

Abstract
Dithiolopyrrolones (DTPs), such as holomycin, are natural products that hold promise as scaffolds for antibiotics as they exhibit inhibitory activity against antibiotic‐resistant pathogens. They consist of a unique bicyclic core containing a disulfide that is crucial for their biological activity. Herein, we establish the DTPs as prochelators. We show that the disulfides are reduced at cellular gluathione levels. This activates the drugs and initiates interactions with targets, particularly metal coordination. In addition, we report an expedient synthesis for the DTPs thiolutin and aureothricin, providing facile access to important natural DTPs and derivatives thereof.
Keywords: Antibiotics, antitumor agents, disulfide, natural products, prodrugs
Natural prochelators: Dithiolopyrrolones are promising antibiotics. They are reduced by glutathione after cell entry. The activated dithiol chelates essential metal ions in cells. We also report an expedient synthesis for natural derivatives.
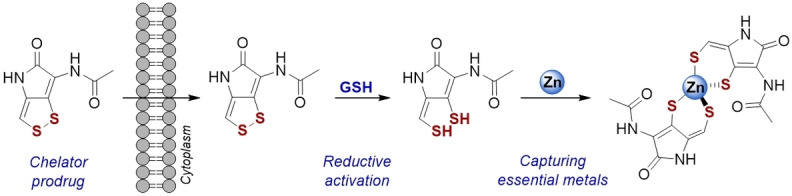
Introduction
The spreading of multi‐resistant pathogens urges the development of new antibiotics. Natural products provide an extensive source for biologically active compounds. [1] They have already been preselected during evolution and are often optimized to pass cellular membranes, even the low‐permeable membranes of Gram‐negative pathogens. Particularly of interest are compounds that act as prodrugs and only become active at the target site. This minimizes side effects and prevents premature inactivation of the drug. One such compound class are the dithiolopyrrolones (DTPs), which are secreted by marine and soil bacteria. They exhibit a broad biological activity against microbes, importantly also against antibiotic‐resistant pathogens. [2] In addition, they are of interest for tumor treatment applications as they also show anti‐cancer activity.
The first reported DTP, aureothricin (Scheme 1), was isolated in 1948. [3] In recent years, mainly the DTPs holomycin and thiolutin have been investigated. All these compounds share the dithiolopyrrolone core containing a cyclic disulfide that appears crucial for their biological activity.[ 2 , 4 ] The DTPs have been proposed to act as prodrugs that are activated in the reductive environment of the cytoplasm under release of dithiols (Scheme 1). Consistently, previous studies showed that a molecular masking of the thiols through methylation significantly attenuates the activity of the DTPs. [4b] However, the cellular reductant of the DTPs has remained unknown.
Scheme 1.

Naturally occurring DTPs, the proposed reductive prodrug activation and modes of action.
The active dithiols are expected to interfere with various processes and targets in cells, which contributes to their broad biological activity. Recently, it was shown that both holomycin and thiolutin act as Zn(II) chelators, inhibiting Zn(II)‐dependent enzymes and disrupting Zn(II) homeostasis in cells.[ 4c , 4d ] Further mechanisms have been proposed, including the controversially discussed inhibition of RNA polymerases, which may require an interaction of the DTPs with Mn(II).[ 2 , 5 ] It is likely that multiple mechanisms are operative in the activity of the DTPs, but further mechanistic investigations will be required for a better understanding.
Despite their high structural similarity, holomycin and thiolutin have been reported to exhibit noticeable differences in their biological activity;[ 2 , 4c , 5 ] but reasons for that remain unknown. Whereas the total synthesis of holomycin is well established, [6] thiolutin is usually isolated from biological sources. A convenient synthesis of thiolutin would ease its availability and also provide access to derivatives.
Herein, we establish the DTPs as prochelators. We show that they are reduced at cellular levels of glutathione (GSH), and that the reductive activation by GSH mediates the chelation of Zn(II). In addition, we present an expedient synthesis for thiolutin and aureothricin based on an established synthetic route for holomycin.
Results and Discussion
Expedient synthetic route for thiolutin and aureothricin
The first synthesis of DTPs, particularly thiolutin and aureothricin, was reported in 1962. [7] Further synthetic routes were published in the following years. [8] However, these syntheses involve many steps. Best established so far is the total synthesis of holomycin; the shortest synthetic route was reported by Nielsen et al. (Scheme 2). [6] It involves the formation of the pyrrolone (6) from a sulfur‐containing acetone precursor (5). Deprotection in TFA yields the trifluorylated precursor 7, which is hydrolyzed and acetylated to yield holomycin (1). This route provides access to various amide derivatives of holomycin, and we assumed that it could also be extended towards the synthesis of thiolutin (2) and aureothricin (3).
Scheme 2.

Total syntheses of holomycin, thiolutin and aureothricin (PMB: p‐methoxybenzyl group).
Thiolutin shares structural identity with holomycin except for a methylation at the endo‐cyclic N. Similarly, aureothricin carries a methyl group at that nitrogen atom, and only differs from thiolutin in the propionyl moiety at the exo‐cyclic N. Initially, we tried to introduce the methyl group during the pyrrolone formation (Scheme 2). However, various attempts reacting MeNH2⋅HCl with the acetone precursor 5 in the presence of different Ti(IV) catalysts (e.g., TiCl4, Ti(OiPr)4), bases and solvents did not yield the methylated pyrrolone 8. A similar approach had been reported before, which, however, did not disclose experimental details. [8e] It employed tert‐butyl protective groups in place of the herein used PMB groups. They may have an impact on the reaction outcome, but require more hazardous conditions for deprotection than the PMB groups.
