

Abstract
Women carry a higher burden of Alzheimer’s disease (AD) compared to men, which is not accounted entirely by differences in lifespan. To identify the mechanisms underlying this effect, we investigated sex-specific differences in the progression of familial AD in humans and in APPswe/PS1ΔE9 mice. Activity dependent protein translation and associative learning and memory deficits were examined in APPswe/PS1ΔE9 mice and wild-type mice. As a human comparator group, progression of cognitive dysfunction was assessed in mutation carriers and non-carriers from DIAN (Dominantly Inherited Alzheimer Network) cohort. Female APPswe/PS1ΔE9 mice did not show recall deficits after contextual fear conditioning until 8 months of age. Further, activity dependent protein translation and Akt1-mTOR signaling at the synapse were impaired in male but not in female mice until 8 months of age. Ovariectomized APPswe/PS1ΔE9 mice displayed recall deficits at 4 months of age and these were sustained until 8 months of age. Moreover, activity dependent protein translation was also impaired in 4 months old ovariectomized APPswe/PS1ΔE9 mice compared with sham female APPswe/PS1ΔE9 mice. Progression of memory impairment differed between men and women in the DIAN cohort as analyzed using linear mixed effects model, wherein men showed steeper cognitive decline irrespective of the age of entry in the study, while women showed significantly greater performance and slower decline in immediate recall (LOGIMEM) and delayed recall (MEMUNITS) than men. However, when the performance of men and women in several cognitive tasks (such as Wechsler’s logical memory) are compared with the estimated year from expected symptom onset (EYO) we found no significant differences between men and women. We conclude that in familial AD patients and mouse models, females are protected, and the onset of disease is delayed as long as estrogen levels are intact.
Subject terms: Molecular neuroscience, Hippocampus
Introduction
Sex related differences have been observed in the progression of Alzheimer’s disease (AD) [1] and the prevalence of AD is greater in women than men in European and American patients [2, 3]. Globally, the number of women living with dementia including AD is more than that of men [4], which has been attributed to longer lifespan (4–5 years) in women [5]. Women (65 years or older) are at a greater risk of developing late-onset AD (LOAD) [6–8]. Cross-sectional and longitudinal studies have revealed faster cognitive decline [9] and widespread atrophy in brain areas in early-onset AD (EOAD) [10–17]. Women EOAD subjects displayed greater cognitive impairment and atrophy than men [18]. Preclinical individuals carrying autosomal dominant AD (ADAD) mutations showed memory deficits [19–21]. Neurological examination findings in AD (AD-NEF) from mutation carriers and non-carriers from the Dominantly Inherited Alzheimer Network (DIAN) revealed that AD-NEF are associated with a rapid cognitive decline and higher hippocampal atrophy [22]. Sex differences in the genetic make-up of resilience and multiple sex-specific molecular mechanisms may underlie resilience to AD pathology [23]. Perimenopausal and postmenopausal women aged 40 to 60 had more AD endophenotype than premenopausal women [24]. Further, women are well protected from stroke [25] and other neurodegenerative diseases relative to men until menopause, however, when the female sex hormone levels decline sharply at menopause, the clinical outcome is worse.
At the epidemiological level, it is crucial to comprehend the sex-specific differences in AD in relation to several elements, including longevity, survival bias, and comorbidities [26]. The risk and development of AD can differ between men and women depending on sociocultural and biological factors [26]. Menopause, oophorectomy, and androgen-deprivation therapy are sex-specific AD risk factors that cause cognitive decline [26]. Further, sex hormones play a critical role in sex specific differences in the brain [27]. However, the molecular mechanisms underlying the role of endogenous estradiol, social behavioral deficits, and the higher burden of AD pathogenesis in women remain unknown. Estrogen is neuroprotective [28] and has a protective effect on the vasculature [29]. Estrogens facilitate hippocampal synaptic plasticity and memory formation [30–33]. Although several animal models have been developed for simulating the state of estrogen depletion and testing the role of estrogen in synaptic plasticity, the ovariectomized (OVX) animals are a well-established model [34–36]. Depletion of estrogen levels in the brain may confer greater susceptibility to age-related neurodegenerative diseases [37]. Further, estrogen levels are significantly decreased in postmortem frontal cortex brain lysates of AD subjects compared with non-AD subjects [38]. In fact, decrease in estrogen levels seen during the transition from perimenopause to menopause coincides with increased β-amyloid (Aβ) deposition [38, 39]. While several studies have attempted to explain the effects of sex differences in AD, the underlying molecular pathways have not been fully explored. Moreover, the potential relationship between the female sex hormone, estrogen and memory impairments associated with AD is yet to be understood.
Akt signaling cascade plays a critical role in neurotransmission and synaptic plasticity [40–42]. The mTOR signaling pathway is the major nutrient-sensitive regulator for cell growth and metabolism [43]. The mTOR signaling cascade is implicated in the regulation of activity dependent mRNA translation at the synapses, which is induced by synaptic activity and required for effective LTP and memory formation [44]. Activation of Akt-mTOR signaling cascade is critical for new protein synthesis at the synapse. Dysregulation of Akt/mTOR has been reported using postmortem brain tissue from AD subjects and in AD animal models [45]. But, how Akt1 and mTOR signaling pathways regulate activity dependent new protein synthesis at synapse has not yet been explored in female AD mouse models.
To investigate this phenomenon, we used a mouse model of AD (APPswe/PS1ΔE9) and examined the molecular mechanisms underlying the pathogenesis and progression of AD. To understand how the findings in the mouse model extrapolate to humans, we analyzed the clinical longitudinal data from the DIAN study, in which participants carry one of the familial mutations in APP, PSEN1 or PSEN2 genes. The rationale for examining findings made in participants carrying familial AD mutations was that these participants show cognitive dysfunctions much earlier and therefore we would have window to look at differences between men and women carrying mutations in their third to fourth decade of life. Non-carriers are siblings of mutation carriers with no mutations considered as a control group. Importantly, this provides us an insight into the progression of the disease in premenopausal women, who have estrogen levels that are relatively intact.
Materials and methods/subjects
Reagents
Analytical grade chemicals and reagents were procured from Sigma-Aldrich Chemical Company (St Louis, MO, USA). L-[35S]-Methionine was purchased from Perkin Elmer Inc, USA. Amytracker 520 was purchased from Ebba Biotech, Sweden. Mouse monoclonal anti-β-tubulin (Cat. No. T4026, RRID: AB_477577) was purchased from Sigma-Aldrich Chemical Company (St Louis, MO, USA). Anti-4E-BP1, phospho (Thr37/Thr46) (Cat. No. 2855, RRID: AB_560835), Rabbit Anti-Akt1 (Cat. No. 2938, RRID: AB_915788), Phospho-Akt1 (Ser473) (Cat. No. 4060, RRID: AB_2315049), Anti-4E-BP1 (53H11) (Cat. No. 9644, RRID: AB_2097841), Phospho-Akt (Thr308) (D25E6) (Cat. No.13038, RRID: AB_262944), Anti-mTOR (L27D4) (Cat. No. 4517, RRID: AB_1904056), Anti-GSK-3β (3D10) (Cat. No. 9832, RRID: AB_10839406), Phospho-GSK-3β (Ser9) (Cat. No. 9336, RRID: AB_331405), Phospho-p70 S6 Kinase (Thr389) (1A5) (Cat. No. 9206, RRID: AB_2285392), p70S6 Kinase (Cat. No. 9202, RRID: AB_331676), Phospho-mTOR (Ser2448) (D9C2) (Cat. No. 5536, RRID: AB_10691552) were purchased from Cell Signaling Technology, Inc, USA. Horseradish peroxidase-conjugated secondary antibodies were purchased from Vector Laboratories, Inc. (Burlingame, CA, USA).
Animals
The APPSwe/PS1ΔE9 (APP/PS1) double transgenic mice on C57BL/6 J background were procured from the Jackson Laboratory, USA (https://www.jax.org/strain/005864; RRID: MMRRC_Stock_No: 034832-JAX). All experiments were conducted with APP/PS1 and littermate wild-type mice. Wild type (WT) and APP/PS1 double transgenic mice were bred at the Institutional Central Animal Facility and all mice were cared by the central animal facility members including veterinary physician and staff. All mice were housed in a temperature-controlled room on a 12 h light/12 h dark cycle and these rooms were maintained under sterile and pathogen-free conditions. All mice had ad libitum access to food and water. All experimental protocols and procedures were approved by the Institutional Animal Care and Use committee and are in accordance with the Guide for the care and Use of Laboratory Animals. All efforts were made to reduce suffering of mice and the number of mice used for our required experiments.
