

Abstract
Cancer of unknown primary (CUP) is a syndrome defined by clinical absence of a primary cancer after standardised investigations. Gene expression profiling (GEP) and DNA sequencing have been used to predict primary tissue of origin (TOO) in CUP and find molecularly guided treatments; however, a detailed comparison of the diagnostic yield from these two tests has not been described. Here, we compared the diagnostic utility of RNA and DNA tests in 215 CUP patients (82% received both tests) in a prospective Australian study. Based on retrospective assessment of clinicopathological data, 77% (166/215) of CUPs had insufficient evidence to support TOO diagnosis (clinicopathology unresolved). The remainder had either a latent primary diagnosis (10%) or clinicopathological evidence to support a likely TOO diagnosis (13%) (clinicopathology resolved). We applied a microarray (CUPGuide) or custom NanoString 18‐class GEP test to 191 CUPs with an accuracy of 91.5% in known metastatic cancers for high–medium confidence predictions. Classification performance was similar in clinicopathology‐resolved CUPs – 80% had high–medium predictions and 94% were concordant with pathology. Notably, only 56% of the clinicopathology‐unresolved CUPs had high–medium confidence GEP predictions. Diagnostic DNA features were interrogated in 201 CUP tumours guided by the cancer type specificity of mutations observed across 22 cancer types from the AACR Project GENIE database (77,058 tumours) as well as mutational signatures (e.g. smoking). Among the clinicopathology‐unresolved CUPs, mutations and mutational signatures provided additional diagnostic evidence in 31% of cases. GEP classification was useful in only 13% of cases and oncoviral detection in 4%. Among CUPs where genomics informed TOO, lung and biliary cancers were the most frequently identified types, while kidney tumours were another identifiable subset. In conclusion, DNA and RNA profiling supported an unconfirmed TOO diagnosis in one‐third of CUPs otherwise unresolved by clinicopathology assessment alone. DNA mutation profiling was the more diagnostically informative assay. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: cancer of unknown primary, gene expression profiling, cancer diagnostic, mutation profiling, targeted therapy, tissue of origin classification
Introduction
Cancer of unknown primary (CUP) is a clinical syndrome representing a heterogeneous group of cancers where the primary tumour evades clinical detection after standardised investigations [1, 2]. Representing 1–3% of all cancer diagnoses [3], CUP patients have a notoriously poor outcome. For instance, despite being the 14th most common diagnosis in Australia, CUP is the sixth most common cause of cancer‐related death [4]. In the absence of a known tissue of origin (TOO), most CUP patients have historically received empirical chemotherapy with limited clinical benefit in most patients [5]. Current guidelines identify a subset (~15%) of patients with a favourable outcome based on clinicopathological features corresponding to more treatment‐responsive cancer types [6]. The improved efficacy of targeted and immunotherapy treatments over chemotherapy in some cancer types, such as non‐small cell lung cancer (NSCLC) and renal cell carcinoma (RCC), has also led to proposed reassignment of some traditionally unfavourable CUP subsets [7, 8, 9]. Improved diagnostic methods to help resolve TOO, combined with more effective precision treatments, are therefore likely to be complementary to improve CUP patient outcomes.
Guidelines for a CUP diagnosis are currently based on standardised gender‐appropriate histopathological and clinical investigations; for example, as described by the European Society of Medical Oncology (ESMO) [6]. TOO classifiers using gene expression profiling (GEP) and DNA methylation testing have also been described. These molecular tests have 83–94% accuracy when tested on known primary and metastatic tumours [10, 11, 12, 13, 14] and are reported to be superior to immunohistochemistry (IHC) [15, 16]. The diagnostic utility of molecular classifiers for CUP has been validated on latent primary CUP, in which a primary tumour becomes known in time or through alignment with IHC and other clinicopathological information [10, 13, 15, 16, 17]. There is evidence that some CUP patients will have molecularly identifiable cancer types that respond better to site‐directed treatments [9, 18, 19, 20]; however, the clinical utility of TOO tests remains in question, based on negative results from two randomised trials [19, 20]. Despite the potential limitations of these trials being that current standard of care was not used for some treatable cancer types, the level of recommendation for molecular testing remains low under current guidelines [6].
DNA mutational profiling has also been explored in CUP [21, 22, 23, 24, 25]. The primary goal of these studies has been to identify clinically actionable mutations to direct targeted therapies [21, 22, 23, 24, 25]. Mutational profiling can also provide insight into the TOO, given that certain mutational processes and the prevalence of cancer driver mutations can be cancer type‐dependent or enriched [21]. Combining GEP and DNA mutational profiling into a single classification model has also been previously demonstrated [26], although these tools and methods are often accessible only through commercial service providers [27]. As the DNA features defining cancer types are realised and comprehensive DNA sequencing becomes more common, such results can be easily interpreted alongside histopathology and clinical picture to resolve diagnostic ambiguities [28].
While GEP and mutation profiling of CUP have been described in previous studies, no attempt has been made to compare the diagnostic value of these methods benchmarked against clinicopathological review. Therefore, we applied GEP and DNA mutation profiling to patients recruited to a national study (Solving Unknown Primary Cancer: SUPER). Based on available clinicopathological data, we firstly curated the CUP cases using the available clinicopathological data to find latent primary CUPs and those cases where a TOO was highly likely given the available clinicopathological evidence, leaving any clinicopathology‐unresolved CUPs, where molecular profiling would be of most benefit. We then compared the diagnostic value of GEP and DNA sequencing in each of these CUP patient groups, identifying recurrent cancer types among the cohort and their molecular features.
Materials and methods
Patient cohort
CUP patients were recruited to the SUPER study from 11 Australian sites with informed patient consent under an approved protocol of the Peter MacCallum Cancer Centre (PMCC) human research ethics committee (HREC protocol: 13/62) per the Declaration of Helsinki 1975, as revised in 1983. Eligibility criteria for patient inclusion and exclusion to the study are described in Supplementary materials and methods.
CUP clinicopathological review
A retrospective review of all histopathology reports and clinical data was performed by a single pathologist (OP) (supplementary material, Tables S1 and S2). A histopathology review of slides was undertaken for a subset of cases from the PMCC (n = 59), including additional IHC staining if necessary if tissue was available (supplementary material, Tables S1 and S2). Further review of the clinical data was done by a medical oncologist (LM) to concur with the pathologist's opinion. Cases were assigned a TOO where possible, or subclassified using a modified version of the Memorial Sloan Kettering Cancer Center (MSKCC) OncoTree classification system for CUPs [29] into undifferentiated malignant neoplasms (UDMN); poorly differentiated carcinoma (PDC); adenocarcinoma, not otherwise specified (ADNOS); neuroendocrine tumours, not otherwise specified (NETNOS); neuroendocrine carcinomas, not otherwise specified (NECNOS); and squamous cell carcinomas, not otherwise specified (SCCNOS). ADNOS were further subdivided based on cytokeratin 7 (CK7) and cytokeratin 20 (CK20) IHC staining; where CK7 was negative and CK20 had positive staining, caudal‐type homeobox 2 (CDX2) was annotated. SCCNOS were subclassified based on p16INK4A (p16) IHC staining positivity (Supplementary materials and methods). The diagnosis for all cases was re‐assessed following the genomic findings.