Given the challenges with an early introduction of the methyl group, we decided to attempt the methylation of holomycin to directly yield thiolutin (Scheme 2). This requires a selective methylation of the endo‐cyclic N, while the exo‐cyclic N should remain unmodified. The fact that both N atoms are located within amide bonds may challenge a selective substitution. Initially, we employed the trifluorylated holomycin analog 7 for the methylation as the CF3 group eases signal assignments during NMR studies.
The reaction with MeI and NaH indeed provided a mono‐methylated product in moderate yield. The use of equimolar amounts of MeI proved crucial. Attempts to increase the yield by increasing the amounts of MeI resulted in the formation of considerable amounts of side products. Unexpectedly, it was necessary to employ an excess of NaH; equimolar amounts of the base did not afford a conversion of the starting material. The use of other bases, including Cs2CO3 and DBU, did not provide the methylated product.
We elucidated the structure of the mono‐methylated product by NMR spectroscopy. Selective 1D‐NOESY revealed an NOE correlation between the CH3 group and the vinyl proton, whereas the NH proton did not show any correlation with the other protons (Figure 1). Given the planar structure of the DTPs, [9] this suggested a spatial proximity of the CH3 group and the dithiole moiety, and thus methylation of the endo‐cyclic N. The structure was further confirmed by a (1H,13C)‐HMBC experiment (Figure S41). It revealed a correlation of the CH3 group with the endo‐cyclic carbonyl carbon, but no correlation with the exo‐cyclic carbonyl carbon. Moreover, it showed a correlation of the NH group with the exo‐cyclic carbonyl carbon, but no correlation with the vinyl carbon. These experiments established the product as the desired trifluorylated analog of thiolutin (9).
Figure 1.

Selective methylation of the endo‐cyclic N. Structure elucidation of 9 by 1H NMR (CDCl3, 400 MHz; bottom) and selective 1D‐NOESY NMR. The introduced CH3 group exhibits an NOE correlation with the vinyl proton. No correlation is observed for the NH proton.
The selective mono‐methylation of precursor 7 suggested a higher acidity and/or accessibility for the endo‐cyclic NH compared to the exo‐cyclic NH, despite the latter is carrying an electron‐withdrawing trifluoroacetyl group. Thus, we assumed that the optimized conditions may work even better when employing holomycin as starting material, where the CF3 is replaced by a CH3 group (Scheme 2). However, thiolutin could only be obtained in low yields from the methylation of holomycin (1). Therefore, we decided to employ the trifluorylated analog 9 as precursor for thiolutin. Hydrolysis and acetylation analogously to the holomycin total synthesis yielded thiolutin (2) in moderate yields. Similarly, employing propionyl chloride for the acylation afforded aureothricin (3). This route highlights compounds 7 and 9 as versatile precursors for holomycin and its methylated analogs, and facile access to various amide derivatives of the DTPs.
Holomycin exhibits unique redox properties
The DTPs have been proposed to be reductively activated after their uptake into cells, but the cellular reductant has remained unknown. Glutathione, the main reductant in cells, was reported to be unable to reduce the DTP disulfides.[ 4a , 4c , 10 ] Thus, we aimed to identify possible reductants by determining the redox potential of holomycin, which had not been reported before. To monitor the reduction of holomycin, we employed UV‐vis spectroscopy and HPLC, which are well suited as the DTPs exhibit a high extinction coefficient. Both methods allow for a clear distinction between the disulfide (Holo) and the dithiol (red‐Holo) (Figure 2A, S16). red‐Holo can be readily formed through reduction with tris(2‐carboxyethyl)phosphine (TCEP), a strong and irreversible reductant (Scheme S1).[ 4a , 4c , 4d ] The titration of Holo with TCEP confirms that under these conditions solely the disulfide bond is reduced (saturation at 1 equiv. TCEP) (Figure 2A). However, the dithiol is highly unstable in the presence of air (Figure S3A). [4a] It is quickly re‐oxidized, likely due to the re‐aromatization of the DTP that takes place upon back formation of the disulfide.
Figure 2.
