

Abstract
Background:
Organophosphate esters (OPEs) are common endocrine-disrupting chemicals, and OPE exposure may be associated with type 2 diabetes (T2D). However, greater knowledge regarding the biomolecular intermediators underlying the impact of OPEs on T2D in humans are needed to understand biological etiology.
Objectives:
We explored the associations between OPE exposure and glycometabolic markers among older Chinese adults 60–69 years of age to elucidate the underlying mechanisms using a multi-omics approach.
Methods:
This was a longitudinal panel study comprising 76 healthy participants 60–69 years of age who lived in Jinan city of northern China. The study was conducted once every month for 5 months, from September 2018 to January 2019. We measured a total of 17 OPEs in the blood, 11 OPE metabolites in urine, and 4 glycometabolic markers (fasting plasma glucose, glycated serum protein, fasting insulin, and homeostatic model assessment for insulin resistance). The blood transcriptome and serum/urine metabolome were also evaluated. The associations between individual OPEs and glycometabolic markers were explored. An adverse outcome pathway (AOP) was established to determine the biomolecules mediating the associations.
Results:
Exposure to five OPEs and OPE metabolites (trimethylolpropane phosphate, triphenyl phosphate, tri-iso-butyl phosphate, dibutyl phosphate, and diphenyl phosphate) was associated with increased levels of glycometabolic markers. The mixture effect analysis further indicated the adverse effect of OPE mixtures. Multi-omics analyses revealed that the endogenous changes in the transcriptional and metabolic levels were associated with OPE exposure. The putative AOPs model suggested that triggers of molecular initiation events (e.g., insulin receptor and glucose transporter type 4) with subsequent key events, including disruptions in signal transduction pathways (e.g., phosphatidylinositol 3-kinase/protein kinase B and insulin secretion signaling) and biological functions (glucose uptake and insulin secretion), may constitute the diabetogenic effects of OPEs.
Discussion:
OPEs are associated with the elevated risk of T2D among older Chinese adults 60–69 years of age. Implementing OPE exposure reduction strategies may help reduce the T2D burden among these individuals, if the relationship is causal. https://doi.org/10.1289/EHP11896
Introduction
Diabetes mellitus, mainly type 2 diabetes (T2D), is one of the four most prevalent noncommunicable diseases.1,2 In 2021, 536.6 (10.5%) million adults 20–79 years of age worldwide were estimated to have diabetes, which resulted in 6.7 million deaths and health expenditures totaling USD $966 billion.3 Diabetes impacts aging societies, affecting an estimated 19.3% of adults 65–99 years of age worldwide.4 Most of the diabetes burden falls on low- and medium-income countries, such as China and India.5 In the past decade, the number of diabetic patients in China increased from 90 (age-adjusted prevalence: 8.8%) to 140 million (10.6%),3 and the number of older individuals ( years of age) with diabetes was 35.5 million, ranking first globally and accounting for 25% of the elderly diabetes patients worldwide.4
The risk of T2D is multifactorial and determined by an interplay of genetic and environmental factors.6 Family history of diabetes, older age, unhealthy diet, physical inactivity, smoking, and overweight and obesity are associated with an increased risk of T2D; of these, overweight and obesity was found to be the strongest risk factor.7 Of note, the age-standardized prevalence of general obesity among Chinese adults, is 14.0%,8 considerably lower than that among American adults (43.3%).9 However, the estimated age-standardized prevalence of diabetes is almost the same among Chinese and American adults (12.4% vs. 14.3%).10,11 Genetic factors, a thrifty phenotype,12–14 and unique dietary patterns (e.g., low intake of whole grains but high intake of refined grains)15 are suspected to be responsible for the increased risk of T2D in the Chinese population. In addition, since industrialization advancements and immense growth took place after the 1980s, chemical pollution has been the leading environmental cause of diseases and deaths in China.16 Emerging pollutants are ever-evolving and are widely used in the industry and for daily necessities worldwide, and their production is gradually shifting to China owing to the increasingly strict regulations in Europe and America. Extensive evidence suggests that disproportionate exposure to environmental pollutants may be an underappreciated contributor to disparities in the incidence of T2D.17
Long-term exposure to environmental pollutants combined with frailty and a decline of immune function makes the elderly population particularly susceptible to T2D.18 Cohort studies have revealed that exposure to endocrine-disrupting chemicals (EDCs) disturbed the onset and progression of T2D among older Swedish and Korean adults.19–21 As a substitute for polybrominated diphenyl ethers, organophosphate esters (OPEs) are commonly used as flame retardants and plasticizers,22 and the global annual consumption of OPEs exceeded 680,000 tons in 2015, with 30% of the usage was reported in China.23 As a result, OPEs are ubiquitously detected in Chinese individuals in the microgram to milliliter range.24 Such high OPE exposure levels adversely impact the reproductive, endocrine, cardiovascular, and nervous systems.25,26 In vivo and in vitro studies have reported that OPEs may cause diabetogenic effects27–29; however, evidence confirming these findings at the population level is relatively scarce,30,31 and the mechanisms underlying the indicated association remain largely unknown. Further research and intervention are required to reduce the individual and societal burden of diabetes among the Chinese elderly population. Such studies should incorporate a comprehensive clarification of modifiable diabetogenic OPEs and the mechanistic standpoints in terms of the etiology of T2D. Furthermore, high-throughput multi-omics (e.g., transcriptomics and metabolomics) approaches can be used to fill in the knowledge gaps between exposure to pollutants and the pathophysiology of T2D.32–34
The present study was part of a well-characterized longitudinal multi-omics study, the China Biomarkers of Air Pollutant Exposure (BAPE) Study, conducted among Chinese individuals 60–69 years of age, with monthly longitudinal monitoring conducted over 5 months.35 The study aimed to a) explore the associations between internal OPE exposure and glycometabolic markers, b) identify the key OPEs associated with glycometabolic markers using mixture effect analysis, and c) elucidate the potential biological perturbations and mechanisms underlying the associations between key OPEs and glucose homeostasis using multi-omics approaches and an adverse outcome pathways (AOPs) model.
Methods
Study Design and Population
This investigation draws on the China BAPE longitudinal panel study that included a visit each month for 5 months to healthy older Chinese adults 60–69 years of age. In brief, 76 healthy adults 60–69 years of age (50% males, 100% Chinese Han nationality) were recruited from within the Jinan megacity, China. Each participant was assessed five times over a 5-month period from 10 September 2018 to 19 January 2019, with a 1-month interval between each assessment. To minimize the potential impact of different dietary patterns, all participants were freely provided three standardized and nutritionally balanced meals (e.g., rice, meat, vegetable, and fruit; in line with local dietary habits) per day for 5 continuous days before bio-sample collection and physical examination. All of the participants were required to finish a questionnaire regarding family information, personal information, and time–activity patterns prior to the physical examination. During the physical examination, fasting venous blood samples and midstream urine samples were collected from the participants at 0700 hours and immediately stored at until further processing. Of the 76 participants, 58 (76%) finished 5 visits; 12 (16%) finished 4 visits; 3 (4%) finished 3 visits; and 3 (4%) finished 2 visits. A total of 353 person-visits were included in our analyses. The study protocol was approved by the ethics committee of the National Institute of Environmental Health (NIEH, Chinese Center for Disease Control and Prevention, No. 201816). Explicit written informed consent was acquired from all the participants. The detailed protocol of the China BAPE Study has been described in a previous publication.35
Characterization of OPE Exposure
Seventeen OPEs were measured in the blood samples (), and 11 OPE metabolites (m-OPEs) were evaluated in the urine samples () (Table S1). Analytical procedures for the measurement of OPEs and the internal concentrations of OPEs were previously reported.36 Urine creatinine (Cr) was measured using a Flex reagent cartridge in a modified kinetic Jaffe assay (model RxL; Dade Behring). Cr-adjusted concentrations of urine m-OPEs were used in further analyses. Concentrations less than the limit of detection (LOD) were imputed using the LOD divided by 2 for each OPE biomarker. The pairwise correlations of the OPE exposures were calculated using the Spearman coefficient. All participants were self-reported nonsmoking participants; thus, plasma cotinine levels were measured using Hypersil GOLD C18 Selectivity HPLC Columns (ThermoFisher Scientific) interfaced with an LC-Q-Exactive Orbitrap Mass Spectrometer (ThermoFisher Scientific) to assess whether the participants were either actively or passively exposed to tobacco.
Assessment of Glycometabolic Markers
Glycometabolic marker levels, including fasting plasma glucose (FPG) and glycated serum protein [GSP, an indicator of short-to-medium–term (latest 2–3 wk) average blood glucose levels], were measured in all blood samples () at Calibra Diagnostics Co. Ltd. using the Cobas 8000 c702 and Cobas 6000 c501 (Roche) modular analyzer series, respectively. Fasting insulin (FINS) levels were determined using a Millipex Human Metabolic Hormone Panel V3 (HMHEMAG-34K-07; Merck) with a fluorescence detection system (Magpix; Luminex Corporation) and the xPonent 4.2 (Luminex Corporation) and Bio-Plex Manager (version 6.1; Bio-Rad) software, according to the manufacturer’s protocol. Homeostatic model assessment for insulin resistance (HOMA-IR) was performed using the following formula: [.37
Transcriptome Analysis
Total RNA was extracted from the leukocytes of venous blood samples (). In brief, leukocytes were isolated from of whole blood. After centrifugation at for 5 minutes, the leukocytes aggregated in the medium were extracted and washed two times with phosphate-buffered saline. The leukocytes were then lysed in TRIzol Reagent (Invitrogen Corp.). Next, the total RNA was assessed for quality and quantified using a NanoDrop ND-2000 (ThermoFisher Scientific) and Agilent 2100 Bioanalyzer. Library preparation with of the extracted RNA was conducted through the TruSeq Stranded Total RNA Library Prep Kit (Illumina, Inc.). Last, the RNA was sequenced using the Illumina Hiseq X Ten System (Novogene). Genes were identified by using HISAT2 software and comparing the data with the human database.38 Gene expression quantification was conducted using the featureCounts read summarization program in the Subread software package.39
Nontargeted Metabolome Assessment
Nontargeted metabolomics analyses were performed in serum () and urine () samples. Sample processing, quality control and data extraction, metabolite identification, data curation, data quantification, and data normalization were conducted according to previously published protocols.40 In brief, the samples were precipitated with methanol for 2 min under severe vibration (GenoGrinder 2000), and then the metabolites were extracted by centrifugation. The supernatant was then separated into five fractions: Two fractions were analyzed by two different reverse phase/ultra-performance liquid chromatography–mass spectrum (RP/UPLC-MS/MS) methods using positive ion mode electrospray ionization (ESI). The remaining three fractions were analyzed by hydrophilic interaction liquid chromatography (HILIC)/UPLC-MS/MS with negative ion mode ESI, RP/UPLC-MS/MS with negative ion mode ESI, or kept for backup, respectively. The samples were put in TurboVap (Zymark) for a short time to remove the organic solvent and then kept at overnight until subsequent analysis.
The metabolomics analysis was conducted using Waters ACQUITY UPLC and Q-Exactive high resolution/accurate mass spectrometer (Thermo Fisher), which were connected to an Orbitrap mass analyzer running at 35,000 mass resolution and a heated ESI source. The first and second aliquots of sample extracts were analyzed following acidic positive ion conditions that were chromatographically optimized for more hydrophilic and hydrophobic compounds, respectively. The third aliquot of sample extracts was gradient eluted from a separate dedicated C18 column and analyzed using basic negative ion–optimized conditions. The fourth aliquot of sample extracts was eluted from a HILIC column and analyzed via negative ionization. The mass spectrum analysis used dynamic exclusion that alternated between MS or data-dependent multistage mass spectrometry (MSn) scans with scan range covering 70–1,000 m/z.
