

Abstract
Organothiocyanates and selenocyanates are valuable compounds, both in terms of functional group interconversion and due to their biological activities. In this contribution, we report the synthesis of a series of these important substances in a mixture of water and dimethyl carbonate (20/1 proportion) using potassium thio- or selenocyanates salts and organic bromides. The key to the effectiveness of the reaction is a chalcogen bond interaction between a selenonium salt catalyst and the organic substrate.
Keywords: selenonium salt, chalcogen bond, organocatalysis, thiocyanate, selenocyanate
1. Introduction
Research on the interaction of organochalcogen compounds with Lewis bases is an emerging field of study. Organochalcogen compounds can interact with electron-rich species due to the presence of regions of positive electrostatic potential on their surface, which are referred to as σ-holes. The non-covalent interaction of the chalcogen electrophilic site with a Lewis base is defined as a chalcogen bond (ChB) [1,2,3,4]. Chalcogenonium salts (chalcogen IV species) are much better chalcogen bond donors compared with chalcogen (II) compounds, due to the depth of their σ-holes and increased Lewis acidity [5,6,7]. In recent years, research has focused on the application of selenonium salts as catalysts in various organic transformations [8,9,10,11,12,13,14,15,16].
Simple organothiocyanates and organoselenocyanates, as depicted in Figure 1, have been reported to display a broad range of biological activities, ranging from chemoprevention [17] and antiproliferative activity against cancer cells [18] to peroxide scavenging [19] and treatment of Chagas disease [20,21,22]. Additionally, these compounds can be easily converted to other functional groups or employed in synthetic transformations [23,24,25,26].
Figure 1.

Structure of biologically active organothiocyanates and organoselenocyanates.
Not surprisingly, the search for methods to prepare these valuable compounds has attracted much attention. They can be prepared, for instance, with the aid of phase transfer catalysts [27,28,29,30,31], in free radical reactions [32,33,34,35], by electrophilic addition to suitable organic substrates [36,37], or in reactions employing ionic liquids as a solvent and the nucleophilic source of chalcogen cyanide [38]. In this contribution, we share our findings on the use of selenonium salts to activate substrates such as benzyl bromides through chalcogen bond interactions, facilitating the displacement reaction with KChCN (Ch = S or Se) using a 20:1 mixture of water and dimethyl carbonate as a solvent. Reactions are fast (10 to 60 min), conducted at room temperature, and deliver products in good to excellent yields.
2. Results and Discussion
Very recently, our group reported the cyanation of benzyl bromides and other organic substrates in water catalyzed by an organoselenide [39]. Our results suggested that the selenonium salt produced throughout the reaction was the alkylating agent, mimicking the behavior of the cofactor S-adenosyl-L-methionine (SAM) in methyltransferase enzymes. Encouraged by these findings, we aimed to extend the protocol to the synthesis of valuable organic thiocyanates and selenocyanates. Initially, we evaluated the thiocyanation of benzyl bromide in anhydrous ethanol using 10 mol% of selenides C1–6 as organocatalysts. Selenide C1 showed minimal activity, only slightly outperforming the control (Table 1, entries 1 and 2). We tested selenides bearing electron-withdrawing C2–3 or electron-donating groups C4–5, among which the electron-rich catalyst C5 displayed the best results, producing the desired product twice as fast as the control reaction (entries 1 and 6). In contrast, selenide C6, which exhibited the best catalytic activity in the cyanation of benzyl bromide, performed poorly in the thiocyanation reaction (entry 7). Ultimately, we found that the reaction could be efficiently performed without any organoselenium catalyst by using hydrated ethanol as the solvent. A broad range of organic thiocyanates and selenocyanates was synthesized under these conditions (results presented in the Supplementary Materials).
Table 1.
Evaluation of selenides C1–6 as catalysts in the thiocyanation of benzyl bromide [a].
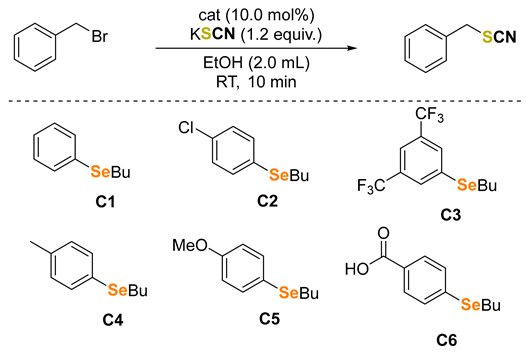
| ||
| Entry | Catalyst | Yield of Benzyl Thiocyanate (%) [b] |
| 1 | - | 17 |
| 2 | C1 | 25 |
| 3 | C2 | 25 |
| 4 | C3 | 20 |
| 5 | C4 | 32 |
| 6 | C5 | 37 |
| 7 | C6 | 28 |
[a] Reaction conditions: benzyl bromide (0.5 mmol), catalyst C1–6 (0.05 mmol), KSCN (0.6 mmol) in dry EtOH (2.0 mL). Ten minutes of reaction at 25 ± 2 °C (water bath) stirring at a constant rate of 360 rpm. [b] Yields for pure and isolated products.
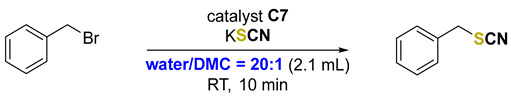
Inspired by recent reports on the use of organochalcogenides, especially organochalcogenium salts, as chalcogen bond activators in organic transformations [8,9,10,11,12,13,14,15,16], we decided to investigate whether selenonium salt C7 could catalyze the desired reaction. We compared its activity against C5, which was the best catalyst tested previously. Additionally, to make the protocol more attractive, we conducted the experiments using water as a solvent. Selenonium salt C7 showed excellent activity, delivering the product at a 53% yield in only 10 min of reaction time, while reactions without a catalyst or with C5 produced hardly any product under the same conditions (Scheme 1). It was possible to reduce the catalyst load to 5 mol %, but the reaction time had to be increased to achieve a 50% yield of the product.
Scheme 1.

Evaluation of selenonium salt C7 as a catalyst.