Gene expression profiling
GEP was performed using a previously described microarray‐based test (CUPGuide) [21] or a custom NanoString nCounter assay (NanoString Technologies Inc., Seattle, WA, USA). A detailed description of nucleic acid extraction, the NanoString assay, and the TOO classifier is given in Supplementary materials and methods. In brief, the NanoString panel included probe sets targeting 225 genes differentially expressed across 18 tumour classes and viral transcripts encoding capsid proteins for HPV16 L1, HPV18 L1, and Merkel cell polyomavirus (VP2) (supplementary material, Table S3). The NanoString classifier was trained on harmonised TCGA RNA‐seq data described previously [14], representing 8,454 samples consolidated into 18 tumour classes (supplementary material, Table S4). The RNA‐seq/NanoString k‐nearest neighbour cross‐platform classifier was validated on an independent test set of 188 metastatic tumours profiled by NanoString (supplementary material, Tables S5 and S6). A probability score was generated for predictions and heuristic thresholds set for classification confidence level (unclassified <0.5, low ≥0.5 and ≤0.7, medium confidence >0.7 and <0.9, high confidence ≥9 probability).
Comprehensive DNA panel sequencing
Targeted enrichment and DNA sequencing were performed on matched blood and tumour DNA, capturing coding regions and exon/intron splice sites of 386 cancer‐related genes (listed in supplementary material, Table S7) using previously described methods [30]. Alignment and variant calling were performed using bcbio‐nextgen cancer somatic variant calling pipelines (https://github.com/bcbio/bcbio-nextgen) and R tools were used for analysis. A detailed description of bioinformatics is given in Supplementary materials and methods.
Reference mutation data and identification of putative diagnostic DNA features
The AACR Project GENIE mutation data for 77,058 tumours (referred to as GENIE) [31] was downloaded from the cBioPortal webpage (https://genie.cbioportal.org/, version 3.7.9) [32, 33]. The frequency of gene‐specific mutations was assessed in tumours annotated as CUP or other cancer types. Genes investigated were restricted to those included in the current study (supplementary material, Table S7). For assessment of cancer driver mutations, GENIE cancer classes with fewer than 50 samples per class were removed from consideration, except for assessing gene fusions, where cancer classes with fewer than 10 samples per cancer class were removed from analysis (supplementary material, Table S8). For identifying DNA features of potential diagnostic utility, the frequency of gene‐wise driver mutations was calculated in 22 pre‐defined cancer classes (supplementary material, Tables S8 and S9). Oncogenes and tumour suppressor genes (TSGs) were annotated using OncoKB [34]. For assessing cancer class mutation frequency in the reference data, only truncating mutations in TSGs or hotspot mutations in both oncogenes and TSGs were used (annotated using the web portal http://www.cancerhotspots.org/ [35]). Oncogenic gene fusions and copy‐number alterations were restricted to a curated set of cancer genes (supplementary material, Table S7). A Fisher's exact test was then performed using the R package stats (https://github.com/arunsrinivasan/cran.stats) to identify genes statistically enriched for DNA alterations in individual cancer classes versus all other cancers. The Holm–Bonferroni method was used for post hoc adjustment to account for multiple testing. Significance was defined as anything with an adjusted P value less than 0.05 and an odds ratio (OR) greater than 1. Significant cancer type diagnostic DNA alterations are summarised in supplementary material, Tables S9 and S10.
Results
Study cohort and subclassification of CUPs
A total of 215 patients were recruited to the SUPER study, and a summary of baseline characteristics is shown in Table 1. Eighty‐nine per cent (191/215) received GEP, 93% (201/215) of patients had comprehensive DNA panel sequencing, and 82% (177/215) received both assays (Table 1). A latent primary diagnosis was reported by the treating clinician during clinical follow‐up in 10% (22/215) of cases based on histopathology, clinical presentation, and/or cancer imaging. In another 13% (27/215) of cases, TOO was assigned after a retrospective review of reported or reviewed morphology, clinical picture, and IHC staining. Notably, among the latent primary and clinicopathology‐resolved CUP cases, there was an enrichment of patients with a prior history of cancer (35%, 17/49), eight of whom had a likely recurrence of their previous disease (supplementary material, Table S1).
Table 1.
Study cohort characteristics.
| Characteristics | Count | Proportion |
|---|---|---|
| Total cohort | 215 | 100% |
| Age, mean (range), years | 61 (20–86) | |
| Sex | ||
| Male | 89 | 41% |
| Female | 107 | 49% |
| Unrecorded | 19 | 9% |
| ECOG 1 grade | ||
| 0 | 67 | 31% |
| 1 | 101 | 47% |
| 2 | 24 | 11% |
| 3 | 1 | 0.5% |
| Not determined | 22 | 10% |
| Previous cancer diagnosis | 58 | 27% |
| ESMO outcome | ||
| Favourable | 31 | 14% |
| Unfavourable | 176 | 82% |
| Not determined | 8 | 4% |
| Gene expression profiling | 191 | 89% |
| NanoString | 172 | 80% |
| CUPGuide | 19 | 9% |
| DNA sequencing | 201 | 93% |
| Both molecular tests* | 177 | 82% |
DNA sequencing and GEP performed successfully.
ECOG: Eastern Cooperative Oncology Group.
Most patients (166/215, 77%) did not have an initial TOO designated (termed clinicopathology‐unresolved) as there was insufficient clinicopathological evidence to support a likely TOO despite an IHC workup according to current ESMO guidelines [6]. Additional IHC stains may have been informative in a small subset (7/166, 4%), but these could not be done owing to tissue availability. The clinicopathology‐unresolved CUPs were classified into histomorphological subtypes using a modified MSKCC OncoTree classification system (see Supplementary materials and methods) (supplementary material, Table S2). The majority of clinicopathology‐unresolved CUPs were adenocarcinomas, not otherwise specified (ADNOS) (51%) or poorly differentiated carcinomas (PDCs) (25%), with minor subsets of squamous cell carcinomas (SCCs) (13%), undifferentiated malignant neoplasms (UDMN) (7%), and neuroendocrine neoplasias (NET/NECNOS) (2%). OncoTree classifications are summarised in supplementary material, Table S11.
GEP classification confidence is lower for clinicopathology‐unresolved CUPs than known metastatic cancers
We used two GEP methods to classify 191/215 CUP tumours, where sufficient RNA was available. A previously described 18‐class microarray‐based classifier (CUPGuide) was used in 20 cases [21], while a novel NanoString classifier was used in the remaining cases (supplementary material, Tables S1 and S2). The NanoString classifier was validated using an independent cohort of 188 metastatic tumours of known origin (supplementary material, Table S6), achieving an overall prediction accuracy of 82.9%, increasing to 91.5% when only high–medium confidence classifications (n = 154) were considered (Figure 1A and supplementary material, Table S5).
