

Abstract
Systemic sclerosis (SSc) is a clinically heterogeneous fibrotic disease with no effective treatment. Myofibroblasts are responsible for unresolving synchronous skin and internal organ fibrosis in SSc, but the drivers of sustained myofibroblast activation remain poorly understood. Using unbiased transcriptome analysis of skin biopsies, we identified downregulation of SPAG17 in multiple independent SSc patient cohorts, and by orthogonal approaches observed significant negative correlation between SPAG17 and fibrotic gene expression. Fibroblasts and endothelial cells explanted from SSc skin biopsies showed reduced chromatin accessibility at the SPAG17 locus. Remarkably, mice lacking Spag17 demonstrated spontaneous skin fibrosis with increased dermal thickness, collagen deposition and stiffness, and altered collagen fiber alignment. Knockdown of SPAG17 in human and mouse fibroblasts and microvascular endothelial cells was accompanied by spontaneous myofibroblast transformation and markedly heightened sensitivity to profibrotic stimuli. These responses were accompanied by constitutive transforming growth factor-β (TGF-β) pathway activation. Thus, we discovered impaired expression of SPAG17 in SSc and identified a new cell-intrinsic role for SPAG17 in negative regulation of fibrotic responses. These findings shed fresh light on the pathogenesis of SSc, and may inform the search for innovative therapies for SSc and other fibrotic conditions via SPAG17 signaling.
Keywords: Systemic sclerosis, SPAG17, myofibroblast, morphogen signaling
INTRODUCTION
Systemic sclerosis (SSc) is an acquired chronic disease of unknown etiology characterized by microvascular changes and fibrosis synchronously affecting the skin and multiple organs (Allanore et al, 2015). There are currently no effective treatments to prevent or reverse fibrosis (Distler et al, 2019; Volkmann and Varga, 2019). Patients with SSc present with protean clinical manifestations and follow diverse temporal trajectories to unpredictable outcomes. Fibrosis in the skin varies from limited to diffuse, and its extent is associated with prognosis and survival (Denton & Khanna, 2017).
The defining feature of SSc is intractable fibrosis affecting the skin, lungs, heart, and muscle (Distler et al, 2017). Fibrosis causing progressive dysfunction in the affected organs underlies the diverse clinical manifestations of SSc and is responsible for its high mortality (Varga & Abraham, 2007). Fibrosis in SSc is characterized by excessive extracellular matrix (ECM) production accompanied by accumulation and persistence of myofibroblasts (Bhattacharyya et al, 2011; Pakshir & Hinz, 2018). Myofibroblasts originate from tissue-resident fibroblasts as well as other stromal cell progenitors through a complex and still incompletely-characterized series of differentiation events. These cells appear transiently in physiological wound repair; why in pathological fibrosis they fail to disappear or re-enter a state of cellular quiescence has been intensively studied, but to date remains unknown (Hinz et al, 2019).
Deploying a comprehensive approach to identify transcripts distinguishing SSc skin biopsies from healthy controls, we identified 526 genes that were downregulated and 1,200 genes that were upregulated in SSc compared to healthy control skin (Roberson et al., 2022). We noted that among these genes, sperm-associated antigen-17 (SPAG17), a functionally poorly characterized gene (Teves et al., 2016), displayed the most significant variation. Experiments presented here revealed a unique role for SPAG17 in modulating myofibroblast differentiation and fibrosis. Thus, SPAG17 appears to play a crucial role in regulating connective tissue homeostasis, and its profound dysregulation in SSc patients may directly contribute to the development or progression of skin fibrosis.
RESULTS
Altered SPAG17 expression in SSc skin biopsies
To uncover molecular markers associated with SSc, skin biopsies from a longitudinal inception cohort of 19 SSc patients and 15 matched healthy controls were studied (Table S1). RNA sequencing showed significant downregulation of SPAG17 mRNA in SSc skin biopsies (FC=−5.1, q=1.95 × 10−15) (Roberson et al., 2022). Reduced SPAG17 expression was seen in SSc patients with both limited (lcSSC; 2.70 × 10−5) and diffuse cutaneous (dcSSc, 1.41 × 10−13) disease (Fig. 1A), with levels lower in dcSSc than lcSSc (2.70 × 10−4, Fig. 1B). Modest negative correlations between modified Rodnan skin scores (MRSS) and SPAG17 expression were noted using both non-parametric Kendall’s tau correlation (tau = −0.32, P = 1.73 × 10−2) and linear regression (MRSS = −0.03, adjusted R2 = 0.23, Fig. 1C and D).
Figure 1:

SPAG17 is significantly downregulated in SSc. (a-c) Skin biopsies from patients with SSc (n=19) and healthy controls (n=15) were subjected to RNA sequencing. (a) Volcano plot showing differentially expressed genes. (b) Expression of SPAG17. The half-violin shows the density distribution. Black circles, baseline biopsies; red triangles, 6 months follow-up biopsies. (c) Correlation of SPAG17 and MRSS. (d) Biopsies stained with hematoxylin and eosin (representative images). Scale bar= 400μm. (e) Skin biopsies from SSc patients (n=22) and healthy controls (n= 18) were subjected to single cell RNA-seq. Bar graph showing percentage of cells expressing SPAG17. (f) Representative ATAC-seq plot demonstrating reduced chromatin accessibility in the SPAG17 locus in SSc (n= 6) vs. healthy control (n=6) skin-derived ECs.
To examine the generalizability of these findings, we analyzed SPAG17 mRNA expression in skin biopsies from an independent cohort of early- stage (mean disease duration 1.3 years) SSc (n= 48) patients and healthy controls (n=33) in the Prospective Registry for Early Systemic Sclerosis (PRESS) cohort (Skaug et al., 2020). Significant down-regulation of SPAG17 in dcSSc skin (fold change=0.34, p<0.001) was confirmed (Figure S1).
