

Abstract
Wild rats can host various zoonotic pathogens. Detection of these pathogens is commonly performed using molecular techniques targeting one or a few specific pathogens. However, this specific way of surveillance could lead to (emerging) zoonotic pathogens staying unnoticed. This problem may be overcome by using broader microbiome‐profiling techniques, which enable broad screening of a sample's bacterial or viral composition. In this study, we investigated if 16S rRNA gene amplicon sequencing would be a suitable tool for the detection of zoonotic bacteria in wild rats. Moreover, we used virome‐enriched (VirCapSeq) sequencing to detect zoonotic viruses. DNA from kidney samples of 147 wild brown rats (Rattus norvegicus) and 42 black rats (Rattus rattus) was used for 16S rRNA gene amplicon sequencing of the V3–V4 hypervariable region. Blocking primers were developed to reduce the amplification of rat host DNA. The kidney bacterial composition was studied using alpha‐ and beta‐diversity metrics and statistically assessed using PERMANOVA and SIMPER analyses. From the sequencing data, 14 potentially zoonotic bacterial genera were identified from which the presence of zoonotic Leptospira spp. and Bartonella tribocorum was confirmed by (q)PCR or Sanger sequencing. In addition, more than 65% of all samples were dominated (>50% reads) by one of three bacterial taxa: Streptococcus (n = 59), Mycoplasma (n = 39) and Leptospira (n = 25). These taxa also showed the highest contribution to the observed differences in beta diversity. VirCapSeq sequencing in rat liver samples detected the potentially zoonotic rat hepatitis E virus in three rats. Although 16S rRNA gene amplicon sequencing was limited in its capacity for species level identifications and can be more difficult to interpret due to the influence of contaminating sequences in these low microbial biomass samples, we believe it has potential to be a suitable pre‐screening method in the future to get a better overview of potentially zoonotic bacteria that are circulating in wildlife.
Keywords: 16S, kidney, rats, surveillance, virome, zoonoses
1. INTRODUCTION
Wild brown rats (Rattus norvegicus) and black rats (Rattus rattus) can host various zoonotic pathogens, including Seoul orthohantavirus, Leptospira spp. and vector‐borne bacteria (Himsworth et al., 2015; Meerburg et al., 2009). Their synanthropic lifestyle increases the risk of pathogen transmission from rats to humans via (in‐)direct contact. Screening of zoonotic pathogens of wild rats gives insight in the potential public health risks. Early detection and identification of these pathogens is crucial to respond faster and more adequate to emerging infectious diseases.
Currently, diagnostic techniques such as (q)PCR and PCR‐based reverse line blot hybridization assays are often used for the detection of zoonotic pathogens (Elbaz et al., 2020). Though they are designed to be highly sensitive and specific (Bird & Mazet, 2018), they have the disadvantage that they can detect only a limited number of specific pathogens per test. As a consequence, certain pathogens can remain undetected, because they are not expected to be found in a particular species and therefore are not targeted (Razzauti et al., 2015). Therefore, it is important to keep optimizing detection methods and to test potential new techniques. In the last decades, huge advances have been made in developing new pathogen identification tools such as (metagenomic) deep sequencing (Bird & Mazet, 2018; Radford et al., 2012). The advantage of such techniques is the ability to broadly screen a sample's bacteriome or virome composition without exact sequence knowledge about the presence or absence of specific bacteria or viruses. This feature could make metagenomic sequencing a suitable tool to screen for zoonotic pathogens in wildlife, and it could facilitate the detection of unexpected or emerging pathogens. Although numerous microbiome‐profiling studies have been performed on humans, such studies performed on wildlife are limited (Egan et al., 2021; Firth et al., 2014; Ge et al., 2018; Raghwani et al., 2022; Razzauti et al., 2015).
In this study, we investigated if 16S rRNA gene amplicon sequencing would be a suitable tool for the detection of zoonotic pathogens in wild rats using kidney samples. These results were compared with qPCR results. In addition, we used VirCapSeq sequencing on liver samples to detect zoonotic viruses. Furthermore, we examined the kidney bacterial composition in more detail and investigated if there were internal (microbial diversity, species, bodyweight/age and sex) or external (trapping location type) factors correlated with zoonotic pathogen carriage in these wild rats. With this information, we aim to improve targeted screening and surveillance of zoonotic pathogens in wild rats to enhance early detection.
2. MATERIALS AND METHODS
2.1. Sample collection
From 2013 to 2018, pest control agencies captured brown and black rats in different municipalities across the Netherlands using live traps and snap traps for various surveillance studies (Maas et al., 2018). From all trapped rats, we included 189 rats in this study based on species and trapping location type (Table S1). Trapping locations were divided into the following categories: urban, rural, agriculture and industry. Locations with <1000 addresses/km2 were defined as rural and locations with >1000 addresses/km2 were defined as urban. When rats were captured on farms, their location was defined as agriculture, and when they were captured in industrial areas, their location was defined as industry. The live trapped rats were anaesthetized using isoflurane and euthanized by cervical dislocation or an isoflurane overdose after which they were dissected. The rats captured using snap traps were stored at −20°C until dissection. During dissection, we collected data on species, sex and bodyweight, as well as kidney and liver samples. Samples were stored at −80°C until DNA extraction. Kidney and liver samples were used for bacteriome and virome analyses, respectively. Bodyweight was used to divide rats into age classes. For males we used: juvenile (<100 g), subadult (101–200 g), and adult (>200 g), and for females we used: juvenile (<100 g), sub‐adult (101–175 g) and adult (>175 g) (Franssen et al., 2016).
2.2. Kidney bacterial composition
2.2.1. DNA extraction and 16S rRNA gene quantification
A small cross section of each kidney was cut and weighed. We adjusted the volume of lysis buffer according to weight and subsequently used equal volumes per sample for DNA extraction. We extracted DNA using the DNeasy Blood and Tissue Kit (Qiagen, Venlo, the Netherlands) according to the manufacturer's protocol, including extraction controls to monitor potential contamination during DNA extraction. We performed a 16S rDNA gene qPCR as previously described (Bogaert et al., 2011) to quantify the amount of 16S rRNA gene copies present in the samples.
2.2.2. Development of a rat mitochondrial 16S rRNA gene targeted blocking primer
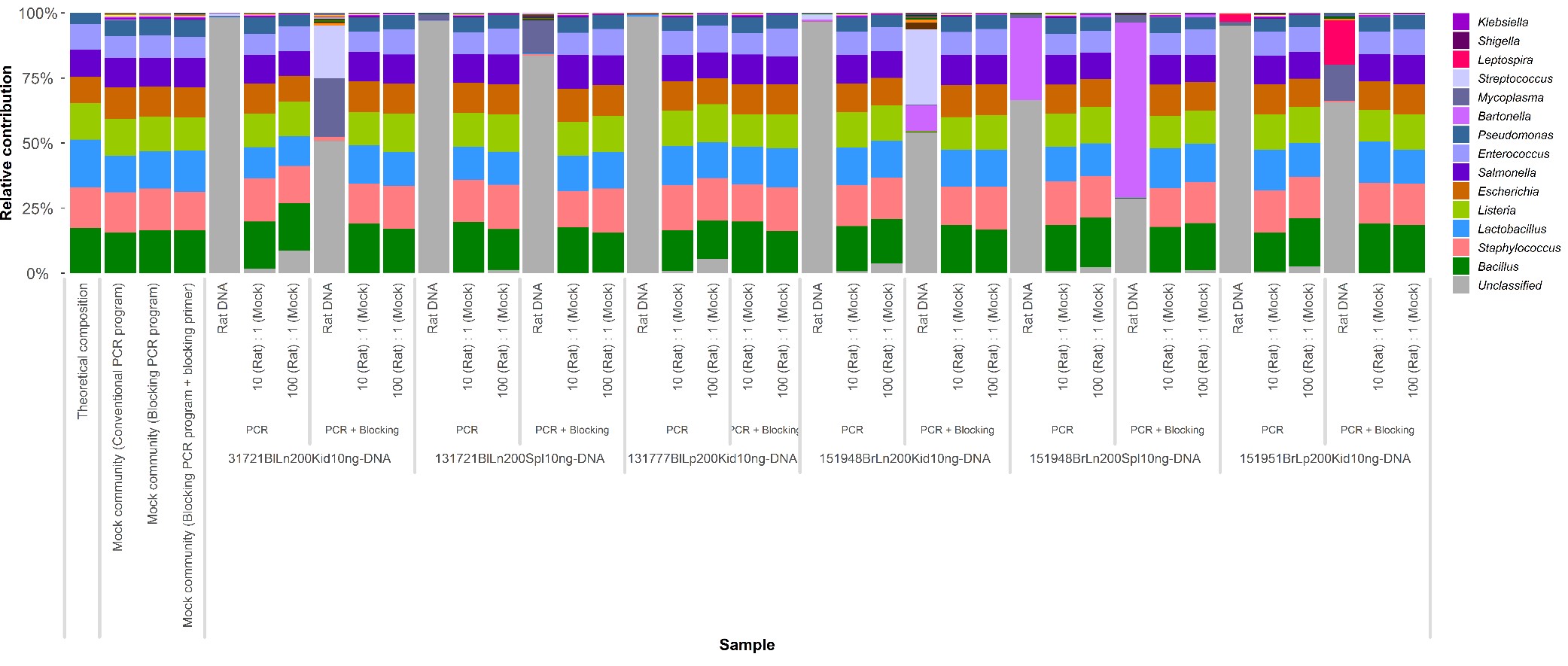
During an initial pilot study where 16S rRNA gene amplicons of the hypervariable V3–V4 region were sequenced, a substantial amount of sequences derived from rat host DNA were obtained. This was probably due to the low microbial biomass of kidney samples and the nonspecific amplification of 16S rat mitochondrial rRNA. To prevent the amplification of the untargeted rat‐derived DNA, peptide nucleic acid (PNA)‐based blocking primers were designed, in collaboration with BaseClear (Leiden, the Netherlands). In short, unique amplicon‐derived sequences obtained during the pilot were aligned against mitochondrial 16S rRNA amplicon sequences from other mammals obtained from DNA databases (Table S2). The sequence alignment was manually inspected for regions of identity. One region was identified from which 15–25‐nt sequences were extracted as candidate blocking primers. Candidate blocking primers were aligned to 16S rRNA gene databases, and primers that showed matches in the database were excluded from further analysis. Ultimately, a single remaining candidate sequence was obtained that matched the rat mitochondrial sequences as well as those from other eukaryotic host species, such as Vulpes vulpes, Meles meles, Martes martes, Mustela nivalis, Cervus elaphus, Dama dama, Sciurus vulgaris, Talpa europaea, Crocidura russula and Lepus europaeus. The resulting PNA blocking primer (5′‐TGGTAAATTTCGTGCCAGCCA‐3′) was synthesized by Eurogentec (Maastricht, the Netherlands) and used at a final concentration of 800 nM. The blocking primer reduced the amount of host DNA sequenced with approximately 15%–45% per sample (Figure S1). Blocking primers were included in the final sequencing PCR to gain more bacteria‐derived sequences from the microorganisms in the samples.