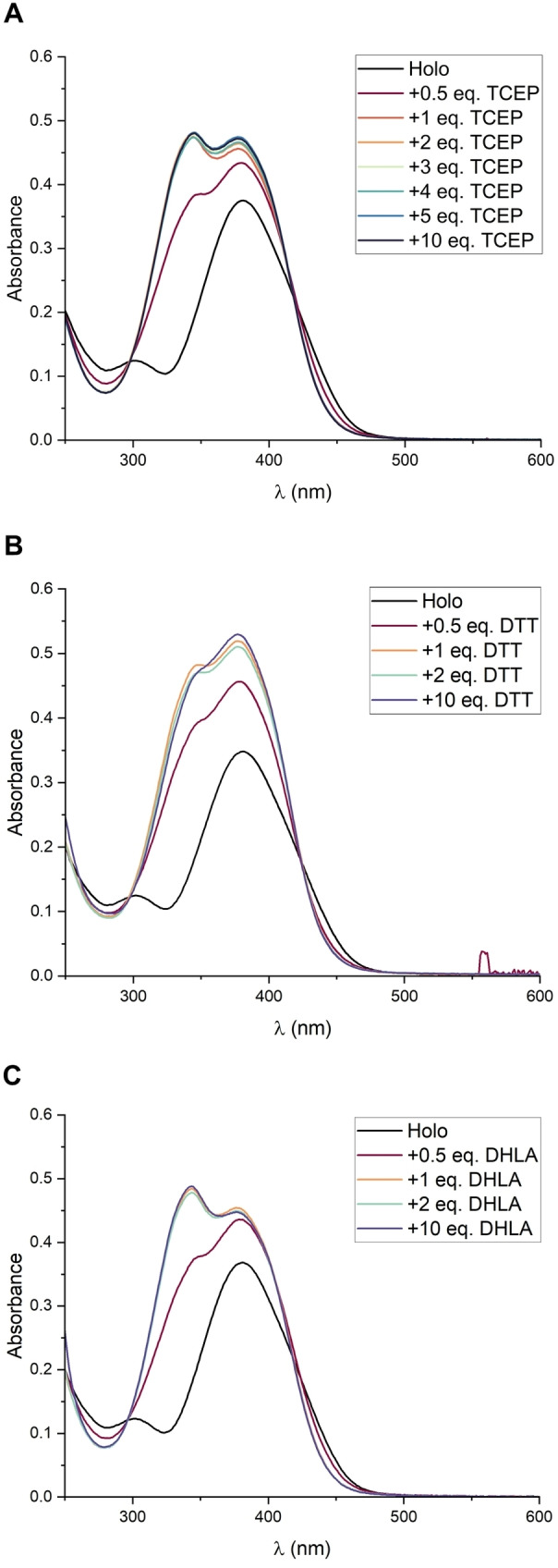
Holomycin is readily reduced by TCEP, DTT and DHLA. (A) Titration with TCEP. (B) Titration with DTT. (C) Titration with DHLA. In all cases, the reduction of holomycin proceeds promptly and quantitatively at 1 equiv. of the reducing agent. The assays were performed by adding the respective amount of TCEP, DTT or DHLA to 50 μM Holo in 75 mM Tris‐HCl, pH 7.4.
The fast re‐oxidation of the dithiol suggested that the redox potential of Holo may be rather low. Moreover, given that Holo was reported not to be reduced by GSH, [10] we expected that its redox potential would be much lower than that of GSH (E°’(GSH/GSSG)=−0.25 V (vs. SHE, pH 7)). [11] Thus, we initiated our redox studies employing dithiothreitol (DTT), a well‐studied reducing agent. In its oxidized form (DTTox), it contains a cyclic disulfide similar to Holo (Scheme S1), and it has a much lower redox potential than GSH (E°’(DTT/DTTox)=−0.33 V).[ 11 , 12 ] The titration of Holo with DTT revealed that Holo is readily and quantitatively reduced by 1 equiv. DTT (Figure 2B). In another experiment, we incubated a mixture of Holo and DTT (1 : 1) under inert conditions for one day to allow for an equilibration between the redox partners (Figure S23). Provided that the E°’ of both compounds do not differ too much, the resulting concentrations of the thiols and disulfides could be used to calculate E°’(Holo) through the Nernst equation.[ 11 , 13 ] But consistent with the titration, the equilibration resulted in a quantitative oxidation of DTT to DTTox, indicating that Holo has a significantly more positive redox potential than DTT. Unfortunately, we could not detect any formed red‐Holo in the reaction mixture. Possibly, the dithiol was not stable enough under the conditions of the quenching and analysis due to its high oxidation susceptibility. The DTT dithiol is more stable and can be detected if formed during the equilibration (see below; Figure S27). The same mixture was obtained when red‐Holo was equilibrated with DTTox (Figure S24), demonstrating that the disulfide DTTox cannot be reduced by the dithiol of Holo. This was confirmed by the observation that the presence of DTTox did not accelerate the oxidation of red‐Holo on air (Figure S3B). Overall, these results indicate that the redox potential of Holo is significantly higher than that of DTT. Despite that, red‐Holo is much faster oxidized on air than DTT, a fact that emphasizes the unique redox properties of the DTPs.
Lipoic acid readily reduces holomycin
Our studies indicated that cellular reductants with a redox potential similar to DTT should be effective reductants for the DTPs. Based on this, we selected dihydrolipoic acid (DHLA) (E°’(DHLA/LA)=−0.29 V),[ 11 , 14 ] a dithiol that is found as cofactor in several enzymes (Scheme S1). Indeed, DHLA readily and quantitatively reduced Holo at stoichiometric amounts (Figure 2C). Similar to DTT, the equilibration with Holo resulted in a quantitative oxidation of DHLA (Figure S25). Consistently, no reduction of lipoic acid (LA) by red‐Holo was observed (Figure S26), and the presence of LA did not accelerate the oxidation of red‐Holo on air (Figure S3C). As during the experiments with DTT, we could only detect oxidized species in the reaction mixtures from the equilibration assay. Hence, we verified our method by equilibrating a mixture of DTT and LA (Figure S27). As expected from the similar redox potentials of the compounds, the equilibration yielded a mixture with similar amounts of each thiol (DTT, DHLA) and disulfide (DTTox, LA). This confirmed that the thiols DTT and DHLA were stable under the assay conditions, and were oxidized if Holo was present. A UV‐vis‐based monitoring further confirmed that DHLA is much less prone to oxidation by air than red‐Holo (Figure S1B). Overall, our studies demonstrate that the redox potential of Holo is significantly higher than that of LA, despite that both compounds share a 5‐membered cyclic disulfide. We expect that LA retains approximately the same E°’ as reported even when tethered to a protein, as the cofactor is attached through a long alkyl linker. Thus, if Holo encounters the cofactor in cells, DHLA can act as potent reductant and activate the DTP prodrug.
Although the HPLC‐based thiol/disulfide interchange assays generally confirm the observations from the UV‐vis‐based experiments, they are not suitable for estimating the redox potential of the DTPs due to their high oxidation susceptibility. Furthermore, although the main peaks in the chromatograms correspond to the disulfides, some additional peaks are observed, which were, however, not further characterized.