After the raw mass spectrum data extraction and peak recognition, the metabolites were identified by comparison with an internal library. The library was based on certified standards that included a) m/z, b) retention time, and c) chromatographic data of all molecules. In addition, metabolite identifications were based on the following three criteria: a) retention time within 100 ms tolerance, b) accurate mass match to the internal library ( mass tolerance), and b) MS/MS fractions based on the ions present in the experimental spectrum compared with the ions present in the library spectrum. To ensure data quality, all peaks were manually checked. Then the area under the curve was used to quantify and all data were normalized before further analysis.
For quality control (QC) purposes, three types of controls were analyzed in concert with the experimental samples: a) process blanks, b) mixed matrix samples ( of each sample), and c) a cocktail of QC standards spiked into samples. All the experimental samples were randomly distributed on the platform and QC samples were also evenly distributed among the injections.
Statistical Analyses
All data were analyzed according to the following pipeline: a) a linear mixed-effects model (LMM) was conducted to explore the associations between OPE exposures and the glycometabolic markers; b) quantile g-computation (qgcomp)41 was used to assess the effects of OPE mixtures on the glycometabolic markers and to identify the most important OPEs with relative positive weights within the OPE mixtures; c) the associations between each key OPE and each biomolecule (transcripts and metabolites obtained using multi-omics profiling) were examined using LMM, and the biomolecules mediating the impact of each key OPE on a specific glycometabolic marker were determined using the causal inference test (CIT)42; and d) Integrative Pathway Analysis (IPA) of the biomolecular intermediators was conducted to investigate the underlying biological mechanisms43 (Figure 1). The detailed processes are described in the following sections.
Figure 1.

Overview of the study design. Diagram of the present study design. Internal OPE exposure of 76 healthy older Chinese adults 60–69 years of age with five monthly longitudinal sample (blood and urine) collections was characterized previously. Glycometabolic markers (FPG, GSP, FINS, and HOMA-IR) were measured, and multi-omics profiling (peripheral blood transcriptome, serum metabolome, and urine metabolome) was conducted. Exposure–health outcome associations and multi-omics integrative analyses were further used to identify the key OPEs and to interpret the biological mechanisms underlying the perturbations of glycometabolic markers, respectively. Note: CIT, causal inference test; FINS, fasting insulin; FPG, fasting plasma glucose; GSP, glycated serum protein; HOMA-IR, homeostatic model assessment for insulin resistance; IPA, Ingenuity Pathway Analysis; LMM, linear mixed-effects model; OPE, organophosphate ester; qgcomp, quantile g-computation.
LMM
The measurement values of certain OPE exposures and glycometabolic markers were logarithm (base 10)-, square root-, power one-third-, square-, or cubic-transformed to approach Gaussian distribution (Table S1). The maximum missing rate for certain exposure variables (blood OPEs) was 0.28% owing to the runout of one blood sample. After imputing the missing data for exposures using a chained equation (mice package with the predictive mean matching method),44 the exposures were standardized through -score normalization to denote a change of 1 standard deviation (SD) in the glycometabolic marker values to facilitate their comparisons. A LMM with participant-specific intercepts and “unstructured” covariance structure was used to independently assess the associations between exposure to each OPE and each glycometabolic marker. Each main model was adjusted for the set of predefined adjustment factors according to previous studies45,46: age (continuous), sex (female/male), body mass index [BMI; continuous; calculated as weight divided by height squared (in kilograms per meter squared)], education level (below primary school, primary school, junior school, senior high school, and university), financial income [continuous; annual household income (in 10,000 CNY)], plasma cotinine concentration (continuous), and other diet (the total frequency of extra food consumption in addition to the provided standardized meals during the 3-d investigation). Among them, information on age, sex, education level, and financial income was assessed at the first visit for all of the participants, whereas other time-varying factors (BMI, plasma cotinine concentration, and other diet) were assessed at all five visits. Other diet information was recorded using a daily time–activity questionnaire at each visit, and the summarized results are shown in Table S2. Stratification analysis of the associations between OPE exposures and glycometabolic markers were conducted by sex. Multiple hypothesis testing-corrected -values were obtained by calculating the false discovery rate (FDR) and the estimated proportion of false discoveries made vs. the number of total discoveries made at a given significance level ().47 In multiple testing corrections, FDR was statistically significant at . estimates and standard errors from the models were converted to percentage change values with the 95% confidence intervals (95% CIs) associated with 1-SD increases in individual OPE concentrations.
qgcomp
The OPEs that were found to be significantly associated with each glycometabolic marker after multiple testing using LMM were packed as a chemical mixture. The qgcomp method was performed to assess the effects of the OPE mixtures on the glycometabolic markers based on parameter inference via the qgcomp package in R. This approach combined the inferential simplicity of weighted quantile sum regression (WQS) with the flexibility of g-computation.41 The advantage of qgcomp was that exposures could interact with outcomes in any directions. Gaussian distributions were specified as link functions, and parameter q was set to four in the linear model. Five hundred bootstrap iterations were performed to calculate the 95% CI for each mixture. With the exception of predefined adjustment factors, visit time was included as an adjusting covariate for qgcomp analysis. OPEs with positive weights of were identified as key components.
CIT
To identify the biomolecular intermediates, the causal relationship inference of OPEs–biomolecular intermediators–glycometabolic markers was assessed using the CIT.42 In brief, each OPE–metabolite/transcript–glycometabolic marker relationship was individually analyzed to classify the components as consequential to/mediated by/independent of expression of gene or metabolite. In this study, causal inference was defined by the following four criteria: a) the OPEs and glycometabolic markers were significantly correlated, b) the OPEs were significantly associated with the expression of biomolecules after adjusting for the glycometabolic markers, c) the expression of biomolecules was significantly associated with the glycometabolic markers after adjusting for OPEs, and d) the OPEs were independent of the glycometabolic markers after adjusting for the expression of biomolecules. In addition to predefined adjustment factors, visit time (month of sample collection) was included as an adjustment covariate for the CIT model. To summarize the -values for the whole CIT, the intersection–union test framework was used as the maximum -value of the four test components.42
IPA
IPA canonical pathway analysis of all the significant biomolecular intermediators (both genes and metabolites) of the causal relationship between each OPE and glycometabolic markers was performed using IPA software (version 68752261; Qiagen; https://digitalinsights.qiagen.com/IPA). The corresponding biological pathways of these transcripts and metabolites were identified for each OPE independently. Canonical pathways with a (Fischer’s exact test) were regarded as statistically significant. IPA helps to uncover new biological insights and interpretations by combining an enormous schemata of existing knowledge from the literature with a massive collection of gene and metabolite expression measurements.
Sensitivity Analysis
Sensitivity analysis was performed to assess various models by controlling for one of the following covariates in the main model: month of sample collection, tea consumption (number of cups over 3 d), and frequency of alcohol consumption (over 3 d). A two-sided in sensitivity analysis indicated statistical significance. All statistical evaluations were conducted using R (R Development Core Team) with the lme4 and qgcomp packages.
Results
Overview of OPE Exposure and Glycometabolic Markers
Participant characteristics are presented in Table 1. Of all participants, 38 (50%) were women, and the participant age was y. The concentrations of the 17 blood OPEs and 11 urinary OPE metabolites over five longitudinal visits are summarized in Figure 2A and Table S1 (see also Excel Table S1). The concentrations of tributyl phosphate (TBP), triphenyl phosphate (TPHP), and tri(1-chloro-2-propyl) phosphate (TCPP) were relatively high in the blood samples, whereas those of bis(2-chloroethyl) phosphate (BCEP), di(2-ethylhexyl) phosphate (DEHP), and bis(1,3-dichloro-2-propyl) phosphate (BDCPP) were relatively high in the urine samples. The pairwise Spearman correlations of the OPE exposures are shown in Figure S1. Meanwhile, four glycometabolic markers (FPG, GSP, FINS, and HOMA-IR) reflecting the status of insulin resistance and glycometabolic homeostasis are shown in Figure 2B (see also Excel Table S2).
Table 1.
Demographics of the study participants in the China BAPE Study 2018–2019 ( with 353 measurements).
| Variables | (%) or |
|---|---|
| Age (y) | |
| Sex (male) | 38 (50.0) |
| BMI () | |
| Highest education level | |
| Primary school or below | 8 (10.5) |
| Junior middle or high school | 54 (71.1) |
| College graduate or beyond | 14 (18.4) |
| Annual financial income ( CNY) | |
| 25 (32.9) | |
| 7–10 | 26 (34.2) |
| 25 (32.9) | |
| Tea consumption (number of cups/3 d) | |
| Alcohol consumption (frequency/3 d) | |
| Plasma cotinine concertation (ng/mL) | |
| Other diet (frequency/3 d) | |
| Month of sampling ( samples) | |
| September | 66 (18.7) |
| October | 74 (21.0) |
| November | 71 (20.1) |
| December | 71 (20.1) |
| January | 71 (20.1) |
Note: BAPE, Biomarkers of Air Pollutant Exposure; BMI, body mass index; SD, standard deviation.
Figure 2.

Characterization of the internal exposure of OPEs and glycometabolic markers as part of this study ( with 353 measurements). (A) Profiling of the internal exposures (blood and urine) of OPEs among healthy older adults over the five monthly longitudinal visits (see also Excel Table S1). (B) Distribution of glycometabolic markers among healthy older adults over the five monthly longitudinal visits (see also Excel Table S2). Note: Cr, creatinine; Dec, December; FINS, fasting insulin; FPG, fasting plasma glucose; GSP, glycated serum protein; HOMA-IR, homeostatic model assessment for insulin resistance; Jan, January; Nov, November; Oct, October; OPE, organophosphate ester; Sep, September.
Association between OPE Exposure and Glycometabolic Markers
The percentage change in estimates and 95% CIs of the glycometabolic markers for a 1-SD increase in the concentration of each OPE are shown in Figure 3A and Excel Table S3. After multiple testing corrections, 6, 7, 3, and 9 OPEs were found to be significantly associated with FPG [TPHP, trimethylolpropane phosphate (TMPP), 2-ethylhexyl diphenyl phosphate (EHDPP), diphenyl phosphate (DPHP), dibutyl phosphate (DBP), and BCEP], GSP [TPHP, TMPP, EHDPP, hydroxyphenyl 2-ethylhexyl diphenyl phosphate (EHDPP-OH), DPHP, DBP, and BCEP], FINS (TPHP, DPHP, and DBP), and HOMA-IR [TPHP, tri--butyl phosphate (TnBP), TMPP, tri-iso-butyl phosphate (TiBP), TBP, EHDPP, DPHP, DBP, and BCEP], respectively. Of these OPEs, blood TPHP and urinary DBP, and DPHP showed an undesirable positive influence on all four glycometabolic markers. The results of the sensitivity analysis were consistent with the findings of the main model (Figure S2 and Excel Table S4). A stratification analysis of effect modification of sex was conducted (Figure S3 and Excel Table S5), and the correlation directions and significance between OPE exposure and glycometabolic marker changes in different sex were generally consistent.
Figure 3.
