

Abstract
Faecalibacterium prausnitzii is a promising biomarker of a healthy human microbiota. However, previous studies reported the heterogeneity of this species and found the presence of several distinct groups at the species level among F. prausnitzii strains. Our recent study revealed that methods previously developed for quantification of F. prausnitzii were not specific to the species level because of the heterogeneity within the F. prausnitzii species and the application of 16S rRNA gene, which is an invalid genetic marker for the species. Therefore, previously available data failed to provide information on different groups, which limits our understanding of the importance of this organism for host health. Here, we propose an alternative gene marker for quantification of F. prausnitzii-related taxa. A total of nine group-specific primer pairs were designed by targeting rpoA gene sequences. The newly developed rpoA-based qPCR successfully quantified targeted groups. Application of the developed qPCR assay in six healthy adults revealed marked differences in abundance and prevalence among the different targeted groups in stool samples. The developed assay will facilitate detailed understanding of the impact of Faecalibacterium populations at the group level on human health and to understand the links between depletion of specific groups in Faecalibacterium and different human disorders.
Keywords: Faecalibacterium, group-specific quantification, qPCR, recA, rpoA, taxonomy
The newly developed rpoA-based qPCR facilitates more detailed understanding of the impact of the different Faecalibacterium spp. populations on human health.
Abbreviations
- MAM
microbial anti-inflammatory molecule
- IBD
inflammatory bowel diseases
- ANI
average nucleotide identity
- qPCR
quantitative PCR
- SD
standard deviation
Introduction
Faecalibacterium prausnitzii produces butyrate from the metabolism of carbohydrates in human gut (Louis and Flint 2009). Butyrate is the major energy source of intestinal epithelial cells and has multiple beneficial properties for host health. It exert beneficial effects by different pathways, including regulation of histone acetylation and mitogen-activated protein kinases (Davie 2003, Kida et al. 2006, Macfarlane and Macfarlane 2012). The colonization of F. prausnitzii in mice is also related to several metabolites, such as salicylic acid and shikimic acid, which potentially exert beneficial effects (Miquel et al. 2015). Moreover, the microbe produces peptides derived from the microbial anti-inflammatory molecule (MAM) that are associated with the decreased activation of the NF-κB pathways in the host (Quevrain et al. 2016, Breyner et al. 2017, Auger et al. 2022). The beneficial properties of these active molecules are associated with the anti-inflammatory properties, maintenance of gut barrier function, gut immune homeostasis, and induction of apoptosis in colorectal cancer cells in host animals (Kinoshita et al. 2002, Furusawa et al. 2013, Donohoe et al. 2014). The depletion of this microbe in the gut was associated with the development and severity of several diseases, including inflammatory bowel diseases (IBD), metabolic disorders, and psychiatric disorders (Sokol et al. 2009, Lopez-Siles et al. 2017, Borkent et al. 2022, Michels et al. 2022). All these facts underline the importance of this microbe for health maintenance.
Recent studies reported a large genomic heterogeneity among F. prausnitzii strains and found eight groups, distinct at the species level being present among the tested strains (Fitzgerald et al. 2018, Tanno et al. 2022). In total, three of the eight groups were very recently reclassified as novel species in the genus Faecalibacterium mainly based on genomic level identities determined by average nucleotide identity (ANI) analysis. They were named Faecalibacterium duncaniae, F. hattorii, and F. longum originated from the human gut (Zou et al. 2021, Sakamoto et al. 2022). Other groups have not been studied for reclassification yet. In addition, Faecalibacterium butyricigenerans, which was out of the previously mentioned eight groups, was also described from the human gut (Zou et al. 2021). Additionally, Faecalibacterium gallinarum from the chicken gut was described (Sakamoto et al. 2022). Previous studies reported distinct populations between different groups of Faecalibacterium in patients with atopic dermatitis and in paediatric IBD (Song et al. 2016, Zhang et al. 2018). Moreover, a recent study reported that MAM derived from different groups of Faecalibacterium displayed distinct anti-inflammatory properties (Auger et al. 2022). These studies suggest that different groups have varying impacts on host health.
The gene encoding 16S rRNA (16S rRNA gene) is the best studied and characterized gene marker for bacterial taxonomy. The species threshold of 16S rRNA gene sequence similarity is around 98.7%–99.0% (Stackebrandt and Ebers 2006), while distinct species sometimes share a similarity of more than 99%. On the other hand, Tanno et al. reported that Faecalibacterium species generally possessed six copies of 16S rRNA gene per genome, and sequence similarities among copies in a single genome/strain were sometimes lower than the species threshold of 98.7% (Tanno et al. 2022). These low similarities are due to considerable nucleotide substitutions, particularly in the V6 region of the 16S rRNA gene, among copies. Moreover, 16S rRNA gene copies sharing low sequence similarities sometimes showed higher sequence similarities with strains in other groups, i.e. other species. Due to the heterogeneity of 16S rRNA gene sequences, certain groups have been divided into two or three clusters by a 16S rRNA gene-based phylogenetic tree (Tanno et al. 2022). These findings clearly indicated that 16S rRNA gene is not a well-suited gene marker for the identification and classification of Faecalibacterium spp. Very often, 16S rRNA gene is used as a target gene for the quantification of Faecalibacterium. A number of primer pairs combined with real-time quantitative PCR (qPCR) have been developed for the quantification of F. prausnitzii, and quantitative data were included in studies on the differing abundance of the microbe between healthy subjects and patients with specific disorders (Bartosch et al. 2004, Rinttilä et al. 2004, Balamurugan et al. 2008, Sokol et al. 2009). However, due to the heterogeneity of 16S rRNA gene sequence, several of the primer pairs used targeted only some of the groups in Faecalibacterium (Tanno et al. 2022). Even if primer pairs cover all groups, the quantified population is a sum of all groups in Faecalibacterium (the former F. prausnitzii sensu lato group), not specific groups. Therefore, individual populations of F. prausnitzii and related groups in the human gut have not yet been characterized, although the species is regarded as a promising biomarker of a healthy microbiota (Lopez-Siles et al. 2017).
In the present study, housekeeping genes were evaluated as a potential new gene marker for the classification of Faecalibacterium spp. The housekeeping genes were also used to design primer pairs specific to each group in Faecalibacterium for specific quantification.
