

Abstract
Objectives:
To reveal the heterogeneity of different cell types of osteoarthritis (OA) synovial tissues at a single-cell resolution, and determine by novel methodology whether bulk-RNA-seq data could be deconvoluted to create in silico scRNA-seq data for synovial tissue analyses.
Methods:
OA scRNA-seq data (102,077 synoviocytes) were provided by 17 patients undergoing total knee arthroplasty; 9 tissues with matched scRNA-seq and bulk RNA-seq data were used to evaluate six in silico gene deconvolution tools. Predicted and observed cell types and proportions were compared to identify the best deconvolution tool for synovium.
Results:
We identified seven distinct cell types in OA synovial tissues. Gene deconvolution identified three (of six) platforms as suitable for extrapolating cellular gene expression from bulk RNA-seq data. Using paired scRNA-seq and bulk RNA-seq data, an “arthritis” specific signature matrix was created and validated to have a significantly better predictive performance for synoviocytes than a default signature matrix. Use of the machine learning tool, Cell-type Identification By Estimating Relative Subsets of RNA Transcripts x (CIBERSORTx), to analyze rheumatoid arthritis (RA) and OA bulk RNA-seq data yielded proportions of T cells and fibroblasts that were similar to the gold standard observations from RA and OA scRNA-seq data, respectively.
Conclusion:
This novel study revealed heterogeneity of synovial cell types in OA and the feasibility of gene deconvolution for synovial tissue.
Keywords: Osteoarthritis, single-cell RNA sequencing, gene deconvolution, synovial tissue, synovitis
Introduction
Osteoarthritis (OA) is a leading cause of joint pain, disability and health-care costs worldwide1. Nowadays, synovitis has been recognized to play an important role in the pathogenesis of OA2. Though many studies have shown that different cell populations, including synovial fibroblasts3, 4 and immune cells5, 6 play critical roles in OA, it is still not clear which cell types drive the pathogenesis. Considering that synovium is a highly heterogenous tissue, single-cell analyses provide great advantages over bulk RNA-seq and facilitate exploration of the unique synovial cell heterogeneities of OA.
The current study performed a comparative analysis of bulk and single cell RNA-seq of paired samples to evaluate the ability of six different in silico gene deconvolution algorithms to extract simulated single-cell level data from bulk RNA data. The cellular heterogeneity of OA synovium was revealed and a specific signature was generated and validated by publicly available datasets.
Methods
Results
Cell-types identified in OA synovia
We profiled 93,208 synovial cells from a total of fourteen individuals with knee OA. Unsupervised clustering with the top 2,000 variable genes revealed great heterogeneity of cell types. Seven distinct cell populations (Figure 1A) were confidently identified on the basis of differentially expressed genes within each cluster (Supplementary Table S1), including (from most to least relative abundance): fibroblasts (59%), antigen presenting cells (APCs) (13.6%), T cells (11.4%), endothelial cells (ECs) (10%), mural cells (3%), B cells (1.8%) and mast cells (1%). The distinct nature of each major cell type was demonstrated by a representative gene (Figure 1B) and the top 5 genes expressed by each cluster (Figure 1C).
