

Abstract
The tetracationic cyclophane, cyclobis(paraquat‐p‐phenylene), also known as the little blue box, constitutes a modular receptor that has facilitated the discovery of many host–guest complexes and mechanically interlocked molecules during the past 35 years. Its versatility in binding small π‐donors in its tetracationic state, as well as forming trisradical tricationic complexes with viologen radical cations in its doubly reduced bisradical dicationic state, renders it valuable for the construction of various stimuli‐responsive materials. Since the first reports in 1988, the little blue box has been featured in over 500 publications in the literature. All this research activity would not have been possible without the seminal contributions carried out by Siegfried Hünig, who not only pioneered the syntheses of viologen‐containing cyclophanes, but also revealed their rich redox chemistry in addition to their ability to undergo intramolecular π‐dimerization. This Review describes how his pioneering research led to the design and synthesis of the little blue box, and how this redox‐active host evolved into the key component of molecular shuttles, switches, and machines.
Keywords: Artificial Molecular Machines, Host-Guest Systems, Mechanically Interlocked Molecules, Molecular Devices, Molecular Recognition
The exploration of the chemistry associated with the little blue box has been a journey characterized by serendipity and fulfillment. We pay homage to Professor Siegfried Hünig, who forged the path to the modern frontiers of viologen chemistry on which we carry out research today.

1. Introduction
Compartmentalization is utilized [1] routinely in biology to provide spatial confinement for molecular and supramolecular entities, such as proteins, genetic materials, and organelles. To sustain complex life processes, nature has evolved strategies to produce confined environments required for specific tasks in relation to, for example, DNA replication, [2] protein synthesis, [3] cargo transport, [4] immune response, [5] and enzyme catalysis. [6] Synthetic chemists have long sought to emulate [7] nature's capacity for constructing nanoconfined environments at the molecular level. The landmark discoveries of crown ethers, [8] cryptands, [9] spherands, [10] carcerands, [11] and hemicarcerands [12] by Pedersen, [13] Lehn, [14] and Cram, [15] who were awarded the Nobel Prize in Chemistry in 1987, has spurred [16] the development of molecular containers. By extending the synthetic possibilities beyond traditional macrocyclic receptors, such as cyclodextrins, [17] calixarenes, [18] cucurbiturils, [19] and pillararenes, [20] the use of cyclophanes, [21] nanorings, [22] boxes, [23] cages, [24] and capsules [25] has expanded significantly the scope of host–guest chemistry [15] that can be performed within nanoconfined spaces. The ability to tune cavity sizes, geometries, and stereoelectronic environments in these structures has led to a rich diversity of binding pockets for molecular recognition, [26] the stabilization of reactive intermediates and molecules, [27] and selective transformations. [28]
We have been inspired [29] by the properties and functionality of a particular molecular receptor—namely, cyclobis(paraquat‐p‐phenylene)—or CBPQT 4+ for short. This tetracationic cyclophane, also known as the little blue box, contains two 4,4′‐bipyridinium (or viologen) units that are held together in a face‐to‐face manner by two bridging p‐xylylene linkers, leaving a cavity in between the two viologen units to accommodate aromatic guests. In its tetracationic state, the electron‐deficient CBPQT 4+ binds [30] small, π‐electron‐rich molecules strongly as a result of favorable donor–acceptor interactions. In contrast, the bisradical dicationic form, CBPQT 2(⋅+), possesses [31] a high affinity for viologen radical cations (V⋅+) as a result of strong radical‐pairing interactions. These two recognition modes of CBPQT are responsible [32] for its broad utility in the syntheses of mechanically interlocked molecules (MIMs) and artificial molecular machines (AMMs).
There is, however, a scientific giant [33] without whom this 35‐year odyssey of exploration would most likely not have occurred: this giant (Figure 1) was Professor Siegfried Hünig. As one of the most renowned organic chemists of the latter half of the 20th century, Hünig not only pioneered [34] the syntheses of viologen‐containing cyclophanes, but also spearheaded [35] the evolution of the rich redox chemistry, for example, π‐dimerization (pimerization), associated with viologen‐containing compounds. In celebration of his longevity, we dedicate this Review to his remarkable achievements in chemistry, as well as to his outstanding contributions to the chemical community. Here, we highlight some of the milestones in our exploration of the little blue box, and how Siegfried Hünig's extraordinary contributions to this field of research had a profound influence on our evolving understanding of the redox properties of this tetracationic cyclophane.
Figure 1.

Pioneering research by Professor Siegfried Hünig (University of Würzburg) on viologen‐containing cyclophanes—namely, the syntheses and characterization of o,o‐, o,m‐, and m,m‐CBPQT 4+, which led to our 35‐year journey of investigating the chemistry of cyclobis(paraquat‐p‐phenylene) (p,p‐CBPQT 4+, which we refer to simply in the Review as CBPQT 4+), also known as the little blue box.
2. The Making of the Little Blue Box: A Tribute to Siegfried Hünig
As one of the most highly creative and versatile chemists to grace the latter half of the 20th century, Hünig is perhaps best known for developing [36] the sterically hindered tertiary amine—namely, N,N‐diisopropylethylamine, which has been called Hünig's base after him. What is not generally known, however, is that he also made a name [37] for himself in the field of organic metals and multistate redox systems in addition to the invention [38] of new methods and reagents for organic synthesis. In fact, in contrast with the only two publications [36] that are related to the study of sterically hindered tertiary amines, Hünig recorded [33] in more than 100 publications the systematic investigation of π‐electron systems that harbor redox properties. Much of this research activity, which centered [39] on bipyridinium salts, was to have a major influence on our own development of the redox chemistry associated with the little blue box.
The first viologen‐containing cyclophanes were synthesized [34] and characterized by Hünig in 1983 at the University of Würzburg. These tetracationic cyclophanes contain (Figure 1) two viologen units connected in a phane‐like manner by either two o‐ or two m‐xylylene bridges (i.e., o,o‐ and m,m‐CBPQT 4+) or by one o‐ and one m‐xylylene bridge, namely, o,m‐CBPQT 4+. Since these cyclophanes are highly strained, rendering them inaccessible synthetically by traditional high‐dilution methods, Hünig [34] devised a novel, stepwise procedure to first of all prepare a dicationic intermediate by reacting xylylene dibromides with an excess of 4,4′‐bipyridine, followed by cyclization with another linker of his choice. High dilution was achieved by carrying out the reaction in a two‐phase organic/aqueous solution. Hünig also employed a mixture of solvents, namely, MeOH, H2O, and 50 % aq EtNH3Cl, to purify the desired cyclophane by silica gel column chromatography. Although the original report [34] focuses mainly on investigating the conformations of these new cyclophanes in solution by 1H NMR spectroscopy, the potential of forming inclusion complexes within their cavities was also recognized.
In 1987, we discovered [40] that crown‐ether‐like molecular receptors, such as bis‐p‐phenylene[34]crown–10 [41] (BPP34C10), which contains π‐electron‐rich hydroquinone (HQ) units, are able to form (Figure 2a) face‐to‐face complexes, on account of favorable donor–acceptor interactions, with methyl viologen (MV 2+), a π‐electron‐deficient dication. It occurred to us that it might be possible to reverse the structural roles of the receptor and the substrate and hence bind (Figure 2b) a π‐electron‐rich diphenol ether, such as 1,4‐dimethoxybenzene, inside CBPQT 4+ in which two viologen units are held about 7 Å apart in a face‐to‐face manner by two p‐xylylene linkers. We dubbed this new receptor the little blue box [42] on account of the fact that its π‐electron‐deficient bipyridinium units (blue) bind[ 29 , 30 ] small π‐electron‐rich donors (red). No sooner had we submitted our findings in two manuscripts[ 29 , 30 ] to Angewandte Chemie, than one (J.F.S.) of us received a letter from Professor Hünig. Although he was full of praise for our research, he was at pains to point out that he had also identified [43] similar host–guest complexes, albeit with different bipyridinium‐containing cyclophanes. In the event, J.F.S. invited Professor Hünig to submit a communication to Angewandte Chemie so that our two back‐to‐back communications on the little blue box ended up being juxtaposed with his communication in the same issue in 1988, which happened to be the Centenary Year of the Journal. This coincidence involving tetracationic cyclophanes marked our first meeting of minds with Professor Hünig.
Figure 2.

The advent of CBPQT 4+. a) Solid‐state and structural representations of the formation of a donor–acceptor complex between the methyl viologen (MV 2+) dication and a BPP34C10 macrocycle (blue inside red) and its binding constant in MeCN at room temperature. b) Solid‐state and structural representation of the formation of a donor–acceptor complex between 1,4‐dimethoxybenzene (1/4‐DMB) and a CBPQT 4+ cyclophane (red inside blue) and its binding constant in MeCN at room temperature.
Although our research program and that of Professor Hünig diverged along very different lines in the following decade, with his focusing on organic metals [44] and ours on synthesizing MIMs, [45] his research continued to provide inspiration for many important discoveries made in our group. It is hard to imagine how our research, as well as that of many others, could have advanced without the foundational synthetic chemistry introduced by Hünig.
3. Synthesis and Solid‐State (Super)Structure of the Little Blue Box
Since we first reported the synthesis of the little blue box in 1988, methods for making CBPQT 4+ have evolved continuously. The synthesis commences (Figure 3) with the preparation of p‐xylylene‐bis(4‐(4‐pyridyl)pyridinium) (XBPP 2+), which is formed by treating one molar equivalent of p‐xylylene dibromide with an excess of 4,4′‐bipyridine, followed by cyclization of XBPP 2+ with another molar equivalent of p‐xylylene dibromide to give CBPQT⋅4PF 6 after purification and counterion exchange with NH4PF6 in H2O. Since there is significant ring strain associated with this rigid tetracationic cyclophane, the cyclization step typically results in a low yield (ca. 12 %) of CBPQT⋅4PF 6 under reflux in MeCN. In the presence of a π‐electron‐rich template, [46] however, the yield can be improved at least threefold, as a result of favorable donor–acceptor and [C−H⋅⋅⋅O] interactions between templates, to which diethylene glycol functions are appended, and the acyclic intermediate resulting from the reaction of XBPP 2+ with p‐xylylene dibromide. This templation, not only directs this SN2 reaction towards ring closure rather than to oligomerization, but also compensates for the strain introduced during cyclophane formation. An excellent yield (Figure 3) of CBPQT⋅4PF 6 can be obtained [47] by tagging the diethylene glycol chains onto a 1,5‐dioxynaphthalene (DN) unit and using this compound as a template for the reaction under ultrahigh pressure, namely, 12 kbar. Although these templates are typically removed by cumbersome liquid–liquid extractions, they can also be exchanged [48] with a pH‐responsive derivative of 1,5‐diaminonaphthalene (DAN), which dissociates easily from CBPQT 4+ following protonation of its amino groups.
Figure 3.

Synthesis of CBPQT 4+. Typically, the synthesis involves a two‐step procedure: 1) the reaction between para‐xylylene dibromide and an excess of 4,4′‐bipyridine, which affords para‐xylylenebis(4‐(4‐pyridyl)pyridinium bis(hexafluorophosphate) (XBPP⋅2PF 6 ) in an almost quantitative yield after counterion exchange and 2) the reaction between XBPP⋅2PF 6 and one equivalent of para‐xylylene dibromide, which produces CBPQT⋅4PF 6 in a wide range of yields after counterion exchange, depending on the reaction conditions—12 % without any additive at 80 °C, 62 % in the presence of an HQ‐derived template under high pressure (12 kbar) at 25 °C, 81 % in the presence of a DN‐derived template under high pressure (12 kbar) at 25 °C and 20 % in the presence of a catalytic amount of tetrabutylammonium iodide (TBAI) at 80 °C.
Despite the high efficiency of these synthetic procedures employing templates, they are not suitable for the large‐scale production of CBPQT⋅4PF 6 , since the templates are expensive and need to be used in excess. In fact, the most practical procedure we employ nowadays does not require a template, but rather relies on the use of tetrabutylammonium iodide (TBAI) as an inexpensive catalyst. This approach enhances [49] (Figure 3) the yield of CBPQT⋅4PF 6 twofold compared to the noncatalyzed reaction. Under these conditions, the SN2 reaction between XBPP⋅2PF6 and p‐xylylene dibromide becomes reversible at high temperatures, thereby conferring a self‐correcting mechanism upon the assembly of the rigid tetracationic cyclophane. TBAI also accelerates the rate of the final ring‐closing SN2 reaction, particularly under high‐dilution conditions. It is worth noting that, while CBPQT⋅4PF 6 is soluble in solvents such as MeCN and MeNO2, CBPQT⋅4Cl is water soluble, establishing the fact that CBPQT 4+ can be investigated in both aqueous and non‐aqueous media depending on the nature of the counterions.
X‐ray crystallography shows that CBPQT 4+ adopts [29] (Figure 4a) a rigid centrosymmetric rectangular box‐like geometry that is considerably strained in the solid state: its two viologen units span a distance of 10.3 Å, supported by two p‐xylylene linkers. The centroid‐to‐centroid distance (Figure 4b) between the two viologen units is 6.8 Å, allowing for near‐perfect co‐facial [π⋅⋅⋅π] stacking interactions between CBPQT 4+ and aromatic π‐electron‐rich guests. The viologen units exhibit (Figure 4b) a 21° torsional angle about their 4C−4′C bond with their long axes tilted (Figure 4c) 82° with respect to the planes defined by the p‐phenylene units of the CBPQT 4+ ring. It turns out that slow evaporation of iPr2O into a MeCN solution of CBPQT⋅4PF 6 leads [48] to two polymorphic crystalline forms of the little blue box—one block‐like and the other needle‐like‐corresponding (Figure 4d and 4 e) to two different superstructures.
Figure 4.

Solid‐state (super)structures of CBPQT 4+ from X‐ray crystallography carried out on single crystals. a) A perspective view of CBPQT 4+. b) A plan view of CBPQT 4+, showing the centroid‐to‐centroid distance between the two viologen units. c) A side‐on view of CBPQT 4+, illustrating the dihedral angle between the viologen and the phenylene units. d) Plan view of CBPQT 4+ cyclophanes present in block‐like crystals, determined by X‐ray crystallography, revealing the superstructure and relative positioning of the PF6 − counterions. e) Plan view of CBPQT 4+ cyclophanes present in needle‐like crystals, determined by X‐ray crystallography, revealing the superstructure and relative positioning of the PF6 − counterions.
4. CBPQT4+ as an π‐Electron‐Deficient Host Molecule
The ability of CBPQT 4+ to bind small π‐donors in a rather promiscuous manner was recognized [30] immediately after its introduction [29] in 1988. The solid‐state superstructures of some representative host–guest complexes based on CBPQT 4+ as the host are portrayed in Figure 5. 1,4‐Dimethoxybenzene (1/4‐DMB) binds [30] (Figure 5a) CBPQT 4+ with a K a value of 17 M−1 in MeCN. 1,5‐Dimethoxynaphthalene (1/5‐DMN), which has more π‐electrons and a larger π‐surface area than 1/4‐DMB, associates [50] (Figure 5b) with CBPQT 4+ much more strongly (K a≈103 M−1 in MeCN). In both host–guest complexes, the protons on the aromatic core of 1/4‐DMB and 1/5‐DMN participate [51] (Figure 5a and 5 b) in [C−H⋅⋅⋅π] interactions with the p‐phenylene units of the CBPQT 4+ ring, in addition to the face‐to‐face [π⋅⋅⋅π] stacking interactions between the host and the guests. The [C−H⋅⋅⋅π] interactions dictate the co‐conformation between the host and the guests. Sulfur‐containing conjugated heterocycles, such as bithiophene (2T) and tetrathiafulvalene (TTF), are also excellent guests [52] for incorporation (Figure 5c,d) into the cavity of CBPQT 4+ (K a=100 and 6900 M−1, respectively in MeCN) as a result of their rich supply of π‐electrons. It is noteworthy that CBPQT 4+ can also be incorporated [53] as a guest molecule into a larger, π‐electron‐rich cyclophane.
Figure 5.

Solid‐state superstructures and binding constants (in MeCN at 25 °C) of host–guest complexes formed between CBPQT 4+ and a) 1/4‐DMB, b) 1/5‐DMN, c) 2T, d) TTF, and e) 1/4‐R2B where R denotes 2‐(2‐(2‐(2‐hydroxyethoxy)ethoxy)ethoxy)ethoxy appendages, illustrating the attractive [C−H⋅⋅⋅π] and [C−H⋅⋅⋅O] interactions between the host and the guest.
Anchoring oligoethylene glycol chains to guests results [54] in substantial increases in their binding constants (K a=2520 M−1 for 1/4‐R2B⊂CBPQT 4+ (where R=2‐(2‐(2‐(2‐hydroxyethoxy)ethoxy)ethoxy)ethoxy loops) with CBPQT 4+ in MeCN, since the polyether loops engage [55] (Figure 5e) in [C−H⋅⋅⋅O] interactions with the acidic α‐protons on the bipyridinium units of the tetracationic cyclophane. These additional stabilizing effects are crucial when it comes to raising the efficiency of template‐assisted syntheses of donor–acceptor catenanes. A large collection of π‐electron‐rich guests derived from HQ, [56] DN, [57] 1,5‐dithianaphthalene (DTN), [58] DAN,[ 48 , 59 ] TTF, [60] bispyrrolotetrathiafulvalene [61] (BPTTF), biphenol, [62] benzidine, [63] azobenzene, [64] pentiptycene‐based crown ethers, [65] triptycene‐based cucurbiturils, [66] amino acids, [67] neurotransmitters, [68] and phenyl glucopyranosides [69] have all been investigated for their ability to complex with the little blue box. [70]
5. Syntheses of Donor–Acceptor Mechanically Interlocked Molecules (MIMs)
Mechanically interlocked molecules [32] (MIMs), including catenanes, [71] rotaxanes, [72] knots, [73] links, [74] daisy chains, [75] Borromean rings, [76] and so forth, are molecules composed of component parts that cannot be separated without breaking constitutive covalent or coordinative bonds. These well‐defined structures, not only possess aesthetically pleasing architectures, but they also open the doors to emergent properties and functions. Despite existing examples in nature—for example, lasso peptides [77] and knotted circular DNA [78] —and statistical [79] as well as covalent‐directed syntheses [80] in laboratory settings, the efficient and selective production of MIMs had remained an open challenge for years. It was not until the introduction of metal templation by Sauvage [81] in the 1980s that MIMs became [82] readily accessible.
Our first venture into the host–guest chemistry of CBPQT 4+ in 1988 left us wondering if it might be possible to have the π‐accepting tetracationic cyclophane interlock mechanically with a π‐donating crown ether, such as BPP34C10. [41] The outcome from the first reaction blew our minds: we were able to isolate [83] (Figure 6a) the first donor–acceptor [2]catenane (C1 4+) in a remarkable 70 % yield simply by stirring XBPP⋅2PF6 and p‐xylylene dibromide at room temperature in MeCN in the presence of 3 molar equivalents of BPP34C10 as the template. The crystal structure, which confirmed unambiguously the making of a [2]catenane, was selected for display on the front cover of the October issue of Angewandte Chemie in 1989. The reaction proceeds by the displacement of one bromide from p‐xylylene dibromide by XBPP⋅2PF6, forming (Figure 6a) a viologen unit that is encircled by BPP34C10 before a second ring‐closing nucleophilic substitution takes place [84] to afford C1 4+. It is clear from the crystal structure that this templating effect arises from donor–acceptor interactions between the viologen units and the two catechol rings in BPP34C10, aided and abetted [55] by the formation of [C−H⋅⋅⋅O] hydrogen bonds. [85] These weak bonding interactions accrued during the template‐directed synthesis “live on” in the [2]catenane afterwards, as indicated by cyclic voltammetry (CV) and 1H NMR spectroscopy.
Figure 6.

a) Synthesis of the first donor–acceptor [2]catenane C1 4+. This high‐yielding catenation is aided and abetted by [C−H⋅⋅⋅O] as well as donor–acceptor and [C−H⋅⋅⋅π] interactions between BPP34C10 and the acyclic intermediate resulting from reaction of XBPP 2+ with para‐xylylene dibromide. b) Synthesis of Olympiadane. The synthesis involves two separate steps: 1) the reaction between bitolyl‐bis(4‐pyridyl)pyridinium dications (BBPP 2+) and 4,4′‐bis(bromomethyl)‐1,1′‐biphenyl (BBB) in the presence of two equivalents of TNP57C15 as templates to afford a [3]catenane, C2 4+ and 2) treatment of C2 4+ with two equivalents of XBPP 2+ and para‐xylylene dibromide, which cyclize around one of the DN units in each of the two TNP57C15 rings to afford Olympiadane. Note that XBPP 2+, BBPP 2+, C1 4+, C2 4+, and Olympiadane are all supported by the requisite number of PF6 − counterions.
The SN2 clipping procedure employed in the synthesis of C1 4+ was later expanded to embrace the preparation of a wide array of donor–acceptor [2]catenanes containing different crown[10] ethers. The performance of the clipping reaction is impacted by a number of factors, including i) the reactant stoichiometry, where an excess of either π‐donating or π‐accepting precursors typically results in an increase of yield, ii) the solvent—higher yields of [2]catenanes were obtained in MeCN than in DMF, presumably because of the higher association constants (K a values) of their corresponding pseudorotaxane precursors in MeCN, iii) pressure—ultrahigh pressures at room temperature in DMF lead to superior reaction performance, a feature characteristic of the Menschutkin reaction, which is associated with a negative change in molar volume, iv) temperature—while association constants decrease with increasing temperature, it was found that heat can accelerate catenation and improve yields in some cases, and v) reaction time. A wide variety of crown ethers can be accommodated, such as those containing HQ,[ 56c , 56d , 58 , 86 ] DN,[ 57b , 87 ] DAN, [88] TTF, [89] azobenzene, [64] anthracene, [90] fluorenone, [91] triazole, [92] and their derivatives, in addition to crown/cyclophane hybrids, [93] cryptands, [94] and oxacalixcrowns. [95] There is no strong correlation between the size and/or π‐donating ability of crown ether's recognition units and the yields as a result of templation.
Despite the detailed characterization of the first donor–acceptor [2]catenane, the community in the early 1990s, was, to some extent, dubious and/or indifferent about the emergence of catenanes and rotaxanes. Such skepticism was resolved unequivocally with the advent of higher order catenanes, [96] which could also be synthesized using template‐directed procedures. One of the most appealing catenanes (Figure 6b) in this category was Olympiadane, [97] a [5]catenane reminiscent of the logo of the Olympic movement, since it consists of five mechanically interlocked rings. The first step in its synthesis relies on the clipping of bitolyl‐bis(4‐pyridyl)pyridinium (BBPP 2+) and 4,4′‐bis(bromomethyl)‐1,1′‐biphenyl (BBB) around two trisnaphthalene‐containing crown ethers, TNP57C15. The approach capitalizes [98] on the propensity of cyclobis(paraquat‐4,4′‐biphenylene) (CBPQB 4+), also known as the Big Blue Box, to host two π‐electron donors in the form of a ternary complex. When the resulting [3]catenane was treated with a large excess of XBPP⋅2PF6 and p‐xylylene dibromide, templation around (Figure 6b) one of the two naphthalene recognition sites of the two TNP57C15 rings leads to the formation of Olympiadane. The solid‐state structure of Olympiadane reveals a vast array of noncovalent bonding interactions, including [C−H⋅⋅⋅π] edge‐to‐face interactions involving aromatic rings and donor–acceptor interactions between them, in addition to multiple [C−H⋅⋅⋅O] hydrogen bonds. The sheer number and wealth of these favorable interactions give rise to strong templation, enabling the synthesis of the high‐order catenane. Olympiadane was subsequently used to make a branched [7]catenane, [99] whose solid‐state structure at the time took two weeks for the collection of data, followed by six months of structural elucidation and refinement.
The synthesis of donor–acceptor catenanes and rotaxanes is, in many cases, kinetically controlled, namely, bond formation is irreversible, which can take a variety of forms in addition to catenation by SN2 substitution. For example, in a conceptual extension of the original donor–acceptor catenane synthesis, we have demonstrated [100] (Figure 7a) the possibility of assembling a degenerate [2]rotaxane, namely, R1 4+, by employing a similar clipping procedure, but with two bulky silyl stoppers attached to the two ends of a polyether chain instead of them being present in a crown ether. Other bulky substituents, such as 4‐tritylphenyl [101] and porphyrin, [102] can also serve as the stoppers. The viologen unit resulting from the reaction between XBPP⋅2PF6 and p‐xylylene dibromide is recognized (Figure 7a) by the two HQ units on the polyether dumbbell, culminating in mechanical bond formation following cyclization to clip on the CBPQT4+ ring. Rotaxanes can also be produced in the presence of a single donor unit, such as HQ, [103] DN, [104] TTF, [105] BPTTF, [106] monopyrrolotetrathiafulvalene, [107] (MPTTF) and anthracene. [108] The reaction yields are sensitive to various structural parameters, such as the nature of the donor unit, linker length, and stopper constitution, as well as reaction conditions. Clipping is not limited to SN2 reactions. With CBPQT 4+ as the host and an alkyne‐tethered, DN‐containing polyether as the guest, catenation [109] (Figure 7b) or rotaxanation [110] can also occur in a highly efficient manner by either an intra‐ or intermolecular Eglinton–Glaser–Hay coupling between the two terminal alkyne groups. It is worth noting that DAN [111] and TTF [112] are both acceptable donors for the construction of [2]catenanes from alkyne‐containing precursors.
Figure 7.

Kinetically controlled syntheses of donor–acceptor MIMs. a) Synthesis of a [2]rotaxane R1 4+ by an SN2 clipping approach. b) Synthesis of a [2]catenane C3 4+ by an Eglinton–Glaser–Hay coupling approach. c) Synthesis of a [2]rotaxane R2 4+ by a stoppering approach. d) Synthesis of a [2]catenane C4 4+ and a [2]rotaxane R3 4+ using “click” chemistry. Note that R1 4+, R2 4+, R3 4+, C3 4+, and C4 4+ are all supported by the requisite number of PF6 − counterions.
An alternative, kinetically controlled approach to the construction of rotaxanes is “stoppering”. One example involves [54c] (Figure 7c) the silylation of a pseudorotaxane's terminal hydroxy functions with triisopropylsilyl groups in the presence of 2,6‐lutidine. In this case, the CBPQT 4+ ring forms a host–guest complex with π‐donors such as HQ, [54c] DN, [113] benzidine,[ 62a , 63 ] biphenol, [62a] or an oligothiophene [114] before suitable stoppers are installed.
Perhaps one of the most transformative and ubiquitous tools for forging mechanical bonds is the copper‐catalyzed azide‐alkyne cycloaddition [115] (CuAAC). The CuAAC reaction was first utilized[ 109a , 116 ] in the construction of donor–acceptor rotaxanes and catenanes in 2006 at a time that coincided with the assembly of MIMs facilitated by transition‐metal [117] and active‐metal [118] templates. The seminal synthesis of the [2]rotaxane R3 4+, featuring the use of a DN recognition unit flanked by two azide‐terminated triethylene glycol chains, was accomplished [116] in 82 % yield in DMF by conjugation with propargylated tetraphenylmethane stoppers in the presence of CuSO4 and ascorbic acid. When one of the two azide groups was replaced with a terminal alkyne group, cyclization occurred [109a] under the same conditions to afford the [2]catenane C4 4+ in 41 % yield. The beauty of click chemistry lies in its robustness and ease of operation in linking two molecular entities—namely, an azide and an alkyne—together without affecting other functional groups on substrates, making it extremely versatile [119] in the preparation of CBPQT4+‐containing molecular switches and machines, as well as MIMs having more complicated topologies and sophisticated architectures.
In contrast to kinetic control, where there is no going back upon completion of a reaction, thermodynamic control [120] is all about the reversibility of bond formation during MIM syntheses. In these cases, the interconversion among all reactive species will eventually result in the generation of the most thermodynamically stable products. One of the most elegant examples in this category is the synthesis of rotaxanes by slippage, for example, the CBPQT 4+ ring can slip [121] over (Figure 8a) the aza[18]crown‐6 (A18C6) stopper at 50 °C in MeCN to form R4 4+. Because of the thermodynamic sink resulting from favorable donor–acceptor interactions between CBPQT4+ and the HQ recognition unit, no deslipping occurred even at 80 °C in MeCN. When the A18C6 stoppers complex with Na+ or K+ ions, however, the kinetic barrier becomes [121] insurmountable for slippage by the CBPQT 4+ ring as a result of increased steric and electrostatic repulsion by the stoppers. The fact that no chemical bond is broken or formed during slippage highlights the nuanced difference between a rotaxane (a kinetically stable molecule) and a pseudorotaxane (a kinetically labile host–guest complex), a topic that has led [122] to considerable discussion with no consensus being reached so far by the chemical community. We take the view that the pseudorotaxane versus rotaxane discussion is dependent on the environment (e.g. solvent, temperature, etc.). Other stoppers, such as MPTTF, [123] β‐d‐glucopyranose, [124] and 2‐methoxy‐1,3‐benzamide‐based oligomer [125] have also been found to allow slippage of the CBPQT 4+ ring.
Figure 8.
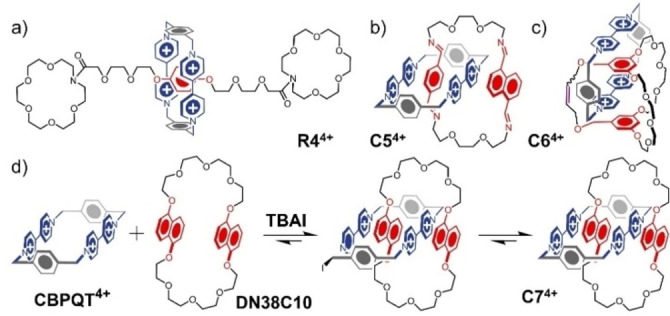
Thermodynamically controlled syntheses of donor–acceptor MIMs. a) Synthesis of a [2]rotaxane R4 4+ by slippage. b) Synthesis of a [2]catenane C5 4+ that relies on dynamic imine chemistry. c) Synthesis of a [2]catenane C6 4+ that relies on olefin metathesis. d) Synthesis of a [2]catenane C7 4+ by an iodide‐mediated dynamic SN2 substitution. Note that R4 4+, C5 4+, C6 4+, and C7 4+ are all supported by the requisite number of PF6 − counterions.
Dynamic covalent chemistry [126] (DCC) is another powerhouse that drives the assembly of MIMs under thermodynamic control. The idea is that by virtue of reversible chemical reactions, mechanically interlocked species can be selected for and amplified under appropriate conditions on account of favorable interactions with templates or other additives according to their free energies of binding. Liu [127] has invented (Figure 8b) a clipping procedure, based on the reversible imine condensation between two diamines and two different dialdehydes appended to phenylene and naphthalene cores, for the self‐assembly of C5 4+ in the presence of CBPQT 4+. The [2]catenane is formed in an almost quantitative (>90 %) yield in MeCN with almost complete (97 %) selectivity for the translational isomer in which the phenylene unit occupies the cavity of the CBPQT4+ ring. Also relying on imine chemistry but with a cryptand as the donor macrobicycle, Yan and Zhou [128] have demonstrated the synthesis of a [2]catenane with multiannulated mechanical bonds by clipping. By harnessing the strong association of the CBPQT 4+ ring with HQ and DN moieties, rotaxanes can also be produced from dumbbells stoppered with dynamic imine bonds, [129] photoisomerizable azobenzene units, [130] or ammonium crown ether host–guest complexes. [131]
Olefin metathesis, [132] now ubiquitous in the world of organic synthesis, constitutes another powerful method for the construction of MIMs. In the presence of a Grubbs catalyst, ring‐closing metathesis (RCM) can convert two terminal olefins into a loop with extrusion of ethylene. Huang [133] has taken advantage of this process to close (Figure 8c) an olefin‐modified m‐phenylene[32]crown‐8 (MP32C8) around one of the viologen units in the CBPQT 4+ ring to prepare the macrocyclic [2]catenane C6 4+.
The discovery of new catalysts has contributed significantly to the growing importance of DCC and its creative use in thermodynamically controlled MIM syntheses. Since CBPQT 4+ is highly strained and its methylene units can be resubjected to substitution chemistry, it was reasoned that a suitably mild nucleophile might transiently open the box and act as a leaving group, thereby rendering the SN2 chemistry dynamic. TBAI was identified [134] as a suitable catalyst (Figure 8d) for SN2‐mediated equilibrations to give donor–acceptor [2]catenanes directly as if by magic from their constituent rings. Remarkably, the two rings are topologically linked in a traceless manner to form [135] selectively [2]‐ and [3]catenanes in high yields, reminiscent of an act of minimally invasive molecular surgery. In addition to DCC in solution, CBPQT 4+ can also be catenated [136] onto a gold surface through dynamic gold‐thiolate bonds.
A diverse family of topologically distinct donor–acceptor MIMs comprised of CBPQT4+ has been made (Figure 9) by employing either kinetic or thermodynamic approaches, including a) rotacatenanes, [137] with the illustrated example consisting [138] of a fused pillar[5]arene/naphtho[36]crown‐10 macrobicyclic core and an imidazolium‐based dumbbell, b) [3]pseudocatenanes or handcuff catenanes, [139] comprising π‐donating macrocyclic polyethers fused to a central TTF core, c) side‐chain poly[2]catenanes, [140] with the illustrated example consisting of polyurethane as the backbone, d) bis[2]catenanes, comprising two CBPQT4+ rings fused to a flexible linker [141] or a central phenylene unit, [142] e) a cyclic bis[2]catenane, with two catenanes connected together by a covalent linker, [143] f) a side‐chain poly[2]rotaxane with polyacrylate as the backbone, [144] g) tripodal [4]rotaxanes, reminiscent of G1 dendrimers,[ 116 , 145 ] h) branched catenanes possessing a combination of linear and radial links,[ 99 , 146 ] i) a [3]catenane, which comprises a fused triple‐torus polyether catenated by two CBPQT4+ rings, [147] j) a pseudo[1]catenane, where the interlocked polyether macrocycle is fused to a phenylene unit in CBPQT4+, [148] k) [1]rotaxanes, [149] in which the rods that thread through CBPQT4+ are fused to the encircling rings, l) a molecular figure‐of‐eight, in which the rod is anchored to opposite ends of an encircling ring, [150] and m) oligorotaxanes [151] in which CBPQT4+ rings thread onto polyether‐bridged DN oligomers, causing the thread to fold into an extended stack of alternating π‐donors and π‐acceptors. Many other donor–acceptor MIMs, such as pretzelanes, [152] polyrotaxanes, [153] and poly[2]catenanes [154] are not discussed in this Review owing to space limitations.
Figure 9.