2.2.3. 16S rRNA gene amplification and sequencing
The extracted DNA was used as template for 16S rRNA gene amplification followed by sequencing of the amplicons by BaseClear (Leiden, the Netherlands) using the Illumina MiSeq platform. In short, amplicons of V3–V4 hypervariable regions of 16S rRNA genes were generated by PCR using a limited number of cycles using the forward primer 341F (5′‐CCTACGGGNGGCWGCAG‐3′), reverse primer 785R (5′‐GACTACHVGGGTATCTAATCC‐3′ (Gommers et al., 2019) and the PNA blocking primer described above, followed by a second PCR to incorporate the Illumina sequencing adaptors. PCR products were purified using a magnetic bead–based protocol, and DNA concentration was measured by fluorometric analysis (Qubit, Thermo Fisher Scientific). Subsequently, PCR amplicons were equimolarly pooled, and samples or controls with negligible amplicon DNA concentrations were added at the maximum allowed volume in the library tagging procedure. Pooled amplicons were sequenced on an Illumina MiSeq run with the paired‐end 300 cycles protocol. The sequencing data was demultiplexed with the Illumina CASAVA pipeline (v1.8.3) based on sample‐specific barcodes. The raw sequencing data was processed by removing the sequence reads of too low quality (only “passing filter” reads were selected) and discarding reads containing adaptor sequences or PhiX control with an in‐house filtering protocol. A quality assessment on the remaining reads was performed using the FastQC quality control tool version 0.10.0.
2.2.4. Data preparation
The data preparation and all analyses were performed in R 4.1.1 (R Core Team, 2021) and RStudio (RStudio Team, 2021). To go from raw reads to community analyses, we mostly followed a predefined workflow (Callahan et al., 2016). The Illumina demultiplexed paired‐end sequence reads were processed using the DADA2 R package. Paired‐end reads were merged, primers were removed (trimLeft (27,31)), sequences were trimmed to 280 bp (forward reads) and 240 bp (reverse reads) and a maximum of two ambiguous nucleotides was used. This was followed by error correction, data pooling, merging of sequences, chimaera filtering and clustering the reads into amplicon sequence variants (ASVs) using the reference database SILVA 16S version 138.1 (Quast et al., 2012). Then we combined the taxonomy counts into a phyloseq R object (McMurdie & Holmes, 2013) containing the ASV counts, taxonomy data and sample metadata. We applied taxonomic filtering to remove non‐bacterial ASVs (almost 70% were eukaryotic ASVs and a single archaeal ASV) and a few ASVs that were unidentifiable at Phylum level. We subjected the 16S rRNA amplicon sequences of all potentially zoonotic genera also to NCBI BLAST against the nr/nt database (date accessed: 10‐Nov‐2021) to identify identical matching species.
2.2.5. Diversity analyses
Alpha diversity was calculated using the Shannon diversity index. Because we wanted to compare only alpha diversity versus number of reads per sample, we did not use rarefied data as this would force us to remove a substantial number of samples with very low read counts (e.g. when rarefied to 1000 reads, almost 50% of samples would be removed). Prior to performing beta‐diversity analysis, we removed ASV singletons and doubletons in each sample to reduce the influence of contamination, and we performed a Hellinger transformation on the data. Beta‐diversity was assessed by principal coordinate analyses (PCoAs) on relative abundance data using Bray–Curtis dissimilarities using the vegan R package (Oksanen et al., 2013). To identify significant differences between community structures, PERMANOVA was performed using adonis (vegan; p = 0.05, 999 permutations and set.seed(100)), and dispersions were tested for homogeneity using betadisper (vegan). To assess the contribution of individual taxa to the observed differences in beta diversity between groups, we used SIMPER analysis (simper_pretty function; Steinberger, 2017). The SIMPER‐identified taxa that contributed most to the observed group differences were tested for significance with the Kruskal–Wallis test (R_krusk function; Steinberger, 2017). Taxa were considered significant if fdr‐adjusted p‐values <0.05.
2.3. Liver virome
2.3.1. DNA and RNA extraction followed by enriched‐metagenome sequencing
A total of 189 frozen liver samples were sliced. Per 0.5‐g liver, we added 1.75‐ml cold PBS with 1% complete EDTA‐free protease inhibitor cocktail (Roche, ref: 11873580001), followed by a 4‐h benzonase treatment at 37°C to remove free (non‐encapsulated) nucleic acids. Samples were pooled in batches of 3–5 samples per pool based on rat species and trapping location. Subsequent DNA purification was performed using the Qiagen QIAamp MinElute Virus Spin Kit (Qiagen, Venlo, the Netherlands) according to the manual prescription. For the RNA fraction extraction, the Zymo Research Direct‐zol kit (BaseClear, Leiden, the Netherlands) was used following the kit instructions with an internal DNase treatment. To increase the amount of RNA and DNA, a pre‐enrichment was performed with a random SISPA amplification (Chen et al., 2011). Shotgun sequence libraries were created from the extracted DNA and reverse transcribed RNA following enrichment using VirCapSeq (according to Roche) and subsequently sequenced using Illumina short‐read sequencing (MiSeq). When one of the pools was found positive for a relevant virus, the individual samples were processed separately.
2.3.2. Data analysis
For downstream analysis, the Illumina raw sequencing data was demultiplexed using the Illumina software (bcl2fastq v2.20.0.422, Illumina Inc) and subsequently polished (trimmed for artefacts and QC (BBMap – Bushnell B. – sourceforge.net/projects/bbmap/). To determine the presence of viruses in the samples, the polished reads were mapped to the present viruses of the NCBI database. The output table was manually inspected and the top scoring accession numbers were downloaded and used in further analyses.
The NCBI database was used to in silico enrich the viral reads per sample. The selected polished reads were used as input for de novo assembly using default settings in spades (SPAdes v3.13.0) (Prjibelski et al., 2020). Blast analysis was performed on the output files of the assembly.
2.4. Zoonotic pathogen identification and confirmation
Pathogen confirmation analyses were performed for Leptospira spp., Bartonella spp., Mycoplasma spp. and Brucella spp. For the first three pathogens, we extracted DNA from kidney samples using the DNeasy Blood and Tissue Kit (Qiagen, Venlo, the Netherlands) according to the manufacturer's protocol. For Brucella, we isolated DNA from Brucella‐suspected colonies by suspending the colony in 200‐μl nuclease‐free water (Sigma‐Aldrich, https://www.sigmaaldrich.com) and subsequent boiling at 100°C for 8 min, followed by centrifugation for 2 min at 20,000 × g.
For Leptospira spp. identification, we used a previously described qPCR specifically targeting pathogenic Leptospira species with forward primer LipgrF2 5′‐CGC‐TGA‐AAT‐GGG‐AGT‐TCG‐TAT‐GAT‐TTC‐C‐3′, reverse primer LipgrR2 5′‐GGC‐ATT‐GAT‐TTT‐TCT‐TCY‐GGG‐GTW‐GCC‐3′ and probe LipgrP1 5′‐Fam‐AGG‐CGA‐AAT‐CGG‐KGA‐RCC‐AGG‐CGA‐YGG‐BHQ1‐3′ (Ahmed et al., 2020). The Leptospira interrogans kantorow strain was used as positive control. Samples with sigmoid melting curves and Ct values <45 were considered positive.
For Bartonella spp. identification, we first performed a qPCR on genus level with forward primer ssrA‐F 5′‐GCTATGGTAATAAATGGACAATGAAATAA‐3′, reverse primer ssrA‐R 5′‐GCTTCTGTTGCCAGGTG‐3′ and probe 5′‐atto520‐ACCCCGCTTAAACCTGCGACG‐3′‐BHQ1 (Diaz et al., 2012). Subsequently, we performed conventional PCRs for Sanger sequencing on the samples detected positive by qPCR. These PCRs targeted two different genes: gltA and rpoB. We first used gltA primers: gltA‐2 (Bhcs.781p fwd: 5′‐GGGGACCAGCTCATGGTGG‐3′ and Bhcs.1137n rev: 5′‐ATTGCAAAAAGAACAGTAAACA‐3′) (Norman et al., 1995). In case results were negative, we also used rpoB primers: rpoB (1400F: 5′‐CGCATTGGCTTACTTCGTATG‐3′ and 2300R 5‐GTAGACTGATTAGAACGCTG‐3′) (Renesto et al., 2001). The strain of Bartonella henselae ATCC 49882 was used as positive control. PCR products were sequenced by BaseClear (Leiden, the Netherlands).
For Mycoplasma spp. identification, we performed a Mycoplasma pulmonis–specific PCR and a Mycoplasma genus PCR. For the M. pulmonis‐specific PCR, we used forward primer MP1 5′‐AGC‐GTT‐TGC‐TTC‐ACT‐TTG‐AA‐3′ and reverse primer MP2 5′‐GGG‐CAT‐TTC‐CTC‐CCT‐AAG‐CT‐3′ (Ferreira et al., 2008). For the Mycoplasma genus PCR, we targeted the 16S rRNA gene using forward primer HemMyco16S‐41s 5′‐GYATGCMTAAYACATGCAAGTCGARCG‐3′ and reverse primer HemMyco16S‐938as 5′‐CTCCACCACTTGTTCAGGTCCCCGTCGTC‐3′ (Maggi et al., 2013). The obtained PCR products were sequenced by BaseClear (Leiden, the Netherlands).
For Brucella spp. identification, we cultured and isolated Brucella spp. using the Castañeda method and selective media according to the OIE protocol (OIE, 2018). Suspected colonies were analysed by matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry on the Bruker MALDI Biotyper (Bruker, https://www.bruker.com) by using an extended in‐house Brucella spp. database (Kroese et al., 2018) and PCR.