Materials and methods
Acquisition of genomic data and grouping of strains based on ANI
The genomic data of 86 strains of F. prausnitzii strains, used in our previous study (Tanno et al. 2022), were included in the present study. These were all complete or draft genome sequences of F. prausnitzii strains at the time of the analysis (January 2020) in the NCBI database after the exclusion of potential incompleteness and contamination strains (excluded, genome sizes of <2.6 Mbp or >3.5 Mbp). Moreover, the genomic data of F. butyricigenerans AF52-21T and F. longum CM04-06T were obtained from the CNGBdb database and included herein, resulting in a list of 88 strains. Genome level identities of the strains were determined by calculating ANI values, and the ANI values were used to prepare a distance matrix to represent the ANI divergence (100% ANI) and for group separation (threshold of ∼94%), as described previously (Tanno et al. 2022).
Phylogenetic analysis based on housekeeping genes
Since the 16S rRNA gene was not a suitable gene marker for the classification and identification of Faecalibacterium spp. at the group level, six housekeeping genes, including atpA, dnak, groEL, pheS, recA, and rpoA, which have been characterized for the classification and identification of other Bacillota (formerly known as Firmicutes) members and related bacteria (Torriani et al. 2001, Naser et al. 2007, Neumann and Rehberger 2009, Muñoz et al. 2017, Liu et al. 2018), were included in the initial screening. However, two (atpA and dnak) out of the six genes were excluded from this analysis due to the presence of two copies in the genomes of completely sequenced strains (data not shown). The remaining four housekeeping genes were present as a single copy in the genomes and were, thus, included in the phylogenetic analysis. The sequences of housekeeping genes were obtained from the genomes of the 88 strains and were aligned and used to construct phylogenetic trees using ClustalW (Larkin et al. 2007). The number of bootstrapping replicates was 1000. Sequence similarities in the housekeeping genes were assessed using Genetyx software ver. 13 (Genetyx, Tokyo, Japan).
Design and evaluation of rpoA-based primer pairs for the specific quantification of each group
The sequences of the rpoA gene were used to design specific primer pairs for the different groups previously found in Faecalibacterium. In the design of primers, all complete and draft genomes of F. prausnitzii and Faecalibacterium sp. (n = 147) containing the rpoA gene sequence, which were deposited in the NCBI database at a time of the analysis (July 2021), were further added to the original 88 strains. This addition was due to the limited number of strains in Groups 2, 5, 7, 8, and 9, originally containing 1, 3, 4, 2, and 1 strain, respectively, when the 88 strains were used. ANI values were determined for the newly added strains, as previously described (Maeno et al. 2016), and the strains were separated into groups based on the values. After this separation, number of strains in Groups 2, 5, 7, 8, and 9 increased to 3, 7, 8, 8, and 4 strains, respectively (data not shown). Moreover, all completely sequenced strains (n = 21) available at the time of the analysis (July 2022) were used to count the copy numbers of 16S rRNA gene per genome.
Primers were designed in consideration of no sequence mismatches with the targeted group, but the presence of sequence mismatches with nontargeted groups as much as possible. The specificity of the primers was evaluated using primer-BLAST to the tested strains and BLASTN to all deposited DNA sequences with default settings. A list of the specific primers designed is shown in Table 1.
Table 1.
Primers used in the present study.
| Primer | Sequence (5’→3’) | Target | References |
|---|---|---|---|
| F-Bact_1369 | CGGTGAATACGTTCCCGG | Total bacteria | Lopez-Siles et al. (2016) |
| R_Prok_1492 | TACGGCTACCTTGTTACGACTT | Total bacteria | Lopez-Siles et al. (2016) |
| Fprau223F | GATGGCCTCGCGTCCGATTAG | Faecalibacterium genus | Bartosch et al. (2004) |
| Fprau420R | CCGAAGACCTTCTTCCTCC | Faecalibacterium genus | Bartosch et al. (2004) |
| Faecali-group1-sp-F | CCTGAGTGGCACATTGCAACT | Group 1 (F. prausnitzii) | This study |
| Faecali-group1-sp-R | TAAATGCTGTCAACGGGAAGG | Group 1 (F. prausnitzii) | This study |
| Faecali-group2-sp-F | CCAAGCTCGTCATGGAGCTC | Group 2 | This study |
| Faecali-group2-sp-R | ATGGTCAGCTTGTCGTAGTCA | Group 2 | This study |
| Faecali-group3-sp-F | AACCTGTCCGATGAGGCAGCC | Group 3 | This study |
| Faecali-group3-sp-R | TCTTCCACCGTGTTGATGCCT | Group 3 | This study |
| Faecali-group4-sp-F | GCCATCATCGAGAAGAATGAC | Group 4 (F. longum) | This study |
| Faecali-group4-sp-R | TGTGCATTGATCGTGCCATCC | Group 4 (F. longum) | This study |
| Faecali-group5-sp-F | GAAATTGTCCTGAACCTGAAA | Group 5 | This study |
| Faecali-group5-sp-R | CCTGTTTGTTGCGCTCAGCC | Group 5 | This study |
| Faecali-group6-sp-F | AAGGGCCGCGGTTATGTGCCT | Group 6 (F. duncaniae) | This study |
| Faecali-group6-sp-R | TAATCGATGGCCTGTCCAACG | Group 6 (F. duncaniae) | This study |
| Faecali-group7-sp-F | CCTGAATGGCACATCGCAACCT | Group 7 (F. hattorii) | This study |
| Faecali-group7-sp-R | ATGCTATCGACGGGAAGCGTA | Group 7 (F. hattorii) | This study |
| Faecali-group8-sp-F | GGTGAATTACAATGTTGAGAA | Group 8 | This study |
| Faecali-group8-sp-R | TCTCGGTGCCAGCGGCCTCA | Group 8 | This study |
| Faecali-group9-sp-F | AATGTCGAGAGCACCCGTGTG | Group 9 (F. butyricigenerans) | This study |
| Faecali-group9-sp-R | GATCTCAGCGCCAGCGGCCTCG | Group 9 (F. butyricigenerans) | This study |
A qPCR-based primer evaluation was conducted to confirm the specificity of the primer pairs designed using three steps. In the first step, the entire rpoA gene sequences (954–969 bp) of 12 strains of Faecalibacterium spp. were synthesized by insertion into the pEX-A2J2 vector by Eurofins Genomics (Tokyo, Japan). These 12 strains included at least one strain each of all groups, and three and two strains were included from Groups 1 and 6, respectively (Fig. 2). qPCR was conducted by the combination of the specific primers and 100 pg ( = 7.43 log10rpoA gene copies) of synthesized DNA. DNA Calculator (https://www.molbiotools.com/dnacalculator.html) was used to determine the number of molecules in 100 pg of the synthesized DNA. FastStart Essential DNA Green Master Mix combined with the LightCycler 96 system (Roche, Basel, Switzerland) was used for qPCR according to the manufacturer’s instructions. The qPCR program consisted of initial denaturation at 95°C for 10 min and 45 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 15 s. Standard curves were produced for each primer pair using the synthesized DNA of each targeted group, while ATCC 27768T and A2165 were used as references for Groups 1 and 6, respectively. A melting curve analysis was conducted to confirm specific amplification. Samples were run in triplicate in the same plate, and the mean and SD were obtained. The detection limit of qPCR was assessed based on the results obtained using serially diluted synthetic DNA.