Next, to characterize the cell populations that express SPAG17, biopsies (HC=18 and SSc=22) were subjected to single-cell RNA-sequencing (scRNA-seq) analysis. SPAG17 expression was detected in multiple cell populations in the skin, with fibroblasts showing the highest levels (about 70%). Although 75% of endothelial cells also expressed SPAG17, levels were lower compared to fibroblasts (Fig. 1E). Minimal SPAG17 expression was also detected in lymphocytes, keratinocytes, and mast cells (Figure S2A). Comparison of healthy and SSc skin biopsies showed a markedly decreased frequency of SPAG17-expressing cells in SSc (Fig. 1E). SPAG17 reduction was most prominent in fibroblasts, with 30% of this cell population no longer expressing the gene. Moreover, 15% of ECs lost SPAG17 gene expression, and the remaining 60% showed reduced SPAG17 expression compared to healthy controls. Fibroblast subclustering showed lowest SPAG17 expression in COL8A1+ fibroblasts in SSc biopsies ( Figure S2C). Notably, this fibroblast subpopulation expressed high levels of ACTA2, identifying these cells as myofibroblasts.
In light of the striking downregulation of SPAG17 mRNA noted in SSc skin biopsies (Fig. 1A), we sought to examine SPAG17 expression in skin biopsies and isolated skin fibroblasts. We found that SPAG17 protein was detectable in fibroblasts and vascular endothelial cells from healthy donors, but markedly reduced in SSc biopsies ( Figure S3A). Examination of SPAG17 in explanted skin fibroblasts showed lower expression in SSc (n=6) compared to healthy controls (n=5) (Figure S4).
To investigate epigenetic mechanisms that might potentially account for reduced cellular levels of SPAG17 in SSc, dermal fibroblasts and microvascular ECs isolated from SSc (n=6) and healthy donor (n=6) skin biopsies were studied by ATAC-seq. Notably, significantly reduced chromatin accessibility in the SPAG17 locus was observed in SSc compared to healthy ECs and fibroblasts (Fig. 1F, Table S2).
Loss of SPAG17 in the mouse is associated with skin fibrosis recapitulating human SSc
To examine the role of SPAG17 in the skin, we used a conditional Spag17 knockout mouse model generated with Sox2-Cre mice (Kazarian et al., 2018). Deletion of the Spag17 gene in the skin was confirmed by RT-PCR (Figure S5B). Mice were assessed for skin phenotype at different ages. Remarkably, Spag17/Sox2-Cre knockout mice showed significant skin changes compared to age- and sex-matched wild-type mice. In particular, increased accumulation of tightly packed collagen-rich ECM in the dermis; reduced thickness of the dermal white adipose tissue (dWAT); and increased dermal thickness were greatest between 3 and 5 months of age (Fig. 2 and Figure S6). Characterization by atomic force microscopy revealed a significant increase in mechanical stiffness, a hallmark of fibrosis, in skin from Spag17/Sox2-Cre KO mice (Fig. 2C and Figure S7). Increased alignment of dermal collagen bundles is a distinct feature of SSc skin accompanying fibrosis (Cao et al., 2017). We noted striking ECM alterations in Spag17 KO mice, with a pattern of highly organized collagen bundles compared to a more random distribution in wild-type mice (Fig. 2D). These initial analyses indicate that loss of SPAG17 in the mouse is associated with skin changes that resemble those seen in SSc skin. However, these studies are limited and focused on a narrow period of age. Further studies are needed to determine disease progression and whether this knockout shows microvascular changes and synchronos multi-organ fibrosis like SSc.
Figure 2:

Loss of Spag17 in the mouse drives skin fibrosis. Wild-type and SPAG17 knockout mice were sacrificed (from 3 to 11 month old) and skin was harvested. (a) Trichrome stain; representative images. Scale bar= 100μm. (b) Quantification of dermal thickness from WT (n=35) and KO (n=42) mice; (five determinations/mouse). (c) Quantification of skin stiffness WT (n=3) and KO (n=4). Scale bar= 50 and 30μm. (d) Quantification of collagen alignment using Second Harmonic Generation (SHG) Photophysics, WT (n=12) and KO (n=37). Scale bar=2003BCm Results are presented as mean±SEM. *p < 0.05. Abbreviations: WT, wild-type; KO, Spag17/Sox2-Cre knockout.
Loss of SPAG17 is associated with fibrotic signaling
To unravel mechanistic links between SPAG17 and fibrosis, we first analyzed the associations of SPAG17 and differentially expressed genes in SSc. Notably, reduced SPAG17 in SSc skin biopsies was associated with upregulation of multiple genes involved in ECM production, growth factors binding, focal adhesion, and other processes implicated in fibrosis (Fig. 3A and Table S3).