Figure 1.
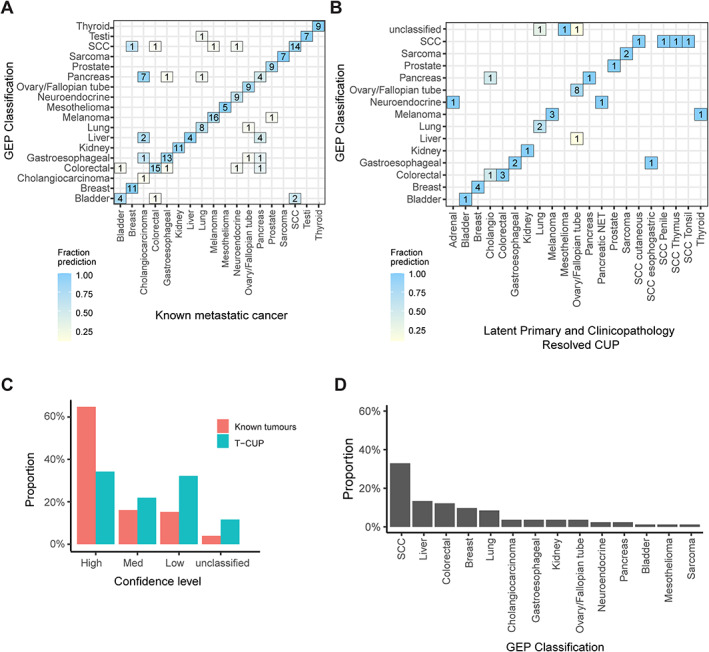
Gene expression profiling (GEP) tissue of origin classification of known metastatic cancers and SUPER cancer of unknown primary (CUP) tumours. (A) The NanoString GEP classifier was tested on 188 known origin metastatic tumours with confusion matrix showing concordance of tissue of origin prediction and known cancer type. (B) The GEP classifier tested on clinicopathology‐resolved CUPs showing concordance between the likely tissue of origin and the predicted cancer type. Latent primary and clinicopathology‐resolved CUPs representing cancer types not represented in the classifier model were removed from the analysis. (C) Fraction of cases within confidence probability score grouping contrasting classification of Clinicopathology‐unresolved CUPs and clinicopathology‐resolved CUPs combined with known metastatic tumours (unclassified <0.5, low ≥0.5 and ≤0.7, medium confidence ≥0.8 and ≤0.9, high confidence = 1). (D) GEP cancer class predictions of all clinicopathology‐unresolved CUPs with high–medium confidence predictions.
GEP TOO classification was possible for 45 clinicopathology‐resolved CUPs (Figure 1B), of which 80% (36/45) had a high–medium confidence classification (Figure 1C). Among high–medium confidence predictions, 94% (31/33) were concordant with their latent primary or histological diagnosis (supplementary material, Table S1). Three cases were considered outside the 18‐class GEP TOO differential, including one rare ampullary tumour (colorectal, high confidence) and two uterine tumours (SCC, medium confidence; ovarian, high confidence). Recurrent high–medium confidence misclassifications were observed among the clinicopathology‐resolved and latent primary CUP cases as well as known metastatic cancers in the validation set that included cholangiocarcinoma (1/1 and 5/11, respectively) and pancreatic adenocarcinomas (3/10 in the known metastatic set) (Figure 1A,B).
GEP TOO classification was performed on 146 clinicopathology‐unresolved CUPs, of which high–medium confidence classifications were made for only 56% (82/146) of cases (Figure 1C). The most frequent high–medium confidence classifications included SCC (32%), liver (13%), colorectal (12%), breast (10%), and lung (8.5%) (Figure 1D). A lower percentage of high–medium confidence classifications among clinicopathology‐unresolved CUPs compared with known metastatic cancers indicates that many CUP tumours either have an atypical transcriptional profile or are potentially enriched for cancer types outside the GEP classifier differential.
The mutation profile of the SUPER CUP cohort is consistent with other CUP cohorts
Comprehensive DNA panel sequencing was performed for 201/215 CUP tumours. We detected mutational features including single nucleotide variants (SNVs), gene fusions, copy number alterations (CNAs), SNV 96 trinucleotide mutational signatures (COSMIC v2) [36], tumour mutation burden (TMB), and off‐target viral DNA sequences (Figure 2). Viral RNA transcripts detected by NanoString also supported viral status for HPV‐positive tumours.
Figure 2.
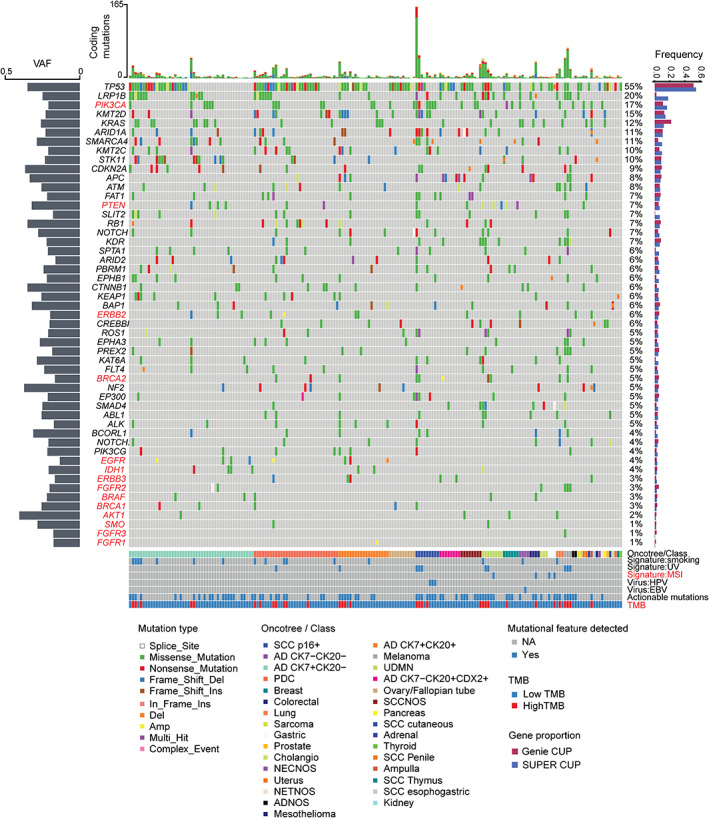
DNA mutational profiling of the SUPER cancer of unknown primary (CUP) cohort. The Oncoplot shows somatic mutations in CUPs in descending order of frequency. Genes and mutational features coloured red are actionable and targeted in the CUPISCO trial. The proportion of mutations in the SUPER CUPs was compared with the AACR Project GENIE CUP cohort (right‐hand bar plot). The left‐hand plot shows the average variant allele frequency (VAF) distribution per gene. The top bar plot shows the number of coding mutations per sample. Annotations include MSKCC OncoTree class for clinicopathology‐unresolved CUPs or the assigned tumour class for latent primary and clinicopathology‐resolved CUPs; detection of COSMIC mutational signatures (V2): smoking signature, ultra‐violet (UV) signature, DNA mismatch repair signature; oncoviruses: human papillomavirus 16 (HPV16) and Epstein–Barr virus (EBV); and tumour mutation burden (TMB) status: high >10 mutations/Mb or low <10 mutations/Mb.