Figure 2.

Relative amplification ratio against the targeted group using the synthetic rpoA gene and rpoA-based group-specific primer pairs. A DNA amount of 100 pg (= 7.43 log10rpoA gene copies) was used in each PCR reaction. (A) to (I) use specific primer pairs to Groups 1–9, respectively. ATCC 27768 and A2165 were used as references in Groups 1 and 6, respectively. Samples were run in triplicate in the same plate, and the mean and SD were obtained. ND, not detected. BDL, below the detection limit.
In the second step of primer specificity confirmation, DNAs isolated from representative strains in each group, which are available in public culture collections or our own collection, were used for qPCR. DNA was isolated from cultures of BCRC 81047T ( = F. prausnitzii ATCC 27768T) in Group 1, CNCM 4541 in Group 2, CNCM 4540 in Group 3, JCM 39211T ( = F. longum CM04-06T) in Group 4, JCM 31915T ( = F. duncaniae A2-165T) in Group 6, JCM 39210T ( = F. hattorii APC922/41–1T) in Group 7, and JCM 39212T ( = F. butyricigenerans AF52-21T) in Group 9, and 10 ng of isolated DNA was used in this study. The BCRC strain and JCM strains were obtained from the Bioresource Collection and Research Center (BCRC) and the Japan Collection of Microorganisms (JCM), respectively. CNCM strains were obtained from our private collection. No strains in Groups 5 or 8 were available in the public culture collection at the time of the analysis (March 2022). qPCR combined with specific primers to each group was conducted using the methods described above. Samples were run in triplicate in the same plate, and the mean and SD were obtained.
In the third step of the primer specificity evaluation, cultures (∼107 cells) of F. prausnitzii BCRC 81047T (Group 1) or F. duncaniae JCM 31915T (Group 6) were added to 200 mg of infantile stool (9 months old), and DNA was extracted from stool samples with or without the addition of bacterial cultures using the QIAamp DNA Stool Mini Kit (Qiagen, Tokyo, Japan). Sample collection was conducted according to the guidelines of the Declaration of Helsinki and was approved by the Ethics Committee of the Tokyo University of Agriculture. Written informed consent was received from the parent of the infant. Isolated DNAs were used for rpoA-based qPCR of the nine groups. The qPCR products were sequenced by the methods as described previously (Endo and Okada 2005). The genus Faecalibacterium was also quantified using the 16S-based primer pair of Fprau223F/Fprau420R designed by Bartosh and coworkers (Bartosch et al. 2004) shown in Table 1 and the synthesized 16S rRNA gene of ATCC 27768T included in a previous study (Tanno et al. 2022) for a standard curve. The primer pair combined with qPCR equally quantified all groups in Faecalibacterium in the previous study (Tanno et al. 2022), and the primer pair had substantial sequence mismatches with the 16S rRNA gene of other members in the family Oscillospiraceae. qPCR conditions were described elsewhere (Tanno et al. 2022). Moreover, the 16S rRNA gene copy number of total bacteria was quantified to assess the relative abundance of the genus Faecalibacterium and each group using the 16S-based bacterial universal primer pair of F-Bact_1369/R_Prok_1492 (Lopez-Siles et al. 2016), as shown in Table 1. The synthesized 16S rRNA gene of ATCC 27768T was used for the standard curve, and the qPCR program consisted of initial denaturation at 95°C for 10 min, and 45 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 15 s. Samples were run in triplicate in the same plate, and the mean and SD were obtained. The detection limit of qPCR was determined based on the results obtained using serially diluted DNA from a F. prausnitzii BCRC 81047T-added stool sample.
Quantification of different Faecalibacterium groups in stool samples of healthy individuals
Volunteers who had not received antibiotics for more than 8 weeks or prebiotics for more than 2 weeks prior to the donation of stool samples were recruited, and fecal samples were collected from six healthy adult men (volunteers A–F, mean ± SD, 26.3 ± 6.7 years old) with a body mass index ranging between 22 and 25 in a previous study (Endo et al. 2020). After defecation, stool samples were immediately placed in anaerobic jars (AnaeroPack-Anaero, Mitsubishi Gas Chemical, Tokyo, Japan), transported to the laboratory, and stored at −80°C within 2 h of collection. Sample collection was conducted according to the guidelines of the Declaration of Helsinki and was approved by the Ethics Committee of the Tokyo University of Agriculture. Written informed consent was received from the volunteers. DNA was extracted from the collected samples within 2 weeks using a previously described method (Takahashi et al. 2014). The quantification of the nine groups in Faecalibacterium (rpoA-based), the genus Faecalibacterium (16S-based), and total bacteria (16S-based) was conducted by the method described above. The relative abundance (%) of each group in Faecalibacterium and the genus Faecalibacterium was calculated using the total bacterial count. The 16S/rpoA ratio was obtained using 16S-based genus Faecalibacterium qPCR and the sum of rpoA-based nine group-specific qPCR. Samples were run in triplicate in the same plate, and the mean and SD were obtained.