A selection of MIMs synthesized by donor–acceptor templation. a) A catenarotoxane. b) A [3]pseudorotaxane or handcuff catenane. c) A side‐chain poly[2]catenane. d) A bis[2]catenane. e) A cyclic bis[2]catenane. f) A side‐chain poly[2]rotaxane. g) A tripodal [4]rotaxane. h) A branched [6]catenane. i) A [3]catenane. j) A pseudo[1]catenane. k) A [1]rotaxane. l) A molecular figure‐of‐eight. m) An oligorotaxane. All these MIMs are positively charged and supported by the requisite number of PF6 − counterions.
6. CBPQT2(⋅+) as a Radical Host: A Homage to Siegfried Hünig
Our exploration of the little blue box brought our chemistry, for the second time, hand‐in‐hand with Siegfried Hünig's in 2010. Some 22 years after the introduction of CBPQT 4+ as a multipurpose π‐donor receptor, we were brought face‐to‐face with yet another cornerstone of Hünig chemistry—that is, what he called intramolecular pimerization of viologen radical cations (V⋅+). It had been known for a long time that the methyl viologen dication (MV 2+) can undergo single‐electron reduction to form the corresponding radical cation (MV⋅+). In fact, the term “viologen” takes its name from the violet color of this radical species in solution, a phenomenon observed by Michaelis [155] back in the 1930s. It was not until 1964, however, that Kosower [156] established that the violet color does not originate from MV⋅+—as it is colored blue by itself—but rather from its π‐dimer, (MV⋅+)2, which is observable in concentrated aqueous solutions. This phenomenon, which is now known as π‐dimerization, [157] or multicentered pancake bonding [158] on account of the spin‐pairing of π‐electrons, has been identified in a wide variety of conjugated organic π‐radicals, such as V⋅+[156, 159] and tetrathiafulvalene [160] (TTF⋅+) radical cations, naphthalene diimide [161] (NDI⋅−), tetracyanoquinodimethane [162] (TCNQ⋅−) and 2,5‐dimethyl‐N,N′‐dicyanoquinonediimine [163] (DCNQI⋅−) radical anions, as well as in neutral phenalenyl radicals. [164] Nevertheless, homodimeric associations between these π‐radicals were generally considered to be very weak in solution, leaving them, for most of the time, to be treated as chemical curiosities.
Hünig's longstanding interest in the redox chemistry of bipyridinium dications prompted him to delve into the phenomena of π‐dimerization. Following his pioneering research [34] on the synthesis of viologen‐containing cyclophanes, he discovered [165] that radical‐pairing interactions between two V⋅+ units can be enhanced significantly when they are connected by a suitably chosen rigid linker. For example, when two viologen units are linked (Figure 10a) by an o‐xylylene bridge (3.0 Å), they experience [165] unusually strong radical‐pairing interactions upon being reduced to their corresponding radical cations. A similar phenomenon was observed [165] (Figure 10b) when the two V⋅+ units are part of an o‐xylylene‐based doubly bridged cyclophane, namely, o,o‐CBPQT 2(⋅+), which exists exclusively as a π‐dimer. When the linker is changed to m‐xylylene, which spans a longer distance (5.0 Å) between its methylene carbon atoms, however, only weak attractive interactions are detected between the two V⋅+ units. These results not only demonstrated the possibility of having strong radical‐pairing interactions between V⋅+ in an intramolecular setting, but also established, for the first time, the optimal pairing distance required for their effective π‐dimerization.
Figure 10.

a) Seminal studies by Professor Siegfried Hünig on the intramolecular π‐dimerization of a pair of viologen radical cations connected by an ortho‐xylylene bridge under reducing conditions. b) Schematic representation of the strong intramolecular π‐dimerization between the two viologen units in the o,o‐CBPQT 2(⋅+) diradical dication under reducing conditions. c) Schematic representation of the strong association between a MV⋅+ radical cation and a CBPQT 2(⋅+) diradical dication in MeCN at 25 °C and the solid‐state superstructure of [MV⊂CBPQT] 3(⋅+). d) Cyclic voltammograms (MeCN, 0.1 M TBAPF6, 200 mV s−1) of CBPQT 4+, V1 2+, and an equimolar mixture of CBPQT 4+ and V1 2+. e) UV/Vis/NIR spectra of CBPQT 2(⋅+), V1 (⋅+), and an equimolar mixture of CBPQT 2(⋅+) and V1 (⋅+).
We did not realize just how important this seminal research by Siegfried Hünig was until, in 2010, former postdoctoral fellow Ali Trabolsi [166] investigated what now seems to be an obvious question in radical recognition, namely, can we design a radical cyclophane, in which the two opposing, parallel V⋅+ units are positioned apart at a distance that would allow strong association with a MV⋅+ radical cation based on radical‐pairing interactions? The answer turned out to be the little blue box! When reduced to its diradical dicationic state, CBPQT 2(⋅+) binds (Figure 10c) MV⋅+ strongly at room temperature in MeCN (K a=7.9×104 M−1), a result that was discovered [31] during a CV experiment performed below 0 V! Formation of the trisradical tricationic complex, namely, [MV⊂CBPQT] 3(⋅+), is rapid [31] (k on=2.1×106 s−1) at room temperature, and it dissociates completely into its individual host–guest components upon oxidation, on account of strong Coulombic repulsions between fully oxidized CBPQT 4+ and MV 2+. The ability of a this host–guest system to switch [167] between strongly associative and strongly repulsive states on redox stimulation offers a facile way to control molecular motion, as well as to drive chemical systems out‐of‐equilibrium. Indeed, this historic discovery in our exploration of the little blue box has fueled an extensive investigation and utilization of the trisradical recognition motif in the elaboration of MIMs [32] and AMMs. [168] It is worth noting that the binding constants of the trisradical tricationic complexes can be finely tuned over a wide range—from 8×102 to 1.8×105 M−1 in MeCN—either by changing [169] the N,N′‐substituents on the guest V⋅+ radical cations, or by swapping [170] the p‐xylylene linker in CBPQT 2(⋅+) for different linkers, such as m‐xylylene, 2,5‐thiophenedimethylene and 2,6‐pyridinedimethylene.
The solid‐state superstructure (Figure 10c) of [MV⊂CBPQT] 3(⋅+) revealed [171] a radical‐pairing distance (3.2 Å) between MV⋅+ and the two V⋅+ units in CBPQT 2(⋅+) that is shorter than the typical [π⋅⋅⋅π] stacking distance (3.4 Å) observed in complexes of CBPQT 4+ with π‐electron rich guests. To accommodate this shorter pairing distance in radical recognition, the V⋅+ centroid‐to‐centroid distance in CBPQT 2(⋅+) decreases from 6.92 to 6.43 Å upon guest incorporation, reminiscent of the induced‐fit model [172] in enzymatic catalysis. The MV⋅+ guest is oriented centrosymmetrically within the CBPQT 2(⋅+) cavity with its long axis tilted at 76° with respect to the mean plane of the cyclophane. Unlike fully oxidized CBPQT 4+, the V⋅+ units are associated with a nearly 0° torsional angle about their 4,4′ C−C bond. Intermolecular cofacial radical stacking between the V⋅+ units on CBPQT 2(⋅+) is also observed in the extended superstructure, which spans a distance of 3.2 Å, characteristic of radical‐pairing interactions. Upon attaching two methoxy groups onto the p‐xylylene linker of CBPQT 2(⋅+), however, the resulting trisradical tricationic host–guest complex packs [173] in a discrete manner rather than as a continuous radical stack. Such a unique arrangement in the solid state provides opportunities for the design of organic magnetic materials based on this paramagnetic host–guest complex.
Evidence of π‐dimerization between CBPQT 2(⋅+) and V⋅+ guests in solution can also be observed by cyclic voltammetry as well as by UV/Vis/NIR spectroscopy, a technique that was originally popularized by Hünig. Compared to the reoxidation peaks of the individual solutions of V1⋅+ (−0.31 V) and CBPQT 2(⋅+) (−0.24 V), the reoxidation peak of an equimolar amount of V1⋅+ and CBPQT 2(⋅+) is shifted [31] (Figure 10d) to more positive potential (−0.19 V) as a result of the stability of the inclusion complex. The solution of an equimolar amount of V1⋅+ and CBPQT 2(⋅+) also exhibits [31] (Figure 10e) a NIR absorption band at around 1075 nm that is absent in the spectrum of either V1⋅+ or CBPQT 2(⋅+), on account of the strong charge‐transfer interaction between the host and the guest in [V1⊂CBPQT] 3(⋅+).
The observation that V⋅+ guests can be encapsulated with high affinity in CBPQT 2(⋅+) as a result of strong radical‐pairing interactions has stimulated (Figure 11) the development of a variety of viologen‐based host–guest systems based on the principle of size‐matched radical recognition. For example, by altering the centroid‐to‐centroid distance between the two V⋅+ units of a bisradical dicationic cyclophane, cyclobis(paraquat‐m‐phenylene) diradical dication ( m CBPQT 2(⋅+)) and CBPQT 2(⋅+) can be recognized [174] (Figure 11a,b) by CBPQB 2(⋅+) and cyclobis(paraquat‐4,4′‐ethynebiphenylene) diradical dication (CBPQE 2(⋅+)), respectively. More recently, binding of a pair of MV⋅+ radical cations has also been achieved [175] (Figure 11c) using a size‐matched radical host, namely, the cyclobis(paraquat‐2,6‐naphthalene) diradical dication (CBPQN 2(⋅+)). In all of these cases, the binding distance between each V⋅+ unit in the solid‐state superstructures of the inclusion complexes is within 3.05–3.25 Å, a range that is critical to achieve strong radical‐pairing interactions between hosts and guests in solution.
Figure 11.

Size‐matched radical recognition. a) The strong association between CBPQB 2(⋅+) and a molecule of m CBPQT 2(⋅+) in MeCN at room temperature, forming a ring‐in‐ring tetrakisradical tetracationic complex [mCBPQT⊂CBPQB] 4(⋅+). b) The strong association between CBPQE 2(⋅+) and a molecule of CBPQT 2(⋅+) in MeCN at room temperature, forming a ring‐in‐ring tetrakisradical tetracationic complex [CBPQT⊂CBPQE] 4(⋅+). c) The strong association between CBPQN 2(⋅+) and two molecules of MV⋅+ in MeCN at room temperature, forming a tetrakisradical tetracationic complex [(MV)2 ⊂CBPQN] 4(⋅+). Note that CBPQB 2(⋅+), m CBPQT 2(⋅+), CBPQE 2(⋅+), CBPQT 2(⋅+), CBPQN 2(⋅+), and MV⋅+ are all supported by the requisite number of PF6 − counterions.
Depending on the reducing potentials of the chemical reductants, CBPQT 4+ can be reduced either to its diradical dicationic state (in the presence of zinc or copper dust) or to its neutral state [176] (in the presence of 4 molar equivalents of CoCp2)—namely, CBPQT 0, also known as the red box. The reduction potentials for these two processes are −0.33 and −0.75 V, respectively, in MeCN versus a SCE reference. CBPQT 0 exhibits very different host–guest chemistry compared with its radical and fully oxidized analogues. Not only can it encapsulate [176] (Figure 12a) π‐electron deficient guests, such as 1,4‐dicyanobenzene, it can also bind [177] (Figure 12b) MV⋅+ in an unusual 2 : 1 host/guest stoichiometry.
Figure 12.

Solid‐state superstructures of a) DCB⊂CBPQT 0, highlighting its 1 : 1 host–guest binding stoichiometry and b) MV⋅+⊂(CBPQT 0 ) 2, featuring its 2 : 1 host–guest binding stoichiometry.
7. Syntheses of Radical MIMs
The ability of CBPQT 2(⋅+) to bind the MV⋅+ radical cation with high affinity immediately spawned a whole new generation of MIMs in which trisradical tricationic recognition motifs were employed as templates. One of the first examples was the template‐directed synthesis of [2]rotaxanes [178] by a click‐chemistry‐based stoppering approach. CBPQT 4+ and an azide‐terminated V 2+ derivative were first of all reduced (Figure 13a) to their corresponding (bis)radical (di)cations by either i) photoinduced charge transfer from the excited state of a Ru(bpy)3 2+ sensitizer under visible‐light irradiation in the presence of triethanolamine as a sacrificial electron donor, or ii) by reduction with zinc dust. Upon formation of the trisradical tricationic complex as a result of host–guest radical‐pairing interactions, the azides react with the stopper precursor, di‐tert‐butylacetylene‐dicarboxylate, to afford (Figure 13a), after oxidation in air, a series of hexacationic [2]rotaxanes R5(n) 6+ in yields of up to 31 %. Because of strong Coulombic repulsion between the V2+ units on CBPQT4+ and the axles, the rings are forced [178] (Figure 13a) to encircle the oligomethylene chains adjacent to the V2+ cores of the dumbbells. The rotaxanes with shorter dumbbells were found [179] to persist longer in their radical states upon exposure to air as a result of increased charge repulsion between fully oxidized V2+ units constrained by the mechanical bonds, raising the kinetic barriers to oxidation of the radical cations while enhancing their thermodynamic stability on account of spin‐pairing interactions.
Figure 13.

a) Synthesis of a [2]rotaxane R5(n) 4+ by radical templation and a click stoppering approach. b) Synthesis of a homo[2]catenane C8 8+ by radical templation and SN2 cyclization. The formation of C8 8+ proceeds through intermediate redox states—namely, C8 4(⋅+), C8 2⋅6+, and C8⋅7+. All the MIMs are positively charged and supported by the requisite number of PF6 − counterions.
What will happen if the viologen guest in the trisradical tricationic complex is appended with two xylylene bromide groups, which can engage in SN2 substitution with another molecule of 4,4′‐bipyridine? This bold idea led [180] (Figure 13b) to the remarkable synthesis of a mechanically and electrostatically constrained homo[2]catenane C8 8+. Its preparation, which is incredibly simple, involves mixing the bis(α‐bromoxylyl)bipyridinium precursor with 4,4′‐bipyridine and zinc dust in MeCN under an inert atmosphere. The key intermediate in this reaction is a tetracationic trisradical inclusion complex, which is converted into the homo[2]catenane C8 4(⋅+) after a final ring‐closing nucleophilic substitution and in situ reduction. The tetrakisradical tetracationic homo[2]catenane can only be oxidized (Figure 13b) to its monoradical heptacationic state, C8⋅7+, which exists in equilibrium with the bisradical hexacation C8 2⋅6+ upon exposure to air. In fact, the full oxidation state of the homo[2]catenane is so energetically demanding that it can only be accessed [181] (Figure 13b) by oxidation with a strong oxidant, such as tris(4‐bromophenyl)aminium hexachloroantimonate (TBPA⋅+). These results showcased the unusual stability of this radical homo[2]catenane as a result of mechanical bonding as well as the otherwise unfavorable interactions between its viologen units in its fully oxidized state. It is noteworthy that the homo[2]catenane can pass [180] through a total of six accessible redox states (C8 8+ C8⋅7+ C8 2⋅6+ C8 4(⋅+) C8 6⋅2+ C8 0) during a voltametric scan, thereby harboring an enormous capacity for storing electrons within a volume of only one cubic nanometer.
Stoppering and cyclization represent [167] (Figure 14a) two general strategies for the synthesis of MIMs using the radical templation approach. The use of CBPQT or one of its close analogues as the macrocycle has led to a number of radical [2]catenanes possessing remarkable stability and unusual electrochemical properties. For example, C9⋅7+, which contains (Figure 14b) a 2,7‐diazapyrenium unit and exists as a stable, persistent monoradical in air, possesses [182] seven redox states, 0, 1+, 2+, 4+, 6+, 7+, and 8+. C10 2⋅6+, which consists of two m‐xylylene linkers as the bridging units in the two mechanically interlocked cyclophanes, exists [183] (Figure 14c) exclusively as a radical dimer under ambient conditions. This bisradical [2]catenane enjoys even higher stability compared with that of C8⋅7+ as a result of the closer distance between viologen units enforced by the m‐xylylene linkers. Consequently, these viologen units experience even stronger Coulombic repulsions once fully oxidized, as indicated by the significantly more positive potentials of the first two reduction peaks for C10 2⋅6+ compared with those for C8⋅7+.
Figure 14.

a) Schematic representation of the stoppering and cyclization approaches to the syntheses of rotaxanes and catenanes using radical templation. b) Graphical representation of an air‐stable monoradical [2]catenane C9⋅7+ synthesized by radical templation. c) Graphical representation of an air‐stable bisradical [2]catenane C10 2⋅6+ synthesized by radical templation. d) Graphical representation of a radial [5]catenane C11 24+ synthesized by radical templation. e) Graphical representation of a [2]rotaxane R6 6+ synthesized by radical templation and a “click” stoppering approach. f) Graphical representation of a [2]catenane C12 6+ synthesized by radical templation and olefin metathesis. All the MIMs are positively charged and supported by the requisite number of PF6 − counterions.
The CuAAC reaction turns out to be a particularly robust tool for the synthesis of radical MIMs since 1) it is compatible with V⋅+ radical cations and 2) the reaction can be carried out in the presence of copper dust, which serves as both a reducing reagent for converting V 2+ into V⋅+ and the catalyst for the cycloaddition upon its in situ oxidation to Cu+. This symbiotic use of copper circumvents the need for additional sacrificial electron donors as well as photosensitizers to reduce V 2+. Apart from making (Figure 14e) [2]catenanes and [2]rotaxanes, [184] for example, R6 6+, we have demonstrated [185] (Figure 14d) the possibility of using this procedure for the construction of a radial [5]catenane, C11 24+, which supports up to 24+ positive charges inside its nanometer‐size cavity.
So far, the vast majority of radical MIMs, featuring the intermediacy of a trisradical tricationic recognition motif, have been synthesized under kinetic control—for example, through irreversible SN2 substitution or click chemistry. A rare example of making radical MIMs under thermodynamic control relies on olefin metathesis. By careful tuning of reaction conditions, it was found that a RCM can proceed [186] (Figure 14f) in the presence of a trisradical tricationic complex and close a V⋅+‐containing, alkene‐terminated oligoethylene glycol chain into a loop, thereby furnishing a [2]catenane C12 6+. The possibility of using DCC to prepare radical MIMs constitutes a research area waiting to be explored.
8. The Mechanostereochemistry of Donor–Acceptor MIMs
One of the most compelling properties of MIMs is the relative motion experienced by their component parts. Whereas conventional molecules experience stretching, bending, and rotation associated with their covalent bonds, MIMs are also subject [187] to the relative motion made possible through mechanical bonding, giving rise to co‐conformational changes. [188] Graphical representations of the donor–acceptor [2]catenane C1 4+, as well as [2]rotaxane R1 4+, have been employed in Figure 15 to illustrate the different kinds of co‐conformational changes in [2]catenanes and [2]rotaxanes. In C1 4+, the BPP34C10 and CBPQT4+ rings can undergo (Figure 15a) a process we have described as “rocking”. This motion [189] is by far the fastest (2×106 s−1 at RT) compared with all the other relative mechanical motions experienced by the rings in C1 4+. Circumrotation [83] of the BPP34C10 ring, for example, through the CBPQT4+ ring (Figure 15b) is much slower (30 s−1 at RT), since it involves rupture of all the noncovalent bonding interactions between the two rings. Pirouetting, [83] which refers to the rotation of the BPP34C10 ring with respect to the CBPQT4+ ring, involves only partial rupture of the noncovalent bonding interactions between the two rings, taking place (Figure 15c) at a faster rate (1800 s−1 at RT). It is worth noting that the [2]catenane C1 4+ exists [190] in the solid state in two enantiomeric co‐conformations that undergo rapid racemization in solution. The two enantiomeric co‐conformations can be distinguished by 1H NMR spectroscopy in the presence of chiral reagents at low temperatures. In rotaxanes, there is another motion called translation, [100] which describes the movement of rings along the axles that they encircle. In R1 4+, the translation of the CBPQT4+ ring between the two HQ recognition sites occurs 2000 s−1 at RT. The rates of these motions in MIMs can be determined by variable‐temperature 1H NMR spectroscopy with the assistance of dynamic 1H NMR line‐shape simulations. An Eyring plot ( vs ) can be used to obtain the ΔH ≠ and ΔS ≠ values, as well as the energy barrier ΔG T ≠ for the relative motion between the CBPQT4+ ring and the dumbbell.
Figure 15.
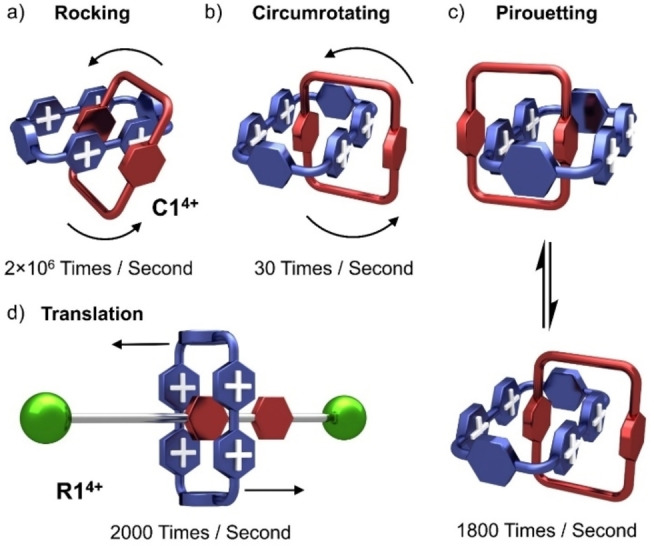
Graphical representations of the relative movements of the component parts of CBPQT‐containing, degenerate MIMs. a) Rocking. b) Circumrotating. c) Pirouetting movements in the donor–acceptor [2]catenane C1 4+. d) Translational movement in the donor–acceptor [2]rotaxane R1 4+.
The term molecular shuttle was introduced [100] in 1991 to describe the shuttling of a CBPQT4+ ring back and forth between two identical HQ recognition sites in the degenerate donor–acceptor [2]rotaxane R1 4+. It was soon envisioned that if the π‐donor recognition sites in a [2]rotaxane are different, their translational isomers will become nondegenerate and thus associated with different free energies of binding with the CBPQT4+ ring. With sufficiently low activation barriers to reach equilibrium, the populations of these isomers are dictated [191] by their energy difference, ΔG°, between the ground state (GSCC) and metastable state (MSCC) co‐conformations. The translational isomerism of these donor–acceptor MIMs was investigated and summarized [192] in a 67‐part “Molecular Meccano” collection of publications in various journals throughout the 1990s and early 2000s. In short, many structural parameters can influence the kinetics and thermodynamics associated with the translational isomerism in MIMs, such as i) the nature of the recognition units and linkers, ii) the ring sizes, (iii) their topology/architecture, and iv) the constitutions [193] of the MIMs. These movements constitute the basis for the operation of molecular switches and machines.
9. Molecular Switches
Precise control over molecular motions [194] is of fundamental and practical importance in physics, chemistry, and biology. In particular, the design of molecular systems in which switching can be accomplished [195] under the influence of an external signal constitutes a major advance towards the realization of molecular electronics. MIMs that incorporate two or more different recognition units are excellent prototypes [196] for the design of molecular switches. With an ever‐increasing number of methods for the making of mechanical bonds, as well as the many molecular recognition motifs available, a number of recognition sites can be incorporated judiciously into MIMs so that their preferential associations with CBPQT4+ rings can be altered in response to external stimuli. In an effort to realize this concept, we first of all embarked [197] on the idea of an acid/base switch based on the protonation/deprotonation of a benzidine unit. When one of the HQ units in the molecular shuttle is replaced by a benzidine unit and the other by a biphenol unit, it was found (Figure 16a) that the CBPQT4+ ring spends 84 % of its time on the benzidine unit and 16 % on the biphenol unit at equilibrium at RT in MeCN. Upon the addition of acid, the nitrogen atoms on the benzidine unit become protonated, inducing, as a result of Columbic repulsion, the CBPQT4+ ring to migrate to the neutral biphenol unit. The switch reverts to its original state upon addition of base. Switching can also be triggered by single‐electron oxidation of the benzidine unit to its radical cationic state, which also generates a repulsive Coulombic force that obliges the CBPQT4+ ring to encircle the alternative biphenol unit. It follows that this donor–acceptor [2]rotaxane R7 4+ is switchable by both redox and pH stimuli. In addition to shuttling, the threading and dethreading of a CBPQT4+ ring‐containing pseudorotaxane has also been subjected [198] to acid/base control.
Figure 16.

The influence of pH, solvents, temperature, pressure, counterions, and mechanical force on the operation of donor–acceptor molecular switches. a) The preferential association of the CBPQT4+ cyclophane with the benzidine and the biphenol units in [2]rotaxane R7 4+ under basic and acidic conditions, respectively. b) The preferential association of the CBPQT4+ cyclophane with the DAN and HQ units in [2]catenane C13 4+ under basic and acidic conditions, respectively. c) The preferential association of the CBPQT4+ cyclophane with the DN and HQ units in [2]catenane C14 4+ in Me2SO and Me2CO, respectively. d) The preferential association of the CBPQT4+ cyclophane with the DN and DTN units in [2]catenane C15 4+ at 243 K and 303 K, respectively. e) The preferential association of the CBPQT4+ cyclophane with the HQ and MPTTF units in [2]rotaxane R8 4+ under low and high pressure during synthesis, respectively. f) The preferential association of the CBPQT4+ cyclophane with the BPTTF and DN units in [2]rotaxane R9 4+ in the presence of PF6 − and TRISPHAT− as the counteranion, respectively. g) The mechanical forces associated with pulling the CBPQT4+ cyclophane in the bistable [2]rotaxane R10 4+ into energetically unfavorable geometries under reducing and oxidizing conditions, respectively.
Acid/base switches can also appear [88] in the form of donor–acceptor [2]catenanes. The [2]catenane C13 4+ exists (Figure 16b) in two co‐conformational isomers in a 78 : 22 ratio in favor of the CBPQT4+ ring encircling the DAN recognition site. When the amino groups on DAN are protonated, the CBPQT4+ ring encapsulates [198] the HQ unit exclusively as a result of Coulombic repulsion between DANH2 2+ and the CBPQT4+ ring.
Solvents, temperature, pressure, and even counterions all play important roles in determining the ratios of translational isomers in donor–acceptor MIMs. For example, in the presence of more polar solvents, such as Me2SO, the CBPQT4+ ring prefers [199] (Figure 16c) to bind the DN unit in the [2]catenane C14 4+, whereas in the presence of less polar solvents, such as Me2CO, it is biased towards encircling the HQ unit. A possible explanation for this observation is the fact that the larger π‐surface of the DN unit can more effectively insulate the exterior surface of CBPQT4+ from unfavorable cation–solvent interactions in less polar solvents. Similar phenomena were observed [200] when changing the solvent from H2O to CHCl3 with a [2]catenane that incorporates DN and DTN as the two competing recognition sites.
A number of donor–acceptor [2]catenanes[ 56d , 58 , 199 ] and [2]rotaxanes [201] have been found to exhibit different populations of translational isomers at low and high temperatures. For example, C15 4+ exists as a mixture of two co‐conformations in a 55 : 45 ratio, favoring (Figure 16d) the complexation of the DN unit by CBPQT4+ at 303 K. Upon lowering the temperature to 243 K, however, the ratio of the two conformations is reversed [58] (40 : 60), with DTN preferentially occupying the cavity of CBPQT4+.
Jeppesen [202] has reported a rare example of a donor–acceptor [2]rotaxane R8 4+ in which the distribution of the two translational isomers can be influenced (Figure 16e) by pressure. Contrary to most other bistable rotaxenes based on HQ and TTF‐derived recognition sites, the CBPQT4+, ring prefers to reside on the HQ unit rather than on the more‐electron‐rich MPTTF unit following mechanical bond formation at ambient pressure. Under high pressure during the synthesis, however, a higher, ratio of the CBPQT4+, ring is found to be associated with the MPTTF unit. It is noteworthy that these two co‐conformations are not interconvertible as a result of the steric barrier imposed by the SEt group on the MPTTF unit.
The counterions associated with CBPQT4+ have been shown [203] (Figure 16f) to influence the co‐conformational isomerism of the bistable [2]rotaxane R9 4+. Whereas the DN and BPTTF recognition sites are almost equally populated in the two translational isomers of R9 4+ in the presence of PF6 − as the counterion, the equilibrium is heavily biased (>95 %) towards the co‐conformation in which CBPQT4+ encircles the BPTTF unit when Lacour's TRISPHAT− anion is employed.
The advent of atomic force microscopy (AFM) has enabled [204] chemists to measure the forces associated with the positioning of the CBPQT4+ ring in energetically unfavorable co‐conformations on a bistable donor–acceptor [2]rotaxane. The rotaxane R10 4+ consists of (Figure 16g) an SiO2‐immobilized dumbbell that contains DN and TTF recognition sites, threaded by a CBPQT4+ ring decorated with a dithiolane tether that can be anchored to a gold‐coated AFM cantilever. Whereas an average of 66 pN of force is required to separate the ring and the dumbbell in the presence of a neutral TTF recognition site, an average of 145 pN is needed to dissociate the ring when TTF is oxidized to its dication, indicating that the Coulombic repulsion between CBPQT4+ and TTF2+ can generate up to 79 pN of force. Similar experiments have also been performed [205] on CBPQT‐containing oligorotaxane foldamers.
Given the ability of the little blue box to switch between multiple recognition sites in different redox states, it is not hard to imagine that the majority of molecular switches based on CBPQT4+ motion are controlled by redox chemistry. For illustration purposes, the recognition sites and the CBPQT4+ ring in these switches are represented schematically. Depending on their mode of motion, these switches can be classified (Figure 17) into three categories: a) translation, b) circumrotation, and c) expansion/contraction.
Figure 17.