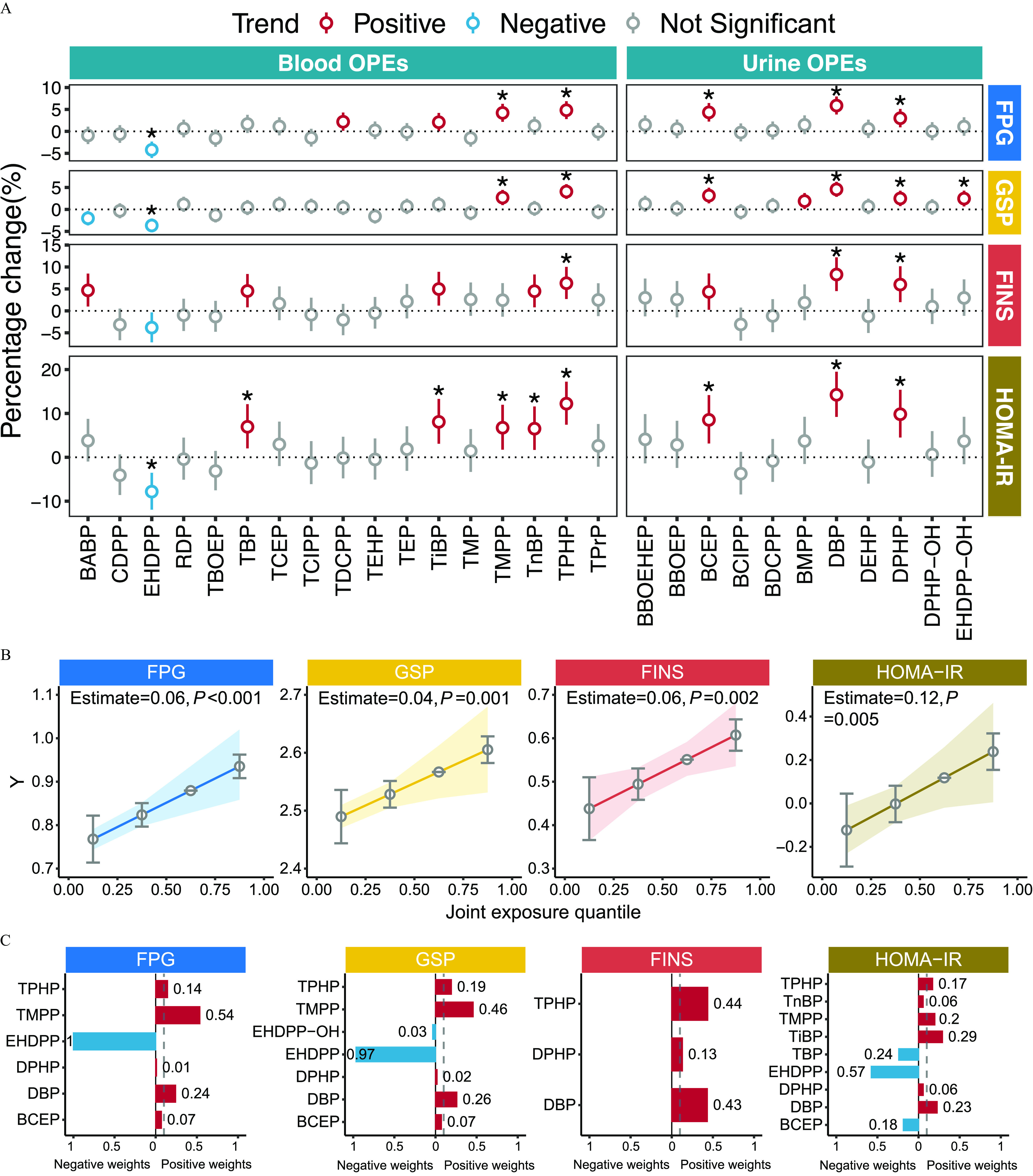
OPE exposure–glycometabolic marker association and qgcomp analyses–determined exposure to the key OPEs among healthy older Chinese adults 60–69 years of age. (A) Forest plots of the results of the LMM between OPE exposure and glycometabolic markers (FPG, GSP, FINS, and HOMA-IR). The FDR-adjusted -values of each predictor are given as *FDR (see also Excel Table S3). (B) Effect diagrams of the changes in the -scores of glycometabolic markers with a quantile increase in the mixture concentration (see also Table S3). (C) Bar plots of the relative weight of each pollutant within four chemical mixtures constructed to assess their effects on glycometabolic markers (see also Table S4). Note: FDR, false discovery rate; FINS, fasting insulin; FPG, fasting plasma glucose; GSP, glycated serum protein; HOMA-IR, homeostatic model assessment for insulin resistance; LMM, linear mixed-effects model; OPE, organophosphate ester; qgcomp, quantile g-computation.
Identification of the Key OPEs
After multiple testing corrections, OPEs that were associated with the four glycometabolic markers were grouped as four different mixtures for subsequent qgcomp analyses. Figure 3B shows the log-transformed concentrations of four OPE mixtures that were positively associated with the -scores of FPG (), GSP (), FINS (), and HOMA-IR (), respectively (see also Table S3). Each quartile increment in the mixture concentration was associated with elevated -scores of 0.06 (95% CI: 0.03, 0.08), 0.04 (95% CI: 0.02, 0.05), 0.06 (95% CI: 0.02, 0.09), and 0.12 (95% CI: 0.04, 0.20) for FPG, GSP, FINS, and HOMA-IR, respectively. In Figure 3C and Table S4, the weight of each OPE reflects the contribution of the correlated components to the overall mixture effect. Within the mixtures, TMPP had the highest weight (54%) for elevated FPG, followed by DBP (24%) and TPHP (14%), whereas exposure to BCEP and DPHP contributed minimally () to the overall mixture positive effect. TMPP contributed to 46% of the overall positive mixture effect of elevated GSP, followed by DBP and TPHP (26% and 19%, respectively). TPHP and TiBP had the highest weights of contribution to FINS and HOMA-IR for positive weight, at 44% and 29%, respectively. For negative weight, EHDPP had the highest weights for FPG, GSP, and HOMA-IR with 100%, 97%, and 57%, respectively. Ultimately, five OPEs (TMPP, TPHP, TiBP, DBP, and DPHP) with positive weights of were considered the key OPEs and were suspected to be responsible for the increased risks of glycometabolic disorder and therefore were included in further analyses.
Longitudinal Correlations of OPEs–Metabolites
The numbers of endogenous metabolites, based on metabolomics classifications in the serum and urine samples, are shown in Figure 4A. The numbers and directions of the associations between the five key OPEs and the respective serum and urine metabolomes are presented in Figure 4B,C. A total of 1,354 associations were found (710 and 644 for the serum and urine metabolomes, respectively; FDR ; Excel Tables S6 and S7). The network diagram shows the exposure–metabolite pairwise associations with FDR for the serum and urine metabolomes (Figure 4D,E; Excel Tables S6 and S7; raw serum and urine metabolome data in Excel Tables S8 and S9). Specifically, for the serum metabolome, the top three OPEs of high connective degree included TPHP, TMPP, and DBP. The top three metabolite classes with positive associations were peptides (20%), cofactors and vitamins (12%), and energy (12%), whereas those with negative associations were peptides (18%), carbohydrates (15%), and nucleotides (12%) (Figure 4F; Table S5). For the urine metabolome, DBP, DPHP, and TPHP were the three OPEs with the highest connective degree and were found to have mostly negative correlations. The top three metabolite classes with positive associations were carbohydrates (10%), secondary metabolites (10%), and cofactors and vitamins (8%), whereas those with negative associations were nucleotides (17%), global metabolites (12%), and cofactors and vitamins (11%) (Figure 4G; Table S6).
Figure 4.

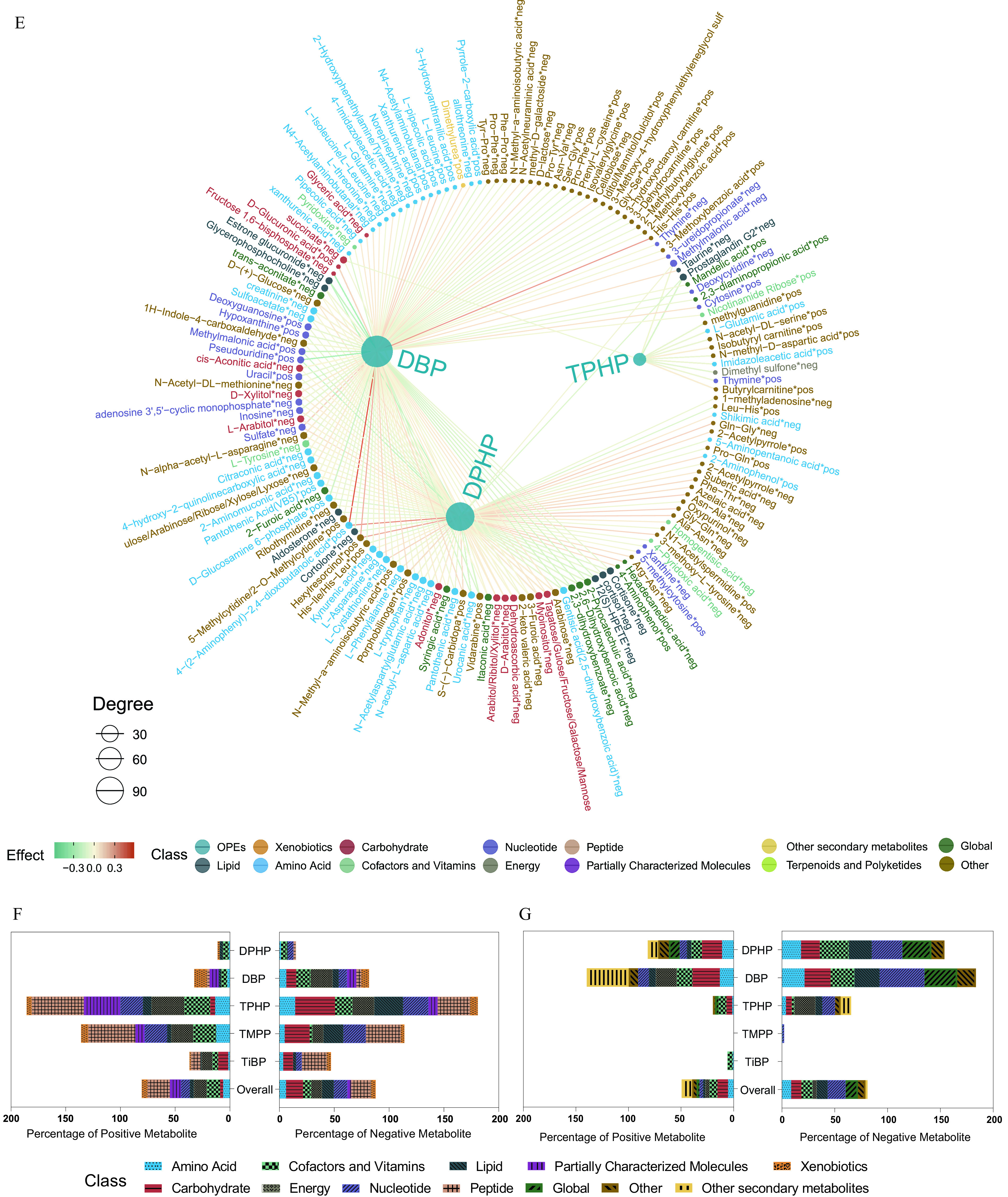
Serum and urine metabolome profiling of exposure to the key OPEs among healthy older Chinese adults 60–69 years of age. (A) The number of endogenous metabolites based on metabolic classifications in the serum and urine. (B,C) Volcano plots of the coefficient estimates of the key OPEs vs. the FDR values in the associations of the exposure–serum metabolome (B; see also Excel Table S6) and exposure–urine metabolome (C; see also Excel Table S7). Coefficient estimates are expressed as percentage changes (%) in FPG, GSP, FINS, and HOMA-IR per 1-SD change in each exposure, which was previously transformed to approach normality. The dashed horizontal line shows where the FDR value equals 0.05. (D,E) Network diagram of the association analysis between exposure to the key OPEs and the serum/urine metabolome (only metabolites with associations of FDR are shown; see also Excel Tables S6 and S7). The size of the node represents the degree of the exposure–metabolite connection, and the color of the edge represents the coefficient estimate of the exposure–metabolite association. (F,G) Stacking histograms of the percentages (%) of positively and negatively associated metabolites within each class, as well as the overall average for the serum and urine metabolomes with an FDR value of (see also Tables S5 and S6). Note: FDR, false discovery rate; FINS, fasting insulin; FPG, fasting plasma glucose; GSP, glycated serum protein; HOMA-IR, homeostatic model assessment for insulin resistance; OPE, organophosphate ester; SD, standard deviation.
