

Abstract
The sex ratio is a key ecological demographic parameter crucial for population viability. However, the epigenetic mechanisms operating during gonadal development regulating gene expression and the sex ratio remain poorly understood. Moreover, there is interest in the development of epigenetic markers associated with a particular phenotype or as sentinels of environmental effects. Here, we profiled DNA methylation and gene expression of 10 key genes related to sex development and stress, including steroidogenic enzymes, and growth and transcription factors. We provide novel information on the sex‐related differences and on the influence of elevated temperature on these genes in zebrafish, a species with mixed genetic and environmental influences on sex ratios. We identified both positive (e.g., amh, cyp11c and hsd11b2) and negative (e.g., cyp11a1 and dmrt1) correlations in unexposed males, and negative correlation (amh) in exposed females between DNA methylation and gene expression levels. Further, we combined DNA methylation analysis with machine learning procedures and found a series of informative CpGs capable not only of correctly identifying sex (based on cyp19a1a DNA methylation levels) but also of identifying whether males and females had been exposed to abnormally elevated temperature when young (based on amh and foxl2a DNA methylation levels, respectively). This was achieved in the absence of conspicuous morphological alterations of the gonads. These DNA methylation‐based epigenetic biomarkers represent molecular resources that can correctly recapitulate past thermal history and pave the way for similar findings in other species to assess potential ecological effects of environmental disturbances in the context of climate change.
Keywords: climate change, DNA methylation, essential epigenetic marks, machine learning, sex differentiation, temperature
1. INTRODUCTION
In contrast to the situation in birds and mammals, where sex is genetically determined, in most reptiles and many fish and amphibians sex is determined by a combination of genetic and environmental influences (Penman & Piferrer, 2008; Valenzuela, 2008). While there is diversity in the sex‐determining genes even in closely related species (Nagahama et al., 2021; Tanaka et al., 2007), the components making up the downstream gene networks appear fairly conserved across vertebrates (Capel, 2017). In recent years, the contribution of epigenetic mechanisms in regulating gene expression during sex determination and differentiation has become progressively clearer (Gunes et al., 2016; Kuroki & Tachibana, 2018; Piferrer, 2013). Because epigenetics integrates both genomic and environmental information and sex is particularly plastic in fish, these animals constitute good models to study the contribution of epigenetic changes during sex differentiation in response to environmental perturbations (Beal et al., 2018; Ortega‐Recalde et al., 2020).
An inverse relationship between epigenetic silencing and gene expression should be expected in genes that are preferentially expressed in males (“pro‐male” genes) and females (“pro‐female” genes) during sex differentiation. Thus, for example, in ovaries the DNA methylation in pro‐female genes should be lower when compared with the level of the same genes in males, while in pro‐male genes DNA methylation should be high in the ovaries (Piferrer et al., 2019). The opposite would apply in developing testes. After reviewing the available data published at the time regarding genes related to sexual development in both gonochoristic and hermaphrodite fish species, cyp19a1a and dmrt1, two key genes for ovary and testis development, respectively, were found to follow this principle. However, in other genes, such as foxl2a and amh, the association was not clear (Piferrer et al., 2019). This led to the formulation of the model of the “conserved epigenetic regulation of sex” (CERS) (Piferrer et al., 2019). This model states that epigenetic and gene expression patterns are more closely associated with the development of a particular gonadal phenotype (e.g., testis differentiation) rather than with the intrinsic or extrinsic causes that lead to the development of this phenotype. Nevertheless, genes that also play important roles in gonad formation, such as those coding for steroidogenic enzymes (Caulier et al., 2015; Fernandino et al., 2012, 2013), have not been evaluated yet under this framework. In addition, to date, only a few studies have examined the effects of temperature on DNA methylation and in relation to gene expression patterns in fish gonads, such as the European seabass (Dicentrarchus labrax) (Anastasiadi, Vandeputte, et al., 2018; Navarro‐Martín et al., 2011), the Nile tilapia (Oreochromis niloticus) (Wang et al., 2019), the half‐smooth tongue sole (Cynoglossus semilaevis) (Shao et al., 2014), fugu (Takifugu rubripes) (Zhou et al., 2019) and zebrafish (Danio rerio) (Hosseini et al., 2022; Pierron et al., 2021).
DNA methylation changes occurring during early development determine lasting gene expression programmes (Bird, 2002; Feng et al., 2010). As in other animals, in fish early developmental periods are particularly vulnerable to external perturbations, which can affect the expression of sex‐specific genes (Shen & Wang, 2014). These changes may remain permanent and detectable in adult gonads, even once the original cue has long disappeared (Anastasiadi, Esteve‐Codina, & Piferrer, 2018). Because of the sensitive nature of DNA methylation marks (Flores et al., 2013), changes indicative of a given physiological state or as a result of an environmental influence can be stable and, if associated with a given phenotype, can be used as marks. Essential epigenetic marks (EEMs) are defined as “the number and identity of informative epigenetic marks that are strictly necessary, albeit perhaps not sufficient, to bring about a specific, measurable, phenotype of interest” (Piferrer, 2019). Thus, epigenetic markers can have many applications. For example, they are used in medicine (Mikeska & Craig, 2014) as a diagnostic tool to identify cancers of unknown primary origin (Moran et al., 2016). An example is the Epi proColon kit, designed to detect aberrant DNA methylation of the v2 of promoter region of the SEPT9 gene in blood‐borne colorectal cancer cells (de Vos et al., 2009; Model et al., 2007). In ecology, DNA methylation changes can also provide reliable information for age estimation in animals, from humans (Horvath, 2013), other mammals and birds (De Paoli‐Iseppi et al., 2019; Polanowski et al., 2014; Thompson et al., 2017) to fish (Anastasiadi & Piferrer, 2020). Epigenetic markers have been also used to identify the presence of pollutants or changes in temperature (Beal et al., 2018). DNA methylation marks were suggested to detect early stress in red blood cells of chickens later in life (Pértille et al., 2017). However, no studies have been conducted with epigenetic biomarkers to develop tools to detect past exposures to abnormal environmental conditions using machine learning procedures with predictive statistical power.
In this study, we used the laboratory zebrafish AB strain, which has a polygenic sex determination system (Liew et al., 2012) and where sex depends on genetic and environmental influences. In zebrafish, sex ratio responses to temperature and density are family specific (Ribas, Liew, et al., 2017; Ribas, Valdivieso, et al., 2017a, 2017b; Valdivieso et al., 2022). Our objectives were to study both sex‐specific differences in DNA methylation in a suite of key sex‐ and stress‐related genes and the effect of environmental influences on DNA methylation in the gonads. The ultimate goal was two‐fold: first, by analysing genes other than cyp19a1a and dmrt1, to expand the catalogue of genes analysed to investigate whether the predictions of the CERS model are fulfilled or not. This information, obtained in animals subjected to different temperatures, is essential to advance our knowledge on the contribution of epigenetic regulatory mechanisms in integrating genomic and environmental information to bring about the phenotype, in this case the sexual phenotype. Second, we aimed to identify both sex‐specific and persistent DNA methylation marks that could be used as EEMs indicative of the sex of a DNA sample obtained from a fish sample with no record of phenotypic sex, and whether fish had been exposed to abnormally elevated temperature. To this end, larvae zebrafish were exposed to elevated temperatures during sex differentiation to induce masculinization (Ribas, Liew, et al., 2017). By using a locus‐specific approach previously developed in our laboratory (Anastasiadi, Vandeputte, et al., 2018), we adapted the method specifically for zebrafish and included the evaluation of the DNA methylation of ten genes including steroidogenic enzymes (cyp19a1a, cyp11a1, cyp11c1, hsd11b2, hsd17b1 and hsd17b3), and growth and transcription factors (amh, dmrt1, dmrt3a and foxl2a). These genes are involved in reproduction and stress responses in fish (Brunner et al., 2001; Fernandino et al., 2012; Guo et al., 2005; Hsu et al., 2009; Lau et al., 2016; Mindnich et al., 2004; Rodríguez‐Marí et al., 2005; Sreenivasan et al., 2008; Wang & Orban, 2007). In addition, we measured the expression of these genes to determine its relationship with DNA methylation.
2. MATERIALS AND METHODS
2.1. Animal rearing conditions
Zebrafish (AB strain; ZFIN ID: ZDB‐GENO‐960809‐7) from the European Zebrafish Resource Center (EZRC, Germany) were housed at the experimental facilities of the Institute of Marine Sciences (ICM‐CSIC, Barcelona). The housing conditions, the physicochemical water parameters and the feeding regime for fish were monitored as described elsewhere (Ribas, Valdivieso, et al., 2017a). Fertilized eggs were obtained after natural spawning and larvae were raised from 6 to 18 days post‐fertilization (dpf) under regular conditions of temperature (28°C) and appropriate density levels (eight fish per litre) to avoid density‐dependent induced masculinization (Ribas, Valdivieso, et al., 2017a). The animal facility used for the present study is licensed by the Bioethical Committee of the Government of Catalonia with reference code 9977.
2.2. Experimental design
The thermal experiment was carried out as previously described (Ribas, Liew, et al., 2017). Briefly, the larvae originating from one female and one male were randomly divided into two groups, each containing an equal number of fish, and were exposed from 18 to 32 dpf to two different temperature treatments: 28°C (control) and 35°C (elevated temperature), which was achieved with the aid of an electric resistance and by a gradual increase until 22 dpf. In the 35°C group, temperature was maintained stable until 32 dpf, at which point it was gradually decreased back to 28°C by 36 dpf. Thereafter, all groups were reared in a recirculating system at 28°C. The experiment finished at 90 dpf and fish were killed by ice‐water immersion. The whole experiment was repeated twice to allow for biological variability by using fish from a different family, also originating from the cross of one male and one female. Phenotypic sex was determined visually with the aid of a dissecting scope, and gonads were classified according to the degree of maturation based on their overall size, shape, colour and texture as immature (type 1), maturing (type 2) or mature (type 3) (Ribas, Liew, et al., 2017). For DNA methylation and gene expression analysis, we selected type 3 gonads and made sure that those selected fulfilled this criterion within and between groups to avoid bias in the results. Gonads were extracted and kept at −80°C for molecular analysis. Detailed information on the total number of samples of each experiment and samples for DNA methylation analysis per each family, sex and temperature are given in Table S1.
2.3. DNA and RNA extractions
Genomic DNA and total RNA were isolated from the same gonad sample to allow comparisons of DNA methylation and gene expression in the same individual. For DNA extraction, gonads were hemisected and one half was treated overnight at 56°C with digestion buffer containing 1 μg of proteinase K to eliminate proteins (P2308; Sigma‐Aldrich). Then, a standard phenol–chloroform–isoamyl alcohol protocol (PCI 25:24:1, by vol.) with 0.5 μg ribonuclease A (12091021; PureLink RNase A, Life Technologies) to eliminate trace RNA was performed. RNA extraction was carried out in the other half of the gonad by Trizol Reagent (T9424; Sigma‐Aldrich) according to the manufacturer's instructions. The quality and quantity of DNA and RNA was measured by a NanoDrop (ND‐1000) spectrophotometer (Thermo Fisher Scientific).
2.4. Gene selection
Ten genes were selected for DNA methylation analysis based on their functions in reproduction and stress in fish. These genes encode steroidogenic enzymes: cyp19a1a, cyp11a1, cyp11c1, hsd11b2, hsd17b1 and hsd17b3, and growth and transcription factors: amh, dmrt1, dmrt3a and foxl2a. The full name of the genes is given in Table S2.
2.5. Primer design for bisulphite converted DNA
The precise genomic regions and sequences were determined using the zebrafish genome (danRer10) obtained from the University of California Santa Cruz (UCSC) genome browser database (Karolchik et al., 2003). Primers were specifically designed for bisulphite‐converted DNA using the methprimer software (Li & Dahiya, 2002). Amplicons targeted the proximal promoter and/or the beginning of the first exon of each gene, encompassing the maximum possible number of CpG sites, given the importance of these elements in the regulation of gene expression (Brenet et al., 2011; Deaton & Bird, 2011). Nextera adapter sequences were then added to the 5′ ends of the region‐specific primers (Table S3) as described in the Illumina's protocol: “16 S metagenomic library preparation.” The genomic coordinates of the amplicons were checked using the Integrated Genome Browser (igv) software (version 2.3) (Robinson et al., 2010; Thorvaldsdottir et al., 2013). Detailed information on the sequences of the primers, the corresponding annealing temperature, their length, and the total number of CpG sites and CpG islands within the amplicon are given in Table S2.