Graphical representations of redox‐active molecular switches that express translocating, circumrotating, and expanding/contracting motions. a) Translocation induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+), respectively, by the DN and V⋅+ units in the [2]rotaxane R11 6+ under oxidizing and reducing conditions, respectively. b) Translocation induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+), respectively, by the DN and NDI⋅− in the [2]rotaxane R12 4+ under oxidizing and reducing conditions, respectively. c) Translocation induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+), by the TTF, the DN, and the V⋅+ units in the [2]rotaxane R13 6+ under neutral, oxidizing, and reducing conditions, respectively; d) Rotation induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+), respectively, by the TTF, the DN, and the V⋅+ units in the [2]catenane C16 6+ under neutral, oxidizing, and reducing conditions, respectively. e) Rotation induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+) by the TTF and DN units, respectively, in the [2]catenane C17 4+ under neutral and oxidizing conditions, respectively. f) Expansion/contraction induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+), respectively, by the triazole and V⋅+ units in [c3]daisy chain R14 18+ under oxidizing and reducing conditions, respectively. g) Expansion/contraction induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+), respectively, by the DN and V⋅+ units in the [c2]daisy chain R15 12+ under oxidizing and reducing conditions, respectively. h) Contraction induced by the preferential binding of CBPQT4+ and CBPQT2(⋅+), respectively, by the V⋅+ units in the [3]rotaxane R16 18+, forming a radical stack under reducing conditions.
One of the earliest examples of molecular switches based on donor–acceptor and radical‐pairing‐induced molecular recognition was made [206] (Figure 17a) by appending a RuII photosensitizer (shaded in yellow) to a [2]rotaxane that contains a DN (shaded in red) and a V2+ (shaded in blue) unit, threaded by a CBPQT4+ ring. Notably, the propensity of CBPQT4+ to accept [207] electrons from different photosensitizers has been well studied. In its fully oxidized state, the CBPQT4+ ring resides primarily on the DN unit as a result of favorable donor–acceptor interactions. Upon photoirradiation, however, the RuII stopper can undergo photoinduced electron transfer and reduce the V2+ components both on the dumbbell and on the cyclophane to their radical cationic states, following a catalytic cycle that employs triethanolamine as the sacrificial electron donor. The resulting CBPQT2(⋅+) ring no longer possesses high affinity for the DN unit and thus moves to the V⋅+ unit, where it participates in strong radical‐pairing interactions. Exposing the [2]rotaxane to air oxidizes the radicals and returns the rotaxane to its ground state. A number of other CBPQT4+‐containing photoswitches,[ 149b , 208 ] including a [2]catenane bearing the same recognition sites and a photosensitizing RuII complex, [209] have also been reported in the literature.
Li [210] found that redox‐driven movement of CBPQT4+ can also occur (Figure 17b) in the presence of DN and NDI (shaded in blue) recognition sites in the [2]rotaxane R12 4+. The idea is predicated on the fact that CBPQT2(⋅+) possesses a strong binding affinity (K a=1.2×105 M−1) for NDI⋅− that correlates with both radical‐pairing interactions and Coulombic attraction between the two radicals with opposite charges. Upon reduction with zinc dust, the CBPQT2(⋅+) ring departs from the π‐electron‐rich DN unit for the highly attractive NDI⋅− unit, while exposing the solution to air reverses the switch because of the negligible affinity between NDI and fully oxidized CBPQT4+. A similar bistable [2]catenane switched by hetero‐radical pairing interactions has also been reported [211] by the same researchers.
The incorporation of more than two orthogonal recognition sites in a [2]rotaxane leads to a multistable switching system, as exemplified [31] (Figure 17c) by the movement of CBPQT4+ to one of three different recognition sites in the [2]rotaxane R13 6+. The recognition sites include a DN (shaded in red), a V2+ (shaded in blue), and a TTF (shaded in green) unit that are linked together by oligoethylene glycol loops using click chemistry. Under ambient conditions, CBPQT4+ spends most of its time on the TTF site rather than on the DN site. Oxidation of the TTF unit to its dicationic form, however, repels the CBPQT4+ ring and causes it to pass over V2+ and reach the DN site, which becomes the most π‐electron‐rich recognition component in the system. When both the CBPQT4+ ring and the V2+ site on the dumbbell are reduced to their (di)radical (di)cationic forms, the ring migrates to the V⋅+ site, forming a strong trisradical tricationic species. It is worth noting that switching between three different recognition sites can also be achieved [212] by controlling the three redox states of the little blue box, namely, CBPQT4+, CBPQT2(⋅+), and CBPQT0. In this case, a different set of recognition sites—namely, a triazole ring for CBPQT4+, a V2+ site for CBPQT2(⋅+) under reducing conditions, and a tetrafluorobenzene site for CBPQT0—must be used for the tristable molecular switch to operate under redox control. Although several other redox‐switchable [2]rotaxanes have also been reported, [213] they are not discussed in this Review.
Whereas a multistable [2]rotaxane can undergo switching as a result of CBPQT4+ movement along its dumbbell, a multistable [2]catenane can experience switching by circumrotational motions between different recognition sites. This idea has been realized [89a] (Figure 17d) by incorporating DN, V2+, and TTF units into a [2]catenane, namely, C16 6+, which operates in a very similar manner to R13 6+. Importantly, because of the high specificity of the interactions between recognition sites and CBPQT in different redox states, the relative positions of the two rings can be controlled precisely by modifying the redox potentials. A similar situation applies when it comes to the circumrotational motions in a bistable [2]catenane with two recognition sites. For example, C17 4+, which contains TTF and DN recognition sites, exists (Figure 17e) predominantly as the co‐conformation in which the CBPQT4+ ring encircles the TTF site under normal conditions. After oxidation of TTF to its dicationic form, charge repulsion between TTF2+ and CBPQT4+ triggers circumrotational motion within the [2]catenane and encirclement of the tetracationic cyclophane around the DN site. A number of other redox‐switchable catenanes, featuring the use of the little blue box, have also been reported[ 89f , 112 , 214 ] in the literature.
An unorthodox type of motion that can arise from switching in higher‐order MIMs is expansion and contraction. A recent example revealed [215] (Figure 17f) the contraction and expansion of a cyclic trimer of a [2]rotaxane, R14 18+, promoted by redox chemistry. This molecular switch draws on the well‐established association between mCBPQT2(⋅+) rings and V⋅+ recognition sites in their radical cationic states. Under reducing conditions, the [c3]daisy chain contracts as a result of radical‐pairing interactions, adopting a tris‐armed star‐shaped co‐conformation, whereas upon oxidation, the [c3]daisy chain expands with the mCBPQT4+ rings obliged to remain distant from the fully oxidized V 2+ units, encircling the triazole rings.
Closely related to this research is a [c2]daisy chain R15 12+ that also exhibits [216] (Figure 17g) similar contractions and expansions in response to redox stimuli. This molecular switch depends on the ability of CBPQT4+ to recognize the DN unit in its oxidized state and the V⋅+ unit under reducing conditions. When R15 12+ is reduced electrochemically to its hexaradical hexacationic state, the molecule expands (Figure 16f) to permit both of its CBPQT2(⋅+) rings to encircle the corresponding V⋅+ units on the dumbbells. It is noteworthy that contraction and expansion have also been explored[ 149c , 217 ] in [c1]daisy chains and molecular lassos.
A highly unconventional type of contraction and expansion was observed [218] (Figure 17h) in the redox‐mediated switching of the oligorotaxane R16 18+. The construction of the molecular accordion, which consists of two CBPQT4+ rings and a p‐xylylene‐bridged pentaviologen axle, was made possible by their effective templation in their singly reduced states on account of radical‐pairing interactions. The oligorotaxane unfolds into an elongated chain in its fully oxidized state as a result of Coulombic repulsions between V2+ units, while its two CBPQT4+ rings encircle the hexamethylene linkers at the termini. Under reducing conditions, however, the V⋅+ units fold and form a π‐radical stack that is intercalated by the two CBPQT2(⋅+) rings to establish favorable radical‐pairing interactions. Folding can also be observed [219] in a TTF‐containing oligopseudorotaxane, in which threading of two CBPQT4+ rings can be controlled by redox chemistry.
Although molecular switches constitute a robust and versatile platform for realizing stimuli‐responsive mechanical movements on the nanoscale, there is no net directionality with respect to their motions following a full cycle of operation. One important reason—as Astumian [220] has pointed out—is the lack of kinetic asymmetry in these systems, preventing their translation into molecular motors and machines. In the next section, we show how it is possible to overcome these constraints by designing chemical building blocks, acting as kinetic barriers, to promote the unidirectional transportation of the little blue box.
10. Molecular Machines
From the use of biomolecular pumps and motors in living systems to the development of engines during the past few centuries, machines are ubiquitous in our everyday lives. Chemists continue to be inspired [221] by the possibility of harvesting energy/work from switchable molecules to create nanomachines in the microscopic and nanoscopic worlds. Despite the daunting challenges associated with designing such intricate (supra)molecular systems, tremendous progress has been made [222] in recent years in the design and syntheses of AMMs. The advent of these AMMs has enabled [168] an unprecedented level of control over the unidirectional motions and transport of molecules thanks to the development of kinetically controlled, away‐from‐equilibrium chemistry.
A key element in the design of AMMs is the introduction of kinetic asymmetry, [220] which endows molecular motions with directionality. A number of elegant approaches have emerged [168] in recent years as a consequence of the incorporation of information and energy ratchets as the symmetry‐breaking elements in AMMs. Our continued focus on the relative motions between components of MIMs motivated us to come up with an array of chemical building blocks that serve as kinetic barriers to promote the unidirectional motion of CBPQT4+ rings along dumbbells and around loops. In 2013, a prototype pump was devised [223] (Figure 18a) to allow, for the first time, the threading and de‐threading of a CBPQT 4+ ring unidirectionally on and off a pseudo‐dumbbell, D1 +. This prototype pump consists of a π‐electron‐rich DN recognition site flanked by a neutral 2‐isopropylphenyl (IPP) group, serving as a steric barrier, and a cationic 3,5‐dimethylpyridinium (3,5‐DMPy+) group, acting as a Coulombic barrier. The strong Coulombic repulsion between the CBPQT 4+ ring and the 3,5‐DMPy+ group creates a kinetic barrier (22.9 kcal mol−1) to threading that is much larger than that (16.9 kcal mol−1) imposed by the IPP group. Consequently, the CBPQT 4+ ring threads almost exclusively from the IPP terminus onto the DN unit of D1 +. Reduction of CBPQT 4+ to its diradical dicationic state is accompanied by a significant loss of its affinity for the DN recognition site as well as its electrostatic repulsion with the 3,5‐DMPy+ group. At the same time, the energy barrier for the radical cyclophane to slip over the IPP terminus is increased because of its smaller cavity compared with that of CBPQT 4+, making this pathway even more energetically demanding (by 2.9 kcal mol−1 according to DFT calculations) than that involving the 3,5‐DMPy+ terminus. Thus, the CBPQT 2(⋅+) ring prefers to leave D1 + by passing over the 3,5‐DMPy+ group. Importantly, the prototype pump establishes a feasible energy ratchet mechanism for the unidirectional transport of CBPQT 4+ rings. The sequence of a steric barrier, a binding site, and a Coulombic barrier constitutes what we term [224] a pumping cassette.
Figure 18.

Graphical representations and, in some cases, the related structural formulas associated with the design and operation of: a) a prototype molecular pump D1 +, which contains a π‐electron‐rich DN recognition unit flanked by a neutral 2‐isopropylphenyl (IPP) group, serving as a steric barrier, and a cationic 3,5‐dimethylpyridinium (3,5‐DMPy+) group, acting as a Coulombic barrier. Under oxidizing conditions, the CBPQT 4+ cyclophane threads onto the DN unit of D1 + over the IPP terminus, whereas upon reduction, the CBPQT2(⋅+) cyclophane departs from D1 + by passing over the 3,5‐DMPy+ group, thus establishing unidirectional motion. b) A one‐stroke molecular pump D2 3+, which contains a 3,5‐DMPy+ Coulombic barrier, a viologen (V⋅+) recognition site, and a long collecting chain terminated by a 2,6‐diisopropylphenyl stopper. Under reducing conditions, the CBPQT 2(⋅+) cyclophane passes over the 3,5‐DMPy+ group and thread onto the V⋅+ unit of D2 3+ as a result of radical‐pairing interactions, whereas upon oxidation, the CBPQT4+ cyclophane is forced onto the collecting chain as a result of strong Coulombic repulsions with the V2+ unit and the 3,5‐DMPy+ group. c) A first‐generation (Mark I) molecular pump D3 3+, which contains an isopropylphenylene (IPP) steric barrier between its viologen recognition site and its collecting chain. The IPP steric barrier prevents the CBPQT cyclophanes on the collecting chain from returning to the V⋅+ recognition site under reducing conditions, making it possible to pump more than one ring using redox chemistry. d) A second‐generation (Mark II) molecular pump D4 3+, in which the 3,5‐DMPy+ group in D3 3+ is replaced by a 2,6‐dimethylpyridinium (2,6‐DMPy+) anchor and a shorter link (one methylene group) between the V2+ and IPP units is used. The Mark II pump features faster operation and increased unidirectionality when compared with D3 3+.
The next question was: how can we pump CBPQT 4+ rings onto a collecting chain rather than into bulk solution? In other words, how can we transform the input redox energy from chemical reagents or electricity into a stored potential energy rather than meaningless thermal release? We realized that significant work had to be done to compensate for the entropy loss following the recruitment of a CBPQT 4+ ring from bulk solution onto a collecting chain. Unfortunately, the binding between CBPQT 4+ and the DN unit is too weak [54d] (K a=768 M−1 in MeCN) to allow such a process to proceed with high efficiency. Radical‐pairing between CBPQT 2(⋅+) and V⋅+, on the other hand, represents [225] a much more potent system for pumping CBPQT 4+ because i) their binding, namely, CBPQT 2(⋅+) to V⋅+, is much stronger (K a>104 M−1 in MeCN) under reducing conditions and ii) the system can switch between a strongly associative and a strongly repulsive state simply by redox stimulation, thus creating [167] a large energy difference (ΔΔG>21 kcal mol−1) between the reduced and oxidized states, a situation which offers more potential for useful work to be done.
Building on this concept, a one‐stroke molecular pump, D2 3+, was synthesized [226] by connecting a 3,5‐DMPy+ Coulombic barrier and a long collecting chain, terminated by a 2,6‐diisopropylphenyl stopper, to a viologen recognition site. Under reducing conditions, the attraction between the CBPQT2(⋅+) ring and the V⋅+ recognition site creates (Figure 18b) a deep thermodynamic well for the ring to pass over the low kinetic barrier imposed by 3,5‐DMPy+ and encircle the dumbbell. Upon oxidation, the repulsion between the resulting CBPQT4+ ring and the V2+ unit forces the ring to thread onto the collecting chain, even though there are few attractive interactions between them. On the other hand, the DMPy+ presents (Figure 18b) such a high kinetic barrier that de‐threading of the CBPQT4+ ring from the dumbbell is completely forbidden. The net result is that work has been done in pumping the CBPQT4+ ring into a high‐energy [2]rotaxane.
One major limitation of this molecular pump, however, is that it is unable to operate in a repetitive manner, simply because the CBPQT4+ ring on the collecting chain will move backwards and occupy the V⋅+ recognition site as soon as it is reduced to its diradical dicationic state, thus inhibiting the collection of a second ring from bulk solution. Clearly, a second kinetic barrier is required to prevent the CBPQT2(⋅+) ring on the collecting chain from being drawn back into the thermodynamic sink induced by the V⋅+ unit. A neat solution to this problem is the introduction of an isopropylphenylene (IPP) steric barrier between the V2+ unit and the collecting chain. This structural modification formed [224] the basis (Figure 18c) for the synthesis of a first‐generation (Mark I) artificial molecular pump (AMP). Following a full cycle of reduction by zinc dust and oxidation by NOPF6, a CBPQT 4+ ring is pumped onto the pumping cassette and resides in between the V2+ and IPP units. In this state, the steric barrier provided by the IPP unit is lower than the Coulombic barriers posed by the V2+ and 3,5‐DMPy+ units. With sufficient thermal energy (i.e. heating), the ring slips over the IPP unit and becomes kinetically trapped on the collecting chain. No de‐slipping of the first ring back to the pumping cassette is observed when the system is reduced to allow uptake of a second ring. By taking two CBPQT 4+ rings from bulk solution towards a higher local concentration, this molecular pump drives the chemical system as a whole out‐of‐equilibrium.
On the back of this investigation, we designed [227] a second‐generation molecular pump (Mark II) in which the original 3,5‐DMPy+ unit is replaced (Figure 18c) by a 2,6‐dimethylpyridinium (2,6‐DMPy+) unit and the bismethylene chain between the V2+ and IPP units is shortened to only one methylene group. With a higher kinetic barrier forged by the new 2,6‐DMPy+ unit and faster kinetics for the CBPQT4+ ring to traverse the IPP unit, the Mark II pump enjoys more rapid operation and greater unidirectionality than the Mark I pump.
The successful implementation of the Mark II pump has stimulated the development of a wide variety of AMPs by taking advantage of its pumping cassette. For example, by attaching (Figure 19a) a pumping cassette to both ends of an oligomethylene chain containing two quaternary ammonium centers, it becomes possible to pump [228] two CBPQT 4+ rings simultaneously onto the collecting chain in a single redox cycle under electrochemical control. When two individual AMPs are linked together in a head‐to‐tail fashion, pumping of a CBPQT 4+ ring from bulk solution takes place [229] (Figure 19b) in a consecutive manner, by forming a [2]rotaxane in one redox cycle, before releasing the ring back into bulk solution following a second redox cycle. Pumping can be controlled, not only by electricity, but also by light. When a photocleavable stopper is used [230] instead of the 2,6‐diisopropylphenyl stopper in the original Mark II pump, the CBPQT4+ ring that has been pumped onto the collecting chain using electricity can be released (Figure 19c) back into bulk solution upon light irradiation. Recently, by placing a pyrene fluorophore at the center of a collecting chain with two pumping cassettes at both ends, it has been demonstrated [231] that the fluorescence of the dumbbell is quenched upon sequestering two CBPQT 4+ rings as a result of their mechanical‐bond‐enforced proximity to the fluorophore.
Figure 19.

Applications of Mark II molecular pumps in the design and synthesis of: a) A dual molecular pump D5 8+, which can pump two CBPQT 4+ cyclophanes simultaneously onto its collecting chain in a single redox cycle under electrochemical control. b) A molecular dual pump, D6 6+, which links two pumping cassettes in a head‐to‐tail fashion so that pumping of a CBPQT 4+ cyclophane can take place in a consecutive manner and involves the intermediacy of a [2]rotaxane which has been isolated and characterized. c) A light‐ and electricity‐responsive pump D7 3+, which can recruit a CBPQT 4+ cyclophane onto a collecting chain using electricity and release it into bulk solution upon light irradiation. d) A polyrotaxane synthesizer, D8 6+, which can collect up to 10 CBPQT4+ cyclophanes onto its PEG chain depending on the number of redox cycles performed.
As a testament to the robustness and durability of the Mark II pump undergoing multiple redox cycles, a polyrotaxane synthesizer has been constructed [232] by grafting pumping cassettes onto both ends of a polyethylene glycol (PEG) chain to afford a dumbbell. Many of the previous methods for the syntheses of polyrotaxanes suffer from a lack of control of the number of threaded rings. The use of molecular pumps and pumping cassettes allows delivery of an exact number of threaded rings according to the number of redox cycles performed on the dumbbell. When the synthesizer is operated either chemically or electrochemically, the CBPQT 4+ rings are transported (Figure 19d) onto the PEG chain in pairs from the two pumping cassettes, affording polyrotaxanes with two, four, six, eight, or ten rings, depending on the number (one, two, three, four, or five) of redox cycles performed. A recent synthesis [233] of daisy‐chain polymers has also benefited from the use of molecular pumps. The key design principle in this case is a self‐complementary monomer that comprises the original one‐stroke molecular pump (D3 3+) and a covalently attached CBPQT4+ ring. Up to 11 degrees of polymerization can be obtained for the daisy‐chain polymers following reduction and rapid oxidation. These two examples demonstrate the potential of AMPs in the fabrication of mechanically interlocked polymers with appealing structures and functions.
11. Molecular Electronics and Porous Materials
Billions of years of evolution have honed nature's extraordinary capability, not only for carrying out life‐sustaining nanoscopic work inside cells, but also in performing macroscopic work (e.g., muscles) by scaling up the mechanical motions of biomolecular machines. With a plethora of molecular switches and AMMs available, the stage is set for us, as well as others, to integrate [234] them into molecular electronics and porous materials that can be put to practical use. The fruitful marriage between MIMs and molecular electronics, in particular, has led [235] to a burgeoning of molecular devices and integrated circuits that include photoswitches and detectors, artificial molecular muscles, molecular mechanical biosensors, electrochromic devices, single‐molecule transistors, random‐access memories, and configurable logic circuits.
Considerable progress has been made [195a] in our own research by manipulating the self‐organization and electrochemical operation of CBPQT4+‐containing, bistable MIMs as a means of storing and processing information, as well as expressing motions on a macroscopic level in molecular devices. Our foray into this area was initiated by a fruitful collaboration at UCLA with Jim Heath, starting in 1999 with the production [236] (Figure 20e) of Langmuir–Blodgett (LB) monolayers of bistable [2]catenanes and [2]rotaxanes and the assembly of them onto crossbar architectures involving two‐dimensional tiled arrays of molecular switch tunnel junctions (MSTJs). Although a bistable [2]catenane was chosen [237] initially as the switching component, it soon became evident that it is much easier to introduce [238] amphiphilicity, a prerequisite for the use of the LB technique [239] in the fabrication of MSTJ crossbars, into bistable [2]rotaxanes. The idea was based on the observation [240] that CBPQT4+ prefers (Figure 20a) to encircle the TTF unit over the DN unit by a factor of 9 : 1 in the amphiphilic [2]rotaxane R17 4+ under ambient conditions (GSCC), whereas upon electrochemical oxidation, the ring moves to encircle the DN unit as a result of its strong repulsion by the TTF2+ dication. The MSCC, which turns out to be more conducting than the GSCC, can be accessed by applying a +2.0 V pulse before returning to near‐zero bias. Application of a −2.0 V potential reduces the TTF2+ dication to its neutral state (MSCC) and restores the GSCC. With the majority (if not all) of molecules synchronized in a particular state in each MSTJ, they can be addressed uniquely and separately in the crossbar architecture.
Figure 20.

Molecular electronic devices based on bistable MIMs. a) Structural formulas of the two translational isomers of an amphiphilic bistable [2]rotaxane in its ground state co‐conformation (GSCC) and metastable state co‐conformation (MSCC) and their equilibrium in MeCN at room temperature (left). Schematic representation (right) of the bistable potential energy surface with potential energy wells corresponding to the GSCC and MSCC (ΔG 0=1.6 kcal mol−1/ΔG ≠=16 kcal mol−1). b) Eyring plots and free energy barrier (ΔG ≠) in solution, on a self‐assembled monolayer, in a polymer gel of high viscosity, and in a molecular switch tunnel junction (MSTJ) device. c) Schematic illustrations showing the relaxation of bistable rotaxanes from their MSCC states (not illustrated) back to their GSCC states in different physical environments. The bistable rotaxanes can be housed in solution, on self‐assembled monolayers formed on a flat Au surface, or on metal nanoparticles, in polymer matrices, and finally in monolayers in MSTJs. d) Schematic representation of the light‐driven actuation of redox‐bistable [2]rotaxanes in graphitic monolayer devices. e) A MIM‐based dynamic random‐access memory circuit. Top left, high‐resolution scanning electron microscope image of the nanowire crossbar circuit. Bottom left, different states of a bistable [2]catenane (top) or a bistable [2]rotaxane (bottom). The GSCC is less conducting than the MSCC. Right, schematic illustration of the integration of MSTJs into a crossbar device. f) The switching operation of this memory device. Top, the pulse sequence used to operate the memory device. Bottom, storing ASCII characters to form the acronym CIT for California Institute of Technology, using binary numbers.
It transpires that amphiphilic [2]rotaxanes can also be introduced (Figure 20c) into highly viscous polymer electrolyte gels, [241] into liquid crystals, [242] as monolayers on gold surfaces, [243] or on metal nanoparticles, [244] as well as being assembled [245] in their hundreds or thousands in MSTJs between two wires (electrodes). Switching is universal in these materials. The activation barrier (ΔG ≠) is raised (Figure 20b) dramatically when the molecules are brought from solution (16 kcal mol−1) and introduced into soft materials all the way up to 21 kcal mol−1 in MSTJs. Temperature also has a profound influence (Figure 20b) on switching in MSTJs, indicating that their operation involves the relative motions of components in bistable MIMs, namely, switching is a molecularly based phenomenon.
Half a decade of unremitting research on optimizing the amphiphilic bistable [2]rotaxanes, from 2002 to 2007, produced integrated circuits based on crossbar‐containing monolayers of increasing complexity, starting with a 4×4 crossbar architecture, [245] followed by an 8×8 one, [246] and ultimately leading to a 400×400 one [247] with wires (electrodes) of only 16 nm in width. With around 100 bistable [2]rotaxane molecules trapped in each MSTJ of the 400×400 crossbar architecture, the 160 000 junctions constitute (Figure 20e) a 160‐kbit memory device patterned at a density of bits per square centimeter. This device is operated by applying different bias voltages (Figure 20f), writing with a +1.5 V pulse, reading at a bias of around 0 V, and erasing with a −1.5 V pulse. A device, based on bistable [2]rotaxane LB films using platinum instead of polysilicon as the bottom electrode, was developed [248] at Hewlett–Packard (HP). Switching, however, was physically based and led to the comment [249] that the HP devices would work even if their MSTJs were coated with butter!
When mounted onto a gold nanodisc array, the host–guest complex between CBPQT 4+ and MPTTF serves as a voltage‐activated molecular logic gate based on resonant surface‐enhanced Raman scattering output. [250] It is also possible to actuate molecular devices under photoirradiation by incorporating photosensitive components into bistable MIMs. [251] Besides serving as logic gates, an array of bistable MIMs can also telescope their molecular motions into forces on a macroscopic scale. By immobilizing trillions of redox‐switchable bistable [3]rotaxane (molecular muscles) on the surface of a thin‐gold‐coated microcantilever, it was shown [252] that the cantilever bends up and down under redox control. Bistable [2]rotaxanes can also be mounted onto electrodes [253] and mesoporous nanoparticles, [254] where they serve as nanovalves for the controlled release of cargos, for example, drugs.
One major limitation that crippled the further development of molecular electronics based on bistable MIMs is their lifetime: they last at most for hundreds of cycles before failure sets in, probably because of degradation of the MIMs. The susceptibility of these devices to structural damage prompted us to turn our attention to investigating more robust platforms in the guise of metal–organic frameworks [255] (MOFs), porous materials that are known to be highly robust on account of their structural rigidity that is reinforced by linkages between metal nodes and organic struts. Coupling the rigidity and periodicity of MOFs with the addressability and mobility of molecular switches and machines holds promise [256] for preparing new materials that are simultaneously both robust and dynamic. In 2009, we (J.F.S.), in collaboration with Omar Yaghi, introduced [257] the concept of “docking” in MOFs by incorporating BPP34C10, capable of binding MV 2+ in solution, within the organic struts of MOF‐1001. Remarkably, the receptor function of BPP34C10 is preserved in the rigid pore of the MOF, transferring a well‐known [41] molecular recognition process from solution to a surface. The success of this experiment, which brought the concept of robust dynamics to fruition, has led [258] (Figure 21a) to the preparation of a copper‐based MOF, MOF‐1011, in which a dicarboxylic‐acid‐containing BPP34C10 ring in a strut is mechanically interlocked with a CBPQT4+ ring to give a [2]catenane (C18 4+). The solid‐state structure of MOF‐1011 reveals that the 2D network is replete with ordered catenanes—one per copper unit, eight per unit cell, and 81 per 100 nm2 of surface—throughout the crystal. Subsequent extension of the dicarboxylic acid strut resulted [259] in MOFs with twofold interpenetrated 3D matrices or gridlike 2D sheets. Unfortunately, the relative movements of the rings in these [2]catenanes are arrested completely as a result of their highly ordered and densely packed environments. It seems that [π⋅⋅⋅π] stacking interactions between the donor–acceptor [2]catenanes play an important role, in addition to the formation of metal–ligand joints, in the internal organization of the networks during their assembly. [260] In a conceptually similar manner, docking of CBPQT4+ has also been achieved [261] in a metal‐organic cage.
Figure 21.