On the isolated DNA, we performed qPCR targeting the IS711 sequences of Brucella spp. (Maio et al., 2014) using forward primer IS711F 5′‐GACCAAGCTGCATGCTGTTG‐3′, reverse primer IS711R 5′‐GCCGGGTGTTGGCTTTATT‐3′ and probe IS711P FAM‐CGATGCTATCGGCCTACCGCTGCG‐BHQ1. Colonies and tissue samples were considered positive after qPCR if the results showed a cycle threshold (Ct) value of <36 (with sigmoid curve), inconclusive if Ct value was >36 but <40 (with inconclusive sigmoid curve), and negative if Ct value was >40 or if there was no Ct detected.
2.5. Statistical analyses
In addition to the diversity analyses, we performed statistical analyses to investigate the correlation between the presence of zoonotic pathogens detected in these rats and specific rat characteristics (species, weight/age, location type and sex). The prevalence of zoonotic pathogens was tested for both qPCR and 16S rRNA amplicon sequencing results separately. The primary outcome variable was infection status (positive versus negative). We included the following explanatory variables: location type (urban, rural, agriculture or industry), species (R. norvegicus or R. rattus), sex (male or female), age class (juvenile, sub‐adult or adult) and bodyweight (g). Trapping area (municipality where the rats were trapped) was included as a random factor. We used generalized linear mixed models (GLMM) with binomial logit link function to examine the relationship between infection status and the explanatory variables using the glmer function (lme4). Variables that were significantly associated with infection status (p < .05) were included in the final model. Rats with missing data for one or more variables were excluded. All statistical analyses were performed in R 4.1.1 and RStudio (R Core Team, 2013). Confidence intervals were computed using the 95% Wald confidence interval. Variables were considered significant when p < 0.05.
3. RESULTS
3.1. 16S rRNA gene amplicon sequencing of rat kidney microbial DNA
Sequencing resulted in 3416,038 16S rRNA amplicon read pairs, of which 3330,609 were derived from Bacteria (97.5%) and 85,429 were derived from Eukarya (rats) (2.5%; Figures S1 and S4). The data showed large variation in the final number of reads per sample, ranging from 12 to 57,798 with a mean of 17,622 and median of 5388 reads per sample (Figure S2). Rarefaction curves indicate that sufficient reads were obtained to capture the variation present in the samples (Figure S3). After filtering the sequence data, we identified a total of 854 unique ASVs of which 504 could be classified to genus level and 229 to species level, resulting in 233 distinct bacterial genera and 173 distinct bacterial species. The most prevalent and abundant genera were Streptococcus (93%), Mycoplasma (81%) and Leptospira (54%; Figure 1). These three genera already comprised almost 90% of taxonomy counts. Many samples were dominated (>50% reads) by one of these three genera, most often by Streptococcus (n = 59), followed by Mycoplasma (n = 39) and Leptospira (n = 25; Figure 1).
FIGURE 1.
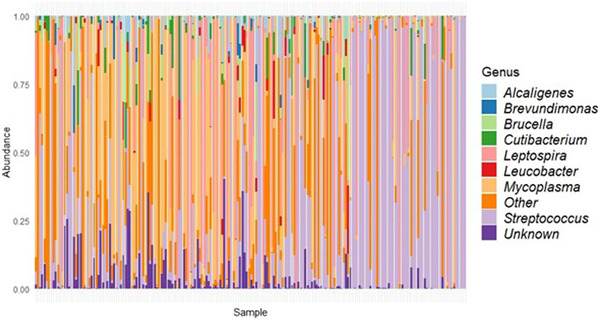
Bar plot displaying the relative abundance of bacteria (y‐axis) per sample (x‐axis) at genus level. Samples are in order of 16S rRNA amplicon concentration from low (left) to high (right).
3.2. Potentially zoonotic bacteria detected using 16S rRNA gene amplicon sequencing
We identified 14 out of 233 bacterial genera as potentially zoonotic (Table 1). The prevalence and total number of reads of these potentially zoonotic bacterial genera varied considerably (Table 1). Streptococcus, Mycoplasma and Leptospira were detected with high prevalence and abundance, whereas other potentially zoonotic genera (Bacillus, Mycobacterium, Chlamydia and Campylobacter) and members of the Erysipelotrichaceae family were detected in very low quantities (<0.1% of total reads), which makes the presence of these genera in the samples uncertain. Most of these potentially zoonotic genera could also be identified to species level, resulting in seven potentially zoonotic bacterial species: Leptospira interrogans, Brucella melitensis, Bartonella vinsonii, Staphylococcus aureus, Escherichia coli, Proteus mirabilis and Bacillus anthracis (Table 1). From the NCBI BLAST, we identified (almost) identical matching zoonotic species for the same seven genera that were identified using the SILVA v138.1 species reference database. For some potentially zoonotic genera, more than one identical matching species was found, belonging to different genera. This was the case for Brucella, E. coli, Bacillus and Erysipelotrichaceae.
TABLE 1.
Potentially zoonotic bacterial genera detected in rat kidneys using 16S rRNA gene amplicon sequencing
| Potentially zoonotic genus | Species according to 16S SILVA v138.1 reference database | Prevalence (%, n) a | Total number of reads (%, n) | Nr of ASVs b | BLAST result c | BLAST identity (%) | Known zoonotic species | References for known zoonotic species |
|---|---|---|---|---|---|---|---|---|
| Streptococcus | >2 species (most prevalent one is S. ruminantium) | 93%, n = 176 | 59%, n = 1,963,767 | 18 | >2 Streptococcus species | >97.05 | S. canis, S. iniae, S. suis and S. equi sub. zooepidemicus | Fulde and Valentin‐Weigand ( 2012 ) |
| Mycoplasma | M. coccoides and M. haemomacaque | 81%, n = 153 | 7.9%, n = 262,265 | 10 | Mycoplasma spp. | >98.95 | M. pulmonis and M. arginini | Piasecki et al. ( 2017 ) and Yechouron et al. ( 1992 ) |
| Leptospira | L. interrogans | 54%, n = 103 | 23%, n = 750,323 | 2 | >2 Leptospira species (including L. interrogans) | All 100 | L. interrogans | Bharti et al. ( 2003 ) |
| Brucella | B. melitensis | 38%, n = 71 | 0.2%, n = 6,175 | 1 | >2 Brucella species (including B. suis and B. melitensis) and Ochrobactrum spp. | All 100 | B. suis, B. melitensis, B. abortus and B. canis | Mortagy and Wells ( 2021 ) |
| Bartonella | B. vinsonii | 17%, n = 33 | 2.4%, n = 81,158 | 2 | >2 zoonotic Bartonella species | All 100 | >14 different species | Guptill ( 2010 ) |
| Staphylococcus | >2 species (most prevalent one is S. aureus) | 17%, n = 32 | 0.3%, n = 8,911 | 10 | >2 Staphylococcus species (including S. aureus) | >99.75 | Methicillin resistant S. aureus and S. pseudointermedius | Somayaji et al. ( 2016 ) and Springer et al. ( 2009 ) |
| Escherichia/Shigella | E. coli | 11%, n = 20 | 0.9%, n = 30,190 | 1 | E. coli, Escherichia spp. and Shigella flexneri | All 100 | ESBL/AmpC producing E. coli | Pitout ( 2012 ) |
| Corynebacterium | >2 species (most prevalent one is C. tuberculostearicum) | 10%, n = 19 | <0.1%, n = 2,066 | 14 | >2 Corynebacterium species | >99.48 | C. ulcerans, C. diphtheriae and C. pseudotuberculosis | Guido et al. ( 1997 ) |
| Proteus | P. mirabilis | 3%, n = 5 | 0.2%, n = 5,615 | 4 | >2 Proteus species (including P. mirabilis) | >99.75 | P. mirabilis | Armbruster et al. ( 2018 ) |
| Bacillus | >2 species (most prevalent one is B. anthracis) | 2%, n = 4 | <0.1%, n = 306 | 4 | >2 Bacillus species (including B. anthracis), Neobacillus spp., Cytobacillus firmus and Priestia spp. | All 100 | B. anthracis | Fasanella et al. ( 2010 ) |
| Mycobacterium | N/A | 1%, n = 1 | <0.1%, n = 70 | 1 | M. wolinskyi, Mycobacterium spp. | All 100 | M. bovis, M. avium | Biet et al. ( 2005 ) |
| Chlamydia | C. muridarum | 1%, n = 1 | <0.1%, n = 32 | 1 | C. muridarum | 100 | C. trachomatis | Rohde et al. ( 2010 ) |
| Campylobacter | C. sputorum | 1%, n = 1 | <0.1%, n = 5 | 1 | C. sputorum | 100 | C. jejuni and C. coli | Bolton ( 2015 ) |
| Erysipelotrichaceae family | N/A | 1%, n = 1 | <0.1%, n = 5 | 1 | Erysipelotrichaceae spp., Erysipelatoclostridium spp., Clostridium spp., Longibaculum spp. and Faecalibacillus spp. | All 100 | E. rhusiopathiae | Wang et al. ( 2010 ) |
Abbreviation: ASV, amplicon sequence variant.
Prevalence: number of rats in which >2 reads of a specific taxa was found with 16S rRNA gene amplicon sequencing, displayed in percentages and number of positive animals (=n).
Multiple ASVs, consisting of different DNA sequences, can be assigned to the same genus.
Shows matching species based on 16S DNA sequence. Identity percentage per BLAST result is shown, all sequences have 100% query cover.
3.3. (Potentially) zoonotic bacteria: species identification and prevalence comparison based on 16S rRNA gene amplicon sequencing and qPCR results
From the 14 potentially zoonotic genera identified earlier, four genera (Leptospira, Bartonella, Mycoplasma and Brucella) were selected to compare the results from 16S rRNA gene amplicon sequencing with those from quantifications using PCR‐based assays and to further identify them to species level. The Leptospira prevalence based on 16S rRNA gene amplicon sequencing reads (with >2 reads per ASV per sample considered positive) was 55% (103/189), and the prevalence based on qPCR (targeting pathogenic Leptospira species) was 46% (86/189; 95%). These two prevalences are not significantly different (binomial GLM; p = .100). In addition, we found a negative correlation between the 16S rRNA amplicon concentration and the qPCR Ct‐values (Figure 2). In total, 133 samples (70%) were positive in both assays, 36 samples (19%) were positive in the 16S rRNA gene amplicon sequencing only and 20 samples (11%) were positive in qPCR only. In a previous study, 22 of the rats used in this study were typed to serovar level and L. interrogans serovar Icterohaemorrhagiae (n = 8; 36%) and serovar Copenhageni (n = 14; 64%) belonging to serogroup Icterohaemorrhagiae were detected (Maas et al., 2018). Both serovars are zoonotic. The identified species (L. interrogans) was the same as the species identified based on the SILVA database and BLAST result.