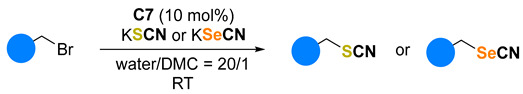
One major issue with the procedure using only water as a solvent is its reproducibility. Much more reliable results were found using a 20:1 mixture of water and dimethyl carbonate (DMC) as the solvent. DMC and benzyl bromide are denser than water, so they produce an organic substrate-rich phase, facilitating the mass transport process. Nevertheless, DMC is recognized as an environmentally friendly solvent [40]. The addition of 10 mol% of C7 to a flask containing benzyl bromide, 1.2 equivalents of KSCN in a 20:1 mixture of water and DMC produced much more product after 10 min than the uncatalyzed reaction (Table 2, entries 1 and 2). Increasing the amount of KSCN to 2.0 equivalents proved optimal, as the result obtained did not change even after extending the reaction time to 1 h (Entries 3 and 4). Catalytic activity was also observed when reducing the amount of C7 to 5.0 and 2.5 mol% (Entries 5 and 6 respectively). However, replacing benzyl bromide with benzyl chloride or iodide did not result in good yields (results not shown).
Table 2.
Optimization of the reaction conditions using catalyst C7. [a].
| |||
| Entry | C7 (mol%) | KSCN (equiv.) | Yield (%) [b] |
| 1 | 0.0 | 1.2 | 5 ± 2 |
| 2 | 10.0 | 1.2 | 41 ± 1 |
| 3 | 10.0 | 2.0 | 86 ± 2 |
| 4 [c] | 10.0 | 2.0 | 85 ± 2 |
| 5 | 5.0 | 2.0 | 37 ± 2 |
| 6 | 2.5 | 2.0 | 13 ± 2 |
[a] Reaction conditions: benzyl bromide (0.5 mmol), catalyst C7 (0.0125 to 0.05 mmol), KSCN (0.6 to 1.0 mmol) in a mixture of water:DMC = 20:1 (2.1 mL). Ten minutes of reaction at 25 ± 2 °C (water bath) stirring at a constant rate of 360 rpm. [b] Yields for pure and isolated products. [c] 60 min of reaction.
Evidence for the activation of benzyl bromide by the Lewis acidity of the selenonium salt C7 through a chalcogen bond interaction (Scheme 2a) was observed by 1H NMR. A small, but noticeable signal shift of hydrogen atoms (Ha) adjacent to the selenonium center was detected when benzyl bromide was added to a solution of C7 in CDCl3. Upon the addition of benzyl bromide, the chemical shift of those hydrogens became lower, as expected, and was directly dependent on the amount of Lewis base added (Scheme 2b). Conversely, the 1H NMR chemical shift observed for the methylene group of benzyl bromide (Hb) increased by the same magnitude due to the interaction with the selenonium salt. The largest shift was detected when 0.5 equivalents of BnBr were added relative to C7. Adding an excess of BnBr resulted in the chemical shift of Hb being almost identical to that of the pure compound. Inconclusive results were obtained from the 77Se NMR experiments. Although a very small chemical shift was detected, prolonged exposure to benzyl bromide resulted in the decomposition of the selenonium salt.
Scheme 2.

(a) Chalcogen bond formation between selenonium salt and benzyl bromide. (b) Correlation of the 1H NMR chemical shift of hydrogens adjacent to selenium and concentration of benzyl bromide.
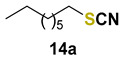
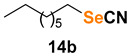
Finally, we turned our efforts to broaden the variety of substrates used in this transformation (Table 3). Representative bromides were converted into thiocyanates or selenocyanates upon treatment with KSCN or KSeCN in a mixture of water and DMC catalyzed by selenonium salt C7. For benzyl bromide and its congeners assembled with electron-withdrawing groups, the corresponding thio- and selenocyanates were prepared with good to excellent yields after 10 min of reaction (entries 1–7). In most cases, the selenocyanates were produced in better yields, and product formation was not drastically affected by the position of the substituent group. On the other hand, for benzyl bromides bearing electron-donating groups, the reaction time had to be extended to 60 min to achieve better results (entries 8–10). Sterically hindered, heteroaromatic, and α-carbonyl bromides were also satisfactorily converted to the desired products (entries 11–13). The only substrate that did not react under these conditions was 1-bromooctane. Even after 24 h of reaction, no product could be detected (entry 14).
Table 3.
Substrate scope for the preparation of thio- and selenocyanates catalyzed by C7. [a].
| |||||
| Entry |
Time
(min) |
Product |
Yield
(%) [b] |
Product |
Yield
(%) [b] |
| 1 | 10 |
|
86 |
|
94 |
| 2 | 10 |
|
86 |
|
97 |
| 3 | 10 |
|
86 |
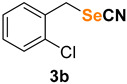
|
87 |
| 4 | 10 |
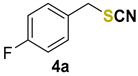
|
84 |
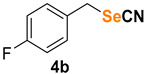
|
74 |
| 5 | 10 |
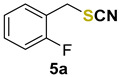
|
61 |
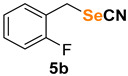
|
82 |
| 6 | 10 |
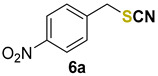
|
76 |
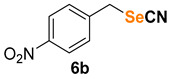
|
52 |
| 7 | 10 |
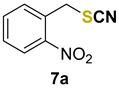
|
68 |
|
82 |
| 8 | 60 |
|
87 |
|
69 |
| 9 | 60 |
|
92 |
|
80 |
| 10 | 60 |
|
60 |
|
81 |
| 11 | 10 |
|
37 |
|
41 |
| 12 | 10 |
|
64 |
|
72 |
| 13 | 10 |
|
60 |
|
70 |
| 14 | 24 h |
|
0 |
|
0 |
[a] Reaction conditions: substrate (0.5 mmol), catalyst C7 (0.05 mmol), KSCN, or KSeCN (1.0 mmol) in a mixture of water/DMC = 20/1 (2.1 mL). Reaction at 25 ± 2 °C (water bath) stirring at a constant rate of 360 rpm. [b] Yields for pure and isolated products.
3. Materials and Methods
3.1. General Remarks
All commercial reagents were used as received. Solvents were analytical grade and purified before use. Moisture-sensitive liquids were transferred using a gas-tight syringe through a rubber septum and stored under argon. Nuclear magnetic resonance (NMR) spectra were determined on Bruker DPX-200 and DRX-400 spectrometers. Chemical shifts (δ) are stated in parts per million (ppm) and coupling constants (J) in Hertz (Hz). Tetramethylsilane (TMS) was used as the internal reference standard for 1H NMR, and CDCl3 for 13C NMR. The following abbreviations are used in the description of NMR data: s = singlet, bs = broad singlet, d = doublet, t = triple, q = quartet, m = multiplet.