Observed power of analysis is known to be inversely proportional to observed p-value. Variability in transgenic APP/PS1 expression across mice results in variation in the observed biochemical parameters. To account for this impact and rule out the possibility of litter-specific effects, 4–8 mice per group were selected from separate litters and processed individually for each biochemical assay. For behavioral studies, the number of animals to be utilized was based on behavioral tests conducted in several labs, and 6–10 mice per genotype were selected as the sample size.
Data inclusion and exclusion. No samples were excluded from the experiments or analysis.
Randomization and blinding. All the animal experiments were designed and followed in compliance with the ARRIVE guidelines and applying double-blinded analysis when possible. In experiments involving wild type and APP/PS1 mice, the animals were assigned randomly to the respective groups based on the genotype.
In some experiments, female mice (2–2.5 months old) were anesthetized by intraperitoneal administration of 100 mg ketamine and 25 mg xylazine/kg body weight. The animal was fixed on surgical board with heating pad to maintain body temperature and an incision of about 2–3 cm was made in the lower abdomen for ovariectomy. Ovaries were removed carefully, and the abdomen was sutured. In the sham-operated mice, a similar incision was made, and the abdomen was sutured without removing ovaries. Ovariectomized and sham-operated mice were used for experimentation after 2 and 6 months. During sacrifice, atrophy of uterus and lack of ovaries were examined to ensure success of ovariectomy.
Contextual fear conditioning
Contextual fear conditioning (cFC) behavior was assessed as reported previously [46] and detailed protocol presented as supplementary methods.
Preparation of synaptosomes
Synaptosomal fractions were isolated as described previously [45] and detailed protocol provided as supplementary methods.
Immunoblotting
Equal amounts of synaptosomes were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane by electroblotting. Immunoblots were blocked in 5% bovine serum albumin for 1 hour at room temperature and immunoblotted with respective primary antibodies and incubated at 4 °C overnight. The following day, all immunoblots were washed and incubated at room temperature in respective secondary antibodies. Immunoreactive bands were detected using enhanced chemiluminescence (Clarity Western ECL blotting substrate, Bio-Rad). Immunoreactive signals were acquired using the Bio-Rad Chemidoc-XRS and analyzed with Imagelab software (Bio-Rad).
Isolation of synaptoneurosomes and L-[35S]-methionine incorporation assay
Isolation of synaptoneurosomes from mouse hippocampal tissue and L-[35S]-methionine incorporation assay were performed as described previously [45] and detailed protocol provided as supplementary methods.
Statistical analyses from mice studies
Statistical analyses were performed using GraphPad Prism (Prism 7.01, GraphPad Software Inc, La Jolla, CA, USA). Continuous variables were checked for normality using Shapiro-Wilk test and then an independent t-test or Mann–Whitney U-test was used appropriately to compare between two groups. Statistical comparisons more than two groups were performed using ANOVA with Tukey’s post hoc test. Results are represented as mean ± standard error of the mean. A p-value of <0.05 was considered statistically significant. No statistical methods were used to predetermine sample sizes.
DIAN data analyses
Both longitudinal and cross-sectional data on psychometric tests were obtained from the DIAN study (DIAN Data Freeze 12). Research involving human participants was done in accordance with the guidelines provided by the Institutional Human Ethics Committee (IHEC; approval# 10/1/2015). The age ranges of mutation carriers are men (19 years to 67 years, SD ± 10.79 years), women (18 years to 67 years, SD ± 10.84 years), and non-carriers are men (18 years to 66 years, SD ± 10.1 years), women (18 years to 69 years, SD ± 11.4 years). The characteristics of DIAN participants are summarized in Supplementary Table 1. To assess cognitive decline, analysis was performed on longitudinal Mini-Mental State Exam (MMSE) data. Analysis was also performed on longitudinal data from tests for episodic memory including immediate and delayed Wechsler’s logical memory test and immediate and delayed word list recall. Linear mixed effects models (lme) were fitted between scores of various cognitive examinations and age for men and women subjects in mutation carriers and non-carrier groups using the statistical computing language, R (lme ‘R’ package). Models were structured as a function of age. The intercept was placed at the average age at study entry. However, as participants enter the study at different ages, to account for between person differences in baseline age, we still controlled the intercept and slope for age at study entry. Intercept and rate of change were adjusted for age at baseline and sex. The linear fit was used to determine the correlation and the rate of changes in cognitive score with aging. We fitted a series of linear mixed effects models to cognitive scores from each of the tests available in the DIAN study.
The equations are as follows:
Here, Yit represents the result of the cognitive test Y for individual i at time t; and αi0 and αi1 individual i’s intercept and linear rate of change per year closer to the EYO. These are modeled as a function of population mean values α0 and α1 respectively, and the individual’s age at study entry and sex. The error term εit represents within-individual random error whereas u0i and u1i are the between-individual random effects that estimate the difference, after controlling for age at study entry and sex, between the population mean intercept and rate of change (α0 and α1) and the individual intercept and rate of change respectively.
Cognitive scores were aligned as a linear function of the estimated year from expected symptom onset (EYO) at each visit, setting the intercept at EYO = 0. The estimated years of symptom onset were calculated as the age of the participant at the time of the study assessment minus the age of the parent at symptom onset [47]. Both the intercept and rate of change were adjusted for baseline age and sex. Independent analyses were conducted for the subsamples of mutation carrier and non-carrier participants, splitting each of the subsamples by CDR status (CDR = 0 and CDR > 0). All models were estimated using the lme function of the nlme package, which uses restricted maximum likelihood estimation and assume missing data are missing at random.
Results
Age dependent impairment of contextual fear memory in APP/PS1 mice
In order to examine potential sex-related differences in learning and memory as a function of age and the presence of amyloidosis, we assessed the recall deficits, if any, on cFC in male and female APP/PS1 mice at different ages (2, 4, 6, 8, 10, and 12 months). At 2 months of age (t = 0.2923; df = 14; p = 0.7744), 4 months of age (p = 0.4255), and 6 months of age (p = 0.1419), APP/PS1 female mice behaved like WT and did not display any recall deficits (Fig. 1A). However, APP/PS1 male mice showed these recall deficits as early as 2 months of age (t = 4.594; df = 14; p = 0.0004) (Fig. 1B) and these recall deficits persisted at 4 months of age (t = 3.168; df = 10; p = 0.01), 6 months of age (p = 0.0317), 8 months of age (p = 0.0002), and 12 months of age (t = 4.824; df = 14; p = 0.0003) (Fig. 1B). In the present study, we did not conduct recall memory task using male mice at 10 months of age. Remarkably, we found that at 8 months of age, APP/PS1 female mice showed significant recall deficits as compared to WT (t = 3.794; df = 14; p = 0.002) (Fig. 1A; 10 months of age (t = 4.538; df = 14; p = 0.0005)), which was sustained until 12 months of age (t = 4.650; df = 14; p = 0.0004) (Fig. 1A). Our results provide evidence that deficits in recall after cFC were not observed in young APP/PS1 female mice but emerged significantly at 8 months of age and later, while in the corresponding male mice the recall deficits were seen from 2 to 12 months of age.
Fig. 1. Impairment in recall memory after contextual fear conditioning depends on sex and age of APP/PS1 mice.