Results
Selection of a potential new gene marker for the classification of Faecalibacterium spp
A total of 88 strains were divided into nine groups based on ANI value-based grouping, i.e. eight groups described in a previous study (Tanno et al. 2022) with an additional ninth group of F. butyricigenerans (Figure S1, Supporting Information). Group 1 was F. prausnitzii sensu stricto, and Groups 4, 6, 7, and 9 were F. longum, F. duncaniae, F. hattorii, and F. butyricigenerans, respectively. Other groups were obviously different from F. prausnitzii at the species level but have not yet been taxonomically reconsidered and separated from this taxon. Phylogenetic trees based on the nucleotide sequences of the genes revealed that the nine groups produced independent clusters on the trees (Fig. 1a; Figure S2, Supporting Information). Of the four genes, recA had the lowest intergroup sequence similarity (Fig. 1b; Figures S1b, S1d, and S1f, Supporting Information). The highest intergroup sequence similarity value of 94.2% on recA gene was recorded between strains in Groups 4 and 9 (Table S1a, Supporting Information), whereas Group 4 strains shared more than 98.5% sequence similarities among the strains. The medians of intragroup recA gene sequence similarities were at least 97.7% in Group 1, whereas those of the intergroup were less than 93.7% recorded between Groups 4 and 9 (Fig. 1b). This result suggests that the recA gene has high discriminatory power and is a suitable gene marker for the classification and identification of Faecalibacterium spp.
Figure 1.
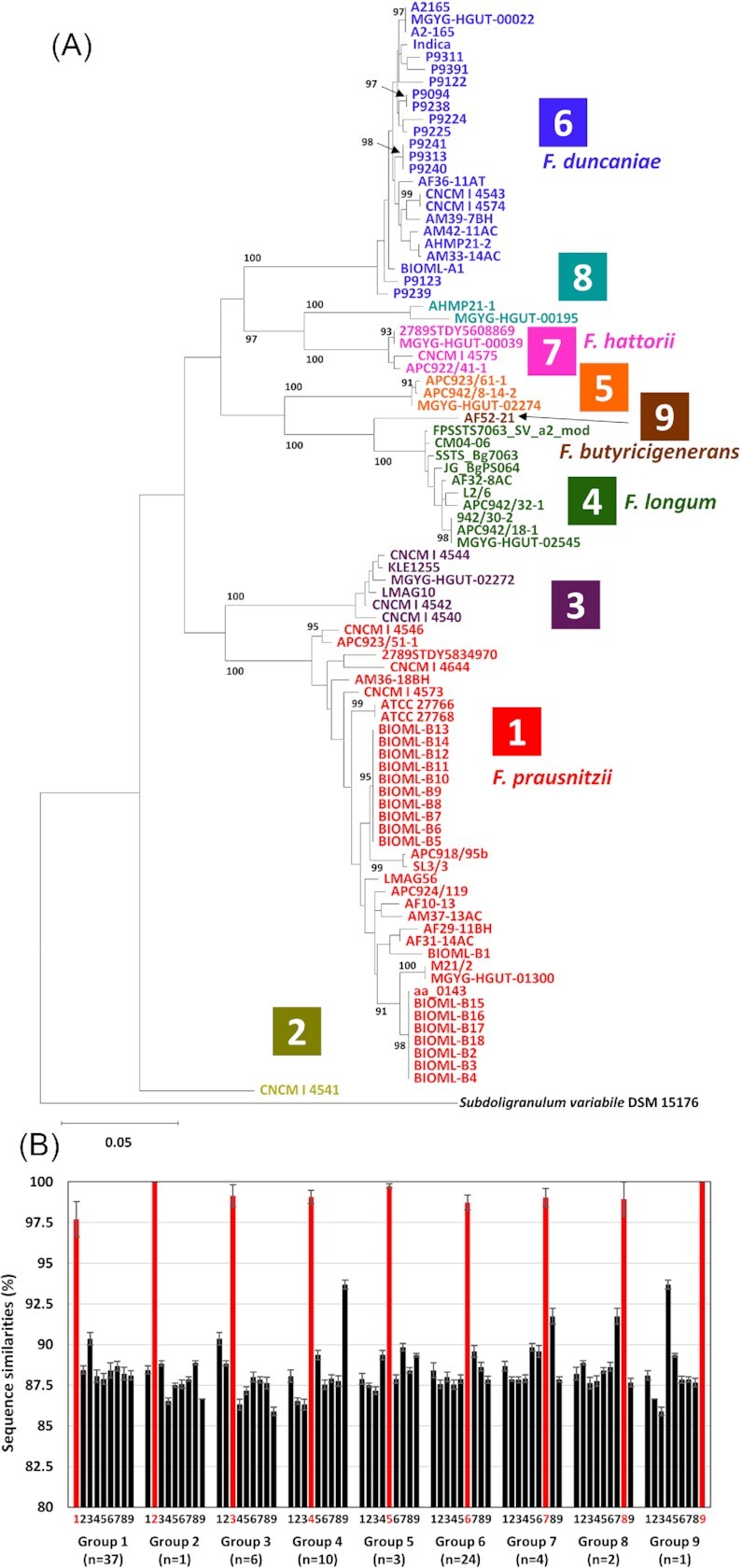
Phylogenetic tree based on recA gene nucleotide sequences of 88 strains of Faecalibacterium spp. (A) and intra- (red bars) and inter- (black bars) group recA gene sequence similarities (B). The maximum-likelihood tree was constructed using the best-fit evolutionary model. The values on the branches are bootstrap support from 1000 rapid bootstrapping replicates and only values higher than 90% are indicated. Subdoligranulum variabile DSM 15176 (GCA_000157955.1) was used as an outgroup. The scale bar means substitution per site. Groups based on ANI-values (Figure S1, Supporting Information) are indicated in different colours, and Groups 1, 4, 6, 7, and 9 are F. prausnitzii sensu stricto, F. longum, F. duncaniae, F. hattorii, and F. butyricigenerans, respectively. In (B), bars numbered 1–9 indicate the medians of sequence similarities to Groups 1–9, respectively, and error bars indicate SD. The numbers of strains in each group are indicated.
Design and evaluation of novel specific primer sets for the selective quantification of each group
Among the four housekeeping genes (groEL, pheS, recA, and rpoA) tested, recA gene has a high power to discriminate groups. However, this gene showed a high level of intragroup sequence divergence, and, thus, designation of group-specific primers targeting recA gene sequences was not possible. In contrast, the rpoA gene sequences contained low levels of intragroup sequence divergence, maintaining group-specific sequence regions, thus allowing the design of group-specific primers. The designed primer pairs contained no sequence mismatches with the rpoA gene sequences of the targeted groups but contained substantial sequence mismatches with the sequences of nontargeted groups (Table 1). BLASTN analysis confirmed that all DNA sequences deposited in the database did not completely match with the primer sequences.
