors of metabolic pathways can have a better effect in preventing the growth and progression of tumor cells due to the simultaneous inhibition of pathways such as the PI3K/AKT/mTOR pathway. However, in some cancers, such as advanced pancreatic neuroendocrine tumors (pNET), using inhibitors of each pathway separately has had a better effect than dual inhibitors. Despite the various advantages, administrating dual inhibitors has multiple challenges and limitations. For example, the mTOR pathway can sometimes trigger anti-tumor immune responses. In these cases, its inhibition may be associated with the suppression of the immune system, and this issue can entirely depend on the tumor type, signal, and stage. For instance, in melanoma, the PI3K/Akt, MyD88, and IKK pathways could be involved in IL-36β-mediated mTORC1 activation, promoting CD8+ T cell activation and inducing antitumor immune responses in vitro and in vivo [180]. Based on the available studies, it appears that combining dual inhibitors with other chemotherapeutic agents (paclitaxel and cisplatin) or other targeted therapies, such as trastuzumab or anti-immune checkpoint blockers (anti-PD-1 and anti-CTLA-4), can increase the effectiveness of the treatment [105,181,182]. However, common toxicities, especially gastrointestinal toxicities and drug dose adjustments, are also essential factors that should be considered in designing a pharmacologic protocol using monotherapy with dual inhibitors of metabolic pathways or combination therapies.
Author Contributions
K.J.: conception, design, and inviting co-authors to participate. M.C., H.L. and S.Y.: writing the original manuscript draft. K.J. and Y.C.: review and editing of the manuscript critically for important intellectual content and provided comments and feedback for the scientific contents of the manuscript. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding Statement
This work was supported by Zhejiang Provincial Science and Technology Projects (grants no. LGF22H160017 to M.C. and LGF22H160046 to H.L.), National Natural Science Foundation of China (grant no. 82104445 to H.L.), and Jinhua Municipal Science and Technology Projects (grants no. 2021-3-040 to K.J. and 2021-3-046 to H.L.).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Boroughs L.K., DeBerardinis R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia L., Oyang L., Lin J., Tan S., Han Y., Wu N., Yi P., Tang L., Pan Q., Rao S. The cancer metabolic reprogramming and immune response. Mol. Cancer. 2021;20:28. doi: 10.1186/s12943-021-01316-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vazquez A., Liu J., Zhou Y., Oltvai Z.N. Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst. Biol. 2010;4:58. doi: 10.1186/1752-0509-4-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapa B., Gonçalves A.C., Jorge J., Alves R., Pires A.S., Abrantes A.M., Coucelo M., Abrunhosa A., Botelho M.F., Nascimento-Costa J.M. Acute myeloid leukemia sensitivity to metabolic inhibitors: Glycolysis showed to be a better therapeutic target. Med. Oncol. 2020;37:72. doi: 10.1007/s12032-020-01394-6. [DOI] [PubMed] [Google Scholar]
- 5.Callao V., Montoya E. Toxohormone-like factor from microorganisms with impaired respiration. Science. 1961;134:2041–2042. doi: 10.1126/science.134.3495.2041. [DOI] [PubMed] [Google Scholar]
- 6.Payen V.L., Mina E., Van Hée V.F., Porporato P.E., Sonveaux P. Monocarboxylate transporters in cancer. Mol. Metab. 2020;33:48–66. doi: 10.1016/j.molmet.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domiński A., Krawczyk M., Konieczny T., Kasprów M., Foryś A., Pastuch-Gawołek G., Kurcok P. Biodegradable pH-responsive micelles loaded with 8-hydroxyquinoline glycoconjugates for Warburg effect based tumor targeting. Eur. J. Pharm. Biopharm. 2020;154:317–329. doi: 10.1016/j.ejpb.2020.07.019. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J., Yang J., Lin C., Liu W., Huo Y., Yang M., Jiang S.-H., Sun Y., Hua R. Endoplasmic Reticulum stress-dependent expression of ERO1L promotes aerobic glycolysis in Pancreatic Cancer. Theranostics. 2020;10:8400. doi: 10.7150/thno.45124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang B., Song B.-l., Xu C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020;2:132–141. doi: 10.1038/s42255-020-0174-0. [DOI] [PubMed] [Google Scholar]
- 10.Chen B., Gao A., Tu B., Wang Y., Yu X., Wang Y., Xiu Y., Wang B., Wan Y., Huang Y. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials. 2020;255:120187. doi: 10.1016/j.biomaterials.2020.120187. [DOI] [PubMed] [Google Scholar]
- 11.Terry S., Engelsen A.S., Buart S., Elsayed W.S., Venkatesh G.H., Chouaib S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett. 2020;492:1–10. doi: 10.1016/j.canlet.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Yan Y., Chang L., Tian H., Wang L., Zhang Y., Yang T., Li G., Hu W., Shah K., Chen G. 1-Pyrroline-5-carboxylate released by prostate Cancer cell inhibit T cell proliferation and function by targeting SHP1/cytochrome c oxidoreductase/ROS Axis. J. Immunother. Cancer. 2018;6:148. doi: 10.1186/s40425-018-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang C.-H., Qiu J., O’Sullivan D., Buck M., Noguchi T., Curtis J., Chen Q., Gindin M., Gubin M., Tonc E. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amirani E., Hallajzadeh J., Asemi Z., Mansournia M.A., Yousefi B. Effects of chitosan and oligochitosans on the phosphatidylinositol 3-kinase-AKT pathway in cancer therapy. Int. J. Biol. Macromol. 2020;164:456–467. doi: 10.1016/j.ijbiomac.2020.07.137. [DOI] [PubMed] [Google Scholar]