2.6. PCR of amplicons
One microgram of DNA per sample was bisulphite converted using the EZ DNA Methylation‐Direct Kit (D5020; Zymo Research) following the manufacturer's protocol. PCR amplifications were performed using 2 μl of bisulphite‐converted DNA (~26–30 ng) as initial template, 4 μl MgCl2 (25 mm), 2 μl dNTPs (R0193,10 mm; Thermo Fisher Scientific), 0.5 μl forward and reverse primers (10 μm; Life Technologies), 5 μl 5× Green GoTaq Flexi Buffer (M7405; Promega), 0.12 μl GoTaq G2 Hot Start polymerase (M7405; Promega) and 10.8 μl MilliQ autoclaved water. The PCR conditions of the thermal cycler were: one step of 95°C for 7 min, followed by 40 cycles of 95°C for 1 min, then specific annealing temperature for each gene for 2 min (Table S2), 65°C for 2 min and a final step of 65°C for 10 min. The presence and the size of each amplicon were confirmed in all samples using 4 μl of PCR amplification product on a 2% agarose gel. In this step, one sample was discarded because no amplifications of any target gene were observed in the gel. Finally, the primers of each target gene were validated by Sanger sequencing using amplicons produced from a pool of three males and three females. The primers used for Sanger sequencing were complementary to the Nextera adapters (Table S3). The Sanger output sequences were checked using the blastn software (version 2.7.1) (Altschul et al., 1990) to ensure the identity of the amplicon.
2.7. Size‐selection and normalization of amplicons
The working solution of magnetic beads (65152105050250, Sera‐mag Speed Beads; GE Healthcare) was prepared following the protocol described in Anastasiadi, Vandeputte, et al. (2018) and adapted from Rohland and Reich (2012). The size‐selection and normalization of DNA quantities across the PCR amplicons were performed following the methodology described in Anastasiadi, Vandeputte, et al. (2018) from a customized version (Hosomichi et al., 2014). Briefly, for size‐selection of the amplicons, 10 μl PCR amplification product from each gene in each sample was transferred to a new tube and 40 μl MilliQ water and 20 μl 2× beads (0.4×) were added. Samples were incubated at room temperature for 5 min, placed on the magnet for 5 min and supernatant was transferred to a new tube. Then, samples were mixed with 42 μl 2× beads (0.6×), incubated for 5 min and placed on magnet discarding the supernatant. Samples were carefully washed on the magnet with 200 μl of freshly prepared 70% ethanol for 10 s and were dried for 10 min. Samples were rehydrated in 22 μl MilliQ water outside the magnet for 5 min, then placed back on the magnet for five additional minutes and finally 20 μl of supernatant was transferred to a new tube. For the normalization step, 40 μl MilliQ water, 20 μl 20‐fold diluted beads (2×) and 20 μl propan‐2‐ol (I9516; Sigma‐Aldrich) were added to the 20 μl supernatant from the previous size‐selection step. Samples were then incubated for 5 min, placed on the magnet for 5 min and a second ethanol wash was performed exactly as described above. Next, samples were rehydrated in 12 μl MilliQ water for 10 min and placed back on the magnet for 10 min. Finally, 10 μl of each gene of each sample were transferred to a new tube.
2.8. Index PCR
Three microlitres from the normalized amplicons of each gene belonging to the same sample were pooled in a new tube. To make libraries, samples were individually labelled by mixing 5 μl of the pool of each gene per each sample with 5 μl index Nextera primers (Nextera XT Index Kit V2 set A, FC‐131–2001; Illumina), 25 μl 2× KAPA HiFi HotStart Ready Mix PCR kit (07958919001; Kapa Byosystems) and 10 μl MilliQ water. The PCR conditions in the thermal cycler were: one step at 95°C for 3 min, eight cycles of 95°C for 30 s, 55°C for 30 s and 72°C for 30 s, with a final step at 72°C for 5 min, according to the “Illumina's protocol for 16 S metagenomic library preparation.” Next, an extra clean‐up step was carried out on the indexed samples, following the previous customized size‐selection and normalization protocol but using 1× beads and eluting the samples in 15 μl MilliQ water. Finally, 3 μl from each indexed sample was pooled in a tube making a single multiplex bisulphite sequencing (MBS) Library. Before sequencing, the DNA concentration of the single tub composed of 70 libraries was measured by a Qubit fluorimeter (Invitrogen) using a Qubit dsDNA HS Assay Kit (Invitrogen) and the distribution of pooled amplicons was checked by a High Sensitivity DNA Assay on a Bioanalyzer system (Agilent 2100). The pooled samples containing the 70 libraries were sequenced on a MiSeq sequencing system (Illumina) using the 250 paired‐end (PE) protocol.
2.9. Bioinformatics analysis
Samples were demultiplexed based on the index codes by the Illumina software. The Nextera adapters were removed using trim galore! software (version 0.4.5) (Krueger, 2015) with parameters: ‐‐nextera, −‐quality 20 and ‐‐phred64. After trimming, the number and quality of the reads were checked using the fastqc software (version 0.11.6) (Andrews, 2010). In parallel, the whole zebrafish genome (danRer10) was bisulphite converted in silico using the function “bismark_genome_preparation.” Alignments of the PE reads were performed using the function “bismark”: ‐‐bowtie2, −‐non‐directional and ‐‐score_min L, 0, − 0.6. The methylation status of all CpG sites was obtained using the function “bismark_methylation_extractor”: ‐‐bedGraph. All three steps were carried out by bismark software (version 16.0) (Krueger & Andrews, 2011). The efficiency of the bisulphite conversion was calculated for each sample as 100% minus the percentage of the cytosines (C) methylated in the CHH context (where H indicates a non‐G nucleotide). In this step, three samples were removed because they had less than 99.0% bisulphite conversion efficiency. The coordinates of all CpG sites were obtained using a specific package for zebrafish, “BSgenome.Drerio.UCSC.danRer10” from bsgenome (Pagés, 2018). We intersected CpG sites of the target gene using the amplicons' boundary coordinates by bed tools (Quinlan & Hall, 2010). Finally, the percentage methylation of the filtered CpG sites was calculated as: . From the targeted CpG sites, only those that showed coverage >5× were retained for subsequent analysis.
2.10. Quantitative real‐time PCR (qPCR)
From each sample, 1 μg RNA was treated with DNAse I Amplification Grade (18068–015; Invitrogen) to remove any genomic DNA. Then, 500–600 ng of RNA was reverse transcribed into cDNA using the SuperScript III Reverse Transcriptase (18080093; Invitrogen) for qPCR. The qPCR primer sequences and the two references genes (eef1a1l1 and rpl13a), both previously validated for zebrafish (Tang et al., 2007), are listed in Table S4. The qPCR primers targeted regions between at least two exons to avoid amplification of possible traces of genomic DNA. The efficiencies of the primers (E = 10−1/slope) were estimated from the slope value derived from the log‐linear regression from a standard curve of five serial cDNA dilutions (1, 1:5, 1:10, 1:50, 1:100, 1:500) of a pool of 1 μl cDNA of all samples. The qPCRs were carried out in triplicate, taking 2 μl of cDNA (1:10), 0.5 μl forward and reverse primer, 2 μl 5× PyroTaq EvaGreen dye (87H24‐001; Cultek) and 5 μl MilliQ water per well for each gene and sample with their corresponding negative controls. The qPCR conditions in the QuantStudio 12 K Flex (Thermo Fisher Scientific) thermal cycler were: 50°C for 2 min, 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min followed by a melting curve to confirm single amplification product.
2.11. Statistical analysis
All statistical analyses were carried out using R software (version 3.0.2) (Team, 2013). Significant results were considered at p < .05. All graphs were generated using the ggplot2 package (version 3.1.0) (Wickham, 2016).
2.11.1. Proportions of males and females
For the analysis of sex ratios, a χ2 test with Yate's correction (Yates, 1934) was applied to compare the number of males and females between the 28°C and 35°C groups.
2.11.2. Methylation data analysis
We calculated the mean DNA methylation across all the CpG sites from each gene for each sample. Finally, we averaged the DNA methylation data from all samples, grouped them per sex and temperature for each family, and then combined all data of the two families. The effects of sex and temperature on DNA methylation were evaluated by two‐way ANOVA followed by a Tukey's HSD test. Normality of the residuals was checked by the Shapiro–Wilk test, and a Levene's test for homogeneity of variance was used. When the assumptions of normality were not met, a logit transformation was applied. In cases when transformed data failed to follow the assumptions, we used two‐way ANOVAs with modified M‐estimators and 5000 bootstraps (function pbad2way) using the wrs2 package (version 0.9.2) (Mair & Wilcox, 2020), as described in Anastasiadi, Vandeputte, et al. (2018).
2.11.3. Gene expression and correlation analysis
The quantification cycle (Cq) data were averaged, after eliminating samples that had more than 0.30 standard deviation between the three technical replicates. Data were normalized applying the formula: ΔCq = Cq (Mean target gene) − Cq (Geometric mean eef1a1l1 and rpl13a). Gene expression was calculated as E (ΔCq), where E is the efficiency of the primer. The ΔCq values were analysed for normality and homogeneity of variances by the Shapiro–Wilk and Levene's test, respectively. The effects of sex and temperature on gene expression were evaluated using two‐way ANOVA followed by a Tukey test. To test the correlation between DNA methylation and gene expression for the same gene, a Spearman's rank correlation (ρ) test was applied using the corrplot package (version 0.84) (Wei & Simko, 2017).
2.11.4. Genes more influenced by sex and elevated temperature in males and females
First, and for each gene, we evaluated the differences between sexes by subtracting the mean DNA methylation of males minus the mean DNA methylation of females using data only from the 28°C group. Next, and for each sex, we evaluated the differences due to elevated temperature by subtracting the mean DNA methylation levels of the 35°C group minus the mean DNA methylation of the 28°C group.
2.11.5. Epigenetic biomarkers: Indicators of phenotypic sex and exposure to thermal stress
Based on the observed sex‐ and temperature‐related differences of DNA methylation levels of the genes examined in this study, we reasoned whether these differences could be used as epigenetic biomarkers. Here, a two‐step approach involving three separate classifications was implemented. In the first classification, we used all fish to predict their sex regardless of the temperature they had been exposed to (N = 70 samples). For the second and third classifications, males (N = 37) and females (N = 33) were used separately. In the latter two classifications, the goal was to determine whether the temperature they had been exposed to during the period of gonad development while juveniles could be predicted from the DNA methylation levels in 90‐day‐old adults. We only used genes that showed significant differences by sex and temperature. If any sample(s) had missing data for the selected genes, the values were imputed by applying the mean value replacement method for the corresponding group in which the sample pertained (Zhang, 2016).
For the classification and prediction, we applied a flexible discriminant analysis (FDA) with repeated K‐fold cross‐validation (10‐fold and five repetitions) using the caret package (version 6.0–82) (Kuhn, 2008, 2020). The efficiency of the prediction accuracy (comparing the prediction result from the model output with the real data) and the Cohen's kappa coefficient test (κ) values were extracted from the caret package. The coefficient κ measures inter‐rater agreement for qualitative (categorical) items and can take values between 1 (complete agreement) and 0 (no agreement). In addition, the level of the agreement classification was assigned as described in McHugh (2012). First, we evaluated each gene using the mean methylation (mean of all the CpGs of the amplicon) as a single variable predictor. Second, for the genes that showed the highest accuracy and κ, we broke down the methylation values to individual CpGs in order to improve the classification. Third, we combined the best predictors between them (those that showed the highest accuracy and κ values) to observe if the addition of more predictors could improve the classification efficiency. We used the ldahist function from the mass package (version 7.3–51.1) (Venables & Ripley, 1999) to make stacked histograms of the discriminant coefficient.
2.11.6. Independent samples for machine learning validation
To validate the results of the sex prediction from the machine learning algorithm, we extracted the methylation data (15 CpGs of the cyp19a1a amplicon) of 20 testes and 20 ovaries from an unrelated offspring obtained from a third family produced as described above and sampled at the age of 90 dpf and raised at 28°C.
3. RESULTS
3.1. Temperature shifted the sex ratio
We tested two independent families, and in both cases the offspring exposed to elevated temperature showed an increase of males (i.e., 42.6% and 20.4% increase for Family 1 and 2, respectively) although the proportions between groups was only significant in Family 1 (Figure S1a). When taking the two families together, sex ratio analysis showed that the group exposed to 35°C had a significantly (χ2 = 31.32; p < .001) higher proportion of males when compared with the 28°C group (Figure S1b), indicating the effectiveness of the temperature treatment in inducing masculinization, as expected (Hosseini et al., 2019; Ribas, Liew, et al., 2017; Valdivieso et al., 2020, 2022). During sampling, visual inspection of the fish subjected to 35°C showed no obvious alterations of the gonads when compared with those of fish exposed to 28°C.
3.2. DNA methylation patterns in the gonads are sex‐ and temperature‐dependent
Regarding the quality of the methylation data, the number (mean ± SEM) of sequenced reads per sample was 469,779 ± 22,188, alignment efficiency 71.9 ± 1.63% and bisulphite conversion rate 99.6 ± 1.36% (see Table S5). The final number of samples for Family 1 and Family 2 was 34 and 36, respectively.