3.2. Synthesis of Catalysts C1–5
A dry 50 mL two-neck round-bottom flask equipped with a reflux condenser under argon was charged with the corresponding diselenide (5.0 mmol) and THF (15.0 mL) and stirred at room temperature for 5 min. Then, NaBH4 (770.0 mg, 20.0 mmol) was added followed by EtOH (3.0 mL). As soon as the reaction color faded away, a solution of 1-bromobutane (648 μL, 6.0 mmol) in THF (5.0 mL) was added dropwise and the reaction mixture was allowed to stir at 50 °C for 15 h. After this, the mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic extracts were washed with water (1 × 20 mL), dried over MgSO4, and solvents were evaporated under reduced pressure.
Butyl(phenyl)selane (C1) [41] was purified by silica gel chromatography column (hexanes) to afford a pale-yellow oil, 0.98 g (46% yield). 1H NMR (CDCl3, 400 MHz) δ = 7.49–7.48 (m, 2H), 7.27–7.21 (m, 2H), 2.91 (t, J = 7.4 Hz, 2H), 1,69 (pentet, J = 7.4 Hz, 2H), 1.42 (sextet, J = 7.4 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ = 132.5, 130.9, 129.1, 126.7, 32.4, 27.7, 23.1, 13.7.
Butyl(4-chlorophenyl)selane (C2) [41] was obtained pure after work-up as a pale-yellow oil, 2.3 g (93% yield). 1H NMR (CDCl3, 400 MHz) δ = 7.39 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 8.5 Hz, 2H), 2.89 (t, J = 7.5 Hz, 2H), 1.66 (pentet, J = 7.5 Hz, 2H), 1.41 (sextet, J = 7.5 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ = 133.9, 132.9, 129.3, 129.0, 32.3, 28.2, 23.1, 13.7.
(3,5-bis(trifluoromethyl)phenyl)(butyl)selane (C3) [41] was purified by silica gel chromatography column (hexanes) to afford a pale-yellow oil, 2.27 g (65% yield). 1H NMR (CDCl3, 400 MHz) δ = 7.85 (s, 2H), 7.69 (s, 2H), 3.03 (t, J = 7.4 Hz, 2H), 1.73 (pentet, J = 7.5 Hz, 2H), 1.46 (sextet, J = 7.4 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ = 134.5, 132.2 (q, J = 32.8 Hz), 131.3 (d, J = 3.07 Hz), 123.3 (q, J = 271.3 Hz), 120.3 (pentet, J = 3.75 Hz), 31.9, 28.0, 23.1, 13.6.
Butyl(p-tolyl)selane (C4) [41] was obtained pure after work-up as a colorless oil, 1.5 g (66% yield). 1H NMR (CDCl3, 400 MHz) δ = 7.38 (d, J = 8.0 Hz, 2H), 7.05 (d, J = 8.0 Hz, 2H), 2.86 (t, J = 7,4 Hz, 2H), 2.30 (s, 3H), 1.66 (pentet, J = 7.5 Hz, 2H), 1.41 (sextet, J = 7.5 Hz, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ = 136.8, 133.1, 129.9, 126.9, 32.5, 28.1, 23.1, 21.2, 13.7.
Butyl(4-methoxyphenyl)selane (C5) [42] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a pale-yellow oil, 1.3 g (54% yield). 1H NMR (CDCl3, 400 MHz) δ = 7.45 (d, J = 8.7 Hz, 2H), 6.79 (d, J = 8.7 Hz, 2H), 3.77 (s, 3H), 2.81 (t, J = 7.5 Hz, 2H), 1.63 (pentet, J = 7.6 Hz, 2H), 1.39 (sextet, J = 7.5 Hz, 2H), 0.88 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ = 159.3, 135.6, 120.4, 114.8, 55.4, 32.5, 28.9, 22.9, 13.7.
3.3. Synthesis of Catalyst 4-(butylselanyl)benzoic Acid (C6)
Prepared as described in reference [39].
3.4. Synthesis of Catalyst Dibutyl(phenyl)selenonium Tetrafluoroborate (C7)
A dry 25 mL one-neck round-bottom flask under argon was charged with Butyl(phenyl)selane C1 (1.07 g, 5.0 mmol) and 1-bromobutane (1.6 mL, 15 mmol). The mixture was stirred until it became homogeneous, and AgBF4 (1.07 g, 5.5 mmol) was added. After stirring for 6 h at room temperature in the dark, dichloromethane (3.0 mL) was added. After 5 min the mixture was filtered through a pad of celite, activated charcoal was added to the solution, and then it was filtered again through a pad of celite. The solvent was evaporated under reduced pressure and the product was washed with diethyl ether (3.0 mL). After decantation, the solvent was removed with the aid of a pipette. This step was repeated three times, and then the resulting product was dried in a high vacuum pump. Catalyst C7 was obtained as a viscous colorless oil, 0.84 g (47% yield) [8]. 1H NMR (CDCl3, 400 MHz) δ = 7.88–7.86 (m, 2H), 7.74–7.67 (m, 3H), 3.86–3.79 (m, 2H), 3.73–3.66 (m, 2H), 1.79–1.59 (m, 4H), 1.49–1.42 (m, 4H), 0.89 (t, J = 7.3 Hz, 6H). 13C NMR (CDCl3, 100 MHz) δ = 133.8, 131.7, 131.4, 122.2, 43.1, 27.5, 22.4, 13.44. 77Se NMR (CDCl3, 76 MHz) δ = 399.6.
3.5. Synthesis of Thio- and Selenocyanates
A test tube was charged with catalyst C7 (18 mg, 0.05 mmol), dimethyl carbonate (0. 1 mL), and the corresponding substrate (0.5 mmol). The mixture was stirred until it became homogeneous, and then KSCN (97.2 mg, 1.0 mmol) or KSeCN (144.1 mg, 1.0 mmol) diluted in water (2.0 mL) was added. The reaction mixture was stirred at 25 ± 2 °C (water bath) at a constant rate of 360 rpm for the time indicated in Table 3. Then, the mixture was extracted with AcOEt (3 × 10.0 mL) and the combined organic phases were washed with water (3 × 10.0 mL) and brine (3 × 10.0 mL), dried over MgSO4, and evaporated under reduced pressure. Purification was performed by a silica gel chromatography column with mixtures of hexanes and AcOEt.