Cyclic voltammetry of holomycin
Given the difficulties with detecting red‐Holo during the equilibration assays, we turned to cyclic voltammetry (CV) to get further insights into the redox potential of Holo. It is important to note that in the case of disulfides, CV only allows to estimate redox potentials; the electrochemical reduction of disulfides is irreversible due to the bond cleavage that results after the electron transfer. To mimic physiological conditions, we performed the CV experiments in a buffer solution at pH 7.4. As expected, we observed an irreversible cathodic peak for Holo (Figure 3), resulting from the reduction of the disulfide and subsequent formation of the dithiolate (Scheme S2). In addition to the reduction peak, two oxidation peaks were detectable under the employed conditions. The oxidation peak at lower potential likely originates from the oxidation of the dithiolate, which results in back formation of the disulfide bond. The second oxidation peak may be assigned to the oxidation of the disulfide yielding a thiosulfinate. [15] Further details of the CV studies are discussed in the Supporting Information. The studies also indicate that the broad reduction peak results from an overlap of the reductions of both the disulfide and the thiosulfinate. It should be noted that the potential values given in Figure 3, which were measured against an Ag/AgCl electrode, are not directly comparable to the literature values mentioned above.
Figure 3.

CV of Holo and GSSG. The reduction of GSSG proceeds at lower potential than the reduction of Holo. The measurements were performed with 1 mM Holo or 5 mM GSSG, respectively, in 1 M KCl, 75 mM Tris‐HCl, pH 7.4 at a glassy carbon electrode. The potential values are given as measured against the employed Ag/AgCl/KClsat reference electrode.
To compare the reduction potential of Holo with that of glutathione, we measured the CV of the glutathione disulfide (GSSG) under the same conditions (Figure 3). Based on the literature reporting that Holo cannot be reduced by GSH,[ 4a , 4c , 10 ] we expected the reduction peak of GSSG to be at higher potential than that of Holo. However, under the employed electrochemical conditions, we observed a lower reduction peak potential for GSSG than for Holo. This observation indicated that GSH should be able to reduce Holo.
For comparison, we also measured the CV of LA (Figure S33). As expected, no reduction peak could be observed under the employed conditions. We assume that the reduction of LA proceeds at a potential lower than the potential range that was accessible without degradation of the buffer. Overall, this observation is consistent with the results from our chemical reduction assays that indicated a much higher redox potential for Holo compared to LA.
GSH and Cys reduce the DTPs
The results from the CV experiments motivated us to revisit the chemical reduction of Holo with GSH. Consistent with the CV, Holo was indeed reduced in the presence of GSH (Figure 4, S4). In contrast to the other tested reducing agents (TCEP, DTT, DHLA), an excess of GSH was required and the reduction proceeded slower; a quantitative reduction was observed in the presence of 5 mM GSH after about 45 min. Likely due to the high oxidation sensitivity of red‐Holo in the presence of air, only partially reduced Holo was observed at lower GSH concentrations and longer incubation time. These observations indicate that Holo possesses a redox potential similar to GSH. This is confirmed by the fact that the presence of GSSG accelerates the oxidation of red‐Holo on air (Figure S3D, S3E). It indicates that red‐Holo can reduce GSSG and that a redox equilibrium between holomycin and glutathione forms (Scheme S1).
Figure 4.

The Holo prodrug is reduced by GSH. Titration of Holo with GSH; Holo is reduced at 5 mM GSH. The assay was performed by adding the specified amount of GSH to 50 μM Holo in 75 mM Tris‐HCl, pH 7.4 (in presence of air). The spectrum of quantitatively reduced Holo (with TCEP) is included for comparison.
Our experiments show that Holo is quantitatively reduced at a ratio of GSH/Holo 100 : 1. Earlier studies only tested ratios of up to GSH/Holo 40 : 1 (2 mM GSH with 50 μM Holo), [4c] which were apparently too low to detect a significant reduction of Holo in the presence of air. Although a large excess of GSH is required for a quantitative reduction of Holo, we expect that Holo gets reduced upon entry into cells. Glutathione is the main component of the redox buffer in most cells. The thiol GSH is present at large excess over the disulfide GSSG, and thereby maintains the reductive conditions in the cytoplasm. [16] Many cell types contain 2 mM to 10 mM GSH. [17] Given that probably less than the here used 50 μM Holo will reach the cytoplasm of cells, even a lower GSH concentration should suffice for the reduction of the DTP. Moreover, compared to the aerobic experimental conditions, the oxygen concentration in cells is likely lower so that competing re‐oxidation of the dithiol should be less pronounced than in the experiments. Furthermore, when the formed red‐Holo interacts with targets, it will be removed from the reaction with GSH, the redox equilibrium will be shifted and further Holo molecules will be reduced (see below).
In addition to GSH, we tested two other thiol‐based reductants with similar redox potentials, namely cysteine (Cys, E°’(Cys/CySS)=−0.19 V) and β‐mercaptoethanol (BME, E°’(BME/BMEox=−0.20 V) (Scheme S1). [18] As expected, Holo was also reduced in the presence of excess Cys and BME (Figure S5, S6). In contrast, when we used ascorbate (Asc), a reductant with significantly higher redox potential (E°’(Asc/DHA)=+0.06 V, two‐electron transfer), [19] Holo was not reduced (Figure S2).