- 15.Kim J., Yang G.S., Lyon D., Kelly D.L., Stechmiller J. Metabolomics: Impact of comorbidities and inflammation on sickness behaviors for individuals with chronic wounds. Adv. Wound Care. 2021;10:357–369. doi: 10.1089/wound.2020.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Araki K., Turner A.P., Shaffer V.O., Gangappa S., Keller S.A., Bachmann M.F., Larsen C.P., Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali E.S., Mitra K., Akter S., Ramproshad S., Mondal B., Khan I.N., Islam M.T., Sharifi-Rad J., Calina D., Cho W.C. Recent advances and limitations of mTOR inhibitors in the treatment of cancer. Cancer Cell Int. 2022;22:284. doi: 10.1186/s12935-022-02706-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viana S.D., Reis F., Alves R. Therapeutic use of mTOR inhibitors in renal diseases: Advances, drawbacks, and challenges. Oxidative Med. Cell. Longev. 2018;2018:3693625. doi: 10.1155/2018/3693625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews D.J., O’Farrell M., James J., Giddens A.C., Rewcastle G.W., Denny W.A. Preclinical characterization of PWT33597, a dual inhibitor of PI3-kinase alpha and mTOR. Cancer Res. 2011;71:4485. doi: 10.1158/1538-7445.AM2011-4485. [DOI] [Google Scholar]
- 20.Herschbein L., Liesveld J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2018;32:235–248. doi: 10.1016/j.blre.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Chen J., Zhao K.-N., Li R., Shao R., Chen C. Activation of PI3K/Akt/mTOR pathway and dual inhibitors of PI3K and mTOR in endometrial cancer. Curr. Med. Chem. 2014;21:3070–3080. doi: 10.2174/0929867321666140414095605. [DOI] [PubMed] [Google Scholar]
- 22.Bhatt A.P., Bhende P.M., Sin S.-H., Roy D., Dittmer D.P., Damania B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood J. Am. Soc. Hematol. 2010;115:4455–4463. doi: 10.1182/blood-2009-10-251082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabbah D.A., Brattain M.G., Zhong H. Dual inhibitors of PI3K/mTOR or mTOR-selective inhibitors: Which way shall we go? Curr. Med. Chem. 2011;18:5528–5544. doi: 10.2174/092986711798347298. [DOI] [PubMed] [Google Scholar]
- 24.Moreno-Sánchez R., Rodríguez-Enríquez S., Marín-Hernández A., Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 25.Mazurek S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Jiang P., Du W., Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. 2014;5:592–602. doi: 10.1007/s13238-014-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amelio I., Cutruzzolá F., Antonov A., Agostini M., Melino G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014;39:191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altman B.J., Stine Z.E., Dang C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q., Luo Q., Halim A., Song G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017;401:39–45. doi: 10.1016/j.canlet.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y., Li P. Fatty acid metabolism and cancer development. Sci. Bull. 2016;61:1473–1479. doi: 10.1007/s11434-016-1129-4. [DOI] [Google Scholar]
- 31.Sun L., Song L., Wan Q., Wu G., Li X., Wang Y., Wang J., Liu Z., Zhong X., He X. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015;25:429–444. doi: 10.1038/cr.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schug Z.T., Vande Voorde J., Gottlieb E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer. 2016;16:708–717. doi: 10.1038/nrc.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schug Z.T., Peck B., Jones D.T., Zhang Q., Grosskurth S., Alam I.S., Goodwin L.M., Smethurst E., Mason S., Blyth K. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mashimo T., Pichumani K., Vemireddy V., Hatanpaa K.J., Singh D.K., Sirasanagandla S., Nannepaga S., Piccirillo S.G., Kovacs Z., Foong C. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng Z., Wang H., Liu J., Deng Y., Zhang N. Comprehensive understanding of anchorage-independent survival and its implication in cancer metastasis. Cell Death Dis. 2021;12:629. doi: 10.1038/s41419-021-03890-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Endo H., Owada S., Inagaki Y., Shida Y., Tatemichi M. Metabolic reprogramming sustains cancer cell survival following extracellular matrix detachment. Redox Biol. 2020;36:101643. doi: 10.1016/j.redox.2020.101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghesquière B., Wong B.W., Kuchnio A., Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511:167–176. doi: 10.1038/nature13312. [DOI] [PubMed] [Google Scholar]
- 38.Thwe P.M., Amiel E. The role of nitric oxide in metabolic regulation of dendritic cell immune function. Cancer Lett. 2018;412:236–242. doi: 10.1016/j.canlet.2017.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williford J.-M., Ishihara J., Ishihara A., Mansurov A., Hosseinchi P., Marchell T.M., Potin L., Swartz M.A., Hubbell J.A. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 2019;5:eaay1357. doi: 10.1126/sciadv.aay1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y., Hwang J.-Y., Park H.-b., Yadav D., Oda T., Jin J.-O. Porphyran isolated from Pyropia yezoensis inhibits lipopolysaccharide-induced activation of dendritic cells in mice. Carbohydr. Polym. 2020;229:115457. doi: 10.1016/j.carbpol.2019.115457. [DOI] [PubMed] [Google Scholar]
- 41.Jeon J.-H., Hong C.-W., Kim E.Y., Lee J.M. Current understanding on the metabolism of neutrophils. Immune Netw. 2020;20:e46. doi: 10.4110/in.2020.20.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearce E.L., Poffenberger M.C., Chang C.-H., Jones R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pearce E., Pearce E. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi T., Lam P.Y., Jiang H., Bednarska K., Gloury R., Murigneux V., Tay J., Jacquelot N., Li R., Tuong Z.K. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood. 2020;136:3004–3017. doi: 10.1182/blood.2020005602. [DOI] [PubMed] [Google Scholar]