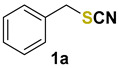
(thiocyanatomethyl)benzene (1a) [33] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a pale-yellow solid, 64.2 mg (86% yield); m.p. = 39.0–40.0 °C; 1H NMR (CDCl3, 400 MHz) δ = 7.39–7.32 (m, 5H), 4.12 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 134.5, 129.2, 129.1, 128.9, 112.1, 38.4.
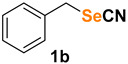
(selenocyanatomethyl)benzene (1b) [35] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a white solid, 92.2 mg (94% yield); m.p. = 67.5–69.0 °C (lit. = 71.0–73.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.37–7.35 (m, 5H), 4.30 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 135.6, 129.3, 129.2, 128.9, 102.0, 32.9.
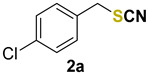
1-chloro-4-(thiocyanatomethyl)benzene (2a) [33] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow oil, 79.0 mg (86% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.36–7.34 (m, 2H), 7.29–7.27 (m, 2H), 4.09 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 130.0, 131.1, 130.4, 129.4, 111.8, 37.6.
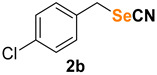
1-chloro-4-(selenocyanatomethyl)benzene (2b) [35] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow solid, 111.8 mg (97% yield); m.p. = 55.0–55.5 °C (lit. = 56.0–58.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.35–7.28 (m, 4H), 4.23 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 134.8, 134.3, 130.5, 129.5, 101.7, 31.9.
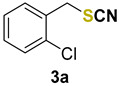
1-chloro-2-(thiocyanatomethyl)benzene (3a) [33] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow oil, 79.0 mg (86% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.44–7.39 (m, 2H), 7.34–7.27 (m, 2H), 4.23 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 134.3, 132.4, 131.3, 130.6, 130.2, 127.6, 36.3.
1-chloro-2-(selenocyanatomethyl)benzene (3b) [43] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow oil, 100.3 mg (87% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.42–7.37 (m, 2H), 7.29–7.25 (m, 2H), 4.31 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 134.1, 133.8, 130.9, 130.3, 130.1, 127.5, 101.8, 30.6.
1-fluoro-4-(thiocyanatomethyl)benzene (4a) [44] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow oil, 70.2 mg (84% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.36–7.32 (m, 2H), 7.09–7.05 (m, 2H), 4,12 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 163.0 (d, J = 246.9 Hz), 130.9 (d, J = 8.6 Hz); 130.4 (d, J = 3.4 Hz), 116.3 (d, J = 21.9 Hz), 119.9, 37.7.
1-fluoro-4-(selenocyanatomethyl)benzene (4b) [45]was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a white solid, 79.2 mg (74% yield); m.p. = 62.4–62.5 °C (lit. = 64.0–65.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.36–7.32 (m, 2H), 7.07–7.03 (m, 2H), 4.26 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 162.8 (d, J = 246.8 Hz), 131.6 (d, J = 3.2 Hz), 130.9 (d, J = 8.5 Hz), 116.2 (d, J = 21.6 Hz), 101.9, 32.0.
1-fluoro-2-(thiocyanatomethyl)benzene (5a) [46] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow oil, 60.0 mg (61% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.39–7.33 (m, 2H), 7.19–7.09 (m, 2H), 4.19 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 160.9 (d, J = 247.7 Hz), 131.2, 131.1 (d, J = 3.1 Hz), 124.9 (d, J = 3.7 Hz), 122.1 (d, J = 14.5 Hz), 116.1 (d, J = 20.7 Hz), 111.8, 31.8 (d, J = 3.5 Hz).
1-fluoro-2-(selenocyanatomethyl)benzene (5b) [47] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a white solid, 87.8 mg (82% yield); m.p. = 60.5–61.5 °C (lit. = 65.0–66.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.38–7.26 (m, 2H), 7.17–7.07 (m, 2H), 4.26 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 160.8 (d, J = 247.8 Hz), 131.0 (d, J = 3.1 Hz), 130.8 (d, J = 8.5 Hz), 124.8 (d, J = 3.8 Hz), 127.4 (d, J = 14.3 Hz), 115.9 (d, J = 20.6 Hz), 101.7, 25.6 (d, J = 3.6 Hz).
1-nitro-4-(thiocyanatomethyl)benzene (6a) [48] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a white solid, 73.8 mg (76% yield); m.p. = 82.2–84.4 °C; 1H NMR (CDCl3, 400 MHz) δ = 8.25 (d, J = 8.7 Hz, 2H), 7.58 (d, J = 8.7 Hz, 2H), 4.23 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 148.1, 141.9, 130.1, 124.4, 11.2, 36.9.
1-nitro-4-(selenocyanatomethyl)benzene (6b) [35] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a yellow solid, 62.7 mg (52% yield); m.p. = 112.9–113.1 °C (lit. = 122.0–124.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 8.24 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.6 Hz, 2H), 4.31 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 148.0, 143.4, 130.1, 124.5, 100.9, 31.1.
1-nitro-2-(thiocyanatomethyl)benzene (7a) [49] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a yellow solid, 66.0 mg (68% yield); m.p. = 67.0–68.0 °C (lit. = 69.0–71.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 8.24–8.22 (m, 1H), 7.74–7.70 (m, 1H), 7.63–7.58 (m, 1H), 7.56–7.54 (m, 1H), 4.46 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 147.1, 134.7, 132.6, 131.2, 130.5, 126.3, 112.2, 36.7.
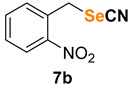
1-nitro-2-(selenocyanatomethyl)benzene (7b) [35] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a yellow solid, 98.9 mg (82% yield); m.p. = 74.0–74.5 °C (lit. = 72.0–74.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 8.23–8.19 (m, 1H), 7.73–7.69 (m, 1H), 7.59–7.54 (m, 2H), 4.46 (s, 2H), 13C NMR (CDCl3, 100 MHz) δ = 146.6, 134.9, 133.3, 132.1, 130.1, 126.2, 102.8, 30.7.