We further confirmed the GSH‐mediated reduction of the DTPs by testing thiolutin and aureothricin. Similar to holomycin, both were reduced at excess GSH (Figure S8, S9). Taken together, our studies demonstrate that the DTPs are reduced at cellular GSH levels and that they should get reduced upon cell entry, confirming the earlier proposed prodrug mechanism.
GSH activates DTPs for metal coordination
After we established that the DTPs are reduced by GSH, we investigated whether GSH would activate the DTPs for interactions with their targets. Previous studies reported that the DTPs inhibit Zn(II)‐dependent enzymes by coordinating to Zn(II).[ 4c , 4d ] The chelation of Zn(II) by holomycin, thiolutin and aureothricin can be observed by UV‐vis spectroscopy upon reduction with TCEP (Figure S10). Consistent with our reduction studies, the complex formation of holomycin also proceeds readily when DHLA is added as reductant (Figure S11), and confirms that DHLA can mediate the interaction of the DTPs with metal‐based targets. Importantly, also the reduction by GSH mediates the chelation of Zn(II) by the DTPs (Figure 5, S12, S13, S14). Notably, a lower GSH concentration was necessary to observe a quantitative complex formation (1 mM GSH) than it was necessary to observe a quantitative reduction (5 mM GSH; Figure 4, S4). This confirms our assumption that the DTPs and GSH form a redox equilibrium (Scheme S1). When metal ions are present, red‐DTPs coordinate and are thereby removed from the redox equilibrium. This causes a re‐adjustment of the equilibrium and the formation of more red‐DTPs. In addition, the coordination to metal ions protects the DTP dithiols from oxidation by air. Overall, our studies demonstrate that GSH can activate the DTP prodrugs and mediate their interaction with metal‐based and other targets in cells, establishing the DTPs as prochelators.
Figure 5.

GSH activates the DTPs for Zn(II) coordination. (A) Formation of the Zn(II) complex of holomycin (1) in the presence of different GSH amounts. (B) Formation of the Zn(II) complex of holomycin (1) in the presence of 1 mM GSH, followed over the specified incubation times. (C) Complex formation with thiolutin (2). (D) Complex formation with aureothricin (3). The assays were performed with 50 μM DTP and 0.5 equiv. Zn(II) in 75 mM Tris‐HCl, pH 7.4. The spectra of the Zn(II) complexes formed upon reduction with TCEP are included for comparison.
Conclusion
Given their broad biological activity, the DTPs are of interest as scaffolds for drug molecules such as antibiotics. Our studies demonstrate the unique redox properties of the DTPs. Despite the structural similarity with LA and DTT, and the conjugated system, the redox potential of Holo seems to be more positive. And still, the dithiol is more labile in the presence of oxygen. We show that the DTPs are reduced at cellular levels of GSH, in line with the earlier proposed mechanism that the DTPs are reduced after cell entry. Moreover, we show that the reduction by GSH mediates the chelation of Zn(II), and should similarly allow interactions of the activated dithiol with other targets in cells.
In addition to GSH, also structurally related thiols, such as bacillithiol and mycothiol, should reduce the DTPs. They are employed by some bacteria in place of GSH and their redox potentials are similar to that of GSH. [20] Depending on the accessibility, the DTPs may also be reduced by Cys residues of other peptides or proteins, as suggested previously. [10] Similarly, the DHLA cofactor could contribute to the reduction of the DTPs. Our results indicate that the redox potential of Holo is similar to that of GSH (E°’=−0.25 V) and more positive than that of DHLA (E°’=−0.29 V). Thus, also the thioredoxins (Trx), small redox‐active proteins that act as oxidoreductases and facilitate the reduction of disulfides in cells, could serve as reductants. For example, the redox potential of Escherichia coli Trx1 is E°’=−0.27 V, [21] and presumably lower than that of Holo and other DTPs. Consistently, thioredoxins have been reported to contribute to the inhibitory activity of thiolutin. [5] Overall, there seem to be multiple possibilities how the DTPs get reduced in cells. These redox reactions may even contribute to their biological activity when important cellular reductants get oxidized.
Our studies also corroborate the similarities between the DTPs and the epipolythiodioxopiperazines (ETPs). These mycotoxins, including gliotoxin and sporidesmin, contain an internal disulfide that bridges a diketopiperazine core. [22] Similar to the DTPs, the ETPs act as prochelators that inhibit Zn(II)‐dependent enzymes. [23] Consistent with our results, also the ETPs were reported to be reduced by stoichiometric amounts of DTT and DHLA, and at 100‐fold excess of GSH and Cys. [24]
In addition to establishing the DTPs as prochelators, we report an expedient route for the synthesis of thiolutin and aureothricin. It involves the selective N‐methylation of a holomycin precursor, and also allows access to related amide derivatives. The facile synthetic access to the DTPs will benefit studies on structure‐activity relationships. And together with our mechanistic insights it contributes to the optimization of the DTP scaffold for therapeutic applications.
Experimental Section
General Materials and Methods: Details on general materials and methods, as well as NMR spectra, UV/Vis spectra, HPLC chromatograms, and cyclic voltammograms are provided as Supporting Information.