- 45.Domka K., Goral A., Firczuk M. cROSsing the line: Between beneficial and harmful effects of reactive oxygen species in B-cell malignancies. Front. Immunol. 2020;11:1538. doi: 10.3389/fimmu.2020.01538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X.-Y., Wei Y., Hu B., Liao Y., Wang X., Wan W.-H., Huang C.-X., Mahabati M., Liu Z.-Y., Qu J.-R. c-Myc-driven glycolysis polarizes functional regulatory B cells that trigger pathogenic inflammatory responses. Signal Transduct. Target. Ther. 2022;7:105. doi: 10.1038/s41392-022-00948-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolb D., Kolishetti N., Surnar B., Sarkar S., Guin S., Shah A.S., Dhar S. Metabolic modulation of the tumor microenvironment leads to multiple checkpoint inhibition and immune cell infiltration. ACS Nano. 2020;14:11055–11066. doi: 10.1021/acsnano.9b10037. [DOI] [PubMed] [Google Scholar]
- 48.Palmer C.S., Ostrowski M., Balderson B., Christian N., Crowe S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015;6:1. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Togo M., Yokobori T., Shimizu K., Handa T., Kaira K., Sano T., Tsukagoshi M., Higuchi T., Yokoo S., Shirabe K. Diagnostic value of 18F-FDG-PET to predict the tumour immune status defined by tumoural PD-L1 and CD8+ tumour-infiltrating lymphocytes in oral squamous cell carcinoma. Br. J. Cancer. 2020;122:1686–1694. doi: 10.1038/s41416-020-0820-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu J., Villa M., Sanin D.E., Buck M.D., O’Sullivan D., Ching R., Matsushita M., Grzes K.M., Winkler F., Chang C.-H. Acetate promotes T cell effector function during glucose restriction. Cell Rep. 2019;27:2063–2074.e5. doi: 10.1016/j.celrep.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sukumar M., Roychoudhuri R., Restifo N.P. Nutrient competition: A new axis of tumor immunosuppression. Cell. 2015;162:1206–1208. doi: 10.1016/j.cell.2015.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harmon C., O’Farrelly C., Robinson M.W. Tumor Microenvironment. Springer; Berlin/Heidelberg, Germany: 2020. The immune consequences of lactate in the tumor microenvironment; pp. 113–124. [DOI] [PubMed] [Google Scholar]
- 53.Kareva I. Metabolism and gut microbiota in cancer immunoediting, CD8/Treg ratios, immune cell homeostasis, and cancer (immuno) therapy: Concise review. Stem Cells. 2019;37:1273–1280. doi: 10.1002/stem.3051. [DOI] [PubMed] [Google Scholar]
- 54.Donahue T.R., Tran L.M., Hill R., Li Y., Kovochich A., Calvopina J.H., Patel S.G., Wu N., Hindoyan A., Farrell J.J. Integrative Survival-Based Molecular Profiling of Human Pancreatic CancerIntegrative Profile of Human Pancreatic Cancer. Clin. Cancer Res. 2012;18:1352–1363. doi: 10.1158/1078-0432.CCR-11-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katso R., Okkenhaug K., Ahmadi K., White S., Timms J., Waterfield M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 56.Hennessy B.T., Smith D.L., Ram P.T., Lu Y., Mills G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 57.Guo H., German P., Bai S., Barnes S., Guo W., Qi X., Lou H., Liang J., Jonasch E., Mills G.B. The PI3K/AKT pathway and renal cell carcinoma. J. Genet. Genom. 2015;42:343–353. doi: 10.1016/j.jgg.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manning B.D., Cantley L.C. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang J., Nie J., Ma X., Wei Y., Peng Y., Wei X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer. 2019;18:26. doi: 10.1186/s12943-019-0954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Masui K., Harachi M., Cavenee W.K., Mischel P.S., Shibata N. mTOR complex 2 is an integrator of cancer metabolism and epigenetics. Cancer Lett. 2020;478:1–7. doi: 10.1016/j.canlet.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Huang K., Fingar D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014;36:79–90. doi: 10.1016/j.semcdb.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Csibi A., Lee G., Yoon S.-O., Tong H., Ilter D., Elia I., Fendt S.-M., Roberts T.M., Blenis J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014;24:2274–2280. doi: 10.1016/j.cub.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Csibi A., Fendt S.-M., Li C., Poulogiannis G., Choo A.Y., Chapski D.J., Jeong S.M., Dempsey J.M., Parkhitko A., Morrison T. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153:840–854. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang X., Liang T., Yang W., Zhang L., Wu S., Yan C., Li Q. Astragalus membranaceus injection suppresses production of interleukin-6 by activating autophagy through the AMPK-mTOR pathway in lipopolysaccharide-stimulated macrophages. Oxidative Med. Cell. Longev. 2020;2020:1364147. doi: 10.1155/2020/1364147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grabiner B.C., Nardi V., Birsoy K., Possemato R., Shen K., Sinha S., Jordan A., Beck A.H., Sabatini D.M. A Diverse Array of Cancer-Associated MTOR Mutations Are Hyperactivating and Can Predict Rapamycin SensitivityCancer-Associated Hyperactivating MTOR Mutations. Cancer Discov. 2014;4:554–563. doi: 10.1158/2159-8290.CD-13-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pilotto S., Simbolo M., Sperduti I., Novello S., Vicentini C., Peretti U., Pedron S., Ferrara R., Caccese M., Milella M. OA06. 06 druggable alterations involving crucial carcinogenesis pathways drive the prognosis of squamous cell lung carcinoma (SqCLC) J. Thorac. Oncol. 2017;12:S266–S267. doi: 10.1016/j.jtho.2016.11.260. [DOI] [Google Scholar]
- 68.Morrison Joly M., Hicks D.J., Jones B., Sanchez V., Estrada M.V., Young C., Williams M., Rexer B.N., Sarbassov D.D., Muller W.J. Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast CancersHER2-Mediated Tumorigenesis Requires mTORC2. Cancer Res. 2016;76:4752–4764. doi: 10.1158/0008-5472.CAN-15-3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mafi S., Mansoori B., Taeb S., Sadeghi H., Abbasi R., Cho W.C., Rostamzadeh D. mTOR-mediated regulation of immune responses in cancer and tumor microenvironment. Front. Immunol. 2022;12:5724. doi: 10.3389/fimmu.2021.774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chalhoub N., Baker S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. Mech. Dis. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lien E.C., Lyssiotis C.A., Cantley L.C. Metabolism in Cancer. Springer; Berlin/Heidelberg, Germany: 2016. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer; pp. 39–72. [DOI] [PubMed] [Google Scholar]