Integration of molecular switches and pumps in metal–organic frameworks (MOFs). a) Schematic diagram of the incorporation of the donor/‐acceptor [2]catenane C18 4+ into the struts of a copper‐based MOF, forming MOF‐1011. b) Schematic diagram of the immobilization of a trisradical tricationic [2]rotaxane in the porous channels of a zirconium‐based MOF following post‐synthetic modification. c) Schematic diagram showing the grafting of Mark II molecular pumps onto the surface of a zirconium‐based 2D MOF and their application in recruiting CBPQT 4+ rings from solution and concentrating them on the surfaces of the MOF.
In a collaboration involving Wasielewski, Snurr, Hupp, and Farha at Northwestern University (NU), we immobilized [262] a trisradical tricationic [2]rotaxane (Figure 21b) post‐synthetically in the porous channels of the zirconium‐based MOF NU‐1000 by solvent‐assisted ligand incorporation (SALI). A semirotaxane, R18 3(⋅+), which is formed firstly in solution on account of radical‐pairing interactions between the CBPQT2(⋅+) ring and the V(⋅+)‐containing dumbbell, binds to coordination sites in the MOF through the carboxylic acid group. Once fixed as side chains within the pores, the [2]rotaxane becomes kinetically trapped, with the MOF serving as the second stopper. By employing a similar post‐synthetic functionalization procedure, a bistable [2]catenane was also anchored [263] inside the pores of NU‐1000. The reversible redox‐switching ability of the bistable [2]catenane is retained in the MOF, thus enabling robust dynamics within a porous material.
AMPs, while highly unidirectional with respect to their motion within a single molecule, operate in solution where they are randomly oriented, causing zero net movement of a population of these machines. By integrating AMPs within highly ordered MOFs, however, they can be organized periodically and precisely in 3D space, allowing [264] their otherwise incoherent motions to be rectified. We, in collaboration with Farha at Northwestern University and Astumian at the University of Maine, have happened upon [265] (Figure 21c) a new type of adsorption phenomenon, namely, mechanisorption, by tethering an AMP, D9 3+, onto the surface of a zirconium‐based 2D MOF. The pump contains a Mark II‐type pumping cassette, a PEG collecting chain, and a carboxylic acid end group that can be attached to zirconium clusters through post‐synthetic modification of the MOF. By carrying out repetitive redox chemical steps, the pumps are able to recruit CBPQT4+ rings from bulk solution and concentrate them on surfaces, thus creating a large chemical potential commensurate with storing energy in a non‐equilibrium state. The rings can be released back into solution upon dissociation of the pumps from the MOF in the presence of acid. By driving chemical systems away from equilibrium, mechanisorption offers a transformative approach for controlling chemistry at surfaces and interfaces.
12. Catalysis
Nanoconfinement [28g] is a unifying strategy adopted by enzymes to accumulate substrates and to stabilize transition states or reaction intermediates in segregated nano‐environments during catalysis. Although tremendous success has been achieved [266] in the construction of nanoconfined catalysts for promoting challenging chemical reactions, it was not until recently that we became equipped with the ability to leverage CBPQT 4+ and its MIMs as a platform for catalysis.
One example lies [267] in the area of photocatalysis (Figure 22a). It is well‐known that a π‐electron‐rich molecule, such as a DN derivative, can experience [268] charge transfer (CT) interactions with the π‐electron‐deficient viologen units of a CBPQT4+ ring when nanoconfined in its cavity. Such complexation narrows the HOMO–LUMO energy gap between the two components, causing their absorption band to become red‐shifted into the visible region of the spectrum. Photoexcitation with visible light should, in principle, induce [269] single‐electron transfer within the complex and generate highly reactive radical ion‐pairs, composed of a V⋅+ and a DN⋅+, that can participate in subsequent reactions. Nevertheless, the association between DN derivatives and CBPQT 4+ is generally weak in solution, particularly at low concentrations, rendering them unable to undergo efficient visible‐light harvesting. By forcing the DN unit to be nanoconfined in the cavity of CBPQT4+ in a [2]catenane, however, stoichiometric CT complexation can be achieved as a result of mechanical bonding. We, in collaboration with Stupp, have shown [267] how it is possible to use the photoinduced electron transfer (PET) ability of a well‐known [2]catenane, C1 4+, to promote 1) the catalytic generation (Figure 22a) of H2 gas in the presence of platinum nanoparticles as a co‐catalyst and tetrakis(methylenephosphonic acid) (EDTP) as a sacrificial electron donor and 2) the catalytic aerobic oxidation (Figure 22b) of l‐methionine. This research highlights the ability of mechanical‐bonding‐induced nanoconfinement to reinforce CT interactions and enable otherwise challenging PET processes in photocatalysis. It is worth noting that CBPQT 4+ has also been used [270] as a catalytic electron‐transfer mediator for the reduction of benzoquinone on account of its propensity for forming inclusion complexes with aromatic substrates.
Figure 22.
a) Schematic representation of the visible‐light‐induced electron‐transfer process in the donor–acceptor [2]catenane C1 4+ in the catalytic generation of H2 gas in the presence of platinum nanoparticles as co‐catalysts and EDTP as a sacrificial reductant. b) Schematic representation of the visible‐light‐induced electron‐transfer process in C1 4+ in the catalytic oxidation of organic sulfides in the presence of O2. c) Schematic representation of the mechanism for an electron‐catalyzed molecular recognition process. The direct (gray dashed arrow) formation of a [D9⊂CBPQT] +3(⋅+) trisradical complex from CBPQT 2⋅+ and D9 +(⋅+) is kinetically forbidden. The catalytic (black solid arrows) complexation comprises four steps, including one initiating step and three propagating steps. Step 1—the injection of an electron. Step 2—a single‐electron reduction of either one of the V⋅+ units in CBPQT 2⋅+ or the V⋅+ unit in D9 +(⋅+). Step 3—the rapid formation of a [D9⊂CBPQT] +2(⋅+) bisradical complex, favored by a decrease in the Coulombic repulsion between the host and guest molecules. Step 4—the oxidation of the [D9⊂CBPQT] +2(⋅+) bisradical complex to the trisradical complex, while releasing an electron to close the catalytic cycle. Steps 1/2/3/4 are portrayed inside grey full moons.
Another example exploits [271] the catalytic ability of an unconventional, while important, type of entity in organic chemistry, namely the electron. Although the possibility of using an electron to catalyze covalent bond formation is well‐known, [272] its ability to catalyze molecular recognition and supramolecular assembly had remained elusive. By using a one‐stroke molecular pump, D10 3+, with 2,6‐DMPy+ as the Coulombic barrier, it was found [271] that molecular recognition between CBPQT 2(⋅+) and the V⋅+ unit on the dumbbell, which is kinetically forbidden under ambient conditions, is accelerated (Figure 22c) substantially upon the addition of catalytic amounts of a chemical electron source. The transient reduction (Figure 22c, step 2) of one of the viologen units on CBPQT 2(⋅+) or on the dumbbell by the electron leads to the rapid formation (Figure 22c, step 3) of a [D10⊂CBPQT]+2(⋅+) bisradical tricationic complex, on account of decreased Coulombic repulsion between the host and guest molecules. This complex can be oxidized subsequently to the trisradical tetracationic complex, [D10⊂CBPQT]+3(⋅+), releasing (Figure 22c, step 4) an electron that can participate in the next molecular recognition process. There are high hopes that this highly collaborative research [271] between Astumian, Goddard, Wasielewski, and ourselves will aid and abet the design of other molecular recognition and supramolecular assembly processes that can be catalyzed by entities as simple as the electron.
13. Conclusions
From the design of a multipurpose viologen‐containing cyclophane to its applications in the synthesis of mechanically interlocked molecules, artificial molecular machines, molecular electronics, and porous materials, we, as well as others, have come a long way in utilizing the little blue box in a wide variety of fields in chemistry and materials science. A number of major advances are summarized chronologically in Figure 23. In retrospect, the year 2010 marks the renaissance of this 35‐year‐long scientific journey, starting in 1988. When Ali Trabolsi [166] opened [31] the door on the radical chemistry of the little blue box, he gave it a new lease of life from which it has so far not looked back. While the fully oxidized form of the little blue box has resulted in numerous donor–acceptor catenanes and rotaxanes, molecular shuttles and switches, as well as molecular electronics, it is its radical cationic form that has unlocked the operation of many artificial molecular machines, which has been a central topic of our research during the past decade. With an increasingly deep understanding of the recognition between cyclobis(paraquat‐p‐phenylene) and viologen radical cations, our repertoire in using the radical chemistry of little blue box has also been expanded into areas other than host–guest chemistry, such as non‐equilibrium chemistry, catalysis, and functional materials. The modularity of the little blue box has also inspired the design [273] of other extended pyridinium cyclophanes and the discovery[ 174a , 175 ] of new host–guest chemistry involving viologen radical cations. Many of these accomplishments in research are the culmination of extensive collaborations with scientific experts all around the world. Going forward, we believe that the uncovering of new phenomena and principles in science using the little blue box will require unfettered imagination, relentless efforts, and above all, intimate collaborations with members of the scientific community.
Figure 23.

A chronological summary of the major advances associated with the little blue box.
There are no firsts in science. We are indebted to Professor Siegfried Hünig, who forged the path to the modern frontier of viologen chemistry on which we tread today. Particularly, we recognize the fundamental breakthroughs he made in the synthesis of viologen‐containing cyclophanes, as well as the redox and π‐dimerization (pimerization) chemistry associated with viologen‐containing compounds. Hünig was a very kind, generous and supportive person who cared deeply about the well‐being of people around him. In fact, after our first report of cyclobis(paraquat‐p‐phenylene) in 1988, one (J.F.S.) of the authors in this Review became a close friend of Siegfried Hünig and had the privilege to be hosted, along with his wife, in style in Würzburg by the entire Hünig family on the occasion of his 80th birthday in 2001. Hünig's legacy will live on in the generations of scientists he trained and all those who have benefited from his research and mentorship. It is truly a great pleasure, as well as honor, to be able to dedicate this Review to a legendary figure in the field of organic chemistry.
Conflict of interest
The authors declare no conflict of interest.
14.
Biographical Information
Xiao‐Yang Chen was born in Guangzhou, China. After obtaining his BS from Fudan University in 2014, he moved to the US to pursue his PhD at Princeton University under the supervision of Professor Erik Sorensen. In 2019, he joined the laboratory of Professor Sir Fraser Stoddart at Northwestern University as a postdoctoral fellow. His research is focused upon two areas: 1) the use of γ‐cyclodextrin metal–organic frameworks as robust nanoreactors for performing selective chemical reactions, and 2) the design of new radical recognition motifs for the construction of mechanically interlocked molecules and artificial molecular machines.

Biographical Information
Hongliang Chen received his PhD in 2016 from the College of Chemistry and Molecular Engineering, Peking University, under the guidance of Professor Xuefeng Guo. From 2016 to 2018, he worked as a research scientist in the Core R&D Department in Dow Chemical Company. From there, he moved to Northwestern University in the United States and worked as a postdoctoral research fellow in Professor Sir Fraser Stoddart's group from 2018 to 2021. He joined Zhejiang University as an assistant professor under ZJU 100‐Talent Program in June 2021. His research interests are focused on supramolecular electronics.

Biographical Information
Sir Fraser Stoddart received all (BSc, PhD, DSc) his degrees from the University of Edinburgh, UK. He currently holds a Board of Trustees Professorship in the Department of Chemistry at Northwestern University, United States. His research focuses on the chemistry and molecular nanotechnology associated with mechanically interlocked molecules. He received the Nobel Prize in Chemistry in 2016 for his contribution to the design and synthesis of molecular machines.

