

Summary
Biodiversity hotspots, such as the Caucasus mountains, provide unprecedented opportunities for understanding the evolutionary processes that shape species diversity and richness. Therefore, we investigated the evolution of Primula sect. Primula, a clade with a high degree of endemism in the Caucasus.
We performed phylogenetic and network analyses of whole‐genome resequencing data from the entire nuclear genome, the entire chloroplast genome, and the entire heterostyly supergene. The different characteristics of the genomic partitions and the resulting phylogenetic incongruences enabled us to disentangle evolutionary histories resulting from tokogenetic vs cladogenetic processes. We provide the first phylogeny inferred from the heterostyly supergene that includes all species of Primula sect. Primula.
Our results identified recurrent admixture at deep nodes between lineages in the Caucasus as the cause of non‐monophyly in Primula. Biogeographic analyses support the ‘out‐of‐the‐Caucasus’ hypothesis, emphasizing the importance of this hotspot as a cradle for biodiversity.
Our findings provide novel insights into causal processes of phylogenetic discordance, demonstrating that genome‐wide analyses from partitions with contrasting genetic characteristics and broad geographic sampling are crucial for disentangling the diversification of species‐rich clades in biodiversity hotspots.
Keywords: admixture, Caucasus, cytonuclear incongruence, hybridization, phylogenomics, Primula, supergene, whole‐genome resequencing
Introduction
Biodiversity hotspots, which are regions with high levels of species diversity and endemism (Myers et al., 2000; Habel et al., 2019), are central to our understanding of how species evolve and diversify through time. These ‘evolutionary hotspots’ often harbor both old and new lineages that played a key role in generating extant species richness, thus representing a reservoir of evolutionary history and genetic diversity (Molina‐Venegas et al., 2017; Zhang et al., 2021). Among the known diversification centers, mountains contribute disproportionately to the abundance of biodiversity on Earth (Rahbek et al., 2019b). Mountain regions represent the centers of diversity for many organisms, and studies of clades with high degrees of endemism in mountains can enhance our ability to reconstruct the evolutionary and biogeographic history of these regions (Dagallier et al., 2020; Perrigo et al., 2020; Chiocchio et al., 2021). This is due, in part, to mountains providing novel and ecologically diverse habitats, functioning as refugia during cycles of glaciations, and acting both as corridors and as barriers of species dispersal (Rahbek et al., 2019a). Furthermore, the repeated upward and downward movements of mountain habitats and climatic zones, particularly with respect to climatic dynamics in the Quaternary Period, lead to rapid range shifts bringing previously isolated taxa into contact and potentially triggering admixture and hybridization (Comes & Kadereit, 1998; Antonelli et al., 2018; Rahbek et al., 2019a). Consequently, admixture and hybridization may be among the key processes responsible for higher plant diversity in mountains (Stebbins, 1959; Soltis & Soltis, 2009; Baack et al., 2015; Yang et al., 2019; Stubbs et al., 2020b).
Recurring admixture and speciation between previously isolated lineages is well documented in several montane plant clades such as Primula sect. Aleuritia (Primulaceae, Guggisberg et al., 2009), Ranunculus (Ranunculaceae, Emadzade et al., 2015), Cupressus (Cupressaceae, Ma et al., 2019), Micranthes (Saxifragaceae, Stubbs et al., 2020a), and Polemonium (Polemoniaceae, Rose et al., 2021). This phenomenon presents a challenge for reconstructing evolutionary relationships as strictly bifurcating hierarchies (Funk, 1985). Thus, admixture and its genomic consequences must be taken into consideration when reconstructing the evolutionary histories of plant clades. For example, recently admixed individuals may exhibit greater genome‐wide similarity with a distantly related introgressive donor lineage than with the population from which they are derived (Runemark et al., 2019). Disentangling network‐like evolutionary histories may require not only sampling many different regions across genomes, as is common for multispecies coalescent (MSC) methods (Xu & Yang, 2016; Sukumaran & Knowles, 2017), but also incorporating information from distinct genomic compartments. For instance, plastomes and regions of suppressed recombination, such as inversions and supergenes, can retain historical evolutionary patterns that have been elsewhere eroded by admixture and recombination (Brandvain et al., 2014; Schumer et al., 2018; Li et al., 2019). However, comparative phylogenetic approaches examining evolutionary history across entire genomes (e.g. whole‐genome resequencing (WGR)) remain scarce.
Primula (primroses, Primulaceae) is ideally suited for studying complex evolutionary processes due to its unique biological and geographic attributes and the extensive scientific knowledge accumulated for this mostly northern and montane clade of c. 550 species (de Vos et al., 2014a). Primroses have been extensively studied in terms of their reproductive, floral, and pollination biology, as well as their ecology, genetics, biogeography, and taxonomy for over a century (Darwin, 1862; Ernst, 1925; Smith & Fletcher, 1947; Valentine, 1952; Richards, 2003; Theodoridis et al., 2013; de Vos et al., 2014b; Cocker et al., 2018; Keller et al., 2021; Mora‐Carrera et al., 2021). This persistent interest in Primula is due, in part, to primroses serving as the primary model for the study of heterostyly, a complex genetic, reproductive, and morphological syndrome that occurs in at least 28 angiosperm families (Ganders, 1979). In primroses, heterostyly is controlled by the S‐locus, or heterostyly supergene, which is composed of five adjacent genes held together via hemizygosity, preventing recombination between them (Nowak et al., 2015; Huu et al., 2016; Li et al., 2016; Potente et al., 2022). The S‐locus is present only in the genome of S‐morph individuals, characterized by flowers with short styles and long anthers, while it is absent in L‐morph individuals, characterized by flowers with the reciprocal spatial arrangement of female and male sexual organs.
Within primroses, the relationships of Primula sect. Primula, a section consisting of seven species and 13 subspecies (all diploid except for Primula grandis) and a high degree of endemism in the Caucasus region, remain unresolved. Importantly, investigating relationships in Primula sect. Primula is of significance because it has been hypothesized that this clade originated in an eastern refugium in the Caucasus and spread westward into Europe (Richards, 2003; Volkova et al., 2020). Although the Caucasus region is considered an important reservoir of biodiversity, studies investigating speciation patterns within this region, its biogeographic role in Eurasia, and overall knowledge of the biodiversity of this area are limited (Myers et al., 2000; Zazanashvili, 2009; Koch et al., 2016; Mumladze et al., 2020).
Previous phylogenetic analysis suggested a lack of reciprocal monophyly and conflict in the evolutionary relationships within this clade among the three widespread species, that is, Primula elatior (P. el.), Primula veris (P. ve.), and Primula vulgaris (Schmidt‐Lebuhn et al., 2012). This study was hampered by three factors: it employed limited geographic sampling and only three genetic markers (rps16‐trnK, trnS‐trnG, and internal transcribed spacer); the relatively young age of Primula sect. Primula (2.46 million years ago (Ma), 95% highest posterior density: 3.268–1.527 Ma; de Vos et al., 2014a) and short branch lengths within this clade, which might imply a recent, rapid radiation and/or incomplete lineage sorting (ILS); and hybridization that has been extensively documented in both natural populations and artificial crosses (Darwin, 1868; Smith & Fletcher, 1947; Valentine, 1947, 1952, 1961; Woodell, 1969; Kálmán et al., 2004; Keller et al., 2016, 2021; Tendal et al., 2018). Overall, it is unclear whether the recovered non‐monophyly was the result of limited sampling, limited genetic resolution, ILS, and/or indicative of reticulate evolution.
In this study, we conducted widespread geographic sampling of Primula sect. Primula across Eurasia (Fig. 1a) and generated novel WGR data for 106 specimens to assess the evolutionary relationships and causal processes that underlie the previously identified non‐monophyly in this clade. We specifically investigated whether the evolution in Primula sect. Primula was shaped by speciation in the Caucasus biodiversity hotspot before westward dispersal into Europe, reflecting an ‘out‐of‐Caucasus’ biogeographic pattern (Richards, 2003; Volkova et al., 2020). Compared to previous research that found the three widespread species to be non‐monophyletic, our analyses with genome‐scale data recovered two of these three species as monophyletic. Our analyses suggest that the remaining non‐monophyly in this section is primarily driven by past admixture between lineages in the Caucasus and confirmed the Caucasus as the center of origin for Primula sect. Primula. The research presented here highlights the importance of this understudied biodiversity hotspot in shaping species phylogenetic history and diversity and demonstrates that dense genomic and extensive geographic sampling can provide resolution in recalcitrant groups.
Fig. 1.
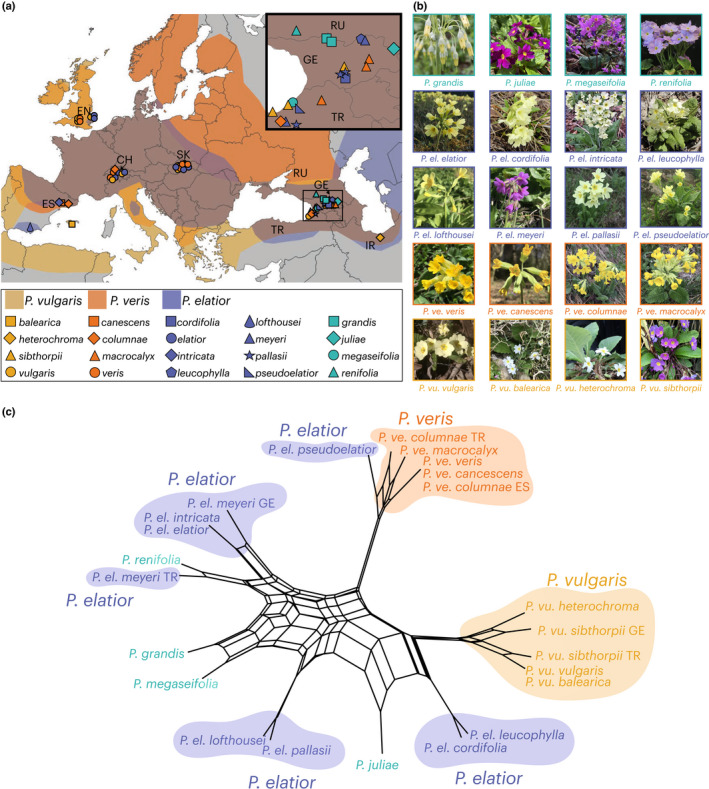
Overview of Primula sect. Primula. (a) Approximate distribution of the three widespread Primula sect. Primula species across Eurasia. Colored shapes indicate sampling localities and correspond to species and subspecies. Inset map shows Caucasus collections. Countries where samples were collected are identified by the two‐letter country code. (b) Species and subspecies used in this study. (c) Neighbor‐Net split graphs based on quartet distances from 3623 genome fragment trees with three widespread species highlighted. Non‐monophyletic subspecies are distinguished by the two‐letter country code. P. ve., P. veris; P. vu., P. vulgaris; P. el., P. elatior; EN, England; CH, Switzerland; SK, Slovakia; GE, Georgia; TR, Turkey; ES, Spain; IR, Iran; RU, Russia.
