

Abstract
Background:
Evidence of the impact of genetic diagnosis on medical management in individuals with previously unexplained epilepsy is lacking in the literature. Our goal was to determine the impact of genetic diagnosis on medical management in a cohort of individuals with early-onset epilepsy.
Methods:
We performed detailed phenotyping of individuals with epilepsy who underwent clinical genetic testing with an epilepsy panel and/or exome sequencing at Boston Children’s Hospital between 2012 and 2019. We assessed the impact of genetic diagnosis on medical management.
Results:
We identified a genetic etiology in 152 of 602 (25%) individuals with infantile- or childhood-onset epilepsy who underwent next-generation sequencing. Diagnosis impacted medical management in at least one category for 72% of patients (110 of 152) and in more than one category in 34%. Treatment was impacted in 45% of individuals, including 36% with impact on antiseizure medication choice, 7% on use of disease-specific vitamin or metabolic treatments, 3% on pathway-driven off-label use of medications, and 10% on discussion of gene-specific clinical trials. Care coordination was impacted in 48% of individuals. Counseling on a change in prognosis was reported in 28% of individuals, and 1% of individuals had a correction of diagnosis. Impact was documented in 13 of 13 individuals with neurotypical development and in 55% of those with epilepsy onset after age two years.
Conclusion:
We demonstrated meaningful impact of genetic diagnosis on medical care and prognosis in over 70% of individuals, including those with neurotypical development and age of epilepsy onset after age two years.
Keywords: Epilepsy, Next-generation sequencing, Whole exome sequencing, Precision treatment, Development
Introduction
Early-onset epilepsies are common neurological disorders that occur in approximately seven of 10,000 infants per year,1,2 and genetic factors are thought to play a major role in cases without a clear structural or metabolic etiology.1,3–6 Single-gene etiologies can be diagnosed in approximately 25% to 30% of individuals with unexplained epilepsy, and in up to 55% to 80% of those with neonatal-onset epilepsy and developmental and epileptic encephalopathies (DEEs).3–9 In the more common epilepsies, heritability data suggest polygenic contribution to risk, but there is also evidence of higher burden of rare variants in known epilepsy genes.10,11
Epilepsy gene panels and exome sequencing (ES) have been increasingly utilized for precision clinical diagnosis in epilepsy. Beyond the benefits of diagnosis and related genetic counseling, specific treatment approaches are indicated for an increasing number of genetic epilepsies.12–18 Genetic diagnosis may also lead to more specific information regarding prognosis, such as rates of sudden unexpected death in epilepsy or a progressive disease course, or the need to screen for associated conditions (e.g., renal or cardiac disease).13,14,19,20 However, evidence of the impact of genetic diagnosis on management of epilepsy in a clinical setting to establish the utility of genetic testing in epilepsy is a critical area of unmet need, highlighted in a systematic review of genetic testing for the epilepsies.9,15,19–21
In this study, we sought to assess the impact of genetic diagnosis on medical management, as well as to determine phenotypic predictors associated with diagnosis and medical management impact.
Materials and Methods
Cohort and genetic testing
This is a retrospective cohort study approved by the Boston Children’s Hospital (BCH) Institutional Review Board. We evaluated a cohort of individuals seen at BCH who had a clinical epilepsy gene panel result from GeneDx, the major commercial laboratory with whom BCH contracted during this time frame, between June 2012 and January 2019. These individuals were identified from a list provided by GeneDx. Inclusion criteria included epilepsy gene panel report and diagnosis of epilepsy22,23 confirmed by retrospective medical record review. A subset of individuals underwent either clinical or research ES. Clinical ES was based on clinician referral, typically following nondiagnostic initial genetic testing. Research exomes through the Children’s Rare Disease Initiative or the BCH Epilepsy Genetics Program were offered for individuals with unexplained epilepsy, including those with a negative clinical genetic evaluation, those for whom insurance did not cover clinical testing, and epilepsies for which clinical genetic testing was not standard of care (e.g., benign epilepsy syndromes).24
Genetic testing classification
Epilepsy panel or ES results were considered diagnostic if variants identified on initial testing or reanalysis were interpreted by the treating team to explain the patient’s epilepsy.24 The majority of diagnostic variants were pathogenic or likely pathogenic based on classification per American College of Medical Genetics (ACMG) Guidelines.25 The exceptions to this rule were as follows: (1) the laboratory classification was variant(s) of unknown significance (VUS) or variant(s) in candidate genes, but literature after the laboratory report led to reclassification as disease-associated and upgrade of the variant(s) to (likely) pathogenic; and (2) the variant remained a VUS but the result was considered diagnostic by expert clinical diagnosis with unique clinical findings not captured by ACMG criteria (Table 1). Epilepsy panels and ES were considered nondiagnostic if they were negative, included VUS other than the aforementioned ones, or identified variants in gene(s) not consistent with the patient’s epilepsy phenotype or the condition’s mode of inheritance.