Acknowledgements
We thank Frank Würthner (University of Würzburg) for encouraging us to write this Review. The writing—for multiple reasons—took much longer than we anticipated in the beginning. We are also grateful to Frank for his reading of the manuscript and his comments that led to an improvement in the quality of the Review. Financial support from Northwestern University and the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (Grant No. SN‐ZJU‐SIAS‐006) is gratefully acknowledged.
Chen X.-Y., Chen H., Fraser Stoddart J., Angew. Chem. Int. Ed. 2023, 62, e202211387; Angew. Chem. 2023, 135, e202211387.
Contributor Information
Xiao‐Yang Chen, Email: xc3@northwestern.edu.
Hongliang Chen, Email: hongliang.chen@zju.edu.cn.
J. Fraser Stoddart, Email: stoddart@northwestern.edu.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1. Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P., Molecular Biology of the Cell , 4th ed., Garland Science, New York, 2002. [Google Scholar]
- 2. Berdis A. J., Chem. Rev. 2009, 109, 2862–2879. [DOI] [PubMed] [Google Scholar]
- 3. Ramakrishnan V., Cell 2002, 108, 557–572. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Lombardi A., Nastri F., Pavone V., Chem. Rev. 2001, 101, 3165–3190; [DOI] [PubMed] [Google Scholar]
- 4b. Kaksonen M., Roux A., Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [DOI] [PubMed] [Google Scholar]
- 5. Van Oss C. J., Mol. Immunol. 1995, 32, 199–211. [DOI] [PubMed] [Google Scholar]
- 6. Lancaster L., Abdallah W., Banta S., Wheeldon I., Chem. Soc. Rev. 2018, 47, 5177–5186. [DOI] [PubMed] [Google Scholar]
- 7. Steed J. W., Atwood J. L., Supramolecular Chemistry , 3rd ed., Wiley, Hoboken, 2022. [Google Scholar]
- 8.
- 8a. Pedersen C. J., J. Am. Chem. Soc. 1967, 89, 2495–2496; [Google Scholar]
- 8b. Pedersen C. J., Frensdorff H. K., Angew. Chem. Int. Ed. Engl. 1972, 11, 16–25; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1972, 84, 16–26. [Google Scholar]
- 9. Dietrich B., Lehn J.-M., Sauvage J.-P., Tetrahedron Lett. 1969, 10, 2889–2892. [Google Scholar]
- 10.
- 10a. Cram D. J., Kaneda T., Helgeson R. C., Lein G. M., J. Am. Chem. Soc. 1979, 101, 6752–6754; [Google Scholar]
- 10b. Cram D. J., Kaneda T., Lein G. M., Helgeson R. C., J. Chem. Soc. Chem. Commun. 1979, 948b–950; [Google Scholar]
- 10c. Cram D. J., Angew. Chem. Int. Ed. Engl. 1986, 25, 1039–1057; [Google Scholar]; Angew. Chem. 1986, 98, 1041–1060. [Google Scholar]
- 11.
- 11a. Cram D. J., Karbach S., Kim Y. H., Baczynskyj L., Kallemeyn G. W., J. Am. Chem. Soc. 1985, 107, 2575–2576; [Google Scholar]
- 11b. Cram D. J., Karbach S., Kim Y. H., Baczynskyj L., Marti K., Sampson R. M., Kalleymeyn G. W., J. Am. Chem. Soc. 1988, 110, 2554–2560; [Google Scholar]
- 11c. Sherman J. C., Cram D. J., J. Am. Chem. Soc. 1989, 111, 4527–4528. [Google Scholar]
- 12.
- 12a. Tanner M. E., Knobler C. B., Cram D. J., J. Am. Chem. Soc. 1990, 112, 1659–1660; [Google Scholar]
- 12b. Cram D. J., Tanner M. E., Thomas R., Angew. Chem. Int. Ed. Engl. 1991, 30, 1024–1027; [Google Scholar]; Angew. Chem. 1991, 103, 1048–1051; [Google Scholar]
- 12c. Cram D. J., Tanner M. E., Knobler C. B., J. Am. Chem. Soc. 1991, 113, 7717–7727. [Google Scholar]
- 13. Pedersen C. J., Angew. Chem. Int. Ed. Engl. 1988, 27, 1021–1027; [Google Scholar]; Angew. Chem. 1988, 100, 1053–1059. [Google Scholar]
- 14. Lehn J.-M., Angew. Chem. Int. Ed. Engl. 1988, 27, 89–112; [Google Scholar]; Angew. Chem. 1988, 100, 91–116. [Google Scholar]
- 15. Cram D. J., Angew. Chem. Int. Ed. Engl. 1988, 27, 1009–1020; [Google Scholar]; Angew. Chem. 1988, 100, 1041–1052. [Google Scholar]
- 16.
- 16a. Amouri H., Desmarets C., Moussa J., Chem. Rev. 2012, 112, 2015–2041; [DOI] [PubMed] [Google Scholar]
- 16b. Liu W., Stoddart J. F., Chem 2021, 7, 919–947. [Google Scholar]
- 17.
- 17a. Szejtli J., Chem. Rev. 1998, 98, 1743–1754; [DOI] [PubMed] [Google Scholar]
- 17b. Stoddart J. F., Carbohydr. Res. 1989, 192, xii–xv; [Google Scholar]
- 17c. Nepogodiev S. A., Stoddart J. F., Chem. Rev. 1998, 98, 1959–1976; [DOI] [PubMed] [Google Scholar]
- 17d. Breslow R., Dong S. D., Chem. Rev. 1998, 98, 1997–2012; [DOI] [PubMed] [Google Scholar]
- 17e. Takahashi K., Chem. Rev. 1998, 98, 2013–2034; [DOI] [PubMed] [Google Scholar]
- 17f. Harada A., Takashima Y., Yamaguchi H., Chem. Soc. Rev. 2009, 38, 875–882; [DOI] [PubMed] [Google Scholar]
- 17g. Crini G., Chem. Rev. 2014, 114, 10940–10975. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Gutsche C. D., Acc. Chem. Res. 1983, 16, 161–170; [Google Scholar]
- 18b. Gutsche C. D., Bauer L. J., J. Am. Chem. Soc. 1985, 107, 6052–6059; [Google Scholar]
- 18c. Gutsche C. D., Rogers J. S., Stewart D., See K.-A., Pure Appl. Chem. 1990, 62, 485–491; [Google Scholar]
- 18d. van Dienst E., Bakker W. I. I., Engbersen J. F. J., Verboom W., Reinhoudt D. N., Pure Appl. Chem. 1993, 65, 387–392; [Google Scholar]
- 18e. Ikeda A., Shinkai S., Chem. Rev. 1997, 97, 1713–1734; [DOI] [PubMed] [Google Scholar]
- 18f. Stewart D. R., Gutsche C. D., J. Am. Chem. Soc. 1999, 121, 4136–4146; [Google Scholar]
- 18g. Prins L. J., De Jong F., Timmerman P., Reinhoudt D. N., Nature 2000, 408, 181–184; [DOI] [PubMed] [Google Scholar]
- 18h. Molenveld P., Engbersen J. F. J., Reinhoudt D. N., Chem. Soc. Rev. 2000, 29, 75–86; [Google Scholar]
- 18i. Matthews S. E., Beer P. D., Supramol. Chem. 2005, 17, 411–435; [Google Scholar]
- 18j. Guo D.-S., Liu Y., Chem. Soc. Rev. 2012, 41, 5907–5921; [DOI] [PubMed] [Google Scholar]
- 18k. Kumar R., Sharma A., Singh H., Suating P., Kim H. S., Sunwoo K., Shim I., Gibb B. C., Kim J. S., Chem. Rev. 2019, 119, 9657–9721; [DOI] [PubMed] [Google Scholar]
- 18l. Pan Y.-C., Hu X.-Y., Guo D.-S., Angew. Chem. Int. Ed. 2021, 60, 2768–2794; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 2800–2828; [Google Scholar]
- 18m. Crowley P. B., Acc. Chem. Res. 2022, 55, 2019–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Freeman W. A., Mock W. L., Shih N. Y., J. Am. Chem. Soc. 1981, 103, 7367–7368; [Google Scholar]
- 19b. Lagona J., Mukhapadhyay P., Chakrabarti S., Isaacs L., Angew. Chem. Int. Ed. 2005, 44, 4844–4870; [DOI] [PubMed] [Google Scholar]
- 19c. Kim K., Selvapalam N., Ko Y. H., Park K. M., Kim D., Kim J., Chem. Soc. Rev. 2007, 36, 267–279; [DOI] [PubMed] [Google Scholar]
- 19d. Isaacs L., Chem. Commun. 2009, 619–629; [DOI] [PubMed] [Google Scholar]
- 19e. Assaf K. I., Nau W. M., Chem. Soc. Rev. 2015, 44, 394–418; [DOI] [PubMed] [Google Scholar]
- 19f. Barrow S. J., Kasera S., Rowland M. J., del Barrio J., Scherman O. A., Chem. Rev. 2015, 115, 12320–12406; [DOI] [PubMed] [Google Scholar]
- 19g. Pazos E., Novo P., Peinador C., Kaifer A. E., García M. D., Angew. Chem. Int. Ed. 2019, 58, 403–416; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 409–422; [Google Scholar]
- 19h. Liu Y.-H., Zhang Y.-M., Yu H.-J., Liu Y., Angew. Chem. Int. Ed. 2021, 60, 3870–3880; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 3914–3924; [Google Scholar]
- 19i. Nie H., Wei Z., Ni X.-L., Liu Y., Chem. Rev. 2022, 122, 9032–9077. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Ogoshi T., Kanai S., Fujinami S., Yamagishi T.-A., Nakamoto Y., J. Am. Chem. Soc. 2008, 130, 5022–5023; [DOI] [PubMed] [Google Scholar]
- 20b. Xue M., Yang Y., Chi X., Zhang Z., Huang F., Acc. Chem. Res. 2012, 45, 1294–1308; [DOI] [PubMed] [Google Scholar]
- 20c. Strutt N. L., Zhang H., Schneebeli S. T., Stoddart J. F., Acc. Chem. Res. 2014, 47, 2631–2642; [DOI] [PubMed] [Google Scholar]
- 20d. Ogoshi T., Yamagishi T.-A., Nakamoto Y., Chem. Rev. 2016, 116, 7937–8002; [DOI] [PubMed] [Google Scholar]
- 20e. Zhang H., Liu Z., Zhao Y., Chem. Soc. Rev. 2018, 47, 5491–5528; [DOI] [PubMed] [Google Scholar]
- 20f. Song N., Kakuta T., Yamagishi T.-A., Yang Y.-W., Ogoshi T., Chem 2018, 4, 2029–2053; [Google Scholar]
- 20g. Li Q., Zhu H., Huang F., Trends Chem. 2020, 2, 850–864; [Google Scholar]
- 20h. Li M.-H., Lou X.-Y., Yang Y.-W., Chem. Commun. 2021, 57, 13429–13447; [DOI] [PubMed] [Google Scholar]
- 20i. Li Z., Yang Y.-W., Acc. Mater. Res. 2021, 2, 292–305; [Google Scholar]
- 20j. Chen Y.-Y., Jiang X.-M., Gong G.-F., Yao H., Zhang Y.-M., Wei T.-B., Lin Q., Chem. Commun. 2021, 57, 284–301; [DOI] [PubMed] [Google Scholar]
- 20k. Wang K., Jordan J. H., Velmurugan K., Tian X., Zuo M., Hu X.-Y., Wang L., Angew. Chem. Int. Ed. 2021, 60, 9205–9214; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 9289–9298. [Google Scholar]
- 21.
- 21a. Diederich F., Angew. Chem. Int. Ed. Engl. 1988, 27, 362–386; [Google Scholar]; Angew. Chem. 1988, 100, 372–396; [Google Scholar]
- 21b. Seel C., Vögtle F., Angew. Chem. Int. Ed. Engl. 1992, 31, 528–549; [Google Scholar]; Angew. Chem. 1992, 104, 542–563; [Google Scholar]
- 21c. Liu Z., Nalluri S. K. M., Stoddart J. F., Chem. Soc. Rev. 2017, 46, 2459–2478. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Jasti R., Bhattacharjee J., Neaton J. B., Bertozzi C. R., J. Am. Chem. Soc. 2008, 130, 17646–17647; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Takaba H., Omachi H., Yamamoto Y., Bouffard J., Itami K., Angew. Chem. Int. Ed. 2009, 48, 6112–6116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6228–6232; [Google Scholar]
- 22c. Yamago S., Watanabe Y., Iwamoto T., Angew. Chem. Int. Ed. 2010, 49, 757–759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 769–771; [Google Scholar]
- 22d. Bols P. S., Anderson H. L., Acc. Chem. Res. 2018, 51, 2083–2092; [DOI] [PubMed] [Google Scholar]
- 22e. Leonhardt E. J., Jasti R., Nat. Chem. Rev. 2019, 3, 672–686; [Google Scholar]
- 22f. Li Y., Kono H., Maekawa T., Segawa Y., Yagi A., Itami K., Acc. Mater. Res. 2021, 2, 681–691; [Google Scholar]
- 22g. Guo Q.-H., Qiu Y., Wang M.-X., Stoddart J. F., Nat. Chem. 2021, 13, 402–419; [DOI] [PubMed] [Google Scholar]
- 22h. Segawa Y., Watanabe T., Yamanoue K., Kuwayama M., Watanabe K., Pirillo J., Hijikata Y., Itami K., Nat. Synth. 2022, 1, 535–541. [Google Scholar]
- 23.
- 23a. Gong H.-Y., Rambo B. M., Karnas E., Lynch V. M., Sessler J. L., Nat. Chem. 2010, 2, 406–409; [DOI] [PubMed] [Google Scholar]
- 23b. Rambo B. M., Gong H.-Y., Oh M., Sessler J. L., Acc. Chem. Res. 2012, 45, 1390–1401; [DOI] [PubMed] [Google Scholar]
- 23c. Dale E. J., Vermeulen N. A., Juríček M., Barnes J. C., Young R. M., Wasielewski M. R., Stoddart J. F., Acc. Chem. Res. 2016, 49, 262–273. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Fujita M., Oguro D., Miyazawa M., Oka H., Yamaguchi K., Ogura K., Nature 1995, 378, 469–471; [Google Scholar]
- 24b. Olenyuk B., Whiteford J. A., Fechtenkötter A., Stang P. J., Nature 1999, 398, 796–799; [DOI] [PubMed] [Google Scholar]
- 24c. Fujita M., Tominaga M., Hori A., Therrien B., Acc. Chem. Res. 2005, 38, 369–378; [DOI] [PubMed] [Google Scholar]
- 24d. Yoshizawa M., Klosterman J. K., Fujita M., Angew. Chem. Int. Ed. 2009, 48, 3418–3438; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3470–3490; [Google Scholar]
- 24e. Tozawa T., Jones J. T. A., Swamy S. I., Jiang S., Adams D. J., Shakespeare S., Clowes R., Bradshaw D., Hasell T., Chong S. Y., Tang C., Thompson S., Parker J., Trewin A., Bacsa J., Slawin A. M. Z., Steiner A., Cooper A. I., Nat. Mater. 2009, 8, 973–978; [DOI] [PubMed] [Google Scholar]
- 24f. Holst J. R., Trewin A., Cooper A. I., Nat. Chem. 2010, 2, 915–920; [DOI] [PubMed] [Google Scholar]
- 24g. Chakrabarty R., Mukherjee P. S., Stang P. J., Chem. Rev. 2011, 111, 6810–6918; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24h. Liu Y., Hu C., Comotti A., Ward M. D., Science 2011, 333, 436–440; [DOI] [PubMed] [Google Scholar]
- 24i. Zhang G., Mastalerz M., Chem. Soc. Rev. 2014, 43, 1934–1947; [DOI] [PubMed] [Google Scholar]
- 24j. Jin Y., Wang Q., Taynton P., Zhang W., Acc. Chem. Res. 2014, 47, 1575–1586; [DOI] [PubMed] [Google Scholar]
- 24k. Cook T. R., Stang P. J., Chem. Rev. 2015, 115, 7001–7045; [DOI] [PubMed] [Google Scholar]
- 24l. Hasell T., Cooper A. I., Nat. Rev. Mater. 2016, 1, 16053; [Google Scholar]
- 24m. Zhang D., Ronson T. K., Nitschke J. R., Acc. Chem. Res. 2018, 51, 2423–2436; [DOI] [PubMed] [Google Scholar]
- 24n. Mastalerz M., Acc. Chem. Res. 2018, 51, 2411–2422; [DOI] [PubMed] [Google Scholar]
- 24o. Mukhopadhyay R. D., Kim Y., Koo J., Kim K., Acc. Chem. Res. 2018, 51, 2730–2738; [DOI] [PubMed] [Google Scholar]
- 24p. Sepehrpour H., Fu W., Sun Y., Stang P. J., J. Am. Chem. Soc. 2019, 141, 14005–14020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24q. Percástegui E. G., Ronson T. K., Nitschke J. R., Chem. Rev. 2020, 120, 13480–13544; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24r. Liu W., Bobbala S., Stern C. L., Hornick J. E., Liu Y., Enciso A. E., Scott E. A., Stoddart J. F., J. Am. Chem. Soc. 2020, 142, 3165–3173; [DOI] [PubMed] [Google Scholar]
- 24s. Liu W., Tan Y., Jones L. O., Song B., Guo Q.-H., Zhang L., Qiu Y., Feng Y., Chen X.-Y., Schatz G. C., Stoddart J. F., J. Am. Chem. Soc. 2021, 143, 0; [DOI] [PubMed] [Google Scholar]
- 24t. McTernan C. T., Davies J. A., Nitschke J. R., Chem. Rev. 2022, 122, 10393–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Kang J., J. Rebek, Jr. , Nature 1996, 382, 239–241; [DOI] [PubMed] [Google Scholar]
- 25b. Kang J., J. Rebek, Jr. , Nature 1997, 385, 50–52; [DOI] [PubMed] [Google Scholar]
- 25c. MacGillivray L. R., Atwood J. L., Nature 1997, 389, 469–472; [Google Scholar]
- 25d. Dalgarno S. J., Power N. P., Atwood J. L., Coord. Chem. Rev. 2008, 252, 825–841; [Google Scholar]
- 25e. Laughrey Z., Gibb B. C., Chem. Soc. Rev. 2011, 40, 363–386; [DOI] [PubMed] [Google Scholar]
- 25f. Dumele O., Trapp N., Diederich F., Angew. Chem. Int. Ed. 2015, 54, 12339–12344; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12516–12521; [Google Scholar]
- 25g. Jordan J. H., Gibb B. C., Chem. Soc. Rev. 2015, 44, 547–585; [DOI] [PubMed] [Google Scholar]
- 25h. Yoshizawa M., Catti L., Acc. Chem. Res. 2019, 52, 2392–2404; [DOI] [PubMed] [Google Scholar]
- 25i. Yang L.-P., Wang X., Yao H., Jiang W., Acc. Chem. Res. 2020, 53, 198–208; [DOI] [PubMed] [Google Scholar]
- 25j. Pullen S., Tessarolo J., Clever G. H., Chem. Sci. 2021, 12, 7269–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Hof F., Craig S. L., Nuckolls C., J. Rebek, Jr. , Angew. Chem. Int. Ed. 2002, 41, 1488–1508; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1556–1578; [Google Scholar]
- 26b. Persch E., Dumele O., Diederich F., Angew. Chem. Int. Ed. 2015, 54, 3290–3327; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3341–3382; [Google Scholar]
- 26c. Busschaert N., Caltagirone C., Van Rossom W., Gale P. A., Chem. Rev. 2015, 115, 8038–8155; [DOI] [PubMed] [Google Scholar]
- 26d. Gropp C., Quigley B. L., Diederich F., J. Am. Chem. Soc. 2018, 140, 2705–2717; [DOI] [PubMed] [Google Scholar]
- 26e. Rizzuto F. J., von Krbek L. K. S., Nitschke J. R., Nat. Chem. Rev. 2019, 3, 204–222; [Google Scholar]
- 26f. Wang D.-X., Wang M.-X., Acc. Chem. Res. 2020, 53, 1364–1380; [DOI] [PubMed] [Google Scholar]
- 26g. Dong J., Davis A. P., Angew. Chem. Int. Ed. 2021, 60, 8035–8048; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 8113–8126; [Google Scholar]
- 26h. Liu W., Das P. J., Colquhoun H. M., Stoddart J. F., CCS Chem. 2022, 4, 755–784. [Google Scholar]
- 27. Galan A., Ballester P., Chem. Soc. Rev. 2016, 45, 1720–1737. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Ramamurthy V., Parthasarathy A., Isr. J. Chem. 2011, 51, 817–829; [Google Scholar]
- 28b. Leenders S. H. A. M., Gramage-Doria R., de Bruin B., Reek J. N. H., Chem. Soc. Rev. 2015, 44, 433–448; [DOI] [PubMed] [Google Scholar]
- 28c. Zhang Q., Catti L., Tiefenbacher K., Acc. Chem. Res. 2018, 51, 2107–2114; [DOI] [PubMed] [Google Scholar]
- 28d. Hong C. M., Bergman R. G., Raymond K. N., Toste F. D., Acc. Chem. Res. 2018, 51, 2447–2455; [DOI] [PubMed] [Google Scholar]
- 28e. Yu Y., J. Rebek, Jr. , Acc. Chem. Res. 2018, 51, 3031–3040; [DOI] [PubMed] [Google Scholar]
- 28f. Yu Y., Yang J.-M., J. Rebek, Jr. , Chem 2020, 6, 1265–1274; [Google Scholar]
- 28g. Grommet A. B., Feller M., Klajn R., Nat. Nanotechnol. 2020, 15, 256–271; [DOI] [PubMed] [Google Scholar]
- 28h. Morimoto M., Bierschenk S. M., Xia K. T., Bergman R. G., Raymond K. N., Toste F. D., Nat. Catal. 2020, 3, 969–984; [Google Scholar]
- 28i. Wang K., Jordan J. H., Hu X.-Y., Wang L., Angew. Chem. Int. Ed. 2020, 59, 13712–13721; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 13816–13825; [Google Scholar]
- 28j. Jiao Y., Chen X.-Y., Stoddart J. F., Chem 2022, 8, 414–438. [Google Scholar]
- 29. Odell B., Reddington M. V., Slawin A. M. Z., Spencer N., Stoddart J. F., Williams D. J., Angew. Chem. Int. Ed. Engl. 1988, 27, 1547–1550; [Google Scholar]; Angew. Chem. 1988, 100, 1605–1608. [Google Scholar]
- 30. Ashton P. R., Odell B., Reddington M. V., Slawin A. M. Z., Stoddart J. F., Williams D. J., Angew. Chem. Int. Ed. Engl. 1988, 27, 1550–1553; [Google Scholar]; Angew. Chem. 1988, 100, 1608–1611. [Google Scholar]
- 31. Trabolsi A., Khashab N., Fahrenbach A. C., Friedman D. C., Colvin M. T., Cotí K. K., Benítez D., Tkatchouk E., Olsen J.-C., Belowich M. E., Carmielli R., Khatib H. A., W. A. Goddard III , Wasielewski M. R., Stoddart J. F., Nat. Chem. 2010, 2, 42–49. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Bruns C. J., Stoddart J. F., The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, Hoboken, 2016; [Google Scholar]
- 32b. Stoddart J. F., Angew. Chem. Int. Ed. 2017, 56, 11094–11125; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11244–11277. [Google Scholar]
- 33. Reissig H.-U., Angew. Chem. Int. Ed. 2021, 60, 9180–9191; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 9264–9275. [Google Scholar]
- 34. Geuder W., Hünig S., Suchy A., Angew. Chem. Int. Ed. Engl. 1983, 22, 489–490; [Google Scholar]; Angew. Chem. 1983, 95, 501–502. [Google Scholar]
- 35.
- 35a. Hünig S., Pure Appl. Chem. 1967, 15, 109–122; [Google Scholar]
- 35b. Horner M., Hünig S., Angew. Chem. Int. Ed. Engl. 1977, 16, 410–411; [Google Scholar]; Angew. Chem. 1977, 89, 424–425. [Google Scholar]
- 36.
- 36a. Hünig S., Kiessel M., J. Prakt. Chem. 1958, 5, 224–232; [Google Scholar]
- 36b. Hünig S., Kiessel M., Chem. Ber. 1958, 91, 380–392. [Google Scholar]
- 37. Deuchert K., Hünig S., Angew. Chem. Int. Ed. Engl. 1978, 17, 875–886; [Google Scholar]; Angew. Chem. 1978, 90, 927–938. [Google Scholar]
- 38.
- 38a. Hünig S., Angew. Chem. Int. Ed. Engl. 1964, 3, 548–560; [Google Scholar]; Angew. Chem. 1964, 76, 400–412; [Google Scholar]
- 38b. Hünig S., Müller H. R., Thier W., Angew. Chem. Int. Ed. Engl. 1965, 4, 271–280; [Google Scholar]; Angew. Chem. 1965, 77, 368–377. [Google Scholar]
- 39. Hünig S., Berneth H., Organic Chemistry, Springer Berlin Heidelberg, Berlin, 1980, pp. 1–44. [Google Scholar]
- 40. Allwood B. L., Spencer N., Shahriari-Zavareh H., Stoddart J. F., Williams D. J., J. Chem. Soc. Chem. Commun. 1987, 1064–1066. [Google Scholar]
- 41. Helgeson R. C., Tarnowski T. L., Timko J. M., Cram D. J., J. Am. Chem. Soc. 1977, 99, 6411–6418. [Google Scholar]
- 42.We decided back in 1988 to use royal blue to identify π-electron-deficient units and molecules containing them and pillar-box red to identify π-electron-rich units and molecules containing them. Although the use of color was a little controversial at that time, it has stood the test of time.
- 43. Bühner M., Geuder W., Gries W.-K., Hünig S., Koch M., Poll T., Angew. Chem. Int. Ed. Engl. 1988, 27, 1553–1556; [Google Scholar]; Angew. Chem. 1988, 100, 1611–1614. [Google Scholar]
- 44. Hünig S., Herberth E., Chem. Rev. 2004, 104, 5535–5564. [DOI] [PubMed] [Google Scholar]
- 45. Amabilino D. B., Stoddart J. F., Chem. Rev. 1995, 95, 2725–2828. [Google Scholar]
- 46.
- 46a. Brown C. L., Philp D., Stoddart J. F., Synlett 1991, 462–464; [Google Scholar]
- 46b. Doddi G., Ercolani G., Mencarelli P., Piermattei A., J. Org. Chem. 2005, 70, 3761–3764. [DOI] [PubMed] [Google Scholar]
- 47. Asakawa M., Dehaen W., L'Abbé G., Menzer S., Nouwen J., Raymo F. M., Stoddart J. F., Williams D. J., J. Org. Chem. 1996, 61, 9591–9595. [Google Scholar]
- 48. Sue C.-H., Basu S., Fahrenbach A. C., Shveyd A. K., Dey S. K., Botros Y. Y., Stoddart J. F., Chem. Sci. 2010, 1, 119–125. [Google Scholar]
- 49. Barnes J. C., Juríček M., Vermeulen N. A., Dale E. J., Stoddart J. F., J. Org. Chem. 2013, 78, 11962–11969. [DOI] [PubMed] [Google Scholar]
- 50. Venturi M., Dumas S., Balzani V., Cao J., Stoddart J. F., New J. Chem. 2004, 28, 1032–1037. [Google Scholar]
- 51. Reddington M. V., Slawin A. M. Z., Spencer N., Stoddart J. F., Vicent C., Williams D. J., J. Chem. Soc. Chem. Commun. 1991, 630–634. [Google Scholar]
- 52.
- 52a. Philp D., Slawin A. M. Z., Spencer N., Stoddart J. F., Williams D. J., J. Chem. Soc. Chem. Commun. 1991, 1584–1586; [Google Scholar]
- 52b. Nielsen M. B., Jeppesen J. O., Lau J., Lomholt C., Damgaard D., Jacobsen J. P., Becher J., Stoddart J. F., J. Org. Chem. 2001, 66, 3559–3563; [DOI] [PubMed] [Google Scholar]
- 52c. Ikeda T., Higuchi M., Kurth D. G., J. Inclusion Phenom. Macrocyclic Chem. 2009, 64, 299–303. [Google Scholar]
- 53.
- 53a. Jiang J., MacLachlan M. J., Chem. Commun. 2009, 5695–5697; [DOI] [PubMed] [Google Scholar]
- 53b. Gong W., Yang X., Zavalij P. Y., Isaacs L., Zhao Z., Liu S., Chem. Eur. J. 2016, 22, 17612–17618; [DOI] [PubMed] [Google Scholar]
- 53c. Kawakami Y., Ogishima T., Kawara T., Yamauchi S., Okamoto K., Nikaido S., Souma D., Jin R.-H., Kabe Y., Chem. Commun. 2019, 55, 6066–6069. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Anelli P. L., Ashton P. R., Spencer N., Slawin A. M. Z., Stoddart J. F., Williams D. J., Angew. Chem. Int. Ed. Engl. 1991, 30, 1036–1039; [Google Scholar]; Angew. Chem. 1991, 103, 1052–1054; [Google Scholar]
- 54b. Ashton P. R., Philip D., Spencer N., Stoddart J. F., J. Chem. Soc. Chem. Commun. 1991, 1677–1679; [Google Scholar]
- 54c. Anelli P. L., Ashton P. R., Ballardini R., Balzani V., Delgado M., Gandolfi M. T., Goodnow T. T., Kaifer A. E., Philp D., J. Am. Chem. Soc. 1992, 114, 193–218; [Google Scholar]
- 54d. Castro R., Nixon K. R., Evanseck J. D., Kaifer A. E., J. Org. Chem. 1996, 61, 7298–7303; [DOI] [PubMed] [Google Scholar]
- 54e. Kristensen R., Andersen S. S., Olsen G., Jeppesen J. O., J. Org. Chem. 2017, 82, 1371–1379. [DOI] [PubMed] [Google Scholar]
- 55. Raymo F. M., Bartberger M. D., Houk K. N., Stoddart J. F., J. Am. Chem. Soc. 2001, 123, 9264–9267. [DOI] [PubMed] [Google Scholar]
- 56.
- 56a. Ashton P. R., Iqbal S., Stoddart J. F., Tinker N. D., Chem. Commun. 1996, 479–481; [Google Scholar]
- 56b. Kimura M., Misawa Y., Yamaguchi Y., Hanabusa K., Shirai H., Chem. Commun. 1996, 2785–2786; [Google Scholar]
- 56c. Ballardini R., Balzani V., Credi A., Brown C. L., Gillard R. E., Montalti M., Philp D., Stoddart J. F., Venturi M., White A. J. P., Williams B. J., Williams D. J., J. Am. Chem. Soc. 1997, 119, 12503–12513; [Google Scholar]
- 56d. Ballardini R., Balzani V., Gandolfi M. T., Gillard R. E., Stoddart J. F., Tabellini E., Chem. Eur. J. 1998, 4, 449–459; [Google Scholar]
- 56e. Shipway A. N., Lahav M., Blonder R., Willner I., Chem. Mater. 1999, 11, 13–15; [Google Scholar]
- 56f. Tootoonchi M. H., Sharma G., Calles J., Prabhakar R., Kaifer A. E., Angew. Chem. Int. Ed. 2016, 55, 11507–11511; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11679–11683. [Google Scholar]
- 57.
- 57a. Asakawa M., Iqbal S., Stoddart J. F., Tinker N. D., Angew. Chem. Int. Ed. Engl. 1996, 35, 976–978; [Google Scholar]; Angew. Chem. 1996, 108, 1053–1056; [Google Scholar]
- 57b. Asakawa M., Ashton P. R., Boyd S. E., Brown C. L., Gillard R. E., Kocian O., Raymo F. M., Stoddart J. F., Tolley M. S., White A. J. P., Williams D. J., J. Org. Chem. 1997, 62, 26–37; [DOI] [PubMed] [Google Scholar]
- 57c. Star A., Liu Y., Grant K., Ridvan L., Stoddart J. F., Steuerman D. W., Diehl M. R., Boukai A., Heath J. R., Macromolecules 2003, 36, 553–560; [Google Scholar]
- 57d. Cooke G., Garety J. F., Hewage S. G., Rabani G., Rotello V. M., Woisel P., Chem. Commun. 2006, 4119–4121; [DOI] [PubMed] [Google Scholar]
- 57e. Northrop B. H., Khan S. J., Stoddart J. F., Org. Lett. 2006, 8, 2159–2162; [DOI] [PubMed] [Google Scholar]
- 57f. Cooke G., Garety J. F., Hewage S. G., Jordan B. J., Rabani G., Rotello V. M., Woisel P., Org. Lett. 2007, 9, 481–484; [DOI] [PubMed] [Google Scholar]
- 57g. Bria M., Cooke G., Cooper A., Garety J. F., Hewage S. G., Nutley M., Rabani G., Woisel P., Tetrahedron Lett. 2007, 48, 301–304; [Google Scholar]
- 57h. Caldwell S. T., Cooke G., Cooper A., Nutley M., Rabani G., Rotello V., Smith B. O., Woisel P., Chem. Commun. 2008, 2650–2652; [DOI] [PubMed] [Google Scholar]
- 57i. Li J., Zhou W., Li Y., Liu H., Ouyang C., Yin X., Zheng H., Zuo Z., Tetrahedron Lett. 2008, 49, 4857–4861; [Google Scholar]
- 57j. Bigot J., Bria M., Caldwell S. T., Cazaux F., Cooper A., Charleux B., Cooke G., Fitzpatrick B., Fournier D., Lyskawa J., Nutley M., Stoffelbach F., Woisel P., Chem. Commun. 2009, 5266–5268; [DOI] [PubMed] [Google Scholar]
- 57k. Louisy J., Delattre F., Lyskawa J., Malfait A., Maclean C. E., Sambe L., Zhu N., Cooke G., Woisel P., Chem. Commun. 2011, 47, 6819–6821; [DOI] [PubMed] [Google Scholar]
- 57l. Xiao T., Feng X., Ye S., Guan Y., Li S.-L., Wang Q., Ji Y., Zhu D., Hu X., Lin C., Pan Y., Wang L., Macromolecules 2012, 45, 9585–9594; [Google Scholar]
- 57m. Xiao T., Li S.-L., Zhang Y., Lin C., Hu B., Guan X., Yu Y., Jiang J., Wang L., Chem. Sci. 2012, 3, 1417–1421; [Google Scholar]
- 57n. Sambe L., de La Rosa V. R., Belal K., Stoffelbach F., Lyskawa J., Delattre F., Bria M., Cooke G., Hoogenboom R., Woisel P., Angew. Chem. Int. Ed. 2014, 53, 5044–5048; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5144–5148; [Google Scholar]
- 57o. McGonigal P. R., Li H., Cheng C., Schneebeli S. T., Frasconi M., Witus L. S., Stoddart J. F., Tetrahedron Lett. 2015, 56, 3591–3594; [Google Scholar]
- 57p. Belal K., Stoffelbach F., Lyskawa J., Fumagalli M., Hourdet D., Marcellan A., Smet L. D., de la Rosa V. R., Cooke G., Hoogenboom R., Woisel P., Angew. Chem. Int. Ed. 2016, 55, 13974–13978; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14180–14184; [Google Scholar]
- 57q. López-Vidal E. M., Prokofjevs A., Gibbs-Hall I. C., Dale E. J., Quintela J. M., Peinador C., Dalton Trans. 2018, 47, 2492–2496; [DOI] [PubMed] [Google Scholar]
- 57r. Xiao T., Xu L., Götz J., Cheng M., Würthner F., Gu J., Feng X., Li Z.-Y., Sun X.-Q., Wang L., Mater. Chem. Front. 2019, 3, 2738–2745; [Google Scholar]
- 57s. Guo H., Hourdet D., Marcellan A., Stoffelbach F., Lyskawa J., de Smet L., Vebr A., Hoogenboom R., Woisel P., Chem. Eur. J. 2020, 26, 1292–1297; [DOI] [PubMed] [Google Scholar]
- 57t. Belal K., Stoffelbach F., Hourdet D., Marcellan A., Lyskawa J., de Smet L., Vebr A., Potier J., Cooke G., Hoogenboom R., Woisel P., Macromolecules 2021, 54, 1926–1933. [Google Scholar]
