

Abstract
The Switch/Sugar Non-Fermenting (SWI/SNF) nucleosome remodeling complexes play important roles in normal development and in the development of various cancers. Core subunits of the SWI/SNF complexes have been shown to have oncogenic roles in acute myeloid leukemia. However, the roles of the unique targeting subunits, including that of Arid2 and Arid1b, in AML leukemogenesis are not well understood. Here, we used conditional knockout mouse models to elucidate their role in MLL-AF9 leukemogenesis. We uncovered that Arid2 has dual roles; enhancing leukemogenesis when deleted during leukemia initiation and yet is required during leukemia maintenance. Whereas, deleting Arid1b in either phase promotes leukemogenesis. Our integrated analyses of transcriptomics and genomic binding data showed that, globally, Arid2 and Arid1b regulate largely distinct sets of genes at different disease stages, respectively, and in comparison, to each other. Amongst the most highly dysregulated transcription factors upon their loss, Arid2 and Arid1b converged on the regulation of Etv4/Etv5, albeit in an opposing manner while also regulating distinct TFs including Gata2,Tcf4, Six4, Irf4 and Hmgn3. Our data demonstrate the differential roles of SWI/SNF subunits in AML leukemogenesis and emphasize that cellular context and disease stage are key in determining their functions during this process.
INTRODUCTION
The Switch/Sugar Non-Fermenting (SWI/SNF) complexes are highly evolutionarily conserved chromatin remodeling complexes1,2. Two of the main human SWI/SNF complexes are BRG1 associated factors, BAF and Polybromo-associated BAF, PBAF. They share a conserved set of nine proteins, including the catalytic ATPase, Brahma-related gene 1 (BRG1) or Brahma (BRM) and are distinguished by unique AT-rich interactive domain-containing (ARID) proteins: ARID1A or ARID1B (BAF250A/B) in the BAF complex and ARID2 (BAF200) in the PBAF complex. Polybromo 1 (PBRM1/BAF180) and bromodomain containing 7 (BRD7) proteins are additionally unique to the PBAF complex3–9. These unique subunits play a role in targeting of the complexes. SWI/SNF complexes remodel chromatin through ATP-dependent disruption of nucleosome position which ultimately affects gene transcription10–12.
SWI/SNF chromatin remodeling complexes play important roles in various normal developmental and differentiation processes. SWI/SNF subunits have been shown to play important roles in hematopoietic stem cell (HSC) self-renewal and lineage differentiation13. Inactivating mutations in SWI/SNF subunits have been found in about 25% of all cancers14,15, including hematological malignancies, and generally suggest a tumor suppressor role14. SWI/SNF mutations in acute myeloid leukemia (AML) are rare. In fact, Brg1 and two other core subunits have been shown to have an oncogenic role in AML maintenance16,17. The roles of the other subunits of the SWI/SNF, including that of the ARID proteins, are not well understood. Arid1b and Arid2 loci have been shown to be targets of the onco-fusion protein, MLL-AF9, and were amongst positive hits in an shRNA screen of MLL-AF9 target genes that are required for leukemia maintenance18. In this study, using conditional knockout models of Arid2 and Arid1b, we showed that deleting Arid2 during leukemia initiation results in leukemia cells with increased leukemogenic potential while it is required for leukemia maintenance. On the other hand, deleting Arid1b during leukemia initiation showed a similar phenotype as that of Arid2, however, during leukemia maintenance its loss also promoted leukemogenesis. We further demonstrated that their differential role in leukemogenesis is mediated by controlling largely unique gene transcription program.
MATERIALS AND METHODS
A full description of mouse experiments, MLL-AF9 leukemia transplantations, flow cytometry and sorting, viral production and transduction, RNA-sequencing, CUT&Tag and ChIP-RTqPCR, colony forming assay, data analysis and statistical methods, list of primers (Table S1), flow cytometry antibodies (Table S2), and CUT&Tag/ChIPqPCR antibodies (Table S3) are included in the supplemental Methods.
RESULTS
ARID1B and ARID2 are mutated in human AML
We mined existing genomic data to examine mutations of ARID2 and ARID1B in human AML. Using AML patient data from the OHSU data set, ARID2 and ARID1B are mutated at a frequency of 0.9% and 0.8%, respectively. ARID2 mutations include frame shift and splice mutations that are predicted by OncoKB to be potential driver mutations, and in-frame insertion and missense mutation with unknown significance. ARID1B mutations include in-frame deletions and missense mutation of unknown significance19 (Figure 1A). Patients with ARID2 or ARID1B mutation have significantly decreased survival (Figure 1B). We additionally analyzed patient survival data from the OSHU and TCGA AML patient cohort after stratifying ARID2 and ARID1B expression (Figure 1C and 1D). We found that patients with low expression level (defined as Z score<−1) of ARID1B have lower overall survival or trend towards lower survival compared to those with high expression level (Z score >1). A similar trend was observed in patients from the TCGA patient cohort with regards to ARID2 expression level, however was not statistically significant in patients from the OSHU cohort. Overall, these data suggest that ARID2 and ARID1B may play a role in human AML.
Figure 1. ARID1B and ARID2 in human AML.
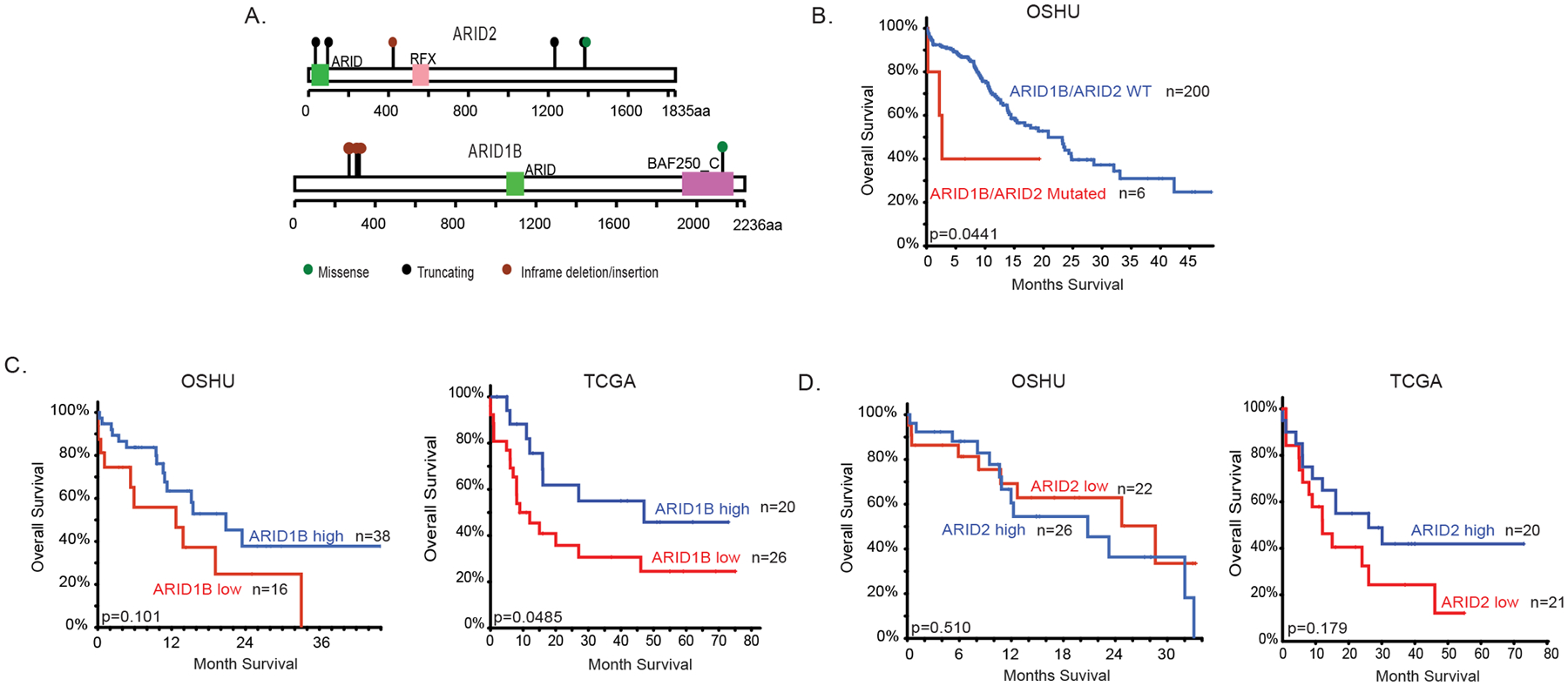
(A) Graphical representation of ARID1B and ARID2 mutations identified in primary AML patient samples in the OSHU AML cohort. (B) Survival curve of de novo non-MRC (Mylodysplasia-Related Changes) AML patients with or without mutation in ARID1B or ARID2 from the OSHU AML cohort. (C,D) Survival curves of de novo non-MRC AML patients in the OSHU AML cohort and primary AML patients from the TCGA AML data set stratified by high and low expression of ARID1B (C) or ARID2 (D). Expression is stratified using Z score from cBioPortal34,35.