Compounds
Previously reported compounds: Holomycin (1) and precursors 4, 5, 6, 7 were synthesized as reported earlier. [6]
Trifluorylated thiolutin (9): Under an N2 atmosphere, precursor 7 (0.40 g; 1.5 mmol; 1.0 equiv.) and NaH (60 % in mineral oil; 0.09 g; 2.2 mmol; 1.5 equiv.) were dissolved in anhydrous DMF (40 mL). CH3I (100 μL; 1.6 mmol; 1.1 equiv.) was added, and the reaction mixture was stirred for 2.5 h at r.t. The mixture was diluted with EtOAc (150 mL), and the solution was washed with 1 M HCl (2×100 mL); the aqueous phase was extracted twice with EtOAc. The combined organic phases were washed with brine, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by flash chromatography (dissolved crude in EtOAc and adsorbed on silica for loading; eluted with EtOAc/n‐heptane 1 : 3→1 : 1) to yield 9 as orange solid (0.18 g, 42 %). TLC R f=0.56 (EtOAc/n‐heptane 3 : 1). 1H NMR (CDCl3, 400 MHz): δ 3.40 (s, 3H, NCH3 ), 6.81 (s, 1H, C=CH), 8.37 (br s, 1H, NH). 1H NMR (DMSO‐d6, 400 MHz): δ 3.30 (s, 3H, NCH3 ), 7.60 (s, 1H, C=CH), 11.68 (br s, 1H, NH). 19F NMR (CDCl3, 235 MHz): δ −75.1 (s, CF3 ). 13C{1H} NMR (DMSO‐d6, 100 MHz): δ 27.7 (s, NCH3), 111.6 (s, C=C−N), 113.4 (s, C=CH), 115.5 (q, 1 J C,F=265 Hz, CF3), 135.8 (s, C=CH), 138.8 (s, S−C=C), 153.8 (q, 2 J C,F=38 Hz, exo‐C=O), 165.7 (s, endo‐C=O). Selective 1D‐NOESY (Figure S37), (1H,13C)‐HSQC (Figure S40) and (1H,13C)‐HMBC (Figure S41) are reported as Supporting Figures.
Thiolutin (2): To a solution of 9 (0.17 g, 0.6 mmol, 1.0 equiv.) in MeOH (20 mL), concentrated HCl (0.8 mL) was added and the mixture was refluxed for 2 h at 75 °C. The solvent was removed in vacuo and the obtained hydrochloride was used for the next step without further purification.
Under an N2 atmosphere, the hydrochloride was suspended in anhydrous THF (35 mL). The solution was cooled to 0 °C. Acetyl chloride (54 μL; 0.8 mmol; 1.2 equiv.) was added dropwise, followed by the dropwise addition of NEt3 (184 μL; 1.3 mmol; 2.1 equiv.). The reaction mixture was stirred for 30 min at r.t. The solvent was removed under reduced pressure. The crude was purified by flash chromatography (dissolved crude in CH2Cl2/MeOH 1 : 1 and adsorbed on silica; eluted with EtOAc/n‐heptane 1 : 3→1 : 1→3 : 1→EtOAc) to yield 2 as yellow solid (0.09 g, 63 %). TLC R f=0.5 (EtOAc/n‐heptane 3 : 1). 1H NMR (DMSO‐d6, 400 MHz): δ 2.02 (s, 1H, COCH3 ), 3.25 (s, 3H, NCH3 ), 7.33 (s, 1H, C=CH), 9.97 (br s, 1H, NH). 13C{1H} NMR (DMSO‐d6, 100 MHz): δ 22.3 (s, NCH3), 27.5 (s, COCH3), 110.8 (s, C=C), 114.8 (s, C=C), 132.3 (s, C=C), 136.0 (s, C=C), 166.1 (s, C=O), 168.8 (s, C=O). HR‐MS (ESI): [M+H]+ m/z calcd. 229.0106, found 229.0088; [M+Na]+ m/z calcd. 250.9925, found 250.9912.
Aureothricin (3): To a solution of 9 (0.15 g, 0.6 mmol, 1.0 equiv.) in MeOH (20 mL), concentrated HCl (0.8 mL) was added and the mixture was refluxed for 2 h at 75 °C. The solvent was removed in vacuo and the obtained hydrochloride was used for the next step without further purification.
Under an N2 atmosphere, the hydrochloride was suspended in anhydrous THF (50 mL). The solution was cooled to 0 °C. Propionyl chloride (55 μL; 0.7 mmol; 1.2 equiv.) was added dropwise, followed by the dropwise addition of NEt3 (165 μL; 1.2 mmol; 2.2 equiv.). The reaction mixture was stirred for 30 min at r.t. The solvent was removed under reduced pressure. The crude was purified by flash chromatography (dissolved crude in CH2Cl2/MeOH 1 : 1 and adsorbed on silica; eluted with EtOAc/n‐heptane 1 : 3→1 : 1→3 : 1→EtOAc) to yield 3 as yellow solid (0.05 g, 40 %). TLC R f=0.17 (EtOAc/n‐heptane 1 : 1). 1H NMR (DMSO‐d6, 400 MHz): δ 1.02 (t, 3 J H,H=7.6 Hz, 3H, CH2CH3 ), 2.35 (q, 3 J H,H=7.6 Hz, 2H, CH2 CH3), 3.25 (s, 3H, NCH3 ), 7.32 (s, 1H, C=CH), 9.89 (br s, 1H, NH). 13C{1H} NMR (DMSO‐d6, 100 MHz): δ 9.5 (s, CH2 CH3), 27.5 (s, NCH3), 28.0 (s, CH2CH3), 110.7 (s, C=C), 114.8 (s, C=C), 132.2 (s, C=C), 136.0 (s, C=C), 166.2 (s, C=O), 172.6 (s, C=O). HR‐MS (ESI): [M+H]+ m/z calculated: 243.0262, found: 243.0245; [M+Na]+ m/z calcd. 265.0082, found 265.0064.