- 72.Buller C.L., Loberg R.D., Fan M.-H., Zhu Q., Park J.L., Vesely E., Inoki K., Guan K.-L., Brosius F.C., III A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008;295:C836–C843. doi: 10.1152/ajpcell.00554.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gordan J.D., Thompson C.B., Simon M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mossmann D., Park S., Hall M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer. 2018;18:744–757. doi: 10.1038/s41568-018-0074-8. [DOI] [PubMed] [Google Scholar]
- 75.Yecies J.L., Zhang H.H., Menon S., Liu S., Yecies D., Lipovsky A.I., Gorgun C., Kwiatkowski D.J., Hotamisligil G.S., Lee C.-H. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hagiwara A., Cornu M., Cybulski N., Polak P., Betz C., Trapani F., Terracciano L., Heim M.H., Rüegg M.A., Hall M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 77.Laplante M., Sabatini D.M. mTOR signaling at a glance. J. Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Driscoll D.R., Karim S.A., Sano M., Gay D.M., Jacob W., Yu J., Mizukami Y., Gopinathan A., Jodrell D.I., Evans T.R.J., et al. mTORC2 Signaling Drives the Development and Progression of Pancreatic Cancer. Cancer Res. 2016;76:6911–6923. doi: 10.1158/0008-5472.CAN-16-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bian Y., Wang Z., Xu J., Zhao W., Cao H., Zhang Z. Elevated Rictor expression is associated with tumor progression and poor prognosis in patients with gastric cancer. Biochem. Biophys. Res. Commun. 2015;464:534–540. doi: 10.1016/j.bbrc.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 80.Zhang F., Zhang X., Li M., Chen P., Zhang B., Guo H., Cao W., Wei X., Cao X., Hao X., et al. mTOR Complex Component Rictor Interacts with PKCζ and Regulates Cancer Cell Metastasis. Cancer Res. 2010;70:9360–9370. doi: 10.1158/0008-5472.CAN-10-0207. [DOI] [PubMed] [Google Scholar]
- 81.Li H., Lin J., Wang X., Yao G., Wang L., Zheng H., Yang C., Jia C., Liu A., Bai X. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast Cancer Res. Treat. 2012;134:1057–1066. doi: 10.1007/s10549-012-2036-2. [DOI] [PubMed] [Google Scholar]
- 82.Gulhati P., Cai Q., Li J., Liu J., Rychahou P.G., Qiu S., Lee E.Y., Silva S.R., Bowen K.A., Gao T., et al. Targeted Inhibition of Mammalian Target of Rapamycin Signaling Inhibits Tumorigenesis of Colorectal Cancer. Clin. Cancer Res. 2009;15:7207–7216. doi: 10.1158/1078-0432.CCR-09-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xie S., Chen M., Yan B., He X., Chen X., Li D. Identification of a Role for the PI3K/AKT/mTOR Signaling Pathway in Innate Immune Cells. PLoS ONE. 2014;9:e94496. doi: 10.1371/journal.pone.0094496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim E.H., Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front. Immunol. 2013;4:20. doi: 10.3389/fimmu.2013.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012;12:325–338. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Delgoffe G.M., Pollizzi K.N., Waickman A.T., Heikamp E., Meyers D.J., Horton M.R., Xiao B., Worley P.F., Powell J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guri Y., Nordmann T.M., Roszik J. mTOR at the Transmitting and Receiving Ends in Tumor Immunity. Front. Immunol. 2018;9:578. doi: 10.3389/fimmu.2018.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Crompton J.G., Sukumar M., Roychoudhuri R., Clever D., Gros A., Eil R.L., Tran E., Hanada K.-i., Yu Z., Palmer D.C., et al. Akt Inhibition Enhances Expansion of Potent Tumor-Specific Lymphocytes with Memory Cell Characteristics. Cancer Res. 2015;75:296–305. doi: 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zheng W., O’Hear C.E., Alli R., Basham J.H., Abdelsamed H.A., Palmer L.E., Jones L.L., Youngblood B., Geiger T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia. 2018;32:1157–1167. doi: 10.1038/s41375-017-0008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kawalekar O.U., O’Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Patel P.R., Guedan S., Scholler J., Keith B., et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 91.Yuan J., Dong X., Yap J., Hu J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020;13:113. doi: 10.1186/s13045-020-00949-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hawley S.A., Pan D.A., Mustard K.J., Ross L., Bain J., Edelman A.M., Frenguelli B.G., Hardie D.G. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 93.Shaw R.J., Kosmatka M., Bardeesy N., Hurley R.L., Witters L.A., DePinho R.A., Cantley L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Woods A., Johnstone S.R., Dickerson K., Leiper F.C., Fryer L.G.D., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 95.Kim Y.K., Chae S.C., Yang H.J., An D.E., Lee S., Yeo M.G., Lee K.J. Cereblon Deletion Ameliorates Lipopolysaccharide-induced Proinflammatory Cytokines through 5’-Adenosine Monophosphate-Activated Protein Kinase/Heme Oxygenase-1 Activation in ARPE-19 Cells. Immune Netw. 2020;20:e26. doi: 10.4110/in.2020.20.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Salminen A., Kauppinen A., Kaarniranta K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019;97:1049–1064. doi: 10.1007/s00109-019-01795-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang S., Lin Y., Xiong X., Wang L., Guo Y., Chen Y., Chen S., Wang G., Lin P., Chen H., et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clin. Cancer Res. 2020;26:4921–4932. doi: 10.1158/1078-0432.CCR-20-0113. [DOI] [PubMed] [Google Scholar]