Arid2 plays dual roles in MLL-AF9 leukemogenesis
To examine the role of Arid2 and Arid1b in AML leukemogenesis, we obtained a genetic floxed mouse model of Arid2 from European Mutant Mouse Consortium20,21 and a previously published genetic floxed model of Arid1b22. We characterized the effect of losing Arid2 and Arid1b on cellular phenotype of MLL-AF9 in-vitro but did not find any differences in apoptosis, differentiation, or proliferation (Figure S1). To study the effect of the loss of Arid2 on leukemogenesis in-vivo, we first assessed whether loss of Arid2 affects leukemic initiation using the VavCre model. In primary transplantation, we did not observe any differences in overall survival of primary recipients receiving Arid2f/f VavCre (Arid2 KO) and VavCre (control) LSKs transduced with MLL-AF9 (Figure 2A). Disease burden in the primary recipient moribund mice, as indicated by percentage of GFP+ MLL-AF9 cells in BM and SPL and SPL weight, was also similar between Arid2 KO and control recipients (Figure S2A). We further examined whether the loss of Arid2 alters leukemogenesis in the VavCre model by performing limiting dilution secondary transplantations using whole BM from moribund primary recipients (Figure 2A, Figure S2B–D). Overall survival of Arid2 null MLL-AF9 BM cells recipient trended to be decreased from control recipients. In the 100cell transplantation, Arid2 KO secondary recipients had significantly increased percentages of GFP+ cell in the BM and SPL (Figure 2B) along with a modest but significant decrease in SPL weight (Figure 2C). The frequency of leukemia initiating cells between the two cohorts showed a two-fold (range 2–50 folds) increase in leukemia initiating cells frequency (Table 1). These data suggest that loss of Arid2 during leukemia initiation increases the leukemogenic potential of the resulting leukemia cells. We further examined the effect of the loss of Arid2 on leukemia maintenance using the inducible MxCre model. We observed no differences in the latency of primary recipients transplanted with Arid2f/f MxCre (Arid2 KO) and MxCre (control) upon pIpC induction or in disease burden (Figure 2D, Figure S2E). However, to our surprise, upon transplanting un-induced primary leukemia into secondary recipients followed by excision of Arid2 with pIpC, we observed significant prolonged survival in the 100 and 1000cell cohorts, with 40–50% of recipient mice never succumbing to disease at end point (Figure 2E). Mice that did succumb to disease showed significantly decreased percent of GFP+ cells in their BM and SPL (Figure 2F, Figure S2F) and a significantly decreased SPL weight (Figure 2G, Figure S2F). Such differences were not observed in the 30,000cell cohort (Figure S2G). Excision frequency of Arid2 in the BM in both primary and secondary recipients averaged 90%, regardless of mouse model (Figure S2H). Overall, these data suggest that Arid2 plays a dual role in leukemogenesis; its deletion during leukemia initiation results in leukemic cells with enhanced leukemogenic potential but is required during leukemia maintenance.
Figure 2. Arid2 has dual roles in MLL-AF9 leukemogenesis.
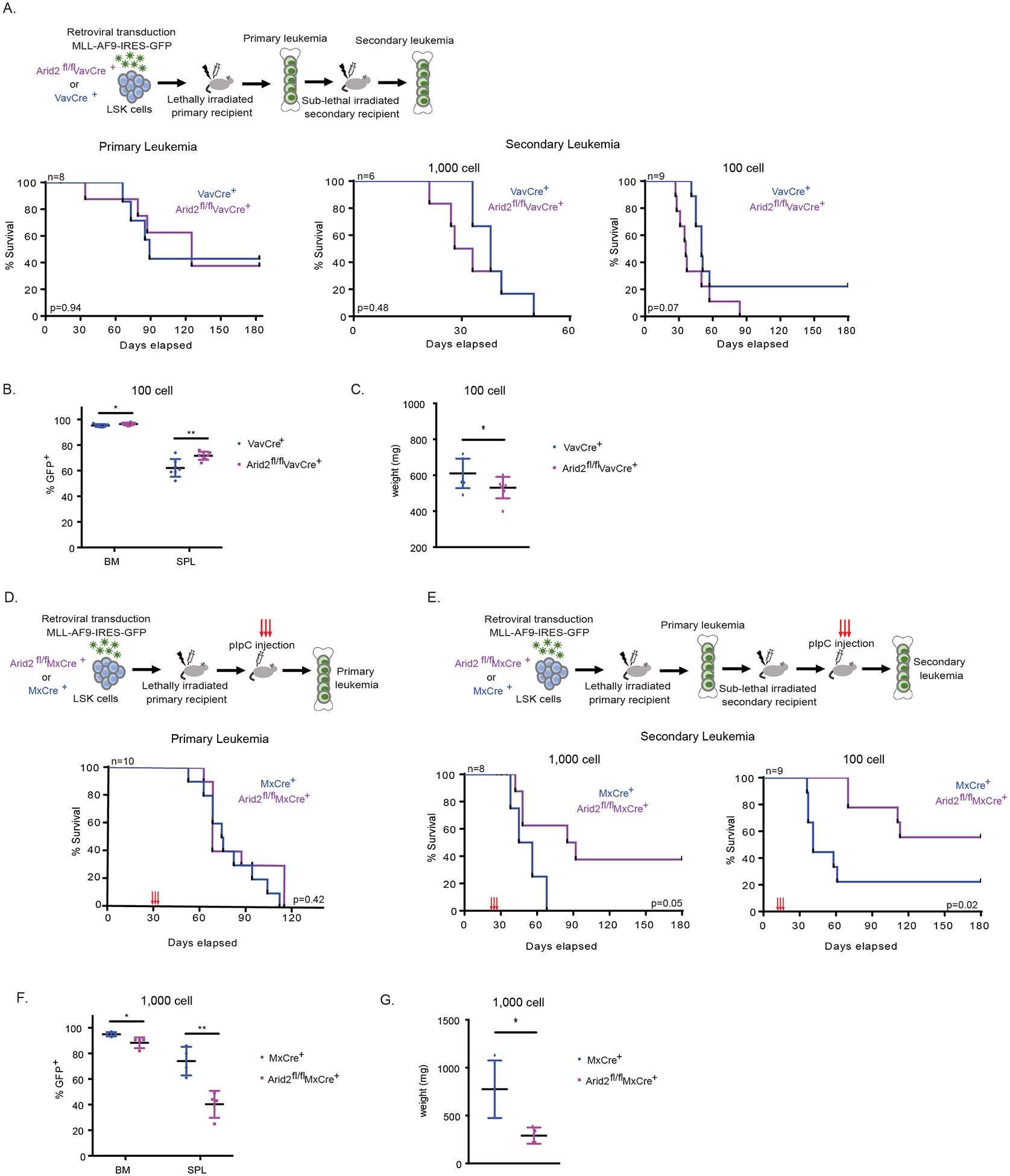
(A-C) In vivo leukemogenesis upon the loss of Arid2 in the VavCre mouse model. (A) Experimental scheme (top). Survival curve of primary recipient mice (bottom, left). Survival curves of secondary recipient mice (bottom, right). (B) Percent of GFP+ cells in the BM and SPL of moribund recipients in the 100 cell secondary cohort. (C) SPL weight of moribund recipients in the 100 cell secondary cohort. (D-G) Leukemia maintenance upon the loss of Arid2 utilizing the MxCre mouse model. (D) Experimental scheme of the primary leukemia (top). Survival curve of primary recipient mice (bottom). pIpC was administered at 30 days post transplantation. (E) Experimental scheme of the secondary leukemia (top). Survival curves of secondary recipient mice (bottom). pIpC was administered at day 21 (1000 cell) and day 14 (100 cell). (F) Percent of GFP+ cells in the BM and SPL of moribund recipients in the 1,000 cell secondary cohort. (G) SPL weight of moribund recipients in the 1,000 cell secondary cohort. n=recipient mice per genotype. VavCre model LSK donors n=2. MxCre model LSK donors n=2. Survival curve p value calculated using log-rank test. p-value *< 0.05, **< 0.01, ns, not significant, un-paired, parametric t-test, error bars indicate s.d.