Figure 3:
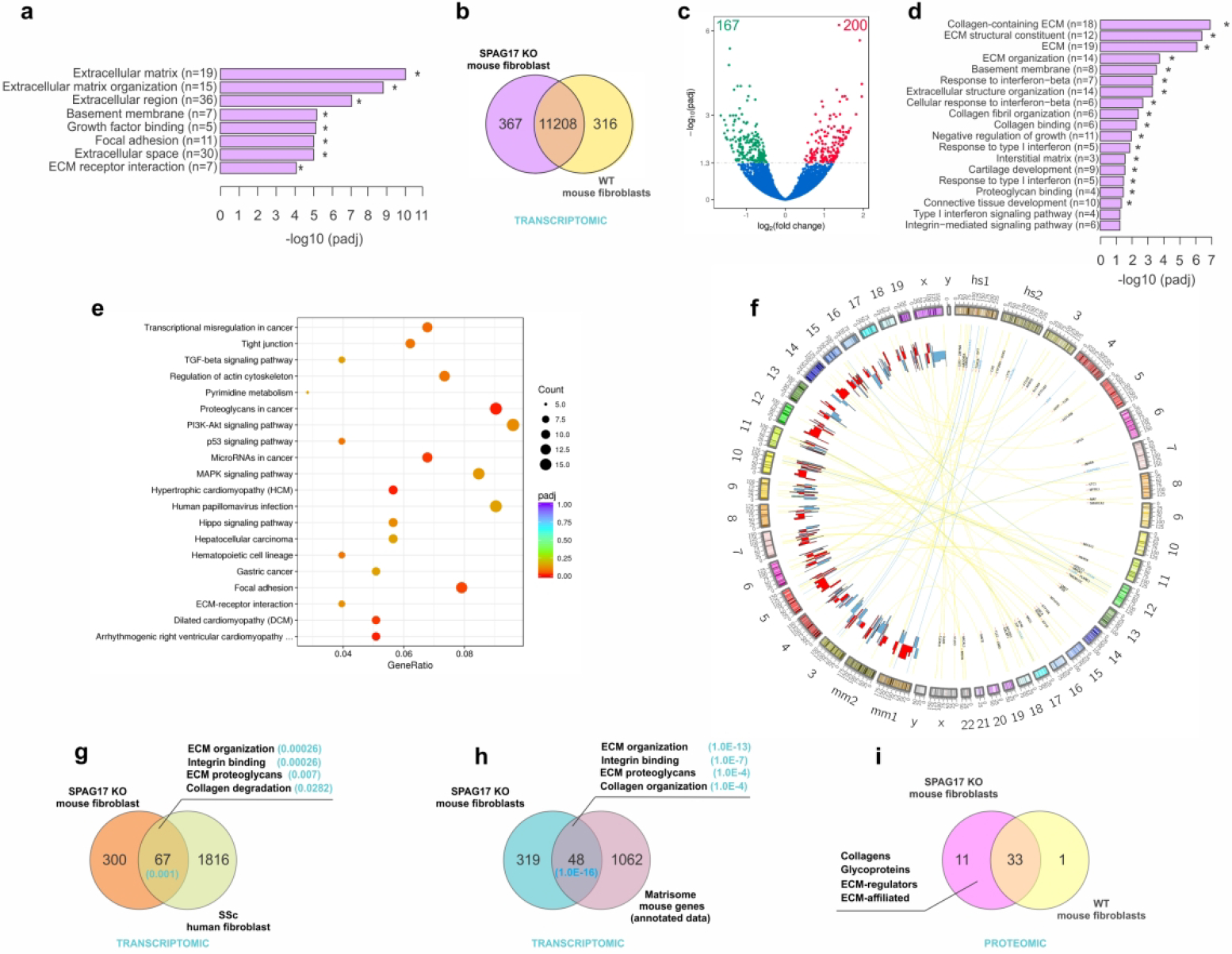
Loss of SPAG17 is associated with upregulation of profibrotic genes. (a) SPAG17 association with differential gene expression from patients with SSc (n=48) and healthy controls (n=33). (b-f) Confluent skin fibroblasts isolated from neonatal WT and KO mice (n=5) were subjected to RNA-seq. (b) Venn diagram and (c) Volcano plot showing differential gene expression. (d) GO pathway analysis. (e) KEGG enriched pathway analysis. (f) Circos diagram showing cross-species analysis comparing fibroblasts from SSc patients and from Spag17 KO mice. (g) Venn diagram comparing overlap of genes from SSc skin fibroblasts and Spag17 KO mouse fibroblasts. (h) Venn diagram showing overlapping of genes from KO fibroblast and matrisome genes from annotated database. (i) Proteomic characterization of ECM produced in vitro by mouse skin fibroblasts.
To analyze how SPAG17 modulates fibrotic responses, we studied skin fibroblasts explanted from Spag17/Sox2-Cre KO mice and wild-ype control mice. Unbiased transcriptome profiling revealed differential expression of 367 genes (167 down-regulated and 200 up-regulated; q<0.0005, Fig. 3B and C). Analysis of differentially expressed genes (DEGs) by GO and KEGG showed significant enrichment in fibrotic programs, including integrin-mediated signaling pathway, ECM organization, collagen fibril organization, focal adhesion, regulation of actin cytoskeleton, TGF-β signaling, and others (Fig. 3D and E). We next sought to evaluate similarities in differential gene expression between Spag17-deficient mouse fibroblasts and SSc skin fibroblasts using human-mouse cross-species transcriptome analysis (Sargent et al., 2016). Comparing transcriptomes from SSc fibroblasts explanted from patients with early diffuse cutaneous disease vs. healthy donors (Chadli et al., 2019) and mouse skin fibroblast explanted from Spag17/Sox2-Cre KO mice vs. wild-type control mice showed significant overlap among differentially expressed fibrotic genes (Figs. 3F, G, Figure S8, and Table S4). Notably, the pathways enriched with DEGs common to both SSc fibroblasts and Spag17/Sox2-Cre KO mouse fibroblasts included ECM organization, proteoglycans, collagen degradation, and integrin binding. We also found an overlap of these genes with the previously defined “matrisome” gene set, which corresponds to the ensemble of genes encoding structural ECM proteins and ECM-associated proteins (Naba et al., 2016) (Fig. 3H). Having shown altered expression of ECM genes in Spag17/Sox2-Cre KO fibroblasts, we next examined protein composition of the ECM produced in vitro by skin fibroblasts isolated from newborn wild-type (n=5), Spag17/Sox2-Cre heterozygous (n=5), or Spag17/Sox2-Cre KO (n=5) mice (Table S5A). Proteomic analysis identified 65 distinct matrisome proteins (Figures S9A, B; Table S5B). Comparison of the list of ECM proteins identified in samples from each group revealed that most proteins were detected in at least 2 or 3 of the five samples for each genotype ( Figure S9B and C, Table S5B). To identify protein changes associated with loss of SPAG17, we further defined the “matrisome” of each genotype as the ensemble of ECM and ECM-associated proteins detected in at least 3 of the five biological replicates for each group (Table S5C). Using this definition, the matrisome of fibroblasts for each genotype comprised 34 proteins (wild-type), 41 proteins (heterozygous), and 44 proteins (Spag17/Sox2-Cre knockout, Table S5C). We found that 33 proteins were produced by wild-type, heterozygous and Spag17/Sox2-Cre KO fibroblasts in similar abundance. This group included fibrillar collagens (Col1a1, Col1a2, Col3a1, Col5a1, Col5a2), fibrillar ECM glycoproteins such as fibronectin (Fn1) and fibrillins 1 and 2 (Fbn1, Fbn2), and the matricellular proteins periostin (Postn), SPARC, and tenascin-C (Tnc) (Table S5B and S5C). Interestingly, 11 matrisome proteins were uniquely detected in SPAG17 knockout but not by wild-type fibroblasts (Fig. 3I and Table S5C). Of note, lack of detection of these proteins in wild-type fibroblasts does not necessarily mean that they are absent since abundance level of these 11 proteins might falls under the limit of detection. Of interest, we found the COL4A2 collagen type IV alpha 2 chain, a type IV collagen associated with laminin, nidogen, and heparan sulfate proteoglycans that constitute a major component of nearly all basement membranes. The matricellular protein thrombospondin 2 (THBS2), a fibrosis marker that is elevated in SSc skin and has been shown to stimulate collagen expression (Kajihara et al., 2012), was also detected in Spag17/Sox2-Cre KO fibroblasts. The metalloproteinases MMP14 and MMP2 and the metalloproteinase inhibitor TIMP3, involved in collagen remodeling and implicated in fibrosis (Giannandrea & Parks, 2014), were more abundant in ECM generated by SPAG17-deficient fibroblasts (Table S5C).