The designed primer pairs were initially evaluated using synthesized DNA. The evaluation revealed that the newly developed rpoA-based qPCR assay accurately quantified the targeted group (∼7.43 log10rpoA gene copies). The relative amplification ratio of the nontargeted groups against the targeted group was generally less than 1%, and over 50% of the nontargeted groups were not detected or were below the detection limit (101.7 molecules/reaction) in total (Fig. 2).
A primer specificity evaluation was also conducted using isolated DNAs from cultures originating from seven out of the nine groups. The rpoA-based qPCR assay accurately quantified the targeted group, but quantified the nontargeted groups less than 1% of the targeted groups (Fig. 3). In total, over 60% of the nontargeted groups were not detected or were below the detection limit.
Figure 3.

Relative amplification ratio against the targeted group using DNA isolated from cultures and rpoA-based group-specific primer pairs. A DNA amount of 10 ng was used in each PCR reaction (A) to (G) use primer pairs specific to Groups 1–9, respectively. Samples were run in triplicate in the same plate, and the mean and SD were obtained. ND, not detected. BDL, below the detection limit.
An evaluation of primer specificity was further conducted by using the addition of cultures to infantile stool (9 months-old infant), which originally contained very low levels (below the detection limit) of Groups 1, 3, 6, and 9 (Table 2). After the addition of F. prausnitzii BCRC 81047T ( = ATCC 27768T, Group 1) culture (∼107 cells) to the stool sample, DNA was isolated and was used for qPCR. The level of Group 1 increased after the culture addition. The relative abundance of Groups 3 and 6 against Group 1 was ∼0.2% (Table 2), while other groups were below the detection limit (105.6 cells/g of feces) or were not detected. When a culture of F. duncaniae JCM 31915T ( = A2-165T, Group 6) was added to the stool sample, similar results were obtained. The level of Group 6 increased following the culture addition, whereas other groups were below the detection limit or were not detected (Table 2). The products in the Group 1-speicific qPCR and Group 6-specific qPCR were 100% matched with rpoA gene sequences of BCRC 81047T (accession no. NZ_PXUP00000000.1, LocusTag C7J97_RS08250) in Group 1 and JCM 31915T in Group 6 (accession number NZ_CP022479.1, LocusTag CG447_RS10630), respectively. Moreover, 16S-based qPCR was applied to quantify the Faecalibacterium genus in the infantile stool samples with or without the culture addition, which was compared with the rpoA-based qPCR results. The infantile stool was free from the genus Faecalibacterium without the culture addition. Cell numbers determined by 16S-based qPCR (as the genus Faecalibacterium) increased following the culture addition. The 16S/rpoA ratio, calculated using the values obtained from 16S-based genus Faecalibacterium qPCR and the sum of rpoA-based group-specific qPCR, was 4.5- to 5.3-fold (data not shown). All completely sequenced Faecalibacterium strains tested (n = 21) possessed six copies of 16S rRNA gene (Table S2, Supporting Information).
Table 2.
Relative abundance (%) of each group in infantile stool with or without the addition of the F. prausnitzii BCRC 81047T (Group 1) or F. duncaniae JCM 31915T (Group 6) culture.
| Group | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| Without addition* | BDL | – | BDL | – | – | BDL | – | – | BDL |
| + BCRC 81047T** | 100 | – | 0.2 | BDL | – | 0.2 | – | – | BDL |
| + JCM 31915T*** | BDL | BDL | BDL | – | BDL | 100 | – | – | BDL |
–, Not detected; BDL, below detection limit (< 105.6 cells/g of feces).
All groups were not detected or BDL in stool sample without culture addition.
Relative abundance (%) to Group 1.
Relative abundance (%) to Group 6.
Quantification of each Faecalibacterium group in healthy adults
The rpoA-based group-specific qPCR assays showed marked differences in prevalence and abundance among the groups in the six healthy adults (Fig. 4a). Group 3 was observed in all subjects and was the most abundant group in three (subjects A, D, and E) out of six subjects (Fig. 4b). Group 4 F. longum and Group 9 F. butyricigenerans were the second most prevalent groups and were detected in five out of six subjects. Group 6 F. duncaniae was found in four out of six subjects and was the most abundant group in two (subjects B and F) subjects. Group 1 F. prausnitzii and Group 5 were quantified in two out of six subjects. Group 2, Group 7 F. hattorii, and Group 8 were not detected or were below the detection limit in all subjects tested. The sum of the relative abundance of the nine groups ranged between 0.152% and 0.759% (median ± standard deviation (SD) = 0.521 ± 0.237%). The relative abundance of the genus Faecalibacterium assessed by 16S-based qPCR ranged between 0.86% and 8.48% (median ± SD = 3.37 ± 2.68%, Fig. 4) in the six subjects. The 16S/rpoA ratio calculated based on qPCR of the genus Faecalibacterium and the sum of the nine specific groups analyzed was between 4.7 and 14.4 (median ± SD = 6.81 ± 3.91).
Figure 4.