Although the effectiveness of temperature‐induced masculinization varied between families, and because we did not have exposed females at 35°C in Family 1 due to 100% masculinization, we analysed DNA methylation profiles by grouping the two families and considering sex and temperature as factors for the analysis. Further, we were interested in finding not family‐specific differences but rather sex‐ and temperature‐specific differences that could be independent of family. However, we calculated the mean methylation of the 10 genes for each family separately (Table S6), and we compared mean methylation values of all 10 genes between these two families. The results showed no significant differences in females at 28°C (Family 1 = 35.13 ± 25.08; Family 2 = 31.05 ± 26.94, t = 0.3324, p = .74), in males at 28°C (Family 1 = 60.65 ± 39.38; Family 2 = 34.74 ± 25.5, t = 1.67, p = .15), or in males at 35°C (Family 1 = 60.24 ± 41.66; Family 2 = 60.69 ± 38.77, t = 0.0226, p = .98). When grouping families, an overall assessment of DNA methylation revealed two main differences between sexes (Figure S2). First, in females the median DNA methylation values of the genes analysed in this study essentially covered the range 0%–100%, whereas in males the DNA methylation values were either usually above 75% or below 10% with no intermediate values (Figure S2a). Furthermore, in males the DNA methylation at 28°C showed high variability among individuals, but this variation was reduced at 35°C (Figure S2b). These results show clear sex‐related differences in DNA methylation in zebrafish and on the effect of temperature on these values.
We examined the DNA methylation of genes coding for six different steroidogenic enzymes (i.e., cyp19a1a, cyp11a1, cyp11c1, hsd11b2, hsd17b1 and hsd17b3), including those leading to the synthesis of 11‐ketotestosterone and oestradiol‐17β, considered the major androgen and oestrogen in fish, respectively, in terms of sex differentiation (Figure 1a–f). We observed significant (p < .001) sex‐dependent differences in DNA methylation values in the 28°C group for all genes. Temperature significantly affected DNA methylation of cyp19a1a, cyp11a1, cyp11c1 and hsd17b3 (Figure 1a–c,f) and only cyp11a1 showed a significant interaction between sex and temperature (Figure 1b). We also examined the DNA methylation of genes coding for key sex‐related growth and transcription factors (i.e., amh, dmrt1, dmrt3a and foxl2a). Sex‐specific differences were also observed for all genes examined (Figure 1g–j). In contrast to amh, DNA methylation levels of dmrt1, dmrt3a and foxl2a were lower in males than in females, although for foxl2a DNA methylation values were very low (Figure 1h–j). Temperature was associated with an increase and decrease of the DNA methylation levels of amh and dmrt1 in males, respectively (Figure 1g,h), the former being the only one of the second group of genes where sex and temperature interaction was significant (Figure 1g).
FIGURE 1.
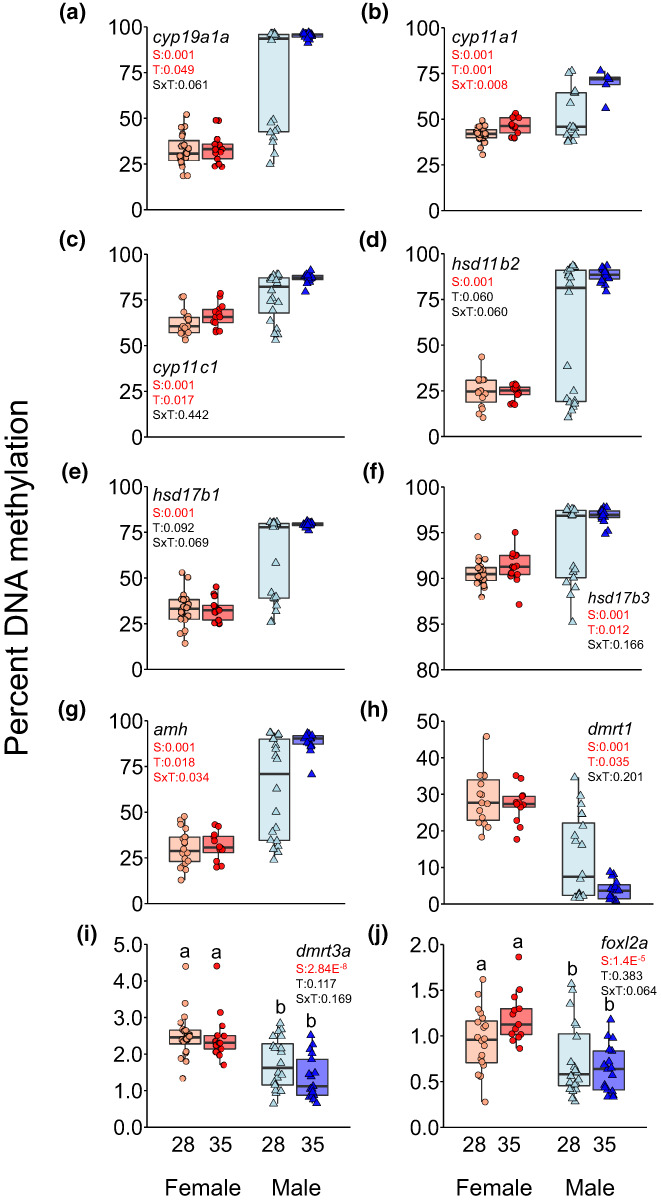
DNA methylation levels according to sex and early temperature. Box plot of percent DNA methylation levels on the promoter region of: (a) cyp19a1a, (b) cyp11a1, (c) cyp11c1, (d) hsd11b2, (e) hsd17b1, (f) hsd17b3, (g) amh, (h) dmrt1, (i) dmrt3a and (j) foxl2a genes from mature gonads of females and males exposed to 28°C and 35°C during sex differentiation (18–32 days post‐fertilization).The p‐values for the factor effects of sex (S), temperature (T) or the interaction of both factors (SxT) are reported for each gene. Two‐way ANOVA followed by post hoc Tukey test was applied in the dmrt3a and foxl2a genes and two‐way ANOVA with robust estimation was applied in the cyp19a1a, cyp11a1, cyp11c1, hsd11b2, hsd17b1, hsd17b3, amh and dmrt1 genes
3.3. Gene expression in the gonads is altered after early heat response
For gene expression analysis we only considered the genes with significant sex‐dependent DNA methylation differences (threshold >10%). We observed sex‐related significant differences in gene expression in the 28°C group in cyp19a1a, cyp11a1, cyp1c, hsd11b2, amh and dmrt1 (Figure 2a–d,f,g) while the hsd17b1 gene remained invariable (Figure 2e). Elevated temperature during gonadal development significantly downregulated the expression of cyp11a1 (Figure 2b) and increased cyp11c1, amh and dmrt1 in testes but not in ovaries (Figure 2c,f,g, respectively).
FIGURE 2.
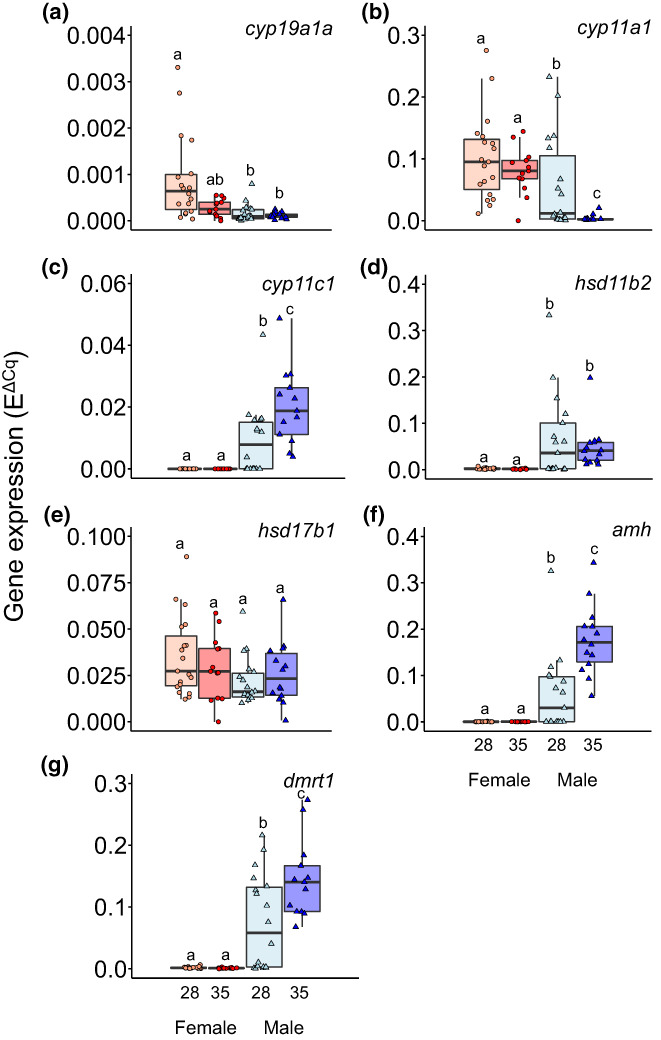
Gene expression levels according to sex and early temperature. Box plot of gene expression levels of (a) cyp19a1a, (b) cyp11a1, (c) cyp11c1, (d) hsd11b2, (e) hsd17b1, (f) amh and (g) dmrt1 genes in mature gonads of females and males exposed to 28°C and 35°C during sex differentiation (18–32 days post‐fertilization). Two‐way ANOVA followed by post hoc Tukey test was applied. Different letters indicate significant differences (p < .05) between sex and temperature
3.4. Correlation analysis between DNA methylation and gene expression
We carried out correlation analyses between DNA methylation and gene expression levels of cyp19a1a, cyp11a1, cyp11c1, hsd11b2, hsd17b1, amh and dmrt1 in gonads of females and males exposed to 28 and 35°C (Figure 3). In females, we found a significant negative correlation in amh (r = −.69; p = .0423) only in the 35°C group. In contrast, in males exposed at 28°C we observed a significant negative correlation in cyp11a1 (r = −.79; p = .006) and dmrt1 (r = −.78; p < .001), and a significant positive correlation in cyp11c1 (r = .80; p = 9.3 × 10−5), hsd11b2 (r = .68; p = .002) and amh (r = .68; p = 4.9 × 10−5) (Figure 3). In males, the effects of 35°C made these relationships less evident when analysing the same genes. However, comparing results between temperature treatments, the genes hsd11b2 and dmrt1 showed the same tendency of the relationship whereas amh showed an opposite correlation. The cyp11c1 gene, due to the low number of samples, could not be compared even though it showed the same negative relationship. In summary, at the control temperature (28°C) we did not observe any significant correlation, positive or negative, between DNA methylation and gene expression in any of the genes examined in females, whereas in males it was significantly positive for three genes (cyp11c1, hsd11b2 and amh) and negative for two (cyp11a1 and dmrt1). On the other hand, elevated temperature provoked a negative correlation for amh in females where there was none before and cancelled all correlations, positive or negative, found in males at the control temperature.
FIGURE 3.
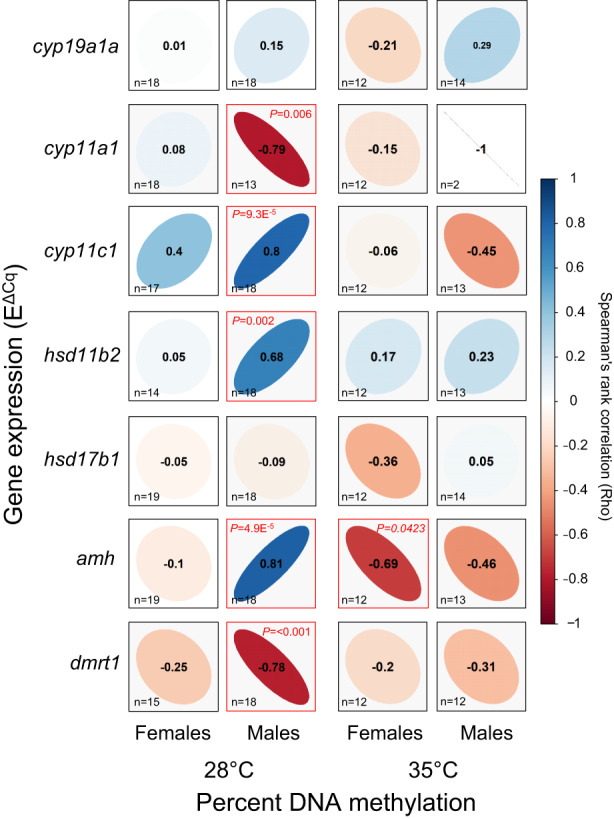
Relationship between DNA methylation and gene expression. Correlations of percentage DNA methylation of the promoter regions and gene expression levels for cyp19a1a, cyp11a1, cyp11a1, hsd11b2, hsd17b1, amh and dmrt1 in the gonads of females and males exposed to 28°C and 35°C during sex differentiation (18–32 days post‐fertilization). Spearman's rank correlation coefficient (ρ) are shown. The direction of the long axis of the ellipses and the colour indicate the type of correlation: Negative is shown in red and positive in blue. The short axis of the ellipse and the intensity of the colour are proportional to the correlation coefficients. Significant correlations are considered when p < .05 (red frame square). The letter n inside the squares indicates the sample size for each correlation
3.5. DNA methylation differed by sex and temperature
We were interested in knowing which genes had the highest sex‐related differences in DNA methylation and which were the most affected by temperature in each sex. These would be ideal candidates to apply predictive machine learning procedures. At control temperature and between sexes, the promoter regions of cyp19a1a, hsd11b2, amh and hsd17b1 showed the highest hypermethylation (>20%) in favour of males, whereas dmrt1 showed the highest hypermethylation (~20%) in favour of females (Figure S3a). Within females, the genes most affected by temperature were cyp11a1, cyp11c1, amh and foxl2a (Figure S3b). Within males, hypermethylation was highest in hsd11b2, amh, cyp19a1a, hsd17b1 and cyp11a1 whereas hypomethylation was observed in dmrt1 (Figure S3c).