At least one protein‐coding mutation was found in 98.5% (198/201) of CUPs, with a median TMB of 4.4 mutations/Mb (range 0.5–149 mutations/Mb). The most frequently mutated genes were TP53 (55%), LRP1B (20%), PIK3CA (17%), KMT2D (15%), KRAS (12%), ARID1A (11%), and SMARCA4 (11%) (Figure 2). The variant allele frequency of these mutations ranged between 16% and 40.5%, consistent with clonal cancer driver events when tumour purity was considered. Additionally, 8% (n = 16) of the cohort had dominant signature 4 (smoking), 5% (n = 11) signature 7 (ultra‐violet light, UV), and 2% (n = 4) signature 6 (microsatellite instability, MSI). HPV16 (DNA and RNA) was detected in five cases and EBV (DNA only) in one case.
The gene‐wise mutation frequency in the CUP cohort was also compared to 2,785 CUPs in the GENIE database. The mutation profile between the SUPER CUP and GENIE CUP cohorts was highly similar with some minor differences, including a higher frequency of KRAS mutations (12% versus 22%) among the GENIE CUPs and a higher frequency of LRP1B (18% versus 2%) mutations in the SUPER cohort, the latter explained by LRP1B not being included in the MSK‐IMPACT panel (Figure 2) [37]. Actionable mutations by reference to the CUPISCO trial criteria, as previously described [25], showed that 86 CUP patients (40%) were matched to either the targeted therapy or immunotherapy arm of CUPISCO (Figure 2) (supplementary material, Table S12). DNA mutation profiling therefore showed that the genomic landscape of our CUP cohort is highly similar to that of other CUP cohorts.
Mutation profiling and GEP can augment histopathology review
We next considered the independent or combined diagnostic value of DNA and RNA features with a histopathological review. Here, we considered driver gene mutations, gene fusions, mutational signatures, oncoviruses, and high–medium confidence GEP classifications. To identify gene mutations and fusions with significant cancer type associations, we referenced the GENIE database of 77,058 tumour samples involving 22 solid cancer types. The cancer type enrichment of a gene feature was determined by comparing one cancer type versus all the others [Fisher exact test adjusted P value <0.05 and odds ratio (OR) >1] (supplementary material, Table S10). A total of 171 genes passed the threshold in one or more cancer types, and 90 genes were significantly enriched in only one cancer type (supplementary material, Figures S1 and S2). Mutational signatures also provided important diagnostic evidence to support likely TOO; for example, signature 7 (UV) is associated with skin cancer or signature 4 (smoking) with cancers of the airways, although not excluding liver cancer [36].
Of the clinicopathology‐resolved and latent primary CUPs, 69% (33/49) had one or more DNA features consistent with the TOO diagnosis: gene mutations (n = 24), mutational signatures (n = 4), CNA (n = 2), gene fusions (n = 1), and oncoviruses (n = 1) (Figure 3A and supplementary material, Figure S3). By comparison, high–medium confidence GEP classifications were diagnostically useful in 51% of this group (n = 25). An example of where DNA and RNA features were consistent with the latent primary and clinicopathology TOO designation included 8/10 (80%) ovarian cases with high‐confidence GEP classification and a TP53 mutation, the latter occurring in more than 96% of high‐grade serous ovarian cancers [38] (supplementary material, Figure S3 and Table S1).
Figure 3.
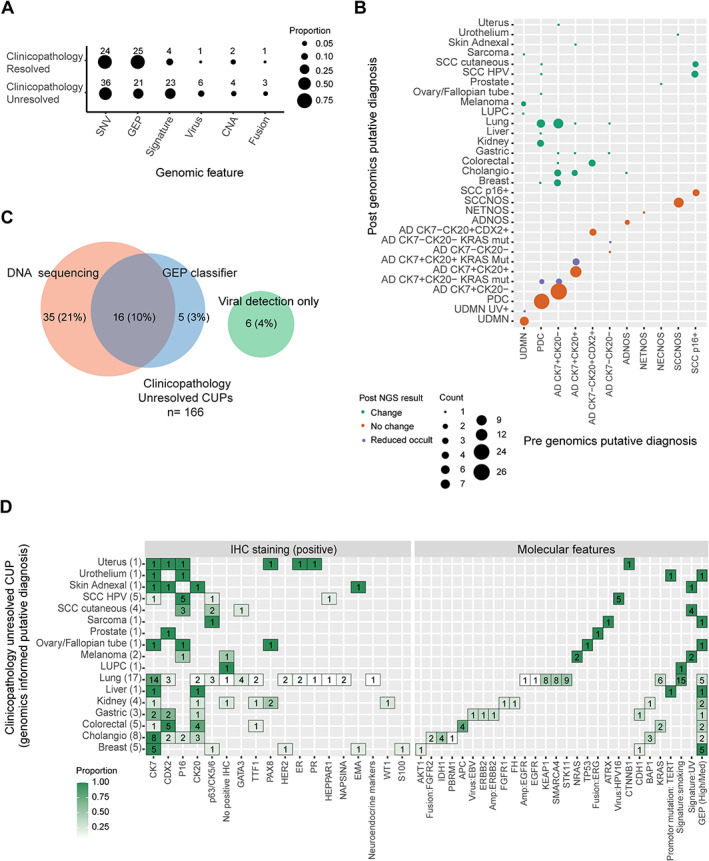
Identification of diagnostically useful DNA features using AACR Project GENIE mutation data and summary of evidence used to support tissue of origin (TOO) diagnosis among SUPER cancer of unknown primary (CUP) cases. (A) Proportion and number of cases where each genomic feature supported a putative TOO for clinicopathology‐unresolved CUPs and clinicopathology‐resolved CUPs. Genomic features included single nucleotide variants (SNV), gene expression profiling (GEP), mutational signatures, oncoviral sequences, copy number alterations (CNAs), and gene fusions. (B) MSKCC OncoTree cancer classification of clinicopathology‐unresolved CUPs before and after genomic analysis. CUPs were either resolved to a putative TOO, had reduced occult diagnosis, or remained clinicopathology‐unresolved. (C) Proportion of cases where DNA and/or GEP classification supported TOO diagnosis among clinicopathology‐unresolved CUPs. (D) Detailed summary of supportive genomic features and IHC staining used to assign a putative TOO for all genomically resolved clinicopathology‐unresolved CUPs.