Longitudinal Correlations of OPEs–Transcripts
The associations between five key OPEs and the blood transcriptome were also explored, and the numbers and directions of the associations are displayed using volcano plots (Figure 5A; Excel Table S10). A total of 16,585 associations (8,673 positive and 7,912 negative) were significant. The network diagram shows the OPE–transcript pairwise associations of the top 100 transcripts selected for absolute effect value, with an FDR of for each OPE (Figure 5B; Excel Table S10). Specifically, the top three OPEs correlated with gene expressions of high connective degree were TPHP (3,875 positive and 3,476 negative associations), DBP (3,056 positive and 2,653 negative associations), and TMPP (1,741 positive and 1,783 negative associations) (Figure 5C).
Figure 5.
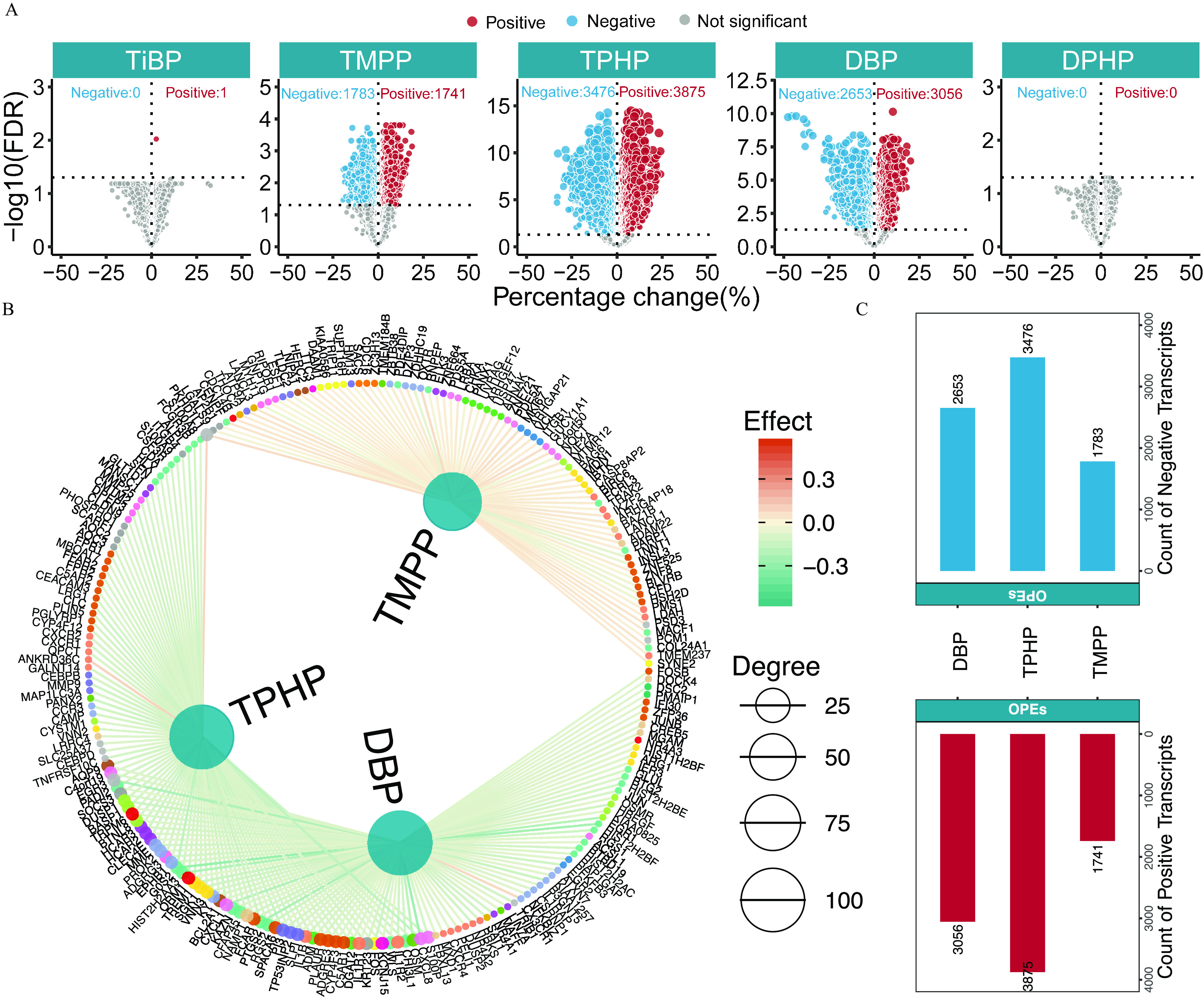
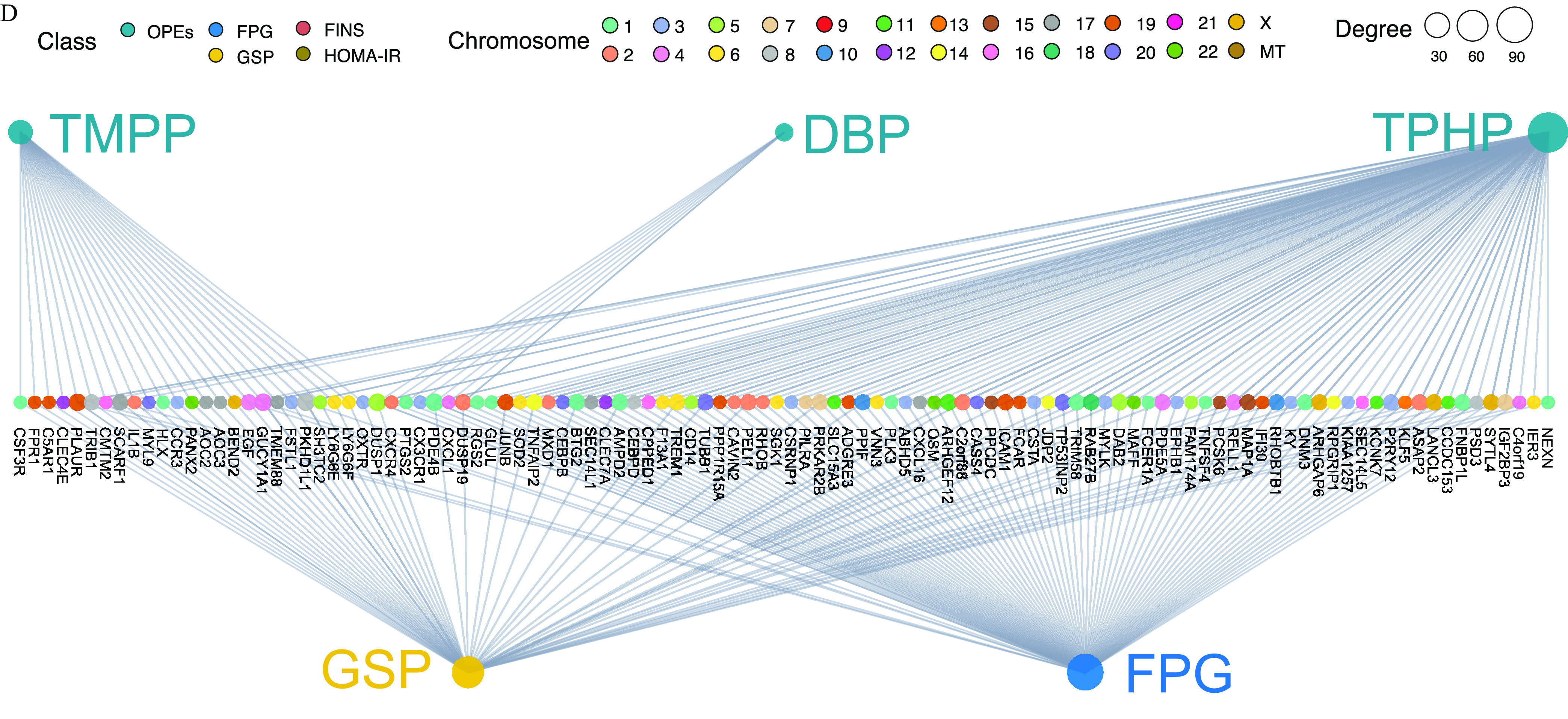
Transcriptome profiling of exposure to the key OPEs among healthy older Chinese adults 60–69 years of age. (A) Volcano plots of the coefficient estimates for exposure to the key OPEs vs. the FDR values among the exposure–transcriptome associations (see also Excel Table S10). Coefficient estimates are expressed as percentage changes (%) of FPG, GSP, FINS, and HOMA-IR per 1-SD change in each exposure, which was previously transformed to approach normality. The dashed horizontal line shows where the FDR value equals 0.05. (B) Network diagram of the association analysis between exposure to the key OPEs and the blood transcriptome (only the top 100 transcripts of each exposure with associations of FDR are shown). The size of the node represents the degree of exposure–transcript connection, and the color of the edge represents the coefficient estimates of exposure–transcript association (see also Excel Table S10). (C) Stacking histograms of the counts of the positively and negatively associated transcripts for each exposure with an FDR value of . (D) Tripartite network of the inferred causal relationships (top 150 genes selected for the absolute effect value for each OPE with an FDR value of ) of exposure to the key OPEs to the outcome (FPG and GSP) through transcriptome mediators (see also Excel Table S12). The size of the node represents the degree of exposure–transcript–outcome connection. (B,D) share a common legend. Note: FDR, false discovery rate; FINS, fasting insulin; FPG, fasting plasma glucose; GSP, glycated serum protein; HOMA-IR, homeostatic model assessment for insulin resistance; OPE, organophosphate ester; SD, standard deviation.
Biomolecules Mediating the Effects of OPEs on Glycometabolic Markers
A tripartite network plot of OPEs–metabolites–glycometabolic markers constructed using all of the significant metabolites and transcripts as potential biomolecular intermediators is presented in Figure 6A (see also Excel Table S11). A total of 106 serum and 29 urinary metabolites were inferred as biomolecular intermediators, with an FDR of . Specifically, 87, 60, 31, and 10 metabolites were inferred as biomolecular intermediators of the effects of TMPP, TPHP, DPHP, and DBP on glycometabolic markers, respectively (the number of common and specific metabolites of each OPE are shown in Figure S4A). In addition, 1,651 transcripts were inferred as biomolecular intermediators, with an FDR of , and a network plot of OPEs–transcript–glycometabolic markers is presented in Figure 5D (top 150 genes selected for the absolute effect value for each OPE; see also Excel Table S12). Specifically, 1,248, 649, and 116 transcripts were inferred as biomolecular intermediators of the effects of TMPP, TPHP, and DBP on glycometabolic markers, respectively (the number of common and specific transcripts of each OPE are shown in Figure S4B).