3.6. Prediction of sex and exposure to elevated temperature
Based on the previous results of DNA methylation differences by sex and temperature, we tested whether these sex‐ and temperature‐related differences in specific genes could be used as reliable indicators of: (i) phenotypic sex (e.g., in a sample of unknown sex) and (ii) whether fish had previously been exposed to high temperatures. To achieve this, we used an FDA with K‐fold cross‐validation to evaluate the predictive model using the DNA methylation values, either using individual CpGs or the mean of CpGs per amplicon. The best predictors (selecting single genes or different combinations of them) based on accuracy and κ values for each purpose are shown in Table 1.
TABLE 1.
Epigenetic biomarkers: Predictors of phenotypic sex and previous thermal exposure
| Objective | Predictor | Accuracy | Cohen's kappa | Interpretation of Cohen's kappa |
|---|---|---|---|---|
| To distinguish males from females at 28°C | cyp19a1a | 0.88 | 0.77 | Substantial |
| dmrt1 | 0.88 | 0.76 | Substantial | |
| amh | 0.87 | 0.75 | Substantial | |
| foxl2a | 0.73 | 0.47 | Substantial | |
| amh + dmrt1 | 0.82 | 0.64 | Substantial | |
| cyp19a1a + amh + dmrt1 | 0.85 | 0.72 | Substantial | |
| cyp19a1a + foxl2a + dmrt1 | 0.88 | 0.76 | Substantial | |
| cyp19a1a (using 15 individual CpGs) | 0.88 | 0.77 | Substantial | |
| To distinguish males at 28°C from males at 35°C | amh | 0.71 | 0.44 | Moderate |
| cyp19a1a | 0.69 | 0.42 | Moderate | |
| dmrt1 | 0.68 | 0.38 | Fair | |
| amh + cyp19a1a | 0.63 | 0.32 | Fair | |
| amh + dmrt1 | 0.68 | 0.39 | Fair | |
| cyp19a1a + dmrt1 | 0.68 | 0.41 | Moderate | |
| amh + cyp19a1a + dmrt1 | 0.71 | 0.40 | Moderate | |
| amh (using 15 individual CpGs) | 0.64 | 0.27 | Fair | |
| To distinguish females at 28°C from females at 35°C | foxl2a | 0.63 | 0.28 | Fair |
| cyp19a1a | 0.27 | −0.29 | No agreement | |
| hsd11b2 | 0.29 | 0.52 | No agreement | |
| foxl2a + cyp19a1a | 0.59 | 0.20 | None to slight | |
| foxl2a (using 26 individual CpGs) | 0.78 | 0.55 | Moderate |
Note: Predictors were used either alone or in combination. The kappa values obtained from each predictor follow the classification and interpretation proposed by McHugh (2012). For all predictors, the mean of all CpGs in that predictor were used. Within each objective, the best predictor was also assayed using the values of all the individual CpGs.
To distinguish males from females at control temperature, the classifications gave good accuracies and κ values (between 0.85–0.88 and 0.72–0.77, respectively), either using the mean methylation levels of cyp19a1a, dmrt1 and amh alone or the combination of all three genes. In contrast, the best sex predictor was cyp19a1a when using the values of the 15 targeted CpGs of the cyp19a1a amplicon. Thus, the DNA methylation values of 15 CpGs of the cyp19a1a zebrafish promoter can clearly distinguish whether a DNA sample comes from a male or a female regardless of previous temperature (Figure 4). However, using the mean methylation of these 15 CpGs of the cyp19a1a gene, the classifications gave similar accuracy and κ values. Accordingly, DNA methylation levels of the cyp19a1a gene in zebrafish gonads were enough to predict sex (Table 1).
FIGURE 4.
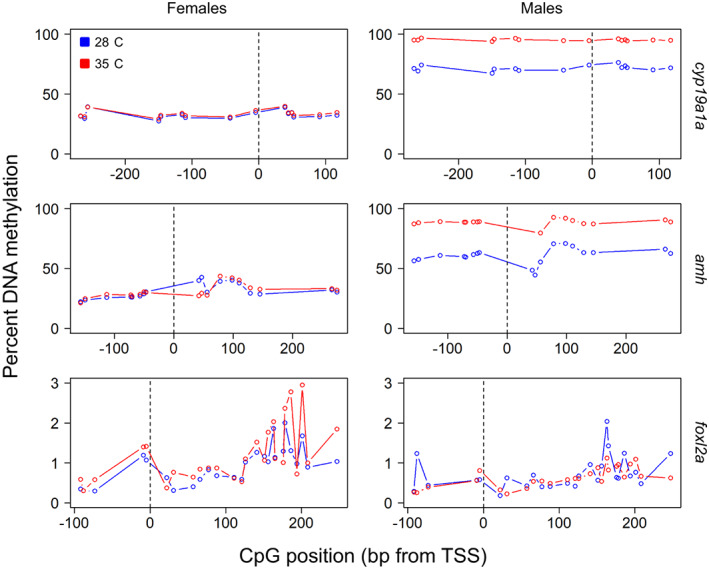
DNA methylation patterns in the adult gonads of females and males of zebrafish subjected to either low (control, blue) or elevated (red) temperature when larvae. Data indicated are for the steroidogenic enzyme cyp19a1a, the growth factor amh and the transcription factor foxl2a. Values on the x‐axis show the distance in base pairs (bp) relative to the transcription start site (TSS) of the gene denoted as 0. Each datapoint corresponds to an individual CpG
Next, we wanted to evaluate whether the prediction of sex was reliable and whether the accuracy of the machine learning procedure obtained from these samples could be repeated with independent samples. First, we extracted the cyp19a1a mean DNA methylation of males and females of an unrelated offspring following the same procedure described earlier and found significantly higher DNA methylation levels in males (Figure S4a). Then, we used the algorithm for sex prediction used previously to classify these 40 independent samples. The accuracy of the classification was 100% and the predicted samples were classified within two clear distinct groups according to their sex (Figure S4b).
To distinguish males previously exposed to high temperature, the best predictors were amh and cyp19a1a (accuracy = 0.71 and 0.69, and κ = 0.44 and 0.42, respectively) (Table 1). These values indicated that the power of classification was categorized as “moderate” according to interpretation of Cohen's kappa values (McHugh, 2012). When methylation values of individual CpGs of amh (18 CpGs) were used, the reliability of the classification (accuracy = 0.64 and κ = 0.27) decreased to “fair.” Thus, to predict past developmental temperature in males, the mean methylation levels of amh in male gonads were sufficient (Figure 4).
Finally, to distinguish previous exposure to elevated temperature in adult females, among all predictors and combinations tested, the best were foxl2a (accuracy = 0.63 and κ = 0.28) and foxl2a + cyp19a1a (accuracy = 0.59 and κ = 0.20), achieving “fair” and “none to slight” reliability, respectively (Table 1). By using the individual CpGs of foxl2a as predictors (26 CpGs) (Figure 4) the classification improved (accuracy = 0.78 and κ = 0.55) and was categorized as “moderate.” Thus, to identify females that had experienced abnormally high temperature during early development, the methylation levels of foxl2a in female gonads can be used (Figure 4).
In summary, the DNA methylation information extracted from promoter regions of cyp19a1a, amh and foxl2a were good epigenetic predictors to discriminate between sex and previous exposure to abnormally high temperature in males and females (Figure 5a,b,c, respectively).
FIGURE 5.
Flexible discriminant analysis (FDA) classifying samples according to sex or early temperature using DNA methylation. Density histogram of prediction values obtained from FDA using (a) cyp19a1a DNA methylation values to distinguish the sex of the fish, (b) amh DNA methylation values to distinguish males at 28°C from males at 35°C, and (c) foxl2a DNA methylation values to distinguish females at 28°C from females at 35°C
4. DISCUSSION
Given the importance of sex ratios as a major population demographic parameter, we studied the influence of the environment on the DNA methylation of a group of genes related to sex differentiation as well as the stress response in vertebrates. In addition, we aimed to identify robust epigenetic biomarkers linked to both phenotypic sex and past thermal insults. To this end, we used a laboratory zebrafish strain (AB), a good model to study the interplay between genetics and the environment (Ribas, Liew, et al., 2017), subjected to two temperatures, 28°C (control) or 35°C, during the critical period of gonadal development (18–32 dpf) and sampled when adults at 90 dpf. Our results on sex ratio response confirmed previous results (Ribas, Liew, et al., 2017; Ribas, Valdivieso, et al., 2017a, 2017b; Valdivieso et al., 2020, 2022) and allowed us to study the integration of environmental information through epigenetics. We were keen to minimize differences in DNA methylation and gene expression due to possible differences in gonad maturation of the 70 samples used, as well as due to effects induced by elevated temperature, although the proportion of germ cells affected is very small in the context of the whole gonad, as shown in a recent study from our laboratory (Valdivieso et al., 2022). Overall, we consider that these differences have little effect on DNA methylation. Apart from the environmental information, the genetic background may influence the DNA methylation variation between individuals. The details of this are far from clear and constitute a relevant topic in environmental epigenetics. In this study, we showed variation in DNA methylation between individuals, indicative of a genetic basis. Interestingly, this variation was more pronounced in males than in females.
Recently, after reviewing the available data on DNA methylation and gene expression in sexual development‐related genes from both gonochoristic and hermaphroditic fish species, we proposed the CERS model, which incorporates the inverse relationship between DNA methylation and gene expression, something that is apparent across species at least for cyp19a1a and dmrt1 (Piferrer et al., 2019). However, such a relationship was in fact the opposite for amh and foxl2a whereas in other genes such as sox9, gsdf, and amhr2, the association between DNA methylation and gene expression was not clear (Piferrer et al., 2019). Here, DNA methylation for a set of ten genes related to sex differentiation, in particular steroidogenic enzymes and growth and transcription factors, have been studied in detail to expand our knowledge with new data.
Steroidogenic enzymes are responsible for the synthesis of sex steroids, which play a key role in vertebrate sex differentiation and gametogenesis (Nakamura et al., 1998). Without exception, all genes coding for steroidogenic enzymes analysed in this study exhibited sex‐dimorphic DNA methylation. In a similar manner, this observation was found previously in two immune‐related genes in adult zebrafish gonads (Caballero‐Huertas et al., 2020). The gene cyp11a1 encodes an enzyme that participates in the conversion of cholesterol to progesterone, a precursor for the synthesis of testosterone and 17β‐oestradiol (Simpson, 1979). The DNA methylation levels for cyp11a1 confirmed that this enzyme shows sexual dimorphic patterns with higher DNA methylation levels and concomitant lower gene expression levels, particularly in males. Thus, cyp11a1 follows the CERS model predictions. Cyp19a1a is a steroidogenic enzyme that catalyses the conversion of androstenedione and testosterone into oestrone and 17β‐oestradiol, respectively. Then, the hsd17b1 steroidogenic enzyme converts oestrone into 17β‐oestradiol (Haller et al., 2010; Mindnich et al., 2004). 17β‐Oestradiol is key for sex differentiation as it is required for ovarian development in all nonmammalian vertebrates (Guiguen et al., 2010; Hinfray et al., 2018; Lau et al., 2016; Piferrer et al., 1994). In the present study, cyp19a1a was hypomethylated in the ovaries while its expression was higher in ovaries than in testis. The DNA methylation patterns found in the two sexes were in agreement with those reported in other fish species, such as the European seabass (Anastasiadi, Vandeputte, et al., 2018; Navarro‐Martín et al., 2011), the half‐smooth tongue sole (Shao et al., 2014), the black porgy (Acanthopagrus schlegelii) (Wu & Chang, 2018) and barramundi (Lates calcarifer) (Domingos et al., 2018).
We also studied the “pro‐male” steroidogenic enzymes, cyp11c1, hsd11b2 and hsd17b3 (Guiguen et al., 2010), which use androstenedione and testosterone as substrates to convert 11β‐hydroxyandrostenedione and 11β‐hydroxytestosterone to 11‐ketotestosterone, a potent androgen in teleost fish (Baroiller et al., 1999; Mindnich et al., 2004). Analysis of DNA methylation of cyp11c1, hsd11b2 and hsd17b3 showed hypermethylation in males compared with females. Expression levels of cyp11c1 and hsd11b2 were higher in males as confirmed in other fish species in which higher expression of these genes is required for testis development (Baker, 2004; Kusakabe et al., 2003; Wang & Orban, 2007). The correlations between DNA methylation and gene expression of both cyp11c1 and hsd11b2 were positive in males but only at the control temperature.