Figure 1. Single-cell transcriptomic cell atlas of human OA synovium.
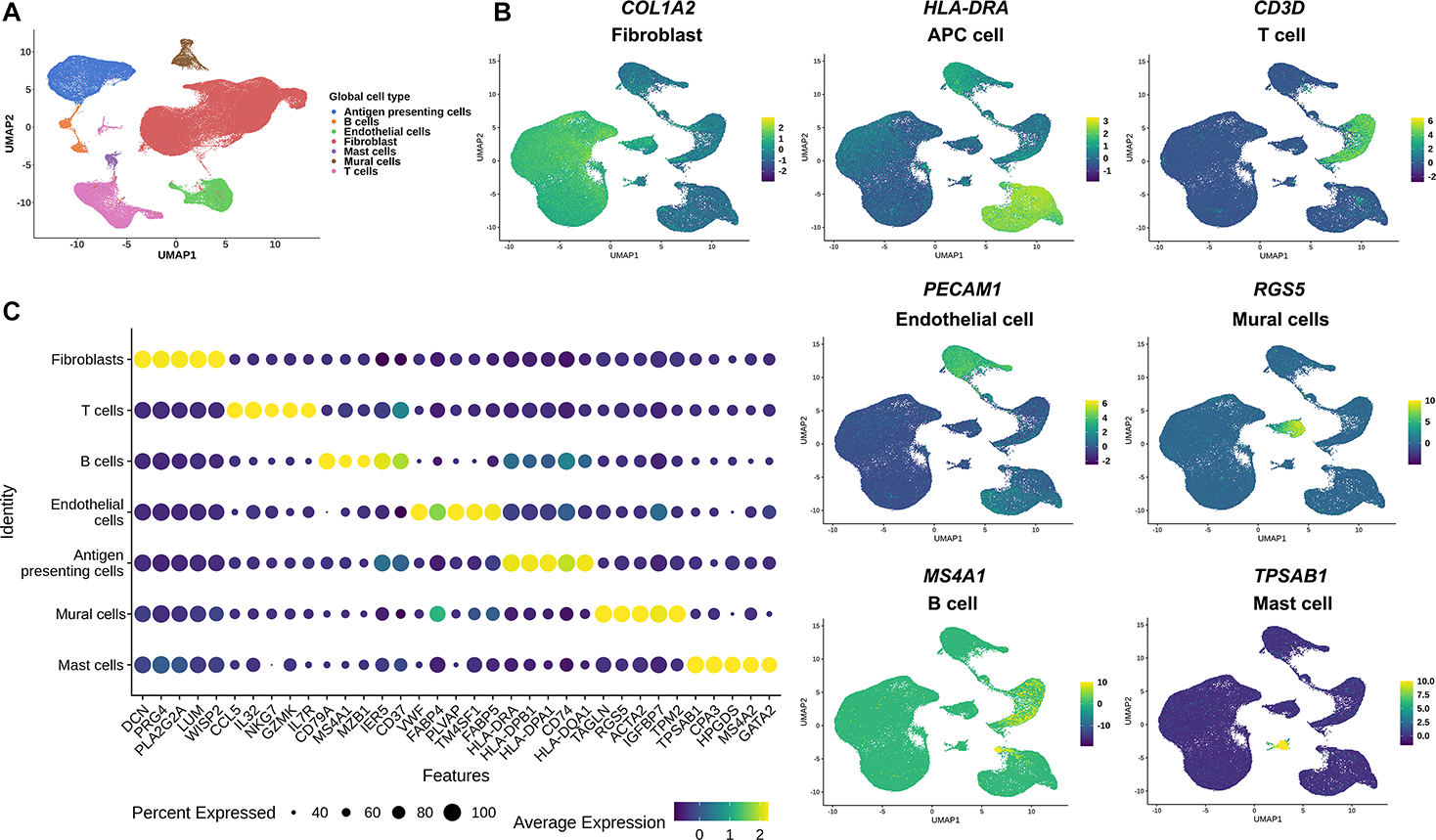
(A) UMAP plot of scRNA-seq showed unsupervised clusters colored according to putative cell types among a total of 93,208 cells in OA synovium. 59%, 13.6%, 11.4%, 10%, 3%, 1.8% and 1% of total acquired cells were fibroblast, APC cells, T cells, endothelial cells, mural cells, B cells and mast cells. (B) Expression of the selected top marker genes for each cell type is shown in UMAP plots. (C) Expression level of the top 5 feature genes in seven distinct cell types is mapped in dot plots (Color stands for the average expression of each gene and the size stands for percent expressed). OA, osteoarthritis; UMAP: Uniform manifold approximation and projection; ST, synovial tissue; APC, antigen presenting cell.
Of the general cell types, we identified a substantial number of T cells, more than reported in our previous study (Supplementary Table S2)7. Cell subtype analysis revealed the presence of both CD4+ and CD8+ T cells (Supplementary Figures S1A&B). Seven different sub-types of CD4+ T cells were identified: activated (5.95%), memory (24.6%), naïve (36.4%), T helper 1 cells (Th1) (23.1%), T helper 2 cells (Th2) (1.20%), T helper 17 cells (Th17) (2.00%) and regulatory T cells (Treg) (6.76%) (Supplementary Figure S1A). Five distinct sub-cluster of CD8+ T cells were identified: cytotoxic (19.3%), naive (16.0%), natural killer cells (NK) (4.99%), proliferating (5.14%), and effector memory T cells (TEM) (54.6%) (Supplementary Figure S1B). The top 5 genes for each sub-type demonstrated their distinctly different nature (Supplementary Figures S1C&D; Supplementary Tables S3&4). Of note, in Th17 cell sub-type, IL-17 was not identified, but several Th17 cell differentiation-related genes (FCER1G, KLRB1, AHR, GATA3, HLA-DRA, IL1R1, IL23R, IL2RA, NFKB1 and NFKBIA) were enriched (Supplementary Figure S1D; Supplementary Table S4). In the trajectory analysis of CD4+ T cells, naïve CD4+ T cells were mainly located on the right side of branch 2 while activated CD4+ T cells and Tregs were mostly located on the left side of branch 3 (Supplementary Figure S1E). Pseudotime analysis revealed that IL-2 and IL17A were highly expressed at the starting point while mature CD4+ T cell-related genes (GATA3, IL4R, FOXP3, IL21R, TBX21 and IL12RB1) were highly expressed at the end point (Supplementary Figure S1G). Trajectory analysis of CD8+ T cells revealed that proliferating CD8+ T cells were mostly allocated at the up-left branch of node 3, well separated from the other sub-types (Supplementary Figure S1F). Pseudotime analysis showed that, at the root node, proliferating CD8+ T cells highly expressed cell proliferation-related genes CTLA4 and IFI27. Along with the pseudotime, the cytotoxic CD8+ T cells (higher expression of GZMB, NKG7) were located in the middle. More memory-related genes (CCL5, IL7R, TCF7) were highly expressed at the ending point (Supplementary Figure S1H).