Table 1. Leukemia initiating cell frequency in the Arid2fl/fl VavCre model.
Leukemia initiating cell (LIC) frequency of the secondary MLL-AF9 VavCre transplant.
| LIC Frequency | Lower | Estimate | Upper |
|---|---|---|---|
| VavCre+ | 149.8 | 66.5 | 29.5 |
| Arid2fl/fl VavCre+ | 79.2 | 1.0 | 1.0 |
Calculated using ELDA software. p=0.08
Arid1b promotes MLL-AF9 leukemogenesis
To study the effect of losing Arid1b on leukemogenesis in vivo, we first used the VavCre model to assess its role in leukemia initiation. We observed a very modest decrease in disease latency in the primary transplantation (Figure 3A). Upon transplantation of whole BM from primary recipients into secondary recipients, at a dosage of 1000 and 100cells, we observed a significant decrease in overall survival of Arid1b null leukemia recipients (Figure 3A). The frequency of leukemia initiating cells between the two cohorts showed a two-fold (range 2–3 folds) increase (Table 2). The percentage of GFP+ MLL-AF9 cells in the BM and SPL, and spleen weight in the primary and secondary transplant recipients were similar (Figure S3A–C). We also assessed MLL-AF9 maintenance using the MxCre model. Our data showed that loss of Arid1b accelerated leukemic progression in primary transplantation (Figure 3B). One-month post induction of Arid1b excision with pIpC, significantly increased percentages of Arid1b null GFP+ MLL-AF9 cells accumulated in the peripheral blood of primary recipients (Figure 3C). Furthermore, when we transplanted un-induced primary leukemia into secondary recipients, and upon pIpC induction, we observed shortened latency in the Arid1b null cohort compared to that of the control cohort, at both a 1,000 and 100cell dose (Figure 3D). Moreover, there was also a higher percentage of GFP+ cells in the SPL of moribund mice in the Arid1b null cohort compared with the control in the 100cell secondary transplantation (Figure 3E), although SPL weight was not significantly different (Figure 3F). The percentage of GFP+ MLL-AF9 cells in the BM and SPL, and spleen weight in the primary and secondary 1,000 cell transplant were not significant (Figure S3D,E). Taken together, our data suggests deleting Arid1b during leukemia initiation promotes leukemogenesis of the resulting leukemia cells and promotes leukemia maintenance.
Figure 3. Loss of Arid1b promotes MLL-AF9 leukemogenesis.
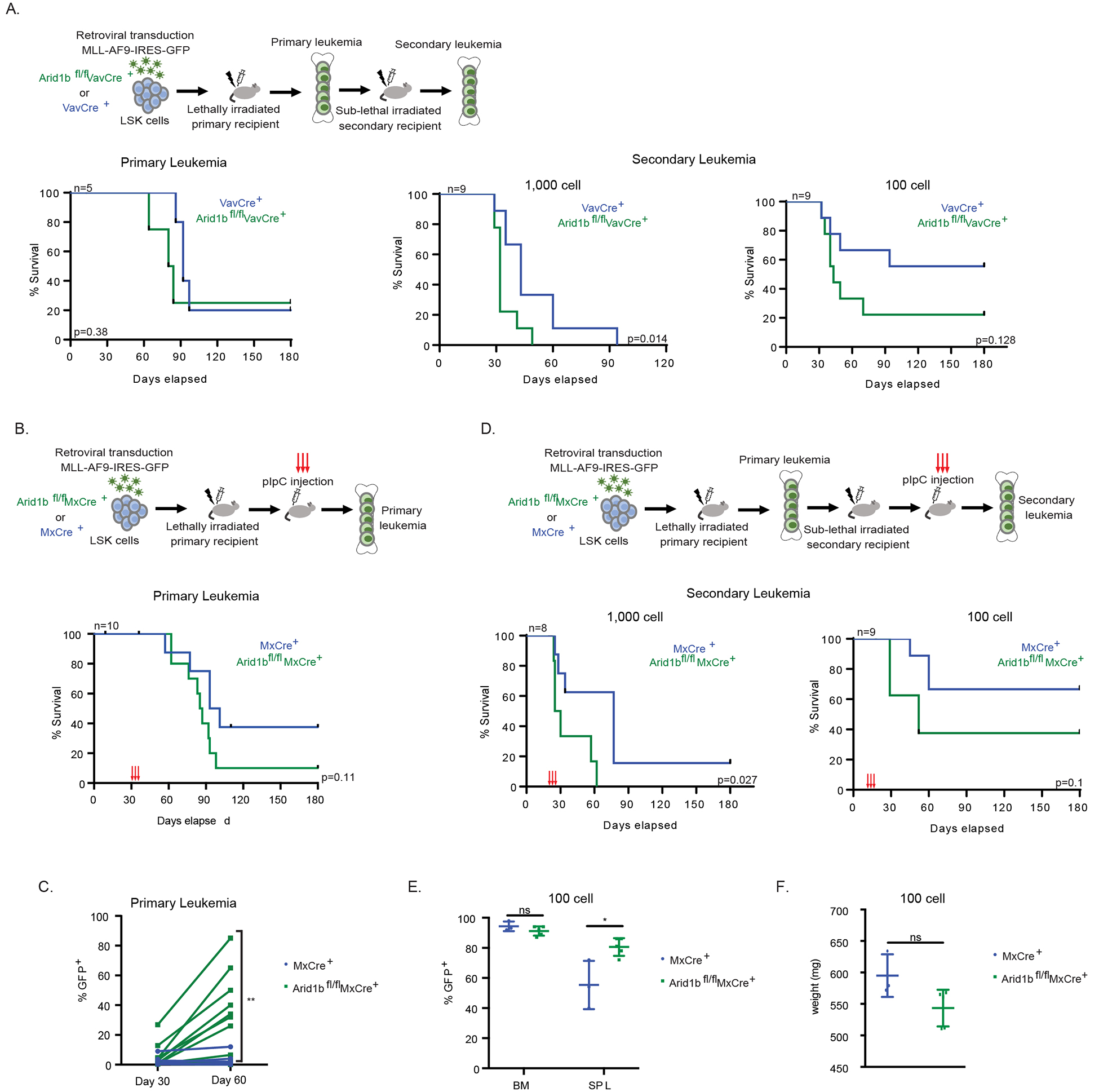
(A) In vivo leukemogenesis upon the loss of Arid1b in the VavCre mouse model. Experimental scheme (top). Survival curve of primary recipient mice (bottom, left). Survival curves of secondary recipient mice (bottom, right). (B-F) Leukemia maintenance upon the loss of Arid1b utilizing the MxCre mouse model. (B) Experimental scheme of the primary leukemia (top). Survival curve of primary recipient mice (bottom). pIpC was administered at 30 days post transplantation. (C) Percent of GFP+ cells in the PB at day 30 (before pIpC) and day 60 (30 days after pIpC) post-transplant in primary leukemia recipients. (D) Experimental scheme of the secondary leukemia (top). Survival curves of secondary recipient mice (bottom). pIpC was administered at day 21 (1000 cell) and day 14 (100 cell). (E) Percent of GFP+ cells in the BM and SPL of moribund recipients in the 100 cell secondary cohort. (F) SPL weight of moribund recipients in the 100 cell secondary cohort. n=recipient mice per genotype. VavCre model LSK donor n=1. MxCre model LSK donors n=2. Survival curve p value calculated using log-rank test. p-value *< 0.05, **< 0.01, ns, not significant, un-paired, parametric t-test, error bars indicate s.d.