The expression of steroidogenic enzymes is regulated by the action of transcription factors (Manna et al., 2003), and some of them are also important for vertebrate sex differentiation (Yang et al., 2017). All the transcription and growth factors analysed in this study (amh, dmrt1, dmrt3a and foxl2a) showed significant sex‐specific differences in DNA methylation. The two genes of the dmrt family, dmrt1 and dmrt3a, located on chromosome 5 of zebrafish (Guo et al., 2005), were hypomethylated in males when compared to females. Our results with dmrt1 are in accordance with previous observations in the European seabass, half‐smooth tongue sole, the Japanese flounder (Paralichthys olivaceus) and Culter alburnus (Anastasiadi, Vandeputte, et al., 2018; Jia et al., 2019; Shao et al., 2014; Wen et al., 2014) suggesting a conserved DNA methylation pattern across species. The dmrt1 gene was significantly more highly expressed in males, and thus an inverse relationship between methylation and gene expression levels was present, conforming to CERS predictions (Piferrer et al., 2019). Further, in this study amh showed higher methylation levels in accordance with other studies in zebrafish (Laing et al., 2018), barramundi (Domingos et al., 2018) and half‐smooth tongue sole (Shao et al., 2014), although in the European seabass the methylation levels were similar between sexes (Anastasiadi & Piferrer, 2020). Gene expression of amh is typically required for testis development in fish (Pfennig et al., 2015) and so higher amh expression levels indicated a positive correlation with DNA methylation levels, thus not following CERS predictions (Piferrer et al., 2019).
Foxl2a is a transcriptional regulator of cyp19a1a and is important for ovarian differentiation in nonmammalian vertebrates (Wang, Bartfai, et al., 2007; Yang et al., 2017; Zhang et al., 2017). The methylation levels of foxl2a were much lower than those of cyp19a1a and were significantly hypermethylated in females when compared with males. These results are in accordance with the methylation levels of foxl2a found in the ovaries of the European seabass (Anastasiadi, Vandeputte, et al., 2018), barramundi (Domingos et al., 2018) and Japanese flounder (Si et al., 2016). In the Japanese flounder, a strong inverse relationship between methylation levels in foxl2a and cyp19a1a with its respective gene expression at different ovarian development stages was observed (Si et al., 2016). In the ovaries of different fish species, there is a positive correlation between the transcriptional levels of foxl2a and cyp19a1a (Baron et al., 2004, 2005). Here, we did not study the expression levels of foxl2a. However, our methylation data for foxl2a and cyp19a1a with gene expression data from cyp19a1a in females, based on previous results in fish (Wang, Kobayashi, et al., 2007), suggest a similar pattern for both genes, involved in female gonadal development (Fan et al., 2017, 2019).
Notably, when we checked the correlation between DNA methylation and gene expression patterns at the control temperature, we detected a strong negative correlation in cyp11a1 and dmrt1 in males and a positive correlation in cyp11c1, hsd11b2 and amh genes only in males. However, the lack of significant correlation in the other genes in females could be due to different reasons. First, the methodology used, MBS, a locus‐specific resolution and high‐coverage technique, has a physical limitation in which the maximum length of DNA to be interrogated is around 500 bp. This allowed us to study only a limited number of CpG sites (ranging from four to 34 in the genes studied), forcibly leaving out other CpG sites that may be relevant in gene regulation. Classically, the most well‐studied targeted region for DNA methylation is the promoter region close to the transcription start site (TSS). However, recently, it has become evident that other genomic regions, such as the first exon and the first intron, have the same or even better correlation with DNA methylation than the promoter (Anastasiadi, Esteve‐Codina, & Piferrer, 2018; Brenet et al., 2011). Further, evidence also exists suggesting that hypermethylation of the promoter may be associated with high transcriptional activity under certain circumstances (Smith et al., 2020). This may explain the lack of some of the expected correlations between DNA methylation and gene expression levels.
We also studied the effects of elevated temperature on the methylation patterns of genes involved in sex differentiation. This was done almost 2 months after the temperature treatment ceased in order to evaluate persistent effects in the adult gonads. In males, temperature was associated with an increase in the methylation levels of cyp19a1a, cyp11a1, cyp11c1, hsd17b3 and amh. In some cases, this increase of methylation in males was also associated with an upregulation of gene expression, such as in amh and in cyp11c1 or, in contrast, with a downregulation of gene expression, as in cyp19a1a. In males, elevated temperature strongly decreased dmrt1 methylation levels, while an upregulation of its expression was found, confirming previous results analysing temperature and gene expression in zebrafish (Ribas, Liew, et al., 2017). Regarding cyp19a1a, no effect was observed in the ovaries of high‐temperature vs. control‐exposed fish as had been observed in the half‐smooth tongue sole (Liu et al., 2019). This may be due to the fact that in zebrafish at 14 dpf, a dimorphic proliferation of primordial germ cells is detected in which an elevated threshold of these cells is required for ovarian development (Tzung et al., 2015). Consequently, this time lapse might have hindered stable epigenetic marks for female differentiation at the time when we subjected fish to high temperatures in our experiment (18–32 dpf). Thus, the effects of temperature on DNA methylation in females could have been stronger if applied earlier. This was shown in the European seabass, in which temperature before sex differentiation was able to alter DNA methylation levels of the genes in ovaries (Anastasiadi, Vandeputte, et al., 2018; Navarro‐Martín et al., 2011). In genotypic female Japanese flounder, elevated temperature during gonad differentiation hypermethylated the promoter of cyp19a1a and its transcriptional activator foxl2a with a concomitant downregulation of their expression levels, resulting in male development (Fan et al., 2017, 2019). These results open new avenues for studying how DNA methylation marks are established before, during and after gonadal development in fish when exposed to environmental stressors.
In recent years, the link between DNA methylation, gene expression and certain phenotypes has prompted the identification of the DNA methylation‐based epigenetic markers (Jin & Liu, 2018). Some of them associate with age, lifestyle or environmental cues (Marsit, 2015), and have been used to predict age in humans and other vertebrates including fish (Anastasiadi & Piferrer, 2020; De Paoli‐Iseppi et al., 2019; Horvath, 2013; Polanowski et al., 2014; Thompson et al., 2017), sex in fish (Anastasiadi, Vandeputte, et al., 2018) and early life stress in chickens (Pértille et al., 2017). In medicine, the methylation status of some genes has been associated with diseases (Jones & Baylin, 2007). Therefore, the methylation levels of specific CpGs have been used to develop tests (Moran et al., 2016) for the diagnosis and prognosis of diseases (Costa‐Pinheiro et al., 2015). For instance, DNA methylation of genes involved in lung and colorectal cancer has been used to develop a test for their early detection (Li et al., 2019; Lind et al., 2011; Model et al., 2007). Nevertheless, there is still a wide range of sensitivity and specificity in such types of tests (20%–90% and 65%–100%, respectively) (Dong & Ren, 2018; Li et al., 2019), indicating that more efforts in developing epigenetic tools for diagnosis are needed. One way to circumvent this has been to combine the methylation levels of CpGs from different genes into a single panel, which has increased the sensitivity and specificity of diagnosis (Lind et al., 2011). In fish, techniques that allow to measure methylation levels for specific CpG contexts facilitate the identification of EEMs for a particular phenotype of interest (Piferrer, 2019). Our results showed that one can reliably infer the sex of an animal, in this case the zebrafish model, examining only a DNA sample. Since sex is a strong covariate influencing many biological variables, being able to infer sex from a DNA sample would be very useful to assign phenotypic sex in DNA samples under a variety of different scenarios: for example, when the sex of the donor individual was not recorded, when the information was lost or, in the case of sex reversals, the phenotypic sex does not match with genotypic sex.
Environmental perturbations, especially occurring during early development, can induce lifelong phenotypic changes mediated by the epigenetic regulation of gene expression. These changes underlie phenotypic plasticity and are thought to contribute to rapid adaptation to new environments (Beal et al., 2018; Jaenisch & Bird, 2003). Thus, epigenetic alterations, including changes to the methylome, constitute a sort of “epigenetic memory” that potentially could allow detection of past exposures to environmental stressors such as pollutants (Wang et al., 2009) or increase of temperature (Mirbahai & Chipman, 2014), the latter being important to understand climate change responses. Examples of epigenetic marks associated with thermal stress have been discovered in marine species. In oyster (Crasssotreas gigas), the relationship between increasing temperature and histone methyl‐marks was associated with delayed development and growth (Fellous et al., 2015). The Antarctic polychaete Spiophanes tcherniani showed an epigenetic acclimation mechanism in which individuals stressed by a slight increase of temperature recovered normal metabolic rates after a month by an increase of DNA methylation (Marsh & Pasqualone, 2014). Another example is found in four wild populations of the ascidian Ciona robusta taken from different habitats around the world. The methylation profiles of CpGs located in the heat‐shock protein 90 (hsp90) and Na+–K+–2Cl− cotransporter (nkcc) genes, with roles in responses to temperature and salinity, allowed the identification of the population of origin, indicating that different geographical locations (in addition to genetics) resulted in characteristic epigenomes (Pu & Zhan, 2017).
Taking into account the possible existence of such an “epigenetic memory” of past environmental stressors, here we used the DNA methylation levels of carefully selected CpGs, not only to infer sex but also to determine what thermal conditions fish had experienced more than 2 months before, when they were larvae. Based only on the methylation of 15 CpGs of the cyp19a1a gene, we were able to classify the sex of fish with 88% accuracy, a remarkable value considering the polygenic nature of sex determination in domesticated zebrafish (Liew et al., 2012). In addition, in the present case, we were able to validate the results with a set of independent samples with 100% success, reinforcing our sex inference model.
In addition to sex, in the present study epigenetic marks have been used to determine past environmental conditions in fish. Strikingly, and for the first time, using the DNA methylation levels of amh in testes and of foxl2a in ovaries, we were able to accurately predict (71% and 78%, respectively) whether fish had been exposed to elevated temperature during early development. Thus, here we present a reliable epigenetic method to infer both sex and past thermal events. Recently, epigenetic marks have been shown to record the long‐term consequences of heatwaves in fish (Anastasiadi et al., 2021), and epigenetic changes due to elevated temperature persist in the unexposed offspring of exposed parents (Valdivieso et al., 2020). Because the zebrafish is a well‐established model for aquaculture (Ribas & Piferrer, 2014), toxicology (Dai et al., 2014; Tanguay, 2018) and environmental issues (Brown et al., 2015), further research should be facilitated by our framework of epigenetic biomarker development presented here that should hold to detect exposure to different environmental disturbances.
We want to point out that a key aspect is whether one could identify sex or past temperature exposure from noninvasive sampling using DNA methylation patterns in blood or fin clip samples. This is not straightforward since methylation levels are tissue specific (Husby, 2020). Determining blood DNA methylation levels in our case was not feasible because the zebrafish is a small fish and although blood can be collected (Zang et al., 2015), the amount would probably not be sufficient to obtain enough DNA for methylation by the targeted approach we used (the MBS technique) and also for gene expression analysis. Although, theoretically, one alternative would be to perform several extractions from the same individual with a few recovering periods, this, in practice, would entail handling stress, which could affect the results, and increase the risk of infections and possibly loss of several individuals, thus reducing the number of replicates. Therefore, our study constitutes a proof of concept that DNA methylation marks not only are sex‐and temperature‐dependent but also that they can be used to infer sex from a DNA sample and past environmental temperature. This paves the way for similar studies with larger species. There, determining whether blood or fin clips can also be used would make more sense. Note also that in routine campaigns for the monitoring of fish populations, including nonmodel species for ecologically oriented research or fisheries, researchers typically kill the fish to assign phenotypic sex. Therefore, the fish is already dead, gonadal sampling is routinely undertaken and gonadal tissue represents the best tissue for analysis. From our results we could infer past temperature exposure in both sexes. The applicability of this study can be envisaged to extend not only for natural abiotic factors but also to pollutants or contaminants that can be reflected in gonads due to disrupted endocrine actions.
5. CONCLUSIONS
This study provides novel information on the DNA methylation dynamics of a suite of 10 genes, including steroidogenic enzymes and transcription and growth factors. In three cases, they represent the first measurement of the DNA methylation in fish gonads (i.e., cyp11c1, hsd17b1 and dmrt3a). Sexually dimorphic patterns were observed in all studied genes. DNA methylation and gene expression relationships were sometimes inverse, allowing us to identify four genes that follow CERS predictions, namely cyp19a1a and dmrt1, confirming previous results, and cyp11a1 and hsd17b1 described here for the first time. Interestingly, elevated temperature affected DNA methylation levels of sex‐related genes (e.g., dmrt1 and dmrt3a) and the steroidogenic pathway (e.g., hsd11b2 and hsd17b1) more in males than in females. We show that analysis of the methylation levels of cyp19a1a alone in a DNA sample is capable of correctly identifying the sex with close to 90% accuracy, reinforcing the idea that sex identification in other species with a similar approach is feasible. Further, we show that analyses of the DNA methylation levels of amh for males and foxl2a for females are sufficient to deduce whether fish had been exposed to suboptimal environmental conditions (temperature in this case) in the absence of conspicuous morphological alterations of the reproductive tissues. Thus, epigenetic biomarkers can be used not only for the identification of key phenotypic traits but also to infer past environmental conditions. These set of CpGs represent EEMs that correctly recapitulate past thermal history. This study paves the way for similar findings in other species that should be particularly useful to assess potential effects of environmental disturbances in the context of a climate change scenario.