A Histograms are showing the freezing response of female mice at different ages. Freezing response was assessed by contextual fear conditioning as percentage of immobility after 24 h of training and no significant difference was observed in 2 M (t = 0.2923; df = 14; p = 0.7744; Hedges’ g (95% CI) 0.14 (−0.79, 1.06)), 4 M (p = 0.4255; Hedges’ g (95% CI) 0.39 (−0.54, 1.32)) and 6 M (p = 0.1419; Hedges’ g (95% CI) −0.37 (−1.37, 0.63)) old APP/PS1 female mice compared with WT female mice. Impairment in freezing response was observed in 8 M (t = 3.794; df = 14; p = 0.0020; Hedges’ g (95% CI) −1.79 (−2.9, −0.63)), 10 M (t = 4.538; df = 14; p = 0.0005; Hedges’ g (95% CI) −2.14 (−3.33, −0.90)), and 12 M (t = 4.65; df = 14; p = 0.0004; Hedges’ g (95% CI) -2.19 (−3.40, −0.94)) old APP/PS1 female mice compared with age matched WT female mice. Statistical analysis: Two-sided t-test or Mann-Whitney U-test was applied to compare WT versus APP/PS1 mouse groups. In all panels, reported values are mean ± s.e.m. *P < 0.05. A, B n = 5–8 mice per group. B Histograms are showing the freezing response of male mice at different ages, and APP/PS1 male mice exhibit significantly decreased freezing response than the WT male from 2 months onwards. WT versus APP/PS1 male mice, 2 M (t = 4.594; df = 14; p = 0.0004; Hedges’ g (95% CI) −2.17 (−3.37, −0.92)), 4 M (t = 3.168; df = 10; p = 0.01; Hedges’ g (95% CI) −1.68 (−2.93, −0.38)), 6 M (p = 0.0317; Hedges’ g (95% CI) −1.90 (−3.32, −0.42)), 8 M (p = 0.0002; Hedges’ g (95% CI) −3.33 (−4.85, −1.78)), and 12 M (t = 4.824; df = 14; p = 0.0003; Hedges’ g (95% CI) −2.28 (−3.50, −1.01)). Statistical analysis: Two-sided t-test or Mann–Whitney U-test was used to compare WT versus APP/PS1 mouse groups. In all panels, reported values are mean ± s.e.m. *P < 0.05. A, B n = 5–8 mice per group. Local activity dependent protein translation at the synapse is significantly affected in APP/PS1 female mice at 8 months but not at 4 months of age. Synaptoneurosomes from WT and APP/PS1 female mice were stimulated with or without KCl in the presence of 50 μCi L-[35S]-methionine at 37 °C for 15 min and newly synthesized proteins (protein translation) were measured by L-[S35]-methionine incorporation assay. C KCl stimulated local protein translation in synaptoneurosomes was not affected between WT and APP/PS1 female mice at 4 months of age (WT-US versus WT-St, t = 6.894; df = 3; p = 0.003; Hedges’ g (95% CI) 4.61 (1.78, 7.40); APP/PS1-US versus APP/PS1-St, t = 3.042; df = 3; p = 0.028; Hedges’ g (95% CI) 2.59 (0.68, 4.42)). Statistical comparisons from each group (unstimulated versus stimulated) were calculated using paired, one-sided, student t-test. n = 4 mice per group. D Stimulation of local protein translation in synaptoneurosomes in the presence of KCl was impaired in APP/PS1 female mice with comparison to WT female mice at 8 months of age (WT-US versus WT-St (t = 2.960; df = 8; p = 0.009; Hedges’ g (95% CI) 0.72 (−0.19, 1.63)); APP/PS1-US versus APP/PS1-St (t = 0.341; df = 7; p = 0.372; Hedges’ g (95% CI) −0.19 (−1.11, 0.74))). Statistical comparisons from each group (unstimulated versus stimulated) were calculated using paired, one-sided, student t-test. All values normalized to unstimulated WT group. Data is represented as mean ± s.e.m. (n = 8–9 mice per group) and *denotes values significantly different from corresponding controls (p < 0.05).
Impairment of synaptic activity dependent protein translation is delayed in hippocampus of APP/PS1 female mice
The activity dependent protein translation at the synapse is essential for learning and memory and synaptic plasticity. Here we sought to test whether activity dependent protein translation is affected in APP/PS1 female mice of different age groups by measuring [35S]-L-methionine incorporation in newly synthesized proteins. Remarkably, we found that incorporation of [35S]-L-methionine by KCl stimulation, which is indicative of protein translation, was unaffected in APP/PS1 female mice at 4 months of age (WT-US versus WT-St (t = 6.894; df = 3; p = 0.003); APP/PS1-US versus APP/PS1-St (t = 3.042; df = 3; p = 0.028)) (Fig. 1C), while this was absent in young APP/PS1 male mice [45] indicating that activity dependent protein translation is intact in APP/PS1 female mice at early age. However, at 8–9 months of age, KCl stimulated [35S]-L-methionine incorporation was affected similarly in both APP/PS1 male [45] and female mice (WT-US versus WT-St (t = 2.960; df = 8; p = 0.009); APP/PS1-US versus APP/PS1-St (t = 0.341; df = 7; p = 0.372)) (Fig. 1D) indicating that no activity dependent translation occurred in the synaptoneurosomes of APP/PS1 mice.
Effect of ovariectomy on recall of contextual fear conditioning in APP/PS1 female mice
To test the effect of ovariectomy on hippocampal dependent learning and memory, WT and APP/PS1 female mice were ovariectomized (OVX) at 2 months of age and then housed for a further period of 2 or 6 months. Next, we evaluated the recall after cFC in WT and APP/PS1 from sham and OVX female mice. At 4 months of age, OVX APP/PS1 female mice showed significant recall deficits as compared to sham APP/PS1 female mice (Interaction: F (1, 36) = 19.64, p < 0.0001; OVX: F (1, 36) = 3.438, p = 0.0719; Genotype: F (1, 36) = 8.732, p = 0.0055) (Fig. 2A), and this observation was similar to that in APP/PS1 male mice [46]. However, recall deficits after cFC were not observed in sham APP/PS1 female mice compared with those in sham WT female mice at this age (Fig. 2A). Further, OVX APP/PS1 female mice exhibited significant recall deficits in comparison to sham or OVX WT female mice (Fig. 2A). Additionally, sham and OVX APP/PS1 female mice displayed significant recall deficits in comparison to sham and OVX WT female mice at 8 months of age (Interaction: F (1, 20) = 4.904, p = 0.0386; OVX: F (1, 20) = 1.991e-005, p = 0.9965; Genotype: F (1, 20) = 46.05, p < 0.0001) (Fig. 2B). Our results indicate that ovariectomy enhances memory impairments in APP/PS1 female mice at early age.
Fig. 2. Ovariectomy persuaded recall deficits and induced impairment of local activity dependent protein translation after contextual fear conditioning in APP/PS1 female mice at 4 months of age.

Sham or ovariectomized WT and APP/PS1 female mice were subjected contextual fear conditioning at 4- and 8-months age and assessed their recall memory as described in methods section. A Impairment in freezing response was observed in OVX APP/PS1 female mice following contextual fear conditioning at 4 months of age with comparison to all other groups as examined. (WT-Sham versus APP/PS1-Sham, p = 0.7251; WT-Sham versus WT-OVX, p = 0.2798; APP/PS1-Sham versus APP/PS1-OVX, p = 0.0005; WT-Sham versus APP/PS1-OVX, p = 0.0086; WT-OVX versus APP/PS1-OVX, p < 0.0001). Partial Eta squared = 0.353. n = 10 mice per group. B Sham or OVX APP/PS1 female mice exhibit significantly decreased freezing response than the sham or OVX WT female mice at 8 months of age. n = 5–7 mice per group. Statistical comparison is performed using two-way ANOVA. (WT-Sham versus APP/PS1-Sham, p < 0.0001; WT-Sham versus WT-OVX, p = 0.3835; APP/PS1-Sham versus APP/PS1-OVX, p = 0.4522; WT-Sham versus APP/PS1-OVX, p = 0.0005; WT-OVX versus APP/PS1-OVX, p = 0.0146). Partial Eta squared=0.197. Data are presented as mean ± s.e.m. Statistical significance indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n = 5–7 mice per group. Protein translation in synaptoneurosomes from sham or OVX WT and APP/PS1 female mice after KCl stimulation was measured as L-[35S]-methionine incorporation. C L-[35S]-methionine incorporation by KCl stimulation was significantly increased in synaptoneurosomes from sham WT or APP/PS1 female mice (4 months) than unstimulated synaptoneurosomes. KCl stimulated protein translation was significantly impaired in synaptoneurosomes from OVX APP/PS1 female mice (4 months) than unstimulated OVX APP/PS1 female mice (4 months). Statistical comparisons from each group (unstimulated versus stimulated) were calculated using paired, one-sided, student t-test. All values normalized to unstimulated WT-sham group. (WT-Sham-US versus WT-Sham-St, t = 5.442; df = 4; p = 0.003; Hedges’ g (95% CI) 2.79 (1.01, 4.51); WT-OVX-US versus WT-OVX-St, t = 2.649; df = 4; p = 0.029; Hedges’ g (95% CI) 0.79 (−0.41, 1.95); APP/PS1-Sham-US versus APP/PS1-Sham-St, t = 6.62; df = 4; p = 0.001; Hedges’ g (95% CI) 1.69 (0.27, 3.04); APP/PS1-OVX-US versus APP/PS1-OVX-St, t = 0.06526; df = 4; p = 0.476; Hedges’ g (95% CI) −0.01 (−1.13, 1.10)). Data are presented as mean ± s.e.m. Statistical significance indicated as *p < 0.05. n = 5 mice per group.