Table 2. Leukemia initiating cell frequency in the Arid1bfl/fl VavCre model.
Leukemia initiating cell (LIC) frequency of the secondary MLL-AF9 VavCre transplant.
| LIC Frequency | Lower | Estimate | Upper |
|---|---|---|---|
| VavCre+ | 406 | 165 | 67.5 |
| Arid1bfl/fl VavCre+ | 150 | 67 | 30 |
Calculated using ELDA software. p=0.15
Arid2 controls differential gene expression programs in VavCre and MxCre models
To gain insight into the mechanism of the dual roles of Arid2 in leukemogenesis, we performed a transcriptomic analysis using RNA-seq. We first investigated the effect of losing Arid2 during leukemia initiation on the resulting leukemia cells. Our RNA-seq results showed that 736 genes were significantly upregulated and 951 genes were significantly downregulated in Arid2f/f VavCre versus VavCre control whole BM leukemia cells from secondary transplant recipients (+/−1.5 fold change; adjusted p<0.05) (Figure 4A, Table S4). We identified GATA transcription factor (TF) motifs to be overrepresented in the top significantly upregulated genes using i-Cis Target analysis (Figure 4B). To gain insight into which differentially expressed genes were directly regulated by Arid2, we preformed and integrated genomic binding data of Arid2 with the RNA-seq data. We were unable to assess the binding of Arid2 in the mouse leukemia BM cells because available mouse ARID2 antibodies were not suitable for the analysis. Instead, we performed CUT&Tag analysis of ARID2 in MOLM13 cells, a human MLL-AF9 leukemia cell line. Approximately 1/4 (419/1687 genes) of all the significant differentially expressed genes were bound by ARID2 in MOLM13 cells (Figure 4C). We focused our analysis on TFs with roles in normal and/or malignant hematopoiesis, which were among the top 50 significantly upregulated genes upon Arid2 loss and were bound by ARID2. We reasoned that these are the TFs whose expression are directly highly regulated by Arid2. This included Six5 (+12.82 fold, adj. p=7.19E−6), Tcf4 (+11.79 fold, adj. p=8.51E−5), and Gata2 (+11.63 fold, adj. p=2.37E−3) (Figure 4D,E, Figure S4A). E2–2 (encoded by Tcf4) and Gata2 have been shown to be required for leukemogenesis of MLL-AF9 and HOXA9/Meis1 AML respectively23,24. GSEA analysis using the differentially expressed genes upon Gata2 knockout in HoxA9/Meis1 leukemia cells identified enrichment of downregulated genes in ARID2 knockout and upregulated genes in control cells24 (Figure 4F, Table S5). Similarly, there is also enrichment of genes that are differentially expressed upon Tcf4 knockdown in mouse MLL-AF9;NRASG12D leukemia cells: downregulated genes upon Tcf4 knockdown are enriched in Arid2 knockout cells, while those upregulated genes are enriched in control cells23 (Figure 4G, Table S5). Additionally, GSEA analysis of upregulated genes upon SIX5 knockout in K562 cells (SIX5 repressed genes) showed enrichment in control but not Arid2 knockout leukemia consistent with upregulation of Six5 in the knockout cells25 (Figure S4B, Table S5). Taken together, these data suggest that loss of Arid2 during MLL-AF9 leukemia initiation leads to the upregulation of Six5, Tcf4 and Gata2 in the resulting leukemia cells in secondary transplantations contributing to their enhanced leukemogenic potential as observed in our limiting diluting transplantation experiment.
Figure 4. Transcriptomic analysis of Arid2 null leukemia in the VavCre model.
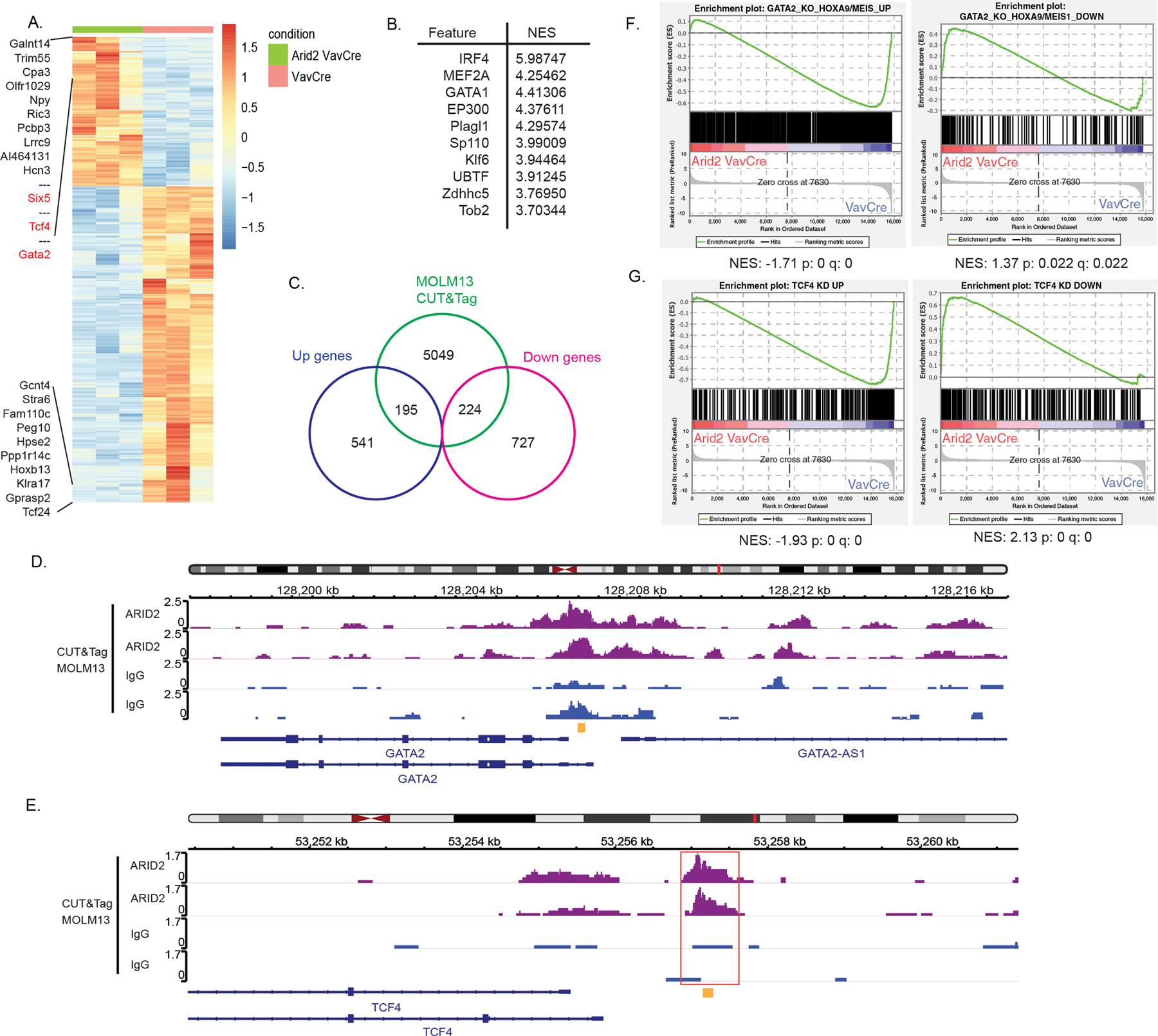
(A) Heatmap of differentially expressed genes between Arid2fl/fl VavCre and VavCre secondary recipient whole BM leukemia cells. Color scale for heatmaps correspond to the gene(row) Z-score. (B) Top 10 overrepresented transcription factor motifs within the top differentially upregulated genes (defined as +1.5 log fold change; adj. p<0.05) (C) Venn diagram showing overlap between significant differentially expressed genes (+/−1.5 fold change; adj. p<0.05) identified in the RNA-seq and peaks called in ARID2 MOLM13 CUT&Tag. (D-E) IGV tracks of ARID2 CUT&Tag in MOLM13 cells at the GATA2(D) and TCF4(E) promoters. Red box indicates called peak location identified by MACS. Yellow indicates ChIP-qPCR amplicon (see Figure 5D). (F-G) GSEA analysis results showing enrichment of differentially expressed genes in Gata2 KO HoxA9/Meis1 LSC (F) and of differentially expressed genes from Tcf4 knockdown (KD) in mouse MLL-AF9;NRASG12D leukemia cells (G).