A single case (1097) thought initially to be colorectal cancer based on histology had contradictory molecular features that resulted in a change in classification to clinicopathology‐unresolved CUP (ADNOS CK7−CK20+CDX2+). In this case, no recurrent gene mutations supported a colorectal origin (e.g. APC, RAS/RAF mutations), while high‐confidence GEP prediction of kidney was made with mutations detected in NF2 and SMARCA4. Confirmatory IHC staining (e.g. for PAX8) may have supported a kidney cancer diagnosis, but no tissue was available to perform the staining.
Molecular features supported a TOO diagnosis consistent with clinicopathological features in 37% (61/166) of clinicopathology‐unresolved CUPs (Figure 3A,B). DNA features supported a diagnosis in 31% (51/166) of such cases, whereas high–medium confidence GEP prediction was useful in only 13% (21/166) and viral detection in 4% (6/166). GEP classification and mutation profiling were informative in 10% (16/166) of cases (Figure 3C). DNA features supporting a diagnosis included driver gene mutations (n = 36), mutational signatures (n = 23), oncoviral nucleic acids (HPV16, n = 5; EBV, n = 1), CNA (n = 4), and gene fusions (n = 3) (Figure 3A). DNA features could also narrow the differential diagnosis in a further 11/166 (7%) cases, although assignment of a single TOO could not be confidently made (Figure 3B). Considering the combined genomics data, the most frequently suspected cancer types among clinicopathology‐unresolved CUPs were lung (n = 18), including a single pleomorphic carcinoma of the lung (LUPC), biliary tract (n = 8), breast (n = 5), colorectal (n = 5), HPV+ SCC (n = 5), and kidney (n = 4) (Figure 3D).
Recurrent CUP types and their molecular and clinicopathological features
Lung‐CUP was the most frequent molecular diagnosis among the clinicopathology‐unresolved CUPs. A dominant smoking mutational signature was found in 14 cases. Driver gene mutations associated with NSCLC included KEAP1 (lung OR = 9.8), STK11 (lung OR = 14.1), SMARCA4 (lung OR = 2.8), and KRAS (lung OR = 2.1) (Figure 3D and supplementary material, Figure S2 and Table S10). Notably, all but one lung‐CUP were negative for TTF1 IHC staining. Among the lung‐CUPs where GEP was possible, a high–medium confidence lung classification was made in only 5/16 cases. Two cases had a high–medium lung GEP classification, but mutation profiling was unsuccessful, and there was insufficient clinicopathological evidence available to support a lung cancer diagnosis. Notably, three lung‐CUPs were CDX2‐positive by IHC but had mutational features consistent with lung carcinoma, including a smoking mutational signature in all three cases. GEP was uninformative in these cases, as one case was classified as colorectal (0.9‐confidence probability) and two were predicted as SCC, a non‐specific classification but potentially in keeping with lung SCC (Figure 3D and supplementary material, Table S2). These CDX2+ lung‐CUPs were favoured to be enteric‐like lung adenocarcinomas [39].
Eight ADNOS tumours were likely to be intrahepatic cholangiocarcinomas supported by mutations in BAP1 (cholangiocarcinoma OR = 7.2) and IDH1 (cholangiocarcinoma OR = 32) (Figure 3D and supplementary material, Table S10). Seven of these tumours presented with liver masses. These tumours lacked KRAS mutations, making pancreatic cancer less likely, given that KRAS mutations occur in approximately 90% of pancreatic adenocarcinomas (supplementary material, Tables S9 and S10) [40]. FGFR2 fusions are also frequent in intrahepatic cholangiocarcinoma (cholangiocarcinoma OR > 100) [41, 42, 43] and therefore supported a cholangiocarcinoma diagnosis in two cases (supplementary material, Tables S2 and S10).
In four clinicopathology‐unresolved CUPs, subsequently diagnosed as kidney cancers using genomic features, two expressed PAX8 by IHC, supported by high PAX8 mRNA expression (z‐score > 2), and in an additional case, PAX8 IHC staining could not be performed, but the tumour had high PAX8 mRNA expression. The fourth case had neither PAX8 IHC staining nor PAX8 mRNA expression. Only two cases were classified as kidney by GEP (Figure 3D). Driver mutations consistent with RCC and detected among kidney‐CUPs included BAP1 (kidney OR = 7.6) and NF2 (kidney OR = 7.2). Another case had a truncating FH mutation consistent with FH‐deficient RCC (kidney OR = 8.1) (supplementary material, Figure S1). No VHL mutations were detected, representing the most common driver gene in clear cell RCC (42.5% RCC in GENIE, OR > 100). Another assigned kidney case was confirmed to be a recurrence of late‐onset adult Wilms’ tumour by detecting a somatic FGFR1 missense mutation in both the original primary tumour and a recurrent metastasis that presented over 20 years after first diagnosis (supplementary material, Table S2 and S10).
Discussion
Consistent with other large retrospective CUP studies, we found that approximately one‐quarter of CUPs may be assigned a likely TOO based on centralised histopathology and clinical review [44, 45]. This is similar to the recent experience of the international CUP clinical trial CUPISCO, where ~20% of patients had a single primary site diagnosis supported by available evidence or strongly suspected TOO [46]. Here, we directly compared the two molecular diagnostic approaches in CUP benchmarked against clinicopathological data. Our study is therefore distinguished from other studies where only a single molecular assay was applied [10, 11, 12, 13, 14] or where DNA and RNA features were algorithmically combined together to make a single TOO prediction [26, 27, 47]. We showed that DNA and RNA tests help to resolve a third of CUP cases where clinicopathological data alone were insufficient to designate a likely TOO diagnosis. Importantly, despite GEP being the most commonly explored molecular diagnostic test for CUP to date, we found that DNA sequencing may be of greater diagnostic value, as many CUP tumours appear to have an atypical transcriptional profile yet retain identifiable and compelling diagnostic mutational features.
GEP classification relies upon the expression of cellular differentiation markers that are often lost or equivocal in CUP tumours [48]. Our observation that fewer CUPs have high–medium confidence GEP classification compared with known metastatic cancers, thus reflecting a poorer classifier performance, is supported by results from other studies. For instance, the CancerTYPE ID classifier, which had extensive multisite validation, showed an overall accuracy of 85% for known cancer metastases. However, in CUP, the concordance of GEP with IHC and clinicopathological evidence was lower, at 75% for latent primary CUPs and 70% compared with the clinical picture only [11]. Similar to our observations, among lung‐CUP cases, GEP classification was even less concordant, with a latent primary tumour corresponding to TOO classification in only 50% of cases [17]. We found that GEP classification accuracy can be low for other cancer types, such as cholangiocarcinomas, as the transcriptional profile is similar to that of pancreatic and upper gastrointestinal neoplasms [14]. Notably, some previous GEP and DNA methylation tests did not include a cholangiocarcinoma class in their models or instead combined them with pancreatic cancers into a single pancreaticobiliary class [13, 49]. We found that DNA mutational profiling may be particularly useful in biliary cancers, given that some gene mutations are highly enriched in cholangiocarcinoma and have both diagnostic and occasionally therapeutic significance, including alterations in IDH1, FGFR2, and BAP1 [41, 42, 43].