Stock solutions: Stock solutions of holomycin (20 mM), thiolutin (5 mM), aureothricin (20 mM), and LA (50 mM) were prepared in DMF, and stored at −20 °C. 50 mM stocks of the reducing and oxidizing agents (TCEP⋅HCl, DTT, DTTox, GSH, GSSG, Cys, BME, NaAsc) in MilliQ‐H2O (0.057 μS/cm) were used and stored at −20 °C. In the case of DHLA, a 10 mM stock was prepared just before the experiment by mixing LA with 1 equiv. TCEP. A 20 mM stock solution of Zn(II) was prepared from ZnCl2 (99 %, Alfa‐Aesar) in MilliQ‐H2O. A buffer stock of 1 M Tris‐HCl, pH 7.4 was used (prepared from Tris Base, Trometamol, >99,9 %, VWR). As blanks for the UV‐vis experiments, the respective amount of solvent for the DTP (DMF) diluted into 75 mM Tris‐HCl, pH 7.4 with MilliQ‐H2O was used.
UV‐vis Assays
General setup: 50 μM of the DTP was diluted into 75 mM Tris‐HCl, pH 7.4. Further reagents were added as stated for the respective experiments. All samples were prepared with a total volume of 1 mL, filled up with MilliQ‐H2O. The samples were incubated for at least 1 min before recording the absorption spectra. All experiments were performed at ambient conditions in the presence of air. As blank, the amount of DMF equivalent to the used amount of the DTP stock was diluted into 75 mM Tris‐HCl, pH 7.4.
Titration with reducing agents: The titrations were not conducted as classical titrations; instead, individual samples were prepared for the different equivalents of the reducing agent (TCEP, DTT, DHLA, GSH, Cys, BME, Asc). The respective equivalents were added to 50 μM DTP in 75 mM Tris‐HCl. and the samples were incubated for the specified time before recording the spectra.
Re‐oxidation on air: Red‐Holo was prepared by adding 1 eq. TCEP to 50 μM Hol in 75 mM Tris‐HCl, and the samples were incubated for 1 min before the recording of the spectra was started. Between the recordings, the samples were left on air in open cuvettes.
Re‐oxidation with oxidizing agents: Red‐Holo was prepared with 1 eq. TCEP. Subsequently, the respective amount of DTTox, LA, or GSSG was added, and the recording of the spectra was started.
Zn(II) coordination. Red‐DTP was prepared by adding 1 eq. TCEP to 50 μM DTP in 75 mM Tris‐HCl and incubating for 1 min. Subsequently, Zn(II) (0.5 equiv.) was added and the samples were incubated for at least another 1 min before the spectra were recorded.
Complex formation in the presence of different reductants: In the case of DHLA, Holo was pre‐incubated with 1 equiv. DHLA, before the metal ions were added. In the case of GSH, the DTPs were mixed with the respective amounts of GSH, Zn(II) (0.5 eq.) was added, and spectra were recorded at the specified incubation times.
Redox Equilibration Assays
Inert conditions: The preparation of the solutions and the incubation was performed under an N2 atmosphere. DMF and 75 mM Tris‐HCl, pH 7.4 were degassed with N2 (bubbled through the solutions for 30 min).
Stocks: All stocks were prepared in degassed solvents. Holo, LA: 20 mM in DMF; DTT, DTTox, TCEP: 20 mM in 75 mM Tris‐HCl; red‐Holo: 10 mM, prepared by mixing Holo (20 mM) with 1 equiv. TCEP (20 mM); DHLA: 10 mM, prepared by mixing LA (20 mM) with 1 equiv. TCEP (20 mM).
Equilibration: The respective reagents were mixed to a final concentration of 5 mM in a total volume of 3 mL, filled up with 75 mM Tris‐HCl. The mixture was stirred for 1 day at room temperature. The reaction was quenched by adding 10 % TFA (final TFA content: 1 %). Immediately after the quench, the reaction mixture was analyzed by RP‐HPLC, operated with gradient‐1.
Cyclic Voltammetry: Cyclic voltammetry was performed using a PalmSens4 potentiostat in a standard three‐electrode setup in a one‐compartment cell. A glassy‐carbon electrode was used as working electrode, a Ag/AgCl/KClsat electrode as pseudo‐reference electrode, and a Pt wire as counter electrode. The working electrode was successively polished with 1.0, 0.3 and 0.1 μm sandpaper and subsequent sonication in acetonitrile for 10 min before the measurement. KCl (1 M) was used as electrolyte in 75 mM Tris‐HCl, pH 7.4. Before each experiment, the electrochemical cell was degassed with Ar for 10 min. The solution was purged with Ar for 10 min, and an Ar atmosphere was maintained in the cell throughout the measurements. The cyclic voltammograms were recorded at a scan rate of 0.1 V/s. Holo (1 mM; from DMF stock), GSSG (5 mM; from MilliQ‐H2O stock), or LA (1 mM; from DMF stock) were added to the listed final concentrations. In the case of red‐Holo, TCEP (1 mM; from MilliQ‐H2O stock) was added to the Holo solution.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
We gratefully acknowledge financial support from the Fonds der Chemischen Industrie (FCI). We thank Prof. N. Metzler‐Nolte for providing laboratory space and equipment; Prof. U.‐P. Apfel for the use of cyclic voltammetry equipment in his lab; Martin Gartmann for support with some of the NMR measurements; Franziska Schmoll for technical assistance with some of the UV‐vis assays; Jacqueline Hausherr for technical assistance with some of the equilibration assays. Open Access funding enabled and organized by Projekt DEAL.
Albini F., Bormann S., Gerschel P., Ludwig V. A., Neumann W., Chem. Eur. J. 2023, 29, e202202567.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.