Cell-type specific analysis of APCs revealed four unique cell subtypes, namely transitional macrophages (T-Mφ) (4.89%), fibrotic immune regulated macrophages (Fibrotic IR-Mφ) (9.12%), interferon stimulated macrophages (IFNS-Mφ) (3.61%) and S100A8/9hi macrophages (S100A8/9hi-Mφ) (4.86%) (Supplementary Figure S2A). Both proinflammatory (CCL3, CCL3L1) and inflammation resolving genes (IGF1, MRC1) were expressed in T-Mφ (Supplementary Figure S2B; Supplementary Table S5). Trajectory analysis showed that T-Mφs were equally distributed in each branch, further confirming that this unique subtype represents a transitional group of macrophages between inflammatory (I-Mφ) and immune regulated macrophages (IR-Mφ) (Supplementary Figure S2C). Fibrosis-related genes (FN1, SPP1)8, 9 were highly expressed in fibrotic IR-Mφ. Interferon-induced genes (EPSTI1, STAT1, MX1, IFI44L, ISG15)10 were highly expressed in IFNS-Mφ, suggesting functions related to inflammation. Genes associated with scavenging of cell (SERPINB2, CD52) and protein fragments (S100A8, S100A9) were highly expressed in S100A8/9hi-Mφs suggesting functions related to autodebridement (“cleanup”) (Supplementary Figure S2B; Supplementary Table S5). Trajectory analysis suggested that dendritic cells (DCs), IFNS-Mφs and S100A8/9hi-Mφs were related to inflammation as they were located after branches 2 and 3, similar to I-Mφ. Fibrotic IR-Mφs were enriched after branch 4, suggesting functions related to immune regulation (Supplementary Figure S2C). Pseudotime analysis revealed that inflammation-related genes such as IL6, IL1A, IL1B, TLR2 and TNF were related to I-Mφs (Supplementary Figure S2D). Inflammation resolving genes (FGCR3A, CD163, MRC1) were highly expressed in relation to IR-Mφs (Supplementary Figure S2D).
Comparison of in silico gene deconvolution algorithms
Using the nine paired bulk RNA-seq and scRNA-seq datasets from nine knee OA patients, we investigated six platforms for deconvolution of bulk RNA-seq into simulated single cell data with default parameter settings (Figure 2A). We observed better correlations of bulk RNA-seq and scRNA-seq data with MCP-counter, QUANTISQ and CIBERSORT than EPIC, TIMER and XCELL (Figure 2B&C). Due to the ability to customize signature matrices with CIBERSORTx and MCP-counter, we chose these two platforms to optimize matching between in silico predicted and measured scRNA-seq data (Supplementary Data 1 for CIBERSORTx and Supplementary Data 2 for MCP-counter). Based on Pearson’s Correlation Coefficients (PCCs), customization significantly improved (p=0.049) the overall correlation of predicted to actual scRNA-seq data in CIBERSORTx: PCC 0.58 (95% confidence interval (CI) [0.30~0.76]) using LM22 (CIBERSORTx default); PCC 0.77 (95% CI [0.72~0.89]) using a customized signature matrix with PCC>0.70 for eight of nine individual cases (Figure 2D&E). No improvement of PCC was observed in MCP-counter (Supplementary Figure S3A), but seven of nine individual cases had PCC>0.70 using the MCP-counter-based customized signature matrix (Supplementary Figure S3B). A low prediction accuracy in Case 3 appeared to be caused by less gene coverage from scRNA-seq, i.e, median genes per cell 1,820 in contrast to >2,000 for other cases. In addition, no significant differences were observed in predicted vs measured cell composition in each case (Figure 2F; Supplementary Figure S3C).
Figure 2. Comparisons of gene deconvolution tools with fix signature matrixes, generation of “synovium” specific signature matrices.