Effect of ovariectomy on synaptic activity dependent protein translation in hippocampus of APP/PS1 female mice
Estrogen can contribute to the regulation of local protein translation and it can rapidly stimulate the phosphorylation of Akt and 4E-BP1 [48]. Here, we sought to determine whether ovariectomy can regulate activity dependent protein translation in APP/PS1 female mice. To test this, we examined the effect of ovariectomy on new protein synthesis as measured by L-[35S]-methionine labeling. We found that stimulation of synaptoneurosomes with KCl from sham or OVX WT female mice displayed significantly increased L-[35S]-methionine incorporation compared with that observed in unstimulated synaptoneurosomes from sham or OVX WT female mice at 4 months of age (WT-Sham-US versus WT-Sham-St, t = 5.442; df = 4; p = 0.003; WT-OVX-US versus WT-OVX-St, t = 2.649; df = 4; p = 0.029) (Fig. 2C). In marked contrast, L-[35S]-methionine incorporation was completely absent in KCl stimulated synaptoneurosomes from OVX APP/PS1 female mice at 4 months of age as compared with that in unstimulated synaptoneurosomes (APP/PS1-Sham-US versus APP/PS1-Sham-St, t = 6.62; df = 4; p = 0.001, APP/PS1-OVX-US versus APP/PS1-OVX-St, t = 0.06526; df = 4; p = 0.476) (Fig. 2C), and these results were comparable to those observed in young APP/PS1 male mice [45]. Further, stimulation of synaptoneurosomes with KCl from sham APP/PS1 female mice showed similar levels of L-[35S]-methionine incorporation compared with stimulated condition of sham WT female mice (Fig. 2C). Altogether, our results provide evidence that ovariectomy regulates synaptic activity dependent protein translation in the hippocampus of APP/PS1 female mice.
Downregulation of synaptosomal Akt1, mTOR phosphorylation and their downstream effectors in aged APP/PS1 female mice
Akt1-mTOR signaling pathway has emerged as an essential regulator of neuronal survival and activity dependent local dendritic protein translation. At 8 months of age, activity dependent protein translation was impaired in APP/PS1 female mice and new protein synthesis is mediated by the Akt1-mTOR pathway. Thus, we aimed to understand the molecular mechanisms that underlie the loss of activity dependent protein translation at synapses by investigating the status of phosphorylation of Akt1 and mTOR and their downstream targets in synaptosomes isolated from APP/PS1 mice.
We assessed the Akt1-mTOR signaling pathway at 4 months of age, we detected phosphorylation of Akt1 (threonine-308 (t = 3.801; df = 14; p = 0.0019) and serine-473 (t = 3.479; df = 14; p = 0.0037), and GSK3β phosphorylation (t = 2.958; df = 14; p = 0.0104)) was significantly upregulated in synaptosomes of APP/PS1 female mice compared with that in synaptosomes of WT female mice (Fig. 3A–C).
Fig. 3. Phosphorylation of Akt1 and GSK3β is increased in synaptosomes of APP/PS1 female mice at 4 months of age while decreased at 8 months of age.

Synaptosomes from 4- and 8- months old WT and APP/PS1 female mouse brain cortex were subjected to SDS-PAGE followed by western transfer and representative immunoblots were probed with (A and D) phospho-Akt1 (threonine-308) antibody, (B and E) phospho-Akt1 (serine-473) antibody, and (C and F) phospho-GSK3β antibody. Subsequently, these blots were stripped and reprobed, sequentially, for Akt1, GSK3β and tubulin, respectively. (A, B, D and E) The data were quantified by measuring phosphorylated Akt1/Akt1 ((A) t = 3.801; df = 14; p = 0.0019; Hedges’ g (95% CI) 1.79 (0.63, 2.91), (B) t = 3.479; df = 14; p = 0.0037; Hedges’ g (95% CI) 1.64 (0.51, 2.72), (D) t = 2.555; df = 14; p = 0.0229; Hedges’ g (95% CI) −1.20 (−2.21, −0.16), (E) t = 2.877; df = 14; p = 0.0122; Hedges’ g (95% CI) −1.36 (−2.39, −0.28)), (C and F) phosphorylated-GSK3β/GSK3β ((C) t = 2.958; df = 14; p = 0.0104; Hedges’ g (95% CI) 1.39 (0.32, 2.43), (F) t = 4.977; df = 14; p = 0.0002; Hedges’ g (95% CI) –2.35 (−3.59, −1.06)) band intensity ratios and Akt1, GSK3β, and tubulin band intensities were quantified using Bio-Rad image lab software 5.1. Statistical analysis: Two-sided, t-test was used for comparison between WT versus APP/PS1 mouse groups. Data are presented as mean ± s.e.m. *p < 0.05; **p < 0.01; ***p < 0.001. n = 8 mice per group.
However, when we assessed at 8 months of age, the phosphorylation of Akt1 at threonine-308 (t = 2.555; df = 14; p = 0.0229) and serine-473 (t = 2.877; df = 14; p = 0.0122) was significantly decreased in synaptosomes compared with WT female mice (Fig. 3D, E). Similarly, phosphorylation of GSK3β (serine-9), a downstream effector molecule in this pathway, was significantly downregulated (t = 4.977; df = 14; p = 0.0002) in synaptosomes of 8 months old APP/PS1 female mice (Fig. 3F).
Mammalian target of rapamycin (mTOR) is a rapamycin sensitive serine/threonine protein kinase and activated by phosphorylation of Akt. Subsequently, activated mTOR stimulates cap-dependent protein synthesis by phosphorylating mRNA translation factors such as eukaryotic initiation factor 4E-binding protein-1 (4E-BP1) and p70 ribosomal S6 kinase (S6K). In this study, phosphorylation of mTOR at serine-2448 (t = 3.367; df = 14; p = 0.0046), phosphorylation of p70S6K (t = 5.908; df = 14; p < 0.0001), and phosphorylation of 4E-BP1 (t = 4.159; df = 14; p = 0.0010) was significantly increased in 4 months old APP/PS1 female mice (Fig. 4A–C) supporting the fact that the Akt1-mTOR pathway was not compromised in females as seen in males [45].
Fig. 4. Phosphorylation of mTOR, p70S6K, and 4E-BP1 is increased in synaptosomes of APP/PS1 female mice at 4 months of age but decreased at 8 months of age.

Synaptosomes from the mouse brain cortex of 4 (A–C) and 8 (D–F) months old WT and APP/PS1 female mice were subjected to western blot analysis for (A and D) phospho-mTOR (serine-2448) antibody, (B and E) phospho-p70S6K (threonine-389) antibody, and (C and F) phospho-4E-BP1 (threonine-37/46). Later, these blots were stripped and reprobed, sequentially, for mTOR, p70S6K, 4E-BP1, and tubulin, respectively. A and D The data were quantified by measuring phosphorylated mTOR/mTOR ((A) t = 3.367; df = 14; p = 0.0046; Hedges’ g (95% CI) 1.59 (0.47, 2.66), (D) t = 4.059; df = 14; p = 0.0012; Hedges’ g (95% CI) −1.91 (−3.06, −0.73)), (B and E) phosphorylated-p70S6K/p70S6K, ((B) t = 5.908; df = 14; p < 0.0001; Hedges’ g (95% CI) 2.79 (1.38, 4.15), (E) t = 6.757; df = 14; p < 0.0001; Hedges’ g (95% CI) −3.19 (−4.66, −1.67)) and (C and F) phosphorylated-4E-BP1/4E-BP1 ((C) t = 4.159; df = 14; p = 0.0010; Hedges’ g (95% CI) 1.96 (0.77, 3.11), (F) t = 2.426; df = 14; p = 0.0294; Hedges’ g (95% CI) −1.14 (−2.14, −0.112)) band intensity ratios were measured. Statistical analysis: Two-sided t-test was conducted to compare between WT versus APP/PS1 mouse groups. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001. n = 8 mice per group.
We observed robust downregulation of mTOR phosphorylation at serine-2448 in synaptosomes of 8 months old APP/PS1 female mice (t = 4.059; df = 14; p = 0.0012) (Fig. 4D). Further, phosphorylation of p70S6K at threonine-389 (t = 6.757; df = 14; p < 0.0001) and phosphorylation of 4E-BP1 (t = 2.426; df = 14; p = 0.0294) was also significantly downregulated in synaptosomes of 8 months old APP/PS1 female mice as compared with age matched WT female mice (Fig. 4E, F). In conclusion, dysregulation of Akt1-mTOR signaling occurring in synaptosomes of APP/PS1 female mice at 8 months of age, which was similar to that observed in male mice at 1–3 months of age, could potentially contribute to the inhibition of activity dependent protein translation only at older age in female than in male mice.
Delayed cognitive decline is observed in women with familial AD mutations
Decline in cognitive performance with increasing age was assessed in men (n = 212) and women (n = 280) DIAN participants. The participants were grouped as carriers versus non-carriers of familial AD mutations in the genes APP, PSEN1 or PSEN2. The characteristics of study participants have been described in Supplementary Table 1. We assessed performance on Wechsler’s logical memory test and word list recall which are tests for episodic memory [49–51], and on the Mini-Mental State Exam (MMSE) [52] using linear mixed effects models.