Materials and Methods
Geographic sampling
Targeted fieldwork was conducted in 2019 to collect accessions covering the taxonomic and geographic diversity of Primula sect. Primula L. (Fig. 1a,b). Every currently recognized species and subspecies of Primula sect. Primula (following Richards, 2003 and Schmidt‐Lebuhn et al., 2012) was obtained from at least one population, and from every population we collected two individuals (Supporting Information Table S1). Additionally, eight outgroup accessions used in previous phylogenetic analyses (Schmidt‐Lebuhn et al., 2012; de Vos et al., 2014a) were obtained from the University of Zurich frozen tissue collection (Table S1). This resulted in 98 ingroup and eight outgroup samples, totaling 106 specimens (see Methods S1 for more details).
DNA extraction and WGR
For all samples, DNA was extracted using the Maxwell Rapid Sample Concentrator 48 and the Maxwell RSC PureFood GMO and Authentication Kit. Manufacturer's instructions were followed with the exclusion of Proteinase K. Library preparation and WGR were conducted at Rapid Genomics (Gainesville, FL, USA; rapid‐genomics.com). Samples were sequenced on the Illumina NovaSeq 6000 S4 (paired‐end 150 bp reads) to an average depth of 20×.
Read alignment, variant calling, and filtering
Raw paired‐end reads were mapped to the chromosome‐scale P. veris L. reference genome (haploid assembly, containing the S‐locus; Potente et al., 2022) using Bwa v.0.7.17 (Li & Durbin, 2009) and the ‐mem algorithm. BAM files were then sorted, marked for duplicates, and indexed using picard 2.7.1 (Broad Institute, 2019) and SAMtools v.1.3 (Li, 2011). Mapping to a high‐quality reference genome allows for an increase in the number of reads used, a more even distribution of mapped reads, and high precision in the location and order of sites and genomic regions of interest, such as repetitive sites (Lowry et al., 2017; Pfeifer, 2017).
Variant calling and filtering followed Ravinet et al. (2021). Variant calling was performed by BCFtools v.1.8 multiallelic caller (Li, 2011; Danecek et al., 2021) and set to output only variant sites. Repetitive sequence regions were masked based on the annotation from Potente et al. (2022). Filtering was performed using Vcftools v.0.1.14 (Danecek et al., 2011) and set to remove indels and include only single nucleotide polymorphisms (SNPs) occurring in at least 80% of individuals, with a minimum site quality of 30, and a mean site depth between 5× and 60×.
Genomic fragment trees
We divided the genome into 50‐kb non‐overlapping genome fragments (GFs) from the 11 chromosomes in the P. veris assembly with custom scripts. We selected every other GF, resulting in 3623 alignments. Iq‐tree v.2.1.2 (Nguyen et al., 2015) was used to estimate individual GF trees. For each alignment, Modelfinder (Kalyaanamoorthy et al., 2017) was used to select the best nucleotide substitution model (adjusted for ascertainment bias) and otherwise default parameters were used. We generated statistical support for branches with 1000 ultrafast bootstraps (BS) (Minh et al., 2013; Chernomor et al., 2016).
Network analyses
We created a species network under the coalescent model with Network inference Algorithm via Neighbour Net Using Quartet distance (Nanuq; Allman et al., 2019) from 3623 GF trees. Nanuq assumes a coalescent process, and therefore accommodates ILS and provides a model‐based interpretation of reticulation that justifies a hybridization network (Allman et al., 2019). Species networks were estimated using the Nanuq algorithm as implemented in the R package mscquartets (Allman et al., 2019). We selected an alpha of 5e−11 to impose a high standard for evidence of hybridization and a beta of 0.05, which requires stronger evidence for any resolution of the four‐taxon network (Methods S2). Using the software Splitstree4 (Huson & Bryant, 2006), a splits graph was inferred from the resulting Nanuq quartet distance matrix.
Phylogenetic inference
Phylogenetic relationships between samples were determined using maximum likelihood (ML). For the ML analysis, due to computational limitations, we randomly selected and concatenated 400 GFs from across the genome, spanning 20 million sites in total. This alignment was used for ML analysis in Iq‐tree with the same parameters as mentioned in the Genomic Fragment Trees section. We also constructed a MSC phylogeny. The MSC phylogeny, which assumes ILS as the primary driver of gene tree discordance, was estimated in the program Astral‐III v.5.7.3 (Zhang et al., 2018; Rabiee et al., 2019) with all 3623 GF trees used as input. As recommended, GF tree branches with support < 10 BS were collapsed before inputting into Astral‐III. Support for each branch was assessed with local posterior probability (LPP) (Sayyari & Mirarab, 2016).
Plastid phylogeny
Phylogenetic incongruence between plant nuclear and organellar genomes can provide evidence of introgression (Folk et al., 2017; Lee‐Yaw et al., 2019; Morales‐Briones et al., 2021; Zhou et al., 2022). To investigate phylogenetic signals from the plastid genome, reads from each individual were mapped to the plastid genome of P. veris NC 031428 (Zhou et al., 2016) using the BWA‐mem algorithm using default parameters. The plastid variant call format (VCF) required different filtering from the nuclear genome due to the plastome being both haploid and present in large copy numbers in each sample (Meleshko et al., 2021). Filtering was performed by Vcftools to keep SNPs occurring in at least 50% of individuals with a minimum quality score of 15 and mean site and genotype depths between 10× and 400×. The filtered VCF retained 147 446 out of 152 682 sites. The plastome phylogeny was estimated using a ML and conducted in IQ‐Tree using the same parameters as for the nuclear analysis.
S‐locus phylogeny
When considering just recombination, the S‐locus is equivalent to both individual nonrecombining loci in the nuclear genome and the entire nonrecombining plastome. However, the S‐locus is larger (c. 300 kb) than most other nonrecombining individual nuclear loci, and therefore the S‐locus can provide far greater genetic resolution than much smaller individual nuclear loci. Additionally, unlike the maternally inherited plastome, the S‐locus can be either maternally or paternally inherited, and thus captures both maternal and paternal evolutionary patterns. Finally, as a result of the increased rate of fixation of mutations by drift in the S‐locus caused by its small effective population size and the decrease in genetic polymorphisms due to linkage between nearly neutral and non‐neutral mutations, the S‐locus is expected to have lower levels of ILS than other parts of the genome (Bergero & Charlesworth, 2009; Willyard et al., 2009; Hobolth et al., 2011; Pease & Hahn, 2013; Martin & Jiggins, 2017; Sackton & Clark, 2019).
We inferred the phylogeny of the S‐locus to provide resolution and insight into the evolution of nonrecombining regions. The location of the S‐locus, containing 255 577 sites, was obtained from the P. veris genome assembly (Potente et al., 2022). We reduced our total alignment to only accessions containing the heterostyly supergene (i.e. plants identified in the field as being S‐morphs) and one outgroup. Then, after trimming the alignment to the location of S‐locus and masking repetitive regions, we removed specimens with a mean depth lower than 5×. We filtered this region to keep SNPs occurring in at least 75% of individuals, with a minimum quality score of 30, and a mean site and genotype depths of between 5× and 30×.
Discordance
Discordance analyses between the nuclear genome, chloroplast, and S‐locus were performed and visualized as tanglegrams using the cophylo function of the R package phytools (Revell, 2012). Additionally, we explored gene and sequence conflict using the gene‐concordance factor (gCF) and site‐concordance factor (sCF) in IQ‐Tree (Nguyen et al., 2015; Minh et al., 2020) based on the GF trees, GF sequence alignments, and all phylogenies. We used default settings, except for increasing the number of sampled quartets for computing sCF to 10 000.
Gene flow and hybrid network analyses
We quantified the contribution of gene flow and hybridization in Primula sect. Primula using D‐statistics. D‐statistic tests (also known as ABBA‐BABA tests) detect gene flow in the presence of ILS through testing for an imbalance in the number of discordant SNPs among quartets of species (Patterson et al., 2012). We used the package Dsuite, which applies D‐statistics in a phylogenetic context (Malinsky et al., 2021). We specifically employed the Fbranch statistic (f b) introduced in Malinsky et al. (2018), which combines the f 4‐ratio with a species phylogeny to assign gene flow evidence to specific branches, with 0 being no admixture and 1 being completely admixed. The f b statistic differs from Patterson's D in that it provides phylogenetically‐based estimates of excess allele sharing. Thus, f b statistics are particularly powerful for complex groups because, unlike Patterson's D, it assigns gene flow evidence to specific branches in a phylogeny (Malinsky et al., 2021; De‐Kayne, 2022). Dsuite was run with default parameters with the entire genomic dataset and the Dtrios and Fbranch commands. One drawback of phylogeny‐based methods for investigating admixture is their reliance on the ‘true’ species tree. Thus, we ran the Dsuite analysis for both the ML and MSC phylogenies. We used D FOIL (Pease & Hahn, 2015) to confirm admixture events identified by the f b statistic and checked for the possibility of ‘ghost’ lineages influencing our results following the methods of Hibbins & Hahn (2021b) (Methods S3).