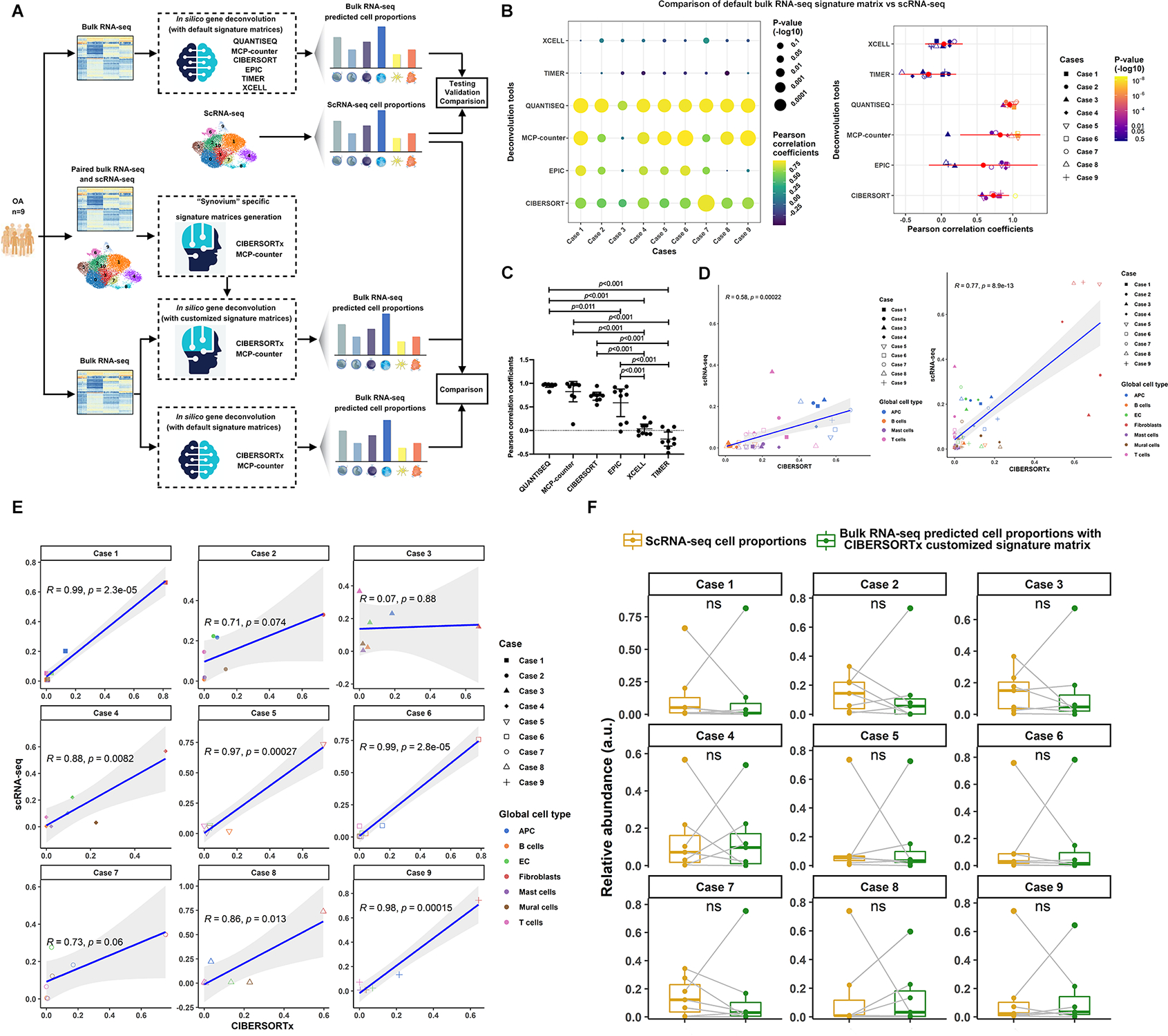
(A) Flowchart showed the experimental strategy for comparison of gene deconvolution tools, generation of “synovium” specific signature matrix and optimization. (B) Comparisons of six different gene deconvolution tools using fix signature matrix in terms of Pearson correlation coefficients. (C) Statistical analysis revealed the satisfying prediction accuracy of QUANTISEQ, MCP-counter and CIBERSORTx compared to EPIC, XCELL and TIMER in cells isolated from OA synovium. Data were compared by the one-way ANOVA with Tukey’s post hoc tests for multiple comparisons and multiple p values were adjusted by the Bonferroni correction. Each bar represents a group (x-axis) with mean and 95% CI (y-axis). (D) Significant improvements in terms of predictive accuracy after using the generated CIBERSORTx-based “synovium” specific signature compared to the default one. The correlation analysis was performed by Pearson correlation. The correlation analysis was performed by Pearson correlation. Comparisons of Pearson correlation coefficients were conducted by one-tail Fisher r-to-z transformation (p=0.049). (E) Satisfying predictive results of relative cell abundance for seven general cell types in six cases of total nine patients. (F) No significant differences were observed between the predicted and measured cell composition in each case. Data comparisons were performed by Kruskal–Wallis test.