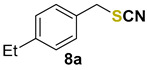
1-ethyl-4-(thiocyanatomethyl)benzene (8a) [50] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a yellow oil, 77.1 mg (87% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.26 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 8.2 Hz, 2H), 4.11 (s, 2H), 2.64 (q, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ = 145.2, 131.6, 129.1, 128.7, 112.3, 38.4, 28.7, 15.5.
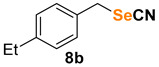
1-ethyl-4-(selenocyanatomethyl)benzene (8b) [35] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a yellow oil, 77.3 mg (69% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.27 (d, J = 7.6 Hz, 2H), 7.18 (d, J = 7.6 Hz, 2H), 4.29 (s, 2H), 2.65 (q, J = 7.6 Hz, 2H), 1.23 (t, J = 7.6 Hz, 2H). 13C NMR (CDCl3, 100 MHz) δ = 145.1, 132.6, 129.2, 128.8, 102.3, 33.0, 28.7, 15.5.
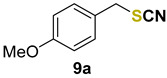
1-methoxy-4-(thiocyanatomethyl)benzene (9a) [33] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow oil, 82.4 mg (92% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.28–7.25 (m, 2H), 6.90–6.88 (m, 2H), 4.12 (s, 2H), 3.79 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ = 160.1, 130.4, 126.4, 114.6, 112.3, 55.4, 38.3.
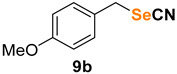
1-methoxy-4-(selenocyanatomethyl)benzene (9b) [35] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow solid, 90.4 mg (80% yield); m.p. = 53.5–55.0 °C (lit. = 52.0–54.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.29–7.26 (m, 2H), 6.89–6.85 (m, 2H), 4.27 (s, 2H), 3.79 (m, 3H). 13C NMR (CDCl3, 100 MHz) δ = 159.9, 130.4, 127.4, 114.6, 102.4, 55.4, 32.9.
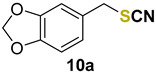
5-(thiocyanatomethyl)benzo[d][1,3]dioxole (10a) [51] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a colorless oil, 58.0 mg (60% yield); 1H NMR (CDCl3, 400 MHz) δ = 6.83–6.77 (m, 3H), 5.97 (s, 2H), 4.09 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 148.3, 127.9, 123.0, 112.2, 109.2, 108.7, 101.6, 38.8.
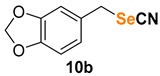
5-(selenocyanatomethyl)benzo[d][1,3]dioxole (10b) [52] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow solid, 97.2 mg (81% yield); m.p. = 70.0–71.5 °C; 1H NMR (CDCl3, 400 MHz) δ = 6.82–6.81 (m, 2H), 6.77–6.75 (m, 1H), 5.96 (s, 2H), 4.23 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 148.2, 148.1, 129.1, 122.9, 109.2, 108.7, 102.2, 101.5, 33.4.
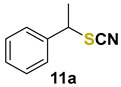
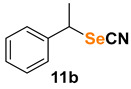
(1-thiocyanatoethyl)benzene (11a) [53] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a yellow oil, 30.2 mg (37% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.39–7.32 (m, 5H), 4.60 (q, J =7.0 Hz, 1H), 1.87 (d, J =7.0, 3H). 13C NMR (CDCl3, 100 MHz) δ = 139.2, 129.3, 129.2, 127.3, 111.9, 48.7, 22.2.
(1-selenocyanatoethyl)benzene (11b) [35] was purified by silica gel chromatography column (hexanes/AcOEt = 95/5) to afford a yellow oil, 43.1 mg (41% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.38–7.30 (m, 5H), 4.90 (q, J = 6,8 Hz, 1H), 2.04 (d, J = 6.9, 3H) 13C NMR (CDCl3, 100 MHz) δ = 139.6, 129.2, 128.9, 127.2, 102.7, 45.7, 22.9.
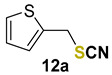
2-(thiocyanatomethyl)thiophene (12a) [33] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow oil, 49.7 mg (64% yield); 1H NMR (CDCl3, 400 MHz) δ = 7.33–7.31 (m, 1H), 7.12–7.11 (m, 1H), 6.99–6.96 (m, 1H), 4.39 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 136.2, 128.8, 127.5, 127.4, 111.8, 33.3.
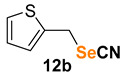
2-(selenocyanatomethyl)thiophene (12b) [54] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow solid, 72.8 mg (72% yield); m.p. = 53.0–55.0 °C (lit. = 48.0–50.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.30–7.29 (m, 1H), 7.12- 7.11 (m, 1H), 6.97- 6.95 (m, 1H), 4.54 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 137.6, 128.7, 127.6, 127.2, 102.1, 27.0.
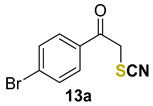
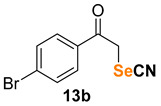
1-(4-bromophenyl)-2-thiocyanatoethan-1-one (13a) [55] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a white crystalline solid, 76.8 mg (60% yield); m.p. = 145.5–146.6 °C (lit. = 148.8–149.2 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.80 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 8.6 Hz, 2H), 4.69 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 190.1, 132.9, 132.8, 130.5, 130.0, 111.7, 42.8.
1-(4-bromophenyl)-2-selenocyanatoethan-1-one (13b) [56] was purified by silica gel chromatography column (hexanes/AcOEt = 9/1) to afford a yellow solid, 106.1 mg (70% yield); m.p. = 138.4–138.6 °C (lit. = 144.0–145.0 °C); 1H NMR (CDCl3, 400 MHz) δ = 7.82 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 8.6 Hz, 2H), 4.86 (s, 2H). 13C NMR (CDCl3, 100 MHz) δ = 192.4, 132.8, 132.7, 130.7, 130.3, 101.7, 38.1.
4. Conclusions
In this study, we developed a simple and efficient protocol to produce a range of thio- and selenocyanates using a sustainable solvent mixture of water and dimethyl carbonate. The reaction was only possible with the activation of substrates using a catalytic amount of selenonium salt. Our experimental evidence showed that selenonium salts are superior catalysts compared with organoselenides and that activation occurred through chalcogen bond interaction, as demonstrated by 1H NMR experiments. We successfully demonstrated the synthesis of thio- and selenocyanates with various electron-withdrawing or electron-donating groups, as well as sterically hindered, heteroaromatic, and α-carbonyl substrates.