Differences in performance between men and women participants on the MMSE [52] were evaluated. MMSE score in both mutation carrier (p = 0.3819) and non-carrier women participants (p = 0.7030) as a function of age showed no statistical significant differences in cognitive decline compared to men participants (Supplementary Figure. 1).
In the Wechsler’s logical memory test, participants are presented with a logically organized story which, they are asked to recall immediately (LOGIMEM) and after a delay of approximately 20 min (MEMUNITS) [50, 53]. Upon performing the linear mixed effects model on the longitudinal data, we observed that among the mutation carriers, men participants showed a significantly greater decline in performance on both immediate (Fig. 5A; p = 0.003) and delayed (Fig. 5C; p = 0.003) test of logical memory as compared to women participants irrespective of age. Women non-carriers showed slower in cognitive decline compared to men (Fig. 5B; p = 0.03) and (Fig. 5D; p = 0.0091).
Fig. 5. Performance on Wechsler’s logical memory test in men mutation carriers as compared to women mutation carriers.

A The linear regression trend lines from linear mixed effects models for longitudinal data on Wechsler’s logical memory test score with age (immediate recall, LOGIMEM) from women and men participants in the mutation and non-carriers groups. Performance in the LOGIMEM test was significantly better in women mutation carriers compared to men mutation carriers suggesting that overall performance of women is significantly higher than men irrespective of the age of entry in the study (men equation, y = −0.306 × age + 10.51; women equation, y = −0.258 × age + 12.266; men vs women, p = 0.003). B Performance in the LOGIMEM test was also significantly higher in women compared to men in non-carriers, but regression lines are different, suggesting that overall performance of women is statistically significant than men subjects irrespective of the age of entry in the study (men equation, y = 0.068 × age + 14.478; women equation, y = −0.016 × age + 15.643; men vs women, p = 0.03) C Performance in the MEMUNITS test was significantly higher in women compared to men mutation carriers suggesting that overall performance of women is significantly higher than men irrespective of the age of entry in the study (men equation, y = −0.289 × age + 9.33; women equation, y = −0.223 × age + 11.163; men vs women, p = 0.003). D Intercept of the performance in the MEMUNITS test was significantly higher in women versus men in non-carriers (men equation, y = 0.083 × age + 13.392; women equation, y = −0.024 × age + 14.899; men vs women, p = 0.0091). The p-value is for the interaction term of age*sex.
We then evaluated cognitive decline as assessed by word list recall in men and women mutation carriers. Word list recall involved the oral presentation of 16 unrelated words to the participants, at a rate of approximately 1 word per second; then, they were asked to recall the list in any order immediately (WORDIM) and after 20−30 min (WORDDEL). We observed no significant age-related difference in performance of men and women in immediate and delayed word list recall in the mutation carrier (WORDIM; p = 0.2414) and (WORDDEL; p = 0.211) and non-carrier groups (WORDIM; p = 0.5602) and (WORDDEL; p = 0.2304). (Supplementary Fig. 2). Altogether, comparison of the slopes obtained from the linear fit showed that sex has influence on the cognitive performance such as Wechsler’s logical memory, in an age dependent fashion, wherein women showed slower decline in all age groups compared to men.
We also analyzed the cognitive performance based on the EYO [47]. As expected, CDR = 0 (cognitively normal) non-carriers perform the best across tests and exhibit the slowest rate of change as individuals approach EYO = 0 (see Supplementary Table. 2 for model results). Symptomatic (CDR > 0) mutation carriers are the individuals who perform the worst and also decline the fastest, although estimates of rate of change did not reach traditional statistical significant thresholds except for MMSE and word immediate recall. Importantly, results from our analyses failed to identify statistically significant sex differences in the absolute scores or rate of change in cognition over time in all groups, (e.g., Wechsler’s logical memory, Supplementary Fig. 3 and Supplementary Table. 2). The one exception is performance on the animal naming test (Fig. 6A, B) (lexical ability and executive control), in this category fluency performance in controlled association the participants are asked to name as many different animals as possible in 1 min, in symptomatic (CDR > 0) mutation carriers where women declined faster than men (ß = −1.14 (SE = 0.44), p-value −0.01) (Fig. 6A).
Fig. 6. Individual longitudinal performance on category fluency (animals) test (lexical ability and executive control) and measures over estimated year from expected symptom onset (EYO) in men and women mutation carriers and non-carriers.
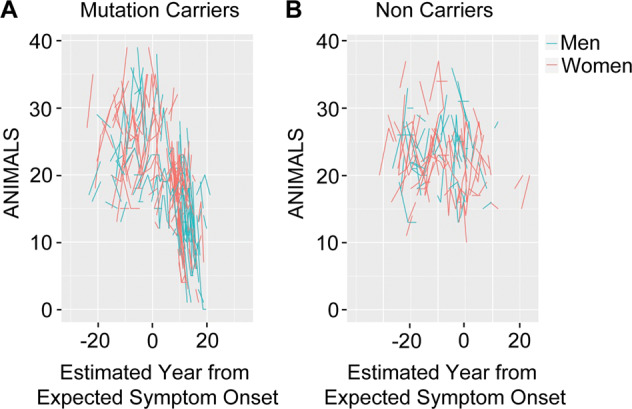
A, B Spaghetti plot of observed category fluency (Animals) test scores as a function of estimated year from expected symptom onset (EYO) from women (red) and men (cyan) mutation carriers and non-carriers, respectively. (Mutation carriers, men vs women, CDR > 0, ß = −1.14 (SE = 0.44), p-value = 0.01; Non-carriers, men vs women, CDR > 0, ß = −0.40 (SE = 0.59), p-value = 0.50). The longitudinal data is estimated using the rate of change from the linear mixed effects models over the years (Supplementary Table. 2).
Discussion
To understand the molecular underpinnings of the sex difference, we examined a mouse model of AD, APPswe/PS1ΔE9 mice, which carry both the APP Swedish mutation and a deletion of exon-9 in PSEN1. Recent studies from our lab have revealed that male APP/PS1 mice at 2 months of age exhibit memory impairments following cFC [46, 54]. We examined the ability to recall after cFC in both male and female mice from 2 months to 12 months. The female mice were able to recall after cFC up to 8 months of age, after which there was significant deficit in their ability to recall. In the males, the recall deficits started at 2 months of age and observed until 12 months of age [46]. Studies suggest that estrogen levels in mice are stable until estropause, whereupon at ~8–10 months of age there is a significant reduction in estrogen levels [55–58]. Ovariectomy reduced sex hormone levels [59], impaired learning and memory in rats [60, 61], and in APP21 rat model [62]. In AD transgenic mouse models, sex and hormone levels affect amyloid load and cognitive functions [36]. To evaluate the potential protective role of estrogen, we analyzed the behavior of ovariectomized WT and APP/PS1 female mice. The ovariectomized mice showed recall deficits, much as males of the same age (Fig. 2A, B) and our findings are supported with other reports [36, 62].
Learning and memory rely on activity dependent protein translation at the synapse [63, 64], which depends on Akt-mTOR signaling [65, 66]. Activity dependent protein translation and Akt-mTOR signaling are disrupted in 1–9-month-old APP/PS1 male mice [45]. However, young APP/PS1 female mice (4 months old) didn’t show activity dependent protein translation deficits until 8 months old (Fig. 1C, D). Female APP/PS1 mice demonstrated upregulation of the Akt-mTOR pathway in synaptosomes at 4 months, but male mice showed downregulation [45]. However, Akt-mTOR pathway upregulation was not found in middle-aged mice, and by 8 months of age, APP/PS1 female mice displayed significant downregulation of Akt-mTOR signaling cascade, comparable to males from 1 month onwards [45, 46]. In AD model systems, increased levels of Akt1 or active pAkt1 explain neuroprotection mediated by neurotrophins [67], estrogen [67], and lipoic acid [68], similar with our results. Further, our data show that the dysregulation of Akt1 and mTOR signaling in APP/PS1 mice may be a factor in behavioral deficits, and its dysregulation is age and sex dependent. Our findings are consistent with other observations that estrogen in ovariectomized mice improves behavioral tasks that dependent on hippocampal memory [69, 70]. Thus, increase in Akt-mTOR signaling at the synapse may be critical for synaptic plasticity throughout life course, particularly in post-estropausal females. In APP/PS1 mice, we detect a remarkable sex difference in disease development, with male animals showing early behavioral impairment that progresses over time, whereas, in female mice, the impairments did not appear until eight months of age, and thereafter progresses rapidly. Therefore, the progression of behavioral deficits in male and female mice is quite different and can be hypothesized that in male mice, since the deficits starts early, compensatory responses would occur in the brain. However, estrogen is a neuroprotective agent and can counteract the pathogenic effects of β-amyloid accumulation until menopause. However, when estrogen levels decline, there is a substantial difference in the progression of disease.