- 1a. Wencewicz T. A., Bioorg. Med. Chem. 2016, 24, 6227–6252; [DOI] [PubMed] [Google Scholar]
- 1b. Wright G. D., Nat. Prod. Rep. 2017, 34, 694–701; [DOI] [PubMed] [Google Scholar]
- 1c. Rossiter S. E., Fletcher M. H., Wuest W. M., Chem. Rev. 2017, 117, 12415–12474; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Lakemeyer M., Zhao W., Mandl F. A., Hammann P., Sieber S. A., Angew. Chem. Int. Ed. Engl. 2018, 57, 14440–14475. [DOI] [PubMed] [Google Scholar]
- 2. Li B., Wever W. J., Walsh C. T., Bowers A. A., Nat. Prod. Rep. 2014, 31, 905–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Umezawa H., Maeda K., Kosaka H., Japan. Med. J. 1948, 1, 512–517. [Google Scholar]
- 4.
- 4a. Li B., Walsh C. T., Biochemistry 2011, 50, 4615–4622; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Li B., Forseth R. R., Bowers A. A., Schroeder F. C., Walsh C. T., ChemBioChem 2012, 13, 2521–2526; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Chan A. N., Shiver A. L., Wever W. J., Razvi S. Z., Traxler M. F., Li B., Proc. Natl. Acad. Sci. USA 2017, 114, 2717–2722; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Lauinger L., Li J., Shostak A., Cemel I. A., Ha N., Zhang Y., Merkl P. E., Obermeyer S., Stankovic-Valentin N., Schafmeier T., Wever W. J., Bowers A. A., Carter K. P., Palmer A. E., Tschochner H., Melchior F., Deshaies R. J., Brunner M., Diernfellner A., Nat. Chem. Biol. 2017, 13, 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qiu C., Malik I., Arora P., Laperuta A. J., Pavlovic E. M., Ugochuckwu S., Naik M., Kaplan C., bioRxiv 2021, 10.1101/2021.05.05.442806 [DOI] [Google Scholar]
- 6. Hjelmgaard T., Givskov M., Nielsen J., Org. Biomol. Chem. 2007, 5, 344–348. [DOI] [PubMed] [Google Scholar]
- 7. Schmidt U., Geiger F., Angew. Chem. 1962, 74, 328–329; [Google Scholar]; Angew. Chem. Int. Ed. 1962, 1, 328–328. [Google Scholar]
- 8.
- 8a. Schmidt U., Geiger F., Justus Liebigs Ann. Chem. 1963, 664, 168–188; [Google Scholar]
- 8b. Schmidt U., Chem. Ber. 1964, 97, 1511–1512; [Google Scholar]
- 8c. Hagio K., Yoneda N., Bull. Chem. Soc. Jpn. 1974, 47, 1484–1489; [Google Scholar]
- 8d. Stachel H. D., Nienaber J., Zoukas T., Liebigs Ann. Chem. 1992, 1992, 473–480; [Google Scholar]
- 8e. Dell I., Godfrey C. R. A., Wadsworth D. J., ACS Symp. Ser. 1992, 504, 384–394. [Google Scholar]
- 9. Jensen B., Acta Crystallogr. 1971, B27, 392–400. [Google Scholar]
- 10. Chan A. N., Wever W. J., Massolo E., Allen S. E., Li B., Chem. Res. Toxicol. 2019, 32, 400–404. [DOI] [PubMed] [Google Scholar]
- 11. Lees W. J., Whitesides G. M., J. Org. Chem. 1993, 58, 642–647. [Google Scholar]
- 12. Cleland W. W., Biochemistry 1964, 3, 480–482. [DOI] [PubMed] [Google Scholar]
- 13. Lukesh J. C., Palte M. J., Raines R. T., J. Am. Chem. Soc. 2012, 134, 4057–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamoureux G. V., Whitesides G. M., J. Org. Chem. 1993, 58, 633–641. [Google Scholar]
- 15. Corduneanu O., Garnett M., Brett A. M. O., Anal. Lett. 2007, 40, 1763–1778. [Google Scholar]
- 16. Morgan B., Ezerina D., Amoako T. N., Riemer J., Seedorf M., Dick T. P., Nat. Chem. Biol. 2013, 9, 119–125. [DOI] [PubMed] [Google Scholar]
- 17. Kosower N. S., Kosower E. M., Int. Rev. Cytol. 1978, 54, 109–160. [DOI] [PubMed] [Google Scholar]
- 18. Keire D. A., Strauss E., Guo W., Noszal B., Rabenstein D. L., J. Org. Chem. 1992, 57, 123–127. [Google Scholar]
- 19. Sapper H., Kang S.-O., Paul H.-H., Lohmann W., Z. Naturforsch. 1982, 37 C, 942–946. [PubMed] [Google Scholar]
- 20. Imber M., Pietrzyk-Brzezinska A. J., Antelmann H., Redox Biol. 2019, 20, 130–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ritz D., Beckwith J., Annu. Rev. Microbiol. 2001, 55, 21–48. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Gardiner D. M., Waring P., Howlett B. J., Microbiology 2005, 151, 1021–1032; [DOI] [PubMed] [Google Scholar]
- 22b. Dolan S. K., O′Keeffe G., Jones G. W., Doyle S., Trends Microbiol. 2015, 23, 419–428. [DOI] [PubMed] [Google Scholar]
- 23. Cook K. M., Hilton S. T., Mecinovic J., Motherwell W. B., Figg W. D., Schofield C. J., J. Biol. Chem. 2009, 284, 26831–26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Munday R., Chem.-Biol. Interact. 1982, 41, 361–374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.