Validation of customized CIBERSORTx signature matrix for synovial bulk RNA-seq deconvolution
To further validate the customized CIBERSORTx-based deconvolution signature matrix, we predicted cell compositions of OA and RA synovia from one large publicly available bulk expression dataset (GSE89408); this was compared to results based on scRNA-seq derived cell compositions (Supplementary Figure S4A). Based on the predicted data, RA synovium had significantly higher proportions of B cells (10.30±13.13% vs. 1.00±1.88%, p<0.001), T cells (0.13±0.65% vs. 0.00±0.00%, p=0.019) and endothelial cells (6.84±5.52% vs. 4.63±3.19%, p=0.009) but fewer fibroblasts (52.47±16.36% vs. 61.77±13.56%, p=0.006) than OA synovium. Based on measured scRNA-seq, RA synovium has significantly higher proportions of T cells (26.70±5.96% vs. 10.87±10.27%, p=0.001), B cells (3.85±0.95% vs. 1.82±2.08%, p=0.007) and fewer mural cells (1.07±0.63% vs. 3.6±3.2%, p=0.006) compared to OA synovium (Supplementary Figure S4B). The relative abundance of fibroblasts (p=0.053, higher in OA) was consistent between predicted and measured scRNA-seq results (Supplementary Figure S4B and Supplementary Table S6). Applying the customized MCP-counter-based deconvolution signature matrix to the same RA validation dataset, only 3 of 7 cell types in the scRNA-seq data were identified (Supplementary Figure S4B).
Discussion
Higher proportions of T cells were identified in the current study compared to the previous one7. However, our finding is consistent with prior results analyzing OA synovium by flow cytometry11. T cell mediated inflammation has been considered of less importance than macrophages mediated inflammation in OA pathogenesis. However, emerging evidence shows that certain subtypes of T cells may play an important role in OA. CD8+ T cell knockout mice have slower OA progression12. We also found Th17 cells in our OA synovium, consistent with Faust et al.’s finding13. These results support a role of T cells in OA pathogenesis. To avoid affecting cell phenotyping, we did not simultaneously stain with CITE-seq antibodies, therefore these results warrant further confirmation by single T-cell receptor sequencing.
Deconvolution of bulk tissue RNA to yield cell composition information has the potential to contribute to a greater understanding of the pathogenesis of multiple diseases14. Methods like fluorescence-activated cell sorting (FACS) or immunohistochemical (IHC) staining were used in the past as the gold standard to estimate cell compositions within a sample15. However, both have their own technical limitations and might not be generally applicable. FACS requires a relatively large amount of tissue and IHC provides an estimate from a single tissue slice, which may not be representative of the heterogenous tissue architecture. Both methods can interrogate only a relatively small number of cell-type-specific markers at one time. More recently, scRNA-seq has come into vogue to characterize cell types and states; yet for the time being, it remains too expensive and laborious for routine use. Thus, the in silico gene deconvolution technique, utilizing machine learning based algorithms to establish models to predict cell proportions from bulk expression data, was introduced in the field16 and widely used on tumor tissue in cancer research14. However, this method has not been validated in synovial tissue, though several studies have applied it in arthritis17. Consistent with our findings, a previous study18 that deconvoluted publicly available OA and RA synovial bulk RNAseq data by CIBERSORT, found a high proportion of T and B cells in RA compared to OA synovial tissues.
To determine if this novel method is suitable for synovial tissue, we used our paired bulk RNA-seq and scRNA-seq data to evaluate six of the most widely used in silico gene deconvolution tools. CIBERSORT, MCP-counter and QUANTISEQ provided satisfactory predictions, suggesting that their default signature matrices can be used to profile cell proportions in synovium. More importantly, using our annotated single-cell transcriptomic data, we generated a “synovium”-specific customized signature matrix for CIBERSORTx, which provided a significantly more accurate estimation of cell composition in synovial samples than what was achievable with the CIBERSORTx default signature matrix (LM22). CIBERSORTx analysis of publicly available OA and RA bulk RNA-seq data yielded proportions of T cells, B cells, APCs, mast cells, mural cells and fibroblasts that were similar to the gold standard yielded from OA and RA scRNA-seq data. There are several possible reasons we did not observe similar results for endothelial cells. First, we had a limited sample size (n=5 for RA synovia, n=17 for OA synovia) with which to generate in silico scRNA-seq data; this might decrease the statistical power, especially for minority cell types. However, this also suggests a strength of using in silico gene deconvolution, namely the affordability of bulk RNA-seq makes it feasible to analyze larger sample sizes, which might reveal more meaningful insights, especially for less abundant cell types, in contrast to scRNA-seq that typically involves relatively small sample sizes due to cost. Second, predicted scRNA-seq cell compositions were generated from publicly available data; the batch effect between bulk expression and the scRNA expression might affect the prediction results. Third, single-cell dissociation efficiencies of measured scRNA-seq might influence the results of cell type proportions, which could influence the outcomes19.