- 58. Asakawa M., Ashton P. R., Dehaen W., L'abbé G., Menzer S., Nouwen J., Raymo F. M., Stoddart J. F., Tolley M. S., Toppet S., White A. J. P., Williams D. J., Chem. Eur. J. 1997, 3, 772–787. [Google Scholar]
- 59. Zhang D., Soto M. A., Lewis L., Hamad W. Y., MacLachlan M. J., Angew. Chem. Int. Ed. 2020, 59, 4705–4710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 4735–4740. [Google Scholar]
- 60.
- 60a. Ashton P. R., Balzani V., Becher J., Credi A., Fyfe M. C. T., Mattersteig G., Menzer S., Nielsen M. B., Raymo F. M., Stoddart J. F., Venturi M., Williams D. J., J. Am. Chem. Soc. 1999, 121, 3951–3957; [Google Scholar]
- 60b. Anelli P.-L., Asakawa M., Ashton P. R., Bissell R. A., Clavier G., Górski R., Kaifer A. E., Langford S. J., Mattersteig G., Menzer S., Philp D., Slawin A. M. Z., Spencer N., Stoddart J. F., Tolley M. S., Williams D. J., Chem. Eur. J. 1997, 3, 1113–1135; [Google Scholar]
- 60c. Devonport W., Blower M. A., Bryce M. R., Goldenberg L. M., J. Org. Chem. 1997, 62, 885–887; [Google Scholar]
- 60d. Asakawa M., Ashton P. R., Balzani V., Credi A., Mattersteig G., Matthews O. A., Montalti M., Spencer N., Stoddart J. F., Venturi M., Chem. Eur. J. 1997, 3, 1992–1996; [Google Scholar]
- 60e. Cooke G., Duclairoir F. M. A., Rotello V. M., Stoddart J. F., Tetrahedron Lett. 2000, 41, 8163–8166; [Google Scholar]
- 60f. Bryce M. R., Cooke G., Devonport W., Duclairoir F. M. A., Rotello V. M., Tetrahedron Lett. 2001, 42, 4223–4226; [Google Scholar]
- 60g. Bryce M. R., Cooke G., Duclairoir F. M. A., Rotello V. M., Tetrahedron Lett. 2001, 42, 1143–1145; [Google Scholar]
- 60h. Nygaard S., Hansen C. N., Jeppesen J. O., J. Org. Chem. 2007, 72, 1617–1626; [DOI] [PubMed] [Google Scholar]
- 60i. Tanabe K., Kato T., Chem. Commun. 2009, 1864–1866; [DOI] [PubMed] [Google Scholar]
- 60j. Olson M. A., Coskun A., Klajn R., Fang L., Dey S. K., Browne K. P., Grzybowski B. A., Stoddart J. F., Nano Lett. 2009, 9, 3185–3190; [DOI] [PubMed] [Google Scholar]
- 60k. Bigot J., Charleux B., Cooke G., Delattre F., Fournier D., Lyskawa J., Sambe L., Stoffelbach F., Woisel P., J. Am. Chem. Soc. 2010, 132, 10796–10801; [DOI] [PubMed] [Google Scholar]
- 60l. Zhang K.-D., Wang G.-T., Zhao X., Jiang X.-K., Li Z.-T., Langmuir 2010, 26, 6878–6882; [DOI] [PubMed] [Google Scholar]
- 60m. Wang X.-J., Xing L.-B., Wang F., Wang G.-X., Chen B., Tung C.-H., Wu L.-Z., Langmuir 2011, 27, 8665–8671; [DOI] [PubMed] [Google Scholar]
- 60n. Sambe L., Stoffelbach F., Lyskawa J., Delattre F., Fournier D., Bouteiller L., Charleux B., Cooke G., Woisel P., Macromolecules 2011, 44, 6532–6538; [Google Scholar]
- 60o. Cao D., Wang C., Giesener M. A., Liu Z., Stoddart J. F., Chem. Commun. 2012, 48, 6791–6793; [DOI] [PubMed] [Google Scholar]
- 60p. Wang X.-J., Xing L.-B., Chen B., Quan Y., Tung C.-H., Wu L.-Z., Org. Biomol. Chem. 2016, 14, 65–68; [DOI] [PubMed] [Google Scholar]
- 60q. Hartlieb K. J., Liu W.-G., Fahrenbach A. C., Blackburn A. K., Frasconi M., Hafezi N., Dey S. K., Sarjeant A. A., Stern C. L., W. A. Goddard III , Stoddart J. F., Chem. Eur. J. 2016, 22, 2736–2745. [DOI] [PubMed] [Google Scholar]
- 61. Andersen S. S., Jensen M., Sørensen A., Miyazaki E., Takimiya K., Laursen B. W., Flood A. H., Jeppesen J. O., Chem. Commun. 2012, 48, 5157–5159. [DOI] [PubMed] [Google Scholar]
- 62.
- 62a. Córdova E., Bissell R. A., Spencer N., Ashton P. R., Stoddart J. F., Kaifer A. E., J. Org. Chem. 1993, 58, 6550–6552; [Google Scholar]
- 62b. Castro R., Berardi M. J., Córdova E., Ochoa de Olza M., Kaifer A. E., Evanseck J. D., J. Am. Chem. Soc. 1996, 118, 10257–10268. [Google Scholar]
- 63.
- 63a. Córdova E., Bissell R. A., Kaifer A. E., J. Org. Chem. 1995, 60, 1033–1038; [Google Scholar]
- 63b. Ikeda T., Aprahamian I., Stoddart J. F., Org. Lett. 2007, 9, 1481–1484. [DOI] [PubMed] [Google Scholar]
- 64. Asakawa M., Ashton P. R., Balzani V., Brown C. L., Credi A., Matthews O. A., Newton S. P., Raymo F. M., Shipway A. N., Spencer N., Quick A., Stoddart J. F., White A. J. P., Williams D. J., Chem. Eur. J. 1999, 5, 860–875. [Google Scholar]
- 65.
- 65a. Cao J., Jiang Y., Zhao J.-M., Chen C.-F., Chem. Commun. 2009, 1987–1989; [DOI] [PubMed] [Google Scholar]
- 65b. Cao J., Lu H.-Y., Xiang J.-F., Chen C.-F., Chem. Commun. 2010, 46, 3586–3588; [DOI] [PubMed] [Google Scholar]
- 65c. Cao J., Guo J.-B., Li P.-F., Chen C.-F., J. Org. Chem. 2011, 76, 1644–1652. [DOI] [PubMed] [Google Scholar]
- 66. Lu X., Samanta S. K., Zavalij P. Y., Isaacs L., Angew. Chem. Int. Ed. 2018, 57, 8073–8078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8205–8210. [Google Scholar]
- 67. Goodnow T. T., Reddington M. V., Stoddart J. F., Kaifer A. E., J. Am. Chem. Soc. 1991, 113, 4335–4337. [Google Scholar]
- 68. Bernardo A. R., Stoddart J. F., Kaifer A. E., J. Am. Chem. Soc. 1992, 114, 10624–10631. [Google Scholar]
- 69. Staley S. A., Smith B. D., Tetrahedron Lett. 1996, 37, 283–286. [Google Scholar]
- 70.Numbers and acronyms are rendered in bold if they represent molecular compounds, which include catenanes and rotaxanes and other mechanically interlocked molecules (MIMs). If an acronym relates to a component part of a MIM or to a unit incorporated with a molecule, then they are set in ordinary type-font.
- 71.
- 71a. Dietrich-Buchecker C. O., Sauvage J.-P., Kern J. M., J. Am. Chem. Soc. 1984, 106, 3043–3045; [Google Scholar]
- 71b. Hunter C. A., J. Am. Chem. Soc. 1992, 114, 5303–5311; [Google Scholar]
- 71c. Fujita M., Ibukuro F., Hagihara H., Ogura K., Nature 1994, 367, 720–723; [Google Scholar]
- 71d. Kidd T. J., Leigh D. A., Wilson A. J., J. Am. Chem. Soc. 1999, 121, 1599–1600; [Google Scholar]
- 71e. Sambrook M. R., Beer P. D., Wisner J. A., Paul R. L., Cowley A. R., J. Am. Chem. Soc. 2004, 126, 15364–15365; [DOI] [PubMed] [Google Scholar]
- 71f. Wang L., Vysotsky M. O., Bogdan A., Bolte M., Böhmer V., Science 2004, 304, 1312–1314; [DOI] [PubMed] [Google Scholar]
- 71g. Sato Y., Yamasaki R., Saito S., Angew. Chem. Int. Ed. 2009, 48, 504–507; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 512–515; [Google Scholar]
- 71h. Hasell T., Wu X., Jones J. T. A., Bacsa J., Steiner A., Mitra T., Trewin A., Adams D. J., Cooper A. I., Nat. Chem. 2010, 2, 750–755; [DOI] [PubMed] [Google Scholar]
- 71i. Tung S.-T., Lai C.-C., Liu Y.-H., Peng S.-M., Chiu S.-H., Angew. Chem. Int. Ed. 2013, 52, 13269–13272; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13511–13514; [Google Scholar]
- 71j. Li H., Zhang H., Lammer A. D., Wang M., Li X., Lynch V. M., Sessler J. L., Nat. Chem. 2015, 7, 1003–1008; [DOI] [PubMed] [Google Scholar]
- 71k. Gil-Ramírez G., Leigh D. A., Stephens A. J., Angew. Chem. Int. Ed. 2015, 54, 6110–6150; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6208–6249; [Google Scholar]
- 71l. Mitra R., Zhu H., Grimme S., Niemeyer J., Angew. Chem. Int. Ed. 2017, 56, 11456–11459; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11614–11617; [Google Scholar]
- 71m. Wu Q., Rauscher P. M., Lang X., Wojtecki R. J., de Pablo J. J., Hore M. J. A., Rowan S. J., Science 2017, 358, 1434–1439; [DOI] [PubMed] [Google Scholar]
- 71n. Denis M., Lewis J. E. M., Modicom F., Goldup S. M., Chem 2019, 5, 1512–1520; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71o. Ronson T. K., Wang Y., Baldridge K., Siegel J. S., Nitschke J. R., J. Am. Chem. Soc. 2020, 142, 10267–10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.
- 72a. Harada A., Li J., Kamachi M., Nature 1992, 356, 325–327; [Google Scholar]
- 72b. Murakami H., Kawabuchi A., Kotoo K., Kunitake M., Nakashima N., J. Am. Chem. Soc. 1997, 119, 7605–7606; [Google Scholar]
- 72c. Hübner G. M., Gläser J., Seel C., Vögtle F., Angew. Chem. Int. Ed. 1999, 38, 383–386; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 395–398; [Google Scholar]
- 72d. Brouwer A. M., Frochot C., Gatti F. G., Leigh D. A., Mottier L., Paolucci F., Roffia S., Wurpel G. W. H., Science 2001, 291, 2124–2128; [DOI] [PubMed] [Google Scholar]
- 72e. MacLachlan M. J., Rose A., Swager T. M., J. Am. Chem. Soc. 2001, 123, 9180–9181; [DOI] [PubMed] [Google Scholar]
- 72f. Stanier C. A., Alderman S. J., Claridge T. D. W., Anderson H. L., Angew. Chem. Int. Ed. 2002, 41, 1769–1772; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1847–1850; [Google Scholar]
- 72g. Wisner J. A., Beer P. D., Drew M. G. B., Sambrook M. R., J. Am. Chem. Soc. 2002, 124, 12469–12476; [DOI] [PubMed] [Google Scholar]
- 72h. Thordarson P., Bijsterveld E. J. A., Rowan A. E., Nolte R. J. M., Nature 2003, 424, 915–918; [DOI] [PubMed] [Google Scholar]
- 72i. Arunkumar E., Forbes C. C., Noll B. C., Smith B. D., J. Am. Chem. Soc. 2005, 127, 3288–3289; [DOI] [PubMed] [Google Scholar]
- 72j. Prikhod'ko A. I., Sauvage J.-P., J. Am. Chem. Soc. 2009, 131, 6794–6807; [DOI] [PubMed] [Google Scholar]
- 72k. Momčilović N., Clark P. G., Boydston A. J., Grubbs R. H., J. Am. Chem. Soc. 2011, 133, 19087–19089; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72l. Lee S., Chen C.-H., Flood A. H., Nat. Chem. 2013, 5, 704–710; [DOI] [PubMed] [Google Scholar]
- 72m. Xue M., Yang Y., Chi X., Yan X., Huang F., Chem. Rev. 2015, 115, 7398–7501; [DOI] [PubMed] [Google Scholar]
- 72n. Zhu K., Baggi G., Loeb S. J., Nat. Chem. 2018, 10, 625–630; [DOI] [PubMed] [Google Scholar]
- 72o. Corra S., de Vet C., Groppi J., La Rosa M., Silvi S., Baroncini M., Credi A., J. Am. Chem. Soc. 2019, 141, 9129–9133; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72p. Kumpulainen T., Panman M. R., Bakker B. H., Hilbers M., Woutersen S., Brouwer A. M., J. Am. Chem. Soc. 2019, 141, 19118–19129; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72q. Zhang M., De Bo G., J. Am. Chem. Soc. 2019, 141, 15879–15883; [DOI] [PubMed] [Google Scholar]
- 72r. Heard A. W., Goldup S. M., Chem 2020, 6, 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.
- 73a. Dietrich-Buchecker C. O., Sauvage J.-P., Angew. Chem. Int. Ed. Engl. 1989, 28, 189–192; [Google Scholar]; Angew. Chem. 1989, 101, 192–194; [Google Scholar]
- 73b. Safarowsky O., Nieger M., Fröhlich R., Vögtle F., Angew. Chem. Int. Ed. 2000, 39, 1616–1618; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 1699–1701; [Google Scholar]
- 73c. Feigel M., Ladberg R., Engels S., Herbst-Irmer R., Fröhlich R., Angew. Chem. Int. Ed. 2006, 45, 5698–5702; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5827–5831; [Google Scholar]
- 73d. Guo J., Mayers P. C., Breault G. A., Hunter C. A., Nat. Chem. 2010, 2, 218–222; [DOI] [PubMed] [Google Scholar]
- 73e. Forgan R. S., Sauvage J.-P., Stoddart J. F., Chem. Rev. 2011, 111, 5434–5464; [DOI] [PubMed] [Google Scholar]
- 73f. Ayme J.-F., Beves J. E., Leigh D. A., McBurney R. T., Rissanen K., Schultz D., Nat. Chem. 2012, 4, 15–20; [DOI] [PubMed] [Google Scholar]
- 73g. Ponnuswamy N., Cougnon F. B. L., Clough J. M., Pantoş G. D., Sanders J. K. M., Science 2012, 338, 783–785; [DOI] [PubMed] [Google Scholar]
- 73h. Prakasam T., Lusi M., Elhabiri M., Platas-Iglesias C., Olsen J.-C., Asfari Z., Cianférani-Sanglier S., Debaene F., Charbonnière L. J., Trabolsi A., Angew. Chem. Int. Ed. 2013, 52, 9956–9960; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10140–10144; [Google Scholar]
- 73i. Danon J. J., Krüger A., Leigh D. A., Lemonnier J.-F., Stephens A. J., Vitorica-Yrezabal I. J., Woltering S. L., Science 2017, 355, 159–162; [DOI] [PubMed] [Google Scholar]
- 73j. Kim D. H., Singh N., Oh J., Kim E.-H., Jung J., Kim H., Chi K.-W., Angew. Chem. Int. Ed. 2018, 57, 5669–5673; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5771–5775; [Google Scholar]
- 73k. Segawa Y., Kuwayama M., Hijikata Y., Fushimi M., Nishihara T., Pirillo J., Shirasaki J., Kubota N., Itami K., Science 2019, 365, 272–276; [DOI] [PubMed] [Google Scholar]
- 73l. Inomata Y., Sawada T., Fujita M., Chem 2020, 6, 294–303; [Google Scholar]
- 73m. Katsonis N., Lancia F., Leigh D. A., Pirvu L., Ryabchun A., Schaufelberger F., Nat. Chem. 2020, 12, 939–944; [DOI] [PubMed] [Google Scholar]
- 73n. Leigh D. A., Schaufelberger F., Pirvu L., Stenlid J. H., August D. P., Segard J., Nature 2020, 584, 562–568. [DOI] [PubMed] [Google Scholar]
- 74.
- 74a. Nierengarten J.-F., Dietrich-Buchecker C. O., Sauvage J.-P., J. Am. Chem. Soc. 1994, 116, 375–376; [DOI] [PubMed] [Google Scholar]
- 74b. Hasenknopf B., Lehn J.-M., Boumediene N., Dupont-Gervais A., Van Dorsselaer A., Kneisel B., Fenske D., J. Am. Chem. Soc. 1997, 119, 10956–10962; [Google Scholar]
- 74c. McArdle C. P., Vittal J. J., Puddephatt R. J., Angew. Chem. Int. Ed. 2000, 39, 3819–3822; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3977–3980; [Google Scholar]
- 74d. Pentecost C. D., Chichak K. S., Peters A. J., Cave G. W. V., Cantrill S. J., Stoddart J. F., Angew. Chem. Int. Ed. 2007, 46, 218–222; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 222–226; [Google Scholar]
- 74e. Peinador C., Blanco V., Quintela J. M., J. Am. Chem. Soc. 2009, 131, 920–921; [DOI] [PubMed] [Google Scholar]
- 74f. Ronson T. K., Fisher J., Harding L. P., Rizkallah P. J., Warren J. E., Hardie M. J., Nat. Chem. 2009, 1, 212–216; [DOI] [PubMed] [Google Scholar]
- 74g. Beves J. E., Campbell C. J., Leigh D. A., Pritchard R. G., Angew. Chem. Int. Ed. 2013, 52, 6464–6467; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6592–6595; [Google Scholar]
- 74h. Leigh D. A., Pritchard R. G., Stephens A. J., Nat. Chem. 2014, 6, 978–982; [DOI] [PubMed] [Google Scholar]
- 74i. Schouwey C., Holstein J. J., Scopelliti R., Zhurov K. O., Nagornov K. O., Tsybin Y. O., Smart O. S., Bricogne G., Severin K., Angew. Chem. Int. Ed. 2014, 53, 11261–11265; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11443–11447; [Google Scholar]
- 74j. Ponnuswamy N., Cougnon F. B. L., Pantoş G. D., Sanders J. K. M., J. Am. Chem. Soc. 2014, 136, 8243–8251; [DOI] [PubMed] [Google Scholar]
- 74k. Wood C. S., Ronson T. K., Belenguer A. M., Holstein J. J., Nitschke J. R., Nat. Chem. 2015, 7, 354–358; [DOI] [PubMed] [Google Scholar]
- 74l. Cougnon F. B. L., Caprice K., Pupier M., Bauzá A., Frontera A., J. Am. Chem. Soc. 2018, 140, 12442–12450; [DOI] [PubMed] [Google Scholar]
- 74m. Song Y. H., Singh N., Jung J., Kim H., Kim E.-H., Cheong H.-K., Kim Y., Chi K.-W., Angew. Chem. Int. Ed. 2016, 55, 2007–2011; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2047–2051; [Google Scholar]
- 74n. Zhang H.-N., Gao W.-X., Lin Y.-J., Jin G.-X., J. Am. Chem. Soc. 2019, 141, 16057–16063. [DOI] [PubMed] [Google Scholar]
- 75.
- 75a. Kraus T., Buděšínský M., Cvačka J., Sauvage J.-P., Angew. Chem. Int. Ed. 2006, 45, 258–261; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 264–267; [Google Scholar]
- 75b. Clark P. G., Day M. W., Grubbs R. H., J. Am. Chem. Soc. 2009, 131, 13631–13633; [DOI] [PubMed] [Google Scholar]
- 75c. Iwaso K., Takashima Y., Harada A., Nat. Chem. 2016, 8, 625–632; [DOI] [PubMed] [Google Scholar]
- 75d. Chang J.-C., Tseng S.-H., Lai C.-C., Liu Y.-H., Peng S.-M., Chiu S.-H., Nat. Chem. 2017, 9, 128–134; [Google Scholar]
- 75e. Goujon A., Lang T., Mariani G., Moulin E., Fuks G., Raya J., Buhler E., Giuseppone N., J. Am. Chem. Soc. 2017, 139, 14825–14828; [DOI] [PubMed] [Google Scholar]
- 75f. Ikejiri S., Takashima Y., Osaki M., Yamaguchi H., Harada A., J. Am. Chem. Soc. 2018, 140, 17308–17315; [DOI] [PubMed] [Google Scholar]
- 75g. Li W.-J., Wang W., Wang X.-Q., Li M., Ke Y., Yao R., Wen J., Yin G.-Q., Jiang B., Li X., Yin P., Yang H.-B., J. Am. Chem. Soc. 2020, 142, 8473–8482. [DOI] [PubMed] [Google Scholar]
- 76.
- 76a. Loren J. C., Yoshizawa M., Haldimann R. F., Linden A., Siegel J. S., Angew. Chem. Int. Ed. 2003, 42, 5702–5705; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 5880–5883; [Google Scholar]
- 76b. Chichak K. S., Cantrill S. J., Pease A. R., Chiu S.-H., Cave G. W. V., Atwood J. L., Stoddart J. F., Science 2004, 304, 1308–1312; [DOI] [PubMed] [Google Scholar]
- 76c. Thorp-Greenwood F. L., Kulak A. N., Hardie M. J., Nat. Chem. 2015, 7, 526–531; [DOI] [PubMed] [Google Scholar]
- 76d. Kim T., Singh N., Oh J., Kim E.-H., Jung J., Kim H., Chi K.-W., J. Am. Chem. Soc. 2016, 138, 8368–8371; [DOI] [PubMed] [Google Scholar]
- 76e. Lu Y., Deng Y.-X., Lin Y.-J., Han Y.-F., Weng L.-H., Li Z.-H., Jin G.-X., Chem 2017, 3, 110–121. [Google Scholar]
- 77. Hegemann J. D., Zimmermann M., Xie X., Marahiel M. A., Acc. Chem. Res. 2015, 48, 1909–1919. [DOI] [PubMed] [Google Scholar]
- 78. Meluzzi D., Smith D. E., Arya G., Annu. Rev. Biophys. 2010, 39, 349–366. [DOI] [PubMed] [Google Scholar]
- 79. Wasserman E., J. Am. Chem. Soc. 1960, 82, 4433–4434. [Google Scholar]
- 80. Schill G., Lüttringhaus A., Angew. Chem. Int. Ed. Engl. 1964, 3, 546–547; [Google Scholar]; Angew. Chem. 1964, 76, 567–568. [Google Scholar]
- 81. Dietrich-Buchecker C. O., Sauvage J.-P., Kintzinger J. P., Tetrahedron Lett. 1983, 24, 5095–5098. [Google Scholar]
- 82. Sauvage J.-P., Angew. Chem. Int. Ed. 2017, 56, 11080–11093; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11228–11242. [Google Scholar]
- 83. Ashton P. R., Goodnow T. T., Kaifer A. E., Reddington M. V., Slawin A. M. Z., Spencer N., Stoddart J. F., Vicent C., Williams D. J., Angew. Chem. Int. Ed. Engl. 1989, 28, 1396–1399; [Google Scholar]; Angew. Chem. 1989, 101, 1404–1408. [Google Scholar]
- 84.
- 84a. Brown C. L., Philp D., Stoddart J. F., Synlett 1991, 459–461; [Google Scholar]
- 84b. Capobianchi S., Doddi G., Ercolani G., Keyes J. W., Mencarelli P., J. Org. Chem. 1997, 62, 7015–7017; [Google Scholar]
- 84c. Capobianchi S., Doddi G., Ercolani G., Mencarelli P., J. Org. Chem. 1998, 63, 8088–8089. [Google Scholar]
- 85.It is noteworthy that [π⋅⋅⋅π] stacking (charge-transfer) interactions between the viologen units and BPP34C10 are relatively weak and thus play a less significant role in bringing about the templating effect compared with their [C−H⋅⋅⋅O] hydrogen-bonding interactions.
- 86.
- 86a. Ashton P. R., Brown C. L., Chrystal E. J. T., Parry K. P., Pietraszkiewicz M., Spencer N., Stoddart J. F., Angew. Chem. Int. Ed. Engl. 1991, 30, 1042–1045; [Google Scholar]; Angew. Chem. 1991, 103, 1058–1061; [Google Scholar]
- 86b. Ashton P. R., Blower M. A., Iqbal S., McLean C. H., Stoddart J. F., Tolley M. S., Williams D. J., Synlett 1994, 1994, 1059–1062; [Google Scholar]
- 86c. Amabilino D. B., Ashton P. R., Brown G. R., Hayes W., Stoddart J. F., Tolley M. S., Williams D. J., J. Chem. Soc. Chem. Commun. 1994, 2479–2482; [Google Scholar]
- 86d. Gillard R. E., Stoddart J. F., White A. J. P., Williams B. J., Williams D. J., J. Org. Chem. 1996, 61, 4504–4505; [DOI] [PubMed] [Google Scholar]
- 86e. Ashton P. R., Diederich F., Gómez-López M., Nierengarten J.-F., Preece J. A., Raymo F. M., Stoddart J. F., Angew. Chem. Int. Ed. Engl. 1997, 36, 1448–1451; [Google Scholar]; Angew. Chem. 1997, 109, 1611–1614; [Google Scholar]
- 86f. Stoddart J. F., Williams D. J., Amabilino D. B., Anelli P.-L., Ashton P. R., Brown G. R., Córdova E., Godinez L. A., Hayes W., J. Am. Chem. Soc. 1995, 117, 11142–11170; [Google Scholar]
- 86g. Amabilino D. B., Ashton P. R., Boyd S. E., Gómez-López M., Hayes W., Stoddart J. F., J. Org. Chem. 1997, 62, 3062–3075; [DOI] [PubMed] [Google Scholar]
- 86h. Asakawa M., Brown C. L., Menzer S., Raymo F. M., Stoddart J. F., Williams D. J., J. Am. Chem. Soc. 1997, 119, 2614–2627; [Google Scholar]
- 86i. Pérez-Alvarez M., Raymo F. M., Rowan S. J., Schiraldi D., Stoddart J. F., Wang Z.-H., White A. J. P., Williams D. J., Tetrahedron 2001, 57, 3799–3808; [Google Scholar]
- 86j. Zhang M., Luo Y., Zheng B., Yan X., Fronczek F. R., Huang F., Eur. J. Org. Chem. 2010, 6798–6803. [Google Scholar]
- 87.
- 87a. Ashton P. R., Brown C. L., Chrystal E. J. T., Goodnow T. T., Kaifer A. E., Parry K. P., Philp D., Slawin A. M. Z., Spencer N., Stoddart J. F., Williams D. J., J. Chem. Soc. Chem. Commun. 1991, 634–639; [Google Scholar]
- 87b. Amabilino D. B., Ashton P. R., Stoddart J. F., Menzer S., Williams D. J., J. Chem. Soc. Chem. Commun. 1994, 2475–2478; [Google Scholar]
- 87c. D'Acerno C., Doddi G., Ercolani G., Mencarelli P., Chem. Eur. J. 2000, 6, 3540–3546; [DOI] [PubMed] [Google Scholar]
- 87d. Tseng H.-R., Vignon S. A., Celestre P. C., Stoddart J. F., White A. J. P., Williams D. J., Chem. Eur. J. 2003, 9, 543–556; [DOI] [PubMed] [Google Scholar]
- 87e. Kang S., Aprahamian I., Stoddart J. F., Isr. J. Chem. 2007, 47, 253–262; [Google Scholar]
- 87f. Fahrenbach A. C., Hartlieb K. J., Sue C.-H., Bruns C. J., Barin G., Basu S., Olson M. A., Botros Y. Y., Bagabas A., Khdary N. H., Stoddart J. F., Chem. Commun. 2012, 48, 9141–9143. [DOI] [PubMed] [Google Scholar]
- 88. Grunder S., McGrier P. L., Whalley A. C., Boyle M. M., Stern C., Stoddart J. F., J. Am. Chem. Soc. 2013, 135, 17691–17694. [DOI] [PubMed] [Google Scholar]
- 89.
- 89a. Asakawa M., Ashton P. R., Balzani V., Credi A., Hamers C., Mattersteig G., Montalti M., Shipway A. N., Spencer N., Stoddart J. F., Tolley M. S., Venturi M., White A. J. P., Williams D. J., Angew. Chem. Int. Ed. 1998, 37, 333–337; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 357–361; [Google Scholar]
- 89b. Brøndsted Nielsen M., Thorup N., Becher J., J. Chem. Soc. Perkin Trans. 1 1998, 1305–1308; [Google Scholar]
- 89c. Balzani V., Credi A., Mattersteig G., Matthews O. A., Raymo F. M., Stoddart J. F., Venturi M., White A. J. P., Williams D. J., J. Org. Chem. 2000, 65, 1924–1936; [DOI] [PubMed] [Google Scholar]
- 89d. Tomcsi M. R., Stoddart J. F., J. Org. Chem. 2007, 72, 9335–9338; [DOI] [PubMed] [Google Scholar]
- 89e. Nygaard S., Hansen S. W., Huffman J. C., Jensen F., Flood A. H., Jeppesen J. O., J. Am. Chem. Soc. 2007, 129, 7354–7363; [DOI] [PubMed] [Google Scholar]
- 89f. Wang C., Dyar S. M., Cao D., Fahrenbach A. C., Horwitz N., Colvin M. T., Carmieli R., Stern C. L., Dey S. K., Wasielewski M. R., Stoddart J. F., J. Am. Chem. Soc. 2012, 134, 19136–19145. [DOI] [PubMed] [Google Scholar]
- 90. Ballardini R., Balzani V., Credi A., Gandolfi M. T., Marquis D., Pérez-García L., Stoddart J. F., Eur. J. Org. Chem. 1998, 81–89. [Google Scholar]
- 91. Lukyanenko N. G., Lyapunov A. Y., Kirichenko T. I., Botoshansky M. M., Simonov Y. A., Fonari M. S., Tetrahedron Lett. 2005, 46, 2109–2112. [Google Scholar]
- 92.
- 92a. Alcalde E., Pérez-García L., Ramos S., Stoddart J. F., Vignon S. A., White A. J. P., Williams D. J., Mendeleev Commun. 2003, 13, 100–102; [Google Scholar]
- 92b. Alcalde E., Pérez-García L., Ramos S., Stoddart J. F., White A. J. P., Williams D. J., Mendeleev Commun. 2004, 14, 233–235; [Google Scholar]
- 92c. Alcalde E., Pérez-García L., Ramos S., Stoddart J. F., White A. J. P., Williams D. J., Chem. Eur. J. 2007, 13, 3964–3979. [DOI] [PubMed] [Google Scholar]
- 93. Cheng P.-N., Huang P.-Y., Li W.-S., Ueng S.-H., Hung W.-C., Liu Y.-H., Lai C.-C., Wang Y., Peng S.-M., Chao I., Chiu S.-H., J. Org. Chem. 2006, 71, 2373–2383. [DOI] [PubMed] [Google Scholar]
- 94.
- 94a. Li S., Liu M., Zhang J., Zheng B., Zhang C., Wen X., Li N., Huang F., Org. Biomol. Chem. 2008, 6, 2103–2107; [DOI] [PubMed] [Google Scholar]
- 94b. Liu M., Li S., Zhang M., Zhou Q., Wang F., Hu M., Fronczek F. R., Li N., Huang F., Org. Biomol. Chem. 2009, 7, 1288–1291; [DOI] [PubMed] [Google Scholar]
- 94c. Liu M., Li S., Hu M., Wang F., Huang F., Org. Lett. 2010, 12, 760–763. [DOI] [PubMed] [Google Scholar]
- 95. Liu H., Li X.-Y., Zhao X.-L., Liu Y. A., Li J.-S., Jiang B., Wen K., Org. Lett. 2014, 16, 5894–5897. [DOI] [PubMed] [Google Scholar]
- 96. Amabilino D. B., Ashton P. R., Reder A. S., Spencer N., Stoddart J. F., Angew. Chem. Int. Ed. Engl. 1994, 33, 433–437; [Google Scholar]; Angew. Chem. 1994, 106, 450–453. [Google Scholar]
- 97. Amabilino D. B., Ashton P. R., Reder A. S., Spencer N., Stoddart J. F., Angew. Chem. Int. Ed. Engl. 1994, 33, 1286–1290; [Google Scholar]; Angew. Chem. 1994, 106, 1316–1319. [Google Scholar]
- 98.
- 98a. Ashton P. R., Menzer S., Raymo F. M., Shimizu G. K. H., Stoddart J. F., Williams D. J., Chem. Commun. 1996, 487–490; [Google Scholar]
- 98b. Asakawa M., Ashton P. R., Menzer S., Raymo F. M., Stoddart J. F., White A. J. P., Williams D. J., Chem. Eur. J. 1996, 2, 877–893; [Google Scholar]
- 98c. Forgan R. S., Wang C., Friedman D. C., Spruell J. M., Stern C. L., Sarjeant A. A., Cao D., Stoddart J. F., Chem. Eur. J. 2012, 18, 202–212. [DOI] [PubMed] [Google Scholar]
- 99. Amabilino D. B., Ashton P. R., Boyd S. E., Lee J. Y., Menzer S., Stoddart J. F., Williams D. J., Angew. Chem. Int. Ed. Engl. 1997, 36, 2070–2072; [Google Scholar]; Angew. Chem. 1997, 109, 2160–2162. [Google Scholar]
- 100. Anelli P. L., Spencer N., Stoddart J. F., J. Am. Chem. Soc. 1991, 113, 5131–5133. [DOI] [PubMed] [Google Scholar]
- 101.
- 101a. Ashton P. R., Bissell R. A., Górski R., Philp D., Spencer N., Stoddart J. F., Tolley M. S., Synlett 1992, 919–922; [Google Scholar]
- 101b. Ashton P. R., Bissell R. A., Spencer N., Stoddart J. F., Tolley M. S., Synlett 1992, 923–926; [Google Scholar]
- 101c. Ashton P. R., Bissell R. A., Spencer N., Stoddart J. F., Tolley M. S., Synlett 1992, 914–918. [Google Scholar]
- 102. Ashton P. R., Johnston M. R., Stoddart J. F., Tolley M. S., Wheeler J. W., J. Chem. Soc. Chem. Commun. 1992, 1128–1131. [Google Scholar]
- 103.
- 103a. Ashton P. R., Grognuz M., Slawin A. M. Z., Stoddart J. F., Williams D. J., Tetrahedron Lett. 1991, 32, 6235–6238; [Google Scholar]
- 103b. Benniston A. C., Harriman A., Lynch V. M., Tetrahedron Lett. 1994, 35, 1473–1476; [Google Scholar]
- 103c. Benniston A. C., Harriman A., Lynch V. M., J. Am. Chem. Soc. 1995, 117, 5275–5291; [Google Scholar]
- 103d. Asakawa M., Ashton P. R., Iqbal S., Quick A., Stoddart J. F., Tinker N. D., White A. J. P., Williams D. J., Isr. J. Chem. 1996, 36, 329–340. [Google Scholar]
- 104.
- 104a. Bravo J. A., Raymo F. M., Stoddart J. F., White A. J. P., Williams D. J., Eur. J. Org. Chem. 1998, 2565–2571; [Google Scholar]
- 104b. Ashton P. R., Bravo J. A., Raymo F. M., Stoddart J. F., White A. J. P., Williams D. J., Eur. J. Org. Chem. 1999, 899–908; [Google Scholar]
- 104c. Nygaard S., Leung K. C. F., Aprahamian I., Ikeda T., Saha S., Laursen B. W., Kim S.-Y., Hansen S. W., Stein P. C., Flood A. H., Stoddart J. F., Jeppesen J. O., J. Am. Chem. Soc. 2007, 129, 960–970; [DOI] [PubMed] [Google Scholar]
- 104d. Yoon I., Benítez D., Zhao Y.-L., Miljanić O. Š., Kim S.-Y., Tkatchouk E., Leung K. C.-F., Khan S. I., W. A. Goddard III , Stoddart J. F., Chem. Eur. J. 2009, 15, 1115–1122; [DOI] [PubMed] [Google Scholar]
- 104e. Zhao Y.-L., Shveyd A. K., Stoddart J. F., Tetrahedron Lett. 2011, 52, 2044–2047. [Google Scholar]
- 105. Jeppesen J. O., Perkins J., Becher J., Stoddart J. F., Org. Lett. 2000, 2, 3547–3550. [DOI] [PubMed] [Google Scholar]
- 106. Nygaard S., Laursen B. W., Hansen T. S., Bond A. D., Flood A. H., Jeppesen J. O., Angew. Chem. Int. Ed. 2007, 46, 6093–6097; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 6205–6209. [Google Scholar]