Table 1.
Race and ethnicity data for 602 individuals with an epilepsy panel with or without exome sequencing, sent through BCH between June 2012 and January 2019.
| Racial Category | Hispanic or Latino | Not Hispanic or Latino | Unknown/Not Reported | Total n (%) |
|---|---|---|---|---|
| American Indian or Alaskan Native | 1 | 2 | 0 | 3 (0.5) |
| Asian | 1 | 40 | 1 | 42 (7.0) |
| Black or African American | 2 | 30 | 8 | 40 (6.6) |
| Native Hawaiian or Other Pacific Islander | 1 | 3 | 0 | 4 (0.7) |
| White (includes both European origin and Middle East and North African origin, per NIH definition) | 31 | 361 | 33 | 425 (70.6) |
| More than one racial category | 2 | 7 | 0 | 9 (1.5) |
| Unknown or not Reported | 33 | 14 | 32 | 79 (13.1) |
| Total n (%) | 71 (11.8) | 457 (75.9) | 74 (12.3) | 602 |
We additionally collected data on chromosomal microarray results and other diagnoses (e.g., hypoxic-ischemic encephalopathy) contributing to epilepsy, both genetic and nongenetic.
Clinical phenotyping
To determine potential clinical predictors of diagnostic yield, we identified clinical features prevalent in DEEs from medical records. We collected seizure data, including epilepsy type, seizure patterns and types, electroclinical syndrome, and electroencephalography (EEG) encephalopathy pattern, according to International League Against Epilepsy standards.22,23 Epileptic encephalopathy patterns as mentioned in EEG clinical reports were categorized as burst suppression, hypsarrhythmia (full or modified), generalized slowing with multifocal and/or generalized epileptiform activity, slow spike and wave ± fast activity, electrical status epilepticus in sleep, and other (Table S1). We additionally collected demographic, developmental, and examination features (Tables 2 and 3, Table S1).
Table 2.
Rationale for diagnosis of 8 individuals with variant(s) of unknown significance (VUS) determined to be causative of epilepsy.
| ID | Gene | Variant | ACMG Classif. | Zygosity | Inheritance | Rationale for Diagnosis |
|---|---|---|---|---|---|---|
| B0257 | ALG11 | c.1402C>T, p.Arg468Cys | VUS | Hom | Both maternally and paternally inherited |
|
| B0272 | GABRG2 | c.1000G>A, p.Ala334Thr | VUS | Het | Maternally inherited |
|
| B0542 |
NHLRC1
NHLRC1 |
c.656G>A, p.Trp219* c.451G>T, p.Val151Phe |
LPATH VUS |
CH | Unknown |
|
| B0046 |
NRXN1
NRXN2 |
c.2686C>T, p.Arg896Trp c.3176G>A, p.Arg1059Gln |
LPATH VUS |
CH | Maternally inherited Paternally inherited |
|
| B0625 |
POLG
POLG |
c.2243G>C, p.Trp748Ser c.3356T>C, p.Leu1119Pro |
PATH VUS |
CH | Maternally inherited Unknown |
|
| B0069 | SCN1A | c.986G>T, p.Gly329Val | VUS | Het | Unknown |
|
| B0225 | SCN1A | c.418A>G, p.Thr140Ala | VUS | Het | Unknown |
|
| B0101 | SLC12A5 | c.983A>G, p.Asn328Ser | VUS | Hom | Both maternally and paternally inherited |
|
Abbreviations: ACMG = American College of Medical Genetics, CH = compound heterozygous, EEG = Electroencephalogram, EIEE = Early Infantile Epileptic Encephalopathy, GTC = generalized tonic clonic seizures, Het = Heterozygous, HGMD = Human Gene Mutation Database, Hom = Homozygous, LPATH = Likely pathogenic, PATH = pathogenic, VUS = variant of uncertain significance.