Conclusion
In summary, using scRNA-seq, we identified seven distinct cell types in OA synovia including unique clusters of APCs and T cells indicating their potential roles in OA pathogenesis. In addition, we identified three tools suitable for in silico gene deconvolution of synovial tissue. Moreover, we generated the first “synovium”-specific deconvolution signature matrix for CIBERSORTx, which can produce more accurate predictions of cell compositions of arthritis-related synovial tissue compared to the use of the default signature matrix of the software. This work has establishes a roadmap to computational cell-type deconvolution suitable for bulk RNA-seq data and provides a successful and stringent proof of this concept applied to analyses of arthritic synovia with high cellular heterogeneity.
Supplementary Material
Supplementary Figure S1. Specific clustering analysis of T cells in OA synovium. (A) UMAP plot of scRNA-seq showed unsupervised clusters colored according to putative cell types of activated CD4+ T cell, memory CD4+ T cells, naïve CD4+ T cells, Th1, Th2, Th17 and Treg. (B) UMAP plot of scRNA-seq showed unsupervised clusters colored according to putative cell types of cytotoxic T cell, naïve CD8+ T cell, NK, proliferating CD8+ T cell and TEM. (C) Expression level of the top 5 feature genes in seven CD4+ T cell subtypes. (D) Expression level of the top 5 feature genes in five CD8+ T cell subtypes. (E) Trajectory analysis revealed distinguished separation of naïve CD4+ T cells and the other mature subtypes. (F) Trajectory analysis revealed distinguished separation of proliferating CD8+ T cells and the other mature subtypes. (G) Key functional gene expression along pseudotime in CD4+ T cells. (H) Key functional gene expression along pseudotime in CD8+ T cells.
Supplementary Figure S2. Specific clustering analysis of APC cells in OA synovium. (A) UMAP plot of scRNA-seq showed unsupervised clusters colored according to putative cell types of DC, I-Mφ, IR-Mφ, T-Mφ, fibrotic IR-Mφ, IFNS-Mφ and S100A8/9hi-Mφ. (B) Expression level of the top 5 feature genes in eight different subtypes (Color stands for the average expression of each gene and the size stands for percent expressed). (C) Trajectory analysis revealed distinguished separation of I-Mφ and IR-Mφ. DC, IFNS-Mφ and S100A8/9hi-Mφ was more I-Mφ like and fibrotic IR-Mφ is more IR-Mφ like. T-Mφ was evenly located between each branch, which stood for a group of macrophages switch between I-Mφ and IR-Mφ. (D) Pseudotime analysis revealed that inflammatory cytokine-related genes (IL6, IL1A, IL1B, TLR2, TNF) were highly expressed near the I-Mφ end. Inflammatory resolving genes (FGCR3A, CD163, MRC1) were highly expressed at the IR-Mφ end. APC, antigen presenting cell; OA, osteoarthritis; UMAP: Uniform manifold approximation and projection; DC, dendritic cell; I-Mφ, inflammatory macrophages; IR-Mφ, immune regulate macrophages; T-Mφ, transitional macrophages; fibrotic IR-Mφ, IFNS-Mφ, interferon stimulated macrophages.
Supplementary Figure S3. Comparisons of gene deconvolution tools with fix signature matrixes, generation of “synovium” specific signature matrix by MCP-counter. (A) No significant improvements in terms of predictive accuracy after using the generated MCP-counter-based “synovium” specific signature compared to the default one. The correlation analysis was performed by Pearson correlation. The correlation analysis was performed by Pearson correlation. Comparisons of Pearson correlation coefficients were conducted by one-tail Fisher r-to-z transformation (p=1.000). (B) Satisfying predictive results of relative cell abundance for seven general cell types in six cases of total nine patients. (C) No significant differences were observed between the predicted and measured cell composition in each case. Data comparisons were performed by Kruskal–Wallis test.