FIGURE 2.
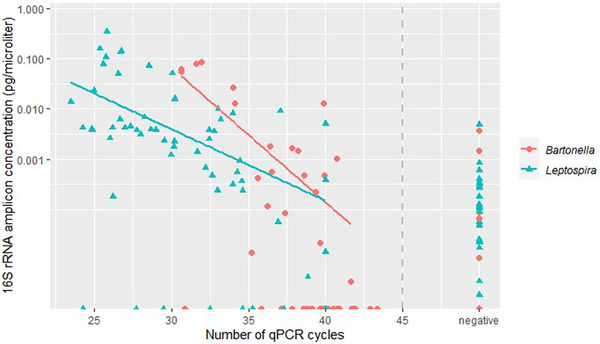
Comparison between the number of qPCR cycles and the 16S rRNA amplicon concentration for both Leptospira and Bartonella. The qPCRs are pathogen‐specific and shows at which cycle number the samples were found positive. The 16S rRNA amplicon concentration was calculated by multiplying the percentage of Leptospira or Bartonella reads per sample with the 16S rRNA gene amplicon concentration per sample. The y‐axis is log‐scaled. Samples with Ct values >45 and/or non‐sigmoidal curves are included in ‘negative’. The qPCR cut‐off value of 45 is represented by a grey dashed line. There is a negative correlation between the 16S rRNA amplicon concentration and the number of qPCR cycles for both Leptospira and Bartonella.
The Bartonella prevalence based on 16S rRNA gene amplicon sequencing reads was 18% (33/189) and the prevalence based on qPCR (targeting a selection of 30+ Bartonella species) was 23% (44/189). These two prevalences are not significantly different (binomial GLM; p = .161). We also found a negative correlation between the 16S rRNA amplicon concentration and the qPCR Ct‐values for Bartonella (Figure 2). In total, 160 samples (85%) were positive in both assays, 9 samples (5%) were positive in the 16S rRNA gene amplicon sequencing only, and 20 samples (11%) were positive in qPCR only. DNA from 22 qPCR‐positive rats was successfully isolated and sequenced. We observed high similarity (97.7%–100%) of all sequences to Bartonella tribocorum (GenBank accession numbers MT741530 (gltA), MG027996 (gltA) and AF165996 (rpoB)). This species is potentially zoonotic (Kosoy et al., 2010). The species B. tribocorum is different from the species identified based on the SILVA database (B. vinsonii).
For Mycoplasma, only a subset of 20 samples positive according to 16S rRNA gene amplicon sequencing was tested by PCR. From these, 19 out of 20 samples were positive in PCR and were further sequenced. From 14 samples, we could obtain good sequences all with high similarity (99.8%–100%) to Candidatus Mycoplasma haemomuris subspecies ratti (GenBank accession number AB758439), which is considered not zoonotic. This species is also different from the species identified based on the SILVA database (Mycoplasma coccoides and Mycoplasma haemomacaque).
Similarly, a subset of 20 samples that were positive for Brucella according to 16S rRNA gene amplicon sequencing results was tested by culture and qPCR. The BLAST result of the 16S Brucella sequence resulted in high similarity with both Brucella (including species Brucella suis and B. melitensis) and Ochrobactrum (Table 1). However, the presence of Brucella spp. could not be confirmed by either qPCR or culture.
3.4. Influence of internal and external factors on carriage of zoonotic bacteria
We investigated the effect of species, sex, bodyweight and location type on Leptospira and Bartonella carriage in wild rats, using the results from both 16S rRNA gene amplicon sequencing and qPCR, using binomial GLMMs. From both 16S rRNA gene amplicon sequencing and qPCR results, we observed that bodyweight was positively correlated with Leptospira carriage (p < .01; Table S3). For Bartonella, we observed a significantly higher prevalence in brown rats compared to black rats from both 16S rRNA gene amplicon sequencing and qPCR results (p < 0.05; Table S3).
3.5. Influence of in‐ and external factors on kidney bacterial composition
We visualized the overall differences in beta diversity in Bray–Curtis dissimilarity ordination (PCoA) plots for the factors species, location type, age and sex (Figure 3). Significant differences were observed for a multi‐variable model, including species, location type and age (PERMANOVA, p < 0.05). The kidney bacterial composition of brown and black rats was also significantly different (PERMANOVA p <0 .05), whereas both dispersions were homogenous (betadisper p > 0.05; Figure 3a). This difference was attributed to significant differences in the abundance of Streptococcus (3 ASVs), Leptospira and Mycoplasma (SIMPER, Kruskal–Wallis p < .05; Table S4), where the abundance of all taxa was higher in brown rats compared to black rats, except for two out of three Streptococcus ASVs.
FIGURE 3.
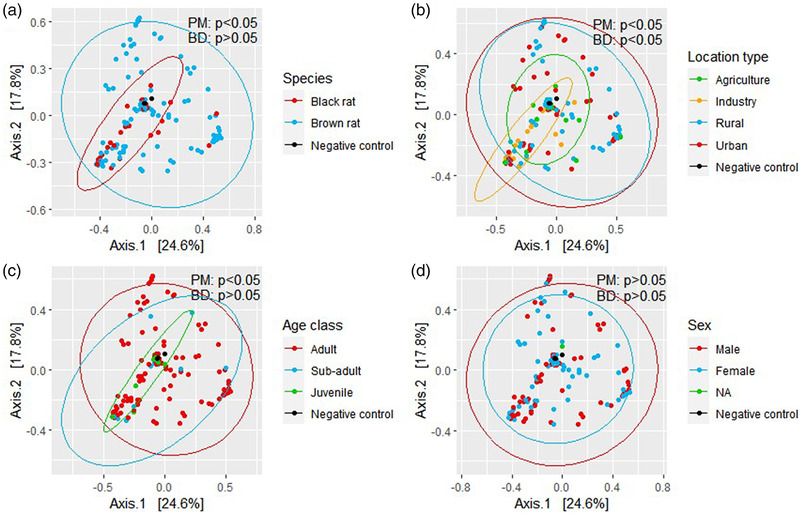
Principal coordinate analysis (PCoA) of wild rats’ kidney bacterial composition based on the Bray–Curtis dissimilarity, visualized per rat species (a), location type (b), age class (c) and sex (d). Ellipses are computed with 95% coverage. BD, betadisper p‐value; PM, PERMANOVA p‐value
Significant differences were also observed for location type (urban, rural, agriculture and industry; PERMANOVA p < 0.05) while dispersions were not homogenous (betadisper p < 0.05), which makes it uncertain whether the observed differences are indeed significant differences in group means or caused by dispersion variation between samples (Figure 3b). Differences between location types were predominantly attributed to differences in the abundance of Streptococcus, Leptospira and Mycoplasma (SIMPER, Kruskal–Wallis p < 0.05; Table S4). A lower mean abundance of Leptospira in industry was observed compared to the other three location types (urban, rural and agriculture).
Significant differences between age classes were observed (PERMANOVA p < 0.05) while dispersions were homogenous (betadisper p > 0.05; Figure 3c). Differences between adult and sub‐adult and between adult and juvenile were both attributed to a difference in the abundance of Leptospira, which was higher in adults compared to sub‐adults and juveniles (SIMPER, Kruskal–Wallis p < 0.05; Table S4). Because age class is based on bodyweight, this result coincides with our earlier finding that Leptospira carriage was positively correlated with bodyweight. For sex, no significant differences in beta diversity were observed (Figure 3d).
As might be expected, the genera mostly contributing to differences in beta diversity (Streptococcus, Leptospira and Mycoplasma) are also the main dominant genera identified earlier. The influence of these dominant genera on differences in beta diversity was visualized in a PCoA plot (Figure 4). Significant differences were observed between Leptospira and all other groups (Mycoplasma, Streptococcus, other and none) and between none and both Streptococcus and Mycoplasma (PERMANOVA p < 0.05). However, there was large variation in the dispersion of the data (betadisper p < 0.05).
FIGURE 4.
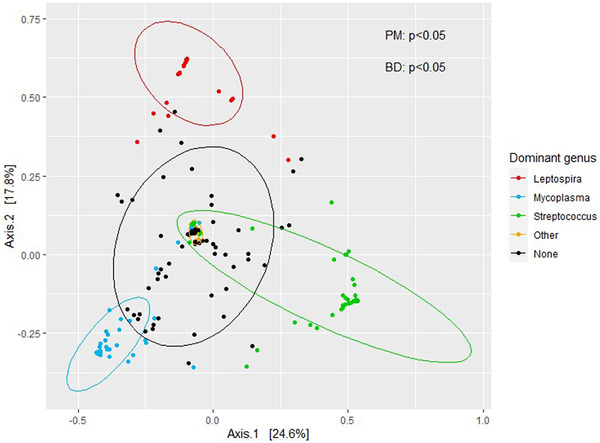
Principal coordinate analysis (PCoA) of wild rats’ kidney bacterial composition with Bray–Curtis distance. Visualized per dominating genus (>50% of total reads) per sample. Category ‘none’ consists of samples without a dominating taxa and ‘other’ consists of samples with a dominating taxa other than Streptococcus, Leptospira or Mycoplasma. BD, betadisper p‐value; PM, PERMANOVA p‐value
3.6. Relation between sequencing depth, alpha diversity and contamination
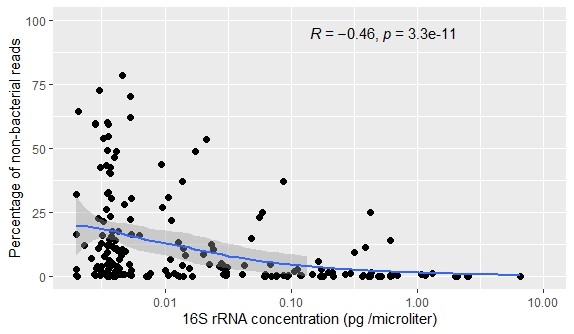
We observed that the Shannon diversity index was negatively correlated with the total number of reads per sample (Spearman correlation R = −0.7; p <0 .05; Figure 5), which implies that samples with higher numbers of reads are less diverse than samples with lower numbers of reads (Figure 5). This coincides with the domination patterns we observed before in these low microbial load samples (Figure 1). This high diversity in samples with low numbers of reads is probably related to contamination, which was detected in sequenced mock communities as well (Figure S1). Besides that, we observed that the percentage of host DNA blocked by the blocking primers varied per sample and that the percentage of eukaryotic reads in the sample were negatively correlated to the 16S rRNA concentration (Figures S1 and S4).