Table 3.
Diagnostic yield by phenotypic features. Frequency of key phenotypic features in relation to yield of epilepsy panel and exome testing (n=602, columns 3–4) and to yield of exome after a non-diagnostic epilepsy panel (n=183, columns 5–6).
| Phenotype | Phenotypic categories | Yield diagnostic panel +/− exome n (row%) |
P value, Chi-square or Fisher’s exact | Yield diagnostic exome after non-diagnostic panel n (row%) |
P value, Chi-square or Fisher’s exact |
|---|---|---|---|---|---|
| Total cohort | n=602$, diagnostic in 152 | n=183$$, diagnostic in 54 | |||
| Sex | Female Male |
71 (25.5) 81 (25) |
0.8793 | 27 (29.7) 27 (29.4) |
0.9619 |
| ASD (if ≥3y) | Yes No |
25 (22.9) 83 (21.9) |
0.8184 | 14 (28.6) 27 (25.2) |
0.6602 |
| ID (if ≥5y) | Yes No |
52 (25.9) 14 (8.3) |
<0.0001* | 25 (34.3) 0 |
<0.0001** |
| Developmental delay (if ≥ 2y) | Global delay Delay in one area None |
105 (31.7) 15 (20.8) 13 (8.8) |
<0.0001 | 45 (38.1) 3 (13.6) 0 |
<0.0001 |
| Developmental regression (if ≥ 3y) | Yes, with epileptic encephalopathy Yes, independent of seizures/change in EEG Yes, unknown or other setting No |
23 (22.6) 7 (36.8) 4 (19.1) 75 (21.4) |
0.4455 | 12 (27.3) 3 (33.3) 3 (30) 23 (24.5) |
0.8691 |
| Head size | Microcephaly Macrocephaly Normal |
26 (30.6) 6 (19.4) 118 (25.2) |
0.4085 | 15 (46.9) 4 (36.4) 35 (25) |
0.0427 |
| Systemic malformations | Yes No |
13 (33.3) 139 (24.7) |
0.2295 | 8 (47.1) 46 (27.7) |
0.0957 |
| Movement Disorder | Yes No |
29 (31.5) 122 (24) |
0.1269 | 14 (43.8) 40 (26.5) |
0.0518 |
| DEE | Yes No |
96 (30.2) 56 (19.9) |
0.0037 | 43 (37.1) 11 (16.4) |
0.0032 |
| Dysmorphic features | Yes No |
27 (32.5) 125 (24.2) |
0.1044 | 16 (59.3) 38 (24.5) |
0.0003 |
| Abnormal metabolic result | Yes No |
22 (42.3) 129 (23.8) |
0.0033 | 12 (60) 41 (25.6) |
0.0015 |
| CVI (if ≥ 2y) | Yes No |
31 (34.1) 96 (21.7) |
0.0114 | 14 (35.9) 32 (24.8) |
0.1735 |
| Muscle tone | Abnormal Normal |
99 (34.7) 53 (16.7) |
<0.0001 | 46 (43) 8 (10.5) |
<0.0001 |
| Epilepsy age of onset ≤ 2y | Yes No |
130 (33.3) 22 (10.5) |
0.0001 | 48 (37.8) 6 (10.7) |
0.0002 |
| Epilepsy type | Focal Generalized Mixed |
54 (27.6) 35 (17.8) 55 (32.7) |
0.0048 | 14 (25.5) 15 (25.4) 19 (34.6) |
0.6501 |
| Family history | Consanguinity Epilepsy, first degree relative Notable extended FH of epilepsy |
13 (23.6) 20 (29.4) 34 (23) |
0.5155# 0.4219 0.5920 |
8 (44.4) 8 (34.8) 9 (20.5) |
0.2772## 0.6055 0.1354 |
DEE = Developmental and epileptic encephalopathy; Y = year(s). Features with missing data >10% noted:
12% missing
14% missing
21% missing
16% missing
Numbers for cohorts with age cutoffs: ≥ 2 years = 544, ≥ 3 years = 498, ≥5 years = 418
Numbers for cohorts with age cutoffs: ≥ 2 years = 171, ≥ 3 years = 158, ≥5 years = 140