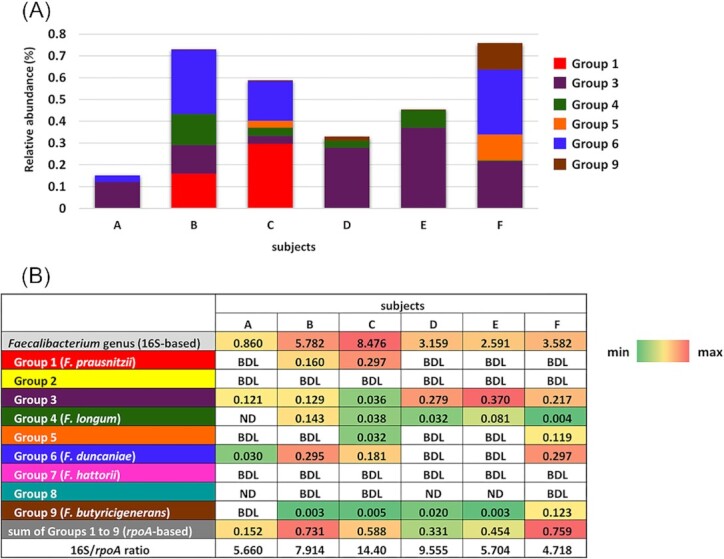
Relative abundance (%) of each group and the Faecalibacterium genus against total bacteria in six healthy adults. Samples were run in triplicate in the same plate, and the mean was obtained. Stacked bars (A) indicating the relative abundance of each group (rpoA-based) in the six subjects. Colours in the stacked bars indicate Groups. Groups 2, 7, and 8 were not indicated, since they were ND or BDL in all subjects tested. In (B), the relative abundance of each group (rpoA-based) and the genus Faecalibacterium (16S-based) are shown as a heat map, and relative abundance values are indicated. The 16S/rpoA ratio was calculated based on the relative abundance of the genus Faecalibacterium and the sum of the relative abundance of Groups 1–9. ND, not detected. BDL, below the detection limit.
Discussion
Even if Faecalibacterium spp. are considered to be of paramount importance for our health status, previous quantification methods of their population contained some issues due to taxonomic concerns and the utilization of a suboptimal genetic marker (i.e. 16S rRNA gene). This fact limited our understanding on the role of these microbes (Tanno et al. 2022). 16S rRNA gene is obviously the best characterized gene for bacterial classification and the study of complex microbiota. It is a reliable marker for more than 97% of bacterial species (Větrovský and Baldrian 2013); however, Faecalibacterium spp. belong to the remaining group (Tanno et al. 2022). Therefore, we studied alternative gene markers for the classification and quantification of Faecalibacterium spp.
Among the housekeeping genes tested, recA gene was the most suitable gene marker for the classification and identification of Faecalibacterium spp. because of the lowest intergroup sequence similarities. The recA gene is one of 120 bacterial marker genes for suitable phylogenetic inference (Parks et al. 2017) and provides accurate differentiation among phylogenetically related microbes (Pietilä et al. 2000, Torriani et al. 2001, Zbinden et al. 2011). On the other hand, recA gene sequences were too divergent within a group to design group-specific primers, whereas rpoA was a suitable gene marker to design these primers. qPCR assays targeting housekeeping genes have been applied to quantify bifidobacteria and lactobacilli in the human gut microbiota (Junick and Blaut 2012, Costa et al. 2014). The rpoA-based primers designed here, combined with qPCR, accurately quantified the targeted groups with very low cross reactivity (less than 1%), indicating that the qPCR assays developed are useful to quantify Faecalibacterium spp. at the group (species) level. Previously reported qPCR assays were unable to quantify Faecalibacterium spp. at the species level and relied on the global quantification of the former F. prausnitzii sensu lato (Tanno et al. 2022).
In culture-added infantile stool, the 16S/rpoA ratio was between 4.5 and 5.3. This was due to different copy numbers between the genes used. A total of six copies of 16S rRNA gene are commonly conserved in a single genome of completely sequenced Faecalibacterium spp., whereas rpoA is a single copy gene. Therefore, relative abundance assessed by 16S-based qPCR is theoretically 6-fold that evaluated by rpoA-based qPCR (the 16S/rpoA ratio). Similar findings were reported by Masco et al. (Masco et al. 2007), who quantified bifidobacteria in probiotic products using 16S-based and recA-based primer pairs. This bias could be removed if we use DNA from cultures for standard curves based on cellular concentration. However, at the time of analysis (March 2022), strains from two out of the nine groups were not available at public culture collections or in our private collections, and, thus, we used synthetic DNA to prepare the standard curve for all groups. DNA extraction from stool samples always includes different extraction efficiencies (Yang et al. 2020), which has significant impacts on quantification of microbes. Standard curves based on synthetic genes are not appropriate to determine absolute cell numbers in stool samples because synthetic genes are free from extraction biases and different amplification efficiencies. However, relative abundance could be evaluated because the values obtained are normalized by the cell numbers of total bacteria.
The rpoA-based qPCR was applied to quantify each group of Faecalibacterium in six healthy Japanese adults. Group 3 was the most prevalent and abundant group and was detected in all subjects with a relative abundance of 0.173 ± 0.121% (median ± SD), although only a limited number of subjects was included in the present study. Despite its predominance, this group has not yet been taxonomically characterized. Future studies are essential to establish the taxonomic position of this group and to study its potential impact on human health. Genomes of Group 1 (F. prausnitzii) and Group 6 (F. duncaniae) were abundant in the public database (Figure S1, Supporting Information), indicating that the two groups have been well-isolated and characterized. Groups 1 and 6 contain the reference strain of ATCC 27768T (type strain of F. prausnitzii) and the most characterized Faecalibacterium strain of A2-165 ( = JCM 31915T, type strain of F. duncaniae), respectively. These two groups were abundant (relative abundance >0.1%) in two or three out of the six adults but were below the detection limit in the other subjects, suggesting that these two groups are abundant in specific adults, but are not prevalent. Since gut microbiota, including a prevalence and abundance of Faecalibacterium, are influenced by several factors, such as age and geography of host (De Filippis et al. 2020, Sang et al. 2022), it would be intriguing to study the population of each group of Faecalibacterium considering these factors.
The 16S/rpoA-ratio of the six subjects was 6.81 ± 3.91 (median ± SD), which was close to the theoretical value of 6, as described above. On the other hand, the ratios of subjects C and D were markedly higher than the theoretical value (Fig. 4b), suggesting that unidentified group(s) of the genus Faecalibacterium were predominant in the subjects. A recent study reported the genomic diversity of Faecalibacterium using reference Faecalibacterium genomes and Faecalibacterium-like metagenome-assembled genomes originating from diverse hosts (De Filippis et al. 2020). This study identified 22 different species-level clades, 11 of which were from humans. The 11 clades included the eight groups detected in the present study, but not Group 9 F. butyricigenerans. They also characterized the prevalence of clades in patients with specific diseases or obese subjects by comparisons with healthy subjects. However, the findings obtained were qualitative, not quantitative. Even if prevalence is important, quantitative data will provide better insights into the importance of each group in Faecalibacterium for human health.