AUTHOR CONTRIBUTIONS
AV performed the temperature experiment, processed the samples, performed the data analysis, interpreted the results and wrote the article. DA performed data analysis and wrote the article. LR conceived the study and wrote the article. FP conceived the study, provided reagents, interpreted results and wrote the article. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
OPEN RESEARCH BADGES
This article has earned an Open Data Badge for making publicly available the digitally‐shareable data necessary to reproduce the reported results. Raw sequence reads are deposited in NCBI (BioProject: PRJNA549930, SRA: SRP202005). Data sets are available at GEO: GSE133088.
BENEFIT‐SHARING STATEMENT
Benefits from this research accrue from the sharing of our data and results on public databases.
Supporting information
Appendix S1
ACKNOWLEDGEMENTS
We thank Sílvia Joly for technical assistance and Gemma Fuster for fish maintenance. This study was supported by the Spanish Ministry of Science grants AGL2016‐787107‐R “Epimark” and PID2019‐108888RB‐I00 “Epipure” to FP. Funding was provided from the Spanish government through the “Severo Ochoa Centre of Excellence” accreditation (CEX2019‐000928‐S). AV was supported by Spanish government scholarships (BES‐2014‐069051), DA by a “Epimark” contract and LR by an AGL2015‐73864‐JIN “Ambisex” contract.
Valdivieso, A. , Anastasiadi, D. , Ribas, L. , & Piferrer, F. (2023). Development of epigenetic biomarkers for the identification of sex and thermal stress in fish using DNA methylation analysis and machine learning procedures. Molecular Ecology Resources, 23, 453–470. 10.1111/1755-0998.13725
Handling Editor: Paul A. Hohenlohe
DATA AVAILABILITY STATEMENT
Raw sequence reads are deposited in NCBI (BioProject: PRJNA549930, SRA: SRP202005). Data sets are available at GEO: GSE133088.
REFERENCES
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Anastasiadi, D. , Esteve‐Codina, A. , & Piferrer, F. (2018). Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics & Chromatin, 11(1), 37. 10.1186/s13072-018-0205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiadi, D. , & Piferrer, F. (2020). A clockwork fish: Age prediction using DNA methylation‐based biomarkers in the European sea bass. Molecular Ecology Resources, 20(2), 387–397. 10.1111/1755-0998.13111 [DOI] [PubMed] [Google Scholar]
- Anastasiadi, D. , Shao, C. , Chen, S. , & Piferrer, F. (2021). Footprints of global change in marine life: Inferring past environment based on DNA methylation and gene expression marks. Molecular Ecology, 30(3), 747–760. 10.1111/mec.15764 [DOI] [PubMed] [Google Scholar]
- Anastasiadi, D. , Vandeputte, M. , Sánchez‐Baizán, N. , Allal, F. , & Piferrer, F. (2018). Dynamic epimarks in sex‐related genes predict gonad phenotype in the European sea bass, a fish with mixed genetic and environmental sex determination. Epigenetics, 13(9), 988–1011. 10.1080/15592294.2018.1529504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute. [Google Scholar]
- Baker, M. E. (2004). Evolutionary analysis of 11β‐hydroxysteroid dehydrogenase‐type 1, −type 2, −type 3 and 17β‐hydroxysteroid dehydrogenase‐type 2 in fish. FEBS Letters, 574(1–3), 167–170. 10.1016/j.febslet.2004.08.023 [DOI] [PubMed] [Google Scholar]
- Baroiller, J.‐F. , Guiguen, Y. , & Fostier, A. (1999). Endocrine and environmental aspects of sex differentiation in fish. Cellular and Molecular Life Sciences, 55(7), 910. 10.1007/s000180050344 [DOI] [Google Scholar]
- Baron, D. , Batista, F. , Chaffaux, S. , Cocquet, J. , Cotinot, C. , Cribiu, E. , De Baeree, E. , Guiguen, Y. , Jaubert, F. , Pailhoux, E. , Pannetier, M. , Vaiman, D. , Vigier, B. , Veitia, R. , & Fellous, M. (2005). Foxl2 gene and the development of the ovary: A story about goat, mouse, fish and woman. Reproduction Nutrition Development, 45(3), 377–382. 10.1051/rnd:2005028 [DOI] [PubMed] [Google Scholar]
- Baron, D. , Cocquet, J. , Xia, X. , Fellous, M. , Guiguen, Y. , & Veitia, R. A. (2004). An evolutionary and functional analysis of FoxL2 in rainbow trout gonad differentiation. Journal of Molecular Endocrinology, 33(3), 705–715. 10.1677/jme.1.01566 [DOI] [PubMed] [Google Scholar]
- Beal, A. , Rodriguez‐Casariego, J. , Rivera‐Casas, C. , Suarez‐Ulloa, V. , & Eirin‐Lopez, J. M. (2018). Environmental epigenomics and its applications in marine organisms. In Oleksiak M. F. & Rajora O. P. (Eds.), Population genomics: Marine organisms (pp. 325–359). Springer International Publishing. 10.1007/13836_2018_28 [DOI] [Google Scholar]
- Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes & Development, 16(1), 6–21. 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- Brenet, F. , Moh, M. , Funk, P. , Feierstein, E. , Viale, A. J. , Socci, N. D. , & Scandura, J. M. (2011). DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One, 6(1), e14524. 10.1371/journal.pone.0014524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, A. R. , Owen, S. F. , Peters, J. , Zhang, Y. , Soffker, M. , Paull, G. C. , Hosken, D. J. , Wahab, M. A. , & Tyler, C. R. (2015). Climate change and pollution speed declines in zebrafish populations. Proceedings of the National Academy of Sciences of the United States of America, 112(11), E1237–E1246. 10.1073/pnas.1416269112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, B. , Hornung, U. , Shan, Z. , Nanda, I. , Kondo, M. , Zend‐Ajusch, E. , Haaf, T. , Ropers, H.‐H. , Shima, A. , Schmid, M. , Kalscheuer, V. M. , & Schartl, M. (2001). Genomic organization and expression of the doublesex‐related gene cluster in vertebrates and detection of putative regulatory regions for DMRT1. Genomics, 77(1–2), 8–17. 10.1006/geno.2001.6615 [DOI] [PubMed] [Google Scholar]
- Caballero‐Huertas, M. , Moraleda‐Prados, J. , Joly, S. , & Ribas, L. (2020). Immune genes, IL1β and casp9, show sexual dimorphic methylation patterns in zebrafish gonads. Fish & Shellfish Immunology, 97, 648–655. 10.1016/j.fsi.2019.12.013 [DOI] [PubMed] [Google Scholar]
- Capel, B. (2017). Vertebrate sex determination: Evolutionary plasticity of a fundamental switch. Nature Reviews Genetics, 18(11), 675–689. 10.1038/nrg.2017.60 [DOI] [PubMed] [Google Scholar]
- Caulier, M. , Brion, F. , Chadili, E. , Turies, C. , Piccini, B. , Porcher, J.‐M. , Guiguen, Y. , & Hinfray, N. (2015). Localization of steroidogenic enzymes and foxl2a in the gonads of mature zebrafish (Danio rerio). Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 188, 96–106. 10.1016/j.cbpa.2015.06.016 [DOI] [PubMed] [Google Scholar]
- Costa‐Pinheiro, P. , Montezuma, D. , Henrique, R. , & Jerónimo, C. (2015). Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics, 7(6), 1003–1015. 10.2217/epi.15.56 [DOI] [PubMed] [Google Scholar]
- Dai, Y. J. , Jia, Y. F. , Chen, N. , Bian, W. P. , Li, Q. K. , Ma, Y. B. , Chen, Y. L. , & Pei, D. S. (2014). Zebrafish as a model system to study toxicology: Zebrafish toxicology monitoring. Environmental Toxicology and Chemistry, 33(1), 11–17. 10.1002/etc.2406 [DOI] [PubMed] [Google Scholar]
- De Paoli‐Iseppi, R. , Deagle, B. E. , Polanowski, A. M. , McMahon, C. R. , Dickinson, J. L. , Hindell, M. A. , & Jarman, S. N. (2019). Age estimation in a long‐lived seabird (Ardenna tenuirostris) using DNA methylation‐based biomarkers. Molecular Ecology Resources, 19(2), 411–425. 10.1111/1755-0998.12981 [DOI] [PubMed] [Google Scholar]
- Deaton, A. M. , & Bird, A. (2011). CpG islands and the regulation of transcription. Genes & Development, 25(10), 1010–1022. 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos, T. , Tetzner, R. , Model, F. , Weiss, G. , Schuster, M. , Distler, J. , Steiger, K. V. , Grützmann, R. , Pilarsky, C. , Habermann, J. K. , Fleshner, P. R. , Oubre, B. M. , Day, R. , Sledziewski, A. Z. , & Lofton‐Day, C. (2009). Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clinical Chemistry, 55(7), 1337–1346. 10.1373/clinchem.2008.115808 [DOI] [PubMed] [Google Scholar]
- Domingos, J. A. , Budd, A. M. , Banh, Q. Q. , Goldsbury, J. A. , Zenger, K. R. , & Jerry, D. R. (2018). Sex‐specific dmrt1 and cyp19a1 methylation and alternative splicing in gonads of the protandrous hermaphrodite barramundi. PLoS One, 13(9), e0204182. 10.1371/journal.pone.0204182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, L. , & Ren, H. (2018). Blood‐based DNA methylation biomarkers for early detection of colorectal cancer. Journal of Proteomics & Bioinformatics, 11(6), 120–126. 10.4172/jpb.1000477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z. , Zou, Y. , Jiao, S. , Tan, X. , Wu, Z. , Liang, D. , Zhang, P. , & You, F. (2017). Significant association of cyp19a promoter methylation with environmental factors and gonadal differentiation in olive flounder Paralichthys olivaceus . Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 208, 70–79. 10.1016/j.cbpa.2017.02.017 [DOI] [PubMed] [Google Scholar]
- Fan, Z. , Zou, Y. , Liang, D. , Tan, X. , Jiao, S. , Wu, Z. , Li, J. , Zhang, P. , & You, F. (2019). Roles of forkhead box protein L2 (foxl2) during gonad differentiation and maintenance in a fish, the olive flounder (Paralichthys olivaceus). Reproduction, Fertility and Development, 31(11), 1742. 10.1071/RD18233 [DOI] [PubMed] [Google Scholar]
- Fellous, A. , Favrel, P. , & Riviere, G. (2015). Temperature influences histone methylation and mRNA expression of the Jmj‐C histone‐demethylase orthologues during the early development of the oyster Crassostrea gigas . Marine Genomics, 19, 23–30. 10.1016/j.margen.2014.09.002 [DOI] [PubMed] [Google Scholar]
- Feng, S. , Jacobsen, S. E. , & Reik, W. (2010). Epigenetic reprogramming in plant and animal development. Science, 330(6004), 622–627. 10.1126/science.1190614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandino, J. I. , Hattori, R. S. , Kishii, A. , Strüssmann, C. A. , & Somoza, G. M. (2012). The cortisol and androgen pathways cross talk in high temperature‐induced masculinization: The 11β‐hydroxysteroid dehydrogenase as a key enzyme. Endocrinology, 153(12), 6003–6011. 10.1210/en.2012-1517 [DOI] [PubMed] [Google Scholar]
- Fernandino, J. I. , Hattori, R. S. , Moreno Acosta, O. D. , Strüssmann, C. A. , & Somoza, G. M. (2013). Environmental stress‐induced testis differentiation: Androgen as a by‐product of cortisol inactivation. General and Comparative Endocrinology, 192, 36–44. 10.1016/j.ygcen.2013.05.024 [DOI] [PubMed] [Google Scholar]
- Flores, K. B. , Wolschin, F. , & Amdam, G. V. (2013). The role of methylation of DNA in environmental adaptation. Integrative and Comparative Biology, 53(2), 359–372. 10.1093/icb/ict019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguen, Y. , Fostier, A. , Piferrer, F. , & Chang, C.‐F. (2010). Ovarian aromatase and estrogens: A pivotal role for gonadal sex differentiation and sex change in fish. General and Comparative Endocrinology, 165(3), 352–366. 10.1016/j.ygcen.2009.03.002 [DOI] [PubMed] [Google Scholar]
- Gunes, S. O. , Mahmutoglu, A. M. , & Agarwal, A. (2016). Genetic and epigenetic effects in sex determination. Birth Defects Research Part C: Embryo Today: Reviews, 108(4), 321–336. 10.1002/bdrc.21146 [DOI] [PubMed] [Google Scholar]
- Guo, Y. , Cheng, H. , Huang, X. , Gao, S. , Yu, H. , & Zhou, R. (2005). Gene structure, multiple alternative splicing, and expression in gonads of zebrafish dmrt1. Biochemical and Biophysical Research Communications, 330(3), 950–957. 10.1016/j.bbrc.2005.03.066 [DOI] [PubMed] [Google Scholar]
- Haller, F. , Moman, E. , Hartmann, R. W. , Adamski, J. , & Mindnich, R. (2010). Molecular framework of steroid/retinoid discrimination in 17β‐hydroxysteroid dehydrogenase type 1 and photoreceptor‐associated retinol dehydrogenase. Journal of Molecular Biology, 399(2), 255–267. 10.1016/j.jmb.2010.04.002 [DOI] [PubMed] [Google Scholar]
- Hinfray, N. , Sohm, F. , Caulier, M. , Chadili, E. , Piccini, B. , Torchy, C. , Porcher, J.‐M. , Guiguen, Y. , & Brion, F. (2018). Dynamic and differential expression of the gonadal aromatase during the process of sexual differentiation in a novel transgenic cyp19a1a‐eGFP zebrafish line. General and Comparative Endocrinology, 261, 179–189. 10.1016/j.ygcen.2017.06.014 [DOI] [PubMed] [Google Scholar]
- Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biology, 14(10), R115. 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosomichi, K. , Mitsunaga, S. , Nagasaki, H. , & Inoue, I. (2014). A bead‐based normalization for uniform sequencing depth (BeNUS) protocol for multi‐samples sequencing exemplified by HLA‐B. BMC Genomics, 15(1), 645. 10.1186/1471-2164-15-645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini, S. , Brenig, B. , Tetens, J. , & Sharifi, A. R. (2019). Phenotypic plasticity induced using high ambient temperature during embryogenesis in domesticated zebrafish, Danio rerio . Reproduction in Domestic Animals, 54(3), 435–444. 10.1111/rda.13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini, S. , Trakooljul, N. , Hirschfeld, M. , Wimmers, K. , Simianer, H. , Tetens, J. , Sharifi, A. R. , & Brenig, B. (2022). Epigenetic regulation of phenotypic sexual plasticity inducing skewed sex ratio in zebrafish. Frontiers in Cell and Developmental Biology, 10, 880779. 10.3389/fcell.2022.880779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, H. J. , Lin, J. C. , & Chung, B. C. (2009). Zebrafish cyp11a1 and hsd3b genes: Structure, expression and steroidogenic development during embryogenesis. Molecular and Cellular Endocrinology, 312(1–2), 31–34. 10.1016/j.mce.2009.07.030 [DOI] [PubMed] [Google Scholar]
- Husby, A. (2020). On the use of blood samples for measuring DNA methylation in ecological epigenetic studies. Integrative and Comparative Biology, 60(6), 1558–1566. 10.1093/icb/icaa123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch, R. , & Bird, A. (2003). Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nature Genetics, 33(S3), 245–254. 10.1038/ng1089 [DOI] [PubMed] [Google Scholar]
- Jia, Y. , Zheng, J. , Chi, M. , Liu, S. , Jiang, W. , Cheng, S. , Gu, Z. , & Chen, L. (2019). Molecular identification of dmrt1 and its promoter CpG methylation in correlation with gene expression during gonad development in Culter alburnus . Fish Physiology and Biochemistry, 45(1), 245–252. 10.1007/s10695-018-0558-1 [DOI] [PubMed] [Google Scholar]
- Jin, Z. , & Liu, Y. (2018). DNA methylation in human diseases. Genes & Diseases, 5(1), 1–8. 10.1016/j.gendis.2018.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. A. , & Baylin, S. B. (2007). The epigenomics of cancer. Cell, 128(4), 683–692. 10.1016/j.cell.2007.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik, D. , Baertsch, R. , Diekhans, M. , Furey, T. S. , Hinrichs, A. , Lu, Y. T. , Roskin, K. M. , Schwartz, M. , Sugnet, C. W. , Thomas, D. J. , Weber, R. J. , Haussler, D. , & Kent, W. J. (2003). The UCSC genome browser database. Nucleic Acids Research, 31(1), 51–54. 10.1093/nar/gkg129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, F. (2015). Trim galore. A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files, 516(517).