The concept of DEE recognizes that “in infants presenting with severe early-onset epilepsy, neurodevelopmental comorbidity may be attributable to both the underlying cause and to the adverse effects of uncontrolled epileptic activity.”22,26,27 In this cohort of individuals with early-onset epilepsy with suspected or confirmed genetic etiology and related developmental impairments in whom other etiologies for epilepsy were excluded, we designated those with a combination of epilepsy onset <18 years, encephalopathy pattern on EEG, and developmental delay (DD) or intellectual disability (ID) as having DEE. We defined status epilepticus as continuous seizure or multiple seizures without recovery, lasting for 30 minutes or longer.28 Cortical visual impairment was defined as “visual dysfunction in the absence of ocular or anterior visual pathway abnormalities.”29
Medical management
The electronic medical records of individuals with positive genetic test results (n = 152) were reviewed by an MD independent of the BCH clinical Epilepsy Genetics Program (I.H.) to establish individualized impact on medical management and verified by a pediatric neurologist with expertise in epilepsy genetics (H.E.O.). We identified four categories of medical management: treatment impact, which includes choice of antiseizure medications (ASMs), gene-specific vitamin/metabolic treatments, pathway-driven off-label use of medications, and disease/gene-specific clinical trials; care coordination, which includes medical management and monitoring for disease-associated features, including specialist referrals (except genetics), surveillance through diagnostic testing, and referrals to disease-specific clinics; change in prognosis; and correction of diagnosis (Table 4).
Table 4.
Impact of genetic diagnosis on individual medical management for 152 individuals with infantile or childhood-onset epilepsy
| Type of impact on medical management | n (%) of 152 individuals with genetic diagnosis | Case examples |
|---|---|---|
| Impact in any category | 110 (72.4) | |
| Impact in more than one category | 51 (33.6) | |
| Treatment impact | 69 (45.4) | |
| Choice of anti-seizure medications | 54 (35.5) |
|
| Vitamin or metabolic treatments, gene-specific (including ketogenic diet) | 10 (6.6) |
|
| Pathway-driven off-label use of medications | 5 (3.3) |
|
| Disease/gene-specific clinical trials or investigational new drug (IND) use | 15 (9.9) |
|
| Care coordination (Medical management and monitoring for disease-associated features) | 73 (48.0) |
|
| Change in prognosis | 42 (27.6) |
|
| Correction of diagnosis, for those with a diagnosis prior to genetic testing | 2 (1.3) |
|
Statistical analysis
We first evaluated the overall diagnostic yield of clinical testing for epilepsy by gene panel with or without ES.
Second, we evaluated diagnostic yield for phenotypic subsets based on clinical features, including those with autism spectrum disorder (ASD), ID, global developmental delay (GDD), neurotypical development (defined as no diagnosis of DD, ASD, or ID), developmental regression, head size, systemic malformations, movement disorders, and those with DEE. Pearson chi-square test or Fisher exact test was performed to examine the distribution of phenotypic features on diagnostic yield.