Rare cancers likely comprise a significant subset of CUP tumours. Perhaps because rare cancers were either not represented or underrepresented in our GEP training data, this may explain lower confidence predictions among CUPs unresolved by histopathology alone. Transdifferentiation or mixed phenotypes can also potentially confuse GEP classification, and in such cases, a DNA profile may be more reliable. For instance, a recent multi‐omics study of lung cancers with mixed histology found that while transcriptomic profiles reflected regional cellular differentiation, the tumour's DNA mutation profile remained remarkably stable [50]. The reliability of mutation profiling over GEP in atypical lung cancers was also captured in our data. For example, we identified pulmonary enteric adenocarcinomas that lacked TTF1 expression but expressed the gastrointestinal marker CDX2 [39]. Importantly, GEP or IHC alone could not resolve such cases, given their atypical transcriptional profile; however, they had DNA features highly suggestive of NSCLC, including a smoking mutational signature and somatic mutations in KRAS, STK11, and SMARCA4. SMARCA4‐deficient lung cancers lack TTF1 expression [51] and are likely to be enriched among the CUP population [22, 23]. It is of high interest that murine SMARCA4‐deficient lung cancers also lose expression of lung differentiation markers and have pro‐metastatic behaviour similar to CUP tumours [52]. Therefore, it is enticing to think that SMARCA4 deficiency may account for the clinical presentation of some lung‐CUP cases.
Kidney cancers are another emerging CUP entity, representing ~4–6% of CUPs [7, 8, 46]. Interestingly, we found somatic mutations in NF2, FH, and BAP1 in some CUPs that were suggestive of kidney cancer when considered with clinicopathological data. Somatic NF2 mutations are characteristic of advanced papillary renal cell tumours and those with biphasic hyalinising psammomatous features [53]. Papillary carcinomas are enriched among other kidney‐CUPs [7, 8] with a mutation profile similar to that of the kidney‐CUPs that we identified in the SUPER cohort [54]. Identifying CUP entities and recurrent therapeutic targets in these groups may help to guide future CUP clinical trials. For instance, while empirical chemotherapy is ineffective in RCC, targeted therapies and immune checkpoint inhibitors are likely more efficacious [7, 55]. Furthermore, detection of NF2 mutations among RCCs could direct targeted treatment of the Hippo pathway using inhibitors of TEAD auto‐palmitoylation [56], now in clinical trials (NCT04665206). TEAD inhibitors therefore add to a growing list of molecular targeted therapies that may be effective in CUP tumours.
In conclusion, we have shown that both DNA and RNA tests can be incorporated into a pathology assessment to improve cancer type diagnosis, identify CUP subtypes, and find treatment targets. Rather than replacing traditional histopathological analysis, molecular testing can augment conventional testing to confirm a suspicion of primary TOO or provide robust diagnostic leads that are not otherwise evident. In practice, in cases where tissue is limited, prioritising genomic testing to guide additional investigations may be more informative before consuming tissue on extended IHC panels. With steady technological improvements and reduced sequencing costs, more comprehensive whole‐genome and transcriptome analysis will likely increase the sensitivity to detect features such as structural variants and mutational signatures that are not reliably detected by panel sequencing [57]. While DNA mutational profiling is not currently recommended in most CUP guidelines, the adoption of comprehensive panel DNA sequencing to assist cancer type diagnosis and detect potential treatment targets would seem of high clinical value for this patient group.
Author contributions statement
RT and LM conceived the study. AP performed the analysis and drafted the figures and tables. OWJP undertook the histopathology review and data analysis. TS and LM reviewed the clinical data. CW, KF and SW collected the data for the study. ADF, NW, CSK, BG, CS, MS, IMC, GR, MW and NK screened patients for eligibility. DE and NT developed the NanoString classifier, and JG and SB validated the classifier. GW, SW, AB, AJG, BJS, DB, SF and RJH contributed samples for training the classifier. AF, HX and SF performed the mutation profiling. APa performed the bioinformatic analysis. AP and RT co‐wrote the manuscript. All the authors critically reviewed the manuscript. PS, DB, LM and RT are the principal investigators and obtained research funding to support the study.
Supporting information
Supplementary materials and methods
Figure S1. Volcano plot showing significantly mutated features by cancer types using the GENIE pan‐cancer cohort
Figure S2. Diagnostically useful mutated features per cancer type using the GENIE pan‐cancer cohort
Figure S3. Supportive genomic features and IHC staining for latent primary and clinicopathology‐resolved CUPs
Table S1. Latent primary and clinicopathology‐resolved CUP clinicopathological and molecular data
Table S2. Clinicopathology‐unresolved CUP clinicopathological and molecular data
Table S3. NanoString custom gene panel
Table S4. Gene expression classifier cancer classes
Table S5. NanoString test set accuracy/specificity/sensitivity
Table S6. GEP classification performance on 188 known origin metastatic cancer samples
Table S7. DNA sequencing SureSelect custom gene panel
Table S8. GENIE grouped cancer classes
Table S9. Frequency of genomic events in GENIE cancer classes
Table S10. GENIE cancer classes significant associated genes
Table S11. Putatively assigned TOO diagnoses for latent primary and clinicopathology‐resolved CUPs and OncoTree classifications of clinicopathology‐unresolved CUPs
Table S12. Actionable genomic events in CUP defined by CUPISCO criteria
Table S13. NanoString gene counts of tissue of origin genes from known origin metastatic tumours (MET) and CUP tumours
Acknowledgements
We acknowledge Cameron Patrick of the Melbourne Statistical Consulting Platform for providing statistical support. We wish to thank Jillian Hung and Niklyn Nevins, SUPER study coordinators at Westmead and Blacktown; Lisa Kay at Nepean; Karin Lyon (ethics and governance); and acknowledge the contributions of the Nepean Cancer Biobank and the Westmead GynBiobank for facilitating the study. We would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. Interpretations are the responsibility of the study authors. We wish to acknowledge the patients who have contributed to this study and the CUP consumer steering committee, Cindy Bryant (chair), Kym Sheehan, Christine Bradford, Clare Brophy, Dale Witton, and Frank Stoss. The study was supported by funding from Cancer Australia (APP1048193, APP1082604) and the Victorian Cancer Agency (TRP13062). RWT was supported by funding from the Victorian Cancer Agency (TP828750). The Westmead, Blacktown, and Nepean study sites were supported by the Cancer Institute NSW 11/TRC/1‐06, 15/TRC/1‐01, and 15/RIG/1‐16. Open access publishing facilitated by The University of Melbourne, as part of the Wiley ‐ The University of Melbourne agreement via the Council of Australian University Librarians.
Conflict of interest statement: Professor Stephen B Fox is an Associate Editor of The Journal of Pathology. No other conflicts of interest were declared.