Ancestral range estimations
Ancestral range estimations (ARE) were employed to test the hypothesis that Primula sect. Primula originated in the Caucasus (Richards, 2003; Volkova et al., 2020). Given that no fossil calibration points are available within our sampled taxa, we conducted an ARE analysis on the ML phylogeny. We chose this approach because multiple studies have found that ancestral state reconstructions are more accurate when performed on phylograms compared with time‐calibrated phylogenies (Litsios & Salamin, 2012; Cusimano & Renner, 2014; Wilson et al., 2022). The ARE analyses were performed with a phylogeny reduced to 19 individuals representing monophyletic groups (Fig. 2). We delineated the following six geographic areas for ARE based on bioclimatic zones, current distribution patterns, and geographic barriers: Alps (A), British Isles (B), Caucasus (C), Iberian Peninsula (I), Carpathians I, and/or Siberia/northern Asia (S).
Fig. 2.
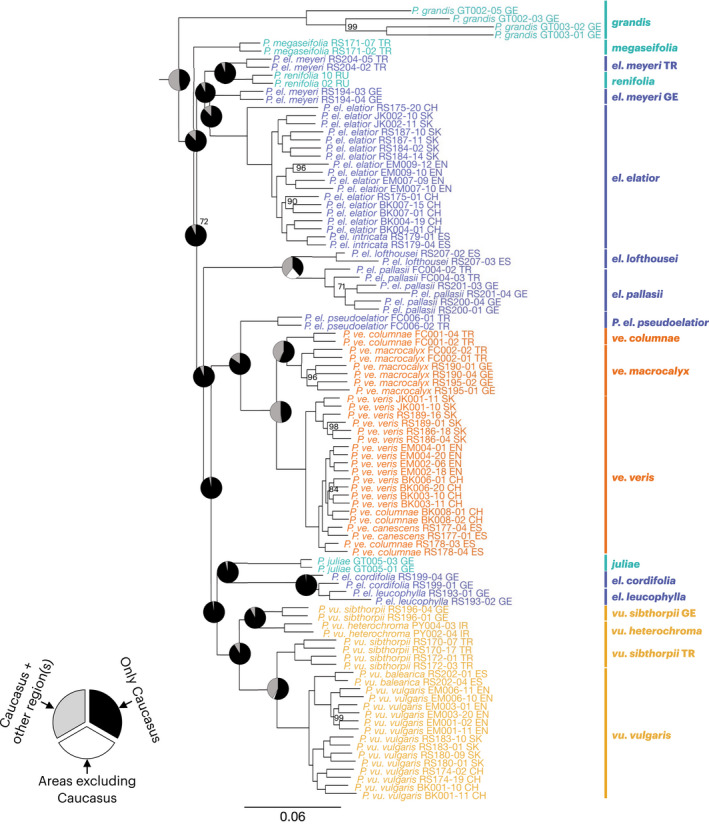
Phylogenomic analysis and ancestral range estimations resolve relationships and recover the Caucasus as the ancestral range of Primula sect. Primula. Topology constructed from the nuclear genome using maximum likelihood (ML). Branches with < 100% bootstrap (BS) are labeled. Pie charts at nodes indicate the probability of that node being recovered in only the Caucasus (black), Caucasus plus the Alps, British Isles, Iberian Peninsula, Carpathians, and/or Siberia/northern Asia (gray), or any areas outside of the Caucasus (white) in the ancestral range estimation analysis. Full pie charts in Supporting Information Fig. S6. Outgroups not shown. P. ve., P. veris; P. vu., P. vulgaris; P. el., P. elatior; EN, England; CH, Switzerland; SK, Slovakia; GE, Georgia; TR, Turkey; ES, Spain; IR, Iran; RU, Russia.
We used the phytools functions make.simmap and fitpolyMk to conduct an ARE analysis under a Bayesian framework. This method was selected because it allows for polymorphic states and can accommodate phylograms. We tested all four models: all transitions occurring at the same rate, backward and forward transitions occurring at the same rate, all transitions occurring at different rates, and the transient model where the acquisition and loss of a state occur at different rates. The best model was selected based on the Akaike information criterion. Stochastic mapping was simulated 1000 times, summarized, and plotted on the phylogram with the posterior probability (PP) of each node occurring in each range. We also investigated whether ARE with a time‐tree recovered similar ancestral ranges as those obtained with a phylogram. We constructed an ultrametric tree through relative dating by putting a tight normal prior on the root of Primula sect. Primula and conducted parallel analyses (Methods S4).
Isolation‐by‐distance
It is expected that populations become less genetically related with increasing geographic distance, that is, a pattern of isolation‐by‐distance (IBD) (Wright, 1943; Hutchison & Templeton, 1999; Twyford et al., 2020). Intraspecific structure can result from IBD because geographically close populations tend to be more genetically similar than populations located farther away (Wright, 1943; Hutchison & Templeton, 1999). We therefore tested for IBD using a Mantel test between the matrix of nuclear genetic distances and a matrix of geographic distances made from collection localities using the R package adegenet v.2.1.5 (Jombart, 2008; Jombart & Ahmed, 2011). If the Mantel test results in a significant P‐value (P < 0.05) and a strong correlation coefficient (r ≥ 0.7), this is suggestive of IBD and the potentially major role of geographic isolation in lineage diversification. We tested for IBD among all populations within P. veris, P. vulgaris Huds., and P. elatior (L.) Hill. Additionally, because the Caucasus endemics were recovered as sister to the Caucasus P. elatior subspecies in the phylogenetic and network analyses (see the Results section), we tested for IBD in a group consisting of P. elatior subspecies plus all four Caucasus endemic species. To visualize the correlation between genetic and geographic distances, densities of distances were plotted using a two‐dimensional kernel density estimation.
Results
Read alignment, variant calling, and filtering
We generated WGR data for 98 Primula sect Primula accessions and eight outgroups (total 106 samples). Paired‐end reads were mapped to the haploid reference genome assembly of P. veris containing the S‐locus (Potente et al., 2022) using Bwa v.0.7.17 (Li & Durbin, 2009). On average, 96.1% ± 1.7 of ingroup reads aligned to the P. veris reference genome (91.8% ± 15.4 with outgroups). The average coverage for each ingroup sample was 20.2× ± 4.5 (19.5× ± 5.0 with outgroups). After base calling, the total dataset had 103 365 340 sites, and after filtering the number of high‐quality variant sites was 15 114 651.
Network analyses
We created a species network under the coalescent model with Nanuq from 3623 GF trees. Unlike other methods employing splits graphs, which should simply be viewed as exploratory devices and cannot be used to identify the underlying biological mechanisms causing network‐like relationships, Nanuq provides a firm model‐based interpretation of reticulation (Allman et al., 2019). The resulting neighbor‐net splits graph showed high levels of reticulation along the backbone of Primula sect. Primula, and less reticulation at shallower nodes (Fig. 1c).
Phylogenetic analysis
The ML and MSC phylogenies were well resolved and received high support (100% BS, 1.0 LPP) at most nodes (Figs 2, S1). Although there was high branch support throughout both nuclear genome phylogenies, there was low gCF and sCF at deeper nodes in both phylogenies (Fig. S2). When both gCF and sCF are low, this is indicative of discordant signal (i.e. ILS and/or introgression), rather than stochastic error from limited information (Lanfear, 2018; Minh et al., 2020). Of note, there is overall a slightly higher gCF and sCF for the nuclear ML topology compared to the MSC topology. The ML and MSC phylogenies varied solely in the relationships of Primula megaseifolia, P. el. lofthousei, and P. el. pallasii (Figs 2, S1). Notably, gCF at these nodes was low for both phylogenies, while sCF was high for the ML topology and low for the MSC topology. Additionally, the branch supporting the sister relationship of P. megaseifolia, P. el. lofthousei, and P. el. pallasii in the MSC phylogeny is the only node along the backbone with < 1.0 LPP (=0.96).
Overall, both analyses were largely congruent and both supported P. veris and P. vulgaris as monophyletic, whereas P. elatior was non‐monophyletic. Furthermore, P. grandis, the one Caucasus endemic species for which we sampled more than one population, was also monophyletic. In addition to the non‐monophyly recovered in P. elatior, non‐monophyly was also recovered at the subspecies level. For instance, the autonymous subspecies (P. el. elatior, P. ve. veris, and P. vu. vulgaris) are all non‐monophyletic in the ML analysis, with the descendants of the most recent common ancestor (MRCA) of each of these widespread subspecies including the narrow endemic subspecies P. el. intricata (ES), P. ve. canescens (ES) plus P. ve. columnae (ES and CH), and P. vu. balearica (ES), respectively (Fig. 2). Consequently, in downstream analyses requiring species monophyly, these narrow endemic subspecies are included in the autonymous subspecies (see Fig. 2).
Chloroplast phylogeny
The phylogenetic analysis of the entire chloroplast genome resulted in a well‐resolved topology, where most branches had BS = 100% (Fig. S3). However, in the chloroplast topology, all three widespread species are recovered as non‐monophyletic (Fig. 3), similar to previous studies with only two plastid spacer regions (Schmidt‐Lebuhn et al., 2012). Furthermore, two specimens of P. el. elatior are recovered among the P. vu. vulgaris samples, and one specimen of P. vu. vulgaris is recovered among P. el. elatior samples (Fig. S3). Additionally, the chloroplast topology is in strong conflict with the nuclear phylogeny, with incongruent relationships being recovered at both deep and shallow relationships (Fig. 3). The gCF and sCF for the plastid topology are extremely low with gCF being zero or close to zero at most nodes (Fig. S2), indicative of the high degree of incongruence between the nuclear and chloroplast topologies.
Fig. 3.
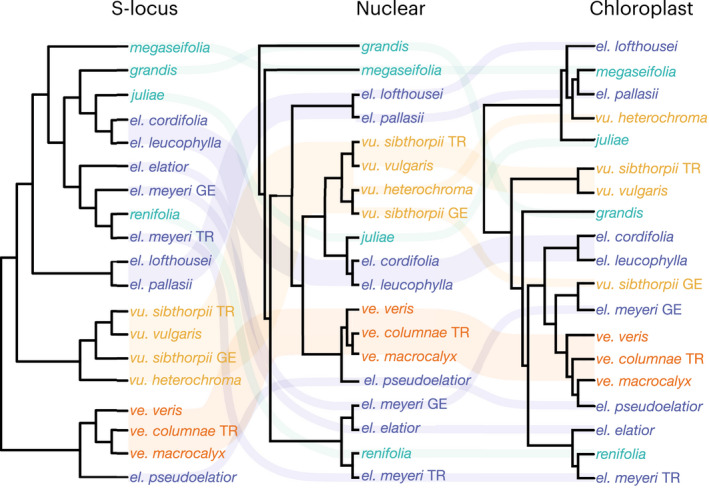
Tanglegram comparing topologies of S‐locus, nuclear genome, and chloroplast cladograms in Primula sect. Primula. Monophyletic groups are reduced to one tip per taxon. Full chloroplast and S‐locus phylogenies in Supporting information Figs S3, S4. P. ve., P. veris; P. vu., P. vulgaris; P. el., P. elatior; GE, Georgia; TR, Turkey.