Numerous studies on sex differences in AD have been limited to clinical diagnoses [71–73]. Some studies revealed a stronger association between AD pathology and clinical AD in women [72, 74], whereas others did not [71, 75, 76]. According to the Framingham Study, a 65-year-old man has a 6.3% lifetime chance of AD and a 10.9% lifetime risk of any dementing disease; while a woman has 12% and 19%, respectively [77]. Estradiol declines following menopause are associated with verbal memory decline [78, 79]. The Colombian Alzheimer’s Prevention Initiative Biomarker Study found no difference in cognitive function (CERAD total score) between men and women PSEN1 mutation carriers (20-56 years) [80]. PSEN1/2 mutant carriers have early-onset dementia, but autosomal dominant and late-onset AD have different behavioral and pathophysiology. To evaluate the translational potential to performance in humans with AD, we examined the sex-specific difference in the rate of cognitive decline in the DIAN cohort [22, 47, 81–85]. We found that men mutation carriers had steeper cognitive decline on immediate and delayed test of logical memory as compared to women mutation carriers (Fig. 5A and C). Perhaps many of the observed sex differences due to post-menopausal sex hormone reduction in women is less of a factor in familial AD given the young ages at onset. AD mouse models mirror the human population, in that they are familial AD mutation carriers and females are resistant to AD-related cognitive decline prior to menopause/estropause. Our findings are consistent where women PSEN1 mutation carriers exhibited better verbal memory than men [80] and the Alzheimer’s Prevention Initiative (API) ADAD Colombia Trial study (30-53 years old) found that women mutation carriers had better delayed recall than men [86, 87]. In women, the average age of menopause onset is between 50−52 years [39, 88]. We find that the rate of change in performance in cognitive tests in mutation carriers showed different trends when the performance was assessed in women versus men as function of age, where men exhibited more rapid cognitive decline than women. We demonstrate that the progression of the disease potentially changes prior to menopause (before 50-52 years) as compared to post-menopause. This is also evident in the sporadic AD, where it has often been shown that women have a rapid cognitive decline in their later years than men [89, 90].
We investigated the intercept and rate of change using linear mixed effects models, taking into consideration clinical dementia rating (CDR) and estimated year from expected symptom onset (EYO) [47]. CDR = 0 non-carriers performed best in all tests and showed the slowest rate of change as individuals approached EYO = 0 (Supplementary Table 2). Symptomatic mutation carriers (CDR > 0) declined the fastest, but estimates of rate of change did not reach statistical significance except for the MMSE test in mutation carriers and word immediate recall in the symptomatic subgroup. In the symptomatic mutation carrier subgroup, women declined faster than men on the animal naming test (lexical ability and executive control) (Fig. 6). The performance on cognitive measures such as immediate and delayed logical memory, analyzed as a function of chronological age, showed that cognitive decline in women was slower as compared to men. Immediate and delayed logical memory tests are not significantly different between men and women when analyzed as a function of EYO. Further, the performances on cognitive tests were not significantly different between men versus women when the analysis was performed in subgroups defined by CDR. Therefore, the results are conflicting and differ depending on whether chronological age, EYO or CDR are considered for assessing the cognitive decline. Nevertheless, our studies suggest potential difference in the trajectory of cognitive decline in men and women, especially when the onset of the disease occurs earlier in life before the age of fifty years.
Our findings indicate that premenopausal women are protected from memory deficits, that hormone replacement therapy may be beneficial, and that the cost-benefit needs to be reevaluated in light of the increasing global burden of AD and the fact that women are disproportionately affected. We conclude that in both humans and mouse models, protection exists so long as estrogen levels are adequate. It protects even in the presence of AD mutations and delays the age of onset. Therefore, estrogen offers substantial benefits and deeper understanding of the signaling pathways is necessary to identify creative approaches to lessen the side effects of hormone replacement treatment.
Supplementary information
Acknowledgements
This work was supported by Tata Trusts (VR). RPK is a Ramalingaswami Fellow of the Department of Biotechnology (BT/RLF/Re-entry/50/2014). This project was funded through the Centre for Brain Research, Indian Institute of Science, India. The authors are grateful to Prof. D.N. Rao for help with the L-[35S]-methionine incorporation assay. Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA), the Alzheimer’s Association (SG-20-690363-DIAN), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Spanish Institute of Health Carlos III (ISCIII), Canadian Institutes of Health Research (CIHR), Canadian Consortium of Neurodegeneration and Aging, Brain Canada Foundation, and Fonds de Recherche du Québec—Santé. This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study.
Author contributions
Conception and design of the study: RPK, AV, LD, VR. Acquisition and analysis of the data: RPK, AV, GM-T, VT, KC, LD, RG, SK, PKM, DIAN consortium. Drafting of the manuscript and figures: RPK, AV, GM-T, VT and VR.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A list of authors and their affiliations appears at the end of the paper.
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-023-02411-8.
References
- 1.Launer LJ, Andersen K, Dewey ME, Letenneur L, Ott A, Amaducci LA, et al. Rates and risk factors for dementia and Alzheimer’s disease: results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups. European studies of dementia. Neurology. 1999;52:78–84. doi: 10.1212/WNL.52.1.78. [DOI] [PubMed] [Google Scholar]
- 2.Beam CR, Kaneshiro C, Jang JY, Reynolds CA, Pedersen NL, Gatz M. Differences between women and men in incidence rates of dementia and Alzheimer’s disease. J Alzheimers Dis. 2018;64:1077–83. doi: 10.3233/JAD-180141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaugler J, James B, Johnson T, Scholz K, Weuve J. Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Nichols E, Szoeke CEI, Vollset SE, Abbasi N, Abd-Allah F, Abdela J, et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18:88–106. doi: 10.1016/S1474-4422(18)30403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48. doi: 10.2147/CLEP.S37929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheyer O, Rahman A, Hristov H, Berkowitz C, Isaacson RS, Diaz Brinton R, et al. Female sex and Alzheimer’s risk: the menopause connection. J Prev Alzheimers Dis. 2018;5:225–30. doi: 10.14283/jpad.2018.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertram L, Lill CM, Tanzi RE. The genetics of alzheimer disease: back to the future. Neuron. 2010;68:270–81. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Rabinovici GD. Late-onset Alzheimer disease. Continuum (Minneap Minn) 2019;25:14–33. doi: 10.1212/CON.0000000000000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wattmo C, Wallin ÅK. Early-versus late-onset Alzheimer’s disease in clinical practice: cognitive and global outcomes over 3 years. Alzheimers Res Ther. 2017;9:70. doi: 10.1186/s13195-017-0294-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meysami S, Raji CA, Merrill DA, Porter VR, Mendez MF. Quantitative MRI differences between early versus late onset Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2021;36:15333175211055325. doi: 10.1177/15333175211055325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aziz AL, Giusiano B, Joubert S, Duprat L, Didic M, Gueriot C, et al. Difference in imaging biomarkers of neurodegeneration between early and late-onset amnestic Alzheimer’s disease. Neurobiol Aging. 2017;54:22–30. doi: 10.1016/j.neurobiolaging.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Harper L, Bouwman F, Burton EJ, Barkhof F, Scheltens P, O’Brien JT, et al. Patterns of atrophy in pathologically confirmed dementias: a voxelwise analysis. J Neurol Neurosurg Psychiatry. 2017;88:908–16. doi: 10.1136/jnnp-2016-314978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Möller C, Vrenken H, Jiskoot L, Versteeg A, Barkhof F, Scheltens P, et al. Different patterns of gray matter atrophy in early- and late-onset Alzheimer’s disease. Neurobiol Aging. 2013;34:2014–22. doi: 10.1016/j.neurobiolaging.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Cho H, Jeon S, Kang SJ, Lee JM, Lee JH, Kim GH, et al. Longitudinal changes of cortical thickness in early- versus late-onset Alzheimer’s disease. Neurobiol Aging. 2013;34:1921.e9–1921.e15. doi: 10.1016/j.neurobiolaging.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Migliaccio R, Agosta F, Possin KL, Canu E, Filippi M, Rabinovici GD, et al. Mapping the progression of atrophy in early- and late-onset Alzheimer’s disease. J Alzheimers Dis. 2015;46:351–64. doi: 10.3233/JAD-142292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parker TD, Slattery CF, Yong KXX, Nicholas JM, Paterson RW, Foulkes AJM, et al. Differences in hippocampal subfield volume are seen in phenotypic variants of early onset Alzheimer’s disease. Neuroimage Clin. 2019;21:101632. doi: 10.1016/j.nicl.2018.101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Contador J, Pérez-Millan A, Guillen N, Tort-Merino A, Balasa M, Falgàs N, et al. Baseline MRI atrophy predicts 2-year cognitive outcomes in early-onset Alzheimer’s disease. J Neurol. 2022;269:2573–83. doi: 10.1007/s00415-021-10851-9. [DOI] [PubMed] [Google Scholar]