Contributor Information
Linda Mileshkin, Email: linda.mileshkin@petermac.org.
Richard W Tothill, Email: rtothill@unimelb.edu.au.
Data availability statement
The datasets used and/or analysed during the current study are available from the corresponding authors on reasonable request. The NanoString count data are provided as supplementary material, Table S13.
References
References 58, 59, 60, 61, 62, 63 are cited only in supplementary material.
- 1. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet 2012; 379: 1428–1435. [DOI] [PubMed] [Google Scholar]
- 2. Greco FA, Hainsworth JD. Cancer of unknown primary site. In DeVita, Hellman, and Rosenberg's Cancer: Principles and Practice of Oncology (8th edn), VT DV Jr., Hellman S, Rosenberg S (eds). Lippincott Williams & Wilkins: Philadelphia, 2008; 2363–2387. [Google Scholar]
- 3. Rassy E, Pavlidis N. The currently declining incidence of cancer of unknown primary. Cancer Epidemiol 2019; 61: 139–141. [DOI] [PubMed] [Google Scholar]
- 4. Australian Institute for Health and Welfare . Cancer data in Australia. Australian Cancer Incidence and Mortality (ACIM) books: cancer of unknown primary site. AIHW: Canberra, 2019. [Google Scholar]
- 5. Massard C, Loriot Y, Fizazi K. Carcinomas of an unknown primary origin – diagnosis and treatment. Nat Rev Clin Oncol 2011; 8: 701–710. [DOI] [PubMed] [Google Scholar]
- 6. Fizazi K, Greco FA, Pavlidis N, et al. Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2015; 26: v133–v138. [DOI] [PubMed] [Google Scholar]
- 7. Greco FA, Hainsworth JD. Renal cell carcinoma presenting as carcinoma of unknown primary site: recognition of a treatable patient subset. Clin Genitourin Cancer 2018; 16: e893–e898. [DOI] [PubMed] [Google Scholar]
- 8. Overby A, Duval L, Ladekarl M, et al. Carcinoma of unknown primary site (CUP) with metastatic renal‐cell carcinoma (mRCC) histologic and immunohistochemical characteristics (CUP‐mRCC): results from consecutive patients treated with targeted therapy and review of literature. Clin Genitourin Cancer 2019; 17: e32–e37. [DOI] [PubMed] [Google Scholar]
- 9. Hainsworth JD, Rubin MS, Spigel DR, et al. Molecular gene expression profiling to predict the tissue of origin and direct site‐specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah Cannon research institute. J Clin Oncol 2013; 31: 217–223. [DOI] [PubMed] [Google Scholar]
- 10. Erlander MG, Ma XJ, Kesty NC, et al. Performance and clinical evaluation of the 92‐gene real‐time PCR assay for tumor classification. J Mol Diagn 2011; 13: 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kerr SE, Schnabel CA, Sullivan PS, et al. Multisite validation study to determine performance characteristics of a 92‐gene molecular cancer classifier. Clin Cancer Res 2012; 18: 3952–3960. [DOI] [PubMed] [Google Scholar]
- 12. Hainsworth JD, Greco FA. Gene expression profiling in patients with carcinoma of unknown primary site: from translational research to standard of care. Virchows Arch 2014; 464: 393–402. [DOI] [PubMed] [Google Scholar]
- 13. Moran S, Martínez‐Cardús A, Sayols S, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 2016; 17: 1386–1395. [DOI] [PubMed] [Google Scholar]
- 14. Zhao Y, Pan Z, Namburi S, et al. CUP‐AI‐dx: a tool for inferring cancer tissue of origin and molecular subtype using RNA gene‐expression data and artificial intelligence. EBioMedicine 2020; 61: 103030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Handorf CR, Kulkarni A, Grenert JP, et al. A multicenter study directly comparing the diagnostic accuracy of gene expression profiling and immunohistochemistry for primary site identification in metastatic tumors. Am J Surg Pathol 2013; 37: 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tothill RW, Shi F, Paiman L, et al. Development and validation of a gene expression tumour classifier for cancer of unknown primary. Pathology 2015; 47: 7–12. [DOI] [PubMed] [Google Scholar]
- 17. Greco FA, Lennington WJ, Spigel DR, et al. Molecular profiling diagnosis in unknown primary cancer: accuracy and ability to complement standard pathology. J Natl Cancer Inst 2013; 105: 782–790. [DOI] [PubMed] [Google Scholar]
- 18. Yoon HH, Foster NR, Meyers JP, et al. Gene expression profiling identifies responsive patients with cancer of unknown primary treated with carboplatin, paclitaxel, and everolimus: NCCTG N0871 (alliance). Ann Oncol 2016; 27: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayashi H, Kurata T, Takiguchi Y, et al. Randomized phase II trial comparing site‐specific treatment based on gene expression profiling with carboplatin and paclitaxel for patients with cancer of unknown primary site. J Clin Oncol 2019; 37: 570–579. [DOI] [PubMed] [Google Scholar]
- 20. Fizazi K, Maillard A, Penel N, et al. A phase III trial of empiric chemotherapy with cisplatin and gemcitabine or systemic treatment tailored by molecular gene expression analysis in patients with carcinomas of an unknown primary (CUP) site (GEFCAPI 04). Ann Oncol 2019; 30(suppl_5): v851–v934. [Google Scholar]
- 21. Tothill RW, Li J, Mileshkin L, et al. Massively‐parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J Pathol 2013; 231: 413–423. [DOI] [PubMed] [Google Scholar]
- 22. Ross JS, Wang K, Gay L, et al. Comprehensive genomic profiling of carcinoma of unknown primary site: new routes to targeted therapies. JAMA Oncol 2015; 1: 40–49. [DOI] [PubMed] [Google Scholar]
- 23. Varghese AM, Arora A, Capanu M, et al. Clinical and molecular characterization of patients with cancer of unknown primary in the modern era. Ann Oncol 2017; 28: 3015–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Löffler H, Pfarr N, Kriegsmann M, et al. Molecular driver alterations and their clinical relevance in cancer of unknown primary site. Oncotarget 2016; 7: 44322–44329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ross JS, Sokol ES, Moch H, et al. Comprehensive genomic profiling of carcinoma of unknown primary origin: retrospective molecular classification considering the CUPISCO study design. Oncologist 2020; 26: e394–e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hayashi H, Takiguchi Y, Minami H, et al. Site‐specific and targeted therapy based on molecular profiling by next‐generation sequencing for cancer of unknown primary site: a nonrandomized phase 2 clinical trial. JAMA Oncol 2020; 6: 1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beaubier N, Bontrager M, Huether R, et al. Integrated genomic profiling expands clinical options for patients with cancer. Nat Biotechnol 2019; 37: 1351–1360. [DOI] [PubMed] [Google Scholar]
- 28. Groschel S, Bommer M, Hutter B, et al. Integration of genomics and histology revises diagnosis and enables effective therapy of refractory cancer of unknown primary with PDL1 amplification. Cold Spring Harb Mol Case Stud 2016; 2: a001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kundra R, Zhang H, Sheridan R, et al. OncoTree: a cancer classification system for precision oncology. JCO Clin Cancer Inform 2021; 5: 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McEvoy CR, Semple T, Yellapu B, et al. Improved next‐generation sequencing pre‐capture library yields and sequencing parameters using on‐bead PCR. Biotechniques 2020; 68: 48–51. [DOI] [PubMed] [Google Scholar]