- 98.Zhu Y.P., Brown J.R., Sag D., Zhang L., Suttles J. Adenosine 5′-Monophosphate–Activated Protein Kinase Regulates IL-10–Mediated Anti-Inflammatory Signaling Pathways in Macrophages. J. Immunol. 2015;194:584–594. doi: 10.4049/jimmunol.1401024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Antonioli L., Pacher P., Vizi E.S., Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Whiteside T., Jackson E. Adenosine and Prostaglandin E2 Production by Human Inducible Regulatory T Cells in Health and Disease. Front. Immunol. 2013;4:212. doi: 10.3389/fimmu.2013.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Umansky V., Sevko A. Overcoming immunosuppression in the melanoma microenvironment induced by chronic inflammation. Cancer Immunol. Immunother. 2012;61:275–282. doi: 10.1007/s00262-011-1164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu Y.-Y., Wu H.-C., Wu J.-E., Huang K.-Y., Yang S.-C., Chen S.-X., Tsao C.-J., Hsu K.-F., Chen Y.-L., Hong T.-M. The dual PI3K/mTOR inhibitor BEZ235 restricts the growth of lung cancer tumors regardless of EGFR status, as a potent accompanist in combined therapeutic regimens. J. Exp. Clin. Cancer Res. 2019;38:282. doi: 10.1186/s13046-019-1282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rosich L., Montraveta A., Xargay-Torrent S., López-Guerra M., Roldán J., Aymerich M., Salaverria I., Beà S., Campo E., Pérez-Galán P. Dual PI3K/mTOR inhibition is required to effectively impair microenvironment survival signals in mantle cell lymphoma. Oncotarget. 2014;5:6788. doi: 10.18632/oncotarget.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hall C.P., Reynolds C.P., Kang M.H. Modulation of Glucocorticoid Resistance in Pediatric T-cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/mTOR Inhibitor BEZ235BEZ235 plus Dexamethasone in ALL. Clin. Cancer Res. 2016;22:621–632. doi: 10.1158/1078-0432.CCR-15-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rodon J., Pérez-Fidalgo A., Krop I.E., Burris H., Guerrero-Zotano A., Britten C.D., Becerra C., Schellens J., Richards D.A., Schuler M. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharmacol. 2018;82:285–298. doi: 10.1007/s00280-018-3610-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Y., Xie C., Li A., Liu X., Xing Y., Shen J., Huo Z., Zhou S., Liu X., Xie Y. PKI-587 enhances chemosensitivity of oxaliplatin in hepatocellular carcinoma through suppressing DNA damage repair pathway (NHEJ and HR) and PI3K/AKT/mTOR pathway. Am. J. Transl. Res. 2019;11:5134. [PMC free article] [PubMed] [Google Scholar]
- 107.Radovich M., Solzak J.P., Wang C.J., Hancock B.A., Badve S.S., Althouse S.K., Bray S.M., Storniolo A.M., Ballinger T.J., Schneider B.P. Initial phase I safety study of gedatolisib plus cofetuzumab pelidotin for patients with metastatic triple-negative breast cancer. Clin. Cancer Res. 2022;28:3235–3241. doi: 10.1158/1078-0432.CCR-21-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xie Y., Liu C., Zhang Y., Li A., Sun C., Li R., Xing Y., Shi M., Wang Q. PKI-587 enhances radiosensitization of hepatocellular carcinoma by inhibiting the PI3K/AKT/mTOR pathways and DNA damage repair. PLoS ONE. 2021;16:e0258817. doi: 10.1371/journal.pone.0258817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gazi M., Moharram S.A., Marhäll A., Kazi J.U. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL) Cancer Lett. 2017;392:9–16. doi: 10.1016/j.canlet.2017.01.035. [DOI] [PubMed] [Google Scholar]
- 110.Schram A.M., Gandhi L., Mita M.M., Damstrup L., Campana F., Hidalgo M., Grande E., Hyman D.M., Heist R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer. 2018;119:1471–1476. doi: 10.1038/s41416-018-0322-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wen P.Y., Omuro A., Ahluwalia M.S., Fathallah-Shaykh H.M., Mohile N., Lager J.J., Laird A.D., Tang J., Jiang J., Egile C. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17:1275–1283. doi: 10.1093/neuonc/nov083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tarantelli C., Gaudio E., Arribas A.J., Kwee I., Hillmann P., Rinaldi A., Cascione L., Spriano F., Bernasconi E., Guidetti F. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination TherapyNovel Dual PI3K/mTOR Inhibitor for Lymphomas. Clin. Cancer Res. 2018;24:120–129. doi: 10.1158/1078-0432.CCR-17-1041. [DOI] [PubMed] [Google Scholar]
- 113.Wicki A., Brown N., Xyrafas A., Bize V., Hawle H., Berardi S., Cmiljanović N., Cmiljanović V., Stumm M., Dimitrijević S. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13) Eur. J. Cancer. 2018;96:6–16. doi: 10.1016/j.ejca.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 114.Brandt C., Hillmann P., Noack A., Römermann K., Öhler L.A., Rageot D., Beaufils F., Melone A., Sele A.M., Wymann M.P. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology. 2018;140:107–120. doi: 10.1016/j.neuropharm.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 115.Wen P.Y., Cloughesy T.F., Olivero A.G., Morrissey K.M., Wilson T.R., Lu X., Mueller L.U., Coimbra A.F., Ellingson B.M., Gerstner E. First-in-Human Phase I Study to Evaluate the Brain-Penetrant PI3K/mTOR Inhibitor GDC-0084 in Patients with Progressive or Recurrent High-Grade GliomaGDC-0084 in Progressive or Recurrent High-Grade Glioma. Clin. Cancer Res. 2020;26:1820–1828. doi: 10.1158/1078-0432.CCR-19-2808. [DOI] [PubMed] [Google Scholar]
- 116.Heffron T.P., Ndubaku C.O., Salphati L., Alicke B., Cheong J., Drobnick J., Edgar K., Gould S.E., Lee L.B., Lesnick J.D. Discovery of clinical development candidate GDC-0084, a brain penetrant inhibitor of PI3K and mTOR. ACS Med. Chem. Lett. 2016;7:351–356. doi: 10.1021/acsmedchemlett.6b00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stumpf A., McClory A., Yajima H., Segraves N., Angelaud R., Gosselin F. Development of an efficient, safe, and environmentally friendly process for the manufacture of GDC-0084. Org. Process Res. Dev. 2016;20:751–759. doi: 10.1021/acs.oprd.6b00011. [DOI] [Google Scholar]
- 118.Ippen F.M., Alvarez-Breckenridge C.A., Kuter B.M., Fink A.L., Bihun I.V., Lastrapes M., Penson T., Schmidt S.P., Wojtkiewicz G.R., Ning J. The Dual PI3K/mTOR Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain MetastasesDual PI3K/mTOR Inhibition in Breast Cancer Brain Metastases. Clin. Cancer Res. 2019;25:3374–3383. doi: 10.1158/1078-0432.CCR-18-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ding L.-t., Zhao P., Yang M.-l., Lv G.-z., Zhao T.-l. GDC-0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem. Biophys. Res. Commun. 2018;503:1941–1948. doi: 10.1016/j.bbrc.2018.07.139. [DOI] [PubMed] [Google Scholar]