S‐locus phylogeny
If ancient admixture occurred in the evolutionary history of Primula sect. Primula, we would expect the nuclear genome and S‐locus phylogenies to be incongruent at deep nodes in the phylogeny. Overall, the S‐locus topology is well resolved and largely aligns with the nuclear phylogeny at shallow nodes (Figs 3, S4). However, we did recover hard incongruence at deeper nodes. One example of deep incongruence pertains to P. megaseifolia. In the S‐locus topology, P. el. pallasii and P. el. lofthousei are sister to a clade containing P. megaseifolia. By contrast, in the nuclear genome topology, P. megaseifolia is sister to a clade containing all other species in the section, excluding P. grandis. Another example of deep incongruence is the relationships of P. vulgaris and P. veris. In the nuclear genome topology, P. vulgaris is sister to a clade consisting of Primula juliae, P. el. cordifolia, and P. el. lecuophylla, which is then sister to P. veris. However, in the phylogenetic analysis of the S‐locus, P. vulgaris is sister to a clade consisting of P. elatior plus the Caucasus species (excluding P. el. pseudoelatior), while P. veris is sister to all other species in the section.
Gene flow analyses
Low f b values were recovered for many species (Fig. 4), corroborating the widespread reticulate evolution at deeper evolutionary time scales recovered in the network analysis (Fig. 1c). However, an excess of shared alleles was seen in multiple species pairs in Primula sect. Primula; therefore, we will focus on the two highest f b values (f b ≥ 0.140), signifying the highest excess of shared alleles (Fig. 4; Table S2). Of note, these same species pairs were also identified by high f b values when using the MSC topology (Fig. S5). The highest f b value was recovered between P. el. pseudoelatior and the ancestor of P. ve. macrocalyx and P. ve. columnae TR (f b = 0.159). The next highest number of shared alleles was recovered between P. megaseifolia and the MRCA of the P. el. meyeri, P. el. elatior, and Primula renifolia clade (f b = 0.140), indicative of admixture between P. megaseifolia and the MRCA and/or the descendants of the renifolia clade (P. el. meyeri, P. el. elatior, and P. renifolia). These admixture events were confirmed by D FOIL (Table S3), and we did not recover any evidence suggesting the impact of a ‘ghost’ lineage on these results (Table S4). Finally, the f 4‐ratio is prone to false positives (Eaton et al., 2015), and admixture fractions are dependent on demography and thus can be skewed by demographic events. It is therefore possible that the recovered admixture results are artifacts of the analysis and/or a complex demographic history. However, it has been shown that the f 4‐ratio, on which the f b statistic is based, is robust under most demographic scenarios (Durand et al., 2011; Patterson et al., 2012). Furthermore, our results remained robust under different combinations of taxa (Fig. S5) and alternative approaches such as network inference (Fig. 1c).
Fig. 4.
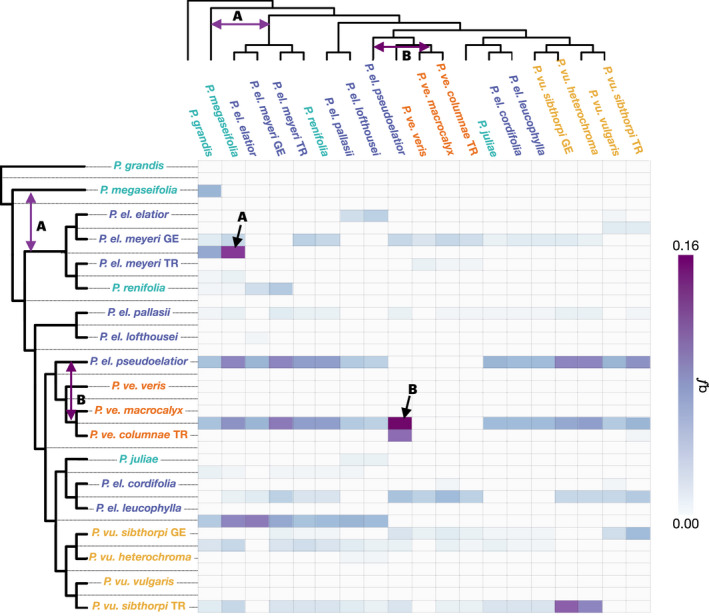
Evidence of gene flow in Primula sect. Primula. Results of the Fbranch (f b) analysis in Dsuite with maximum likelihood (ML) nuclear genome phylogeny. The values in the matrix signify excess allele sharing between the branch identified on the tree on the y‐axis, which includes internal branches, and the species identified on the x‐axis, relative to its sister branch. The light violet to dark purple gradient in the matrix refers to excess allele sharing, and white squares correspond to 0, nonsignificant values, or correspond to tests that are not consistent with the phylogeny. The two squares marking the highest number of shared alleles are designated with A and B. The corresponding admixture event signified by that square is drawn onto each tree. (A) Excess of shared alleles between Primula megaseifolia and the ancestor of P. el. elatior, P. el. meyeri, and P. renifolia. (B) Excess of shared alleles between P. el. pseudoelatior and the ancestor of P. ve. macrocalyx and P. columnae. P. ve., P. veris, P. vu., P. vulgaris; P. el., P. elatior; GE, Georgia; TR, Turkey.
Ancestral range estimation
The ARE analysis suggests that there is strong support for the origin of Primula sect. Primula in the Caucasus (Figs 2, S6, S7; Table S5). The transient model was selected as the best fit to the data in the ARE analysis. This analysis supported an origin in the Caucasus for the MRCA of Primula sect. Primula, with the Caucasus being recovered with the highest PP of 0.482. The next most likely range, albeit with a much lower probability, also included the Caucasus and consisted of the Caucasus plus Iberian Peninsula (PP = 0.077) (Table S5). Additionally, all nodes along the backbone of the phylogeny identified the Caucasus as the ancestral range with the highest probability (Fig. 2). The chronogram analysis recovered congruent results and inferred the Caucasus as the ancestral range (Fig. S7).
Isolation‐by‐distance
Our analysis revealed strong IBD patterns within both P. vulgaris and P. veris, but not within P. elatior (Fig. 5). The IBD analyses for both P. vulgaris and P. veris recovered significant and strong correlations between genetic and geographic distance. Primula vulgaris showed a single high‐density cloud suggesting a continuous cline of differentiation. However, the IBD analysis with P. veris showed two distinct high‐density clouds, suggesting that these are differentiated populations. These discontinuities may be an artifact of our sampling or could be indicative of intraspecific structure. Overall, both P. vulgaris and P. veris conform to the expectation of IBD. By contrast, both analyses with P. elatior recovered three distinct high‐density regions and low correlation (r < 0.4) (Fig. 5). Additionally, both IBD analyses with the P. elatior subspecies indicated a patched pattern of differentiation among populations. Due to the non‐monophyly of P. elatior (Fig. 2), this result is not surprising. However, we evaluated IBD for P. elatior with the Caucasus endemic species expecting to recover a stronger signal of IBD due to the close phylogenetic relationships and geographic overlap of these species. In fact, we found the opposite pattern, whereby there was a lack of significance and a much weaker correlation in the P. elatior analyses that included the Caucasus endemics. This suggests that geographically proximal populations of these species in the Caucasus are genetically distant (Fig. 4, high‐density cloud in upper left corner of plot) and supports the role of biogeographic processes in species diversification.
Fig. 5.
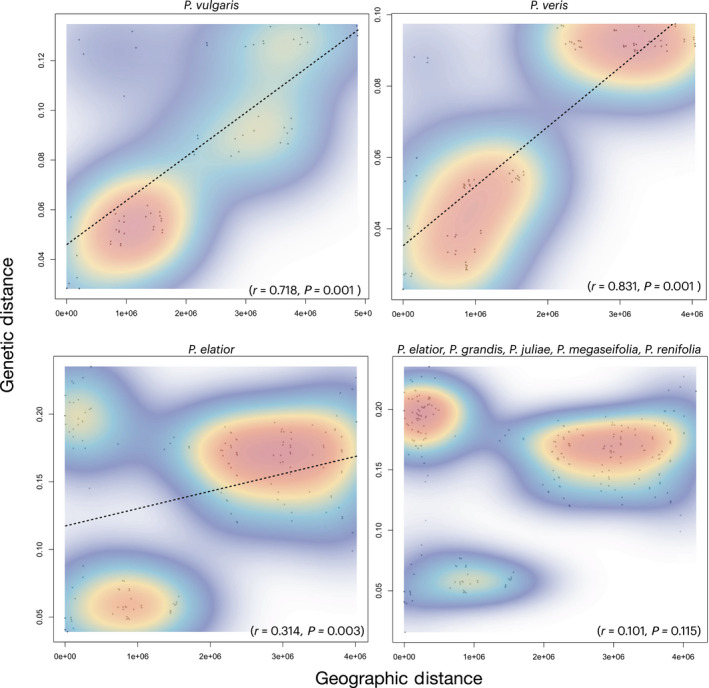
Scatterplots with two‐dimensional kernel density estimation illustrating the relationship between genetic and geographic distance. We tested for isolation‐by‐distance (IBD) using a Mantel test. When correlations are significant (Mantel test, P < 0.05), regression lines are drawn. If the Mantel test results in a significant P‐value and a strong correlation coefficient (r ≥ 0.7), this is suggestive of IBD. A single high‐density cloud suggests a continuous cline of differentiation, while multiple discontinuous high‐density regions suggest differentiated populations. A significant and strong positive correlation between geographic and genetic distance is recovered for both Primula vulgaris and Primula veris, supporting a pattern of intraspecific IBD. By contrast, there was a (very) weak correlation (r < 0.4) between genetic and geographic distances for Primula elatior. Furthermore, despite the close phylogenetic relationships of P. elatior and the Caucasus endemics, there was a lack of significance and a very weak correlation in the analysis combining P. elatior, Primula grandis, Primula juliae, Primula megaseifolia, and Primula renifolia.