FIGURE 5.
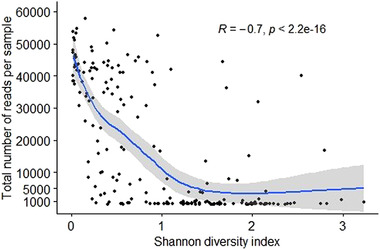
Shannon diversity index per total number of reads per sample. Each dot represents a sample. Correlation was tested using Spearman correlation coefficients (R = −.7; p < 2.2e−16).
3.7. Zoonotic viruses detected in rat liver
Rat liver samples were used for the detection of DNA and RNA viruses. Besides very low levels of different virus sequences, we detected only two viruses at levels of infecting agents: Minute virus of mice (Rodent protoparvovirus 1) and rat Hepatitis E virus (rat HEV) (also referred to as Orthohepevirus C). Of these two viruses, only rat HEV is potentially zoonotic. In total, 3/189 rats (6.3%) tested positive for rat HEV with both VirCapSeq sequencing and qPCR. The rat HEV sequence was detected in two black rats trapped on a pig farm in the southern province of Noord‐Brabant (2016) and in one brown rat trapped on an industrial location close to the harbour in Amsterdam (2014). One positive sample was further analysed, and the genome assemblies demonstrated that an almost full genome could be regenerated (ON644869). The sequence had most identity (87.96% 1–2517 bp and 87.43% 2619–6942 bp) with sequence KM516906 from the United States, followed by sequence MW795566 from a wild brown rat from Hungary captured in 2010 (86.82% 1–2517 bp and 87.01% 2619–6941 bp) (Niendorf et al., 2021), and sequence GU345043 (86.39% 1–2516 bp and 86.88% 2631–6945 bp) from a wild brown rat from Germany captured in 2009 (Johne et al., 2010). When we aligned the previously mentioned sequences with our sequences, we observed a gap of 66 bp from 2522 to 2587 bp.
4. DISCUSSION
In this study, we investigated if 16S rRNA gene amplicon sequencing would be a suitable tool for the detection of zoonotic bacteria in wild rats. These results were compared with (q‐)PCR results. In addition, we used VirCapSeq‐enriched sequencing on liver samples to detect zoonotic viruses. We also examined if there were internal or external factors correlated with zoonotic pathogen carriage in these wild rats.
4.1. Zoonotic bacteria detected in rat kidney
Using 16S rRNA gene amplicon sequencing, we identified 14 potentially zoonotic bacterial genera and 7 potentially zoonotic bacterial species in rat kidneys. We selected four highly prevalent genera for confirmation of results and to be further tested to species level. The presence of zoonotic Leptospira species (L. interrogans serovar Icterohaemorrhagiae and Copenhageni (Maas et al., 2018)) and a potentially zoonotic Bartonella species (B. tribocorum) were confirmed. B. tribocorum has been detected previously in both R. norvegicus and R. rattus (Himsworth et al., 2015; Lin et al., 2008). The Mycoplasma sequence was identified as Candidatus M. haemomuris subspecies ratti, which is considered not zoonotic. However, the Brucella sequences identified by 16S rRNA gene amplicon sequencing could not be confirmed by either culture or qPCR. BLAST results from the Brucella 16S DNA sequence resulted in a match for both Brucella and Ochrobactrum species. Therefore, although it was taxonomically assigned to Brucella, the DNA sequence probably belonged to Ochrobactrum.
The identification to species level based on the 16S SILVA database and qPCR sequencing resulted in both similar (for Leptospira) and different species (for Bartonella, Mycoplasma and Brucella). This emphasizes the need to confirm 16S rRNA gene amplicon sequencing results by other established detection methods and to interpret these results with caution, especially in the case of zoonotic bacteria. Overall, the 16S rRNA gene amplicon sequencing results did not indicate the presence of emerging or unexpected zoonotic bacteria in these wild rats. However, in this study, we tested only rat kidney tissue, likely leading to an underestimation of the number of zoonotic bacteria present in these rats. Therefore, the inclusion and comparison of multiple tissues per animal may improve future studies.
4.2. Comparing 16S rRNA gene amplicon sequencing with qPCR results
The results generated by 16S rRNA gene amplicon sequencing and qPCR showed 70% correlation for Leptospira and 85% for Bartonella. This resulted in non‐significantly different pathogen prevalence estimates based on the threshold values used in this study (>2 reads considered positive and pathogen‐specific Ct‐value cut‐offs). We also observed a negative correlation between the number of qPCR cycles and the 16S rRNA amplicon concentration per zoonotic bacterium, which shows the resemblance of the results obtained with both methods, and which also has been observed previously (Sillanpää et al., 2017). In addition, both methods resulted in similar significant correlations between Leptospira carriage and weight, and between Bartonella carriage and rat species. Therefore, 16S rRNA gene amplicon sequencing could also be used to generate population prevalence estimates.
The observed differences in results from both detection methods can be caused by various factors, including the choice of threshold values, the primers used for amplicon sequencing, the specificity of the qPCRs and the influence of contamination in these low microbial biomass samples. Especially for samples that were negative in qPCR and positive in 16S rRNA gene amplicon sequencing, the species detection range of the qPCRs we used could have limited the detection of certain species. However, standardized experiments using dilutions of known samples in different bacterial combinations should be performed to correctly compare the sensitivity and specificity of these two methods and to potentially define positive infection thresholds for specific bacteria when using 16S rRNA gene amplicon sequencing (Razzauti et al., 2015).
4.3. Influence of in‐ and external factors on zoonotic pathogen carriage and kidney bacterial composition
We investigated the relation between internal (species, sex and bodyweight/age) and external factors (location type) and zoonotic pathogen carriage. We observed a significant positive correlation between Leptospira carriage and rat bodyweight, indicating that heavier rats, and thereby most likely also older rats, are more likely to carry Leptospira. This result is in‐line with previous research (Costa et al., 2014; Himsworth et al., 2013). For Bartonella, we observed a significant difference between rat species, with a higher prevalence of Bartonella in brown rats (29.3%) compared to black rats (2.4%), which agrees with results reported in other studies (Ellis et al., 1999; Hsieh et al., 2010; Peterson et al., 2017). Some studies explained this difference in prevalence by a similar difference in ectoparasite infestation levels (Peterson et al., 2017), but that alone could not always fully explain the observed difference (Brettschneider et al., 2012). In light of risk surveillance, these results suggest the surveillance of zoonotic pathogens in wild rats should focus on adult brown rats.
We also investigated the influence of in‐ and external factors in a broader perspective, by looking at their correlation with the total bacterial diversity of a sample instead of with only the presence or absence of Leptospira and Bartonella. We found significant bacterial diversity differences associated with species and age, which could be mainly attributed to differences in the abundance of Streptococcus, Leptospira and Mycoplasma, the three most dominant bacteria identified. Differences in bacterial diversity associated with species and age have also been observed in previous studies on rodent gut microbiome (Anders et al., 2021; Langille et al., 2014). Streptococcus was divided over three ASVs of which one ASV was more abundant in brown rats and the other two ASVs were more abundant in black rats. Therefore, we suspect there are multiple Streptococcus species present with different host specificities. Mycoplasma abundance was higher in brown rats, but the reason behind this is unclear.
4.4. Bacterial domination in rat kidneys
We observed that almost 70% of all samples were dominated (>50% reads) by one genus, mainly Streptococcus, Leptospira or Mycoplasma. The presence of dominating taxa is in‐line with previous studies looking at the bacterial composition of urine (Brubaker & Wolfe, 2017; Kramer et al., 2018). In human female urine, these dominating taxa mostly consist of Lactobacillus, Streptococcus, Corynebacterium, Bifidobacterium, Staphylococcus and Prevotella (Brubaker & Wolfe, 2017; Thomas‐White et al., 2017). These bacteria were also detected in the rat kidney samples. In the past, this domination was linked to the presence of disease, but recent studies suggest more complex interactions where domination can be linked to both disease susceptible and protective effects (Brubaker & Wolfe, 2016). For example, a protective effect could be that commensal bacteria outcompete pathogenic bacteria for nutrients, produce antimicrobial substances or stimulate the host immune system (Bao et al., 2017). Lactobacillus, Streptococcus and Bifidobacterium have been identified as human commensals of the urine microbiome (Neugent et al., 2020). The following bacteria have been related to the kidney microbiome of healthy human subjects and also occur in rat kidneys from this study: Microbacterium, Pelomonas, Staphylococcus, Streptococcus, Leuconostoc, Corynebacterium, Anaerococcus and Thermicanus (Heidler et al., 2020). Therefore, these bacteria might be considered commensals of the rat kidney. Streptococcus was present in 93% of all rat kidney samples and did not match with known zoonotic Streptococcus species. We, therefore, suspect Streptococcus to be a commensal of the rat kidney as well. This could also be the case for Mycoplasma (present in 81% of all samples and identified as non‐zoonotic), but more research is needed to identify the true bacterial community composition of the rat kidney.