- 120.Tinkle C., Huang J., Campagne O., Pan H., Onar-Thomas A., Chiang J., Klimo P., Boop R., Patay Z., Shulkin B. CTNI-27. first-in-pediatrics phase I study of GDC-0084 (PAXALISIB), a CNS-penetrant PI3K/mTOR inhibitor, in newly diagnosed diffuse intrinsic pontine glioma (DIPG) or other diffuse midline glioma (DMG) Neuro Oncol. 2020;22:ii48. doi: 10.1093/neuonc/noaa215.194. [DOI] [Google Scholar]
- 121.Basu D., Salgado C.M., Bauer B., Khakoo Y., Patel J.R., Hoehl R.M., Bertolini D.M., Zabec J., Brzozowski M.R., Reyes-Múgica M. The dual PI3K/mToR inhibitor omipalisib/GSK2126458 inhibits clonogenic growth in oncogenically-transformed cells from neurocutaneous melanocytosis. Cancer Genom. Proteom. 2018;15:239–248. doi: 10.21873/cgp.20082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tseng C.-Y., Kuo C.-Y., Fu Y.-H., Hou H.-A., Tien H.-F., Lin L.-I. P422: Omipalisib, a dual PI3K/MTOR inhibitor, targets mitochondria and impairs oxidative phosphorylation in acute myeloid leukemia. HemaSphere. 2022;6:322–323. doi: 10.1097/01.HS9.0000844576.50131.5c. [DOI] [Google Scholar]
- 123.Lin L.-I., Chiyang T., Fu Y.-H., Hou H.-A., Chou W.-C., Tien H.-F. Metabolic Profiling Reveals Cellular Reprogramming of Acute Myeloid Leukemia By Omipalisib through Serine Synthesis Pathway. Blood. 2021;138:3296. doi: 10.1182/blood-2021-149200. [DOI] [Google Scholar]
- 124.Qin A.-C., Li Y., Zhou L.-N., Xing C.-G., Lu X.-S. Dual PI3K-BRD4 inhibitor SF1126 inhibits colorectal cancer cell growth in vitro and in vivo. Cell. Physiol. Biochem. 2019;52:758–768. doi: 10.33594/000000053. [DOI] [PubMed] [Google Scholar]
- 125.Mahadevan D., Chiorean E., Harris W., Von Hoff D., Stejskal-Barnett A., Qi W., Anthony S., Younger A., Rensvold D., Cordova F. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer. 2012;48:3319–3327. doi: 10.1016/j.ejca.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Joshi S., Singh A.R., Durden D.L. Pan-PI-3 kinase inhibitor SF1126 shows antitumor and antiangiogenic activity in renal cell carcinoma. Cancer Chemother. Pharmacol. 2015;75:595–608. doi: 10.1007/s00280-014-2639-x. [DOI] [PubMed] [Google Scholar]
- 127.Li Q., Li Z., Luo T., Shi H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol. Biomed. 2022;3:47. doi: 10.1186/s43556-022-00110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shang X., Na X., Wang L., Yang Z., Ren P. Evaluation of a Dual PI3K/mTOR Inhibitor PF-04691502 against Bladder Cancer Cells. Evid. Based Complement. Altern. Med. 2022;2022:8110796. doi: 10.1155/2022/8110796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chow Z., Johnson J., Chauhan A., Izumi T., Cavnar M., Weiss H., Townsend Jr C.M., Anthony L., Wasilchenko C., Melton M.L. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells. 2021;10:1261. doi: 10.3390/cells10051261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stoll J.R., Willner J., Oh Y., Pulitzer M., Moskowitz A., Horwitz S., Myskowski P., Noor S.J. Primary cutaneous T-cell lymphomas other than mycosis fungoides and Sézary syndrome. Part I: Clinical and histologic features and diagnosis. J. Am. Acad. Dermatol. 2021;85:1073–1090. doi: 10.1016/j.jaad.2021.04.080. [DOI] [PubMed] [Google Scholar]
- 131.Hong D.S., Moore K.N., Bendell J.C., Karp D.D., Wang J.S., Ulahannan S.V., Jones S., Wu W., Donoho G.P., Ding Y. Preclinical Evaluation and Phase Ib Study of Prexasertib, a CHK1 Inhibitor, and Samotolisib (LY3023414), a Dual PI3K/mTOR InhibitorPreclinical/Phase Ib Evaluation of Prexasertib+ Samotolisib. Clin. Cancer Res. 2021;27:1864–1874. doi: 10.1158/1078-0432.CCR-20-3242. [DOI] [PubMed] [Google Scholar]
- 132.Sweeney C.J., Babu S., Cultrera J.L., Mehlhaff B.A., Goodman O.B., Morris D.S., Schnadig I.D., Albany C., Shore N.D., Sieber P.R. Phase 1b/2 Study of Enzalutamide with Samotolisib (LY3023414) or Placebo in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022;28:2237–2247. doi: 10.1158/1078-0432.CCR-21-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bendell J.C., Varghese A.M., Hyman D.M., Bauer T.M., Pant S., Callies S., Lin J., Martinez R., Wickremsinhe E., Fink A. A first-in-human phase 1 study of LY3023414, an oral PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin. Cancer Res. 2018;24:3253–3262. doi: 10.1158/1078-0432.CCR-17-3421. [DOI] [PubMed] [Google Scholar]
- 134.Gunder L.C., Moyer T.H., Johnson H.R., Auyeung A.S., Leverson G.E., Zhang W., Matkowskyj K.A., Carchman E.H. Anal Cancer Prevention Through the Topical Use of Single or Dual PI3K/mTOR Inhibitors. J. Surg. Res. 2023;282:137–146. doi: 10.1016/j.jss.2022.09.025. [DOI] [PubMed] [Google Scholar]
- 135.O’Farrell M., Ventura R., Tai A., Matthews D.J. The PI3K/mTOR inhibitor PWT33597 regresses 786-0 renal xenografts. Cancer Res. 2012;72:3737. doi: 10.1158/1538-7445.AM2012-3737. [DOI] [Google Scholar]
- 136.Jang D.K., Lee Y.G., Chae Y.C., Lee J.K., Paik W.H., Lee S.H., Kim Y.-T., Ryu J.K. GDC-0980 (apitolisib) treatment with gemcitabine and/or cisplatin synergistically reduces cholangiocarcinoma cell growth by suppressing the PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2020;529:1242–1248. doi: 10.1016/j.bbrc.2020.06.011. [DOI] [PubMed] [Google Scholar]
- 137.Omeljaniuk W.J., Krętowski R., Ratajczak-Wrona W., Jabłońska E., Cechowska-Pasko M. Novel dual PI3K/mTOR inhibitor, Apitolisib (GDC-0980), inhibits growth and induces apoptosis in human glioblastoma cells. Int. J. Mol. Sci. 2021;22:11511. doi: 10.3390/ijms222111511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fang X., Yuan M., Dai J., Lin Q., Lin Y., Wang W., Jiang Y., Wang H., Zhao F., Wu J. Dual inhibition of glycolysis and oxidative phosphorylation by aptamer-based artificial enzyme for synergistic cancer therapy. Nano Res. 2022;15:6278–6287. doi: 10.1007/s12274-022-4237-2. [DOI] [Google Scholar]