Figure 6.
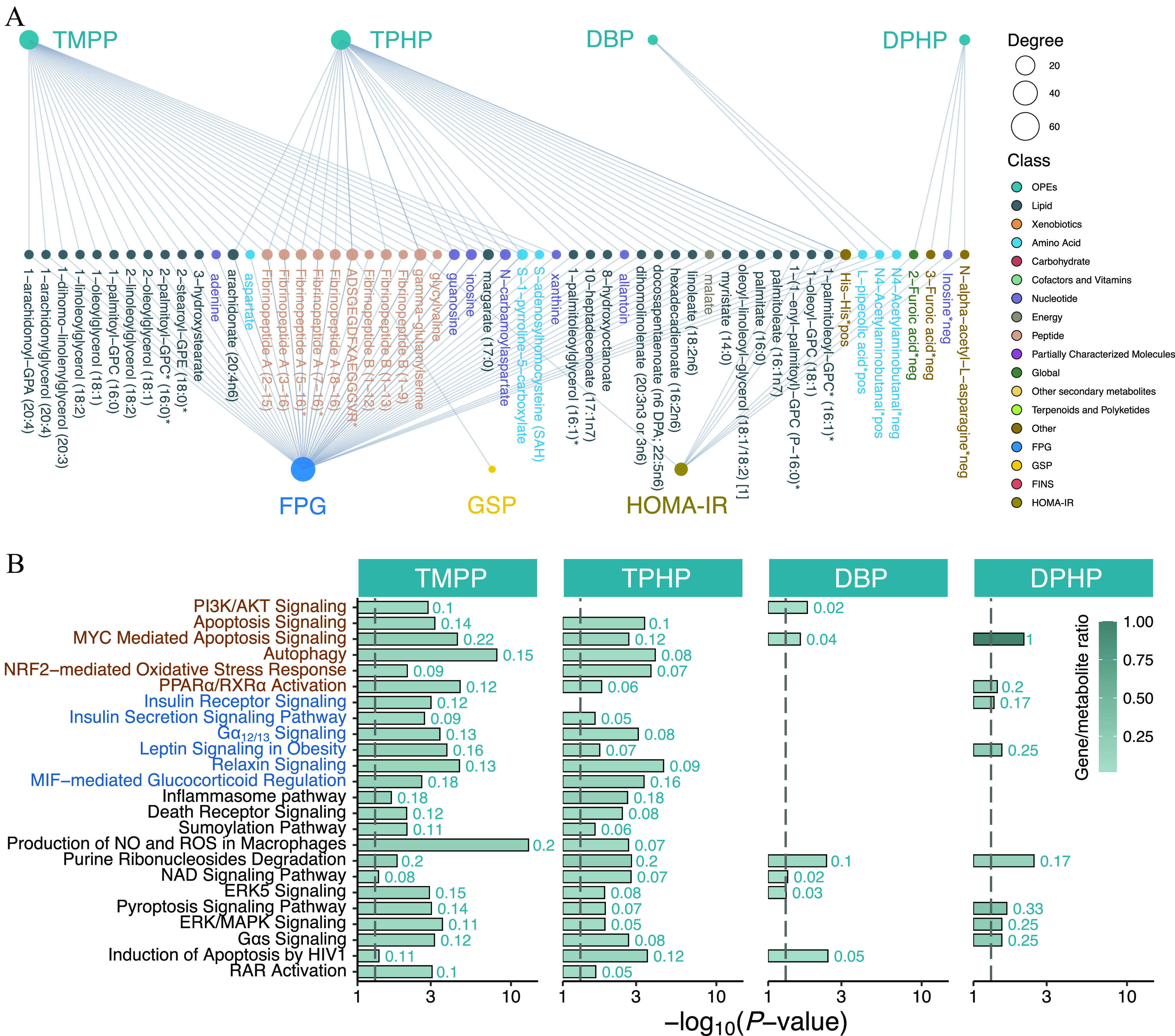
Inferred biomolecular intermediators of insulin resistance and glycometabolic disorders and the results of Integrative Pathway Analysis among healthy older Chinese adults 60–69 years of age. (A) A tripartite network of the inferred causal relationships (causal inference analysis with an FDR value of ) between exposure to the key OPEs and the outcomes (FPG, GSP, FINS, and HOMA-IR) through serum and urine metabolite mediators. The size of the node represents the degree of the exposure–metabolite–outcome connection (see also Excel Table S11). (B) Bar plots of the representative canonical pathways of the statistically significant biomolecular intermediators (genes and serum/urine metabolites) for TMPP, TPHP, DBP, and DPHP (, see also Excel Table S13). The negative logarithm of the -value is displayed on the x-axis, and the color of the bar represents the gene/metabolite ratio. The dashed vertical line shows where . The red and blue texts represent the pathways previously reported in experimental studies and reported for the first time, respectively. Note: DBP, dibutyl phosphate; DPHP, diphenyl phosphate; FINS, fasting insulin; FPG, fasting plasma glucose; GSP, glycated serum protein; HOMA-IR, homeostatic model assessment for insulin resistance; OPE, organophosphate ester; TMPP, trimethylolpropane phosphate; TPHP, triphenyl phosphate.
IPA for Biomolecular Intermediators
Biomolecular intermediators of the diabetogenic effects of TPHP, TMPP, DBP, and DPHP were included in the IPA analysis. Representative canonical pathways () shared by at least two OPEs are presented in Figure 6B and Excel Table S13. Among these, several pathways [e.g., peroxisome proliferator-activated receptor alpha ()/retinoid X receptor alpha () activation pathway, leptin signaling in obesity, G-protein alpha-s () signaling, myelocytomatosis viral oncogene (MYC)-mediated apoptosis signaling, and pyroptosis signaling] simultaneously appeared in the analysis results of at least three key OPEs. Core perturbed genes and metabolites within the abovementioned representative pathways were integrated to propose putative AOPs for impaired glucose homeostasis after OPE exposure (Figure 7A). These AOPs began with possible molecular initiation events (MIEs): activation of first apoptosis signal ligand (FasL) and protease-activated receptor (PAR), as well as inhibition of tumor necrosis factor (TNF), TNF receptor (TNFR), insulin receptor (IR), lysophosphatidic acid receptor (LPAR), and glucose transporter type 4 (GLUT4). Alterations of these ligands and membrane receptors caused a series of key events (KEs) at the molecular/cellular levels, such as KE-1, those involved in aberrant expression of kinases [phosphatidylinositol 3-kinase (PI3K), protein kinase B (AKT), and glycogen synthase kinase-3 (GSK3)], the apoptosis/autophagy regulator Bcl2-antagonist of cell death protein (BAD), the transcription factor nuclear factor kappa B (), metabolic enzymes (e.g., citrate synthase and succinate dehydrogenase), and tricarboxylic acid (TCA) cycle–related metabolites (e.g., citrate and succinate). The downstream KEs at the organ/system levels (KE-2; e.g., those involved in glucose uptake, glycogen synthesis, insulin secretion, energy metabolism, oxidative stress, and inflammation) were subsequently altered and were responsible for perturbations in glucose homeostasis eventually leading to T2D. In general, the up-regulation or down-regulation trends of most MIEs and KEs were consistent among the key OPEs, except for certain molecules (e.g., PAR, , BAD, and GSK3) for urinary DBP and DPHP.
Figure 7.

AOP linking OPE exposure and adverse outcome and schematic of the putative biological mechanisms of OPEs exposure–induced type 2 diabetes outcome. (A) An AOP diagram depicting the MIEs in response to exposure to the key OPEs and the subsequent series of KEs, for example, multiple signal transduction and metabolic pathway perturbations (KE-1 at the molecular/cellular level) and impaired biological functions (KE-2 at the organ/system level), which ultimately induced glycometabolic disorder-related adverse outcomes. Alterations of genes (blue) and metabolites (purple) are represented with colored boxes. (B) Schematic of the putative biological mechanisms that mediate the linkages between OPEs exposure and apical type 2 diabetes outcome. Note: AOP, adverse outcome pathway; KEs, key events; MIEs, molecular initiating events; OPE, organophosphate ester; T2DM, type 2 diabetes mellitus.
Discussion
OPEs are widely used as a substitute for polybrominated diphenyl ethers and are ubiquitously detectable in environmental matrices and human bodies, which has caused considerable concern in terms of their adverse effects worldwide. The present study, for the first time, comprehensively profiled the associations between OPE exposure and insulin resistance and glycometabolic homeostasis among healthy older Chinese adults 60–69 years of age and systematically explored the underlying biological mechanisms using multi-omics profiling.
Identification of the Key OPEs
In this study, exposure to five key OPEs and m-OPEs (TMPP, TPHP, TiBP, DBP, and DPHP) was found to be associated with elevated levels of glycometabolic markers, independent of the traditional risk factors for T2D (e.g., age, sex, BMI, and diet). To the best of our knowledge, the observed associations were first reported at the population level; moreover, some associations had, to varying extents, a priori credibility based on the previous experimental literature (Table S7).28,29,48–62 Specifically, although TMPP, TPHP, and DPHP were widely detected in the Chinese population at concentration ranges of ,24,63,64 to the best of our knowledge, our study is the first to reveal the adverse correlations between blood TPHP and its metabolite DPHP in urine and glucose homeostasis within humans. These findings are in line with those of previous experimental studies reporting the diabetogenic effects of TPHP and DPHP in vivo and in vitro. For instance, exposure to TPHP results in a higher glucose level and HOMA-IR index and inhibits the level of adiponectin, an insulin-sensitizing hormone, in adult male mice49 and pubertal mice.27 Likewise, numerous in vitro studies have confirmed the diabetogenic effects of TPHP and DPHP on 3T3-L1 adipose and HepG2 cells.59–61 However, no previous studies have yet reported the relationships between exposure to TMPP, TiBP, and DBP and abnormal glucose metabolism. Thus, further health hazard assessments through epidemiological studies or in vivo and in vitro experiments are warranted. In addition, it is noteworthy that the dominant pollutants differed between specific glycometabolic markers. For instance, TMPP and TPHP were the leading contributors of elevated FPG and FINS levels, respectively, possibly because of their different diabetogenic potential in triggering MIEs or downstream KEs. Therefore, subsequent CIT and multi-omics analyses were conducted to determine the biomolecular intermediators and obtain mechanistic insights into specific exposure–outcome associations.