- Krueger, F. , & Andrews, S. R. (2011). Bismark: A flexible aligner and methylation caller for bisulfite‐seq applications. Bioinformatics, 27(11), 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, M. (2008). Building predictive models in R using the caret package. Journal of Statistical Software, 28(5), 1–26.27774042 [Google Scholar]
- Kuhn, M. (2020). caret: Classification and regression training ([Version 6.0–86]). https://CRAN.R‐project.org/package=caret [Google Scholar]
- Kuroki, S. , & Tachibana, M. (2018). Epigenetic regulation of mammalian sex determination. Molecular and Cellular Endocrinology, 468, 31–38. 10.1016/j.mce.2017.12.006 [DOI] [PubMed] [Google Scholar]
- Kusakabe, M. , Nakamura, I. , & Young, G. (2003). 11β‐hydroxysteroid dehydrogenase complementary deoxyribonucleic acid in rainbow trout: Cloning, sites of expression, and seasonal changes in gonads. Endocrinology, 144(6), 2534–2545. 10.1210/en.2002-220446 [DOI] [PubMed] [Google Scholar]
- Laing, L. V. , Viana, J. , Dempster, E. L. , Uren Webster, T. M. , van Aerle, R. , Mill, J. , & Santos, E. M. (2018). Sex‐specific transcription and DNA methylation profiles of reproductive and epigenetic associated genes in the gonads and livers of breeding zebrafish. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 222, 16–25. 10.1016/j.cbpa.2018.04.004 [DOI] [PubMed] [Google Scholar]
- Lau, E. S.‐W. , Zhang, Z. , Qin, M. , & Ge, W. (2016). Knockout of zebrafish ovarian aromatase gene (cyp19a1a) by TALEN and CRISPR/Cas9 leads to all‐male offspring due to failed ovarian differentiation. Scientific Reports, 6(1), 37357. 10.1038/srep37357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Fu, K. , Zhou, W. , & Snyder, M. (2019). Applying circulating tumor DNA methylation in the diagnosis of lung cancer. Precision Clinical Medicine, 2(1), 45–56. 10.1093/pcmedi/pbz003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. C. , & Dahiya, R. (2002). MethPrimer: Designing primers for methylation PCRs. Bioinformatics, 18(11), 1427–1431. 10.1093/bioinformatics/18.11.1427 [DOI] [PubMed] [Google Scholar]
- Liew, W. C. , Bartfai, R. , Lim, Z. , Sreenivasan, R. , Siegfried, K. R. , & Orban, L. (2012). Polygenic sex determination system in zebrafish. PLoS One, 7(4), e34397. 10.1371/journal.pone.0034397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind, G. E. , Danielsen, S. A. , Ahlquist, T. , Merok, M. A. , Andresen, K. , Skotheim, R. I. , Hektoen, M. , Rognum, T. O. , Meling, G. I. , Hoff, G. , Bretthauer, M. , Thiis‐Evensen, E. , Nesbakken, A. , & Lothe, R. A. (2011). Identification of an epigenetic biomarker panel with high sensitivity and specificity for colorectal cancer and adenomas. Molecular Cancer, 10(1), 85. 10.1186/1476-4598-10-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Liu, X. , Jin, C. , Du, X. , He, Y. , & Zhang, Q. (2019). Transcriptome profiling insights the feature of sex reversal induced by high temperature in tongue sole Cynoglossus semilaevis . Frontiers in Genetics, 10, 522. 10.3389/fgene.2019.00522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair, P. , & Wilcox, R. (2020). Robust statistical methods in R using the WRS2 package. Behavior Research Methods, 52(2), 464–488. 10.3758/s13428-019-01246-w [DOI] [PubMed] [Google Scholar]
- Manna, P. R. , Wang, X. J. , & Stocco, D. M. (2003). Involvement of multiple transcription factors in the regulation of steroidogenic acute regulatory protein gene expression. Steroids, 68(14), 1125–1134. [DOI] [PubMed] [Google Scholar]
- Marsh, A. G. , & Pasqualone, A. A. (2014). DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai . Frontiers in Physiology, 5, 173. 10.3389/fphys.2014.00173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit, C. J. (2015). Influence of environmental exposure on human epigenetic regulation. Journal of Experimental Biology, 218(1), 71–79. 10.1242/jeb.106971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh, M. L. (2012). Interrater reliability: The kappa statistic. Biochemia Medica, 22(3), 276–282. 10.11613/BM.2012.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikeska, T. , & Craig, J. (2014). DNA methylation biomarkers: Cancer and beyond. Genes, 5(3), 821–864. 10.3390/genes5030821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindnich, R. , Deluca, D. , & Adamski, J. (2004). Identification and characterization of 17β‐hydroxysteroid dehydrogenases in the zebrafish, Danio rerio . Molecular and Cellular Endocrinology, 215(1–2), 19–30. 10.1016/j.mce.2003.11.010 [DOI] [PubMed] [Google Scholar]
- Mirbahai, L. , & Chipman, J. K. (2014). Epigenetic memory of environmental organisms: A reflection of lifetime stressor exposures. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 764–765, 10–17. 10.1016/j.mrgentox.2013.10.003 [DOI] [PubMed] [Google Scholar]
- Model, F. , Osborn, N. , Ahlquist, D. , Gruetzmann, R. , Molnar, B. , Sipos, F. , Galamb, O. , Pilarsky, C. , Saeger, H.‐D. , Tulassay, Z. , Hale, K. , Mooney, S. , Lograsso, J. , Adorjan, P. , Lesche, R. , Dessauer, A. , Kleiber, J. , Porstmann, B. , Sledziewski, A. , & Lofton‐Day, C. (2007). Identification and validation of colorectal neoplasia‐specific methylation markers for accurate classification of disease. Molecular Cancer Research, 5(2), 153–163. 10.1158/1541-7786.MCR-06-0034 [DOI] [PubMed] [Google Scholar]
- Moran, S. , Martínez‐Cardús, A. , Sayols, S. , Musulén, E. , Balañá, C. , Estival‐Gonzalez, A. , Moutinho, C. , Heyn, H. , Diaz‐Lagares, A. , de Moura, M. C. , Stella, G. M. , Comoglio, P. M. , Ruiz‐Miró, M. , Matias‐Guiu, X. , Pazo‐Cid, R. , Antón, A. , Lopez‐Lopez, R. , Soler, G. , Longo, F. , … Esteller, M. (2016). Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. The Lancet Oncology, 17(10), 1386–1395. 10.1016/S1470-2045(16)30297-2 [DOI] [PubMed] [Google Scholar]
- Nagahama, Y. , Chakraborty, T. , Paul‐Prasanth, B. , Ohta, K. , & Nakamura, M. (2021). Sex determination, gonadal sex differentiation, and plasticity in vertebrate species. Physiological Reviews, 101(3), 1237–1308. 10.1152/physrev.00044.2019 [DOI] [PubMed] [Google Scholar]
- Nakamura, M. , Kobayashi, T. , Chang, X.‐T. , & Nagahama, Y. (1998). Gonadal sex differentiation in teleost fish. Journal of Experimental Zoology, 281(5), 362–372. [DOI] [Google Scholar]
- Navarro‐Martín, L. , Viñas, J. , Ribas, L. , Díaz, N. , Gutiérrez, A. , Di Croce, L. , & Piferrer, F. (2011). DNA methylation of the gonadal aromatase (cyp19a) promoter is involved in temperature‐dependent sex ratio shifts in the European sea bass. PLoS Genetics, 7(12), e1002447. 10.1371/journal.pgen.1002447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega‐Recalde, O. , Goikoetxea, A. , Hore, T. A. , Todd, E. V. , & Gemmell, N. J. (2020). The genetics and epigenetics of sex change in fish. Annual Review of Animal Biosciences, 8(1), 47–69. 10.1146/annurev-animal-021419-083634 [DOI] [PubMed] [Google Scholar]
- Pagés, H. (2018). BSgenome: Software infrastructure for efficient representation of full genomes and their SNPs. R Package Version 1.48. 0. https://bioconductor.org/packages/BSgenome [Google Scholar]
- Penman, D. J. , & Piferrer, F. (2008). Fish gonadogenesis. Part I: Genetic and environmental mechanisms of sex determination. Reviews in Fisheries Science, 16(sup1), 16–34. 10.1080/10641260802324610 [DOI] [Google Scholar]
- Pértille, F. , Brantsæter, M. , Nordgreen, J. , Coutinho, L. L. , Janczak, A. M. , Jensen, P. , & Guerrero‐Bosagna, C. (2017). DNA methylation profiles in red blood cells of adult hens correlate with their rearing conditions. The Journal of Experimental Biology, 220(19), 3579–3587. 10.1242/jeb.157891 [DOI] [PubMed] [Google Scholar]
- Pfennig, F. , Standke, A. , & Gutzeit, H. O. (2015). The role of amh signaling in teleost fish – Multiple functions not restricted to the gonads. General and Comparative Endocrinology, 223, 87–107. 10.1016/j.ygcen.2015.09.025 [DOI] [PubMed] [Google Scholar]
- Pierron, F. , Lorioux, S. , Héroin, D. , Daffe, G. , Etcheverria, B. , Cachot, J. , Morin, B. , Dufour, S. , & Gonzalez, P. (2021). Transgenerational epigenetic sex determination: Environment experienced by female fish affects offspring sex ratio. Environmental Pollution, 277, 116864. 10.1016/j.envpol.2021.116864 [DOI] [PubMed] [Google Scholar]
- Piferrer, F. (2013). Epigenetics of sex determination and gonadogenesis. Developmental Dynamics, 242(4), 360–370. 10.1002/dvdy.23924 [DOI] [PubMed] [Google Scholar]
- Piferrer, F. (2019). Epigenetics of sex determination and differentiation in fish. In Wang H. P., Piferrer F., & Chen S. L. (Eds.), Sex control in aquaculture (pp. 65–83). John Wiley & Sons, Ltd. 10.1002/9781119127291.ch3 [DOI] [Google Scholar]
- Piferrer, F. , Anastasiadi, D. , Valdivieso, A. , Sánchez‐Baizán, N. , Moraleda‐Prados, J. , & Ribas, L. (2019). The model of the conserved epigenetic regulation of sex. Frontiers in Genetics, 10, 857. 10.3389/fgene.2019.00857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piferrer, F. , Zanuy, S. , Carrillo, M. , Solar, I. I. , Devlin, R. H. , & Donaldson, E. M. (1994). Brief treatment with an aromatase inhibitor during sex differentiation causes chromosomally female salmon to develop as normal, functional males. Journal of Experimental Zoology, 270(3), 255–262. 10.1002/jez.1402700304 [DOI] [Google Scholar]
- Polanowski, A. M. , Robbins, J. , Chandler, D. , & Jarman, S. N. (2014). Epigenetic estimation of age in humpback whales. Molecular Ecology Resources, 14, 976–987. 10.1111/1755-0998.12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu, C. , & Zhan, A. (2017). Epigenetic divergence of key genes associated with water temperature and salinity in a highly invasive model ascidian. Biological Invasions, 19(7), 2015–2028. 10.1007/s10530-017-1409-1 [DOI] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas, L. , Liew, W. C. , Díaz, N. , Sreenivasan, R. , Orbán, L. , & Piferrer, F. (2017). Heat‐induced masculinization in domesticated zebrafish is family‐specific and yields a set of different gonadal transcriptomes. Proceedings of the National Academy of Sciences of the United States of America, 114(6), E941–E950. 10.1073/pnas.1609411114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas, L. , & Piferrer, F. (2014). The zebrafish (Danio rerio) as a model organism, with emphasis on applications for finfish aquaculture research. Reviews in Aquaculture, 6(4), 209–240. 10.1111/raq.12041 [DOI] [Google Scholar]