Numerous studies reported an inverse association of F. prausnitzii with several disorders, and, thus, it is considered to be a promising biomarker of a healthy microbiota in human (Lopez-Siles et al. 2017). However, the species is no longer a single species and was recently divided into several species (Zou et al. 2021, Sakamoto et al. 2022), making its role as a biomarker unclear. Here, we established a new assay to quantify the nine groups of Faecalibacterium in the complex human gut microbiota. Assays for the quantification of the genus Faecalibacterium had already been developed (Bartosch et al. 2004, Ramirez-Farias et al. 2009) for the quantification of ‘F. prausnitzii sensu lato’. The combination of these assays will provide a more detailed understanding of the impact of the Faecalibacterium genus and the specific groups on human health. For instance, application of the assays will support to identify deficiencies on specific group(s) in diseased patients, including Crohn's disease and type 2 diabetes. It is also important to reset a real active biomarker of a healthy gut microbiota.
Supplementary Material
Acknowledgements
Hiroki Tanno was a recipient of a Grant-in-Aid for JSPS Fellows. The computational analysis was performed on the NIG supercomputer at ROIS.
Contributor Information
Hiroki Tanno, Department of Food, Aroma and Cosmetic Chemistry, Faculty of Bioindustry, Tokyo University of Agriculture, 196 Yasaka, Abashiri, 099-2493 Hokkaido, Japan.
Jean-Marc Chatel, Université Paris-Saclay, INRAE, AgroParisTech, Micalis Institute, 78350, Jouy-en-Josas, France.
Rebeca Martin, Université Paris-Saclay, INRAE, AgroParisTech, Micalis Institute, 78350, Jouy-en-Josas, France.
Denis Mariat, Université Paris-Saclay, INRAE, AgroParisTech, Micalis Institute, 78350, Jouy-en-Josas, France.
Mitsuo Sakamoto, Microbe Division/Japan Collection of Microorganisms, RIKEN BioResource Research Center, 3-1-1 Kouyadai, Tsukuba, 305-0074 Ibaraki, Japan.
Masao Yamazaki, Department of Food, Aroma and Cosmetic Chemistry, Faculty of Bioindustry, Tokyo University of Agriculture, 196 Yasaka, Abashiri, 099-2493 Hokkaido, Japan.
Seppo Salminen, Functional Food Forum, University of Turku, Itäinen Pitkäkatu 4a, 20014 Turku, Finland.
Miguel Gueimonde, IPLA-CSIC, Department of Microbiology and Biochemistry of Dairy Products, Paseo Rio Linares s/n, 33300 Villaviciosa, Spain.
Akihito Endo, Department of Food, Aroma and Cosmetic Chemistry, Faculty of Bioindustry, Tokyo University of Agriculture, 196 Yasaka, Abashiri, 099-2493 Hokkaido, Japan; Department of Nutritional Science and Food Safety, Faculty of Applied Bioscience, Tokyo University of Agriculture, 1-1-1 Sakuragaoka, Setagaya, 156-8502 Tokyo, Japan.
Author contributions
Hiroki Tanno (Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – review & editing), Jean-Marc Chatel (Investigation, Methodology, Resources, Validation, Writing – review & editing), Rebeca Martin (Investigation, Methodology, Resources, Validation, Writing – review & editing), Denis Mariat (Investigation, Methodology, Resources, Validation, Writing – review & editing), Mitsuo Sakamoto (Methodology, Resources, Supervision, Validation, Writing – review & editing), Masao Yamazaki (Project administration, Resources, Supervision, Validation, Writing – review & editing), Seppo Salminen (Conceptualization, Investigation, Supervision, Validation, Writing – review & editing), Miguel Gueimonde (Investigation, Methodology, Supervision, Validation, Writing – review & editing), and Akihito Endo (Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization Writing – original draft, Writing – review & editing)
Conflicts of interest
None declared.
Funding
The present study was supported by JSPS KAKENHI (grant numbers JP20K05792 and JP21J11100).
Data availability
All genomic data used in the present study were obtained from the NCBI and CNGBdb databases.
References
- Auger S, Kropp C, Borras-Nogues Eet al. . Intraspecific diversity of microbial anti-inflammatory molecule (MAM) from Faecalibacterium prausnitzii. IJMS. 2022;23:1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balamurugan R, Rajendiran E, George Set al. . Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J Gastroenterol Hepatol. 2008;23:1298–303. [DOI] [PubMed] [Google Scholar]
- Bartosch S, Fite A, Macfarlane GTet al. . Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl Environ Microbiol. 2004;70:3575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkent J, Ioannou M, Laman JDet al. . Role of the gut microbiome in three major psychiatric disorders. Psychol Med. 2022;52:1222–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyner NM, Michon C, de Sousa CSet al. . Microbial anti-inflammatory molecule (MAM) from Faecalibacterium prausnitzii shows a protective effect on DNBS and DSS-induced colitis model in mice through inhibition of NF-κb pathway. Front Microbiol. 2017;8:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa GN, Marcelino-Guimarães FC, Vilas-Bôas GTet al. . Potential fate of ingested Lactobacillus plantarum and its occurrence in human feces. Appl Environ Microbiol. 2014;80:1013–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133:2485s–93s. [DOI] [PubMed] [Google Scholar]
- De Filippis F, Pasolli E, Ercolini D. Newly explored Faecalibacterium diversity is connected to age, lifestyle, geography, and disease. Curr Biol. 2020;30:4932–43.e4. [DOI] [PubMed] [Google Scholar]
- Donohoe DR, Holley D, Collins LBet al. . A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014;4:1387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo A, Hirano K, Ose Ret al. . Impact of kestose supplementation on the healthy adult microbiota in in vitro fecal batch cultures. Anaerobe. 2020;61:102076. [DOI] [PubMed] [Google Scholar]
- Endo A, Okada S. Monitoring the lactic acid bacterial diversity during shochu fermentation by PCR-denaturing gradient gel electrophoresis. J Biosci Bioeng. 2005;99:216–21. [DOI] [PubMed] [Google Scholar]
- Fitzgerald CB, Shkoporov AN, Sutton TDSet al. . Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genomics. 2018;19:931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda Set al. . Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–50. [DOI] [PubMed] [Google Scholar]