Discussion
We used WGR in conjunction with widespread geographic sampling to reconstruct a highly resolved Primula sect. Primula phylogeny and investigate the biogeographic patterns and role of the Caucasus biodiversity hotspot in the evolution of this section. Our results provide an updated evolutionary framework for Primula sect. Primula and identify the Caucasus as the center of origin for this clade. Using an array of analytical approaches, we found substantial evidence of reticulate evolution primarily driven by deep lineage admixture in the Caucasus (Figs 1, 2, 3). Overall, our results are consistent with the studies of other montane plants that find widespread reticulate evolution in clades occurring in mountain regions (Guggisberg et al., 2009; Emadzade et al., 2015; Rose et al., 2021) and highlight the importance of studying centers of diversification in resolving species evolutionary history.
Genome‐scale data resolve the evolutionary history of Primula sect. Primula and provide insight into the biogeography of Caucasus biodiversity hotspot
In previous analyses of Primula sect. Primula, both deep and shallow relationships were uncertain (Schmidt‐Lebuhn et al., 2012). By contrast, both the ML and the MSC phylogenies from this study were well resolved and supported highly similar topologies (Figs 2, S1). However, the network analysis implies widespread reticulate evolution at deeper evolutionary time scales in this clade (Fig. 1c). Although all phylogenies are well resolved, both the ML and the MSC phylogenies have extremely short branch lengths along the backbone (Figs 2, S1). Due to the young age of this section, this could be indicative of a recent, rapid radiation, as seen in other groups of European Primulaceae (Soldanella, Comes & Kadereit, 2003; Primula sect. Auricula; Zhang et al., 2004; Androsace, Roquet et al., 2013). Consequently, ILS, a well‐documented product of rapid radiations, likely played an important role in the evolutionary history of Primula sect. Primula, in addition to hybridization, a conclusion further supported by the low gCF and sCF values estimated throughout the phylogeny (Fig. S2). It is notoriously difficult to distinguish the signals of ILS and introgression (Cai et al., 2021; Hibbins & Hahn, 2021a; Esquerré et al., 2022), but by employing three programs that account for ILS (i.e. Nanuq, Astral, Dsuite), our results suggest that both introgression and ILS have played a significant role in shaping the inferred evolutionary relationships in this clade. Overall, the complexity of cladogenetic and tokogenetic processes within this clade is not well represented in a bifurcating topology.
Despite the fact that the reticulate processes shaping lineage evolution may not be adequately captured in the phylogenetic analysis, at shallower nodes the network and phylogenetic analyses support congruent results. One key finding concerning shallower relationships is whether the three widespread Primula sect. Primula species are monophyletic or not. In our analyses, based on the entire nuclear genome, we found that P. veris and P. vulgaris are monophyletic, while P. elatior is non‐monophyletic. This differs from previous research that found non‐monophyly in all three species (Schmidt‐Lebuhn et al., 2012); it is therefore likely that the previously‐recovered non‐monophyly of P. veris and P. vulgaris was an artifact of limited phylogenetic information, caused by insufficient genomic data. By contrast, the non‐monophyly of P. elatior recovered in both studies seems indicative of reticulate evolution. Consequently, the evolutionary history of P. elatior is more complex, differing considerably from P. vulgaris and P. veris (see ‘Cytonuclear discordance is indicative of admixture in P. elatior ’ in the Discussion section).
It has been hypothesized, but never tested, that the Primula sect. Primula clade originated in an eastern refugia in the Caucasus and Asia and spread westward into Europe (Richards, 2003; Volkova et al., 2020). Our study provides multiple lines of evidence confirming this hypothesis. First, in the ARE analysis, the Caucasus region is identified as the ancestral range of Primula sect. Primula. Second, both P. veris and P. vulgaris show strong declining genetic similarity with increasing geographic distance, that is, IBD, correlated to the increased genetic distances between the Caucasian and the western European populations (Fig. 5). Taken together, these results support an ‘out‐of‐Caucasus’ model of evolution for the ancestral lineages of Primula sect. Primula and for P. veris and P. vulgaris. These findings are consistent with other studies, which have highlighted the role of the Caucasus biodiversity hotspot as a source for European populations (Hewitt, 2000; Grimm & Denk, 2014; Volkova et al., 2020). However, the model of evolution for P. elatior is less straightforward, and processes of reticulation can be invoked to explain the evolution of this species. Specifically, our results imply that the non‐monophyly of P. elatior can be attributed, in large part, to admixture between the Caucasian P. elatior lineages and Caucasus endemic species (Figs 1c–4).
Cytonuclear discordance is indicative of admixture in P. elatior
Many of our results point toward gene flow as the cause of the non‐monophyly of P. elatior. Specifically, as cytonuclear discordance is indicative of hybridization and/or introgression (Chan et al., 2021; Wang et al., 2021), the incongruences between the nuclear and chloroplast topologies highlight multiple examples of reticulate evolution (Fig. 3). We will focus on three of the most striking cases of strongly supported (> 90% BS) incongruences and draw upon our other analyses to highlight putative cases of reticulate evolution.
Perhaps the most conspicuous example of cytonuclear discordance is seen in P. vu. sibthorpii and P. el. meyeri. In the nuclear phylogeny, both the Georgian and Turkish populations of each species are recovered in clades with their respective conspecifics (Figs 2, 3). However, in the chloroplast phylogeny, the Georgian populations of P. vu. sibthorpii and P. el. meyeri are recovered as sister taxa, while Turkish populations of each subspecies maintain congruent relationships to those seen in the nuclear topology (Fig. 3). We were unable to detect any other strong evidence of gene flow between P. vu. sibthorpii and P. el. meyeri, which aligns with studies showing organelle capture aligning with geography and occurring in the absence of detectable nuclear introgression (Rieseberg & Soltis, 1991; Stegemann et al., 2012). Therefore, the surprising sister relationship of the Georgian populations of P. vu. sibthorpii and P. el. meyeri in the chloroplast topology is strongly indicative of plastome introgression between these subspecies.
Another hard incongruence between the nuclear and chloroplast topologies concerns the relations of the Caucasus‐endemic P. megaseifolia (Fig. 3). In the nuclear ML analysis, P. megaseifolia is sister to all of Primula sect. Primula, excluding P. grandis, while in the plastid phylogeny, P. megaseifolia is sister to P. el. pallasii (Fig. 3). In both nuclear phylogenies, P. megaseifolia is on a very short branch, which is indicative of ILS or introgression plus ILS. Due to the ML analysis being sensitive to ILS, it is quite possible that the recovery of P. megaseifolia as sister to the rest of the section is an artifact of ILS. However, the history of reticulate evolution suggested by topological incongruence recovered in the MSC, chloroplast, and S‐locus phylogenies was supported by other analyses. First, D‐statistics recovered a high level of shared alleles between P. megaseifolia and the MRCA of the renifolia clade (Figs 4, S5). Second, in the network analysis, which accommodates ILS, P. megaseifolia is recovered as an intermediate between P. el. pallasii and P. el. lofthousei and the renifolia clade (Fig. 1c).
All together, we have strong evidence for genetic exchange between the lineages consisting of P. megaseifolia, P. el. Pallasii, and P. el. lofthousei, and the renifolia clade. Although we cannot infer directionality from our methods, due to P. megaseifolia varying throughout our analyses between sister relationships with P. el. pallasii and P. el. lofthousei and the renifolia clade (Figs 1c, 3, 4), it is possible that the ancestor of P. megaseifolia was of hybrid origin between these two lineages. Of note, one requirement of speciation is reproductive isolation (Wang et al., 2021), and P. megaseifolia is the earliest flowering species within Primula sect. Primula (Richards, 2003), a well‐established form of reproductive isolation in angiosperms (Lowry et al., 2008; Baack et al., 2015).
A final example of reticulate evolution in Primula sect. Primula is P. el. pseudoelatior, a subspecies of P. elatior endemic to the Caucasus region. In every phylogenetic analysis, P. el. pseudoelatior was recovered as sister to P. veris, and in the Dsuite analysis, P. el. pseudoelatior had the highest amount of introgressed alleles of any species (Fig. 4), a result corroborated by D FOIL (Table S3). Although both f b statistics and D‐statistics explicitly attempt to distinguish ILS from introgression, the f b considers admixture in a phylogenetic context. In our case, this allowed us to deduce that the introgressed alleles in P. el. pseudoelatior are shared with the Caucasus P. veris subspecies. The high gCF and sCF at this node (Fig. S2) and long branch lengths in both the ML and MSC analyses (Figs 2, S2) suggest that ILS alone cannot account for this discrepancy. Taken together, our analyses imply that the ancestor of P. el. pseudoelatior has undergone gene flow with the ancestor of the Caucasus P. veris subspecies. Overall, we have strong evidence from multiple analyses suggesting a prominent role for reticulate evolution in Primula sect. Primula and specifically P. elatior.
Nonrecombining genomic region recovers nuclear incongruence
We have established that admixture played a significant role in the evolution of Primula sect. Primula, which has resulted in a mosaic of phylogenomic signals in this clade. However, it has been demonstrated that the most accurate reconstruction of ancient branching events will be maintained within regions of the nuclear genome that undergo reduced recombination (Schumer et al., 2018; Li et al., 2019). Specifically, with complete lineage sorting and without gene flow between taxa, the nuclear and S‐locus phylogenies should recover highly concordant relationships, while incongruence between the phylogenies of these genomes would be evidence of ILS and/or hybridization (Pease & Hahn, 2013; Meleshko et al., 2021; Rose et al., 2021). We therefore provided the first phylogeny ever inferred from the heterostyly supergene including all species of Primula sect. Primula. Overall, we found the S‐locus phylogeny to be well resolved and largely congruent with the nuclear genome topology at shallow nodes. However, there was strong incongruence between the S‐locus and the nuclear genome topologies in the backbone relationships of Primula sect. Primula (Fig. 3). One potential cause of this incongruence is ILS. The S‐locus, with suppressed recombination and lower effective population size, generally exhibits less ILS, as seen in sex chromosomes and other regions of low recombination (Willyard et al., 2009; Pease & Hahn, 2013; Rifkin et al., 2019; Sackton & Clark, 2019). The incongruence between the S‐locus and nuclear genome topologies could also be due to S‐locus introgression or to the nuclear genome including high‐recombination regions that are enriched for signals of gene flow, as documented in studies with both empirical (Brandvain et al., 2014; Li et al., 2019) and simulated (Schierup & Hein, 2000, p. 200; Leaché et al., 2014) data. The latter explanation would suggest admixture at deeper evolutionary time scales in Primula sect. Primula, aligning with the results from the network analysis (Fig. 1c).