Acknowledgments
The authors acknowledge the financial support provided by Fundação de Amparo à Pesquisa do Estado de Minas Gerais—FAPEMIG, Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq (A.Y.B.Á. fellowship) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Capes (P.R.O.C. fellowship). The authors also thank Laboratório de Ressonância Magnética de Alta Resolução (LAREMAR) for granting permission to use their NMR spectrometers.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28073056/s1. Result for uncatalyzed reactions and 1H. 13C NMR spectra for products.
Author Contributions
Conceptualization, E.E.A. and A.Y.B.Á.; investigation, A.Y.B.Á. and P.R.O.C.; writing—review and editing, E.E.A., A.Y.B.Á. and P.R.O.C.; supervision, E.E.A.; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of catalysts C1–7 are available from the authors.
Funding Statement
This research was funded by Fundação de Amparo à Pesquisa do Estado de Minas Gerais—FAPEMIG. Grant number APQ-00349-22.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Clark T., Hennemann M., Murray S.J., Politzer P. Halogen bonding: The σ-hole. J. Mol. Model. 2007;13:291. doi: 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- 2.Wonner P., Vogel L., Duser M., Gomes L., Kniep F., Mallick B., Werz D.B., Huber S.M. Carbon–Halogen Bond Activation by Selenium-Based Chalcogen Bonding. Angew. Chem. Int. Ed. 2017;56:12009. doi: 10.1002/anie.201704816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Politzer P., Murray J.S., Clark T., Resnati G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017;19:32166. doi: 10.1039/C7CP06793C. [DOI] [PubMed] [Google Scholar]
- 4.Aakeroy C.B., Bryce D.L., Desiraju G.R., Frontera A., Legon A.C., Nicotra F., Rissanen K., Scheiner S., Terraneo G., Metrangolo P., et al. Definition of the chalcogen bond (IUPAC Recommendations 2019) Pure Appl. Chem. 2019;91:1889. doi: 10.1515/pac-2018-0713. [DOI] [Google Scholar]
- 5.Zhou B., Gabbaï F.P. Redox-controlled chalcogen-bonding at tellurium: Impact on Lewis acidity and chloride anion transport properties. Chem. Sci. 2020;11:7495. doi: 10.1039/D0SC02872J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou B., Gabbaï F.P. Lewis Acidic Telluronium Cations: Enhanced Chalcogen-Bond Donor Properties and Application to Transfer Hydrogenation Catalysis. Organometallics. 2021;40:2371. doi: 10.1021/acs.organomet.1c00279. [DOI] [Google Scholar]
- 7.Novikov A.S., Bolotin D.S. Halonium, chalconium, and pnictonium salts as noncovalent organocatalysts: A computational study on relative catalytic activity. Org. Biomol. Chem. 2022;20:7632. doi: 10.1039/D2OB01415G. [DOI] [PubMed] [Google Scholar]
- 8.Lenardão E.J., Mendes S.R., Ferreira P.C., Perin G., Silveira C.C., Jacob R.G. Selenium- and tellurium-based ionic liquids and their use in the synthesis of octahydroacridines. Tetrahedron Lett. 2006;47:7439. doi: 10.1016/j.tetlet.2006.08.049. [DOI] [Google Scholar]
- 9.Lenardão E.J., Borges E.L., Mendes S.R., Perin G., Jacob R.G. Selenonium ionic liquid as an efficient catalyst for the synthesis of thioacetals under solvent-free conditions. Tetrahedron Lett. 2008;49:1919. doi: 10.1016/j.tetlet.2008.01.096. [DOI] [Google Scholar]
- 10.Lenardão E.J., Feijó J.O., Thurow S., Perin G., Jacob R.G., Silveira C.C. Selenonium ionic liquid as efficient catalyst for the Baylis–Hillman reaction. Tetrahedron Lett. 2009;50:5215. doi: 10.1016/j.tetlet.2009.06.132. [DOI] [Google Scholar]
- 11.He X., Wang X., Tse Y.-L., Ke Z., Yeung Y.-Y. Applications of Selenonium Cations as Lewis Acids in Organocatalytic Reactions. Angew. Chem. Int. Ed. 2018;57:12869. doi: 10.1002/anie.201806965. [DOI] [PubMed] [Google Scholar]
- 12.He X., Wang X., Tse Y.-L., Ke Z., Yeung Y.-Y. Bis-selenonium Cations as Bidentate Chalcogen Bond Donors in Catalysis. ACS Catal. 2021;11:12632. doi: 10.1021/acscatal.1c03622. [DOI] [Google Scholar]
- 13.Okuno K., Nakamura T., Shirakawa S. Asymmetric Catalysis of Chiral Bifunctional Selenides and Selenonium Salts Bearing a Urea Group. Asian J. Org. Chem. 2021;10:655. doi: 10.1002/ajoc.202100001. [DOI] [Google Scholar]
- 14.Il’in M.V., Novikov A.S., Bolotin D.S. Sulfonium and Selenonium Salts as Noncovalent Organocatalysts for the Multicomponent Groebke–Blackburn–Bienaymé Reaction. J. Org. Chem. 2022;87:10199. doi: 10.1021/acs.joc.2c01141. [DOI] [PubMed] [Google Scholar]
- 15.Liao L., Zhao X. Modern Organoselenium Catalysis: Opportunities and Challenges. Synlett. 2021;32:1262. doi: 10.1055/a-1506-5532. [DOI] [Google Scholar]
- 16.Ángel A.Y.B., Campos P.R.O., Alberto E.E. Synthetic application of chalcogenonium salts: Beyond sulfonium. Org. Biomol. Chem. 2023;21:223. doi: 10.1039/D2OB01822E. [DOI] [PubMed] [Google Scholar]
- 17.Rao C.V., Wang C.-Q., Simi B., Rodriguez J.G., Cooma I., El-Bayoumy K., Reddy B.S. Chemoprevention of Colon Cancer by a Glutathione Conjugate of 1,4-Phenylenebis(methylene)selenocyanate, a Novel Organoselenium Compound with Low Toxicity. Cancer Res. 2001;61:3647. [PubMed] [Google Scholar]