Determination of Biomolecular Intermediators and Relevant Pathways
In this study, we comprehensively characterized the biomolecular changes in genes and metabolites induced by the key OPEs in healthy older adults for the first time. Some endogenous changes were first unveiled at the population level, including PI3K/AKT signaling [PI3K, AKT, and mitogen-activated protein kinase kinase kinase 8 (MAP3K8) inhibition], IR signaling [IR and growth factor receptor-bound protein 2 (GRB2) inhibition and IR substrate activation], signaling (protein kinase A and activation, and and inhibition), insulin secretion signaling [eukaryotic initiation factor 2 (EIF2) activation], and TCA cycle processes (citrate synthase, succinate dehydrogenase, and citrate inhibition). Our results were consistent with those of a previous in vitro study that reported that PI3K, AKT, and mRNAs were significantly down-regulated after TPHP treatment of L02 cells.65 Dysregulation in PI3K/AKT signaling, especially AKT activity inhibition, increases insulin resistance among patients with T2D.66 Besides, an in vivo study conducted in mice, showed that TPHP reduces the abundance of citrate and succinate within the TCA cycle52; moreover, the down-regulation of succinate dehydrogenase can impair glucose metabolism in islet cells in T2D.67 Similar to our findings, previous toxicological studies have reported that TPHP could induce cell apoptosis,68,69 autophagy,70 oxidative stress,71 and glucocorticoid regulation disruption72–74 (Table S8).48,60,68,75–93 Alterations of some other pathways (e.g., leptin signaling,94 relaxin signaling,95 and signaling96) are also closely related to insulin resistance and glucose metabolism, although they have not yet been reported to induce diabetogenic effects upon OPE exposure. In addition, to a certain extent, blood TMPP and TPHP share the most common biomolecular intermediators and pathways. In contrast, urinary DBP and DPHP have relatively unique molecular characteristics, suggesting differential modes of action and diabetogenic potentials, which are perhaps caused by their different chemical structures.
Putative AOPs Linking OPE Exposure and the Outcomes of T2D
AOPs can address the existing gaps in biological knowledge and provide evidence-based mechanistic insights into the associations between environmental exposure and health outcomes. To the best of our knowledge, our study is the first to present a putative network of AOPs linking OPE exposure with possible MIEs to downstream KEs that ultimately manifest as outcomes of T2D. The AOPs presented herein revealed that OPE exposure could trigger various MIEs, for example, aberrant alterations of ligands and membrane receptors (TNF/FasL, IR, TNFR, and GLUT4) that can subsequently disturb the downstream signal transduction and metabolic pathways [e.g., PI3K-AKT signaling, IR signaling, activation, and TCA cycle (as KE-1)] and biological processes [e.g., glucose metabolism (as KE-2)]. These pathways primarily communicate through the PI3K-AKT signaling hub, which is a well-recognized major effector of metabolic insulin action. Specifically, IRs stimulate a signaling cascade, leading to the phosphorylation/activation of IR substrates and the subsequent activation and phosphorylation of the PI3K cascade and AKT, respectively.97 This further regulates GSK3 activation98 and GLUT4 expression on the cell membrane,99 contributing to glycogen synthesis and glucose transportation, respectively. Moreover, we observed broad perturbations in the downstream events of AKT phosphorylation, with abnormal pathophysiological functions (e.g., apoptosis, autophagy, oxidative stress, and inflammation), resulting in the outcome of T2D.100 Our findings are also in line with those of previous in silico and in vitro analyses showing that TPHP exposure can trigger the PI3K/AKT pathway as a KE,65 highlighting its potential role as a promising preventive intervention or pharmacological target for OPE-associated T2D.
Strengths and Limitations
The main strength of this study is the multidimensional integration of the transcriptome and metabolome using two different biological markers (i.e., blood and urine), providing a reliable assessment of OPE exposure and endogenous biomolecular perturbations while considering the critical aging window.101 Besides, the five longitudinal repeated measurements helped avoid biases introduced by exposure measurement errors of one-time sampling and presented the molecular scenarios of the effects of differential exposure at individual levels, with the potential to explore causal associations. In addition, the availability of detailed and longitudinal data pertaining to participant demographics and time–activity helped us adjust for confounding factors in the statistical analyses and minimize the effects of other risk factors on our findings.
This study also has some limitations. First, the complete exclusion of potential uncontrolled confounders was impossible because reliance on uncontrolled longitudinal associations limits the ability to make causal determinations. Second, the population representation and sample size of our study could be inadequate, warranting further evaluations among different ethnic groups and larger population sizes with a nested case–control design or validations in toxicological research in vivo. Third, as potential EDCs, OPEs may have sexually dimorphic effects on glucose metabolism. Given the relatively small sample size and complex mixture of OPE exposures, it was underpowered to assess sex-stratified analyses in our exploratory study statistically. Future studies with a larger sample size may evaluate the sex-specific effects of these OPEs on glucose metabolism. Last, owing to the observational design of this study and the high dimensional feature of multi-omics data, caution is warranted with regard to whether these associations are causal.
Conclusions and Implications
The present exploratory study suggests that several OPEs may be independently and jointly associated with abnormal glycometabolism. High production volumes and the broad applications of OPEs in China, combined with the persistence and mobility characteristics, have led to the ever-increasing bioaccumulation of OPEs, possibly contributing to the high risk of T2D among the Chinese population. The putative AOPs linking OPE exposure to T2D, including the activation of a series of MIEs (e.g., IR, TNFR, and PAR) and the disruption of multiple downstream KEs (e.g., PI3K/AKT, activation, and insulin secretion), were also established (Figure 7B). Our findings advance the existing knowledge on the environmental bases of the etiology of T2D among older Chinese adults 60–69 years of age and shed light on the evidence linking chemical safety and noncommunicable disease in public health.
Supplementary Material
Acknowledgments
We thank all the participants of the China Biomarkers of Air Pollutant Exposure (BAPE) Study, the Dianliu Community, the Ankang Community Hospital, the Shandong Center for Disease Control and Prevention (CDC), the Jinan CDC, the China BAPE Study team, and the Metabolomics Facilities in Tsinghua University Protein Research Center, as well as Hangzhou Calibra Diagnostics Ltd., Dian Diagnostics, and Metabolon.
This study was financially supported by the National Research Program for Key Issues in Air Pollution Control of China (DQGG0401), the National Natural Science Foundation of China (82025030, 81941023, and 92043301), and the National Key Research and Development Program of China (2016YFC0206500 and 2022YFC3702700) to X.S.
References
- 1.Weil AR. 2015. The growing burden of noncommunicable diseases. Health Aff (Millwood) 34(9):1439, PMID: , 10.1377/hlthaff.2015.0974. [DOI] [PubMed] [Google Scholar]
- 2.WHO (World Health Organization). 2017. Noncommunicable Diseases Progress Monitor 2017. https://www.who.int/publications/i/item/9789241513029 [accessed May 14, 2022].
- 3.Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. 2022. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 183:109119, PMID: , 10.1016/j.diabres.2021.109119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinclair A, Saeedi P, Kaundal A, Karuranga S, Malanda B, Williams R. 2020. Diabetes and global ageing among 65–99-year-old adults: findings from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract 162:108078, PMID: , 10.1016/j.diabres.2020.108078. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P, Gregg E. 2017. Global economic burden of diabetes and its implications. Lancet Diabetes Endocrinol 5(6):404–405, PMID: , 10.1016/S2213-8587(17)30100-6. [DOI] [PubMed] [Google Scholar]
- 6.Stančáková A, Laakso M. 2016. Genetics of type 2 diabetes. Endocr Dev 31:203–220, PMID: , 10.1159/000439418. [DOI] [PubMed] [Google Scholar]
- 7.Kahn SE, Hull RL, Utzschneider KM. 2006. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444(7121):840–846, PMID: , 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Zhang M, Zhao Z, Huang Z, Deng Q, Li Y, et al. 2020. Geographic variation in prevalence of adult obesity in China: results from the 2013–2014 National Chronic Disease and Risk Factor Surveillance. Ann Intern Med 172(4):291–293, PMID: , 10.7326/M19-0477. [DOI] [PubMed] [Google Scholar]
- 9.Liu B, Du Y, Wu Y, Snetselaar LG, Wallace RB, Bao W. 2021. Trends in obesity and adiposity measures by race or ethnicity among adults in the United States 2011–2018: population based study. BMJ 5(1):21–27, PMID: , 10.1136/bmj.n365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Peng W, Zhao Z, Zhang M, Shi Z, Song Z, et al. 2021. Prevalence and treatment of diabetes in China, 2013–2018. JAMA 326(24):2498–2506, PMID: , 10.1001/jama.2021.22208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Li X, Wang Z, Bancks MP, Carnethon MR, Greenland P, et al. 2021. Trends in prevalence of diabetes and control of risk factors in diabetes among US adults, 1999–2018. JAMA 326(8):1–13, PMID: , 10.1001/jama.2021.9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hales CN, Barker DJ. 1992. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35(7):595–601, PMID: , 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, He Y, Qi L, Jaddoe VW, Feskens EJM, Yang X, et al. 2010. Exposure to the Chinese famine in early life and the risk of hyperglycemia and type 2 diabetes in adulthood. Diabetes 59(10):2400–2406, PMID: , 10.2337/db10-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, et al. 2017. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med 376(20):1933–1942, PMID: , 10.1056/NEJMoa1609581. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Wang DD, Ley SH, Vasanti M, Howard AG, He Y, et al. 2017. Time trends of dietary and lifestyle factors and their potential impact on diabetes burden in China. Diabetes Care 40(12):1685–1694, PMID: , 10.2337/dc17-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landrigan PJ, Fuller R, Acosta NJR, Adeyi O, Arnold R, Basu NN, et al. 2018. The Lancet Commission on pollution and health. Lancet 391(10119):462–512, PMID: , 10.1016/S0140-6736(17)32345-0. [DOI] [PubMed] [Google Scholar]