SPAG17 regulates myofibroblast transition
Accumulation of myofibroblasts originating from quiescent stromal precursor cells, and their persistence within lesional tissues, is a canonical hallmark of SSc that underlies intractable fibrosis in affected organs (Hinz & Lagares, 2020). To examine the impact of SPAG17 on myofibroblast transition, we first analyzed the expression of SPAG17 and the myofibroblast marker ACTA2 by scRNA-seq. Remarkably, SPAG17 was virtually undetectable in activated myofibroblasts in SSc skin biopsies, with SPAG17 and ACTA2 showing a striking anticorrelation (Figure S2B).
Next, we investigated how loss of SPAG17 impacted in vitro myofibroblast transition in human and mouse fibroblasts and endothelial cells in culture. First, we used human skin fibroblasts with SPAG17 deleted using CRISPR/Cas9 technology ( Figures S10 and S11A), and skin fibroblasts explanted from Spag17/Sox2-Cre KO mice (Figure S11B). Notably, both human and mouse fibroblasts lacking SPAG17 showed increased ASMA expression compared to control fibroblasts (Fig. 4A and C). Moreover, loss of SPAG17 in fibrtoblasts was associated with markedly increased in vitro collagen production (Fig. 4B, D, E and Figure S11C–E). Pursuing a similar experimental strategy to examine SPAG17 regulation of cell fates in human and mouse endothelial cells, we found that CRISPR/Cas9-mediated deletion of SPAG17 in human microvascular endothelial cells (HMECs) was associated with increased ASMA expression (Fig. 4F and Figure S12A), altered cadherin 5 localization, and reduced focal adhesions (Fig. 4G and Figure S12B). Moreover, lung endothelial cells explanted from Spag17/Sox2-Cre KO mice showed evidence of spontaneous EndoMT, with increased ASMA and reduced CD31 and VE-cadherin compared to wild-type ECs (Fig. 4H–J and Figure S12C and D).
Figure 4:

Loss of SPAG17 triggers myofibroblast transition and stimulates collagen production. Quantification of fluorescence intensity was perfomed for all the experiments in 30 cells/sample using ImageJ. (a and b) Human skin fibroblasts were transfected with CRISPR/Cas9 and immunolabelled with antibodies to ASMA or procollagen I (n=3). (c-e) Confluent fibroblasts isolated from WT and KO mice were immunolabelled with ASMA, procollagen I or collagen I, (n=6). (f and g) Human microvascular endothelial cells were transfected with SPAG17 CRISPR and virus control, and immunolabelled with antibodies to ASMA, Cadherin5 or SPAG17, (n=3). (h-j) Primary lung endothelial cells from WT and KO mice were immunolabelled with antibodies to ASMA, VE-cadherin or CD31, (n=3). Results are means ± SEM). *p<0.05.
A near-universal mechanism underlying fibrotic myofibroblast transition involves Smad-dependent canonical TGF-β signaling (Lafyatis, 2014, Mori et al., 2003). Because we found that TGF-β signaling was altered in Spag17/Sox2-Cre KO fibroblasts (Fig. 3E), we sought to investigate SPAG17 regulation of TGF-β signaling in myofibroblast transition. Mouse fibroblasts lacking SPAG17 showed increased expression of TGFBRII (Fig. 5A and Figure S13B). Remarkably, we consistently observed increased nuclear accumulation and activation of Smad2 and Smad3 in Spag17/Sox2-Cre KO fibroblasts, even in the absence of exogenous TGF-β (Fig. 5B–C and Figure S13C–D). Next, we investigated whether pharmacological inhibition of TGF-β signaling would reverse the profibrotic phenotype. Remarkably, we noted reduced ASMA, procollagen I and nuclear accumulation of Smad2/3 in Spag17/Sox2-Cre KO fibroblasts treated with distinct inhibitors (Figure S14). Taken together, these ex vivo studies with explanted human and mouse cells provide robust evidence that loss of SPAG17 is accompanied by spontaneous profibrotic reprogramming with myofibroblast transdifferentiation driven in part through canonical TGF-β signaling pathways.