4.5. Bacterial community or contamination?
In this study, rat kidney samples were analysed, which are typically low microbial biomass samples in which only few bacteria are expected to be found, unless the animals were heavily infected. There was large variation in the total number of reads per sample with almost 40% of all samples having <1000 reads in total, which is in‐line with the low measured 16S rRNA amplicon concentration in the samples. Similar data was obtained in a study investigating the urine microbiome of cats, where more than 50% of samples had <500 reads (Kim et al., 2021). We also observed a negative correlation between the total number of reads per sample and the Shannon diversity index, which indicates that samples with low numbers of reads (roughly <1000 reads) show profiles that are indicative of contamination or that those bacteria are present in very low concentrations. This negative correlation is the opposite result of studies performed on the gut microbiome (Willis, 2019) but in‐line with other studies performed on low microbial biomass samples (Karstens et al., 2019; Krawczyk, 2021). Although low numbers of reads are to be expected in low microbial biomass samples such as kidney, the low number of reads complicates distinguishing between DNA from bacteria truly present in the sample (although in low numbers) and DNA from contaminants. Therefore, results with low numbers of reads should be interpreted with caution. In this study, we used only three control samples. Using a larger number of control and also mock samples in future studies will facilitate the identification of possible contaminants. This is especially important for epidemiological studies focusing on zoonotic bacterial genera that are also considered common contaminants (Razzauti et al., 2015).
Cross amplification of mitochondrial mammalian DNA during sequencing in low microbial biomass samples can also negatively affect the amount of bacterial DNA sequenced (Bao et al., 2017). To reduce the interference of host DNA during sequencing, we designed blocking primers. Though the percentage of host DNA blocked varied per sample, it was negatively correlated with the 16S rRNA gene concentration, which implies that the blocking primers are less efficient in blocking host DNA in samples that contain only very few bacteria. To further reduce host DNA contamination, specific DNA isolation kits could be used that maximize the isolation of bacterial DNA (Bao et al., 2017). Although low microbial biomass samples such as kidney bring new challenges regarding interpretation and accuracy, they also have an advantage compared to high microbial biomass samples: Low microbial biomass samples have less commensal/background bacteria, which makes it relatively easier to identify zoonotic bacteria.
4.6. Virus detection in rat liver
Only one potentially zoonotic virus was detected at levels of infecting agents: rat HEV. The whole genome could be sequenced except for the part between 2522 and 2587 bp, which could indicate a deletion in our sequence of 66 bp. This virus was detected in two black rats trapped on a pig farm and in one brown rat trapped on an industrial location, which, to our knowledge, is the first time that rat HEV has been detected in wild rats in the Netherlands. Rat HEV has been detected in both brown and black rats in other European countries (Ryll et al., 2017) and has recently also been found in humans (Rivero‐Juarez et al., 2022). The rat HEV‐positive rats were trapped in 2014 and 2016. Therefore, it would be interesting to include rat HEV in current surveillance studies to investigate the infection prevalence in rats and the risk for public health.
5. CONCLUSION
Using 16S rRNA gene amplicon sequencing, we identified possible zoonotic bacteria in rat kidney samples, and we obtained a better overview of the rat kidney bacterial composition and the apparent domination of certain bacteria. Prevalence estimates and subsequent correlation analyses using both 16S rRNA gene amplicon sequencing and qPCR data were not significantly different, indicating that 16S rRNA gene amplicon sequencing could also be a suitable tool to give indications of population pathogen prevalence and correlations. Moreover, using VirCapSeq‐enriched metagenomic sequencing, we detected rat HEV in wild rats.
Although 16S rRNA gene amplicon sequencing has the advantage to detect multiple pathogens at once and certainly has the potential to be a suitable tool for detection of new zoonotic pathogens and potentially pathogen surveillance, there are several limitations that should be considered when using this method. First, 16S rRNA gene amplicon sequencing can in most cases reliably identify bacteria to genus level only, so subsequent identification or confirmation to species level should be performed when identifying potentially zoonotic bacteria. Second, the low numbers of reads from low microbial biomass samples make it more difficult to distinguish between bacteria present in the sample and contaminating bacteria. Therefore, it is important to use sufficient control and mock samples in future studies to be able to better distinguish them. Future studies could also investigate the effects of using pooled samples (either from multiple animals or from multiple tissues per animal) to decrease laboratory effort and costs. Currently, we recommend to still use more established methods such as (q)PCR for pathogen detection and to use 16S rRNA gene amplicon sequencing as a complementary or pre‐screening method. Especially when only a limited number of pathogens are of interest, qPCR is currently still less time‐consuming and less expensive. When more reliable identification to species level would be possible in the future, 16S rRNA gene amplicon sequencing could be a very promising technique for the surveillance of zoonotic bacteria.
AUTHOR CONTRIBUTIONS
Marieke de Cock, Miriam Maas and Hein Sprong designed this study with practical input from Bartholomeus van den Bogert. Bartholomeus van den Bogert performed the 16S rRNA amplicon sequencing for the bacteria, Renate Hakze‐van der Honing and Wim van der Poel performed the VirCapSeq sequencing for the viruses and Ad Koets performed the Brucella analyses. Marieke de Cock, Ankje de Vries and Manoj Fonville performed all other laboratory analyses. Marieke de Cock analysed the data with guidance from Alex Bossers. All authors helped with the interpretation of the data and revising the manuscript. All authors approved the publication of this paper.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to and the appropriate ethical review committee approval has been received. This study involved the targeted collection and examination of wild rats and was approved by the Animal Ethics Committee of the RIVM (project numbers: 201200208, 201500089 and 2016‐0053).
Supporting information
Figure S1 Relative contribution of the blocking primer to the blocking of host DNA and increased yield of bacterial 16S rRNA amplicon sequences. Individual rat samples are described here as ‘Rat DNA’.
{kind=link}
Figure S2 Raw number of reads per sample. Each dot (blue) represents one sample. Negative control samples are depicted in red.
{kind=link}
Figure S3 Rarefaction curves showing the number of detected ASVs per number of reads sampled per sample
{kind=link}
Figure S4 Percentage of non‐bacterial reads per sample versus the 16S rRNA gene concentration (pg/μl) after the use of blocking primers. Correlation was tested using Spearman correlation coefficients (R = −.46; p = 3.3e−11).
{kind=link}
Table S1 Characteristics of the wild rats used in this study
†Black rats average weight was calculated based on 29/42 rats because the weight from 13 adult rats was not measured. Therefore, the average weight for black rats in our sample is likely underestimated.
Table S2 Mitochondrial 16S rRNA amplicon sequences from other mammals used during blocking primer development
Table S3 Prevalence estimates of Leptospira and Bartonella carriage based on 16S rRNA gene amplicon sequencing and qPCR results, shown per internal or external factor. More than two reads was considered positive for 16S rRNA gene amplicon sequencing. An asterisk (*) depicts a significant difference within or correlation with a factor using the 16S rRNA gene amplicon sequencing or qPCR dataset analysed using GLMMs; *p < .05, **p < .01 and ***p < .001.
Table S4 Results from SIMPER beta‐diversity comparisons that were significantly different between groups according to Kruskal–Wallis tests using fdr‐adjusted p‐values >.05
ACKNOWLEDGEMENTS
We thank all pest control officers for the collection of rats, Aleksandra Krawczyk for her input and guidance during the first phase of this study and Mei Ling Chu and James Groot for performing the 16S qPCR on these samples. From Wageningen Bioveterinary Research, we thank Dré Kampfraath, Heather Graham and Dylano Suanes Lopez for their input and work regarding the detection of Brucella, Stéphanie Vastenhouw for her help with preparing the rat liver samples and Frank Harders for his help with completing the rat Hepatitis E sequence. We thank Edwin Bargeman and Sara Wijburg for their help with development of R scripts. This work was supported by funding from the European Union's Horizon 2020 Research and Innovation programme for the DESIRE project under grant agreement No 773830: One Health European Joint Programme.