Supplementary Figure S4. Application of gene deconvolution with “synovium” specific signature matrices using CIBERSORTx and MCP-counter. (A) Flowchart showed the application of in silico gene deconvolution using the “synovium” specific signature matrices by CIBERSORTx and MCP-counter. Briefly, publicly available bulk microarray data were input into CIBERSORTx and MCP-counter with the “synovium” specific signature matrices, the predicted cell proportions were compared between OA and RA. (B) Application of in silico gene deconvolution with customized the “synovium” specific signature matrix by CIBERSORTx showed similar results in terms of APC, B cells, mural cells and T cells between public acquired microarray data and our scRNA-seq data. Customized MCP-counter-based signature showed similar results in terms of APC, B cells and T cells between public acquired microarray data and our scRNA-seq data. Data comparisons between groups were performed by independent t test. The median values are shown in the middle line. The lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The upper whisker extends from the hinge to the largest value no further than 1.5 times of inter-quartile range (IQR) from the hinge. The lower whisker extends from the hinge to the smallest value at most 1.5 times IQR of the hinge. Data beyond the end of the whiskers are called “outlying” points and plotted individually.
Acknowledgements
We would like to thank all the patients who donated the specimens for the research in the current study. Dr. ZYH would like to thank Dr. Weihua Guo from the Department of Immuno-Oncology, Beckman Research Institute, City of Hope and Drs. John Martin and Ga I Ban from the Department of Orthopaedic Surgery, School of Medicine, Duke University for their valuable suggestions.
Role of the funding source
This work was supported by grants from the National Natural Science Foundation of China (NSFC) to ZYH (92049101; 81972097; 81702185) and ZZK (81672135, 81873987). This research was also supported by “1.3.5 Project for Disciplines of Excellence”, West China Hospital, SiChuan University to ZZK (ZYJC18026), SiChuan Science and Technology Program to ZYH (No. 2018HH0071; No. 22GJHZ0208) and a Claude D. Pepper Older Americans Independence Centers grant (P30 AG028716 from NIH/NIA to VBK). ZYH and VBK would like to thank the following grants which help to generate RA scRNA-seq data (phs001529.v1.p1): UH2AR067691, K01AR066063, R35NS097404, 5UH2AR067691, R21HG009748, R21HG009748, DGE1342536 and DP2HG009623.
Footnotes
Competing interests
None declared.
References
- 1.Murphy LB, Cisternas MG, Pasta DJ, Helmick CG, Yelin EH. Medical Expenditures and Earnings Losses Among US Adults With Arthritis in 2013. Arthritis Care Res (Hoboken) 2018; 70: 869–876. [DOI] [PubMed] [Google Scholar]
- 2.Xie JW, Wang Y, Xiao K, Xu H, Luo ZY, Li L, et al. Alpha defensin-1 attenuates surgically induced osteoarthritis in association with promoting M1 to M2 macrophage polarization. Osteoarthritis Cartilage 2021. [DOI] [PubMed] [Google Scholar]
- 3.Revell PA, al-Saffar N, Fish S, Osei D. Extracellular matrix of the synovial intimal cell layer. Ann Rheum Dis 1995; 54: 404–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najm A, Masson FM, Preuss P, Georges S, Ory B, Quillard T, et al. MicroRNA-17–5p Reduces Inflammation and Bone Erosions in Mice With Collagen-Induced Arthritis and Directly Targets the JAK/STAT Pathway in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol 2020; 72: 2030–2039. [DOI] [PubMed] [Google Scholar]
- 5.Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med 2020; 26: 1295–1306. [DOI] [PubMed] [Google Scholar]
- 6.Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res 2020; 30: 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou CH, Jain V, Gibson J, Attarian DE, Haraden CA, Yohn CB, et al. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci Rep 2020; 10: 10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behmoaras J, Diaz AG, Venda L, Ko JH, Srivastava P, Montoya A, et al. Macrophage epoxygenase determines a profibrotic transcriptome signature. J Immunol 2015; 194: 4705–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J 2019; 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan W, Jiao P, Zhang H, Chen T, Zhou X, Qi Y, et al. Inhibition of African Swine Fever Virus Replication by Porcine Type I and Type II Interferons. Front Microbiol 2020; 11: 1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsueh MF, Zhang X, Wellman SS, Bolognesi MP, Kraus VB. Synergistic Roles of Macrophages and Neutrophils in Osteoarthritis Progression. Arthritis Rheumatol 2021; 73: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh JL, Shiau AL, Lee CH, Yang SJ, Lee BO, Jou IM, et al. CD8+ T cell-induced expression of tissue inhibitor of metalloproteinses-1 exacerbated osteoarthritis. Int J Mol Sci 2013; 14: 19951–19970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faust HJ, Zhang H, Han J, Wolf MT, Jeon OH, Sadtler K, et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J Clin Invest 2020; 130: 5493–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019; 37: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petitprez F, Sun CM, Lacroix L, Sautes-Fridman C, de Reynies A, Fridman WH. Quantitative Analyses of the Tumor