Figure 5:

Loss of SPAG17 in fibroblasts is associated with aberrant intracellular TGF-β signaling. Confluent skin fibroblasts from WT and KO mice were incubated in media with and without 10 ng/ml TGF-β for 72 h, and immunolabelled using antibodies to TGFBRII, SMAD2/3 and pSMAD2/3. (a) Quantification of TGFBRII fluorescence intensity (30 cells/sample) (n=5). (b-c) Quantification of nuclear and cytoplasmic levels of (b) SMAD2/3 (n=6). (c) pSMAD2/3 (n=3). Results are means ± SEM. *p<0.05.
DISCUSSION
The pathogenesis of SSc remains poorly understood (Allanore et al, 2015). In particular, how fibrosis develops synchronously in multiple organs in SSc is an unanswered question with fundamental treatment implications. Myofibroblast accumulation and sustained activation appear to play fundamental roles in fibrosis in SSc (Bhattacharyya et al, 2011). Myofibroblasts in the fibrotic skin arise through reprogramming of quiescent progenitor cells via cell-autonomous epigenetic alterations and altered metabolic states, as well as transient transcriptional changes triggered in response to TGF-β, Hedgehog, Wnts and other morphogens. Here, we report that expression of SPAG17 is markedly downregulated in SSc skin biopsies and skin fibroblasts, and loss of SPAG17 in mice is associated with skin fibrosis phenocopying human SSc. Taken together with our results using isolated human and mouse fibroblasts and endothelial cells, these observations support a previously unrecognized cell-autonomous function for SPAG17 as a regulator of myofibroblast differentiation and fibrotic responses.
The pleiotropic gene SPAG17 is associated with both motile cilia (Teves et al, 2013; Andjelkovic et al, 2018; Abdelhamed et al, 2020) and primary cilia (Teves et al, 2016). In the mouse, global deletion of Spag17 impairs primary cilia formation, resulting in developmental defects and early neonatal death (Teves et al, 2015), while its conditional deletion leads to male infertility (Kazarian et al, 2018). In humans, homozygous null mutations in SPAG17 appear to be exceedingly rare, but were described in children with developmental defects (Córdova-Fletes et al, 2017) and associated with male infertility (Xu et al, 2018), consistent with phenotypes in knockout mice (Teves et al, 2015; Kazarian et al, 2018). Genome-wide association studies have revealed association of the SPAG17 locus with short stature (Weedon & Frayling, 2008; Weedon et al, 2008; Takeuchi et al, 2009; Kim et al, 2010; Zhao et al, 2010; N’Diaye et al, 2011; Wood et al, 2014; van der Valk et al, 2015). Interestingly, upregulation of SPAG17 has been described in several cancers (breast, colon, gastric, lung, and melanoma) (Silina et al, 2011). While these associations of SPAG17 with a variety of pathological conditions suggest important biological roles (Teves et al, 2016), the pleiotropic function and mechanisms of action of SPAG17 remain poorly understood.
We observed reduced SPAG17 expression in SSc skin biopsies, and an inverse correlation with skin involvement. This finding was replicated in independent patient cohorts. Remarkably, reduced SPAG17 expression in the skin was associated with the upregulation of genes important for ECM organization, focal adhesion, and growth factors. Moreover, loss of Spag17 in the mouse was associated with spontaneous skin fibrosis phenocopying the changes seen in patients with SSc.
We further demonstrated reduced SPAG17 expression in fibroblasts explanted from SSc skin biopsies, suggesting that the loss of SPAG17 in SSc is an inherent and durable feature of these fibroblasts. Indeed, genome-wide transcriptome analysis of SSc skin fibroblasts demonstrated a 4-fold reduced expression of SPAG17 compared to healthy fibroblasts (Chadli et al, 2019).
Loss-of-function experiments with explanted human and mouse fibroblasts and ECs demonstrated upregulation of profibrotic gene signatures, suggesting a powerful influence of SPAG17 in modulating cellular fibrotic responses. The mechanisms accounting for reduced SPAG17 in SSc skin are currently unknown. While we found no evidence for the association of SSc with hypomorphic genetic variants of SPAG17 in GWAS databases (unpublished), results from ATAC-seq analysis suggest that cell-type-specific epigenetic mechanisms leading to altered chromatin landscape at the SPAG17 locus might be responsible for reduced SPAG17 expression in SSc fibroblasts and ECs. Further studies are needed to determine whether this is due to DNA methylation or histone modifications, mechanisms that are increasingly linked to the pathogenesis of SSc (Tsou et al, 2021; Tsou et al., 2021b). Reprogramming of metabolically quiescent mesenchymal progenitor cells into activated profibrotic myofibroblasts, a fundamental step in the evolution of all forms of pathological fibrosis, is triggered by profibrotic growth factors, cytokines and morphogens, as well as biomechanical cues, mediated through a variety of receptors and intracellular signaling pathways (Pakshir et al, 2020). We report here that loss of function of SPAG17 leads to constitutive activation of canonical TGF-β signaling, which might account for the observed myofibroblast transitions and pathological fibrosis (Fig. 6). Further studies at the molecular level will be necessary to dissect the SPAG17-interacting protein partners and elucidate their interaction with components of TGF-β signaling pathways.
Figure 6:

Proposed mechanism linking SPAG17 downregulation and SSc pathogenesis. The mechanistic model implicates an epigenetic downregulation in the SPAG17 gene leading to constitutive activation of TGF-β signaling followed by myofibroblast transition and consequently expression of fibrotic genes underlying pathological fibrosis.
The present studies identify an entirely newl cell-autonomous role for SPAG17 in the negative regulation of fibrotic responses. The mechanisms regulating SPAG17 expression are currently unknown, as are the factors accounting for reduced SPAG17 mRNA in SSc skin. Profiling differences between skin biopsy-derived healthy and SSc mesenchymal cells indicated altered chromatin landscape at the SPAG17 locus, suggesting that epigenetic mechanisms might be responsible for reduced SPAG17 expression in SSc, and could represent a possible therapeutic target using epigenetic modifier drugs. Based on the present observations, we propose here a distinct paradigm for SSc pathogenesis implicating SPAG17 loss as a driver of fibrosis. Further studies on the regulation, expression and role of SPAG17 in fibrosis and the underlying mechanisms may inform the search for innovative therapies for SSc and other fibrotic conditions.