Third, we used multivariate logistic regression modeling to assess (1) clinical factors predictive of genetic diagnosis by gene panel, ES, or a combination; (2) clinical factors predictive of diagnostic ES after negative epilepsy gene panel; and (3) clinical factors predictive of impact of diagnosis on medical management. Statistical analysis was done using SAS, version 9.4 (SAS Institute Inc., Cary, NC, USA). The outcome variable for the first analysis was diagnostic panel and/or ES, and in the second analysis, diagnostic ES. Early age of epilepsy onset (particularly in the neonatal and infantile period up to two years) and developmental disorder are previously described predictors of genetic diagnosis5,16,30–32 and were considered in the multivariate model as possible predictors of impact of genetic diagnosis on medical management. Based on this prior literature and consistent with the International League Against Epilepsy age cutoff of two years for the neonatal and infantile period,26 we used epilepsy onset less than or equal to two years in the model. Predictor variables were analyzed using univariate testing of Wald tests, as well as box-and-whisker plot and Wilcoxon rank-sum test for epilepsy age of onset. Each predictor was examined alone in a univariate logistic model. A multivariate logistic regression model was then built, keeping age of epilepsy onset as a key predictor in each model based on preliminary data and prior literature.5,16,30–33 A threshold P value of 0.25 was used to select candidate predictor variables, and we then used a backward selection approach including variables with a P value of < 0.05 in the final model.
Results
Our cohort consists of 602 individuals (278 female) with early-onset epilepsy (seizure onset median 1.0 years; interquartile range [IQR] 4 months, 3.0 years), who had a clinical epilepsy gene panel result at BCH between June 2012 and January 2019. The most commonly used gene panels included the comprehensive epilepsy panel (n = 411) and infantile epilepsy panel (n = 129) (Table S1). A subset with nondiagnostic panel results subsequently underwent clinical or research ES (n = 183) (Fig 1). Our cohort includes 318 of 600 individuals (53.0%, 2 unknown) meeting criteria for DEE,22,26,27 defined as age of epilepsy onset <18 years, encephalopathy pattern on EEG, and DD and/or ID. Epilepsy was generalized in 32.8%, focal in 32.6%, mixed in 28.0%, and unknown in 6.7%. Of individuals with diagnosed electroclinical syndromes (n = 190), the most common included West syndrome (n = 36, 18.9%) and Lennox-Gastaut syndrome (n = 29, 15.3%). Median age at last follow-up was 8.2 years (IQR 4.2 years, 12.9 years). Our cohort is 70.6% white and 75.9% non-Hispanic (Table 2).