- 18.Banerjee K., Padmavathi G., Bhattarcherjee D., Saha S., Kunnumakkara A.B., Bhabak K.P. Potent anti-proliferative activities of organochalcogenocyanates towards breast cancer. Org. Biomol. Chem. 2018;16:8769. doi: 10.1039/C8OB01891J. [DOI] [PubMed] [Google Scholar]
- 19.Banerjee K., Bhattarcherjee D., Mahato S.K., Sufian A., Bhabak K.P. Selenocyanates: Ionic Organoselenium Compounds with Efficient Peroxide Scavenging Activities. Inorg. Chem. 2021;60:12984. doi: 10.1021/acs.inorgchem.1c01410. [DOI] [PubMed] [Google Scholar]
- 20.Szajnman S.H., Yan W., Bailey B.N., Docampo R., Elhalem E., Rodriguez J.B. Design and Synthesis of Aryloxyethyl Thiocyanate Derivatives as Potent Inhibitors of Trypanosoma cruzi Proliferation. J. Med. Chem. 2000;43:1826. doi: 10.1021/jm9905007. [DOI] [PubMed] [Google Scholar]
- 21.Alcolea V., Pérez-Silanes S. Selenium as an interesting option for the treatment of Chagas disease: A review. Eur. J. Med. Chem. 2020;206:112673. doi: 10.1016/j.ejmech.2020.112673. [DOI] [PubMed] [Google Scholar]
- 22.Rubio-Hernández M., Alcolea V., Pérez-Silanes S. Potential of sulfur-selenium isosteric replacement as a strategy for the development of new anti-chagasic drugs. Acta Tropica. 2022;233:106547. doi: 10.1016/j.actatropica.2022.106547. [DOI] [PubMed] [Google Scholar]
- 23.Castanheiro T., Suffert J., Donnard M., Gulea M. Recent advances in the chemistry of organic thiocyanates. Chem. Soc. Rev. 2016;45:494. doi: 10.1039/C5CS00532A. [DOI] [PubMed] [Google Scholar]
- 24.Zhang M., Lin J.-H., Xiao J.-C. HCF2Se/HCF2S Installation by Tandem Substitutions from Alkyl Bromides. J. Org. Chem. 2021;86:13153. doi: 10.1021/acs.joc.1c01718. [DOI] [PubMed] [Google Scholar]
- 25.Karmaker P.G., Huo F. Organic Selenocyanates: Rapid Advancements and Applicationsin the Field of Organic Chemistry. Asian J. Org. Chem. 2022;11:e202200226. doi: 10.1002/ajoc.202200226. [DOI] [Google Scholar]
- 26.Alfuth J., Jeannin O., Fourmigué M. Topochemical, Single-Crystal-to-Single-Crystal [2+2] Photocycloadditions Driven by Chalcogen-Bonding Interactions. Angew. Chem. Int. Ed. 2022;61:e202206249. doi: 10.1002/anie.202206249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azaroon M., Kiasat A.R. Crown ether functionalized magnetic hydroxyapatite as eco-friendly microvessel inorganic-organic hybrid nanocatalyst in nucleophilic substitution reactions: An approach to benzyl thiocyanate, cyanide, azide and acetate derivatives. Appl. Organomet. Chem. 2018;32:e4046. doi: 10.1002/aoc.4046. [DOI] [Google Scholar]
- 28.Goodajdar B.M., Akbari F., Davarpanah J. PEG-DIL-based MnCl42−: A novel phase transfer catalyst for nucleophilic substitution reactions of benzyl halides. Appl. Organomet. Chem. 2018;33:4647. doi: 10.1002/aoc.4647. [DOI] [Google Scholar]
- 29.Shi X.-L., Chen Y., Hu Q., Meng H., Duan P. Fiber-Supported Poly(quaternaryammonium Bromide)s as Supported-Phase Transfer Catalysts in the Spinning Basket Reactor. Ind. Eng. Chem. Res. 2018;57:7450. doi: 10.1021/acs.iecr.8b01302. [DOI] [Google Scholar]
- 30.Shen J.-C., Jiang W.-L., Guo W.-D., Qi Q.-Y., Ma D.-L., Lou X., Shen M., Hu B., Yang H.-B., Zhao X. A rings-in-pores net: Crown ether-based covalent organic frameworks for phase-transfer catalysis. Chem. Commun. 2020;56:595. doi: 10.1039/C9CC07639E. [DOI] [PubMed] [Google Scholar]
- 31.Melillo A., Kiani A., Schettini R., Acocella M.R. Carbon black intercalation compound as catalyst for unprecedent phase-transfer-catalyzed nucleophilic substitution (SN2) in water. Mol. Catal. 2023;537:112951. doi: 10.1016/j.mcat.2023.112951. [DOI] [Google Scholar]
- 32.Guo W., Tan W., Zhao M., Zheng L., Tao K., Chen D., Fan X. Direct Photocatalytic S–H Bond Cyanation with Green “CN” Source. J. Org. Chem. 2018;83:6580. doi: 10.1021/acs.joc.8b00887. [DOI] [PubMed] [Google Scholar]
- 33.Wu D., Duan Y., Liang K., Yin H., Chen F.-X. AIBN-initiated direct thiocyanation of benzylic sp3 C–H with N-thiocyanatosaccharin. Chem. Commun. 2021;57:9938. doi: 10.1039/D1CC04302A. [DOI] [PubMed] [Google Scholar]
- 34.Zhao X., Ji L., Gao Y., Sun T., Qiao J., Li A., Lu K. Visible-Light-Promoted Selenocyanation of Cyclobutanone Oxime Esters Using Potassium Selenocyanate. J. Org. Chem. 2021;86:11399. doi: 10.1021/acs.joc.1c00893. [DOI] [PubMed] [Google Scholar]
- 35.Yu F., Li C., Wang C., Zhang H., Cao Z.-Y. (1-Selenocyanatoethyl)benzene: A Selenocyanation Reagent for Site-Selective Selenocyanation of Inert Alkyl C(sp3)–H Bonds. Org. Lett. 2021;23:7156. doi: 10.1021/acs.orglett.1c02564. [DOI] [PubMed] [Google Scholar]
- 36.Tao S., Jiang L., Du Y. Electrophilic Oxythio/selenocyanation of o-Alkenyl Benzoic Acids: Divergent Synthesis of Thio/selenocyanated Isobenzofuranones and Isocoumarins. Asian J. Org. Chem. 2022;11:e202200595. doi: 10.1002/ajoc.202200595. [DOI] [Google Scholar]