- 139.Dasgupta A., Sierra L., Tsang S.V., Kurenbekova L., Patel T., Rajapakse K., Shuck R.L., Rainusso N., Landesman Y., Unger T. Targeting PAK4 inhibits Ras-mediated signaling and multiple oncogenic pathways in high-risk Rhabdomyosarcoma. Cancer Res. 2021;81:199–212. doi: 10.1158/0008-5472.CAN-20-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xiang S., Huang D., He Q., Li J., Tam K.Y., Zhang S.-L., He Y. Development of dual inhibitors targeting pyruvate dehydrogenase kinases and human lactate dehydrogenase A: High-throughput virtual screening, synthesis and biological validation. Eur. J. Med. Chem. 2020;203:112579. doi: 10.1016/j.ejmech.2020.112579. [DOI] [PubMed] [Google Scholar]
- 141.Holshouser S.L. Ph.D Thesis. Medical University of South Carolina; Charleston, SC, USA: 2018. Dual Inhibitors of KDM1A and Spermine Oxidase: A Novel Approach to Antitumor Therapy. [Google Scholar]
- 142.Dai X.-J., Liu Y., Xue L.-P., Xiong X.-P., Zhou Y., Zheng Y.-C., Liu H.-M. Reversible lysine specific demethylase 1 (LSD1) inhibitors: A promising wrench to impair LSD1. J. Med. Chem. 2021;64:2466–2488. doi: 10.1021/acs.jmedchem.0c02176. [DOI] [PubMed] [Google Scholar]
- 143.Wu X., Xu Y., Liang Q., Yang X., Huang J., Wang J., Zhang H., Shi J. Recent Advances in Dual PI3K/mTOR Inhibitors for Tumour Treatment. Front. Pharmacol. 2022;13:875372. doi: 10.3389/fphar.2022.875372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bharti R., Dey G., Mandal M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016;375:51–61. doi: 10.1016/j.canlet.2016.02.048. [DOI] [PubMed] [Google Scholar]
- 145.Rich J.N., Bao S. Chemotherapy and cancer stem cells. Cell Stem Cell. 2007;1:353–355. doi: 10.1016/j.stem.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 146.Wise-Draper T.M., Moorthy G., Salkeni M.A., Karim N.A., Thomas H.E., Mercer C.A., Beg M.S., O’Gara S., Olowokure O., Fathallah H. A phase Ib study of the dual PI3K/mTOR inhibitor dactolisib (BEZ235) combined with everolimus in patients with advanced solid malignancies. Target. Oncol. 2017;12:323–332. doi: 10.1007/s11523-017-0482-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Salazar R., Garcia-Carbonero R., Libutti S.K., Hendifar A.E., Custodio A., Guimbaud R., Lombard-Bohas C., Ricci S., Klümpen H.J., Capdevila J. Phase II study of BEZ235 versus everolimus in patients with mammalian target of rapamycin inhibitor-naïve advanced pancreatic neuroendocrine tumors. Oncologist. 2018;23:766-e790. doi: 10.1634/theoncologist.2017-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brown J.R., Hamadani M., Hayslip J., Janssens A., Wagner-Johnston N., Ottmann O., Arnason J., Tilly H., Millenson M., Offner F. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: An open-label, phase 2 trial. Lancet Haematol. 2018;5:e170–e180. doi: 10.1016/S2352-3026(18)30030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhao H., Chen G., Liang H. Dual PI3K/mTOR Inhibitor, XL765, suppresses glioblastoma growth by inducing ER stress-dependent apoptosis. OncoTargets Ther. 2019;12:5415. doi: 10.2147/OTT.S210128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Johnson F.M., Janku F., Gouda M.A., Tran H.T., Kawedia J.D., Schmitz D., Streefkerk H., Lee J.J., Andersen C.R., Deng D. Inhibition of the Phosphatidylinositol-3 Kinase Pathway Using Bimiralisib in Loss-of-Function NOTCH1-Mutant Head and Neck Cancer. Oncologist. 2022;27:1004-e1926. doi: 10.1093/oncolo/oyac185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhu D.-s., Dong J.-y., Xu Y.-y., Zhang X.-t., Fu S.-b., Liu W. Omipalisib inhibits esophageal squamous cell carcinoma growth through inactivation of phosphoinositide 3-Kinase (PI3K)/AKT/Mammalian target of rapamycin (mTOR) and ERK signaling. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020;26:e927106. doi: 10.12659/MSM.927106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Garlich J.R., De P., Dey N., Su J.D., Peng X., Miller A., Murali R., Lu Y., Mills G.B., Kundra V. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;68:206–215. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 153.Chow Z. Synergistic Cytotoxic Effect of a Novel mTOR/PI3K Dual Inhibitor PF-04691502 and Radiation Therapy on Neuroendocrine Tumor Cells. Int. J. Radiat. Oncol. Biol. Phys. 2020;108:e527. doi: 10.1016/j.ijrobp.2020.07.1650. [DOI] [Google Scholar]
- 154.Kwiatkowski D.J., Choueiri T.K., Fay A.P., Rini B.I., Thorner A.R., De Velasco G., Tyburczy M.E., Hamieh L., Albiges L., Agarwal N. Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell CarcinomamTOR Pathway Mutations and Response to Rapalogs in RCC. Clin. Cancer Res. 2016;22:2445–2452. doi: 10.1158/1078-0432.CCR-15-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Powles T., Lackner M.R., Oudard S., Escudier B., Ralph C., Brown J.E., Hawkins R.E., Castellano D., Rini B.I., Staehler M.D. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2016;34:1660. doi: 10.1200/JCO.2015.64.8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ganapathy-Kanniappan S., Geschwind J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer. 2013;12:152. doi: 10.1186/1476-4598-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ashton T.M., McKenna W.G., Kunz-Schughart L.A., Higgins G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018;24:2482–2490. doi: 10.1158/1078-0432.CCR-17-3070. [DOI] [PubMed] [Google Scholar]