- 107. Flood A. H., Nygaard S., Laursen B. W., Jeppesen J. O., Stoddart J. F., Org. Lett. 2006, 8, 2205–2208. [DOI] [PubMed] [Google Scholar]
- 108. Ballardini R., Balzani V., Dehaen W., Dell'Erba A. E., Raymo Françisco M., Stoddart J. F., Venturi M., Eur. J. Org. Chem. 2000, 591–602. [Google Scholar]
- 109.
- 109a. Miljanić O. Š., Dichtel W. R., Mortezaei S., Stoddart J. F., Org. Lett. 2006, 8, 4835–4838; [DOI] [PubMed] [Google Scholar]
- 109b. Miljanić O. Š., Dichtel W. R., Khan S. I., Mortezaei S., Heath J. R., Stoddart J. F., J. Am. Chem. Soc. 2007, 129, 8236–8246. [DOI] [PubMed] [Google Scholar]
- 110. Yoon I., Miljanić O. Š., Benítez D., Khan S. I., Stoddart J. F., Chem. Commun. 2008, 4561–4563. [DOI] [PubMed] [Google Scholar]
- 111. Fang L., Basu S., Sue C.-H., Fahrenbach A. C., Stoddart J. F., J. Am. Chem. Soc. 2011, 133, 396–399. [DOI] [PubMed] [Google Scholar]
- 112. Spruell J. M., Paxton W. F., Olsen J.-C., Benítez D., Tkatchouk E., Stern C. L., Trabolsi A., Friedman D. C., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2009, 131, 11571–11580. [DOI] [PubMed] [Google Scholar]
- 113. Bruns C. J., Liu H., Francis M. B., J. Am. Chem. Soc. 2016, 138, 15307–15310. [DOI] [PubMed] [Google Scholar]
- 114.
- 114a. Ikeda T., Higuchi M., Sato A., Kurth D. G., Org. Lett. 2008, 10, 2215–2218; [DOI] [PubMed] [Google Scholar]
- 114b. Ikeda T., Higuchi M., Kurth D. G., Chem. Eur. J. 2009, 15, 4906–4913; [DOI] [PubMed] [Google Scholar]
- 114c. Tsuge A., Nakamura K., Moriguchi T., Araki K., Chem. Lett. 2012, 41, 880–882. [Google Scholar]
- 115.
- 115a. Hein J. E., Fokin V. V., Chem. Soc. Rev. 2010, 39, 1302–1315; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115b. Hänni K. D., Leigh D. A., Chem. Soc. Rev. 2010, 39, 1240–1251. [DOI] [PubMed] [Google Scholar]
- 116. Dichtel W. R., Miljanić O. Š., Spruell J. M., Heath J. R., Stoddart J. F., J. Am. Chem. Soc. 2006, 128, 10388–10390. [DOI] [PubMed] [Google Scholar]
- 117. Mobian P., Collin J.-P., Sauvage J.-P., Tetrahedron Lett. 2006, 47, 4907–4909. [Google Scholar]
- 118. Aucagne V., Hänni K. D., Leigh D. A., Lusby P. J., Walker D. B., J. Am. Chem. Soc. 2006, 128, 2186–2187. [DOI] [PubMed] [Google Scholar]
- 119.
- 119a. Miljanić O. Š., Dichtel W. R., Aprahamian I., Rohde R. D., Agnew H. D., Heath J. R., Stoddart J. F., QSAR Comb. Sci. 2007, 26, 1165–1174; [Google Scholar]
- 119b. Braunschweig A. B., Dichtel W. R., Miljanić O. Š., Olson M. A., Spruell J. M., Khan S. I., Heath J. R., Stoddart J. F., Chem. Asian J. 2007, 2, 634–647. [DOI] [PubMed] [Google Scholar]
- 120. Dichtel W. R., Miljanić O. Š., Zhang W., Spruell J. M., Patel K., Aprahamian I., Heath J. R., Stoddart J. F., Acc. Chem. Res. 2008, 41, 1750–1761. [DOI] [PubMed] [Google Scholar]
- 121. Asakawa M., Ashton P. R., Iqbal S., Stoddart J. F., Tinker N. D., White A. J. P., Williams D. J., Chem. Commun. 1996, 483–486. [Google Scholar]
- 122. Ashton P. R., Baxter I., Fyfe M. C. T., Raymo F. M., Spencer N., Stoddart J. F., White A. J. P., Williams D. J., J. Am. Chem. Soc. 1998, 120, 2297–2307. [Google Scholar]
- 123.
- 123a. Jeppesen J. O., Becher J., Stoddart J. F., Org. Lett. 2002, 4, 557–560; [DOI] [PubMed] [Google Scholar]
- 123b. Jeppesen J. O., Vignon S. A., Stoddart J. F., Chem. Eur. J. 2003, 9, 4611–4625. [DOI] [PubMed] [Google Scholar]
- 124. Ashton P. R., Everitt S. R. L., Gómez-López M., Jayaraman N., Stoddart J. F., Tetrahedron Lett. 1997, 38, 5691–5694. [Google Scholar]
- 125.
- 125a. Zhang K.-D., Zhao X., Wang G.-T., Liu Y., Zhang Y., Lu H.-J., Jiang X.-K., Li Z.-T., Angew. Chem. Int. Ed. 2011, 50, 9866–9870; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10040–10044; [Google Scholar]
- 125b. Zhang K.-D., Zhao X., Wang G.-T., Liu Y., Zhang Y., Lu H.-J., Jiang X.-K., Li Z.-T., Tetrahedron 2012, 68, 4517–4527; [Google Scholar]
- 125c. Wang W.-K., Xu Z.-Y., Zhang Y.-C., Wang H., Zhang D.-W., Liu Y., Li Z.-T., Chem. Commun. 2016, 52, 7490–7493. [DOI] [PubMed] [Google Scholar]
- 126. Rowan S. J., Cantrill S. J., Cousins G. R. L., Sanders J. K. M., Stoddart J. F., Angew. Chem. Int. Ed. 2002, 41, 898–952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 938–993. [Google Scholar]
- 127. Koshkakaryan G., Cao D., Klivansky L. M., Teat S. J., Tran J. L., Liu Y., Org. Lett. 2010, 12, 1528–1531. [DOI] [PubMed] [Google Scholar]
- 128. Li J., Wei P., Wu X., Xue M., Yan X., Zhou Q., RSC Adv. 2013, 3, 21289–21293. [Google Scholar]
- 129.
- 129a. Cantrill S. J., Rowan S. J., Stoddart J. F., Org. Lett. 1999, 1, 1363–1366; [Google Scholar]
- 129b. Rowan S. J., Stoddart J. F., Org. Lett. 1999, 1, 1913–1916. [Google Scholar]
- 130. Yin Y.-F., Yun M.-Y., Wu L., Duan H.-Y., Jiang X.-M., Zhan T.-G., Cui J., Liu L.-J., Zhang K.-D., Angew. Chem. Int. Ed. 2019, 58, 12705–12710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12835–12840. [Google Scholar]
- 131. Soto M. A., MacLachlan M. J., Org. Lett. 2019, 21, 1744–1748. [DOI] [PubMed] [Google Scholar]
- 132. Ogba O. M., Warner N. C., O'Leary D. J., Grubbs R. H., Chem. Soc. Rev. 2018, 47, 4510–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Li S., Liu M., Zheng B., Zhu K., Wang F., Li N., Zhao X.-L., Huang F., Org. Lett. 2009, 11, 3350–3353. [DOI] [PubMed] [Google Scholar]
- 134. Miljanić O. Š., Stoddart J. F., Proc. Natl. Acad. Sci. USA 2007, 104, 12966–12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Patel K., Miljanić O. Š., Stoddart J. F., Chem. Commun. 2008, 1853–1855. [DOI] [PubMed] [Google Scholar]
- 136. Lu T., Zhang L., Gokel G. W., Kaifer A. E., J. Am. Chem. Soc. 1993, 115, 2542–2543. [Google Scholar]
- 137. Forgan R. S., Gassensmith J. J., Cordes D. B., Boyle M. M., Hartlieb K. J., Friedman D. C., Slawin A. M. Z., Stoddart J. F., J. Am. Chem. Soc. 2012, 134, 17007–17010. [DOI] [PubMed] [Google Scholar]
- 138. Hu W.-B., Hu W.-J., Zhao X.-L., Liu Y. A., Li J.-S., Jiang B., Wen K., Chem. Commun. 2015, 51, 13882–13885. [DOI] [PubMed] [Google Scholar]
- 139.
- 139a. Li Z.-T., Stein P. C., Svenstrup N., Lund K. H., Becher J., Angew. Chem. Int. Ed. Engl. 1995, 34, 2524–2528; [Google Scholar]; Angew. Chem. 1995, 107, 2719–2723; [Google Scholar]
- 139b. Li Z.-T., Stein P. C., Becher J., Jensen D., Mørk P., Svenstrup N., Chem. Eur. J. 1996, 2, 624–633; [Google Scholar]
- 139c. Li Z.-T., Becher J., Chem. Commun. 1996, 639–640. [Google Scholar]
- 140.
- 140a. Simone D. L., Swager T. M., J. Am. Chem. Soc. 2000, 122, 9300–9301; [Google Scholar]
- 140b. Olson M. A., Braunschweig A. B., Fang L., Ikeda T., Klajn R., Trabolsi A., Wesson P. J., Benítez D., Mirkin C. A., Grzybowski B. A., Stoddart J. F., Angew. Chem. Int. Ed. 2009, 48, 1792–1797; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1824–1829; [Google Scholar]
- 140c. Olson M. A., Coskun A., Fang L., Basuray A. N., Stoddart J. F., Angew. Chem. Int. Ed. 2010, 49, 3151–3156; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3219–3224. [Google Scholar]
- 141. Ashton P. R., Preece J. A., Stoddart J. F., Tolley M. S., Synlett 1994, 789–792. [Google Scholar]
- 142.
- 142a. Ashton P. R., Reder A. S., Spencer N., Stoddart J. F., J. Am. Chem. Soc. 1993, 115, 5286–5287; [Google Scholar]
- 142b. Hartlieb K. J., Blackburn A. K., Schneebeli S. T., Forgan R. S., Sarjeant A. A., Stern C. L., Cao D., Stoddart J. F., Chem. Sci. 2014, 5, 90–100. [Google Scholar]
- 143. Liu Y., Bonvallet P. A., Vignon S. A., Khan S. I., Stoddart J. F., Angew. Chem. Int. Ed. 2005, 44, 3050–3055; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3110–3115. [Google Scholar]
- 144. Takata T., Hasegawa T., Kihara N., Furusho Y., Polym. J. 2004, 36, 927–932. [Google Scholar]
- 145.
- 145a. Spruell J. M., Dichtel W. R., Heath J. R., Stoddart J. F., Chem. Eur. J. 2008, 14, 4168–4177; [DOI] [PubMed] [Google Scholar]
- 145b. Aprahamian I., Olsen J.-C., Trabolsi A., Stoddart J. F., Chem. Eur. J. 2008, 14, 3889–3895. [DOI] [PubMed] [Google Scholar]
- 146.
- 146a. Amabilino D. B., Ashton P. R., Brown C. L., Córdova E., Godinez L. A., Goodnow T. T., Kaifer A. E., Newton S. P., Pietraszkiewicz M., J. Am. Chem. Soc. 1995, 117, 1271–1293; [Google Scholar]
- 146b. Amabilino D. B., Ashton P. R., Balzani V., Boyd S. E., Credi A., Lee J. Y., Menzer S., Stoddart J. F., Venturi M., Williams D. J., J. Am. Chem. Soc. 1998, 120, 4295–4307; [Google Scholar]
- 146c. Ashton P. R., Baldoni V., Balzani V., Claessens C. G., Credi A., Hoffmann H. D. A., Raymo F. M., Stoddart J. F., Venturi M., White A. J. P., Williams D. J., Eur. J. Org. Chem. 2000, 1121–1130. [Google Scholar]
- 147. Ashton P. R., Horn T., Menzer S., Preece J. A., Spencer N., Stoddart J. F., Williams D. J., Synthesis 1997, 480–488. [Google Scholar]
- 148. Wolf R., Asakawa M., Ashton P. R., Gómez-López M., Hamers C., Menzer S., Parsons I. W., Spencer N., Stoddart J. F., Tolley M. S., Williams D. J., Angew. Chem. Int. Ed. 1998, 37, 975–979; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1018–1022. [Google Scholar]
- 149.
- 149a. Ashton P. R., Gómez-López M., Iqbal S., Preece J. A., Stoddart J. F., Tetrahedron Lett. 1997, 38, 3635–3638; [Google Scholar]
- 149b. Ashton P. R., Ballardini R., Balzani V., Boyd S. E., Credi A., Gandolfi M. T., Gómez-López M., Iqbal S., Philp D., Preece J. A., Prodi L., Ricketts H. G., Stoddart J. F., Tolley M. S., Venturi M., Venturi M., White A. J. P., Williams D. J., Chem. Eur. J. 1997, 3, 152–170; [Google Scholar]
- 149c. Liu Y., Saha S., Vignon S. A., Flood A. H., Stoddart J. F., Synthesis 2005, 3437–3445; [Google Scholar]
- 149d. Liu Y., Flood A. H., Moskowitz R. M., Stoddart J. F., Chem. Eur. J. 2005, 11, 369–385. [DOI] [PubMed] [Google Scholar]
- 150.
- 150a. Boyle M. M., Forgan R. S., Friedman D. C., Gassensmith J. J., Smaldone R. A., Stoddart J. F., Sauvage J.-P., Chem. Commun. 2011, 47, 11870–11872; [DOI] [PubMed] [Google Scholar]
- 150b. Boyle M. M., Gassensmith J. J., Whalley A. C., Forgan R. S., Smaldone R. A., Hartlieb K. J., Blackburn A. K., Sauvage J.-P., Stoddart J. F., Chem. Eur. J. 2012, 18, 10312–10323. [DOI] [PubMed] [Google Scholar]
- 151.
- 151a. Mason P. E., Parsons I. W., Tolley M. S., Angew. Chem. Int. Ed. Engl. 1996, 35, 2238–2241; [Google Scholar]; Angew. Chem. 1996, 108, 2405–2408; [Google Scholar]
- 151b. Basu S., Coskun A., Friedman D. C., Olson M. A., Benítez D., Tkatchouk E., Barin G., Yang J., Fahrenbach A. C., W. A. Goddard III , Stoddart J. F., Chem. Eur. J. 2011, 17, 2107–2119; [DOI] [PubMed] [Google Scholar]
- 151c. Franco I., Ratner M. A., Schatz G. C., J. Phys. Chem. B 2011, 115, 2477–2484; [DOI] [PubMed] [Google Scholar]
- 151d. Zhu Z., Li H., Liu Z., Lei J., Zhang H., Botros Y. Y., Stern C. L., Sarjeant A. A., Stoddart J. F., Colquhoun H. M., Angew. Chem. Int. Ed. 2012, 51, 7231–7235; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7343–7347; [Google Scholar]
- 151e. Zhu Z., Bruns C. J., Li H., Lei J., Ke C., Liu Z., Shafaie S., Colquhoun H. M., Stoddart J. F., Chem. Sci. 2013, 4, 1470–1483. [Google Scholar]
- 152.
- 152a. Liu Y., Vignon S. A., Zhang X., Houk K. N., Stoddart J. F., Chem. Commun. 2005, 3927–3929; [DOI] [PubMed] [Google Scholar]
- 152b. Liu Y., Vignon S. A., Zhang X., Bonvallet P. A., Khan S. I., Houk K. N., Stoddart J. F., J. Org. Chem. 2005, 70, 9334–9344; [DOI] [PubMed] [Google Scholar]
- 152c. Zhao Y.-L., Trabolsi A., Stoddart J. F., Chem. Commun. 2009, 4844–4846. [DOI] [PubMed] [Google Scholar]
- 153.
- 153a. Asakawa M., Ashton P. R., Hayes W., Stoddart J. F., Brown G. R., Menzer S., White A. J. P., Williams D. J., Adv. Mater. 1996, 8, 37–41; [Google Scholar]
- 153b. Asakawa M., Ashton P. R., Hayes W., Janssen H. M., Meijer E. W., Menzer S., Pasini D., Stoddart J. F., White A. J. P., Williams D. J., J. Am. Chem. Soc. 1998, 120, 920–931; [Google Scholar]
- 153c. Mason P. E., Bryant W. S., Gibson H. W., Macromolecules 1999, 32, 1559–1569; [Google Scholar]
- 153d. Zhang W., Dichtel W. R., Stieg A. Z., Benítez D., Gimzewski J. K., Heath J. R., Stoddart J. F., Proc. Natl. Acad. Sci. USA 2008, 105, 6514–6519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153e. Ikeda T., Higuchi M., Kurth D. G., J. Am. Chem. Soc. 2009, 131, 9158–9159; [DOI] [PubMed] [Google Scholar]
- 153f. Yu L., Li M., Zhou X.-P., Li D., Inorg. Chem. 2013, 52, 10232–10234; [DOI] [PubMed] [Google Scholar]
- 153g. Savastano M., Bazzicalupi C., Gellini C., Bianchi A., Chem. Commun. 2020, 56, 551–554. [DOI] [PubMed] [Google Scholar]
- 154.
- 154a. Hamers C., Raymo F. M., Stoddart J. F., Eur. J. Org. Chem. 1998, 2109–2117; [Google Scholar]
- 154b. Menzer S., White A. J. P., Williams D. J., Bělohradský M., Hamers C., Raymo F. M., Shipway A. N., Stoddart J. F., Macromolecules 1998, 31, 295–307. [Google Scholar]
- 155. Michaelis L., Hill E. S., J. Gen. Physiol. 1933, 16, 859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kosower E. M., Cotter J. L., J. Am. Chem. Soc. 1964, 86, 5524–5527. [Google Scholar]
- 157.
- 157a. Hünig S., Groß J., Tetrahedron Lett. 1968, 9, 2599–2604; [Google Scholar]
- 157b. Itoh M., Kosower E. M., J. Am. Chem. Soc. 1968, 90, 1843–1849; [Google Scholar]
- 157c. Kosower E. M., Hajdu J., J. Am. Chem. Soc. 1971, 93, 2534–2535. [Google Scholar]
- 158.
- 158a. Preuss K. E., Polyhedron 2014, 79, 1–15; [Google Scholar]
- 158b. Kertesz M., Chem. Eur. J. 2019, 25, 400–416. [DOI] [PubMed] [Google Scholar]
- 159.
- 159a. Evans A. G., Evans J. C., Baker M. W., J. Am. Chem. Soc. 1977, 99, 5882–5884; [Google Scholar]
- 159b. Evans A. G., Evans J. C., Baker M. W., J. Chem. Soc. Perkin Trans. 2 1977, 1787–1789; [Google Scholar]
- 159c. Furue M., Nozakura S.-I., Chem. Lett. 1980, 9, 821–824; [Google Scholar]
- 159d. Mayhew S. G., Müller F., Biochem. Soc. Trans. 1982, 10, 176–177; [Google Scholar]
- 159e. Neta P., Richoux M.-C., Harriman A., J. Chem. Soc. Faraday Trans. 2 1985, 81, 1427–1443; [Google Scholar]
- 159f. Adar E., Degani Y., Goren Z., Willner I., J. Am. Chem. Soc. 1986, 108, 4696–4700; [Google Scholar]
- 159g. Bockman T. M., Kochi J. K., J. Org. Chem. 1990, 55, 4127–4135; [Google Scholar]
- 159h. Park J. W., Choi N. H., Kim J. H., J. Phys. Chem. 1996, 100, 769–774; [Google Scholar]
- 159i. Monk P. M. S., Hodgkinson N. M., Ramzan S. A., Dyes Pigm. 1999, 43, 207–217; [Google Scholar]
- 159j. Tian F., Jiao D., Biedermann F., Scherman O. A., Nat. Commun. 2012, 3, 1207; [DOI] [PubMed] [Google Scholar]
- 159k. Wadhwa K., Nuryyeva S., Fahrenbach A. C., Elhabiri M., Platas-Iglesias C., Trabolsi A., J. Mater. Chem. C 2013, 1, 2302–2307; [Google Scholar]
- 159l. Buck A. T., Paletta J. T., Khindurangala S. A., Beck C. L., Winter A. H., J. Am. Chem. Soc. 2013, 135, 10594–10597; [DOI] [PubMed] [Google Scholar]
- 159m. Stoffelen C., Voskuhl J., Jonkheijm P., Huskens J., Angew. Chem. Int. Ed. 2014, 53, 3400–3404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3468–3472; [Google Scholar]
- 159n. Berville M., Karmazin L., Wytko J. A., Weiss J., Chem. Commun. 2015, 51, 15772–15775; [DOI] [PubMed] [Google Scholar]
- 159o. Geraskina M. R., Dutton A. S., Juetten M. J., Wood S. A., Winter A. H., Angew. Chem. Int. Ed. 2017, 56, 9435–9439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9563–9567. [Google Scholar]
- 160.
- 160a. Bozio R., Zanon I., Girlando A., Pecile C., J. Chem. Phys. 1979, 71, 2282–2293; [Google Scholar]
- 160b. Torrance J. B., Scott B. A., Welber B., Kaufman F. B., Seiden P. E., Phys. Rev. B 1979, 19, 730–741; [Google Scholar]
- 160c. Rosokha S. V., Kochi J. K., J. Am. Chem. Soc. 2007, 129, 828–838. [DOI] [PubMed] [Google Scholar]
- 161.
- 161a. Penneau J. F., Stallman B. J., Kasai P. H., Miller L. L., Chem. Mater. 1991, 3, 791–796; [Google Scholar]
- 161b. Penneau J.-F., Miller L. L., Angew. Chem. Int. Ed. Engl. 1991, 30, 986–987; [Google Scholar]; Angew. Chem. 1991, 103, 1002–1003; [Google Scholar]
- 161c. Andric G., Boas J. F., Bond A. M., Fallon G. D., Ghiggino K. P., Hogan C. F., Hutchison J. A., Lee M. A.-P., Langford S. J., Pilbrow J. R., Troup G. J., Woodward C. P., Aust. J. Chem. 2004, 57, 1011–1019; [Google Scholar]
- 161d. Wu Y., Frasconi M., Gardner D. M., McGonigal P. R., Schneebeli S. T., Wasielewski M. R., Stoddart J. F., Angew. Chem. Int. Ed. 2014, 53, 9476–9481; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9630–9635. [Google Scholar]
- 162.
- 162a. Boyd R. H., Phillips W. D., J. Chem. Phys. 1965, 43, 2927–2929; [Google Scholar]
- 162b. Lü J.-M., Rosokha S. V., Kochi J. K., J. Am. Chem. Soc. 2003, 125, 12161–12171. [DOI] [PubMed] [Google Scholar]
- 163. Aumüller A., Erk P., Klebe G., Hünig S., von Schütz J. U., Werner H.-P., Angew. Chem. Int. Ed. Engl. 1986, 25, 740–741; [Google Scholar]; Angew. Chem. 1986, 98, 759–761. [Google Scholar]
- 164.
- 164a. Goto K., Kubo T., Yamamoto K., Nakasuji K., Sato K., Shiomi D., Takui T., Kubota M., Kobayashi T., Yakusi K., Ouyang J., J. Am. Chem. Soc. 1999, 121, 1619–1620; [Google Scholar]
- 164b. Takano Y., Taniguchi T., Isobe H., Kubo T., Morita Y., Yamamoto K., Nakasuji K., Takui T., Yamaguchi K., J. Am. Chem. Soc. 2002, 124, 11122–11130; [DOI] [PubMed] [Google Scholar]
- 164c. Small D., Zaitsev V., Jung Y., Rosokha S. V., Head-Gordon M., Kochi J. K., J. Am. Chem. Soc. 2004, 126, 13850–13858; [DOI] [PubMed] [Google Scholar]
- 164d. Small D., Rosokha S. V., Kochi J. K., Head-Gordon M., J. Phys. Chem. A 2005, 109, 11261–11267; [DOI] [PubMed] [Google Scholar]
- 164e. Zaitsev V., Rosokha S. V., Head-Gordon M., Kochi J. K., J. Org. Chem. 2006, 71, 520–526; [DOI] [PubMed] [Google Scholar]
- 164f. Suzuki S., Morita Y., Fukui K., Sato K., Shiomi D., Takui T., Nakasuji K., J. Am. Chem. Soc. 2006, 128, 2530–2531; [DOI] [PubMed] [Google Scholar]
- 164g. Cui Z.-H., Lischka H., Beneberu H. Z., Kertesz M., J. Am. Chem. Soc. 2014, 136, 5539–5542. [DOI] [PubMed] [Google Scholar]
- 165. Geuder W., Hünig S., Suchy A., Tetrahedron 1986, 42, 1665–1677. [Google Scholar]
- 166. Trabolsi A., Nat. Chem. Rev. 2021, 5, 442–443. [DOI] [PubMed] [Google Scholar]
- 167. Cai K., Zhang L., Astumian R. D., Stoddart J. F., Nat. Chem. Rev. 2021, 5, 447–465. [DOI] [PubMed] [Google Scholar]
- 168.
- 168a. Qiu Y., Feng Y., Guo Q.-H., Astumian R. D., Stoddart J. F., Chem 2020, 6, 1952–1977; [Google Scholar]
- 168b. Feng Y., Ovalle M., Seale J. S. W., Lee C. K., Kim D. J., Astumian R. D., Stoddart J. F., J. Am. Chem. Soc. 2021, 143, 5569–5591. [DOI] [PubMed] [Google Scholar]
- 169. Cheng C., Cheng T., Xiao H., Krzyaniak M. D., Wang Y., McGonigal P. R., Frasconi M., Barnes J. C., Fahrenbach A. C., Wasielewski M. R., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2016, 138, 8288–8300. [DOI] [PubMed] [Google Scholar]
- 170. Cai K., Shi Y., Cao C., Vemuri S., Cui B., Shen D., Wu H., Zhang L., Qiu Y., Chen H., Jiao Y., Stern C. L., Alsubaie F. M., Xiao H., Li J., Stoddart J. F., Chem. Sci. 2020, 11, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Fahrenbach A. C., Barnes J. C., Lanfranchi D. A., Li H., Coskun A., Gassensmith J. J., Liu Z., Benítez D., Trabolsi A., W. A. Goddard III , Elhabiri M., Stoddart J. F., J. Am. Chem. Soc. 2012, 134, 3061–3072. [DOI] [PubMed] [Google Scholar]
- 172.
- 172a. D. E. Koshland, Jr. , Proc. Natl. Acad. Sci. USA 1958, 44, 98–104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172b. D. E. Koshland, Jr. , Angew. Chem. Int. Ed. Engl. 1994, 33, 2375–2378; [Google Scholar]; Angew. Chem. 1994, 106, 2468–2472. [Google Scholar]
- 173. Anamimoghadam O., Jones L. O., Cooper J. A., Beldjoudi Y., Nguyen M. T., Liu W., Krzyaniak M. D., Pezzato C., Stern C. L., Patel H. A., Wasielewski M. R., Schatz G. C., Stoddart J. F., J. Am. Chem. Soc. 2021, 143, 163–175. [DOI] [PubMed] [Google Scholar]
- 174.
- 174a. Lipke M. C., Cheng T., Wu Y., Arslan H., Xiao H., Wasielewski M. R., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2017, 139, 3986–3998; [DOI] [PubMed] [Google Scholar]
- 174b. Lipke M. C., Wu Y., Roy I., Wang Y., Wasielewski M. R., Stoddart J. F., ACS Cent. Sci. 2018, 4, 362–371; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174c. Cai K., Lipke M. C., Liu Z., Nelson J., Cheng T., Shi Y., Cheng C., Shen D., Han J.-M., Vemuri S., Feng Y., Stern C. L., W. A. Goddard III , Wasielewski M. R., Stoddart J. F., Nat. Commun. 2018, 9, 5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Chen X.-Y., Mao H., Feng Y., Cai K., Shen D., Wu H., Zhang L., Zhao X., Chen H., Song B., Jiao Y., Wu Y., Stern C. L., Wasielewski M. R., Stoddart J. F., Angew. Chem. Int. Ed. 2021, 60, 25454–25462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 25658–25666. [Google Scholar]
- 176. Frasconi M., Fernando I. R., Wu Y., Liu Z., Liu W.-G., Dyar S. M., Barin G., Wasielewski M. R., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2015, 137, 11057–11068. [DOI] [PubMed] [Google Scholar]
- 177. Liu Z., Frasconi M., Liu W.-G., Zhang Y., Dyar S. M., Shen D., Sarjeant A. A., W. A. Goddard III , Wasielewski M. R., Stoddart J. F., J. Am. Chem. Soc. 2018, 140, 9387–9391. [DOI] [PubMed] [Google Scholar]
- 178. Li H., Fahrenbach A. C., Dey S. K., Basu S., Trabolsi A., Zhu Z., Botros Y. Y., Stoddart J. F., Angew. Chem. Int. Ed. 2010, 49, 8260–8265; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8436–8441. [Google Scholar]
- 179. Li H., Zhu Z., Fahrenbach A. C., Savoie B. M., Ke C., Barnes J. C., Lei J., Zhao Y.-L., Lilley L. M., Marks T. J., Ratner M. A., Stoddart J. F., J. Am. Chem. Soc. 2013, 135, 456–467. [DOI] [PubMed] [Google Scholar]
- 180. Barnes J. C., Fahrenbach A. C., Cao D., Dyar S. M., Frasconi M., Giesener M. A., Benítez D., Tkatchouk E., Chernyashevskyy O., Shin W. H., Li H., Sampath S., Stern C. L., Sarjeant A. A., Hartlieb K. J., Liu Z., Carmieli R., Botros Y. Y., Choi J. W., Slawin A. M. Z., Ketterson J. B., Wasielewski M. R., W. A. Goddard III , Stoddart J. F., Science 2013, 339, 429–433. [DOI] [PubMed] [Google Scholar]
- 181. Barnes J. C., Frasconi M., Young R. M., Khdary N. H., Liu W.-G., Dyar S. M., McGonigal P. R., Gibbs-Hall I. C., Diercks C. S., Sarjeant A. A., Stern C. L., W. A. Goddard III , Wasielewski M. R., Stoddart J. F., J. Am. Chem. Soc. 2014, 136, 10569–10572. [DOI] [PubMed] [Google Scholar]
- 182. Sun J., Liu Z., Liu W.-G., Wu Y., Wang Y., Barnes J. C., Hermann K. R., W. A. Goddard III , Wasielewski M. R., Stoddart J. F., J. Am. Chem. Soc. 2017, 139, 12704–12709. [DOI] [PubMed] [Google Scholar]
- 183. Cai K., Mao H., Liu W.-G., Qiu Y., Shi Y., Zhang L., Shen D., Chen H., Jiao Y., Wu H., Liu Z., Feng Y., Stern C. L., Wasielewski M. R., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2020, 142, 7190–7197. [DOI] [PubMed] [Google Scholar]
- 184. Wang Y., Sun J., Liu Z., Nassar M. S., Botros Y. Y., Stoddart J. F., Angew. Chem. Int. Ed. 2016, 55, 12387–12392; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12575–12580. [Google Scholar]
- 185. Nguyen M. T., Ferris D. P., Pezzato C., Wang Y., Stoddart J. F., Chem 2018, 4, 2329–2344. [Google Scholar]
- 186. Gibbs-Hall I. C., Vermeulen N. A., Dale E. J., Henkelis J. J., Blackburn A. K., Barnes J. C., Stoddart J. F., J. Am. Chem. Soc. 2015, 137, 15640–15643. [DOI] [PubMed] [Google Scholar]
- 187. Fyfe M. C. T., Stoddart J. F., Acc. Chem. Res. 1997, 30, 393–401. [Google Scholar]
- 188.
- 188a. Vignon S. A., Stoddart J. F., Collect. Czech. Chem. Commun. 2005, 70, 1493–1576; [Google Scholar]
- 188b. Neal E. A., Goldup S. M., Chem. Commun. 2014, 50, 5128–5142. [DOI] [PubMed] [Google Scholar]
- 189. Ashton P. R., Preece J. A., Stoddart J. F., Tolley M. S., White A. J. P., Williams D. J., Synthesis 1994, 1344–1352. [Google Scholar]
- 190. Vignon S. A., Wong J., Tseng H.-R., Stoddart J. F., Org. Lett. 2004, 6, 1095–1098. [DOI] [PubMed] [Google Scholar]
- 191.
- 191a. Fahrenbach A. C., Bruns C. J., Cao D., Stoddart J. F., Acc. Chem. Res. 2012, 45, 1581–1592; [DOI] [PubMed] [Google Scholar]
- 191b. Fahrenbach A. C., Bruns C. J., Li H., Trabolsi A., Coskun A., Stoddart J. F., Acc. Chem. Res. 2014, 47, 482–493. [DOI] [PubMed] [Google Scholar]
- 192. Stoddart J. F., Colquhoun H. M., Tetrahedron 2008, 64, 8231–8263. [Google Scholar]
- 193.
- 193a. Hmadeh M., Fahrenbach A. C., Basu S., Trabolsi A., Benítez D., Li H., Albrecht-Gary A.-M., Elhabiri M., Stoddart J. F., Chem. Eur. J. 2011, 17, 6076–6087; [DOI] [PubMed] [Google Scholar]
- 193b. Li H., Zhao Y.-L., Fahrenbach A. C., Kim S.-Y., Paxton W. F., Stoddart J. F., Org. Biomol. Chem. 2011, 9, 2240–2250. [DOI] [PubMed] [Google Scholar]
- 194.
- 194a. Balzani V., Credi A., Raymo F. M., Stoddart J. F., Angew. Chem. Int. Ed. 2000, 39, 3348–3391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3484–3530; [Google Scholar]
- 194b. Abendroth J. M., Bushuyev O. S., Weiss P. S., Barrett C. J., ACS Nano 2015, 9, 7746–7768; [DOI] [PubMed] [Google Scholar]
- 194c. Kay E. R., Leigh D. A., Angew. Chem. Int. Ed. 2015, 54, 10080–10088; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10218–10226; [Google Scholar]
- 194d. Lancia F., Ryabchun A., Katsonis N., Nat. Chem. Rev. 2019, 3, 536–551; [Google Scholar]
- 194e. Dattler D., Fuks G., Heiser J., Moulin E., Perrot A., Yao X., Giuseppone N., Chem. Rev. 2020, 120, 310–433. [DOI] [PubMed] [Google Scholar]
- 195.
- 195a. Coskun A., Spruell J. M., Barin G., Dichtel W. R., Flood A. H., Botros Y. Y., Stoddart J. F., Chem. Soc. Rev. 2012, 41, 4827–4859; [DOI] [PubMed] [Google Scholar]
- 195b. Coskun A., Banaszak M., Astumian R. D., Stoddart J. F., Grzybowski B. A., Chem. Soc. Rev. 2012, 41, 19–30. [DOI] [PubMed] [Google Scholar]
- 196. Pease A. R., Jeppesen J. O., Stoddart J. F., Luo Y., Collier C. P., Heath J. R., Acc. Chem. Res. 2001, 34, 433–444. [DOI] [PubMed] [Google Scholar]
- 197. Bissell R. A., Córdova E., Kaifer A. E., Stoddart J. F., Nature 1994, 369, 133–137. [Google Scholar]
- 198. Matthews O. A., Raymo F. M., Stoddart J. F., White A. J. P., Williams D. J., New J. Chem. 1998, 22, 1131–1134. [Google Scholar]
- 199. Ashton P. R., Blower M., Philp D., Spencer N., Stoddart J. F., Tolley M. S., Ballardini R., Ciano M., Balzani V., Gandolfi M. T., Prodi L., McLean C. H., New J. Chem. 1993, 17, 689–695. [Google Scholar]
- 200. Raymo F. M., Houk K. N., Stoddart J. F., J. Org. Chem. 1998, 63, 6523–6528. [Google Scholar]
- 201. Jeppesen J. O., Perkins J., Becher J., Stoddart J. F., Angew. Chem. Int. Ed. 2001, 40, 1216–1221; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1256–1261. [Google Scholar]
- 202. Sørensen A., Andersen S. S., Flood A. H., Jeppesen J. O., Chem. Commun. 2013, 49, 5936–5938. [DOI] [PubMed] [Google Scholar]
- 203. Laursen B. W., Nygaard S., Jeppesen J. O., Stoddart J. F., Org. Lett. 2004, 6, 4167–4170. [DOI] [PubMed] [Google Scholar]
- 204. Brough B., Northrop B. H., Schmidt J. J., Tseng H.-R., Houk K. N., Stoddart J. F., Ho C.-M., Proc. Natl. Acad. Sci. USA 2006, 103, 8583–8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.