Conclusions
Genome‐scale data allow us to rigorously test hypotheses previously generated from analyses utilizing limited genetic datasets. In this study, we provided the first phylogenetic analysis for primroses using sequence data from the entire nuclear genome, the entire chloroplast genome, and the entire S‐locus, the latter representing the first ever clade‐wide phylogenetic analysis based on this supergene. Future work focusing on species demographic responses to Quaternary climate fluctuations would provide a deeper understanding of the timing and mode of lineage admixture and diversification, and thus identify specific biogeographic processes shaping the evolutionary history of this section (Theodoridis et al., 2017; Folk et al., 2019).
Author contributions
RLS, ST, BK and EC designed the research. RLS, BK, EM‐C, NY, EL‐B, GT, FC and JK collected the data. RLS analyzed the data. RLS, ST, BK, EM‐C, GP, EL‐B, DARE, NY and EC interpreted the data. All authors contributed to the writing of the manuscript. RLS, ST and EC contributed equally to this work.
Supporting information
Fig. S1 Multispecies coalescent phylogeny of Primula sect. Primula made in Astral‐III.
Fig. S2 gCF and sCF for all phylogenies.
Fig. S3 Chloroplast phylogeny of Primula sect. Primula.
Fig. S4 S‐locus phylogeny of Primula sect. Primula.
Fig. S5 Fbranch (f b) analysis in Dsuite with multispecies coalescent topology.
Fig. S6 Ancestral range estimation under a Bayesian framework with phylogram.
Fig. S7 Ancestral range estimation under a Bayesian framework with chronogram.
Methods S1 Geographic sampling.
Methods S2 Species networks estimation with Nanuq.
Methods S3 Supplementary D‐statistic analyses.
Methods S4 Ancestral range estimation.
Table S1 Primula specimens used in the study.
Table S2 Fbranch values from Dsuite.
Table S3 Results from D FOIL.
Table S4 D‐statistics, f4‐ratios, Z‐score, and f4‐ratio from Dsuite for all species trios.
Table S5 Posterior probabilities for every state in ancestral state estimation with phylogram.
Please note: Wiley is not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This work was supported by the Swiss National Science Foundation (grant number: 175556 to EC). The authors are grateful to Hüseyin Baykal, Bryan Drew, Marie‐Ève Garon‐Labrecque, Monika Janišová, Dimitri Khuskivadze, D. Blaine Marchant, Stina Weststrand, and Parvin Yousefi for fieldwork assistance and sample collection. In Fig. 1, all photographs by authors except Primula renifolia by Stina Weststrand and P. vuheterochroma by Parvin Yousefi. We thank Shalva Sikharulidze, Manana Khutsishvili, and Tamar Jugheli for assistance in Georgia with permits and herbarium specimens. We thank Hector Baños, Mark Ravinet, and Joana Meier for guidance with analyses. We used the PNWColors palette (github.com/jakelawlor/PNWColors) in Fig. 5. Finally, we thank two anonymous reviewers for their helpful feedback on this manuscript.
Contributor Information
Rebecca L. Stubbs, Email: stubbsrl@gmail.com.
Spyros Theodoridis, Email: spytheodor@hotmail.com.
Data availability
New sequences are deposited in NCBI's sequence read archive (https://www.ncbi.nlm.nih.gov/sra); accession numbers: SAMN31083866–SAMN31083971 (BioProject ID: PRJNA885283). Sequence alignments, gene trees, and species trees are deposited in the Dryad Digital Repository: 10.5061/dryad.b2rbnzsgz.
References
- Allman ES, Baños H, Rhodes JA. 2019. Nanuq: a method for inferring species networks from gene trees under the coalescent model. Algorithms for Molecular Biology 14: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli A, Kissling WD, Flantua SG, Bermúdez MA, Mulch A, Muellner‐Riehl AN, Kreft H, Linder HP, Badgley C, Fjeldså J. 2018. Geological and climatic influences on mountain biodiversity. Nature Geoscience 11: 718–725. [Google Scholar]
- Baack E, Melo MC, Rieseberg LH, Ortiz‐Barrientos D. 2015. The origins of reproductive isolation in plants. New Phytologist 207: 968–984. [DOI] [PubMed] [Google Scholar]
- Bergero R, Charlesworth D. 2009. The evolution of restricted recombination in sex chromosomes. Trends in Ecology & Evolution 24: 94–102. [DOI] [PubMed] [Google Scholar]
- Brandvain Y, Kenney AM, Flagel L, Coop G, Sweigart AL. 2014. Speciation and introgression between Mimulus nasutus and Mimulus guttatus . PLoS Genetics 10: e1004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute . 2019. Picard toolkit. [WWW document] URL http://broadinstitute.github.io/picard/ [accessed 1 August 2021]. [Google Scholar]
- Cai L, Xi Z, Lemmon EM, Lemmon AR, Mast A, Buddenhagen CE, Liu L, Davis CC. 2021. The perfect storm: gene tree estimation error, incomplete lineage sorting, and ancient gene flow explain the most recalcitrant ancient angiosperm clade, Malpighiales. Systematic Biology 70: 491–507. [DOI] [PubMed] [Google Scholar]
- Chan KO, Hutter CR, Wood PL, Su Y‐C, Brown RM. 2021. Gene flow increases phylogenetic structure and inflates cryptic species estimations: a case study on widespread Philippine puddle frogs (Occidozyga laevis). Systematic Biology 71: 40–57. [DOI] [PubMed] [Google Scholar]
- Chernomor O, von Haeseler A, Minh BQ. 2016. Terrace aware data structure for phylogenomic inference from supermatrices. Systematic Biology 65: 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocchio A, Jan WA, Martínez‐Solano I, de Vries W, Bisconti R, Pezzarossa A, Maiorano L, Canestrelli D. 2021. Reconstructing hotspots of genetic diversity from glacial refugia and subsequent dispersal in Italian common toads (Bufo bufo). Scientific Reports 11: 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocker JM, Wright J, Li J, Swarbreck D, Dyer S, Caccamo M, Gilmartin PM. 2018. Primula vulgaris (primrose) genome assembly, annotation and gene expression, with comparative genomics on the heterostyly supergene. Scientific Reports 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comes HP, Kadereit JW. 1998. The effect of Quaternary climatic changes on plant distribution and evolution. Trends in Plant Science 3: 432–438. [Google Scholar]
- Comes HP, Kadereit JW. 2003. Spatial and temporal patterns in the evolution of the flora of the European Alpine system. Taxon 52: 451–462. [Google Scholar]
- Cusimano N, Renner SS. 2014. Ultrametric trees or phylograms for ancestral state reconstruction: does it matter? Taxon 63: 721–726. [Google Scholar]
- Dagallier L‐PMJ, Janssens SB, Dauby G, Blach‐Overgaard A, Mackinder BA, Droissart V, Svenning J‐C, Sosef MSM, Stévart T, Harris DJ et al. 2020. Cradles and museums of generic plant diversity across tropical Africa. New Phytologist 225: 2196–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST et al. 2011. The variant call format and VCFtools . Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM et al. 2021. Twelve years of SAMtools and BCFtools . GigaScience 10: giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin C. 1862. On the two forms, or dimorphic condition, in the species of Primula, and on their remarkable sexual relations. Botanical Journal of the Linnean Society 6: 77–96. [Google Scholar]
- Darwin C. 1868. On the specific difference between Primula veris, Brit. Fl. (var. officinalis of Linn.), P. vulgaris, Brit. Fl. (var. acaulis, Linn.), and P. elatior, Jacq.; and on the hybrid nature of the common Oxlip. With supplementary remarks on naturally‐produced hybrids in the genus Verbascum . Botanical Journal of the Linnean Society 10: 437–454. [Google Scholar]
- De‐Kayne R. 2022. Genomic architecture of adaptive radiation and hybridization in Alpine whitefish. Nature Communications 13: 4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand EY, Patterson N, Reich D, Slatkin M. 2011. Testing for ancient admixture between closely related populations. Molecular Biology and Evolution 28: 2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DAR, Hipp AL, González‐Rodríguez A, Cavender‐Bares J. 2015. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69: 2587–2601. [DOI] [PubMed] [Google Scholar]
- Emadzade K, Lebmann MJ, Hoffmann MH, Tkach N, Lone FA, Hörandl E. 2015. Phylogenetic relationships and evolution of high mountain buttercups (Ranunculus) in North America and Central Asia. Perspectives in Plant Ecology, Evolution and Systematics 17: 131–141. [Google Scholar]
- Ernst A. 1925. Zur Kenntnis des Artbastardes Primula variabilis Goupil (Pr. vulgaris × veris) und seiner Nachkommenschaft. Zeitschrift für Induktive Abstammungs Und Verbungslehre 27: 233–235. [Google Scholar]
- Esquerré D, Keogh JS, Demangel D, Morando M, Avila LJ, Sites JW, Ferri‐Yáñez F, Leaché AD. 2022. Rapid radiation and rampant reticulation: phylogenomics of south American Liolaemus lizards. Systematic Biology 71: 286–300. [DOI] [PubMed] [Google Scholar]
- Folk RA, Mandel JR, Freudenstein JV. 2017. Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Systematic Biology 66: 320–337. [DOI] [PubMed] [Google Scholar]
- Folk RA, Stubbs RL, Mort ME, Cellinese N, Allen JM, Soltis PS, Soltis DE, Guralnick RP. 2019. Rates of niche and phenotype evolution lag behind diversification in a temperate radiation. Proceedings of the National Academy of Sciences, USA 116: 10874–10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk VA. 1985. Phylogenetic patterns and hybridization. Annals of the Missouri Botanical Garden 72: 681–715. [Google Scholar]
- Ganders FR. 1979. The biology of heterostyly. New Zealand Journal of Botany 17: 607–635. [Google Scholar]
- Grimm GW, Denk T. 2014. The Colchic region as refuge for relict tree lineages: cryptic speciation in field maples. Turkish Journal of Botany 38: 1050–1066. [Google Scholar]
- Guggisberg A, Mansion G, Conti E. 2009. Disentangling reticulate evolution in an arctic‐alpine polyploid complex. Systematic Biology 58: 55–73. [DOI] [PubMed] [Google Scholar]
- Habel JC, Rasche L, Schneider UA, Engler JO, Schmid E, Rödder D, Meyer ST, Trapp N, Sos del Diego R, Eggermont H. 2019. Final countdown for biodiversity hotspots. Conservation Letters 12: e12668. [Google Scholar]