We then aimed to understand whether the same or different gene programs were underlying the requirement of Arid2 during MLL-AF9 leukemia maintenance using similar transcriptomic assessment. 179 genes were upregulated and 178 genes were downregulated in Arid2f/f MxCre versus MxCre control whole BM cells from secondary transplant recipients (+/−1.5 fold change; adjusted p<0.05) (Figure 5A, Table S6). When we examined the overlap between ARID2 bound genes and the differential expressed genes, almost 1/4 (82/357 genes) of all the significant differentially expressed genes were bound by ARID2 in MOLM13 cells (Figure 5B). The TF Etv5 (−6.82 fold, adj. p=1.64E−4) was among the top 50 significantly downregulated genes in Arid2 MxCre and was bound by ARID2 (Figure 5C). Consistent with our observed phenotype, Etv5 depletion has been shown to promote myeloid differentiation and reduce colony forming activity in AML13. However, no ETV5 gene signature in AML is available for GSEA analysis. Nonetheless, the highly significant downregulation of ETV5 upon Arid2 knockout, its being a direct target of ARID2 and its requirement in maintaining AML leukemia cells as published previously strongly suggest that ETV5 plays an important role in mediating the oncogenic role of Arid2 in MLL-AF9 leukemia maintenance. Jun TF motifs are overrepresented in the top differentially downregulated genes and JUN is a direct target of ARID2 (Figure S4C,D) however, GSEA analysis of JUN knockdown gene signatures in human MLL leukemia cell line, THP1, did not show any enrichment26 (Table S5). None of the TFs that are important for mediating the role in the VavCre model (SIX5, E2–2 and GATA2) are significantly differentially regulated in MxCre model and vice versa. Binding of ARID2 to the promoters of the downstream target genes was further confirmed by ChIP-RTqPCR (Figure 5D). Furthermore, there were very little overlap in the differentially expressed genes between the two (Figure 5E), suggesting that Arid2’s roles in leukemogenesis are mediated by distinct mechanisms.
Figure 5. Transcription regulation by Arid2 in MxCre leukemia.
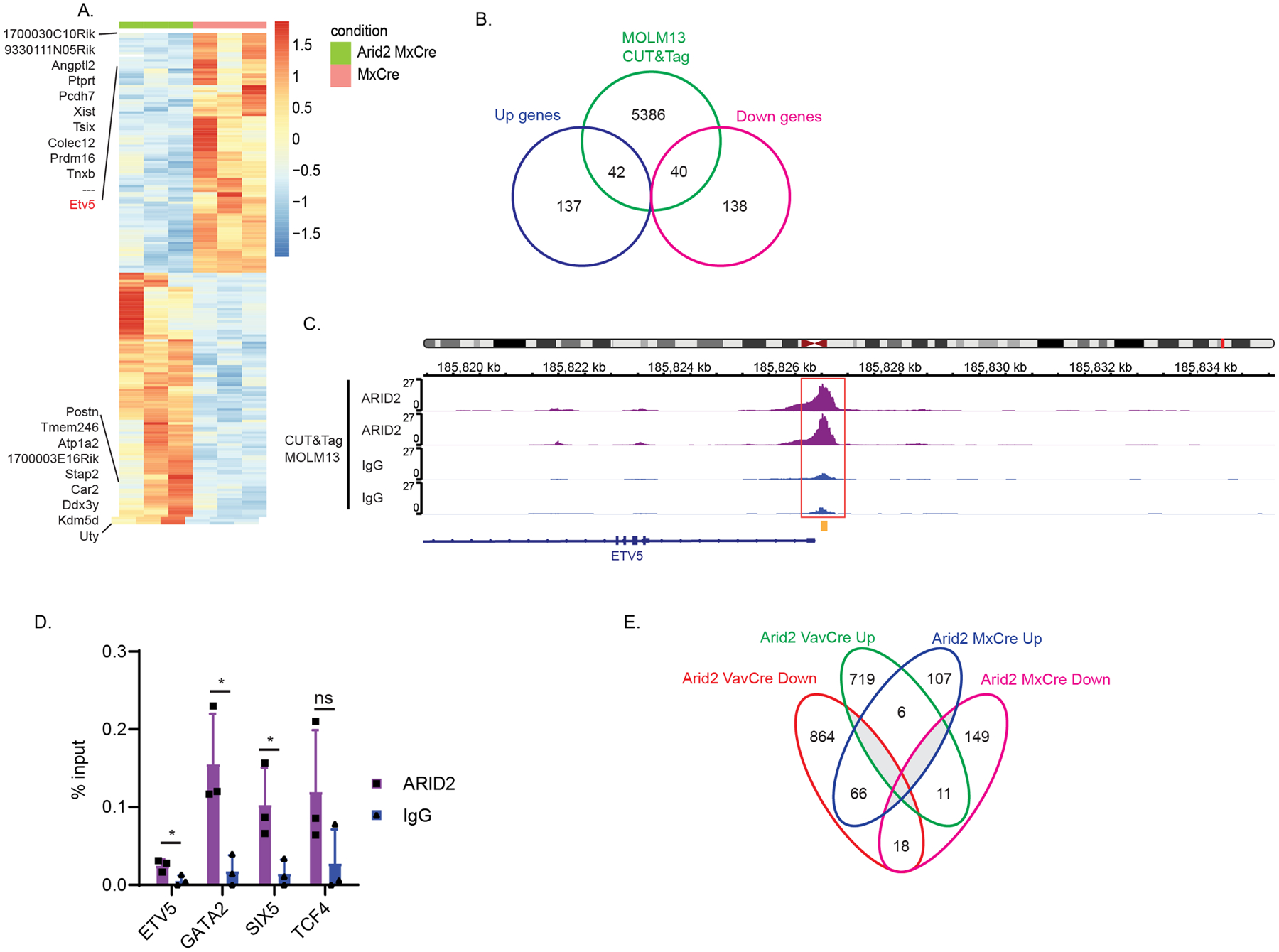
(A) Heatmap of differentially expressed genes between Arid2fl/fl MxCre and MxCre secondary recipient whole BM leukemia cells. Color scale for heatmaps correspond to the gene(row) Z-score. (B) Venn diagram showing overlap between significant differentially expressed genes (+/−1.5 fold change; adj. p<0.05) identified in the RNA-seq and peaks called in ARID2 MOLM13 CUT&Tag. (C) IGV tracks of ARID2 CUT&Tag in MOLM13 cells at the ETV5 promoter locus. Red box indicates called peak location identified by MACS. Yellow indicates ChIP-qPCR amplicon in D. (D) ChIP-RTqPCR of ARID2 or IgG control in MOLM13 cells. p-value *< 0.05, ns, not significant, un-paired, parametric t-test, error bars indicate s.d. (E) Venn diagram showing overlap of the significant differentially expressed genes (+/−1.5 fold change, adj. p<0.05) between Arid2 VavCre and Arid2 MxCre.