de Cock, M. , Fonville, M. , de Vries, A. , Bossers, A. , van den Bogert, B. , Hakze‐van der Honing, R. , Koets, A. , Sprong, H. , van der Poel, W. , & Maas, M. (2022). Screen the unforeseen: Microbiome‐profiling for detection of zoonotic pathogens in wild rats. Transboundary and Emerging Diseases, 69, 3881–3895. 10.1111/tbed.14759
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Ahmed, A. A. , Goris, M. G. , & Meijer, M. C. (2020). Development of lipL32 real‐time PCR combined with an internal and extraction control for pathogenic Leptospira detection. PLoS One, 15(11), e0241584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster, C. E. , Mobley, H. L. T. , & Pearson, M. M. (2018). Pathogenesis of Proteus mirabilis infection. EcoSal Plus, 8(1), 0009–2017. 10.1128/ecosalplus.ESP-0009-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, J. L. , Moustafa, M. A. M. , Mohamed, W. M. A. , Hayakawa, T. , Nakao, R. , & Koizumi, I. (2021). Comparing the gut microbiome along the gastrointestinal tract of three sympatric species of wild rodents. Scientific Reports, 11(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, Y. , Al, K. F. , Chanyi, R. M. , Whiteside, S. , Dewar, M. , Razvi, H. , Reid, G. , & Burton, J. P. (2017). Questions and challenges associated with studying the microbiome of the urinary tract. Annals of Translational Medicine, 5(2), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti, A. R. , Nally, J. E. , Ricaldi, J. N. , Matthias, M. A. , Diaz, M. M. , Lovett, M. A. , Levett, P. N. , Gilman, R. H. , Willig, M. R. , & Gotuzzo, E. (2003). Leptospirosis: A zoonotic disease of global importance. The Lancet Infectious Diseases, 3(12), 757–771. [DOI] [PubMed] [Google Scholar]
- Biet, F. , Boschiroli, M. L. , Thorel, M. F. , & Guilloteau, L. A. (2005). Zoonotic aspects of Mycobacterium bovis and Mycobacterium avium‐intracellulare complex (MAC). Veterinary Research, 36(3), 411–436. [DOI] [PubMed] [Google Scholar]
- Bird, B. H. , & Mazet, J. A. (2018). Detection of emerging zoonotic pathogens: An integrated one health approach. Annual Review of Animal Biosciences, 6, 121–139. [DOI] [PubMed] [Google Scholar]
- Bogaert, D. , Keijser, B. , Huse, S. , Rossen, J. , Veenhoven, R. , Van Gils, E. , Bruin, J. , Montijn, R. , Bonten, M. , & Sanders, E. (2011). Variability and diversity of nasopharyngeal microbiota in children: A metagenomic analysis. PLoS One, 6(2), e17035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton, D J. (2015). Campylobacter virulence and survival factors. Food Microbiology, 48, 99–108. [DOI] [PubMed] [Google Scholar]
- Brettschneider, H. , Anguelov, R. , Chimimba, C. T. , & Bastos, A. D. (2012). A mathematical epidemiological model of gram‐negative Bartonella bacteria: Does differential ectoparasite load fully explain the differences in infection prevalence of Rattus rattus and Rattus norvegicus? Journal of Biological Dynamics, 6(2), 763–781. [DOI] [PubMed] [Google Scholar]
- Brubaker, L. , & Wolfe, A. (2016). The urinary microbiota: A paradigm shift for bladder disorders? Current Opinion in Obstetrics & Gynecology, 28(5), 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker, L. , & Wolfe, A. J. (2017). The female urinary microbiota, urinary health and common urinary disorders. Annals of Translational Medicine, 5(2), 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B. J. , Sankaran, K. , Fukuyama, J. A. , McMurdie, P. J. , & Holmes, S. P. (2016). Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research, 5, 1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, E. C. , Miller, S. A. , DeRisi, J. L. , & Chiu, C. Y. (2011). Using a pan‐viral microarray assay (Virochip) to screen clinical samples for viral pathogens. JoVE (Journal of Visualized Experiments), 27(50), e2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, F. , Porter, F. H. , Rodrigues, G. , Farias, H. , de Faria, M. T. , Wunder, E. A. , Osikowicz, L. M. , Kosoy, M. Y. , Reis, M. G. , & Ko, A. I. (2014). Infections by Leptospira interrogans, Seoul virus, and Bartonella spp. among Norway rats (Rattus norvegicus) from the urban slum environment in Brazil. Vector‐Borne and Zoonotic Diseases, 14(1), 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, M. H. , Bai, Y. , Malania, L. , Winchell, J. M. , & Kosoy, M. Y. (2012). Development of a novel genus‐specific real‐time PCR assay for detection and differentiation of Bartonella species and genotypes. Journal of Clinical Microbiology, 50(5), 1645–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, S. L. , Taylor, C. L. , Banks, P. B. , Northover, A. S. , Ahlstrom, L. A. , Ryan, U. M. , Irwin, P. J. , & Oskam, C. L. (2021). The bacterial biome of ticks and their wildlife hosts at the urban–wildland interface. Microbial Genomics, 7(12), 000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz, E. , Moustafa, M. A. M. , Lee, K. , Ching, A. L. C. , Shimozuru, M. , Sashika, M. , Nakao, R. , El‐Khodery, S. A. , & Tsubota, T. (2020). Utilizing attached hard ticks as pointers to the risk of infection by Babesia and Theileria species in sika deer (Cervus nippon yesoensis), in Japan. Experimental and Applied Acarology, 82(3), 411–429. [DOI] [PubMed] [Google Scholar]
- Ellis, B. , Regnery, R. , Beati, L. , Bacellar, F. , Rood, M. , Glass, G. , Marston, E. , Ksiazek, T. , Jones, D. , & Childs, J. (1999). Rats of the genus Rattus are reservoir hosts for pathogenic Bartonella species: An old world origin for a new world disease? The Journal of Infectious Diseases, 180(1), 220–224. [DOI] [PubMed] [Google Scholar]
- Fasanella, A. , Galante, D. , Garofolo, G. , & Jones, M. H. (2010). Anthrax undervalued zoonosis. Veterinary Microbiology, 140(3–4), 318–331. [DOI] [PubMed] [Google Scholar]
- Ferreira, J. , Yamaguti, M. , Marques, L. , Oliveira, R. , Neto, R. , Buzinhani, M. , & Timenetsky, J. (2008). Detection of Mycoplasma pulmonis in laboratory rats and technicians. Zoonoses and Public Health, 55(5), 229–234. [DOI] [PubMed] [Google Scholar]
- Firth, C. , Bhat, M. , Firth, M. A. , Williams, S. H. , Frye, M. J. , Simmonds, P. , Conte, J. M. , Ng, J. , Garcia, J. , & Bhuva, N. P. (2014). Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. MBio, 5(5), e01933–01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen, F. , Swart, A. , van Knapen, F. , & van der Giessen, J. (2016). Helminth parasites in black rats (Rattus rattus) and brown rats (Rattus norvegicus) from different environments in the Netherlands. Infection Ecology & Epidemiology, 6(1), 31413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulde, M. , & Valentin‐Weigand, P. (2012). Epidemiology and pathogenicity of zoonotic streptococci. Host‐Pathogen Interactions in Streptococcal Diseases, 368, 49–81. [DOI] [PubMed] [Google Scholar]
- Guido, F. , von Graevenitz, A. , Clarridge, J. E., 3rd , & Bernard, K. A. (1997). Clinical microbiology of coryneform bacteria. Clinical Microbiology Reviews, 10(1), 125–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, Y. , Guo, G. , Ge, B. , Yin, H. , & Yin, H. (2018). The spleen microbiota of small wild mammals reveals distinct patterns with tick‐borne bacteria. PLoS Neglected Tropical Diseases, 12(7), e0006499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommers, L. M. , Ederveen, T. H. , van der Wijst, J. , Overmars‐Bos, C. , Kortman, G. A. , Boekhorst, J. , Bindels, R. J. , de Baaij, J. H. , & Hoenderop, J. G. (2019). Low gut microbiota diversity and dietary magnesium intake are associated with the development of PPI‐induced hypomagnesemia. The FASEB Journal, 33(10), 11235–11246. [DOI] [PubMed] [Google Scholar]
- Guptill, L. (2010). Bartonellosis. Veterinary Microbiology, 140(3–4), 347–359. [DOI] [PubMed] [Google Scholar]
- Heidler, S. , Lusuardi, L. , Madersbacher, S. , & Freibauer, C. (2020). The microbiome in benign renal tissue and in renal cell carcinoma. Urologia Internationalis, 104(3–4), 247–252. [DOI] [PubMed] [Google Scholar]
- Himsworth, C. G. , Bai, Y. , Kosoy, M. Y. , Wood, H. , DiBernardo, A. , Lindsay, R. , Bidulka, J. , Tang, P. , Jardine, C. , & Patrick, D. (2015). An investigation of Bartonella spp., Rickettsia typhi, and Seoul hantavirus in rats (Rattus spp.) from an inner‐city neighborhood of Vancouver, Canada: Is pathogen presence a reflection of global and local rat population structure? Vector‐Borne and Zoonotic Diseases, 15(1), 21–26. [DOI] [PubMed] [Google Scholar]
- Himsworth, C. G. , Bidulka, J. , Parsons, K. L. , Feng, A. Y. , Tang, P. , Jardine, C. M. , Kerr, T. , Mak, S. , Robinson, J. , & Patrick, D. M. (2013). Ecology of Leptospira interrogans in Norway rats (Rattus norvegicus) in an inner‐city neighborhood of Vancouver, Canada. PLOS Neglected Tropical Diseases, 7(6), e2270. 10.1371/journal.pntd.0002270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, J. W. , Tung, K. C. , Chen, W. C. , Lin, J. W. , Chien, L. J. , Hsu, Y. M. , Wang, H. C. , Chomel, B. B. , & Chang, C. C. (2010). Epidemiology of Bartonella infection in rodents and shrews in Taiwan. Zoonoses and Public Health, 57(6), 439–446. [DOI] [PubMed] [Google Scholar]
- Johne, R. , Heckel, G. , Plenge‐Bönig, A. , Kindler, E. , Maresch, C. , Reetz, J. , Schielke, A. , & Ulrich, R. G. (2010). Novel hepatitis E virus genotype in Norway rats, Germany. Emerging Infectious Diseases, 16(9), 1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karstens, L. , Asquith, M. , Davin, S. , Fair, D. , Gregory, W. T. , Wolfe, A. J. , Braun, J. , & McWeeney, S. (2019). Controlling for contaminants in low‐biomass 16S rRNA gene sequencing experiments. mSystems, 4(4), e00290–00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. , Carrai, M. , Leung, M. H. , Chin, J. , Li, J. , Lee, P. K. , Beatty, J. A. , Pfeiffer, D. U. , & Barrs, V. R. (2021). Dysbiosis of the urinary bladder microbiome in cats with chronic kidney disease. mSystems, 6(4), e00510–00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosoy, M. , Bai, Y. , Sheff, K. , Morway, C. , Baggett, H. , Maloney, S. A. , Boonmar, S. , Bhengsri, S. , Dowell, S. F. , & Sitdhirasdr, A. (2010). Identification of Bartonella infections in febrile human patients from Thailand and their potential animal reservoirs. The American Journal of Tropical Medicine and Hygiene, 82(6), 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, H. , Kuffel, G. , Thomas‐White, K. , Wolfe, A. J. , Vellanki, K. , Leehey, D. J. , Bansal, V. K. , Brubaker, L. , Flanigan, R. , & Koval, J. (2018). Diversity of the midstream urine microbiome in adults with chronic kidney disease. International Urology and Nephrology, 50(6), 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk, A. I. (2021). Questing microbioticks: Interactions of microbes, ticks, vertebrates, and the environment. Wageningen University. [Google Scholar]