- 18.Contador J, Pérez-Millan A, Guillén N, Sarto J, Tort-Merino A, Balasa M, et al. Sex differences in early-onset Alzheimer’s disease. Eur J Neurol. 2022;29:3623–32. doi: 10.1111/ene.15531. [DOI] [PubMed] [Google Scholar]
- 19.Petok JR, Myers CE, Pa J, Hobel Z, Wharton DM, Medina LD, et al. Impairment of memory generalization in preclinical autosomal dominant Alzheimer’s disease mutation carriers. Neurobiol Aging. 2018;65:149–57. doi: 10.1016/j.neurobiolaging.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weston PSJ, Nicholas JM, Henley SMD, Liang Y, Macpherson K, Donnachie E, et al. Accelerated long-term forgetting in presymptomatic autosomal dominant Alzheimer’s disease: a cross-sectional study. Lancet Neurol. 2018;17:123–32. doi: 10.1016/S1474-4422(17)30434-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckles VD, Xiong C, Bateman RJ, Hassenstab J, Allegri R, Berman SB, et al. Different rates of cognitive decline in autosomal dominant and late-onset Alzheimer disease. Alzheimers Dement. 2022;18:1754–64. doi: 10.1002/alz.12505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vöglein J, Franzmeier N, Morris JC, Dieterich M, McDade E, Simons M, et al. Pattern and implications of neurological examination findings in autosomal dominant Alzheimer disease. Alzheimers Dement. 2023;19:632–45. doi: 10.1002/alz.12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eissman JM, Dumitrescu L, Mahoney ER, Smith AN, Mukherjee S, Lee ML, et al. Sex differences in the genetic architecture of cognitive resilience to Alzheimer’s disease. Brain. 2022;145:2541–54. doi: 10.1093/brain/awac177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosconi L, Berti V, Quinn C, McHugh P, Petrongolo G, Varsavsky I, et al. Sex differences in Alzheimer risk: Brain imaging of endocrine vs chronologic aging. Neurology. 2017;89:1382–90. doi: 10.1212/WNL.0000000000004425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy SJ, McCullough LD, Smith JM. Stroke in the female: role of biological sex and estrogen. ILAR J. 2004;45:147–59. doi: 10.1093/ilar.45.2.147. [DOI] [PubMed] [Google Scholar]
- 26.Nebel RA, Aggarwal NT, Barnes LL, Gallagher A, Goldstein JM, Kantarci K, et al. Understanding the impact of sex and gender in Alzheimer’s disease: a call to action. Alzheimers Dement. 2018;14:1171–83. doi: 10.1016/j.jalz.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McEwen BS, Milner TA. Understanding the broad influence of sex hormones and sex differences in the brain. J Neurosci Res. 2017;95:24–39. doi: 10.1002/jnr.23809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brinton RD. Cellular and molecular mechanisms of estrogen regulation of memory function and neuroprotection against Alzheimer’s disease: recent insights and remaining challenges. Learn Mem. 2001;8:121–33. doi: 10.1101/lm.39601. [DOI] [PubMed] [Google Scholar]
- 29.Xing D, Nozell S, Chen YF, Hage F, Oparil S. Estrogen and mechanisms of vascular protection. Arterioscler Thromb Vasc Biol. 2009;29:289–95. doi: 10.1161/ATVBAHA.108.182279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foy MR, Baudry M, Diaz Brinton R, Thompson RF. Estrogen and hippocampal plasticity in rodent models. J Alzheimers Dis. 2008;15:589–603. doi: 10.3233/JAD-2008-15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walf AA, Frye CA. A review and update of mechanisms of estrogen in the hippocampus and amygdala for anxiety and depression behavior. Neuropsychopharmacology. 2006;31:1097–111. doi: 10.1038/sj.npp.1301067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koss WA, Frick KM. Sex differences in hippocampal function. J Neurosci Res. 2017;95:539–62. doi: 10.1002/jnr.23864. [DOI] [PubMed] [Google Scholar]
- 33.Taxier LR, Gross KS, Frick KM. Oestradiol as a neuromodulator of learning and memory. Nat Rev Neurosci. 2020;21:535–50. doi: 10.1038/s41583-020-0362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Frick KM. Distinct effects of estrogen receptor antagonism on object recognition and spatial memory consolidation in ovariectomized mice. Psychoneuroendocrinology. 2017;85:110–4. doi: 10.1016/j.psyneuen.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 35.Day M, Good M. Ovariectomy-induced disruption of long-term synaptic depression in the hippocampal CA1 region in vivo is attenuated with chronic estrogen replacement. Neurobiol Learn Mem. 2005;83:13–21. doi: 10.1016/j.nlm.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Dubal DB, Broestl L, Worden K. Sex and gonadal hormones in mouse models of Alzheimer’s disease: what is relevant to the human condition? Biol Sex Differ. 2012;3:24. doi: 10.1186/2042-6410-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coffey CE, Lucke JF, Saxton JA, Ratcliff G, Unitas LJ, Billig B, et al. Sex differences in brain aging: a quantitative magnetic resonance imaging study. Arch Neurol. 1998;55:169–79. doi: 10.1001/archneur.55.2.169. [DOI] [PubMed] [Google Scholar]
- 38.Yue X, Lu M, Lancaster T, Cao P, Honda SI, Staufenbiel M, et al. Brain estrogen deficiency accelerates Aβ plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci USA. 2005;102:19198–203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brinton RD, Yao J, Yin F, Mack WJ, Cadenas E. Perimenopause as a neurological transition state. Nat Rev Endocrinol. 2015;11:393–405. doi: 10.1038/nrendo.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–27. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 41.O’Neill C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol. 2013;48:647–53. doi: 10.1016/j.exger.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 42.Levenga J, Wong H, Milstead RA, Keller BN, Laplante LE, Hoeffer CA. AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity. Elife. 2017;6:e30640. doi: 10.7554/eLife.30640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–76. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kindler S, Kreienkamp HJ. Dendritic mRNA targeting and translation. Adv Exp Med Biol. 2012;970:285–305. doi: 10.1007/978-3-7091-0932-8_13. [DOI] [PubMed] [Google Scholar]
- 45.Ahmad F, Singh K, Das D, Gowaikar R, Shaw E, Ramachandran A, et al. Reactive oxygen species-mediated loss of synaptic Akt1 signaling leads to deficient activity-dependent protein translation Early in Alzheimer’s disease. Antioxid Redox Signal. 2017;27:1269–80. doi: 10.1089/ars.2016.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kommaddi RP, Das D, Karunakaran S, Nanguneri S, Bapat D, Ray A, et al. Aβ mediates F-actin disassembly in dendritic spines leading to cognitive deficits in Alzheimer’s disease. J Neurosci. 2018;38:1085–99. doi: 10.1523/JNEUROSCI.2127-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akama KT, McEwen BS. Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J Neurosci. 2003;23:2333–9. doi: 10.1523/JNEUROSCI.23-06-02333.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hodges JR. Memory in the dementias. In: Tulving E, Craik FIM, editors. The oxford handbook of memory. USA: Oxford University Press; 2000. p. 441–64.
- 50.Tromp D, Dufour A, Lithfous S, Pebayle T, Després O. Episodic memory in normal aging and Alzheimer disease: Insights from imaging and behavioral studies. Ageing Res Rev. 2015;24:232–62. doi: 10.1016/j.arr.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Gavett BE, Gurnani AS, Saurman JL, Chapman KR, Steinberg EG, Martin B, et al. Practice effects on story memory and list learning tests in the neuropsychological assessment of older adults. PLoS One. 2016;11:e0164492. doi: 10.1371/journal.pone.0164492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Folstein MF, Folstein SE, McHugh PR. Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 53.Wechsler DA. Manual for the Wechsler memory scale-revised. San Antonio: The Psychological Corporation; 1987.