Arid1b controls tumorigenic gene expression programs in MLL-AF9 leukemia
To gain insight into the mechanism of the tumor promoting roles of Arid1b in leukemogenesis, we performed a similar transcriptomic analysis as in Arid2. In Arid1bf/f VavCre versus VavCre, 1,146 genes were significantly upregulated and 657 genes were significantly downregulated in whole BM cells from secondary transplant recipients (+/−1.5 fold change; adjusted p<0.05) (Figure 6A, Table S7). We identified IRF4 and ETS family TF motifs to be overrepresented in the top significantly upregulated genes using i-Cis Target analysis (Figure 6B). Additionally, we preformed and integrated genomic binding data of Arid1b bound loci with the RNA-seq data. Similar to Arid2, we were unable to assess the binding of Arid1b in the mouse leukemia BM cells because of antibody issues. Nonetheless, we performed CUT&Tag analysis of ARID1B in MOLM13 cells. 1/4 (452/1803 genes) of all the significant differentially expressed genes in Arid1b VavCre were bound by ARID1B in MOLM13 (Figure 6C). In Arid1bf/f MxCre versus MxCre, 86 genes were significantly upregulated and 103 genes were significantly downregulated in whole BM cells from secondary transplant recipients (+/−1.5 fold change; adjusted p<0.05) (Figure 6D, Table S8). We identified Smarcb1, a core catalytic subunit of SWI/SNF, and ETS family TF motifs to be overrepresented in the top significantly upregulated genes using i-Cis Target analysis (Figure 6E). Similarly, to the VavCre model, almost 1/4 (43/189 genes) of all the significant differentially expressed genes in Arid1b MxCre were bound by ARID1B in MOLM13 cells (Figure 6F). Ultimately, we focused our analysis on TFs with roles in normal and/or malignant hematopoiesis which were among the top 50 significantly upregulated genes and bound by ARID1B. Etv4 was significantly upregulated in both Arid1b VavCre (+38.85 fold, adj. p=5.38E−11) and Arid1b MxCre (+53.08 fold, adj. p=4.78E−7) and had ARID1B binding sites near the promoter (Figure 6G). Unfortunately, gene signatures of Etv4 in a relevant cell type were unavailable for testing in GSEA analysis. Additional TFs that were uniquely upregulated in Arid1b VavCre included Hmgn3 (+23.92 fold, 5.48E−7) and Irf4 (+23.59 fold, adj. p=1.31E−10) (Figure S5A,C). High expression of IRF4 has been identified in childhood AML cases and correlated with relapse suggesting an oncogenic role27. HMGN3 is a member of the high mobility group nucleosomal binding protein. Its related family member, HMGN1, has been shown to facilitate AML-ET9a leukemogenesis28,29. Their upregulation in Arid1b knockout leukemia is consistent with the role of Arid1b in promoting leukemogenesis. Gene signatures of Irf4 in a relevant cell type was unavailable, however GSEA analysis of downregulated genes upon HMGN3 knockout in K562 showed enrichment in Arid1b VavCre leukemia cells25 (Figure S5B, Table S5). Though not within the top50 overexpressed genes, Etv5 was also upregulated (+13.83 fold, adj. p=1.61E−5), along with Etv4, in Arid1b VavCre knockout cells and is bound by ARID1B (Figure S5D). Binding of ARID1B to the promoters of the downstream target genes was confirmed by ChIP-RTqPCR (Figure 6H). Despite similar regulation of ETV4/5, overall, there is very little overlap between differentially expressed genes in the two models (Figure 6I). Nonetheless, the upregulation of Etv5 was confirmed to play a role in leukemia promotion by Arid1b as CRISPR knockdown of Etv5 in wildtype and Arid1b null MLL-AF9 leukemia cells decreased their colony forming and replating ability (Figure 6J). Taken together, this suggests that overlapping and unique transcriptional regulation underlies the role of Arid1b in promoting MLL-AF9 leukemogenesis.
Figure 6. Transcription factors regulated by Arid1b in leukemogenesis.
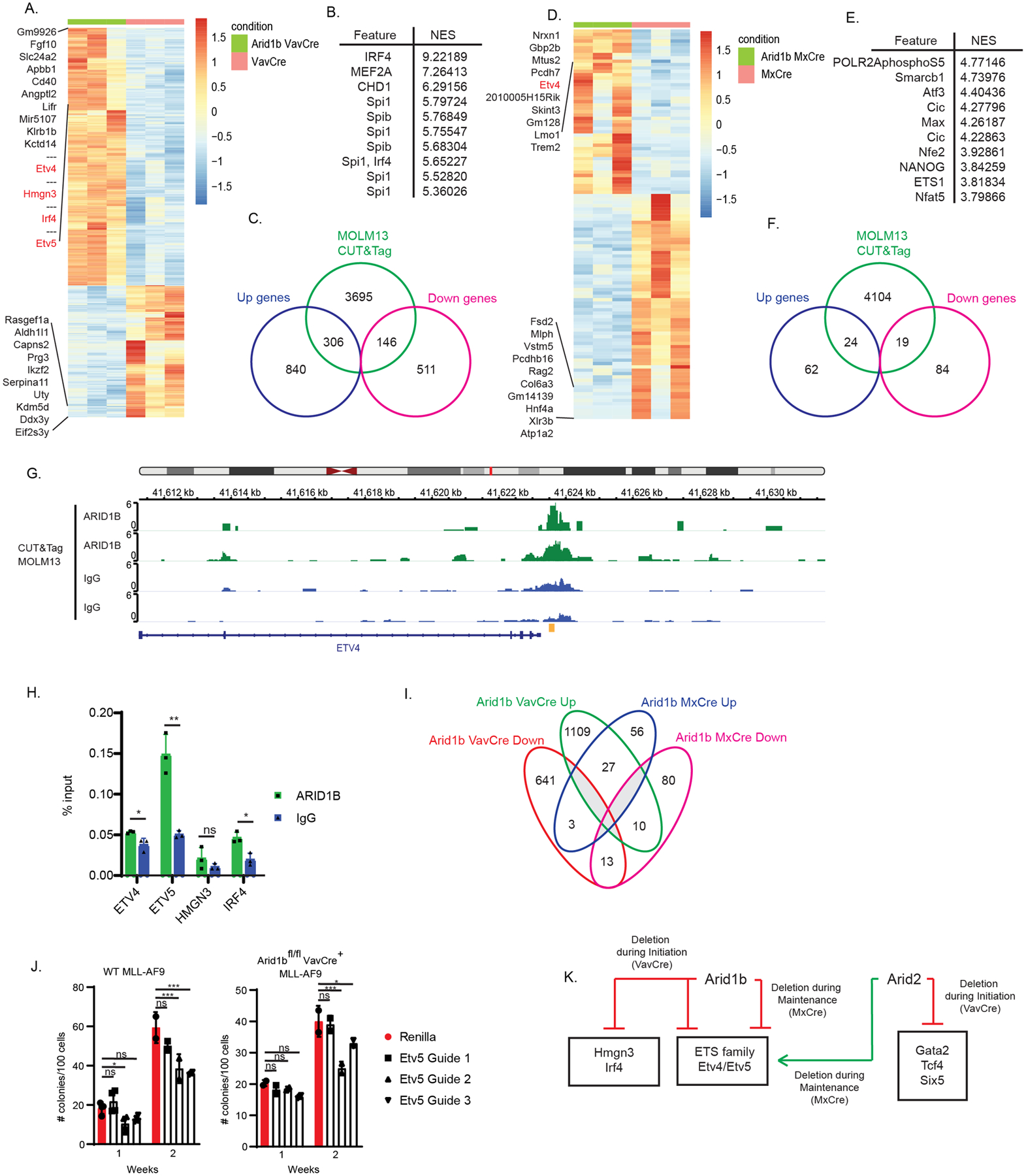
(A) Heatmap of differentially expressed genes between Arid1bfl/fl VavCre and VavCre secondary recipient whole BM leukemia cells. Color scale for heatmaps correspond to the gene(row) Z-score. (B) Top 10 overrepresented transcription factor motifs within the top differentially up regulated genes (+1.5 log fold change; adj. p<0.05) (C) Venn diagram showing overlap between significant differentially expressed genes (+/−1.5 fold change; adj. p<0.05) identified in the Arid1bfl/fl VavCre RNA-seq and peaks called in ARID1B MOLM13 CUT&Tag. (D) Heatmap of differentially expressed genes between Arid1bfl/fl MxCre and MxCre secondary recipient whole BM leukemia cells. Color scale for heatmaps correspond to the gene(row) Z-score. (E) Top 10 overrepresented transcription factor motifs within the top differentially up regulated genes (+1.5 log fold change; adj. p<0.05) (F) Venn diagram showing overlap between significant differentially expressed genes (+/−1.5 fold change; adj. p<0.05) identified in the Arid1bfl/fl MxCre RNA-seq and peaks called in ARID1B MOLM13 CUT&Tag. (G) IGV tracks of ARID1B CUT&Tag in MOLM13 cells at the ETV4 promoter locus. Yellow indicates ChIP-qPCR amplicon in H. (H) ChIP-RTqPCR of ARID1B or IgG control in MOLM13 cells. p-value *< 0.05, **<0.01, ns, not significant, un-paired, parametric t-test, error bars indicate s.d. (I) Venn diagram showing overlap of the significant differentially expressed genes (+/−1.5 fold change, adj. p<0.05) between Arid1b VavCre and Arid1b MxCre. (J) MLL-AF9 colony formation of wildtype cas9 expressing MLL-AF9 single cell clone (left) and Arid1bfl/flVavCre+ MLL-AF9 cells from secondary transplant recipient in methylcellulose transduced with Renilla (control) or CRISPR guides targeting the ETS domain of Etv536. Colonies were counted 7 days after plated (week 1) followed by replating (week 2). p-value *< 0.05, ***<0.001, ns, not significant, two-way ANOVA with multiple comparisons, error bars indicate s.d. (K) Model. Prominent transcription factor networks regulated by Arid2 and Arid1b in MLL-AF9 leukemogenesis. Boxed transcription factors are bound by Arid2 and Arid1b respectively.