- 17.Ruiz D, Becerra M, Jagai JS, Ard K, Sargis RM. 2018. Disparities in environmental exposures to endocrine-disrupting chemicals and diabetes risk in vulnerable populations. Diabetes Care 41(1):193–205, PMID: , 10.2337/dc16-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yehuda AB, Zinger A, Durso S. 2014. The older patient with diabetes: a practical approach. Diabetes Metab Res Rev 30(2):88–95, PMID: , 10.1002/dmrr.2485. [DOI] [PubMed] [Google Scholar]
- 19.Lind L, Zethelius B, Salihovic S, van Bavel B, Lind PM. 2014. Circulating levels of perfluoroalkyl substances and prevalent diabetes in the elderly. Diabetologia 57(3):473–479, PMID: , 10.1007/s00125-013-3126-3. [DOI] [PubMed] [Google Scholar]
- 20.Lind PM, Zethelius B, Lind L. 2012. Circulating levels of phthalate metabolites are associated with prevalent diabetes in the elderly. Diabetes Care 35(7):1519–1524, PMID: , 10.2337/dc11-2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi YH, Kim JH, Hong YC. 2015. Sex-dependent and body weight-dependent associations between environmental PAHs exposure and insulin resistance: Korean urban elderly panel. J Epidemiol Community Health 69(7):625–631, PMID: , 10.1136/jech-2014-204801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blum A, Behl M, Birnbaum L, Diamond ML, Phillips A, Singla V, et al. 2019. Organophosphate ester flame retardants: are they a regrettable substitution for polybrominated diphenyl ethers? Environ Sci Technol Lett 6(11):638–649, PMID: , 10.1021/acs.estlett.9b00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Z, Yin L, Wen X, Jiang C, Long Y, Zhang J, et al. 2021. Organophosphate esters in China: fate, occurrence, and human exposure. Toxics 9(11):310, PMID: , 10.3390/toxics9110310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu L, Yu M, Li Y, Liu L, Li X, Song L, et al. 2022. Association of exposure to organophosphate esters with increased blood pressure in children and adolescents. Environ Pollut 295:118685, PMID: , 10.1016/j.envpol.2021.118685. [DOI] [PubMed] [Google Scholar]
- 25.Hu W, Gao P, Wang L, Hu J. 2023. Endocrine disrupting toxicity of aryl organophosphate esters and mode of action. Crit Rev Environ Sci Technol 53(1):1–18, 10.1080/10643389.2022.2050147. [DOI] [Google Scholar]
- 26.Yang Y, Chen P, Ma S, Lu S, Yu Y, An T. 2022. A critical review of human internal exposure and the health risks of organophosphate ester flame retardants and their metabolites. Crit Rev Environ Sci Technol 52(9):1528–1560, 10.1080/10643389.2020.1859307. [DOI] [Google Scholar]
- 27.Wang C, Le Y, Lu D, Zhao M, Dou X, Zhang Q, et al. 2020. Triphenyl phosphate causes a sexually dimorphic metabolism dysfunction associated with disordered adiponectin receptors in pubertal mice. J Hazard Mater 388:121732, PMID: , 10.1016/j.jhazmat.2019.121732. [DOI] [PubMed] [Google Scholar]
- 28.Du Z, Zhang Y, Wang G, Peng J, Wang Z, Gao S. 2016. TPhP exposure disturbs carbohydrate metabolism, lipid metabolism, and the DNA damage repair system in zebrafish liver. Sci Rep 6:21827, PMID: , 10.1038/srep21827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green AJ, Graham JL, Gonzalez EA, La Frano MR, Petropoulou SSE, Park JS, et al. 2017. Perinatal triphenyl phosphate exposure accelerates type 2 diabetes onset and increases adipose accumulation in UCD-type 2 diabetes mellitus rats. Reprod Toxicol 68:119–129, PMID: , 10.1016/j.reprotox.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bo Y, Zhu Y. 2022. Organophosphate esters exposure in relation to glucose homeostasis and type 2 diabetes in adults: a national cross-sectional study from the National Health and Nutrition Survey. Chemosphere 301:134669, PMID: , 10.1016/j.chemosphere.2022.134669. [DOI] [PubMed] [Google Scholar]
- 31.Luo K, Aimuzi R, Wang Y, Nian M, Zhang J. 2020. Urinary organophosphate esters metabolites, glucose homeostasis and prediabetes in adolescents. Environ Pollut 267:115607, PMID: , 10.1016/j.envpol.2020.115607. [DOI] [PubMed] [Google Scholar]
- 32.Wang S, Yong H, He XD. 2021. Multi-omics: opportunities for research on mechanism of type 2 diabetes mellitus. World J Diabetes 12(7):1070–1080, PMID: , 10.4239/wjd.v12.i7.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beulens JWJ, Pinho MGM, Abreu TC, den Braver NR, Lam TM, Huss A, et al. 2022. Environmental risk factors of type 2 diabetes—an exposome approach. Diabetologia 65(2):263–274, PMID: , 10.1007/s00125-021-05618-w. [DOI] [PubMed] [Google Scholar]
- 34.Price EJ, Vitale CM, Miller GW, David A, Barouki R, Audouze K, et al. 2022. Merging the exposome into an integrated framework for “omics” sciences. iScience 25(3):103976, PMID: , 10.1016/j.isci.2022.103976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang S, Li T, Fang J, Chen R, Cha Y, Wang Y, et al. 2021. The exposome in practice: an exploratory panel study of biomarkers of air pollutant exposure in Chinese people aged 60–69 years (China BAPE Study). Environ Int 157:106866, PMID: , 10.1016/j.envint.2021.106866. [DOI] [PubMed] [Google Scholar]
- 36.Hou M, Fang J, Shi Y, Tang S, Dong H, Liu Y, et al. 2021. Exposure to organophosphate esters in elderly people: relationships of OPE body burdens with indoor air and dust concentrations and food consumption. Environ Int 157:106803, PMID: , 10.1016/j.envint.2021.106803. [DOI] [PubMed] [Google Scholar]
- 37.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. 1985. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28(7):412–419, PMID: , 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 38.Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360, PMID: , 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7):923–930, PMID: , 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 40.Jia X, Jin Q, Fang J, Shi Y, Hou M, Dong H, et al. 2022. Emerging and legacy per- and polyfluoroalkyl substances in an elderly population in Jinan, China: the exposure level, short-term variation, and intake assessment. Environ Sci Technol 56(12):7905–7916, PMID: , 10.1021/acs.est.2c00381. [DOI] [PubMed] [Google Scholar]
- 41.Keil AP, Buckley JP, O’Brien KM, Ferguson KK, Zhao S, White AJ, et al. 2020. A quantile-based g-computation approach to addressing the effects of exposure mixtures. Environ Health Perspect 128(4):47004, PMID: , 10.1289/EHP5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Millstein J, Zhang B, Zhu J, Schadt EE. 2009. Disentangling molecular relationships with a causal inference test. BMC Genet 10:23, PMID: , 10.1186/1471-2156-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krämer A, Green J, Pollard J Jr, Tugendreich S. 2014. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30(4):523–530, PMID: , 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White IR, Royston P, Wood AM. 2011. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 30(4):377–399, PMID: , 10.1002/sim.4067. [DOI] [PubMed] [Google Scholar]
- 45.Boyle M, Buckley JP, Quirós-Alcalá L. 2019. Associations between urinary organophosphate ester metabolites and measures of adiposity among U.S. children and adults: NHANES 2013–2014. Environ Int 127:754–763, PMID: , 10.1016/j.envint.2019.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ospina M, Jayatilaka NK, Wong LY, Restrepo P, Calafat AM. 2018. Exposure to organophosphate flame retardant chemicals in the U.S. general population: data from the 2013–2014 National Health and Nutrition Examination Survey. Environ Int 110:32–41, PMID: , 10.1016/j.envint.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Storey JD. 2002. A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodol 64(3):479–498, 10.1111/1467-9868.00346. [DOI] [Google Scholar]
- 48.Liu X, Zhao X, Wang Y, Hong J, Shi M, Pfaff D, et al. 2020. Triphenyl phosphate permeates the blood brain barrier and induces neurotoxicity in mouse brain. Chemosphere 252:126470, PMID: , 10.1016/j.chemosphere.2020.126470. [DOI] [PubMed] [Google Scholar]
- 49.Wang D, Yan S, Yan J, Teng M, Meng Z, Li R, et al. 2019. Effects of triphenyl phosphate exposure during fetal development on obesity and metabolic dysfunctions in adult mice: impaired lipid metabolism and intestinal dysbiosis. Environ Pollut 246:630–638, PMID: , 10.1016/j.envpol.2018.12.053. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Huang X, Laserna AKC, Li SFY. 2018. Untargeted metabolomics reveals transformation pathways and metabolic response of the earthworm Perionyx excavatus after exposure to triphenyl phosphate. Sci Rep 8(1):16440, PMID: , 10.1038/s41598-018-34814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Philbrook NA, Restivo VE, Belanger CL, Winn LM. 2018. Gestational triphenyl phosphate exposure in C57Bl/6 mice perturbs expression of insulin-like growth factor signaling genes in maternal and fetal liver. Birth Defects Res 110(6):483–494, PMID: , 10.1002/bdr2.1185. [DOI] [PubMed] [Google Scholar]
- 52.Wang D, Zhu W, Chen L, Yan J, Teng M, Zhou Z, et al. 2018. Neonatal triphenyl phosphate and its metabolite diphenyl phosphate exposure induce sex- and dose-dependent metabolic disruptions in adult mice. Environ Pollut 237:10–17, PMID: , 10.1016/j.envpol.2018.01.047. [DOI] [PubMed] [Google Scholar]
- 53.Adams S, Wiersielis K, Yasrebi A, Conde K, Armstrong L, Guo GL, et al. 2020. Sex- and age-dependent effects of maternal organophosphate flame-retardant exposure on neonatal hypothalamic and hepatic gene expression. Reprod Toxicol 94:65–74, PMID: , 10.1016/j.reprotox.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walley SN, Krumm EA, Yasrebi A, Kwiecinski J, Wright V, Baker C, et al. 2021. Maternal organophosphate flame-retardant exposure alters offspring energy and glucose homeostasis in a sexually dimorphic manner in mice. J Appl Toxicol 41(4):572–586, PMID: , 10.1002/jat.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vail GM, Walley SN, Yasrebi A, Maeng A, Conde KM, Roepke TA. 2020. The interactions of diet-induced obesity and organophosphate flame retardant exposure on energy homeostasis in adult male and female mice. J Toxicol Environ Health A 83(11–12):438–455, PMID: , 10.1080/15287394.2020.1777235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krumm EA, Patel VJ, Tillery TS, Yasrebi A, Shen J, Guo GL, et al. 2018. Organophosphate flame-retardants alter adult mouse homeostasis and gene expression in a sex-dependent manner potentially through interactions with ERα. Toxicol Sci 162(1):212–224, PMID: , 10.1093/toxsci/kfx238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vail GM, Walley SN, Yasrebi A, Maeng A, Degroat TJ, Conde KM, et al. 2022. Implications of peroxisome proliferator-activated receptor gamma (PPARY) with the intersection of organophosphate flame retardants and diet-induced obesity in adult mice. J Toxicol Environ Health A 85(9):381–396, PMID: , 10.1080/15287394.2021.2023716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vail GM, Walley SN, Yasrebi A, Maeng A, Degroat TJ, Conde KM, et al. 2022. Implications of estrogen receptor alpha (ERa) with the intersection of organophosphate flame retardants and diet-induced obesity in adult mice. J Toxicol Environ Health A 85(10):397–413, PMID: , 10.1080/15287394.2022.2026849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Le Y, Xu M, Wang W, Chen H, Zhang Q, et al. 2022. Remodeling on adipocytic physiology of organophosphorus esters in mature adipocytes. Environ Pollut 305:119287, PMID: , 10.1016/j.envpol.2022.119287. [DOI] [PubMed] [Google Scholar]