- 54.Kommaddi RP, Tomar DS, Karunakaran S, Bapat D, Nanguneri S, Ray A, et al. Glutaredoxin1 diminishes amyloid beta-mediated oxidation of F-actin and reverses cognitive deficits in an Alzheimer’s disease mouse model. Antioxid Redox Signal. 2019;31:1321–38. doi: 10.1089/ars.2019.7754. [DOI] [PubMed] [Google Scholar]
- 55.Yan Y, Cheng L, Chen X, Wang Q, Duan M, Ma J, et al. Estrogen deficiency is associated with hippocampal morphological remodeling of early postmenopausal mice. Oncotarget. 2017;8:21892–902. doi: 10.18632/oncotarget.15702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nilsson ME, Vandenput L, Tivesten Å, Norlén AK, Lagerquist MK, Windahl SH, et al. Measurement of a comprehensive sex steroid profile in rodent serum by high-sensitive gas chromatography-tandem mass spectrometry. Endocrinology. 2015;156:2492–502. doi: 10.1210/en.2014-1890. [DOI] [PubMed] [Google Scholar]
- 57.Frick KM. Estrogens and age-related memory decline in rodents: What have we learned and where do we go from here? Horm Behav. 2009;55:2–23. doi: 10.1016/j.yhbeh.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cousins SW, Marin-Castaño ME, Espinosa-Heidmann DG, Alexandridou A, Striker L, Elliot S. Female gender, estrogen loss, and sub-RPE deposit formation in aged mice. Invest Ophthalmol Vis Sci. 2003;44:1221–9. doi: 10.1167/iovs.02-0285. [DOI] [PubMed] [Google Scholar]
- 59.Iivonen S, Heikkinen T, Puoliväli J, Helisalmi S, Hiltunen M, Soininen H, et al. Effects of estradiol on spatial learning, hippocampal cytochrome P450 19, and estrogen alpha and beta mRNA levels in ovariectomized female mice. Neuroscience. 2006;137:1143–52. doi: 10.1016/j.neuroscience.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 60.Su J, Sripanidkulchai K, Hu Y, Wyss JM, Sripanidkulchai B. The effect of ovariectomy on learning and memory and relationship to changes in brain volume and neuronal density. Int J Neurosci. 2012;122:549–59. doi: 10.3109/00207454.2012.690795. [DOI] [PubMed] [Google Scholar]
- 61.Wang F, Song YF, Yin J, Liu ZH, Mo XD, Wang DG, et al. Spatial memory impairment is associated with hippocampal insulin signals in ovariectomized rats. PLoS One. 2014;9:e104450. doi: 10.1371/journal.pone.0104450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agca C, Klakotskaia D, Stopa EG, Schachtman TR, Agca Y. Ovariectomy influences cognition and markers of Alzheimer’s disease. J Alzheimers Dis. 2020;73:529–41. doi: 10.3233/JAD-190935. [DOI] [PubMed] [Google Scholar]
- 63.Leal G, Comprido D, Duarte CB. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology. 2014;76 Pt C:639–56. doi: 10.1016/j.neuropharm.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 64.Pfeiffer BE, Huber KM. Current advances in local protein synthesis and synaptic plasticity. J Neurosci. 2006;26:7147–50. doi: 10.1523/JNEUROSCI.1797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Showkat M, Beigh MA, Andrabi KI. mTOR signaling in protein translation regulation: implications in cancer genesis and therapeutic interventions. Mol Biol Int. 2014;2014:686984. doi: 10.1155/2014/686984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006;21:362–9. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- 67.Suwanna N, Thangnipon W, Soi-ampornkul R. Neuroprotective effects of diarylpropionitrile against β-amyloid peptide-induced neurotoxicity in rat cultured cortical neurons. Neurosci Lett. 2014;578:44–49. doi: 10.1016/j.neulet.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 68.Sancheti H, Akopian G, Yin F, Brinton RD, Walsh JP, Cadenas E. Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer’s disease. PLoS One. 2013;8:e69830. doi: 10.1371/journal.pone.0069830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richter JD, Klann E. Making synaptic plasticity and memory last: Mechanisms of translational regulation. Genes Dev. 2009;23:1–11. doi: 10.1101/gad.1735809. [DOI] [PubMed] [Google Scholar]
- 70.Wang DO, Martin KC, Zukin RS. Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 2010;33:173–82. doi: 10.1016/j.tins.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ganguli M, Chandra V, Kamboh MI, Johnston JM, Dodge HH, Thelma BK, et al. Apolipoprotein E polymorphism and Alzheimer disease: the Indo-US cross-national dementia study. Arch Neurol. 2000;57:824–30. doi: 10.1001/archneur.57.6.824. [DOI] [PubMed] [Google Scholar]
- 72.Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry. 2005;62:685–91. doi: 10.1001/archpsyc.62.6.685. [DOI] [PubMed] [Google Scholar]
- 73.Malpetti M, Ballarini T, Presotto L, Garibotto V, Tettamanti M, Perani D. Gender differences in healthy aging and Alzheimer’s Dementia: a 18 F-FDG-PET study of brain and cognitive reserve. Hum Brain Mapp. 2017;38:4212–27. doi: 10.1002/hbm.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oveisgharan S, Arvanitakis Z, Yu L, Farfel J, Schneider JA, Bennett DA. Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathol. 2018;136:887–900. doi: 10.1007/s00401-018-1920-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hebert LE, Scherr PA, McCann JJ, Beckett LA, Evans DA. Is the risk of developing Alzheimer’s disease greater for women than for men? Am J Epidemiol. 2001;153:132–6. doi: 10.1093/aje/153.2.132. [DOI] [PubMed] [Google Scholar]
- 76.Sinforiani E, Citterio A, Zucchella C, Bono G, Corbetta S, Merlo P, et al. Impact of gender differences on the outcome of Alzheimer’s disease. Dement Geriatr Cogn Disord. 2010;30:147–54. doi: 10.1159/000318842. [DOI] [PubMed] [Google Scholar]
- 77.Seshadri S, Wolf PA, Beiser A, Au R, McNulty K, White R, et al. Lifetime risk of dementia and Alzheimer’s disease. The impact of mortality on risk estimates in the Framingham Study. Neurology. 1997;49:1498–504. doi: 10.1212/WNL.49.6.1498. [DOI] [PubMed] [Google Scholar]
- 78.Jacobs EG, Weiss BK, Makris N, Whitfield-Gabrieli S, Buka SL, Klibanski A, et al. Impact of sex and menopausal status on episodic memory circuitry in early midlife. J Neurosci. 2016;36:10163–73. doi: 10.1523/JNEUROSCI.0951-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rentz DM, Weiss BK, Jacobs EG, Cherkerzian S, Klibanski A, Remington A, et al. Sex differences in episodic memory in early midlife: impact of reproductive aging. Menopause. 2017;24:400–8. doi: 10.1097/GME.0000000000000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vila-Castelar C, Guzmán-Vélez E, Pardilla-Delgado E, Buckley RF, Bocanegra Y, Baena A, et al. Examining sex differences in markers of cognition and neurodegeneration in autosomal dominant Alzheimer’s disease: preliminary findings from the Colombian Alzheimer’s prevention initiative biomarker study. J Alzheimers Dis. 2020;77:1743–53. doi: 10.3233/JAD-200723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–70. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benzinger TLS, Blazey T, Jack CR, Koeppe RA, Su Y, Xiong C, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci USA. 2013;110:E4502–4509. doi: 10.1073/pnas.1317918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–60. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McDade E, Wang G, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91:e1295–e1306. doi: 10.1212/WNL.0000000000006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang M, Ryman DC, McDade E, Jasielec MS, Buckles VD, Cairns NJ, et al. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: a comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN-OBS) Lancet Neurol. 2016;15:1317–25. doi: 10.1016/S1474-4422(16)30229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vila-Castelar C, Tariot PN, Sink KM, Clayton D, Langbaum JB, Thomas RG, et al. Sex differences in cognitive resilience in preclinical autosomal-dominant Alzheimer’s disease carriers and non-carriers: baseline findings from the API ADAD Colombia trial. Alzheimers Dement. 2022;18:2272–82. doi: 10.1002/alz.12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vila-Castelar C, Tariot PN, Sink KM, Clayton D, Langbaum JB, Thomas RG, et al. Sex differences in neurodegeneration and memory performance in preclinical autosomal dominant Alzheimer’s disease: baseline findings from the API ADAD trial. Alzheimers Dement. 2020;16:e041225. doi: 10.1002/alz.041225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nelson HD. Menopause. Lancet. 2008;371:760–70. doi: 10.1016/S0140-6736(08)60346-3. [DOI] [PubMed] [Google Scholar]
- 89.Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–67. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 90.Villeneuve S, Vogel JW, Gonneaud J, Binette AP, Rosa-Neto P, Gauthier S, et al. Proximity to parental symptom onset and Amyloid-β burden in sporadic Alzheimer disease. JAMA Neurol. 2018;75:608–19. doi: 10.1001/jamaneurol.2017.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.