- 31. The AACR Project GENIE Consortium . AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov 2017; 7: 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chakravarty D, Gao J, Phillips S, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017; 1: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang MT, Bhattarai TS, Schram AM, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov 2018; 8: 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering‐Integrated Mutation Profiling of Actionable Cancer Targets (MSK‐IMPACT): a hybridization capture‐based next‐generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015; 17: 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol 2010; 221: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H, Cao W. Pulmonary enteric adenocarcinoma: a literature review. J Thorac Dis 2020; 12: 3217–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. The Cancer Genome Atlas Research Network . Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017; 32: 185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014; 59: 1427–1434. [DOI] [PubMed] [Google Scholar]
- 42. Javle MM, Murugesan K, Shroff RT, et al. Profiling of 3,634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J Clin Oncol 2019; 37: 4087. [Google Scholar]
- 43. Silverman IM, Murugesan K, Lihou CF, et al. Comprehensive genomic profiling in FIGHT‐202 reveals the landscape of actionable alterations in advanced cholangiocarcinoma. J Clin Oncol 2019; 37: 4080. [Google Scholar]
- 44. Greco FA, Oien K, Erlander M, et al. Cancer of unknown primary: progress in the search for improved and rapid diagnosis leading toward superior patient outcomes. Ann Oncol 2012; 23: 298–304. [DOI] [PubMed] [Google Scholar]
- 45. Horlings HM, van Laar RK, Kerst JM, et al. Gene expression profiling to identify the histogenetic origin of metastatic adenocarcinomas of unknown primary. J Clin Oncol 2008; 26: 4435–4441. [DOI] [PubMed] [Google Scholar]
- 46. Pauli C, Bochtler T, Mileshkin L, et al. A challenging task: identifying patients with cancer of unknown primary (CUP) according to ESMO guidelines: the CUPISCO trial experience. Oncologist 2021; 26: e769–e779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abraham J, Heimberger AB, Marshall J, et al. Machine learning analysis using 77,044 genomic and transcriptomic profiles to accurately predict tumor type. Transl Oncol 2021; 14: 101016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pavlidis N. Optimal therapeutic management of patients with distinct clinicopathological cancer of unknown primary subsets. Ann Oncol 2012; 23: 282–285. [DOI] [PubMed] [Google Scholar]
- 49. Monzon FA, Lyons‐Weiler M, Buturovic LJ, et al. Multicenter validation of a 1,550‐gene expression profile for identification of tumor tissue of origin. J Clin Oncol 2009; 27: 2503–2508. [DOI] [PubMed] [Google Scholar]
- 50. Tang M, Abbas HA, Negrao MV, et al. The histologic phenotype of lung cancers is associated with transcriptomic features rather than genomic characteristics. Nat Commun 2021; 12: 7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herpel E, Rieker RJ, Dienemann H, et al. SMARCA4 and SMARCA2 deficiency in non‐small cell lung cancer: immunohistochemical survey of 316 consecutive specimens. Ann Diagn Pathol 2017; 26: 47–51. [DOI] [PubMed] [Google Scholar]
- 52. Concepcion CP, Ma S, LaFave LM, et al. Smarca4 inactivation promotes lineage‐specific transformation and early metastatic features in the lung. Cancer Discov 2022; 12: 562–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yakirevich E, Perrino C, Necchi A, et al. NF2 mutation‐driven renal cell carcinomas (RCC): a comprehensive genomic profiling (CGP) study. J Clin Oncol 2020; 38: 726. [Google Scholar]
- 54. Wei EY, Chen YB, Hsieh JJ. Genomic characterisation of two cancers of unknown primary cases supports a kidney cancer origin. BMJ Case Rep 2015; 2015: bcr2015212685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Escudier B. Combination therapy as first‐line treatment in metastatic renal‐cell carcinoma. N Engl J Med 2019; 380: 1176–1178. [DOI] [PubMed] [Google Scholar]
- 56. Tang TT, Konradi AW, Feng Y, et al. Small molecule inhibitors of TEAD auto‐palmitoylation selectively inhibit proliferation and tumor growth of NF2‐deficient mesothelioma. Mol Cancer Ther 2021; 20: 986–998. [DOI] [PubMed] [Google Scholar]
- 57. Priestley P, Baber J, Lolkema MP, et al. Pan‐cancer whole‐genome analyses of metastatic solid tumours. Nature 2019; 575: 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wong SQ, Fellowes A, Doig K, et al. Assessing the clinical value of targeted massively parallel sequencing in a longitudinal, prospective population‐based study of cancer patients. Br J Cancer 2015; 112: 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nakken S, Fournous G, Vodák D, et al. Personal cancer genome reporter: variant interpretation report for precision oncology. Bioinformatics 2018; 34: 1778–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017; 19: 4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shen R, Seshan VE. FACETS: allele‐specific copy number and clonal heterogeneity analysis tool for high‐throughput DNA sequencing. Nucleic Acids Res 2016; 44: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cameron DL, Schröder J, Penington JS, et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res 2017; 27: 2050–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mayakonda A, Lin DC, Assenov Y, et al. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 2018; 28: 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Volcano plot showing significantly mutated features by cancer types using the GENIE pan‐cancer cohort
Figure S2. Diagnostically useful mutated features per cancer type using the GENIE pan‐cancer cohort
Figure S3. Supportive genomic features and IHC staining for latent primary and clinicopathology‐resolved CUPs
Table S1. Latent primary and clinicopathology‐resolved CUP clinicopathological and molecular data
Table S2. Clinicopathology‐unresolved CUP clinicopathological and molecular data
Table S3. NanoString custom gene panel
Table S4. Gene expression classifier cancer classes
Table S5. NanoString test set accuracy/specificity/sensitivity
Table S6. GEP classification performance on 188 known origin metastatic cancer samples
Table S7. DNA sequencing SureSelect custom gene panel
Table S8. GENIE grouped cancer classes
Table S9. Frequency of genomic events in GENIE cancer classes
Table S10. GENIE cancer classes significant associated genes
Table S11. Putatively assigned TOO diagnoses for latent primary and clinicopathology‐resolved CUPs and OncoTree classifications of clinicopathology‐unresolved CUPs
Table S12. Actionable genomic events in CUP defined by CUPISCO criteria
Table S13. NanoString gene counts of tissue of origin genes from known origin metastatic tumours (MET) and CUP tumours
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding authors on reasonable request. The NanoString count data are provided as supplementary material, Table S13.