- 37.Wang J., Wang Y.-Z., Liu Y.-J., Yan X.-X., Yan Y.-H., Chao S.-J., Shang X., Ni T., Zhou P.-X. Synthesis of Isoquinolylselenocyanates and Quinolylselenocyanates via Electrophilic Selenocyanogen Cyclization Induced by Pseudohalogen (SeCN)2 Generated in situ. Adv. Synth. Catal. 2022;364:187. doi: 10.1002/adsc.202101169. [DOI] [Google Scholar]
- 38.Banliat A.D., Grollier K., Damond A., Billard T., Dagousset G., Magnier E., Pégot B. Solvent free nucleophilic selenocyanation with [bmim][SeCN]. Direct access to perfluoroalkylselenide compounds. Tetrahedron. 2021;101:132507. doi: 10.1016/j.tet.2021.132507. [DOI] [Google Scholar]
- 39.Martins N.S., Ángel A.Y.B., Anghinoni J.M., Lenardão E.J., Barcellos T., Alberto E.E. From Stoichiometric Reagents to Catalytic Partners: Selenonium Salts as Alkylating Agents for Nucleophilic Displacement Reactions in Water. Adv. Synth. Catal. 2021;364:87. doi: 10.1002/adsc.202100797. [DOI] [Google Scholar]
- 40.Alder C.M., Hayler J.D., Henderson R.K., Redman A.M., Shukla L., Shuster L.E., Sneddon H.F. Updating and further expanding GSK’s solvent sustainability guide. Green Chem. 2016;18:3879. doi: 10.1039/C6GC00611F. [DOI] [Google Scholar]
- 41.Chen Q., Wang P., Yan T., Cai M. A highly efficient heterogeneous ruthenium(III)-catalyzed reaction of diaryl diselenides with alkyl halides leading to unsymmetrical diorganyl selenides. J. Organomet. Chem. 2017;840:38. doi: 10.1016/j.jorganchem.2017.04.004. [DOI] [Google Scholar]
- 42.Saba S., Botteselle G.V., Godoi M., Frizon T.E.A., Galetto F.Z., Rafique J., Braga A.L. Copper-catalyzed synthesis of unsymmetrical diorganyl chalcogenides (Te/Se/S) from boronic acids under solvent-free conditions. Molecules. 2017;22:1367. doi: 10.3390/molecules22081367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Redon S., Vanelle P. Nucleophilic Selenocyanation from Selenium Dioxide. Synthesis. 2023;55:510. doi: 10.1055/a-1938-2443. [DOI] [Google Scholar]
- 44.Jiang C., Chen P., Liu G. Copper-catalyzed benzylic C–H bond thiocyanation: Enabling late-stage diversifications. CCS Chem. 2021;3:1884. doi: 10.31635/ccschem.020.202000435. [DOI] [Google Scholar]
- 45.Nasim M.J., Witek K., Kincses A., Abdin A.Y., Zeslawska E., Marc M.A., Gajdacs M., Spengler G., Nitek W., Latacz G., et al. Pronounced activity of aromatic selenocyanates against multidrug resistant ESKAPE bacteria. New J. Chem. 2019;43:6021. doi: 10.1039/C9NJ00563C. [DOI] [Google Scholar]
- 46.Kiasat A.R., Badri R., Sayyahi S. A facile and convenient method for synthesis of alkyl thiocyanates under homogeneous phase transfer catalyst conditions. Chin. Chem. Lett. 2008;19:1301. doi: 10.1016/j.cclet.2008.07.019. [DOI] [Google Scholar]
- 47.Iwaoka M., Katsuda T., Komatsu H., Tomoda S. Experimental and theoretical studies on the nature of weak nonbonded interactions between divalent selenium and halogen atoms. J. Org. Chem. 2005;70:321. doi: 10.1021/jo048436a. [DOI] [PubMed] [Google Scholar]
- 48.Bound D.J., Bettadaiah B.K., Srinivas P. Microwave-assisted synthesis of alkyl thiocyanates. Synth. Commun. 2013;43:1138. doi: 10.1080/00397911.2011.622848. [DOI] [Google Scholar]
- 49.Mokhtari B., Azadi R., Rahmani-Nezhad S. In situ-generated N-thiocyanatosuccinimide (NTS) as a highly efficient reagent for the one-pot thiocyanation or isothiocyanation of alcohols. Tetrahedron Lett. 2009;50:6588. doi: 10.1016/j.tetlet.2009.09.066. [DOI] [Google Scholar]
- 50.Priestap H.A. Effect of some benzyl thiocyanate analogs on tetracycline production. J. Antibiot. 1987;40:1341. doi: 10.7164/antibiotics.40.1341. [DOI] [PubMed] [Google Scholar]
- 51.Drabek J. The synthesis of pyrethrum’s synergists with methylene-and ethylenedioxybenzene. Chem. Pap. 1956;10:357–367. [Google Scholar]
- 52.Begines P., Sevilla-Horrillo L., Puerta A., Puckett R., Bayort S., Lagunes I., Maya I., Padrón J.M., López O., Fernández-Bolaños J.G. Masked phenolic-selenium conjugates: Potent and selective antiproliferative agents overcoming P-gp resistance. Pharmaceuticals. 2020;13:1. doi: 10.3390/ph13110358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rad M.N.S. A simple, one-pot and phosphine-free procedure for thiocyanation of alcohols Using N-(p-toluenesulfonyl) imidazole (TsIm) J. Chem. Res. 2016;40:583. doi: 10.3184/174751916X14736925997854. [DOI] [Google Scholar]
- 54.Lam L.K.T., Ahmed N. Organoselenium Compounds for Cancer Chemoprevention. 2002/01652.15A1. U.S. Patent. 2002 November 7;
- 55.Muthyala M.K., Choudhary S., Kumar A. Synthesis of ionic liquid-supported hypervalent iodine reagent and its application as a ‘catch and release’ reagent for α-substituted acetophenones. RSC Adv. 2014;4:14297. doi: 10.1039/c4ra00063c. [DOI] [Google Scholar]
- 56.Xiao J.-A., Li Y.-C., Cheng X.-L., Chen W.-Q., Cui J.-G., Huang Y.-M., Huang J., Xiao Q., Su W., Yang H. Selenocyanobenziodoxolone: A practical electrophilic selenocyanation reagent and its application for solid-state synthesis of α-carbonyl selenocyanates. Org. Chem. Front. 2019;6:1967. doi: 10.1039/C9QO00358D. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are contained within the article and Supplementary Materials.