MATERIALS AND METHODS
A graphic outline of the orthogonal experimental strategies employed in this study is shown in Supplementary Fig. 15.
Human Subjects
All human studies were approved by Northwestern University Institutional Review Board and subjects provided their written, informed consent. Strategic Pharma-Academic Research Consortium (SPARC) cohort. Patients fulfilling American College of Rheumatology classification criteria for SSc (van den Hoogen et al, 2013) enrolled in the SPARC longitudinal observational cohort underwent regular follow-up evaluations and were used for these studies (Table S1) (Roberson et al, 2022).
RNA isolation and analysis
Skin biopsy-derived transcriptome libraries were prepared and analyzed following standard procedures (Roberson et al, 2022; Skaug et al, 2020). Detailed information is provided in Supplementary material.
Single cell RNA-seq
Skin biopsies from SSc (n=22) patients and healthy (n=18) volunteers were processed for single cell RNA-seq (Davis et al, 2021). Additional details are provided in Supplementary material.
Assay for transposase-accessible chromatin using sequencing (ATAC-Seq)
Participants were recruited from the University of Michigan Scleroderma Program. The study was approved by the institutional review board. Upon obtaining informed consent, subjects (6 patients with dcSSc and 6 healthy controls matched for age, sex, and ethnicity) underwent skin biopsies, and cultures of skin fibroblast and microvascular endothelial cells were established. Confluent cells were subjected to ATAC-seq as previously described (Tsou et al, 2021). Additional details are provided in Supplementary material.
SPAG17 knockout mice
All animal studies were conducted in accordance with protocol AM10297 approved by the VCU Institutional Animal Care and Use Committee. Spag17-mutant mice were generated by introducing a deletion in exon 5 that leads to a premature stop codon and absent SPAG17 expression (Kazarian et al., 2018). Skin fibroblasts and lung endothelial cells were obtained from Spag17/Sox2-Cre line, epiblast-derived specific conditional mutant mice. Additional detailed information is provided in Supplementary material.
Biomechanical stiffness
Stiffness in skin from wild-type and knockout mice was determined by atomic force microscopy as previously described (Sicard et al, 2017). Additional details are provided in Supplementary material.
Collagen alignment
Collagen alignment was determined by Second Harmonic Generation (SHG) microscopy in skin mouse samples as previously described (Chen et al., 2012). Additional details are provided in Supplementary material.
SPAG17 CRISPR/Cas9 knockout cells
Human dermal fibroblasts (ATCC® PCS-201-012™) and human microvascular endothelial cells (HMEC, ATCC® CRL-3243™) were cultured following manufacturer’s recommendations and transfected with CRIPR/Cas9 vectors (Santa Cruz sc410482, Santa Cruz sc418922 or Lenti-U6-SPAG17-EPCG-VSVG, Figure S16A) following standar methods. Detailed procedures are provided in Supplementary material.
Immunocytochemistry
Cells were fixed in 4% formalin (Sigma Aldrich, St. Louis, MO) and processed for immunolabelling (Teves et al, 2015) using the indicated primary antibodies (Table S7). Detailed procedures are provided in Supplementary material.
RNA-seq studies in mouse fibroblasts
Confluent skin fibroblasts from neonate wild-type and knockout (Spag17/Sox2-Cre) mice were subjected to RNA-seq using the Illumina platform. Alignment was performed to the mouse reference genome (NCBI/mm10). Analysis was performed using standard methods as indicated in Supporting material.
Preparation of cell-derived ECM for proteomic analysis
Mouse primary dermal fibroblasts were obtained from the back skin from five mice per genotype at P0 and P1 postnatal age for each genotype. Both female and male mice were used. Fibroblasts were cultured and processed as previously reported (Franco-Barraza et al, 2016;). Additional detailed information is provided in Supplementary material.
Data availability
The mass spectrometry proteomics data can be found at http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD019869, hosted at the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al, 2019) with the dataset identifier PXD019869. The RNA-seq data from cultured fibroblasts have been deposited to the BioSample database with accession PRJNA776825 and can be found at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA776825&o=acc_s%3Aa and https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA776825.
Statistical analysis
GraphPad Prism 8 software was used for statistical analysis. Data are presented as means ± standard error. Data were analyzed comparing the means from two groups by Student’s t-test. Samples were considered significantly different when the p-value was < 0.05. Statistics for large data used analysis in R. Detailed information for each method is provided in Supplementary material.
Supplementary Material
ACKNOWLEDGMENT
We thank Drs. Hui Chen from the Mass Spectrometry Core and George Chlipala from the Research Informatics Core at the University of Illinois at Chicago for their technical assistance, and Martin Davis from the Naba lab for technical assistance with the decellularization of cell-derived ECMs and Dr. Ljuba Lyass from Skin Tissue Engineering and Morphology (STEM) Core at Feinberg School of Medicine for technical assistance. MET and JV are supported by a research award from the Scleroderma Foundation and the Rheumatology Research Foundation. ER is partially supported by the Washington University in St. Louis Institute for Clinical and Translational Science, funded, in part, by grant #UL1 TR000448 and by the WUSTL Rheumatic-diseases research resource-based center (NIH P30-AR073752). SA was supported by a grant from Scleroderma Foundation and is supported by grant from the NIAMS/NIH R01AR073284. AHS was supported by NIH- NIAID R01AI097134. JEG is supported by NIH-P30-AR075043. JFS was supported by the NIH-HD-37416. AN and JV were supported by a Catalyst Award from the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust (C-088). Proteomics services were provided by the UIC Research Resources Center Mass spectrometry Core which was established in part by a grant from The Searle Funds at the Chicago Community Trust to the Chicago Biomedical Consortium. Bioinformatic analyses were performed by the UIC Research Informatics Core, supported in part by the National Center for Advancing Translational Sciences (NCATS, Grant UL1TR002003).