Lastly, we assessed the similarities and differences of the gene expression profiles between each of our knockout leukemia models: Arid1b VavCre, Arid1b MxCre, Arid2 VavCre, and Arid2 MxCre. We used a correlation analysis of the significant differentially expressed genes in each knockout leukemia sample between our models (Figure S6A). In general, the samples within each model clustered together and showed that the gene expression profiles of Arid1b VavCre, Arid2 VavCre, and Arid1b MxCre correlate most similarly to each other, consistent with their shared leukemia promoting phenotypes. Arid2 MxCre has the least correlation in gene expression profile with the other samples indicative of the unique phenotype of Arid2’s requirement in MLL-AF9 leukemogenesis in the MxCre model (Figure S6A). GSEA analysis (Table S5, Table S9) of each of the gene signatures, as well as a heatmap of the significant differentially expressed genes across all four models (Figure S6B) further support that gene expression patterns cluster based on disease stage/model (MxCre vs VavCre) and that Arid2 and Arid1b regulate largely distinct set of genes at the global level. We also observed distinct binding patterns between ARID2 and ARID1B within the significant differentially expressed genes between all four models, supporting Arid1b and Arid2 regulate unique sets of genes (Figure S6C).
DISCUSSION
SWI/SNF components have widespread roles in hematopoiesis and function as tumor suppressors in various malignancies. Shared subunits of SWI/SNF complexes have been characterized to be oncogenes in AML17,30, however the exact roles of the unique targeting subunits of PBAF and BAF, Arid2 and Arid1b, respectively, were not characterized. Here we used hematopoietic specific mouse models to demonstrate that Arid2 promotes leukemogenesis of the resulting cells when deleted during leukemia initiation yet is required for leukemia maintenance. Whereas, Arid1b promotes leukemogenesis in both phases.
ARID2 and ARID1B mutations are found in primary AML patients and suggest a role in promoting leukemogenesis (Figure 1). Although the loss of Arid2 or Arid1b had no effect on cellular phenotypes in-vitro (Figure S1), our data did show effects on leukemogenesis in-vivo. Arid2 VavCre knockout leukemia showed decreased survival in limiting dilution transplantation and increased LSC frequency. Surprisingly however, using inducible excision of Arid2 by MxCre in established MLL-AF9 secondary recipients to model leukemia maintenance, the opposite was true. We observed prolonged survival of the recipient mice with Arid2 null leukemia (Figure 2 and Figure S2). These data suggest Arid2 enhances the leukemogenic potential of the resulting leukemia when it is deleted during leukemia initiation yet is required for leukemia maintenance. A finding that perhaps explains the lack of statistical significance in overall survival in patients with low ARID2 expression comparing to those with high expression. This finding also resembles that of EZH2 which plays an oncogenic role for leukemia maintenance but acts as a tumor suppressor during AML initiation. It was found that EZH2 does this by mediating the de-repression of distinct transcriptional programs during each disease state31. Similarly, to that of EZH2, ARID2 seems to be regulating different sets of target genes during these processes. Our integrated transcriptomic and genomic binding analysis showed that, in the VavCre model, ARID2 directly regulates GATA2, E2–2 and SIX5 expression. Loss of Arid2 results in concomitant changes in their expression level, as well as changes in their downstream gene signatures (Figure 4, Figure S4). At the same time we were undergoing these studies, consistent with our finding, a study published that Arid2 accelerates progression of MLL-AF9 leukemogenesis using the Vav-iCre model32. Using targeted RT-qPCR of known hematological malignancy genes, they showed a similar significant upregulation of Gata2. We additionally showed enrichment of Gata2 gene signatures in our data set, as well as the potential involvement of two other TFs, Tcf4 and Six5, in driving the gene expression programs. The transcriptional network underlying the oncogenic role of Arid2 in leukemia maintenance in MxCre model is clearly different from that in the VavCre model. None of the TFs that we identified to be important in the VavCre model are differentially regulated in MxCre model. Instead, we identified the downregulation of a characterized oncogene in AML13 and a direct ARID2 target, the Ets family TF, Etv5, upon Arid2 knockout as potentially the mediator of the oncogenic phenotype of Arid2 in leukemia maintenance. On a global scale, there was little overlap between the differentially expressed genes of the two models, further supporting that distinct transcriptional regulation underlies the differential phenotypes (Figure 5E). At the level of PBAF complexes, the loss of ARID2 seems to result in subsequent loss of other PBAF specific subunits, such as PBRM1 and BRD7, disrupting the formation of the complex33. In light of this, the opposing phenotypes we observed upon Arid2 knockout in VavCre and MxCre models of leukemia may be a result of an adaptation to the loss of PBAF in the VavCre model as deletion happens during embryogenesis, while acute loss of PBAF in the MxCre model results in impairment of leukemia cells.
Unlike Arid2, which demonstrates dual roles, our data indicated that Arid1b promotes leukemogenesis in both leukemia disease phases. In the VavCre and MxCre model, loss of Arid1b led to reduced latency in primary and secondary transplantation (Figure 3 and Figure S3). This role of Arid1b is consistent with SWI/SNF tumor suppressor roles in other cancers and is in line with primary AML patient data suggesting that decreased level of Arid1b correlates with poorer survival. However, it is inconsistent with the core components of BAF having been shown to play oncogenic roles in AML17,30. One possible explanation is that BRG1 and ARID1B have opposite effects on chromatin accessibility. It has been shown that BRG1 knockout leads to an overall loss in chromatin accessibility, while ARID1B knockout presents an overall gain of accessibility with accompanying differences in gene expressions33. In addition, at the complex level, knockout of any one subunit of the BAF complex does not result in disruption of the complex because of the existence, and sometimes increased incorporation, of paralogous proteins into the complex such as ARID1A-ARID1B-ARID233. These may explain the contrasting effects of the loss of Arid1b on MLL-AF9 leukemogenesis compared to that of Brg1 loss.