- Junick J, Blaut M. Quantification of human fecal bifidobacterium species by use of quantitative real-time PCR analysis targeting the groEL gene. Appl Environ Microbiol. 2012;78:2613–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida Y, Shimizu T, Kuwano K. Sodium butyrate up-regulates cathelicidin gene expression via activator protein-1 and histone acetylation at the promoter region in a human lung epithelial cell line, EBC-1. Mol Immunol. 2006;43:1972–81. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Suzuki Y, Saito Y. Butyrate reduces colonic paracellular permeability by enhancing ppargamma activation. Biochem Biophys Res Commun. 2002;293:827–31. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NPet al. . Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. [DOI] [PubMed] [Google Scholar]
- Liu XS, Li WG, Zhang WZet al. . Molecular characterization of Clostridium difficile isolates in China from 2010 to 2015. Front Microbiol. 2018;9:845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Siles M, Duncan SH, Garcia-Gil LJet al. . Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J. 2017;11:841–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Siles M, Martinez-Medina M, Suris-Valls Ret al. . Changes in the abundance of Faecalibacterium prausnitzii phylogroups I and II in the intestinal mucosa of inflammatory bowel disease and patients with colorectal cancer. Inflamm Bowel Dis. 2016;22:28–41. [DOI] [PubMed] [Google Scholar]
- Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. [DOI] [PubMed] [Google Scholar]
- Macfarlane GT, Macfarlane S. Bacteria, colonic fermentation, and gastrointestinal health. J AOAC Int. 2012;95:50–60. [DOI] [PubMed] [Google Scholar]
- Maeno S, Tanizawa Y, Kanesaki Yet al. . Genomic characterization of a fructophilic bee symbiont Lactobacillus kunkeei reveals its niche-specific adaptation. Syst Appl Microbiol. 2016;39:516–26. [DOI] [PubMed] [Google Scholar]
- Masco L, Vanhoutte T, Temmerman Ret al. . Evaluation of real-time PCR targeting the 16S rRNA and recA genes for the enumeration of bifidobacteria in probiotic products. Int J Food Microbiol. 2007;113:351–7. [DOI] [PubMed] [Google Scholar]
- Michels N, Zouiouich S, Vanderbauwhede Bet al. . Human microbiome and metabolic health: an overview of systematic reviews. Obes Rev. 2022;23:e13409. [DOI] [PubMed] [Google Scholar]
- Miquel S, Leclerc M, Martin Ret al. . Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. Mbio. 2015;6.e00300–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz M, Ríos-Chaparro DI, Patarroyo MAet al. . Determining Clostridium difficile intra-taxa diversity by mining multilocus sequence typing databases. BMC Microbiol. 2017;17:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naser SM, Dawyndt P, Hoste Bet al. . Identification of lactobacilli by pheS and rpoA gene sequence analyses. Int J Syst Evol Microbiol. 2007;57:2777–89. [DOI] [PubMed] [Google Scholar]
- Neumann AP, Rehberger TG. MLST analysis reveals a highly conserved core genome among poultry isolates of Clostridium septicum. Anaerobe. 2009;15:99–106. [DOI] [PubMed] [Google Scholar]
- Parks DH, Rinke C, Chuvochina Met al. . Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2:1533–42. [DOI] [PubMed] [Google Scholar]
- Pietilä J, He Q, Oksi Jet al. . Rapid differentiation of Borrelia garinii from Borrelia afzelii and Borrelia burgdorferi sensu stricto by LightCycler fluorescence melting curve analysis of a PCR product of the recA gene. J Clin Microbiol. 2000;38:2756–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevrain E, Maubert MA, Michon Cet al. . Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut. 2016;65:415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Farias C, Slezak K, Fuller Zet al. . Effect of inulin on the human gut microbiota: stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br J Nutr. 2009;101:541–50. [DOI] [PubMed] [Google Scholar]
- Rinttilä T, Kassinen A, Malinen Eet al. . Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97:1166–77. [DOI] [PubMed] [Google Scholar]
- Sakamoto M, Sakurai N, Tanno Het al. . Genome-based, phenotypic and chemotaxonomic classification of Faecalibacterium strains: proposal of three novel species Faecalibacterium duncaniae sp. nov., Faecalibacterium hattorii sp. nov. and Faecalibacterium gallinarum sp. nov. Int J Syst Evol Microbiol. 2022;72:35416766. [DOI] [PubMed] [Google Scholar]
- Sang J, Zhuang D, Zhang Tet al. . Convergent and divergent age patterning of gut microbiota diversity in humans and nonhuman primates. Msystems. 2022;7:e0151221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Seksik P, Furet JPet al. . Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–9. [DOI] [PubMed] [Google Scholar]
- Song H, Yoo Y, Hwang Jet al. . Faecalibacterium prausnitzii subspecies-level dysbiosis in the human gut microbiome underlying atopic dermatitis. J Allergy Clin Immunol. 2016;137:852–60. [DOI] [PubMed] [Google Scholar]
- Stackebrandt E, Ebers J. Taxonomic parameters revisited: tarnished gold standards. Microbiol Tod. 2006;33:152–5. [Google Scholar]
- Takahashi S, Tomita J, Nishioka Ket al. . Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PLoS ONE. 2014;9:e105592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno H, Maeno S, Salminen Set al. . 16S rRNA gene sequence diversity in Faecalibacterium prausnitzii-complex taxa has marked impacts on quantitative analysis. FEMS Microbiol Ecol. 2022;98:fiac004. [DOI] [PubMed] [Google Scholar]
- Torriani S, Felis GE, Dellaglio F. Differentiation of Lactobacillus plantarum, L. pentosus, and L. paraplantarum by recA gene sequence analysis and multiplex PCR assay with recA gene-derived primers. Appl Environ Microbiol. 2001;67:3450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Větrovský T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE. 2013;8:e57923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Sun J, Luo Het al. . Assessment of fecal DNA extraction protocols for metagenomic studies. Gigascience. 2020;9:giaa071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbinden A, Köhler N, Bloemberg GV. recA-based PCR assay for accurate differentiation of Streptococcus pneumoniae from other viridans streptococci. J Clin Microbiol. 2011;49:523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Deeke SA, Ning Zet al. . Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat Commun. 2018;9:2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Lin X, Xue Wet al. . Characterization and description of Faecalibacterium butyricigenerans sp. nov. and F. longum sp. nov., isolated from human faeces. Sci Rep. 2021;11:11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic data used in the present study were obtained from the NCBI and CNGBdb databases.