Footnotes
CONFLICT OF INTEREST
The authors declare that Drs. Naba, Varga and Teves receive research support unrelated to this work from Boehringer-Ingelheim.
REFERENCES
- Abdelhamed Z, Lukacs M, Cindric S, Ali S, Omran H, Stottmann RW. A novel hypomorphic allele of Spag17 causes primary ciliary dyskinesia phenotypes in mice. Dis Model Mech 2020;13:dmm045344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, Varga J, Systemic sclerosis. Nat Rev Dis Primers 2015;1:15002. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Minic P, Vreca M, Stojiljkovic M, Skakic A, Sovtic A, et al. Genomic profiling supports the diagnosis of primary ciliary dyskinesia and reveals novel candidate genes and genetic variants. PLoS One 2018;13:e0205422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol 2011;8:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Lafyatis R, Burkly LC. Increased dermal collagen bundle alignment in systemic sclerosis is associated with a cell migration signature and role of Arhgdib in directed fibroblast migration on aligned ECMs. PLoS One 2019;29;12(6):e0180751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadli L, Sotthewes B, Li K, Andersen SN, Cahir-McFarland E, Cheung M, et al. Identification of regulators of the myofibroblast phenotype of primary dermal fibroblasts from early diffuse systemic sclerosis patients. Sci Rep 2019;9:4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat. Protoc 2012;7:654–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Córdova-Fletes C, Becerra-Solano LE, Rangel-Sosa MM, Rivas-Estilla AM, Alberto Galán-Huerta K, Ortiz-López R, et al. Uncommon runs of homozygosity disclose homozygous missense mutations in two ciliopathy-related genes (SPAG17 and WDR35) in a patient with multiple brain and skeletal anomalies. Eur J Med Genet 2017;61:161–67. [DOI] [PubMed] [Google Scholar]
- Davis FM, Tsoi LC, Melvin WJ, denDekker A, Wasikowski R, Joshi AD, et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J Exp Med 2021;218: e20201839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390:1685–99. [DOI] [PubMed] [Google Scholar]
- Distler JH, Feghali-Bostwick C, Soare A, Asano Y, Distler O, Abraham DJ. Review: Frontiers of Antifibrotic Therapy in Systemic Sclerosis. Arthritis Rheumatol 2017;69: 257–67. [DOI] [PubMed] [Google Scholar]
- Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol 2019;15:705–30. [DOI] [PubMed] [Google Scholar]
- Franco-Barraza J, Beacham DA, Amatangelo MD, Cukierman E. Preparation of Extracellular Matrices Produced by Cultured and Primary Fibroblasts. Curr Protoc Cell Bio 2016;71:10.9.1–10.9.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech 2014;7:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, McCulloch CA, Coelho NM. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp Cell Res 2019;379:119–28. [DOI] [PubMed] [Google Scholar]
- Hinz B, Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol 2020;16:11–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajihara I, Jinnin M, Yamane K, Makino T, Honda N, Igata T, et al. Increased accumulation of extracellular thrombospondin-2 due to low degradation activity stimulates type I collagen expression in scleroderma fibroblasts. Am J Pathol 2012;180:703–14. [DOI] [PubMed] [Google Scholar]
- Kazarian E, Son H, Sapao P, Li W, Zhang Z, Strauss JF, et al. SPAG17 Is Required for Male Germ Cell Differentiation and Fertility. Int J Mol Sci 2018;19:1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee HI, Park T, Kim K, Lee JE, Cho NH, et al. Identification of 15 loci influencing height in a Korean population. J Hum Genet 2010;55:27–31. [DOI] [PubMed] [Google Scholar]
- Lafyatis R Transforming growth factor β―at the centre of systemic sclerosis. Nat Rev Rheumatol 2014;10:706–19. [DOI] [PubMed] [Google Scholar]
- Mori Y, Chen SJ, Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum 2003;48:1964–78. [DOI] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: Tools and insights for the “omics” era. Matrix Bio 2016;49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’Diaye A, Chen GK, Palmer CD, Ge B, Tayo B, Mathias RA, et al. Identification, replication, and fine-mapping of Loci associated with adult height in individuals of african ancestry. PLoS Genet 2011;7: e1002298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 2033;75,4646–58. [DOI] [PubMed] [Google Scholar]
- Pakshir P, Hinz B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol 2018;68–69:81–93. [DOI] [PubMed] [Google Scholar]
- Pakshir P, Noskovicova N, Lodyga M, Son DO, Schuster R, Goodwin A, et al. The myofibroblast at a glance. J Cell Sci 2020;133:jcs227900. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019;47:D442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson EDO, Carns M, Cao L, Aren K, Goldberg IA, Morales-Heil DJ, et al. RNA-Seq analysis identifies alterations of the primary cilia gene SPAG17 and SOX9 locus non-coding RNAs in systemic sclerosis. Arthritis Rheumatol 2022; 10.1002/art.42281. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent JL, Li Z, Aliprantis AO, Greenblatt M, Lemaire R, Wu MH, et al. Identification of Optimal Mouse Models of Systemic Sclerosis by Interspecies Comparative Genomics. Arthritis Rheumatol 2016;68:2003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silina K, Zayakin P, Kalniņa Z, Ivanova L, Meistere I, Endzeliņš E, et al. Sperm-associated Antigens as Targets for Cancer Immunotherapy: Expression Pattern and Humoral Immune Response in Cancer Patients. J Immunother 2011;34:28–44. [DOI] [PubMed] [Google Scholar]
- Skaug B, Khanna D, Swindell WR, Hinchcliff ME, Frech TM, Steen VD, et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis 2020;79:379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard D, Fredenburgh LE, Tschumperlin DJ. Measured pulmonary arterial tissue stiffness is highly sensitive to AFM indenter dimensions J Mech Behav Biomed Mater 2017;74:118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi F, Nabika T, Isono M, Katsuya T, Sugiyama T, Yamaguchi S, et al. Evaluation of genetic loci influencing adult height in the Japanese population. J Hum Genet 2009;54:749–52. [DOI] [PubMed] [Google Scholar]