Microenvironment Composition and Orientation in the Era of Precision Medicine. Front Oncol 2018; 8: 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Park J, Susztak K, Zhang NR, Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun 2019; 10: 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z, Ma Y, Li X, Deng Z, Zheng M, Zheng Q. The Immune Cell Landscape in Different Anatomical Structures of Knee in Osteoarthritis: A Gene Expression-Based Study. Biomed Res Int 2020; 2020: 9647072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W, Wang L, Gulko PS, Zhu J. Computational deconvolution of synovial tissue cellular composition: presence of adipocytes in synovial tissue decreased during arthritis pathogenesis and progression. Physiol Genomics 2019; 51: 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018; 24: 1277–1289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Specific clustering analysis of T cells in OA synovium. (A) UMAP plot of scRNA-seq showed unsupervised clusters colored according to putative cell types of activated CD4+ T cell, memory CD4+ T cells, naïve CD4+ T cells, Th1, Th2, Th17 and Treg. (B) UMAP plot of scRNA-seq showed unsupervised clusters colored according to putative cell types of cytotoxic T cell, naïve CD8+ T cell, NK, proliferating CD8+ T cell and TEM. (C) Expression level of the top 5 feature genes in seven CD4+ T cell subtypes. (D) Expression level of the top 5 feature genes in five CD8+ T cell subtypes. (E) Trajectory analysis revealed distinguished separation of naïve CD4+ T cells and the other mature subtypes. (F) Trajectory analysis revealed distinguished separation of proliferating CD8+ T cells and the other mature subtypes. (G) Key functional gene expression along pseudotime in CD4+ T cells. (H) Key functional gene expression along pseudotime in CD8+ T cells.
Supplementary Figure S2. Specific clustering analysis of APC cells in OA synovium. (A) UMAP plot of scRNA-seq showed unsupervised clusters colored according to putative cell types of DC, I-Mφ, IR-Mφ, T-Mφ, fibrotic IR-Mφ, IFNS-Mφ and S100A8/9hi-Mφ. (B) Expression level of the top 5 feature genes in eight different subtypes (Color stands for the average expression of each gene and the size stands for percent expressed). (C) Trajectory analysis revealed distinguished separation of I-Mφ and IR-Mφ. DC, IFNS-Mφ and S100A8/9hi-Mφ was more I-Mφ like and fibrotic IR-Mφ is more IR-Mφ like. T-Mφ was evenly located between each branch, which stood for a group of macrophages switch between I-Mφ and IR-Mφ. (D) Pseudotime analysis revealed that inflammatory cytokine-related genes (IL6, IL1A, IL1B, TLR2, TNF) were highly expressed near the I-Mφ end. Inflammatory resolving genes (FGCR3A, CD163, MRC1) were highly expressed at the IR-Mφ end. APC, antigen presenting cell; OA, osteoarthritis; UMAP: Uniform manifold approximation and projection; DC, dendritic cell; I-Mφ, inflammatory macrophages; IR-Mφ, immune regulate macrophages; T-Mφ, transitional macrophages; fibrotic IR-Mφ, IFNS-Mφ, interferon stimulated macrophages.
Supplementary Figure S3. Comparisons of gene deconvolution tools with fix signature matrixes, generation of “synovium” specific signature matrix by MCP-counter. (A) No significant improvements in terms of predictive accuracy after using the generated MCP-counter-based “synovium” specific signature compared to the default one. The correlation analysis was performed by Pearson correlation. The correlation analysis was performed by Pearson correlation. Comparisons of Pearson correlation coefficients were conducted by one-tail Fisher r-to-z transformation (p=1.000). (B) Satisfying predictive results of relative cell abundance for seven general cell types in six cases of total nine patients. (C) No significant differences were observed between the predicted and measured cell composition in each case. Data comparisons were performed by Kruskal–Wallis test.
Supplementary Figure S4. Application of gene deconvolution with “synovium” specific signature matrices using CIBERSORTx and MCP-counter. (A) Flowchart showed the application of in silico gene deconvolution using the “synovium” specific signature matrices by CIBERSORTx and MCP-counter. Briefly, publicly available bulk microarray data were input into CIBERSORTx and MCP-counter with the “synovium” specific signature matrices, the predicted cell proportions were compared between OA and RA. (B) Application of in silico gene deconvolution with customized the “synovium” specific signature matrix by CIBERSORTx showed similar results in terms of APC, B cells, mural cells and T cells between public acquired microarray data and our scRNA-seq data. Customized MCP-counter-based signature showed similar results in terms of APC, B cells and T cells between public acquired microarray data and our scRNA-seq data. Data comparisons between groups were performed by independent t test. The median values are shown in the middle line. The lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The upper whisker extends from the hinge to the largest value no further than 1.5 times of inter-quartile range (IQR) from the hinge. The lower whisker extends from the hinge to the smallest value at most 1.5 times IQR of the hinge. Data beyond the end of the whiskers are called “outlying” points and plotted individually.