- Ribas, L. , Valdivieso, A. , Díaz, N. , & Piferrer, F. (2017a). Appropriate rearing density in domesticated zebrafish to avoid masculinization: Links with the stress response. Journal of Experimental Biology, 220(6), 1056–1064. 10.1242/jeb.144980 [DOI] [PubMed] [Google Scholar]
- Ribas, L. , Valdivieso, A. , Díaz, N. , & Piferrer, F. (2017b). Response to “the importance of controlling genetic variation – Remarks on ‘appropriate rearing density in domesticated zebrafish to avoid masculinization: Links with the stress response’”. Journal of Experimental Biology, 220(21), 4079–4080. 10.1242/jeb.167437 [DOI] [PubMed] [Google Scholar]
- Robinson, M. D. , McCarthy, D. J. , & Smyth, G. K. (2010). Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Marí, A. , Yan, Y.‐L. , BreMiller, R. A. , Wilson, C. , Cañestro, C. , & Postlethwait, J. H. (2005). Characterization and expression pattern of zebrafish anti‐Müllerian hormone (amh) relative to sox9a, sox9b, and cyp19a1a, during gonad development. Gene Expression Patterns, 5(5), 655–667. 10.1016/j.modgep.2005.02.008 [DOI] [PubMed] [Google Scholar]
- Rohland, N. , & Reich, D. (2012). Cost‐effective, high‐throughput DNA sequencing libraries for multiplexed target capture. Genome Research, 22(5), 939–946. 10.1101/gr.128124.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, C. , Li, Q. , Chen, S. , Zhang, P. , Lian, J. , Hu, Q. , Sun, B. , Jin, L. , Liu, S. , & Wang, Z. (2014). Epigenetic modification and inheritance in sexual reversal of fish. Genome Research, 24(4), 604–615. 10.1101/gr.162172.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Z. G. , & Wang, H. P. (2014). Molecular players involved in temperature‐dependent sex determination and sex differentiation in teleost fish. Genetics Selection Evolution, 46(1), 26. 10.1186/1297-9686-46-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si, Y. , Ding, Y. , He, F. , Wen, H. , Li, J. , Zhao, J. , & Huang, Z. (2016). DNA methylation level of cyp19a1a and foxl2 gene related to their expression patterns and reproduction traits during ovary development stages of Japanese flounder (Paralichthys olivaceus). Gene, 575(2), 321–330. 10.1016/j.gene.2015.09.006 [DOI] [PubMed] [Google Scholar]
- Simpson, E. R. (1979). Cholesterol side‐chain cleavage, cytochrome P450, and the control of steroidogenesis. Molecular and Cellular Endocrinology, 13(3), 213–227. 10.1016/0303-7207(79)90082-0 [DOI] [PubMed] [Google Scholar]
- Smith, J. , Sen, S. , Weeks, R. J. , Eccles, M. R. , & Chatterjee, A. (2020). Promoter DNA hypermethylation and paradoxical gene activation. Trends in Cancer, 6(5), 392–406. 10.1016/j.trecan.2020.02.007 [DOI] [PubMed] [Google Scholar]
- Sreenivasan, R. , Cai, M. , Bartfai, R. , Wang, X. , Christoffels, A. , & Orban, L. (2008). Transcriptomic analyses reveal novel genes with sexually dimorphic expression in the zebrafish gonad and brain. PLoS One, 3(3), e1791. 10.1371/journal.pone.0001791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, K. , Takehana, Y. , Naruse, K. , Hamaguchi, S. , & Sakaizumi, M. (2007). Evidence for different origins of sex chromosomes in closely related Oryzias fishes: Substitution of the master sex‐determining gene. Genetics, 177(4), 2075–2081. 10.1534/genetics.107.075598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, R. , Dodd, A. , Lai, D. , Mcnabb, W. C. , & Love, D. R. (2007). Validation of zebrafish (Danio rerio) reference genes for quantitative real‐time RT‐PCR normalization. Acta Biochimica et Biophysica Sinica, 39(5), 384–390. 10.1111/j.1745-7270.2007.00283.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanguay, R. L. (2018). The rise of zebrafish as a model for toxicology. Toxicological Sciences, 163(1), 3–4. 10.1093/toxsci/kfx295 [DOI] [PubMed] [Google Scholar]
- Team, R. C . (2013). R: A language and environment for statistical computing. http://www.R‐project.org/ [Google Scholar]
- Thompson, M. J. , von Holdt, B. , Horvath, S. , & Pellegrini, M. (2017). An epigenetic aging clock for dogs and wolves. Aging, 9(3), 1055–1068. 10.18632/aging.101211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir, H. , Robinson, J. T. , & Mesirov, J. P. (2013). Integrative genomics viewer (IGV): High‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzung, K.‐W. , Goto, R. , Saju, J. M. , Sreenivasan, R. , Saito, T. , Arai, K. , Yamaha, E. , Hossain, M. S. , Calvert, M. E. K. , & Orbán, L. (2015). Early depletion of primordial germ cells in zebrafish promotes testis formation. Stem Cell Reports, 4(1), 61–73. 10.1016/j.stemcr.2014.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivieso, A. , Ribas, L. , Monleón‐Getino, A. , Orbán, L. , & Piferrer, F. (2020). Exposure of zebrafish to elevated temperature induces sex ratio shifts and alterations in the testicular epigenome of unexposed offspring. Environmental Research, 186, 109601. 10.1016/j.envres.2020.109601 [DOI] [PubMed] [Google Scholar]
- Valdivieso, A. , Wilson, C. A. , Amores, A. , da Silva Rodrigues, M. , Nóbrega, R. H. , Ribas, L. , Postlethwait, J. H. , & Piferrer, F. (2022). Environmentally‐induced sex reversal in fish with chromosomal vs. Polygenic sex determination. Environmental Research, 213, 113549. 10.1016/j.envres.2022.113549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela, N. (2008). Sexual development and the evolution of sex determination. Sexual Development, 2(2), 64–72. 10.1159/000129691 [DOI] [PubMed] [Google Scholar]
- Venables, W. N. , & Ripley, B. D. (1999). Modern applied statistics with S‐PLUS. Springer. http://www.springerlink.com/content/978‐1‐4757‐3121‐7 [Google Scholar]
- Wang, D.‐S. , Kobayashi, T. , Zhou, L.‐Y. , Paul‐Prasanth, B. , Ijiri, S. , Sakai, F. , Okubo, K. , Morohashi, K. , & Nagahama, Y. (2007). Foxl2 up‐regulates aromatase gene transcription in a female‐specific manner by binding to the promoter as well as interacting with ad4 binding protein/steroidogenic factor 1. Molecular Endocrinology, 21(3), 712–725. 10.1210/me.2006-0248 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Liu, Y. , Jiang, S. , Li, W. , Gui, L. , Zhou, T. , Zhai, W. , Lin, Z. , Lu, J. , & Chen, L. (2019). Transcriptomic and epigenomic alterations of Nile tilapia gonads sexually reversed by high temperature. Aquaculture, 508, 167–177. 10.1016/j.aquaculture.2019.04.073 [DOI] [Google Scholar]
- Wang, X. G. , Bartfai, R. , Sleptsova‐Freidrich, I. , & Orban, L. (2007). The timing and extent of ‘juvenile ovary’ phase are highly variable during zebrafish testis differentiation. Journal of Fish Biology, 70(sa), 33–44. 10.1111/j.1095-8649.2007.01363.x [DOI] [Google Scholar]
- Wang, X. G. , & Orban, L. (2007). Anti‐müllerian hormone and 11 β‐hydroxylase show reciprocal expression to that of aromatase in the transforming gonad of zebrafish males. Developmental Dynamics, 236(5), 1329–1338. 10.1002/dvdy.21129 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Wang, C. , Zhang, J. , Chen, Y. , & Zuo, Z. (2009). DNA hypomethylation induced by tributyltin, triphenyltin, and a mixture of these in Sebastiscus marmoratus liver. Aquatic Toxicology, 95(2), 93–98. 10.1016/j.aquatox.2009.06.008 [DOI] [PubMed] [Google Scholar]
- Wei, T. , & Simko, V. (2017). R package ‘corrplot’: Visualization of a correlation matrix ([Version 0.84]). https://cran.r‐project.org/web/packages/corrplot/index.html [Google Scholar]
- Wen, A. Y. , You, F. , Sun, P. , Li, J. , Xu, D. D. , Wu, Z. H. , Ma, D. Y. , & Zhang, P. J. (2014). CpG methylation of dmrt1 and cyp19a promoters in relation to their sexual dimorphic expression in the Japanese flounder Paralichthys olivaceus . Journal of Fish Biology, 84(1), 193–205. 10.1111/jfb.12277 [DOI] [PubMed] [Google Scholar]
- Wickham, H. (2016). Ggplot2: Elegant graphics for data analysis. Springer. [Google Scholar]
- Wu, G. C. , & Chang, C. F. (2018). Primary males guide the femaleness through the regulation of testicular dmrt1 and ovarian cyp19a1a in protandrous black porgy. General and Comparative Endocrinology, 261, 198–202. 10.1016/j.ygcen.2017.01.033 [DOI] [PubMed] [Google Scholar]
- Yang, Y. J. , Wang, Y. , Li, Z. , Zhou, L. , & Gui, J. F. (2017). Sequential, divergent, and cooperative requirements of foxl2a and foxl2b in ovary development and maintenance of zebrafish. Genetics, 205(4), 1551–1572. 10.1534/genetics.116.199133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates, F. (1934). Contingency tables involving small numbers and the χ2 test. Supplement to the Journal of the Royal Statistical Society, 1(2), 217–235. 10.2307/2983604 [DOI] [Google Scholar]
- Zang, L. , Shimada, Y. , Nishimura, Y. , Tanaka, T. , & Nishimura, N. (2015). Repeated blood collection for blood tests in adult zebrafish. Journal of Visualized Experiments, 102, 53272. 10.3791/53272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Li, M. , Ma, H. , Liu, X. , Shi, H. , Li, M. , & Wang, D. (2017). Mutation of foxl2 or cyp19a1a results in female to male sex reversal in XX Nile tilapia. Endocrinology, 158(8), 2634–2647. 10.1210/en.2017-00127 [DOI] [PubMed] [Google Scholar]
- Zhang, Z. (2016). Missing data imputation: Focusing on single imputation. Annals of Translational Medicine, 4(1), 9. 10.3978/j.issn.2305-5839.2015.12.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Zhuang, Z. X. , Sun, Y. Q. , Chen, Q. , Zheng, X.‐Y. , Liang, Y. T. , Mahboob, S. , Wang, Q. , Zhang, R. , Al‐Ghanim, K. A. , Shao, C. W. , & Li, Y.‐J. (2019). Changes in DNA methylation during epigenetic‐associated sex reversal under low temperature in Takifugu rubripes . PLoS One, 14(8), e0221641. 10.1371/journal.pone.0221641 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Raw sequence reads are deposited in NCBI (BioProject: PRJNA549930, SRA: SRP202005). Data sets are available at GEO: GSE133088.