- Teves ME, Zhang Z, Costanzo RM, Henderson SC, Corwin FD, Zweit J, et al. Sperm-associated antigen-17 gene is essential for motile cilia function and neonatal survival. Am J Respir Cell Mol Biol 2013;48:765–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves ME, Sundaresan G, Cohen DJ, Hyzy SL, Kajan I, Maczis M, et al. Spag17 deficiency results in skeletal malformations and bone abnormalities. PLoS One 2015;10:e0125936–e0125936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves ME, Nagarkatti-Gude DR, Zhang Z, Strauss JF 3rd. Mammalian axoneme central pair complex proteins: Broader roles revealed by gene knockout phenotypes. Cytoskeleton 2016;73:3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou PS, Palisoc PJ, Ali M, Khanna D, Sawalha AH. Genome-wide reduction in chromatin accessibility and unique transcription factor footprints in endothelial cells and fibroblasts in scleroderma skin. Arthritis Rheumatol 2021;73:1501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou PS, Varga J, O’Reilly S. Advances in epigenetics in systemic sclerosis: molecular mechanisms and therapeutic potential. Nat Rev Rheumatol 2021;17:596–607. [DOI] [PubMed] [Google Scholar]
- van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of rheumatology/European League against rheumatism collaborative initiative. Ann Rheum Dis 2013;72:1747–55. [DOI] [PubMed] [Google Scholar]
- van der Valk RJ, Kreiner-Møller E, Kooijman MN, Guxens M, Stergiakouli E, Sääf A, et al. A novel common variant in DCST2 is associated with length in early life and height in adulthood. Hum Mol Gen 2015;24:1155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 2007;117:557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann ER, Varga J. Emerging targets of disease-modifying therapy for systemic sclerosis. Nat Rev Rheumatol 2019;15:208–224. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Frayling TM. Reaching new heights: insights into the genetics of human stature. Trends in Genetics 2008;24:595–603. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet 2008;40:575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014;46:1173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Sha YW, Mei LB, Ji ZY, Qiu PP, Ji H,. A familial study of twins with severe asthenozoospermia identified a homozygous SPAG17 mutation by whole-exome sequencing. Clin Genet 2018;93:345–49. [DOI] [PubMed] [Google Scholar]
- Zhao J, Li M, Bradfield JP, Zhang H, Mentch FD, Wang K, et al. The role of height-associated loci identified in genome wide association studies in the determination of pediatric stature. BMC Med Genet 2010;11: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018; 36: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong WH, Tan YC, Yap SJ, Ng KP. ClicO FS: an interactive web-based service of Circos. Bioinformatics 2015; 31: 3685–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc 2012;7:654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Tsoi LC, Melvin WJ, denDekker A, Wasikowski R, Joshi AD, et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J Exp Med 2021;218: e20201839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 2020; 15: 1484–1506. [DOI] [PubMed] [Google Scholar]
- Franco-Barraza J, Beacham DA, Amatangelo MD, Cukierman E. Preparation of Extracellular Matrices Produced by Cultured and Primary Fibroblasts. Curr Protoc Cell Bio 2016; 71: 10.9.1–10.9.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frech TM, Shanmugam VK, Shah AA, Assassi S, Gordon JK, Hant FN, et al. Treatment of early diffuse systemic sclerosis skin disease. Clin Exp Rheumatol 2013;31:166–71. [PMC free article] [PubMed] [Google Scholar]
- Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol 2013;133:1979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 2008;9:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni R, Teves ME, Han AX, McAllister JM, Strauss JF 3rd. Colocalization of Polycystic Ovary Syndrome Candidate Gene Products in Theca Cells Suggests Novel Signaling Pathways. J Endocr Soc 2019;3:2204–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med 2017;13:9(420):eaal3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550.3. F. M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 2012;40:4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hynes RO. Enrichment of Extracellular Matrix Proteins from Tissues and Digestion into Peptides for Mass Spectrometry Analysis. J Vis Exp 2015;101:e53057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Pearce OMT, Del Rosario A, Ma D, Ding H, Rajeeve V, et al. Characterization of the Extracellular Matrix of Normal and Diseased Tissues Using Proteomics. J Proteome Res 2017;16:3083–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 2033;75,4646–58. [DOI] [PubMed] [Google Scholar]
- Roberson EDO, Carns M, Cao L, Aren K, Goldberg IA, Morales-Heil DJ, et al. RNA-Seq analysis identifies alterations of the primary cilia gene SPAG17 and SOX9 locus non-coding RNAs in systemic sclerosis. Arthritis Rheumatol 2022; 10.1002/art.42281. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug B, Khanna D, Swindell WR, Hinchcliff ME, Frech TM, Steen VD, et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis 2020;79:379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves ME, Sundaresan G, Cohen DJ, Hyzy SL, Kajan I, Maczis M, et al. Spag17 deficiency results in skeletal malformations and bone abnormalities. PLoS One 2015;10:e0125936–e0125936.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou PS, Palisoc PJ, Ali M, Khanna D, Sawalha AH. Genome-wide reduction in chromatin accessibility and unique transcription factor footprints in endothelial cells and fibroblasts in scleroderma skin. Arthritis Rheumatol 2021;73:1501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchierelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014; 32: 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data can be found at http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD019869, hosted at the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al, 2019) with the dataset identifier PXD019869. The RNA-seq data from cultured fibroblasts have been deposited to the BioSample database with accession PRJNA776825 and can be found at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA776825&o=acc_s%3Aa and https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA776825.