In both, Arid1b VavCre and Arid1b MxCre knockout, leukemia models, we discovered that ETS family TF, Etv4, was a direct target of ARID1B and was highly significantly upregulated upon Arid1b knockout (Figure 6). Etv5 was also significantly upregulated in the VavCre model, is a target of ARID1B, and its downregulation impairs colony forming ability of MLL-AF9 leukemia cells (Figure 6J). Despite the similarity of Arid1b in regulating Etv4/5-Lmo1 in both VavCre and MxCre models, Arid1b also appears to regulate additional unique gene programs in the VavCre model. In support of this, TFs, Irf4 and Hmgn3, are direct targets of ARID1B and uniquely upregulated in Arid1b VavCre but not Arid1b MxCre leukemia (Figure 6H, Figure S5). In addition, there is little overlap of differentially expressed genes between the two models (Figure 6I).
Ultimately, we showed that Arid1b and Arid2 bind to and regulate, largely, unique sets of genes in MLL-AF9 leukemogenesis at the global level (Figure S6). Amongst the most highly regulated transcription factors, Arid2 uniquely regulates Gata2, Tcf4 and Six5, while Arid1b uniquely regulates Irf4 and Hmgn3 in the VavCre leukemia model. Whereas they converge on the transcriptional regulation of ETS family members, Etv4/5, albeit Arid1b as a negative regulator in both the VavCre and MxCre model of leukemia, while Arid2 is a positive regulator in the MxCre model. (Figure 6K). In terms of transcriptional regulation, this is consistent with the observation that knockouts of individual paralogous subunits affect overlapping, as well as unique sets of genes33. Such specificity is probably the result of cellular context (e.g.TF presence and abundance) and characteristics of individual subunits (DNA binding specificity). Further investigation is necessary to assess the functional relevance of the transcriptional networks identified upon Arid2 and Arid1b loss in MLL-AF9 leukemogenesis.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Benedetta Bonacci for assistance with cell sorting, Angela Cui for assistance with cloning vectors and Hao Zhu for the gift of the Arid1bfl/fl mouse.
This work was supported by the Versiti Blood Research Institute Foundation and in part by Institutional Research Grant IRG #16-183-31 from the American Cancer Society and the Medical College of Wisconsin Cancer Center.
Footnotes
CONFLICTS OF INTEREST
The authors declare no competing interests.
REFERENCES
- 1.Neigeborn L, Carlson M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 1984; 108: 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang L, Nogales E, Ciferri C. Structure and Function of SWI/SNF Chromatin Remodeling Complexes and Mechanistic Implications for Transcription. Prog Biophys Mol Biol 2010; 102: 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imbalzano AN, Kwon H, Green MR, Kingston RE. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 1994; 370: 481–485. [DOI] [PubMed] [Google Scholar]
- 4.Kwon H, Imbalzano AN, Khavari PA, Kingston RE, Green MR. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994; 370: 477–481. [DOI] [PubMed] [Google Scholar]
- 5.Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 1999; 3: 247–253. [DOI] [PubMed] [Google Scholar]
- 6.Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J 1996; 15: 5370–5382. [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Nagl NG, Wilsker D, Van Scoy M, Pacchione S, Yaciuk P et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J 2004; 383: 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaeser MD, Aslanian A, Dong M-Q, Yates JR, Emerson BM. BRD7, a Novel PBAF-specific SWI/SNF Subunit, Is Required for Target Gene Activation and Repression in Embryonic Stem Cells. J Biol Chem 2008; 283: 32254–32263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A et al. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 2005; 19: 1662–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol 2006; 7: 437–447. [DOI] [PubMed] [Google Scholar]
- 11.Wilson BG, Roberts CWM. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011; 11: 481–492. [DOI] [PubMed] [Google Scholar]
- 12.Lorch Y, Maier-Davis B, Kornberg RD. Mechanism of chromatin remodeling. Proc Natl Acad Sci USA 2010; 107: 3458–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeda R, Asada S, Park S-J, Yokoyama A, Becker HJ, Kanai A et al. HHEX promotes myeloid transformation in cooperation with mutant ASXL1. Blood 2020; 136: 1670–1684. [DOI] [PubMed] [Google Scholar]
- 14.Shain AH, Pollack JR. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLOS ONE 2013; 8: e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013; 45: 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe J-S et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev 2013; 27: 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cruickshank VA, Sroczynska P, Sankar A, Miyagi S, Rundsten CF, Johansen JV et al. SWI/SNF Subunits SMARCA4, SMARCD2 and DPF2 Collaborate in MLL-Rearranged Leukaemia Maintenance. PLoS One 2015; 10: e0142806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu N, Chen M, Eng R, DeJong J, Sinha AU, Rahnamay NF et al. MLL-AF9– and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J Clin Invest 2016; 126: 997–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology 2017; : 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.INFRAFRONTIER Consortium. INFRAFRONTIER--providing mutant mouse resources as research tools for the international scientific community. Nucleic Acids Res 2015; 43: D1171–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raess M, de Castro AA, Gailus-Durner V, Fessele S, Hrabě de Angelis M, INFRAFRONTIER Consortium. INFRAFRONTIER: a European resource for studying the functional basis of human disease. Mamm Genome 2016; 27: 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Celen C, Chuang J-C, Luo X, Nijem N, Walker AK, Chen F et al. Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. Elife 2017; 6. doi: 10.7554/eLife.25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghisi M, Kats L, Masson F, Li J, Kratina T, Vidacs E et al. Id2 and E Proteins Orchestrate the Initiation and Maintenance of MLL-Rearranged Acute Myeloid Leukemia. Cancer Cell 2016; 30: 59–74. [DOI] [PubMed] [Google Scholar]
- 24.Menendez-Gonzalez JB, Vukovic M, Abdelfattah A, Saleh L, Almotiri A, Thomas L et al. Gata2 as a Crucial Regulator of Stem Cells in Adult Hematopoiesis and Acute Myeloid Leukemia. Stem Cell Reports 2019; 13: 291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou C, Martinez E, Di Marcantonio D, Solanki-Patel N, Aghayev T, Peri S et al. JUN is a key transcriptional regulator of the unfolded protein response in acute myeloid leukemia. Leukemia 2017; 31: 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adamaki M, Lambrou GI, Athanasiadou A, Tzanoudaki M, Vlahopoulos S, Moschovi M. Implication of IRF4 aberrant gene expression in the acute leukemias of childhood. PLoS One 2013; 8: e72326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catez F, Brown DT, Misteli T, Bustin M. Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep 2002; 3: 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cabal-Hierro L, van Galen P, Prado MA, Higby KJ, Togami K, Mowery CT et al. Chromatin accessibility promotes hematopoietic and leukemia stem cell activity. Nat Commun 2020; 11: 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buscarlet M, Krasteva V, Ho L, Simon C, Hébert J, Wilhelm B et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood 2014; 123: 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basheer F, Giotopoulos G, Meduri E, Yun H, Mazan M, Sasca D et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med 2019; 216: 966–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Wan X, Zhou P, Zhou X, Zhang W, Hui X et al. The chromatin remodeling subunit Baf200 promotes normal hematopoiesis and inhibits leukemogenesis. J Hematol Oncol 2018; 11: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schick S, Rendeiro AF, Runggatscher K, Ringler A, Boidol B, Hinkel M et al. Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat Genet 2019; 51: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Izaguirre-Carbonell J, Christiansen L, Burns R, Schmitz J, Li C, Mokry RL et al. Critical role of Jumonji domain of JMJD1C in MLL-rearranged leukemia. Blood Adv 2019; 3: 1499–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.