- 205a. Sluysmans D., Devaux F., Bruns C. J., Stoddart J. F., Duwez A.-S., Proc. Natl. Acad. Sci. USA 2018, 115, 9362–9366; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205b. Sluysmans D., Hubert S., Bruns C. J., Zhu Z., Stoddart J. F., Duwez A.-S., Nat. Nanotechnol. 2018, 13, 209–213. [DOI] [PubMed] [Google Scholar]
- 206. Li H., Fahrenbach A. C., Coskun A., Zhu Z., Barin G., Zhao Y.-L., Botros Y. Y., Sauvage J.-P., Stoddart J. F., Angew. Chem. Int. Ed. 2011, 50, 6782–6788; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6914–6920. [Google Scholar]
- 207.
- 207a. Seiler M., Duerr H., Willner I., Joselevich E., Doron A., Stoddart J. F., J. Am. Chem. Soc. 1994, 116, 3399–3404; [Google Scholar]
- 207b. Kropf M., Joselevich E., Dürr H., Willner I., J. Am. Chem. Soc. 1996, 118, 655–665; [Google Scholar]
- 207c. Fathalla M., Barnes J. C., Young R. M., Hartlieb K. J., Dyar S. M., Eaton S. W., Sarjeant A. A., Co D. T., Wasielewski M. R., Stoddart J. F., Chem. Eur. J. 2014, 20, 14690–14697; [DOI] [PubMed] [Google Scholar]
- 207d. David E., Born R., Kaganer E., Joselevich E., Dürr H., Willner I., J. Am. Chem. Soc. 1997, 119, 7778–7790. [Google Scholar]
- 208.
- 208a. Ballardini R., Balzani V., Gandolfi M. T., Prodi L., Venturi M., Philp D., Ricketts H. G., Stoddart J. F., Angew. Chem. Int. Ed. Engl. 1993, 32, 1301–1303; [Google Scholar]; Angew. Chem. 1993, 105, 1362–1364; [Google Scholar]
- 208b. Benniston A. C., Harriman A., Angew. Chem. Int. Ed. Engl. 1993, 32, 1459–1461; [Google Scholar]; Angew. Chem. 1993, 105, 1553–1555; [Google Scholar]
- 208c. Chia S., Cao J., Stoddart J. F., Zink J. I., Angew. Chem. Int. Ed. 2001, 40, 2447–2451; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2513–2517; [Google Scholar]
- 208d. Abraham W., Grubert L., Grummt U. W., Buck K., Chem. Eur. J. 2004, 10, 3562–3568; [DOI] [PubMed] [Google Scholar]
- 208e. Abraham W., Grubert L., Schmidt-Schäffer S., Buck K., Eur. J. Org. Chem. 2005, 374–386; [Google Scholar]
- 208f. Saha S., Johansson E., Flood A. H., Tseng H.-R., Zink J. I., Stoddart J. F., Chem. Eur. J. 2005, 11, 6846–6858; [DOI] [PubMed] [Google Scholar]
- 208g. Saha S., Johansson L. E., Flood A. H., Tseng H.-R., Zink J. I., Stoddart J. F., Small 2005, 1, 87–90; [DOI] [PubMed] [Google Scholar]
- 208h. Schmidt-Schäffer S., Grubert L., Grummt U. W., Buck K., Abraham W., Eur. J. Org. Chem. 2006, 378–398; [Google Scholar]
- 208i. Abraham W., Buck K., Orda-Zgadzaj M., Schmidt-Schäffer S., Grummt U.-W., Chem. Commun. 2007, 3094–3096; [DOI] [PubMed] [Google Scholar]
- 208j. Abraham W., Wlosnewski A., Buck K., Jacob S., Org. Biomol. Chem. 2009, 7, 142–154; [DOI] [PubMed] [Google Scholar]
- 208k. Coskun A., Friedman D. C., Li H., Patel K., Khatib H. A., Stoddart J. F., J. Am. Chem. Soc. 2009, 131, 2493–2495; [DOI] [PubMed] [Google Scholar]
- 208l. Vetter A., Abraham W., Org. Biomol. Chem. 2010, 8, 4666–4681; [DOI] [PubMed] [Google Scholar]
- 208m. Duo Y., Jacob S., Abraham W., Org. Biomol. Chem. 2011, 9, 3549–3559; [DOI] [PubMed] [Google Scholar]
- 208n. Scarpantonio L., Tron A., Destribats C., Godard P., McClenaghan N. D., Chem. Commun. 2012, 48, 3981–3983; [DOI] [PubMed] [Google Scholar]
- 208o. Zhan T.-G., Yun M.-Y., Lin J.-L., Yu X.-Y., Zhang K.-D., Chem. Commun. 2016, 52, 14085–14088. [DOI] [PubMed] [Google Scholar]
- 209. Sun J., Wu Y., Liu Z., Cao D., Wang Y., Cheng C., Chen D., Wasielewski M. R., Stoddart J. F., J. Phys. Chem. A 2015, 119, 6317–6325. [DOI] [PubMed] [Google Scholar]
- 210. Zheng X., Zhang Y., Cao N., Li X., Zhang S., Du R., Wang H., Ye Z., Wang Y., Cao F., Li H., Hong X., Sue A. C. H., Yang C., Liu W.-G., Li H., Nat. Commun. 2018, 9, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Zhang Y., Chen Q., Wang Y., Zheng X., Wang H., Cao F., Sue A. C. H., Li H., Chem. Commun. 2020, 56, 11887–11890. [DOI] [PubMed] [Google Scholar]
- 212. Wang Y., Cheng T., Sun J., Liu Z., Frasconi M., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2018, 140, 13827–13834. [DOI] [PubMed] [Google Scholar]
- 213.
- 213a. Credi A., Montalti M., Balzani V., Langford S. J., Raymo F. M., Stoddart J. F., New J. Chem. 1998, 22, 1061–1065; [Google Scholar]
- 213b. Tseng H.-R., Vignon S. A., Stoddart J. F., Angew. Chem. Int. Ed. 2003, 42, 1491–1495; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 1529–1533; [Google Scholar]
- 213c. Kang S., Vignon S. A., Tseng H.-R., Stoddart J. F., Chem. Eur. J. 2004, 10, 2555–2564; [DOI] [PubMed] [Google Scholar]
- 213d. Nygaard S., Laursen B. W., Flood A. H., Hansen C. N., Jeppesen J. O., Stoddart J. F., Chem. Commun. 2006, 144–146; [DOI] [PubMed] [Google Scholar]
- 213e. Saha S., Flood A. H., Stoddart J. F., Impellizzeri S., Silvi S., Venturi M., Credi A., J. Am. Chem. Soc. 2007, 129, 12159–12171; [DOI] [PubMed] [Google Scholar]
- 213f. Aprahamian I., Dichtel W. R., Ikeda T., Heath J. R., Stoddart J. F., Org. Lett. 2007, 9, 1287–1290; [DOI] [PubMed] [Google Scholar]
- 213g. Zhao Y.-L., Aprahamian I., Trabolsi A., Erina N., Stoddart J. F., J. Am. Chem. Soc. 2008, 130, 6348–6350; [DOI] [PubMed] [Google Scholar]
- 213h. Ye T., Kumar A. S., Saha S., Takami T., Huang T. J., Stoddart J. F., Weiss P. S., ACS Nano 2010, 4, 3697–3701; [DOI] [PubMed] [Google Scholar]
- 213i. Trabolsi A., Fahrenbach A. C., Dey S. K., Share A. I., Friedman D. C., Basu S., Gasa T. B., Khashab N. M., Saha S., Aprahamian I., Khatib H. A., Flood A. H., Stoddart J. F., Chem. Commun. 2010, 46, 871–873; [DOI] [PubMed] [Google Scholar]
- 213j. Fahrenbach A. C., Barnes J. C., Li H., Benítez D., Basuray A. N., Fang L., Sue C.-H., Barin G., Dey S. K., W. A. Goddard III , Stoddart J. F., Proc. Natl. Acad. Sci. USA 2011, 108, 20416–20421; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213k. Fahrenbach A. C., Zhu Z., Cao D., Liu W.-G., Li H., Dey S. K., Basu S., Trabolsi A., Botros Y. Y., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2012, 134, 16275–16288; [DOI] [PubMed] [Google Scholar]
- 213l. Barnes J. C., Fahrenbach A. C., Dyar S. M., Frasconi M., Giesener M. A., Zhu Z., Liu Z., Hartlieb K. J., Carmieli R., Wasielewski M. R., Stoddart J. F., Proc. Natl. Acad. Sci. USA 2012, 109, 11546–11551; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213m. Andersen S. S., Share A. I., Poulsen B. L. C., Kørner M., Duedal T., Benson C. R., Hansen S. W., Jeppesen J. O., Flood A. H., J. Am. Chem. Soc. 2014, 136, 6373–6384; [DOI] [PubMed] [Google Scholar]
- 213n. Manoni R., Romano F., Casati C., Franchi P., Mezzina E., Lucarini M., Org. Chem. Front. 2014, 1, 477–483; [Google Scholar]
- 213o. Witus L. S., Hartlieb K. J., Wang Y., Prokofjevs A., Frasconi M., Barnes J. C., Dale E. J., Fahrenbach A. C., Stoddart J. F., Org. Biomol. Chem. 2014, 12, 6089–6093; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213p. Romano F., Manoni R., Franchi P., Mezzina E., Lucarini M., Chem. Eur. J. 2015, 21, 2775–2779; [DOI] [PubMed] [Google Scholar]
- 213q. Wu Y., Frasconi M., Liu W.-G., Young R. M., W. A. Goddard III , Wasielewski M. R., Stoddart J. F., J. Am. Chem. Soc. 2020, 142, 11835–11846; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213r. Jensen M., Kristensen R., Andersen S. S., Bendixen D., Jeppesen J. O., Chem. Eur. J. 2020, 26, 6165–6175. [DOI] [PubMed] [Google Scholar]
- 214.
- 214a. Asakawa M., Ashton P. R., Balzani V., Boyd S. E., Credi A., Mattersteig G., Menzer S., Montalti M., Raymo F. M., Ruffilli C., Stoddart J. F., Venturi M., Williams D. J., Eur. J. Org. Chem. 1999, 985–994; [Google Scholar]
- 214b. Ballardini R., Balzani V., Fabio A. D., Gandolfi M. T., Becher J., Lau J., Nielsen M. B., Stoddart J. F., New J. Chem. 2001, 25, 293–298; [Google Scholar]
- 214c. Deng W.-Q., Flood A. H., Stoddart J. F., W. A. Goddard III , J. Am. Chem. Soc. 2005, 127, 15994–15995; [DOI] [PubMed] [Google Scholar]
- 214d. Ikeda T., Saha S., Aprahamian I., Leung K. C.-F., Williams A., Deng W.-Q., Flood A. H., W. A. Goddard III , Stoddart J. F., Chem. Asian J. 2007, 2, 76–93; [DOI] [PubMed] [Google Scholar]
- 214e. Fang L., Wang C., Fahrenbach A. C., Trabolsi A., Botros Y. Y., Stoddart J. F., Angew. Chem. Int. Ed. 2011, 50, 1805–1809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1845–1849; [Google Scholar]
- 214f. Zhu Z., Fahrenbach A. C., Li H., Barnes J. C., Liu Z., Dyar S. M., Zhang H., Lei J., Carmieli R., Sarjeant A. A., Stern C. L., Wasielewski M. R., Stoddart J. F., J. Am. Chem. Soc. 2012, 134, 11709–11720; [DOI] [PubMed] [Google Scholar]
- 214g. Wang C., Cao D., Fahrenbach A. C., Grunder S., Dey S. K., Sarjeant A. A., Stoddart J. F., Chem. Commun. 2012, 48, 9245–9247. [DOI] [PubMed] [Google Scholar]
- 215. Cai K., Cui B., Song B., Wang H., Qiu Y., Jones L. O., Liu W., Shi Y., Vemuri S., Shen D., Jiao T., Zhang L., Wu H., Chen H., Jiao Y., Wang Y., Stern C. L., Li H., Schatz G. C., Li X., Stoddart J. F., Chem 2021, 7, 174–189. [Google Scholar]
- 216. Bruns C. J., Frasconi M., Iehl J., Hartlieb K. J., Schneebeli S. T., Cheng C., Stupp S. I., Stoddart J. F., J. Am. Chem. Soc. 2014, 136, 4714–4723. [DOI] [PubMed] [Google Scholar]
- 217.
- 217a. Liu Y., Flood A. H., Stoddart J. F., J. Am. Chem. Soc. 2004, 126, 9150–9151; [DOI] [PubMed] [Google Scholar]
- 217b. Wang Y., Sun J., Liu Z., Nassar M. S., Botros Y. Y., Stoddart J. F., Chem. Sci. 2017, 8, 2562–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Wang Y., Frasconi M., Liu W.-G., Sun J., Wu Y., Nassar M. S., Botros Y. Y., W. A. Goddard III , Wasielewski M. R., Stoddart J. F., ACS Cent. Sci. 2016, 2, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Zhan T.-G., Wu L., Zhao Z., Zhou Z.-B., Yun M.-Y., Wei J., Zheng S.-T., Yin H.-H., Zhang K.-D., Chem. Commun. 2017, 53, 5396–5399. [DOI] [PubMed] [Google Scholar]
- 220.
- 220a. Cheng C., McGonigal P. R., Stoddart J. F., Astumian R. D., ACS Nano 2015, 9, 8672–8688; [DOI] [PubMed] [Google Scholar]
- 220b. Pezzato C., Cheng C., Stoddart J. F., Astumian R. D., Chem. Soc. Rev. 2017, 46, 5491–5507; [DOI] [PubMed] [Google Scholar]
- 220c. Astumian R. D., Proc. Natl. Acad. Sci. USA 2018, 115, 9405–9413; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220d. Astumian R. D., Nat. Commun. 2019, 10, 3837; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220e. Astumian R. D., Pezzato C., Feng Y., Qiu Y., McGonigal P. R., Cheng C., Stoddart J. F., Mater. Chem. Front. 2020, 4, 1304–1314. [Google Scholar]
- 221.
- 221a. Browne W. R., Feringa B. L., Nat. Nanotechnol. 2006, 1, 25–35; [DOI] [PubMed] [Google Scholar]
- 221b. Balzani V., Credi A., Venturi M., Chem. Soc. Rev. 2009, 38, 1542–1550; [DOI] [PubMed] [Google Scholar]
- 221c. Erbas-Cakmak S., Leigh D. A., McTernan C. T., Nussbaumer A. L., Chem. Rev. 2015, 115, 10081–10206; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221d. Kassem S., van Leeuwen T., Lubbe A. S., Wilson M. R., Feringa B. L., Leigh D. A., Chem. Soc. Rev. 2017, 46, 2592–2621; [DOI] [PubMed] [Google Scholar]
- 221e. Feringa B. L., Angew. Chem. Int. Ed. 2017, 56, 11060–11078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11206–11226; [Google Scholar]
- 221f. van Leeuwen T., Lubbe A. S., Štacko P., Wezenberg S. J., Feringa B. L., Nat. Chem. Rev. 2017, 1, 0096; [Google Scholar]
- 221g. Baroncini M., Silvi S., Credi A., Chem. Rev. 2020, 120, 200–268; [DOI] [PubMed] [Google Scholar]
- 221h. García-López V., Liu D., Tour J. M., Chem. Rev. 2020, 120, 79–124; [DOI] [PubMed] [Google Scholar]
- 221i. Aprahamian I., ACS Cent. Sci. 2020, 6, 347–358; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221j. Heard A. W., Goldup S. M., ACS Cent. Sci. 2020, 6, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.
- 222a. Koumura N., Zijlstra R. W. J., van Delden R. A., Harada N., Feringa B. L., Nature 1999, 401, 152–155; [DOI] [PubMed] [Google Scholar]
- 222b. Leigh D. A., Wong J. K. Y., Dehez F., Zerbetto F., Nature 2003, 424, 174–179; [DOI] [PubMed] [Google Scholar]
- 222c. Hernández J. V., Kay E. R., Leigh D. A., Science 2004, 306, 1532–1537; [DOI] [PubMed] [Google Scholar]
- 222d. Serreli V., Lee C.-F., Kay E. R., Leigh D. A., Nature 2007, 445, 523–527; [DOI] [PubMed] [Google Scholar]
- 222e. Ragazzon G., Baroncini M., Silvi S., Venturi M., Credi A., Nat. Nanotechnol. 2015, 10, 70–75; [DOI] [PubMed] [Google Scholar]
- 222f. Wilson M. R., Solà J., Carlone A., Goldup S. M., Lebrasseur N., Leigh D. A., Nature 2016, 534, 235–240; [DOI] [PubMed] [Google Scholar]
- 222g. Erbas-Cakmak S., Fielden S. D. P., Karaca U., Leigh D. A., McTernan C. T., Tetlow D. J., Wilson M. R., Science 2017, 358, 340–343. [DOI] [PubMed] [Google Scholar]
- 223. Li H., Cheng C., McGonigal P. R., Fahrenbach A. C., Frasconi M., Liu W.-G., Zhu Z., Zhao Y., Ke C., Lei J., Young R. M., Dyar S. M., Co D. T., Yang Y.-W., Botros Y. Y., W. A. Goddard III , Wasielewski M. R., Astumian R. D., Stoddart J. F., J. Am. Chem. Soc. 2013, 135, 18609–18620. [DOI] [PubMed] [Google Scholar]
- 224. Cheng C., McGonigal P. R., Schneebeli S. T., Li H., Vermeulen N. A., Ke C., Stoddart J. F., Nat. Nanotechnol. 2015, 10, 547–553. [DOI] [PubMed] [Google Scholar]
- 225. Wang Y., Frasconi M., Stoddart J. F., ACS Cent. Sci. 2017, 3, 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Cheng C., McGonigal P. R., Liu W.-G., Li H., Vermeulen N. A., Ke C., Frasconi M., Stern C. L., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2014, 136, 14702–14705. [DOI] [PubMed] [Google Scholar]
- 227. Pezzato C., Nguyen M. T., Cheng C., Kim D. J., Otley M. T., Stoddart J. F., Tetrahedron 2017, 73, 4849–4857. [Google Scholar]
- 228. Pezzato C., Nguyen M. T., Kim D. J., Anamimoghadam O., Mosca L., Stoddart J. F., Angew. Chem. Int. Ed. 2018, 57, 9325–9329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9469–9473. [Google Scholar]
- 229. Qiu Y., Zhang L., Pezzato C., Feng Y., Li W., Nguyen M. T., Cheng C., Shen D., Guo Q.-H., Shi Y., Cai K., Alsubaie F. M., Astumian R. D., Stoddart J. F., J. Am. Chem. Soc. 2019, 141, 17472–17476. [DOI] [PubMed] [Google Scholar]
- 230. Guo Q.-H., Qiu Y., Kuang X., Liang J., Feng Y., Zhang L., Jiao Y., Shen D., Astumian R. D., Stoddart J. F., J. Am. Chem. Soc. 2020, 142, 14443–14449. [DOI] [PubMed] [Google Scholar]
- 231. Li X., David A. H. G., Zhang L., Song B., Jiao Y., Sluysmans D., Qiu Y., Wu Y., Zhao X., Feng Y., Mosca L., Stoddart J. F., J. Am. Chem. Soc. 2022, 144, 3572–3579. [DOI] [PubMed] [Google Scholar]
- 232. Qiu Y., Song B., Pezzato C., Shen D., Liu W., Zhang L., Feng Y., Guo Q.-H., Cai K., Li W., Chen H., Nguyen M. T., Shi Y., Cheng C., Astumian R. D., Li X., Stoddart J. F., Science 2020, 368, 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Cai K., Shi Y., Zhuang G.-W., Zhang L., Qiu Y., Shen D., Chen H., Jiao Y., Wu H., Cheng C., Stoddart J. F., J. Am. Chem. Soc. 2020, 142, 10308–10313. [DOI] [PubMed] [Google Scholar]
- 234. Flood A. H., Stoddart J. F., Steuerman D. W., Heath J. R., Science 2004, 306, 2055–2056. [DOI] [PubMed] [Google Scholar]
- 235. Chen H., Stoddart J. F., Nat. Rev. Mater. 2021, 6, 804–828. [Google Scholar]
- 236.
- 236a. Collier C. P., Wong E. W., Belohradský M., Raymo F. M., Stoddart J. F., Kuekes P. J., Williams R. S., Heath J. R., Science 1999, 285, 391–394; [DOI] [PubMed] [Google Scholar]
- 236b. Wong E. W., Collier C. P., Běhloradský M., Raymo F. M., Stoddart J. F., Heath J. R., J. Am. Chem. Soc. 2000, 122, 5831–5840; [Google Scholar]
- 236c. Brown C. L., Jonas U., Preece J. A., Ringsdorf H., Seitz M., Stoddart J. F., Langmuir 2000, 16, 1924–1930. [Google Scholar]
- 237.
- 237a. Collier C. P., Mattersteig G., Wong E. W., Luo Y., Beverly K., Sampaio J., Raymo F. M., Stoddart J. F., Heath J. R., Science 2000, 289, 1172–1175; [DOI] [PubMed] [Google Scholar]
- 237b. Asakawa M., Higuchi M., Mattersteig G., Nakamura T., Pease A. R., Raymo F. M., Shimizu T., Stoddart J. F., Adv. Mater. 2000, 12, 1099–1102; [Google Scholar]
- 237c. Huang T. J., Tseng H.-R., Sha L., Lu W., Brough B., Flood A. H., Yu B.-D., Celestre P. C., Chang J. P., Stoddart J. F., Ho C.-M., Nano Lett. 2004, 4, 2065–2071. [Google Scholar]
- 238.
- 238a. Collier C. P., Jeppesen J. O., Luo Y., Perkins J., Wong E. W., Heath J. R., Stoddart J. F., J. Am. Chem. Soc. 2001, 123, 12632–12641; [DOI] [PubMed] [Google Scholar]
- 238b. Lee I. C., Frank C. W., Yamamoto T., Tseng H.-R., Flood A. H., Stoddart J. F., Jeppesen J. O., Langmuir 2004, 20, 5809–5828. [DOI] [PubMed] [Google Scholar]
- 239. Talham D. R., Chem. Rev. 2004, 104, 5479–5502. [DOI] [PubMed] [Google Scholar]
- 240.
- 240a. Jeppesen J. O., Nielsen K. A., Perkins J., Vignon S. A., Di Fabio A., Ballardini R., Gandolfi M. T., Venturi M., Balzani V., Becher J., Stoddart J. F., Chem. Eur. J. 2003, 9, 2982–3007; [Google Scholar]
- 240b. Steuerman D. W., Tseng H.-R., Peters A. J., Flood A. H., Jeppesen J. O., Nielsen K. A., Stoddart J. F., Heath J. R., Angew. Chem. Int. Ed. 2004, 43, 6486–6491; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 6648–6653; [Google Scholar]
- 240c. Tseng H.-R., Vignon S. A., Celestre P. C., Perkins J., Jeppesen J. O., Di Fabio A., Ballardini R., Gandolfi M. T., Venturi M., Balzani V., Stoddart J. F., Chem. Eur. J. 2004, 10, 155–172; [DOI] [PubMed] [Google Scholar]
- 240d. Jeppesen J. O., Nygaard S., Vignon S. A., Stoddart J. F., Eur. J. Org. Chem. 2005, 196–220; [Google Scholar]
- 240e. Nørgaard K., Laursen B. W., Nygaard S., Kjaer K., Tseng H.-R., Flood A. H., Stoddart J. F., Bjørnholm T., Angew. Chem. Int. Ed. 2005, 44, 7035–7039; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7197–7201. [Google Scholar]
- 241. Choi J. W., Flood A. H., Steuerman D. W., Nygaard S., Braunschweig A. B., Moonen N. N. P., Laursen B. W., Luo Y., DeIonno E., Peters A. J., Jeppesen J. O., Xu K., Stoddart J. F., Heath J. R., Chem. Eur. J. 2006, 12, 261–279. [DOI] [PubMed] [Google Scholar]
- 242.
- 242a. Aprahamian I., Yasuda T., Ikeda T., Saha S., Dichtel W. R., Isoda K., Kato T., Stoddart J. F., Angew. Chem. Int. Ed. 2007, 46, 4675–4679; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 4759–4763; [Google Scholar]
- 242b. Yasuda T., Tanabe K., Tsuji T., Coti K. K., Aprahamian I., Stoddart J. F., Kato T., Chem. Commun. 2010, 46, 1224–1226. [DOI] [PubMed] [Google Scholar]
- 243.
- 243a. Tseng H.-R., Wu D., Fang N. X., Zhang X., Stoddart J. F., ChemPhysChem 2004, 5, 111–116; [DOI] [PubMed] [Google Scholar]
- 243b. Jang S. S., Jang Y. H., Kim Y.-H., W. A. Goddard III , Flood A. H., Laursen B. W., Tseng H.-R., Stoddart J. F., Jeppesen J. O., Choi J. W., Steuerman D. W., DeIonno E., Heath J. R., J. Am. Chem. Soc. 2005, 127, 1563–1575; [DOI] [PubMed] [Google Scholar]
- 243c. Zheng Y. B., Yang Y.-W., Jensen L., Fang L., Juluri B. K., Flood A. H., Weiss P. S., Stoddart J. F., Huang T. J., Nano Lett. 2009, 9, 819–825. [DOI] [PubMed] [Google Scholar]
- 244.
- 244a. Klajn R., Fang L., Coskun A., Olson M. A., Wesson P. J., Stoddart J. F., Grzybowski B. A., J. Am. Chem. Soc. 2009, 131, 4233–4235; [DOI] [PubMed] [Google Scholar]
- 244b. Coskun A., Wesson P. J., Klajn R., Trabolsi A., Fang L., Olson M. A., Dey S. K., Grzybowski B. A., Stoddart J. F., J. Am. Chem. Soc. 2010, 132, 4310–4320. [DOI] [PubMed] [Google Scholar]
- 245. Luo Y., Collier C. P., Jeppesen J. O., Nielsen K. A., DeIonno E., Ho G., Perkins J., Tseng H.-R., Yamamoto T., Stoddart J. F., Heath J. R., ChemPhysChem 2002, 3, 519–525. [DOI] [PubMed] [Google Scholar]
- 246. Chen Y., Jung G.-Y., Ohlberg D. A. A., Li X., Stewart D. R., Jeppesen J. O., Nielsen K. A., Stoddart J. F., Williams R. S., Nanotechnology 2003, 14, 462–468. [Google Scholar]
- 247. Green J. E., Choi J. W., Boukai A., Bunimovich Y., Johnston-Halperin E., DeIonno E., Luo Y., Sheriff B. A., Xu K., Shin Y. S., Tseng H.-R., Stoddart J. F., Heath J. R., Nature 2007, 445, 414–417. [DOI] [PubMed] [Google Scholar]
- 248. Stewart D. R., Ohlberg D. A. A., Beck P. A., Chen Y., Williams R. S., Jeppesen J. O., Nielsen K. A., Stoddart J. F., Nano Lett. 2004, 4, 133–136. [Google Scholar]
- 249.This comment was made by Norma Stoddart towards the end of her life when her husband was explaining the difference between the UCLA/CIT and HP devices.
- 250.
- 250a. Witlicki E. H., Andersen S. S., Hansen S. W., Jeppesen J. O., Wong E. W., Jensen L., Flood A. H., J. Am. Chem. Soc. 2010, 132, 6099–6107; [DOI] [PubMed] [Google Scholar]
- 250b. Witlicki E. H., Johnsen C., Hansen S. W., Silverstein D. W., Bottomley V. J., Jeppesen J. O., Wong E. W., Jensen L., Flood A. H., J. Am. Chem. Soc. 2011, 133, 7288–7291. [DOI] [PubMed] [Google Scholar]
- 251.
- 251a. Avellini T., Li H., Coskun A., Barin G., Trabolsi A., Basuray A. N., Dey S. K., Credi A., Silvi S., Stoddart J. F., Venturi M., Angew. Chem. Int. Ed. 2012, 51, 1611–1615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1643–1647; [Google Scholar]
- 251b. Jia C., Li H., Jiang J., Wang J., Chen H., Cao D., Stoddart J. F., Guo X., Adv. Mater. 2013, 25, 6752–6759. [DOI] [PubMed] [Google Scholar]
- 252.
- 252a. Huang T. J., Brough B., Ho C.-M., Liu Y., Flood A. H., Bonvallet P. A., Tseng H.-R., Stoddart J. F., Baller M., Magonov S., Appl. Phys. Lett. 2004, 85, 5391–5393; [Google Scholar]
- 252b. Liu Y., Flood A. H., Bonvallet P. A., Vignon S. A., Northrop B. H., Tseng H.-R., Jeppesen J. O., Huang T. J., Brough B., Baller M., Magonov S., Solares S. D., W. A. Goddard III , Ho C.-M., Stoddart J. F., J. Am. Chem. Soc. 2005, 127, 9745–9759; [DOI] [PubMed] [Google Scholar]
- 252c. Juluri B. K., Kumar A. S., Liu Y., Ye T., Yang Y.-W., Flood A. H., Fang L., Stoddart J. F., Weiss P. S., Huang T. J., ACS Nano 2009, 3, 291–300. [DOI] [PubMed] [Google Scholar]
- 253.
- 253a. Katz E., Lioubashevsky O., Willner I., J. Am. Chem. Soc. 2004, 126, 15520–15532; [DOI] [PubMed] [Google Scholar]
- 253b. Katz E., Sheeney-Haj-Ichia L., Willner I., Angew. Chem. Int. Ed. 2004, 43, 3292–3300; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3354–3362. [Google Scholar]
- 254.
- 254a. Hernandez R., Tseng H.-R., Wong J. W., Stoddart J. F., Zink J. I., J. Am. Chem. Soc. 2004, 126, 3370–3371; [DOI] [PubMed] [Google Scholar]
- 254b. Nguyen T. D., Tseng H.-R., Celestre P. C., Flood A. H., Liu Y., Stoddart J. F., Zink J. I., Proc. Natl. Acad. Sci. USA 2005, 102, 10029–10034; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254c. Saha S., Leung K. C.-F., Nguyen T. D., Stoddart J. F., Zink J. I., Adv. Funct. Mater. 2007, 17, 685–693; [Google Scholar]
- 254d. Nguyen T. D., Leung K. C.-F., Liong M., Liu Y., Stoddart J. F., Zink J. I., Adv. Funct. Mater. 2007, 17, 2101–2110; [Google Scholar]
- 254e. Nguyen T. D., Liu Y., Saha S., Leung K. C. F., Stoddart J. F., Zink J. I., J. Am. Chem. Soc. 2007, 129, 626–634; [DOI] [PubMed] [Google Scholar]
- 254f. Li Z., Barnes J. C., Bosoy A., Stoddart J. F., Zink J. I., Chem. Soc. Rev. 2012, 41, 2590–2605. [DOI] [PubMed] [Google Scholar]
- 255. Furukawa H., Cordova K. E., O'Keeffe M., Yaghi O. M., Science 2013, 341, 1230444. [DOI] [PubMed] [Google Scholar]
- 256. Deng H., Olson M. A., Stoddart J. F., Yaghi O. M., Nat. Chem. 2010, 2, 439–443. [DOI] [PubMed] [Google Scholar]
- 257. Li Q., Zhang W., Miljanić O. Š., Sue C.-H., Zhao Y.-L., Liu L., Knobler C. B., Stoddart J. F., Yaghi O. M., Science 2009, 325, 855–859. [DOI] [PubMed] [Google Scholar]
- 258. Li Q., Zhang W., Miljanić O. Š., Knobler C. B., Stoddart J. F., Yaghi O. M., Chem. Commun. 2010, 46, 380–382. [DOI] [PubMed] [Google Scholar]
- 259.
- 259a. Li Q., Sue C.-H., Basu S., Shveyd A. K., Zhang W., Barin G., Fang L., Sarjeant A. A., Stoddart J. F., Yaghi O. M., Angew. Chem. Int. Ed. 2010, 49, 6751–6755; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6903–6907; [Google Scholar]
- 259b. Cao D., Juríček M., Brown Z. J., Sue A. C.-H., Liu Z., Lei J., Blackburn A. K., Grunder S., Sarjeant A. A., Coskun A., Wang C., Farha O. K., Hupp J. T., Stoddart J. F., Chem. Eur. J. 2013, 19, 8457–8465. [DOI] [PubMed] [Google Scholar]
- 260. Sue A. C.-H., Mannige R. V., Deng H., Cao D., Wang C., Gándara F., Stoddart J. F., Whitelam S., Yaghi O. M., Proc. Natl. Acad. Sci. USA 2015, 112, 5591–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Bruns C. J., Fujita D., Hoshino M., Sato S., Stoddart J. F., Fujita M., J. Am. Chem. Soc. 2014, 136, 12027–12034. [DOI] [PubMed] [Google Scholar]
- 262. McGonigal P. R., Deria P., Hod I., Moghadam P. Z., Avestro A.-J., Horwitz N. E., Gibbs-Hall I. C., Blackburn A. K., Chen D., Botros Y. Y., Wasielewski M. R., Snurr R. Q., Hupp J. T., Farha O. K., Stoddart J. F., Proc. Natl. Acad. Sci. USA 2015, 112, 11161–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Chen Q., Sun J., Li P., Hod I., Moghadam P. Z., Kean Z. S., Snurr R. Q., Hupp J. T., Farha O. K., Stoddart J. F., J. Am. Chem. Soc. 2016, 138, 14242–14245. [DOI] [PubMed] [Google Scholar]
- 264. Krause S., Feringa B. L., Nat. Chem. Rev. 2020, 4, 550–562. [Google Scholar]
- 265. Feng L., Qiu Y., Guo Q.-H., Chen Z., Seale J. S. W., He K., Wu H., Feng Y., Farha O. K., Astumian R. D., Stoddart J. F., Science 2021, 374, 1215–1221. [DOI] [PubMed] [Google Scholar]
- 266. Mitschke B., Turberg M., List B., Chem 2020, 6, 2515–2532. [Google Scholar]
- 267. Jiao Y., Đorđević L., Mao H., Young R. M., Jaynes T., Chen H., Qiu Y., Cai K., Zhang L., Chen X.-Y., Feng Y., Wasielewski M. R., Stupp S. I., Stoddart J. F., J. Am. Chem. Soc. 2021, 143, 8000–8010. [DOI] [PubMed] [Google Scholar]
- 268.
- 268a. Benniston A. C., Harriman A., Philp D., Stoddart J. F., J. Am. Chem. Soc. 1993, 115, 5298–5299; [Google Scholar]
- 268b. Das A., Ghosh S., Angew. Chem. Int. Ed. 2014, 53, 2038–2054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2068–2084. [Google Scholar]
- 269.
- 269a. Hilinski E. F., Masnovi J. M., Amatore C., Kochi J. K., Rentzepis P. M., J. Am. Chem. Soc. 1983, 105, 6167–6168; [Google Scholar]
- 269b. Flamigni L., Talarico A. M., Serroni S., Puntoriero F., Gunter M. J., Johnston M. R., Jeynes T. P., Chem. Eur. J. 2003, 9, 2649–2659; [DOI] [PubMed] [Google Scholar]
- 269c. Stephenson R. M., Wang X., Coskun A., Stoddart J. F., Zink J. I., Phys. Chem. Chem. Phys. 2010, 12, 14135–14143. [DOI] [PubMed] [Google Scholar]
- 270. Noguchi H., Tsutsumi H., Komiyama M., Chem. Commun. 2000, 2455–2456. [Google Scholar]
- 271. Jiao Y., Qiu Y., Zhang L., Liu W.-G., Mao H., Chen H., Feng Y., Cai K., Shen D., Song B., Chen X.-Y., Li X., Zhao X., Young R. M., Stern C. L., Wasielewski M. R., Astumian R. D., W. A. Goddard III , Stoddart J. F., Nature 2022, 603, 265–270. [DOI] [PubMed] [Google Scholar]
- 272. Studer A., Curran D. P., Nat. Chem. 2014, 6, 765–773. [DOI] [PubMed] [Google Scholar]
- 273.
- 273a. Barnes J. C., Juríček M., Strutt N. L., Frasconi M., Sampath S., Giesener M. A., McGrier P. L., Bruns C. J., Stern C. L., Sarjeant A. A., Stoddart J. F., J. Am. Chem. Soc. 2013, 135, 183–192; [DOI] [PubMed] [Google Scholar]
- 273b. Juríček M., Barnes J. C., Dale E. J., Liu W.-G., Strutt N. L., Bruns C. J., Vermeulen N. A., Ghooray K. C., Sarjeant A. A., Stern C. L., Botros Y. Y., W. A. Goddard III , Stoddart J. F., J. Am. Chem. Soc. 2013, 135, 12736–12746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.