- Hewitt G. 2000. The genetic legacy of the Quaternary ice ages. Nature 405: 907–913. [DOI] [PubMed] [Google Scholar]
- Hibbins MS, Hahn MW. 2021a. Phylogenomic approaches to detecting and characterizing introgression. Genetics 220: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbins MS, Hahn MW. 2021b. The effects of introgression across thousands of quantitative traits revealed by gene expression in wild tomatoes. PLOS Genetics 17: e1009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobolth A, Dutheil JY, Hawks J, Schierup MH, Mailund T. 2011. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Research 21: 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Hutchison DW, Templeton AR. 1999. Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53: 1898–1914. [DOI] [PubMed] [Google Scholar]
- Huu CN, Kappel C, Keller B, Sicard A, Takebayashi Y, Breuninger H, Nowak MD, Bäurle I, Himmelbach A, Burkart M et al. 2016. Presence versus absence of CYP734A50 underlies the style‐length dimorphism in primroses. eLife 5: e17956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24: 1403–1405. [DOI] [PubMed] [Google Scholar]
- Jombart T, Ahmed I. 2011. adegenet 1.3‐1: new tools for the analysis of genome‐wide SNP data. Bioinformatics 27: 3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kálmán K, Medvegy A, Mihalik E. 2004. Pattern of the floral variation in the hybrid zone of two distylous Primula species. Flora ‐ Morphology, Distribution, Functional Ecology of Plants 199: 218–227. [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. Modelfinder: fast model selection for accurate phylogenetic estimates. Nature Methods 14: 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller B, de Vos JM, Schmidt‐Lebuhn AN, Thomson JD, Conti E. 2016. Both morph‐ and species‐dependent asymmetries affect reproductive barriers between heterostylous species. Ecology and Evolution 6: 6223–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller B, Ganz R, Mora‐Carrera E, Nowak MD, Theodoridis S, Koutroumpa K, Conti E. 2021. Asymmetries of reproductive isolation are reflected in directionalities of hybridization: integrative evidence on the complexity of species boundaries. New Phytologist 229: 1795–1809. [DOI] [PubMed] [Google Scholar]
- Koch EL, Neiber MT, Walther F, Hausdorf B. 2016. Presumable incipient hybrid speciation of door snails in previously glaciated areas in the Caucasus. Molecular Phylogenetics and Evolution 97: 120–128. [DOI] [PubMed] [Google Scholar]
- Lanfear R. 2018. Calculating and interpreting gene‐ and site‐concordance factors in phylogenomics. The Lanfear Lab @ANU Molecular Evolution and Phylogenetics. [WWW document] URL http://www.robertlanfear.com/blog/files/concordance_factors.html [accessed 15 November 2021].
- Leaché AD, Wagner P, Linkem CW, Böhme W, Papenfuss TJ, Chong RA, Lavin BR, Bauer AM, Nielsen SV, Greenbaum E et al. 2014. A hybrid phylogenetic–phylogenomic approach for species tree estimation in African Agama lizards with applications to biogeography, character evolution, and diversification. Molecular Phylogenetics and Evolution 79: 215–230. [DOI] [PubMed] [Google Scholar]
- Lee‐Yaw JA, Grassa CJ, Joly S, Andrew RL, Rieseberg LH. 2019. An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytologist 221: 515–526. [DOI] [PubMed] [Google Scholar]
- Li G, Figueiró HV, Eizirik E, Murphy WJ. 2019. Recombination‐aware phylogenomics reveals the structured genomic landscape of hybridizing cat species. Molecular Biology and Evolution 36: 2111–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cocker JM, Wright J, Webster MA, McMullan M, Dyer S, Swarbreck D, Caccamo M, van Oosterhout C, Gilmartin PM. 2016. Genetic architecture and evolution of the S locus supergene in Primula vulgaris . Nature Plants 2: 1–7. [DOI] [PubMed] [Google Scholar]
- Litsios G, Salamin N. 2012. Effects of phylogenetic signal on ancestral state reconstruction. Systematic Biology 61: 533–538. [DOI] [PubMed] [Google Scholar]
- Lowry DB, Hoban S, Kelley JL, Lotterhos KE, Reed LK, Antolin MF, Storfer A. 2017. Breaking RAD: an evaluation of the utility of restriction site‐associated DNA sequencing for genome scans of adaptation. Molecular Ecology Resources 17: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Modliszewski JL, Wright KM, Wu CA, Willis JH. 2008. The strength and genetic basis of reproductive isolating barriers in flowering plants. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 363: 3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Wang J, Hu Q, Li J, Sun Y, Zhang L, Abbott RJ, Liu J, Mao K. 2019. Ancient introgression drives adaptation to cooler and drier mountain habitats in a cypress species complex. Communications Biology 2: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinsky M, Matschiner M, Svardal H. 2021. Dsuite‐Fast D‐statistics and related admixture evidence from VCF files. Molecular Ecology Resources 21: 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinsky M, Svardal H, Tyers AM, Miska EA, Genner MJ, Turner GF, Durbin R. 2018. Whole‐genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nature Ecology & Evolution 2: 1940–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Jiggins CD. 2017. Interpreting the genomic landscape of introgression. Current Opinion in Genetics & Development 47: 69–74. [DOI] [PubMed] [Google Scholar]
- Meleshko O, Martin MD, Korneliussen TS, Schröck C, Lamkowski P, Schmutz J, Healey A, Piatkowski BT, Shaw AJ, Weston DJ et al. 2021. Extensive genome‐wide phylogenetic discordance is due to incomplete lineage sorting and not ongoing introgression in a rapidly radiated bryophyte genus. Molecular Biology and Evolution 38: 2750–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Hahn MW, Lanfear R. 2020. New methods to calculate concordance factors for phylogenomic datasets. Molecular Biology and Evolution 37: 2727–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Nguyen MAT, von Haeseler A. 2013. Ultrafast approximation for phylogenetic bootstrap. Molecular Biology and Evolution 30: 1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina‐Venegas R, Aparicio A, Lavergne S, Arroyo J. 2017. Climatic and topographical correlates of plant palaeo‐ and neoendemism in a Mediterranean biodiversity hotspot. Annals of Botany 119: 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora‐Carrera E, Stubbs RL, Keller B, Léveillé‐Bourret É, de Vos JM, Szövényi P, Conti E. 2021. Different molecular changes underlie the same phenotypic transition: origins and consequences of independent shifts to homostyly within species. Molecular Ecology 31: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Briones DF, Kadereit G, Tefarikis DT, Moore MJ, Smith SA, Brockington SF, Timoneda A, Yim WC, Cushman JC, Yang Y. 2021. Disentangling sources of gene tree discordance in phylogenomic data sets: testing ancient hybridizations in Amaranthaceae s.l. Systematic Biology 70: 219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumladze L, Japoshvili B, Anderson EP. 2020. Faunal biodiversity research in the Republic of Georgia: a short review of trends, gaps, and needs in the Caucasus biodiversity hotspot. Biologia 75: 1385–1397. [Google Scholar]
- Myers N, Mittermeier RA, Mittermeier CG, Da Fonseca GA, Kent J. 2000. Biodiversity hotspots for conservation priorities. Nature 403: 853–858. [DOI] [PubMed] [Google Scholar]
- Nguyen L‐T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ‐Tree: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution 32: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MD, Russo G, Schlapbach R, Huu C, Lenhard M, Conti E. 2015. The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biology 16: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. 2012. Ancient admixture in human history. Genetics 192: 1065–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease JB, Hahn MW. 2013. More accurate phylogenies inferred from low‐recombination regions in the presence of incomplete lineage sorting: accurate phylogenies in low‐recombination regions. Evolution 67: 2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease JB, Hahn MW. 2015. Detection and polarization of introgression in a five‐taxon phylogeny. Systematic Biology 64: 651–662. [DOI] [PubMed] [Google Scholar]
- Perrigo A, Hoorn C, Antonelli A. 2020. Why mountains matter for biodiversity. Journal of Biogeography 47: 315–325. [Google Scholar]
- Pfeifer SP. 2017. From next‐generation resequencing reads to a high‐quality variant data set. Heredity 118: 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente G, Léveillé‐Bourret É, Yousefi N, Choudhury RR, Keller B, Diop SI, Duijsings D, Pirovano W, Lenhard M, Szövényi P et al. 2022. Comparative genomics elucidates the origin of a supergene controlling floral heteromorphism. Molecular Biology and Evolution 39: msac035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabiee M, Sayyari E, Mirarab S. 2019. Multi‐allele species reconstruction using Astral . Molecular Phylogenetics and Evolution 130: 286–296. [DOI] [PubMed] [Google Scholar]
- Rahbek C, Borregaard MK, Antonelli A, Colwell RK, Holt BG, Nogues‐Bravo D, Rasmussen CMØ, Richardson K, Rosing MT, Whittaker RJ et al. 2019a. Building mountain biodiversity: geological and evolutionary processes. Science 365: 1114–1119. [DOI] [PubMed] [Google Scholar]
- Rahbek C, Borregaard MK, Colwell RK, Dalsgaard B, Holt BG, Morueta‐Holme N, Nogues‐Bravo D, Whittaker RJ, Fjeldså J. 2019b. Humboldt's enigma: what causes global patterns of mountain biodiversity? Science 365: 1108–1113. [DOI] [PubMed] [Google Scholar]
- Ravinet M, Kume M, Ishikawa A, Kitano J. 2021. Patterns of genomic divergence and introgression between Japanese stickleback species with overlapping breeding habitats. Journal of Evolutionary Biology 34: 114–127. [DOI] [PubMed] [Google Scholar]
- Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods in Ecology and Evolution 3: 217–223. [Google Scholar]
- Richards AJ. 2003. Primula. London, UK: Batsford. [Google Scholar]
- Rieseberg LH, Soltis DE. 1991. Phylogenetic consequences of cytoplasmic gene flow in plants. Evolutionary Trends in Plants 5: 65–84. [Google Scholar]
- Rifkin JL, Castillo AS, Liao IT, Rausher MD. 2019. Gene flow, divergent selection and resistance to introgression in two species of morning glories (Ipomoea). Molecular Ecology 28: 1709–1729. [DOI] [PubMed] [Google Scholar]
- Roquet C, Boucher FC, Thuiller W, Lavergne S. 2013. Replicated radiations of the alpine genus Androsace (Primulaceae) driven by range expansion and convergent key innovations. Journal of Biogeography 40: 1874–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JP, Toledo CAP, Lemmon EM, Lemmon AR, Sytsma KJ. 2021. Out of sight, out of mind: widespread nuclear and plastid‐nuclear discordance in the flowering plant genus Polemonium (Polemoniaceae) suggests widespread historical gene flow despite limited nuclear signal. Systematic Biology 70: 162–180. [DOI] [PubMed] [Google Scholar]
- Runemark A, Vallejo‐Marin M, Meier JI. 2019. Eukaryote hybrid genomes. PLoS Genetics 15: e1008404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackton TB, Clark N. 2019. The perils of intralocus recombination for inferences of molecular convergence. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 374: 20190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari E, Mirarab S. 2016. Fast coalescent‐based computation of local branch support from quartet frequencies. Molecular Biology and Evolution 33: 1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup MH, Hein J. 2000. Consequences of recombination on traditional phylogenetic analysis. Genetics 156: 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt‐Lebuhn AN, de Vos JM, Keller B, Conti E. 2012. Phylogenetic analysis of Primula section Primula reveals rampant non‐monophyly among morphologically distinct species. Molecular Phylogenetics and Evolution 65: 23–34. [DOI] [PubMed] [Google Scholar]
- Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, Sankararaman S, Andolfatto P, Rosenthal GG et al. 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360: 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Fletcher HR. 1947. The genus Primula: section vernales pax. Transactions of the Botanical Society of Edinburgh 34: 402–468. [Google Scholar]
- Soltis PS, Soltis DE. 2009. The role of hybridization in plant speciation. Annual Review of Plant Biology 60: 561–588. [DOI] [PubMed] [Google Scholar]
- Stebbins GL. 1959. The role of hybridization in evolution. Proceedings of the American Philosophical Society 103: 231–251. [Google Scholar]
- Stegemann S, Keuthe M, Greiner S, Bock R. 2012. Horizontal transfer of chloroplast genomes between plant species. Proceedings of the National Academy of Sciences, USA 109: 2434–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs RL, Folk RA, Soltis DE, Cellinese N. 2020a. Diversification in the Arctic: biogeography and systematics of the north American Micranthes (Saxifragaceae). Systematic Botany 45: 802–811. [Google Scholar]
- Stubbs RL, Folk RA, Xiang C‐L, Chen S, Soltis DE, Cellinese N. 2020b. A phylogenomic perspective on evolution and discordance in the alpine‐arctic plant clade micranthes (Saxifragaceae). Frontiers in Plant Science 10: 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran J, Knowles LL. 2017. Multispecies coalescent delimits structure, not species. Proceedings of the National Academy of Sciences, USA 114: 1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tendal K, Ørgaard M, Larsen B, Pedersen C. 2018. Recurrent hybridisation events between Primula vulgaris, P. veris and P. elatior (Primulaceae, Ericales) challenge the species boundaries: using molecular markers to re‐evaluate morphological identifications. Nordic Journal of Botany 36: 1–15. [Google Scholar]
- Theodoridis S, Patsiou TS, Randin C, Conti E. 2017. Forecasting range shifts of a cold‐adapted species under climate change: are genomic and ecological diversity within species crucial for future resilience? Ecography 41: 1357–1369. [Google Scholar]
- Theodoridis S, Randin C, Broennimann O, Patsiou T, Conti E. 2013. Divergent and narrower climatic niches characterize polyploid species of European primroses in Primula sect. Aleuritia . Journal of Biogeography 40: 1278–1289. [Google Scholar]
- Twyford AD, Wong ELY, Friedman J. 2020. Multi‐level patterns of genetic structure and isolation by distance in the widespread plant Mimulus guttatus . Heredity 125: 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine D. 1952. Studies in British Primulas. III. Hybridization between Primula elatior (L.) Hill and P. veris L. New Phytologist 50: 383–399. [Google Scholar]
- Valentine D. 1961. Evolution in the genus Primula . In: Wanstall PJ, ed. A Darwin centenary. London, UK: Botanical Society of the British Isles, 71–87. [Google Scholar]
- Valentine DH. 1947. Studies in British Primulas. I. Hybridization between primrose and Oxlip (Primula vulgaris Huds. and P. elatior Schreb.). New Phytologist 46: 229–253. [Google Scholar]
- Volkova P, Laczkó L, Demina O, Schanzer I, Sramkó G. 2020. Out of Colchis: the colonization of Europe by Primula vulgaris Huds. (Primulaceae). Acta Societatis Botanicorum Poloniae 89: 1–15. [Google Scholar]
- de Vos J, Hughes C, Schneeweiss G, Moore B, Conti E. 2014a. Heterostyly accelerates diversification via reduced extinction in primroses. Proceedings of the Royal Society B: Biological Sciences 281: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos J, Wüest R, Conti E. 2014b. Small and ugly? Phylogenetic analyses of the ‘selfing syndrome’ reveal complex evolutionary fates of monomorphic primrose flowers. Evolution 68: 1042–1057. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang Y, Bi H, Lu Z, Ma Y, Yang X, Chen N, Tian B, Liu B, Mao X et al. 2021. Hybrid speciation via inheritance of alternate alleles of parental isolating genes. Molecular Plant 14: 208–222. [DOI] [PubMed] [Google Scholar]
- Willyard A, Cronn R, Liston A. 2009. Reticulate evolution and incomplete lineage sorting among the ponderosa pines. Molecular Phylogenetics and Evolution 52: 498–511. [DOI] [PubMed] [Google Scholar]
- Wilson JD, Mongiardino Koch N, Ramírez MJ. 2022. Chronogram or phylogram for ancestral state estimation? Model‐fit statistics indicate the branch lengths underlying a binary character's evolution. Methods in Ecology and Evolution 13: 1–11. [Google Scholar]
- Woodell SR. 1969. Natural hybridization in Britain between Primula vulgaris Huds (the primrose) and P. elatior (L.) Hill (the oxlip). Watsonia 7: 115–127. [Google Scholar]
- Wright S. 1943. Isolation by distance. Genetics 28: 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Yang Z. 2016. Challenges in species tree estimation under the multispecies coalescent model. Genetics 204: 1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Folk R, Zhang N, Gong X. 2019. Homoploid hybridization of plants in the Hengduan mountains region. Ecology and Evolution 9: 8399–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zazanashvili N. 2009. The Caucasus hotspot. In: Zazanashvili N, Mallon D, eds. Status and protection of globally threatened species in the Caucasus. Tbilisi, GE, USA: CEPF, WWF. Contour Ltd, 1–14. [Google Scholar]
- Zhang C, Rabiee M, Sayyari E, Mirarab S. 2018. Astral‐III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19: 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Rheindt FE, She H, Cheng Y, Song G, Jia C, Qu Y, Alström P, Lei F. 2021. Most Genomic loci misrepresent the phylogeny of an avian radiation because of ancient gene flow. Systematic Biology 70: 961–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Comes HP, Kadereit JW. 2004. The temporal course of quaternary diversification in the European high mountain endemic Primula sect. Auricula (Primulaceae). International Journal of Plant Sciences 165: 191–207. [Google Scholar]
- Zhou B‐F, Yuan S, Crowl AA, Liang Y‐Y, Shi Y, Chen X‐Y, An Q‐Q, Kang M, Manos PS, Wang B. 2022. Phylogenomic analyses highlight innovation and introgression in the continental radiations of Fagaceae across the Northern Hemisphere. Nature Communications 13: 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Zhao J, Chen C, Meng X, Zhao G. 2016. Characterization of the complete chloroplast genome sequence of Primula veris (Ericales: Primulaceae). Conservation Genetics Resources 8: 455–458. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Multispecies coalescent phylogeny of Primula sect. Primula made in Astral‐III.
Fig. S2 gCF and sCF for all phylogenies.
Fig. S3 Chloroplast phylogeny of Primula sect. Primula.
Fig. S4 S‐locus phylogeny of Primula sect. Primula.
Fig. S5 Fbranch (f b) analysis in Dsuite with multispecies coalescent topology.
Fig. S6 Ancestral range estimation under a Bayesian framework with phylogram.
Fig. S7 Ancestral range estimation under a Bayesian framework with chronogram.
Methods S1 Geographic sampling.
Methods S2 Species networks estimation with Nanuq.
Methods S3 Supplementary D‐statistic analyses.
Methods S4 Ancestral range estimation.
Table S1 Primula specimens used in the study.
Table S2 Fbranch values from Dsuite.
Table S3 Results from D FOIL.
Table S4 D‐statistics, f4‐ratios, Z‐score, and f4‐ratio from Dsuite for all species trios.
Table S5 Posterior probabilities for every state in ancestral state estimation with phylogram.
Please note: Wiley is not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
New sequences are deposited in NCBI's sequence read archive (https://www.ncbi.nlm.nih.gov/sra); accession numbers: SAMN31083866–SAMN31083971 (BioProject ID: PRJNA885283). Sequence alignments, gene trees, and species trees are deposited in the Dryad Digital Repository: 10.5061/dryad.b2rbnzsgz.