- 158.Heske C.M. Beyond energy metabolism: Exploiting the additional roles of NAMPT for cancer therapy. Front. Oncol. 2020;9:1514. doi: 10.3389/fonc.2019.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Abu Aboud O., Chen C.-H., Senapedis W., Baloglu E., Argueta C., Weiss R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer GrowthInhibition of PAK4 and NAMPT Decreases RCC Growth. Mol. Cancer Ther. 2016;15:2119–2129. doi: 10.1158/1535-7163.MCT-16-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li M., Kirtane A.R., Kiyokawa J., Nagashima H., Lopes A., Tirmizi Z.A., Lee C.K., Traverso G., Cahill D.P., Wakimoto H. Local targeting of NAD+ salvage pathway alters the immune tumor microenvironment and enhances checkpoint immunotherapy in glioblastoma. Cancer Res. 2020;80:5024–5034. doi: 10.1158/0008-5472.CAN-20-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nitiss J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer. 2009;9:327–337. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Skok Z.i., Zidar N., Kikelj D., Ilaš J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J. Med. Chem. 2019;63:884–904. doi: 10.1021/acs.jmedchem.9b00726. [DOI] [PubMed] [Google Scholar]
- 163.Zhang X., Wang X., Wu T., Yin W., Yan J., Sun Y., Zhao D. Therapeutic potential of targeting LSD1/KDM1A in cancers. Pharmacol. Res. 2022;175:105958. doi: 10.1016/j.phrs.2021.105958. [DOI] [PubMed] [Google Scholar]
- 164.Furbish A.B., Alford A.S., Burger P., Peterson Y.K., Murray-Stewart T., Casero R.A., Jr., Woster P.M. Identification and Characterization of Novel Small-Molecule SMOX Inhibitors. Med. Sci. 2022;10:47. doi: 10.3390/medsci10030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hu T., Sun D., Zhang J., Xue R., Janssen H.L., Tang W., Dong L. Spermine oxidase is upregulated and promotes tumor growth in hepatocellular carcinoma. Hepatol. Res. 2018;48:967–977. doi: 10.1111/hepr.13206. [DOI] [PubMed] [Google Scholar]
- 166.Arooj M., Sakkiah S., Cao G.P., Lee K.W. An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLoS ONE. 2013;8:e60470. doi: 10.1371/journal.pone.0060470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Janku F., Yap T.A., Meric-Bernstam F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018;15:273–291. doi: 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- 168.Isaacsson Velho P.H., Castro G., Jr., Chung C.H. Targeting the PI3K pathway in head and neck squamous cell carcinoma. Am. Soc. Clin. Oncol. Educ. Book. 2015;35:123–128. doi: 10.14694/EdBook_AM.2015.35.123. [DOI] [PubMed] [Google Scholar]
- 169.Jung K., Kang H., Mehra R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC) Cancers Head Neck. 2018;3:3. doi: 10.1186/s41199-018-0030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Langdon S.P., Kay C., Um I.H., Dodds M., Muir M., Sellar G., Kan J., Gourley C., Harrison D.J. Evaluation of the dual mTOR/PI3K inhibitors Gedatolisib (PF-05212384) and PF-04691502 against ovarian cancer xenograft models. Sci. Rep. 2019;9:18742. doi: 10.1038/s41598-019-55096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.André F., Ciruelos E., Juric D., Loibl S., Campone M., Mayer I., Rubovszky G., Yamashita T., Kaufman B., Lu Y.-S. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2–negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021;32:208–217. doi: 10.1016/j.annonc.2020.11.011. [DOI] [PubMed] [Google Scholar]
- 172.Park Y.L., Kim H.P., Cho Y.W., Min D.W., Cheon S.K., Lim Y.J., Song S.H., Kim S.J., Han S.W., Park K.J. Activation of WNT/β-catenin signaling results in resistance to a dual PI3K/mTOR inhibitor in colorectal cancer cells harboring PIK3CA mutations. Int. J. Cancer. 2019;144:389–401. doi: 10.1002/ijc.31662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Markman B., Tao J.J., Scaltriti M. PI3K pathway inhibitors: Better not left alone. Curr. Pharm. Des. 2013;19:895–906. doi: 10.2174/138161213804547213. [DOI] [PubMed] [Google Scholar]
- 174.Masui K., Tanaka K., Ikegami S., Villa G.R., Yang H., Yong W.H., Cloughesy T.F., Yamagata K., Arai N., Cavenee W.K. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc. Natl. Acad. Sci. USA. 2015;112:9406–9411. doi: 10.1073/pnas.1511759112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ventura-Aguiar P., Campistol J.M., Diekmann F. Safety of mTOR inhibitors in adult solid organ transplantation. Expert Opin. Drug Saf. 2016;15:303–319. doi: 10.1517/14740338.2016.1132698. [DOI] [PubMed] [Google Scholar]
- 176.Shi Z., Tiwari A.K., Shukla S., Robey R.W., Singh S., Kim I.-W., Bates S.E., Peng X., Abraham I., Ambudkar S.V. Sildenafil reverses ABCB1-and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011;71:3029–3041. doi: 10.1158/0008-5472.CAN-10-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wu C.-P., Hung C.-Y., Lusvarghi S., Huang Y.-H., Tseng P.-J., Hung T.-H., Yu J.-S., Ambudkar S.V. Overexpression of ABCB1 and ABCG2 contributes to reduced efficacy of the PI3K/mTOR inhibitor samotolisib (LY3023414) in cancer cell lines. Biochem. Pharmacol. 2020;180:114137. doi: 10.1016/j.bcp.2020.114137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kovarik J.M., Beyer D., Schmouder R.L. Everolimus drug interactions: Application of a classification system for clinical decision making. Biopharm. Drug Dispos. 2006;27:421–426. doi: 10.1002/bdd.524. [DOI] [PubMed] [Google Scholar]
- 179.Moorthy G. Clinical Pharmacokinetics of the Novel Combination of BEZ235, PI3K/mTOR Inhibitor, and Everolimus, mTOR Inhibitor: Phase I Clinical Studies and Non-Clinical Mechanistic Assessment. University of Cincinnati; Cincinnati, OH, USA: 2015. [Google Scholar]
- 180.Zhao X., Chen X., Shen X., Tang P., Chen C., Zhu Q., Li M., Xia R., Yang X., Feng C. IL-36β promotes CD8+ T cell activation and antitumor immune responses by activating mTORC1. Front. Immunol. 2019;10:1803. doi: 10.3389/fimmu.2019.01803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Choi H.J., Heo J.H., Park J.Y., Jeong J.Y., Cho H.J., Park K.S., Kim S.H., Moon Y.W., Kim J.S., An H.J. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019;153:135–148. doi: 10.1016/j.ygyno.2019.01.012. [DOI] [PubMed] [Google Scholar]
- 182.Yan C., Yang J., Saleh N., Chen S.-C., Ayers G.D., Abramson V.G., Mayer I.A., Richmond A. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. Int. J. Mol. Sci. 2021;22:5207. doi: 10.3390/ijms22105207. [DOI] [PMC free article] [PubMed] [Google Scholar]