- 60.Gu J, Su F, Hong P, Zhang Q, Zhao M. 2019. 1H NMR-based metabolomic analysis of nine organophosphate flame retardants metabolic disturbance in Hep G2 cell line. Sci Total Environ 665:162–170, PMID: , 10.1016/j.scitotenv.2019.02.055. [DOI] [PubMed] [Google Scholar]
- 61.Cano-Sancho G, Smith A, La Merrill MA. 2017. Triphenyl phosphate enhances adipogenic differentiation, glucose uptake and lipolysis via endocrine and noradrenergic mechanisms. Toxicol In Vitro 40:280–288, PMID: , 10.1016/j.tiv.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu W, Kang Q, Zhang C, Ma H, Xu C, Wan Y, et al. 2020. Triphenyl phosphate modulated saturation of phospholipids: induction of endoplasmic reticulum stress and inflammation. Environ Pollut 263(pt A):114474, PMID: , 10.1016/j.envpol.2020.114474. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Li Y, Dong S, Han L, Guo R, Fu Y, et al. 2021. The risk and impact of organophosphate esters on the development of female-specific cancers: comparative analysis of patients with benign and malignant tumors. J Hazard Mater 404(pt B):124020, PMID: , 10.1016/j.jhazmat.2020.124020. [DOI] [PubMed] [Google Scholar]
- 64.Ya M, Yu N, Zhang Y, Su H, Tang S, Su G. 2019. Biomonitoring of organophosphate triesters and diesters in human blood in Jiangsu Province, eastern China: occurrences, associations, and suspect screening of novel metabolites. Environ Int 131:105056, PMID: , 10.1016/j.envint.2019.105056. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Wang L, Li F, Teng Y, Ji C, Wu H. 2022. Toxicity pathways of lipid metabolic disorders induced by typical replacement flame retardants via data-driven analysis, in silico and in vitro approaches. Chemosphere 287(pt 4):132419, PMID: , 10.1016/j.chemosphere.2021.132419. [DOI] [PubMed] [Google Scholar]
- 66.Batista TM, Jayavelu AK, Wewer Albrechtsen NJ, Iovino S, Lebastchi J, Pan H, et al. 2020. A cell-autonomous signature of dysregulated protein phosphorylation underlies muscle insulin resistance in type 2 diabetes. Cell Metab 32(5):844–859.e5, PMID: , 10.1016/j.cmet.2020.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sacco F, Seelig A, Humphrey SJ, Krahmer N, Volta F, Reggio A, et al. 2019. Phosphoproteomics reveals the GSK3-PDX1 axis as a key pathogenic signaling node in diabetic islets. Cell Metab 29(6):1422–1432.e3, PMID: , 10.1016/j.cmet.2019.02.012. [DOI] [PubMed] [Google Scholar]
- 68.Yu X, Yin H, Peng H, Lu G, Liu Z, Dang Z. 2019. OPFRs and BFRs induced A549 cell apoptosis by caspase-dependent mitochondrial pathway. Chemosphere 221:693–702, PMID: , 10.1016/j.chemosphere.2019.01.074. [DOI] [PubMed] [Google Scholar]
- 69.Meng Y, Xu X, Niu D, Xu Y, Qiu Y, Zhu Z, et al. 2022. Organophosphate flame retardants induce oxidative stress and Chop/Caspase 3-related apoptosis via Sod1/p53/Map3k6/Fkbp5 in NCI-1975 cells. Sci Total Environ 819:153160, PMID: , 10.1016/j.scitotenv.2022.153160. [DOI] [PubMed] [Google Scholar]
- 70.Deng Q, Jiang L, Mao L, Song XH, He CQ, Li XL, et al. 2020. The role of protein kinase C alpha in tri-ortho-cresyl phosphate-induced autophagy in human neuroblastoma SK-N-SH cells. J Appl Toxicol 40(11):1480–1490, PMID: , 10.1002/jat.3999. [DOI] [PubMed] [Google Scholar]
- 71.Chen R, Hou R, Hong X, Yan S, Zha J. 2019. Organophosphate flame retardants (OPFRs) induce genotoxicity in vivo: a survey on apoptosis, DNA methylation, DNA oxidative damage, liver metabolites, and transcriptomics. Environ Int 130:104914, PMID: , 10.1016/j.envint.2019.104914. [DOI] [PubMed] [Google Scholar]
- 72.de Guia RM, Rose AJ, Herzig S. 2014. Glucocorticoid hormones and energy homeostasis. Horm Mol Biol Clin Investig 19(2):117–128, PMID: , 10.1515/hmbci-2014-0021. [DOI] [PubMed] [Google Scholar]
- 73.Kuo T, McQueen A, Chen TC, Wang JC. 2015. Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol 872:99–126, PMID: , 10.1007/978-1-4939-2895-8_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kang S, Tsai LT, Zhou Y, Evertts A, Xu S, Griffin MJ, et al. 2015. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat Cell Biol 17(1):44–56, PMID: , 10.1038/ncb3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Umamaheswari S, Karthika P, Suvenitha K, Kadirvelu K, Ramesh M. 2021. Dose-dependent molecular responses of Labeo rohita to triphenyl phosphate. Chem Res Toxicol 34(12):2500–2511, PMID: , 10.1021/acs.chemrestox.1c00281. [DOI] [PubMed] [Google Scholar]
- 76.Ramesh M, Angitha S, Haritha S, Poopal RK, Ren Z, Umamaheswari S. 2020. Organophosphorus flame retardant induced hepatotoxicity and brain AChE inhibition on zebrafish (Danio rerio). Neurotoxicol Teratol 82:106919, PMID: , 10.1016/j.ntt.2020.106919. [DOI] [PubMed] [Google Scholar]
- 77.Canbaz D, Logiantara A, van Ree R, van Rijt LS. 2017. Immunotoxicity of organophosphate flame retardants TPHP and TDCIPP on murine dendritic cells in vitro. Chemosphere 177:56–64, PMID: , 10.1016/j.chemosphere.2017.02.149. [DOI] [PubMed] [Google Scholar]
- 78.Van den Eede N, Cuykx M, Rodrigues RM, Laukens K, Neels H, Covaci A, et al. 2015. Metabolomics analysis of the toxicity pathways of triphenyl phosphate in HepaRG cells and comparison to oxidative stress mechanisms caused by acetaminophen. Toxicol In Vitro 29(8):2045–2054, PMID: , 10.1016/j.tiv.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 79.Mennillo E, Cappelli F, Arukwe A. 2019. Biotransformation and oxidative stress responses in rat hepatic cell-line (H4IIE) exposed to organophosphate esters (OPEs). Toxicol Appl Pharmacol 371:84–94, PMID: , 10.1016/j.taap.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 80.Chen G, Zhang S, Jin Y, Wu Y, Liu L, Qian H, et al. 2015. TPP and TCEP induce oxidative stress and alter steroidogenesis in TM3 Leydig cells. Reprod Toxicol 57:100–110, PMID: , 10.1016/j.reprotox.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 81.Wang X, Li F, Liu J, Ji C, Wu H. 2020. Transcriptomic, proteomic and metabolomic profiling unravel the mechanisms of hepatotoxicity pathway induced by triphenyl phosphate (TPP). Ecotoxicol Environ Saf 205:111126, PMID: , 10.1016/j.ecoenv.2020.111126. [DOI] [PubMed] [Google Scholar]
- 82.Schang G, Robaire B, Hales BF. 2016. Organophosphate flame retardants act as endocrine-disrupting chemicals in MA-10 mouse tumor Leydig cells. Toxicol Sci 150(2):499–509, PMID: , 10.1093/toxsci/kfw012. [DOI] [PubMed] [Google Scholar]
- 83.An J, Hu J, Shang Y, Zhong Y, Zhang X, Yu Z. 2016. The cytotoxicity of organophosphate flame retardants on HepG2, A549 and Caco-2 cells. J Environ Sci Health A Tox Hazard Subst Environ Eng 51(11):980–988, PMID: , 10.1080/10934529.2016.1191819. [DOI] [PubMed] [Google Scholar]
- 84.Chen G, Jin Y, Wu Y, Liu L, Fu Z. 2015. Exposure of male mice to two kinds of organophosphate flame retardants (OPFRs) induced oxidative stress and endocrine disruption. Environ Toxicol Pharmacol 40(1):310–318, PMID: , 10.1016/j.etap.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 85.Yuan S, Zhu K, Ma M, Zhu X, Rao K, Wang Z. 2020. In vitro oxidative stress, mitochondrial impairment and G1 phase cell cycle arrest induced by alkyl-phosphorus-containing flame retardants. Chemosphere 248:126026, PMID: , 10.1016/j.chemosphere.2020.126026. [DOI] [PubMed] [Google Scholar]
- 86.Yao Y, Li M, Pan L, Duan Y, Duan X, Li Y, et al. 2021. Exposure to organophosphate ester flame retardants and plasticizers during pregnancy: thyroid endocrine disruption and mediation role of oxidative stress. Environ Int 146:106215, PMID: , 10.1016/j.envint.2020.106215. [DOI] [PubMed] [Google Scholar]
- 87.Wang Y, Hong J, Shi M, Guo L, Liu L, Tang H, et al. 2021. Triphenyl phosphate disturbs the lipidome and induces endoplasmic reticulum stress and apoptosis in JEG-3 cells. Chemosphere 275:129978, PMID: , 10.1016/j.chemosphere.2021.129978. [DOI] [PubMed] [Google Scholar]
- 88.Hong J, Jiang M, Guo L, Lin J, Wang Y, Tang H, et al. 2022. Prenatal exposure to triphenyl phosphate activated PPARγ in placental trophoblasts and impaired pregnancy outcomes. Environ Pollut 301:119039, PMID: , 10.1016/j.envpol.2022.119039. [DOI] [PubMed] [Google Scholar]
- 89.Wang X, Li F, Liu J, Li Q, Ji C, Wu H. 2021. New insights into the mechanism of hepatocyte apoptosis induced by typical organophosphate ester: an integrated in vitro and in silico approach. Ecotoxicol Environ Saf 219:112342, PMID: , 10.1016/j.ecoenv.2021.112342. [DOI] [PubMed] [Google Scholar]
- 90.Bowen C, Childers G, Perry C, Martin N, McPherson CA, Lauten T, et al. 2020. Mitochondrial-related effects of pentabromophenol, tetrabromobisphenol A, and triphenyl phosphate on murine BV-2 microglia cells. Chemosphere 255:126919, PMID: , 10.1016/j.chemosphere.2020.126919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Q, Wang J, Zhu J, Liu J, Zhao M. 2017. Potential glucocorticoid and mineralocorticoid effects of nine organophosphate flame retardants. Environ Sci Technol 51(10):5803–5810, PMID: , 10.1021/acs.est.7b01237. [DOI] [PubMed] [Google Scholar]
- 92.Kojima H, Takeuchi S, Van den Eede N, Covaci A. 2016. Effects of primary metabolites of organophosphate flame retardants on transcriptional activity via human nuclear receptors. Toxicol Lett 245:31–39, PMID: , 10.1016/j.toxlet.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 93.Kojima H, Takeuchi S, Itoh T, Iida M, Kobayashi S, Yoshida T. 2013. In vitro endocrine disruption potential of organophosphate flame retardants via human nuclear receptors. Toxicology 314(1):76–83, PMID: , 10.1016/j.tox.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 94.Wang B, Chandrasekera PC, Pippin JJ. 2014. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev 10(2):131–145, PMID: , 10.2174/1573399810666140508121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bani D, Pini A, Yue SKS. 2012. Relaxin, insulin and diabetes: an intriguing connection. Curr Diabetes Rev 8(5):329–335, PMID: , 10.2174/157339912802083487. [DOI] [PubMed] [Google Scholar]
- 96.Yang YM, Kuen DS, Chung Y, Kurose H, Kim SG. 2020. Gα12/13 signaling in metabolic diseases. Exp Mol Med 52(6):896–910, PMID: , 10.1038/s12276-020-0454-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cartee GD. 2015. Mechanisms for greater insulin-stimulated glucose uptake in normal and insulin-resistant skeletal muscle after acute exercise. Am J Physiol Endocrinol Metab 309(12):E949–E959, PMID: , 10.1152/ajpendo.00416.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shieh JM, Wu HT, Cheng KC, Cheng JT. 2009. Melatonin ameliorates high fat diet-induced diabetes and stimulates glycogen synthesis via a PKCζ-Akt-GSK3β pathway in hepatic cells. J Pineal Res 47(4):339–344, PMID: , 10.1111/j.1600-079X.2009.00720.x. [DOI] [PubMed] [Google Scholar]
- 99.Saltiel AR, Kahn CR. 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414(6865):799–806, PMID: , 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 100.Huang X, Liu G, Guo J, Su Z. 2018. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci 14(11):1483–1496, PMID: , 10.7150/ijbs.27173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lehallier B, Gate D, Schaum N, Nanasi T, Lee SE, Yousef H, et al. 2019. Undulating changes in human plasma proteome profiles across the lifespan. Nat Med 25(12):1843–1850, PMID: , 10.1038/s41591-019-0673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.