- Kroese, M. V. , Beckers, L. , Bisselink, Y. J. , Brasseur, S. , van Tulden, P. W. , Koene, M. G. , Roest, H. I. , Ruuls, R. C. , Backer, J. A. , & Ijzer, J. (2018). Brucella pinnipedialis in grey seals (Halichoerus grypus) and harbor seals (Phoca vitulina) in the Netherlands. Journal of Wildlife Diseases, 54(3), 439–449. [DOI] [PubMed] [Google Scholar]
- Langille, M. G. , Meehan, C. J. , Koenig, J. E. , Dhanani, A. S. , Rose, R. A. , Howlett, S. E. , & Beiko, R. G. (2014). Microbial shifts in the aging mouse gut. Microbiome, 2(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J.‐W. , Chen, C.‐Y. , Chen, W.‐C. , Chomel, B. B. , & Chang, C.‐C. (2008). Isolation of Bartonella species from rodents in Taiwan including a strain closely related to ‘Bartonella rochalimae’ from Rattus norvegicus . Journal of Medical Microbiology, 57(12), 1496–1501. [DOI] [PubMed] [Google Scholar]
- Maas, M. , De Vries, A. , Reusken, C. , Buijs, J. , Goris, M. , Hartskeerl, R. , Ahmed, A. , Van Tulden, P. , Swart, A. , Pijnacker, R. , Koene, M. , Lundkvist, A. , Heyman, P. , Rockx, B. , & Van Der Giessen, J. (2018). Prevalence of Leptospira spp. and Seoul hantavirus in brown rats (Rattus norvegicus) in four regions in the Netherlands, 2011–2015. Infection Ecology & Epidemiology, 8(1), 1490135. 10.1080/20008686.2018.1490135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi, R. G. , Compton, S. M. , Trull, C. L. , Mascarelli, P. E. , Mozayeni, B. R. , & Breitschwerdt, E. B. (2013). Infection with hemotropic Mycoplasma species in patients with or without extensive arthropod or animal contact. Journal of Clinical Microbiology, 51(10), 3237–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maio, E. , Begeman, L. , Bisselink, Y. , van Tulden, P. , Wiersma, L. , Hiemstra, S. , Ruuls, R. , Gröne, A. , Willemsen, P. , & van der Giessen, J. (2014). Identification and typing of Brucella spp. in stranded harbour porpoises (Phocoena phocoena) on the Dutch coast. Veterinary Microbiology, 173(1–2), 118–124. [DOI] [PubMed] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One, 8(4), e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerburg, B. G. , Singleton, G. R. , & Kijlstra, A. (2009). Rodent‐borne diseases and their risks for public health. Critical Reviews in Microbiology, 35(3), 221–270. [DOI] [PubMed] [Google Scholar]
- Mortagy, M. , & Wells, K. (2021). Brucella: An overview. Journal of Alternative Medicine Research, 13(2), 139–142. [Google Scholar]
- Neugent, M. L. , Hulyalkar, N. V. , Nguyen, V. H. , Zimmern, P. E. , & De Nisco, N. J. (2020). Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. MBio, 11(2), e00218–00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niendorf, S. , Harms, D. , Hellendahl, K. F. , Heuser, E. , Böttcher, S. , Jacobsen, S. , Bock, C.‐T. , & Ulrich, R. G. (2021). Presence and diversity of different enteric viruses in wild Norway rats (Rattus norvegicus). Viruses, 13(6), 992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman, A. , Regnery, R. , Jameson, P. , Greene, C. , & Krause, D. (1995). Differentiation of Bartonella‐like isolates at the species level by PCR‐restriction fragment length polymorphism in the citrate synthase gene. Journal of Clinical Microbiology, 33(7), 1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Organisation for Animal Health founded as OIE . (2018). Brucellosis. Retrieved 03‐12‐21 from https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.01.04_BRUCELLOSIS.pdf
- Oksanen, J. , Blanchet, F. G. , Kindt, R. , Legendre, P. , Minchin, P. R. , O'hara, R. , Simpson, G. L. , Solymos, P. , Stevens, M. H. H. , & Wagner, H. (2013). Package ‘vegan. Community ecology package, version (Vol. 2(9), pp. 1–295). [Google Scholar]
- Peterson, A. C. , Ghersi, B. M. , Alda, F. , Firth, C. , Frye, M. J. , Bai, Y. , Osikowicz, L. M. , Riegel, C. , Lipkin, W. I. , & Kosoy, M. Y. (2017). Rodent‐borne Bartonella infection varies according to host species within and among cities. EcoHealth, 14(4), 771–782. [DOI] [PubMed] [Google Scholar]
- Piasecki, T. , Chrzastek, K. , & Kasprzykowska, U. (2017). Mycoplasma pulmonis of rodents as a possible human pathogen. Vector‐Borne and Zoonotic Diseases, 17(7), 475–477. [DOI] [PubMed] [Google Scholar]
- Pitout, J. (2012). Extraintestinal pathogenic Escherichia coli: A combination of virulence with antibiotic resistance. Frontiers in Microbiology, 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prjibelski, A. , Antipov, D. , Meleshko, D. , Lapidus, A. , & Korobeynikov, A. (2020). Using SPAdes de novo assembler. Current Protocols in Bioinformatics, 70(1), e102. [DOI] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , Peplies, J. , & Glöckner, F. O. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web‐based tools. Nucleic Acids Research, 41(D1), D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford, A. D. , Chapman, D. , Dixon, L. , Chantrey, J. , Darby, A. C. , & Hall, N. (2012). Application of next‐generation sequencing technologies in virology. The Journal of General Virology, 93(Pt 9), 1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghwani, J. , Faust, C. L. , Francois, S. , Nguyen, D. , Marsh, K. , Raulo, A. , Hill, S. C. , Parag, K. V. , Simmonds, P. , & Knowles, S. C. (2022). Seasonal dynamics of the wild rodent faecal virome. bioRxiv. 10.1101/2022.02.09.479684 [DOI] [PMC free article] [PubMed]
- Razzauti, M. , Galan, M. , Bernard, M. , Maman, S. , Klopp, C. , Charbonnel, N. , Vayssier‐Taussat, M. , Eloit, M. , & Cosson, J.‐F. (2015). A comparison between transcriptome sequencing and 16S metagenomics for detection of bacterial pathogens in wildlife. PLOS Neglected Tropical Diseases, 9(8), e0003929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renesto, P. , Gouvernet, J. , Drancourt, M. , Roux, V. , & Raoult, D. (2001). Use of rpoB gene analysis for detection and identification of Bartonella species. Journal of Clinical Microbiology, 39(2), 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero‐Juarez, A. , Frias, M. , Perez, A. B. , Pineda, J. A. , Reina, G. , Fuentes‐Lopez, A. , Freyre‐Carrillo, C. , Ramirez‐Arellano, E. , Alados, J. C. , & Rivero, A. (2022). Orthohepevirus C infection as an emerging cause of acute hepatitis in Spain: First report in Europe. Journal of Hepatology, 77, 326–331. [DOI] [PubMed] [Google Scholar]
- Rohde, G. , Straube, E. , Essig, A. , Reinhold, P. , & Sachse, K. (2010). Chlamydial zoonoses. Deutsches Arzteblatt International, 107(10), 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryll, R. , Bernstein, S. , Heuser, E. , Schlegel, M. , Dremsek, P. , Zumpe, M. , Wolf, S. , Pepin, M. , Bajomi, D. , Muller, G. , Heiberg, A. C. , Spahr, C. , Lang, J. , Groschup, M. H. , Ansorge, H. , Freise, J. , Guenther, S. , Baert, K. , Ruiz‐Fons, F. , … Ulrich, R. G. (2017). Detection of rat hepatitis E virus in wild Norway rats (Rattus norvegicus) and Black rats (Rattus rattus) from 11 European countries. Veterinary Microbiology, 208, 58–68. 10.1016/j.vetmic.2017.07.001 [DOI] [PubMed] [Google Scholar]
- Sillanpää, S. , Kramna, L. , Oikarinen, S. , Sipilä, M. , Rautiainen, M. , Aittoniemi, J. , Laranne, J. , Hyöty, H. , & Cinek, O. (2017). Next‐generation sequencing combined with specific PCR assays to determine the bacterial 16S rRNA gene profiles of middle ear fluid collected from children with acute otitis media. mSphere, 2(2), e00006–00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somayaji, R. , Priyantha, M. A. R. , Rubin, J. E. , & Church, D. (2016). Human infections due to Staphylococcus pseudintermedius, an emerging zoonosis of canine origin: Report of 24 cases. Diagnostic Microbiology and Infectious Disease, 85(4), 471–476. [DOI] [PubMed] [Google Scholar]
- Springer, B. , Orendi, U. , Much, P. , Höger, G. , Ruppitsch, W. , Krziwanek, K. , Metz‐Gercek, S. , & Mittermayer, H. (2009). Methicillin‐resistant Staphylococcus aureus: A new zoonotic agent? Wiener klinische Wochenschrift, 121(3), 86–90. [DOI] [PubMed] [Google Scholar]
- Steinberger . (2017). simper_pretty.R and R_krusk.R scripts. 12‐11‐2020. Retrieved 03‐12‐21 from https://github.com/asteinberger9/seq_scripts
- R Core Team . (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing. [Google Scholar]
- Thomas‐White, K. J. , Kliethermes, S. , Rickey, L. , Lukacz, E. S. , Richter, H. E. , Moalli, P. , Zimmern, P. , Norton, P. , Kusek, J. W. , & Wolfe, A. J. (2017). Evaluation of the urinary microbiota of women with uncomplicated stress urinary incontinence. American Journal of Obstetrics and Gynecology, 216(1), 55.e1–55.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Chang, B. J. , & Riley, T. V. (2010). Erysipelothrix rhusiopathiae. Veterinary Microbiology, 140(3–4), 405–417. [DOI] [PubMed] [Google Scholar]
- Willis, A. D. (2019). Rarefaction, alpha diversity, and statistics. Frontiers in Microbiology, 10, 2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yechouron, A. , Lefebvre, J. , Robson, H. G. , Rose, D. L. , & Tully, J. G. (1992). Fatal septicemia due to Mycoplasma arginini: A new human zoonosis. Clinical Infectious Diseases, 15(3), 434–438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Relative contribution of the blocking primer to the blocking of host DNA and increased yield of bacterial 16S rRNA amplicon sequences. Individual rat samples are described here as ‘Rat DNA’.
Figure S2 Raw number of reads per sample. Each dot (blue) represents one sample. Negative control samples are depicted in red.
Figure S3 Rarefaction curves showing the number of detected ASVs per number of reads sampled per sample
Figure S4 Percentage of non‐bacterial reads per sample versus the 16S rRNA gene concentration (pg/μl) after the use of blocking primers. Correlation was tested using Spearman correlation coefficients (R = −.46; p = 3.3e−11).
Table S1 Characteristics of the wild rats used in this study
†Black rats average weight was calculated based on 29/42 rats because the weight from 13 adult rats was not measured. Therefore, the average weight for black rats in our sample is likely underestimated.
Table S2 Mitochondrial 16S rRNA amplicon sequences from other mammals used during blocking primer development
Table S3 Prevalence estimates of Leptospira and Bartonella carriage based on 16S rRNA gene amplicon sequencing and qPCR results, shown per internal or external factor. More than two reads was considered positive for 16S rRNA gene amplicon sequencing. An asterisk (*) depicts a significant difference within or correlation with a factor using the 16S rRNA gene amplicon sequencing or qPCR dataset analysed using GLMMs; *p < .05, **p < .01 and ***p < .001.
Table S4 Results from SIMPER beta‐diversity comparisons that were significantly different between groups according to Kruskal–Wallis tests using fdr‐adjusted p‐values >.05
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.