FIGURE 1.
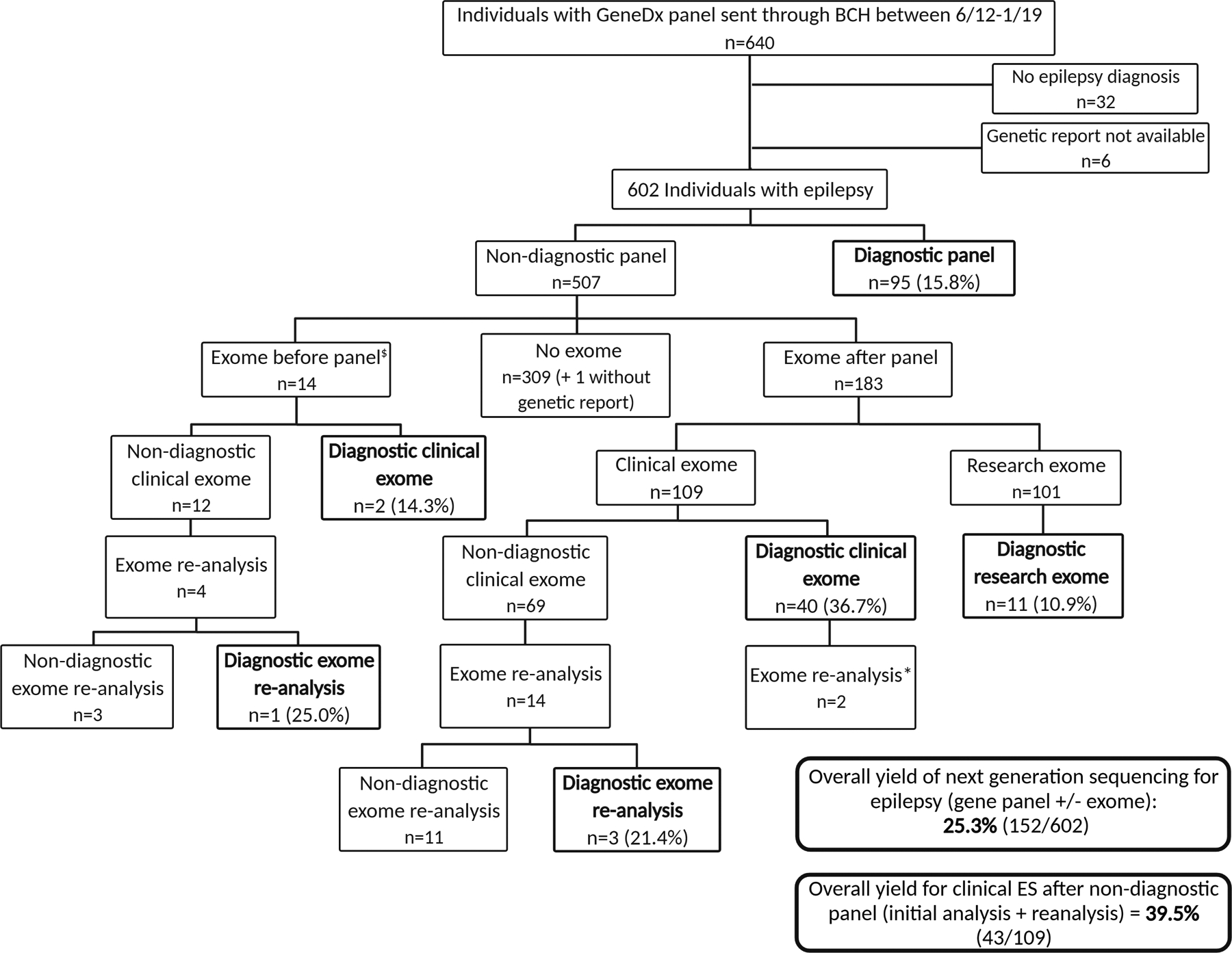
Study flowchart. Inclusion/exclusion criteria, genetic testing algorithm, and results of our cohort. Figure created with BioRender.com. *For two individuals, exome reanalysis was done after clinical exomes with diagnostic findings in AGO1 and CHAT to look for further contribution to epilepsy phenotypes that were more severe than expected. No additional findings were identified. $In 13 of 14, panel was done to expand genetic evaluation when exome was nondiagnostic or did not fully explain the phenotype, including full coverage of updated epilepsy genes and copy number variant analysis. In one additional patient, rapid exome was sent while panel was pending due to severity of the medical condition.
The overall diagnostic yield of clinical testing for epilepsy by gene panel with or without ES was 25.3% (152 of 602 individuals) (Fig 1). This included 15.8% (95 of 602) yield for epilepsy gene panel, with highest yield for the STAT panel (five of 14) and infantile panel (39 of 129) and no definite increase from 2012 to 2019; 36.7% (40 of 109) yield for clinical ES initial analysis after nondiagnostic panel; 21.4% (three of 14) yield for clinical ES reanalysis after non-diagnostic clinical ES; and 10.9% (11 of 101) diagnosed by clinical confirmation of research ES result after initial negative clinical testing (Fig 1). Causative variants were pathogenic or likely pathogenic according to ACMG classification except for those of eight individuals for whom genetic variants remained VUS after ACMG reclassification (Table 1). Eight individuals (1.3%) had a partial or whole gene deletion or duplication that would not have been identified by ES in the time frame of the study.
Of the 152 individuals with a genetic diagnosis, genes in which causative variants were most commonly identified on both panels and exomes included SCN1A (n = 19), KCNQ2 (n = 12), PRRT2 (n = 8), SCN2A (n = 7), CDKL5 (n = 6), CHD2 (n = 5), TSC2 (n = 4),STXBP1 (n = 4), and SCN8A (n = 4) (Fig 2). Of the 54 individuals with a diagnostic exome (clinical or research-based) following a non-diagnostic gene panel, causative variants were reported in the following genes in more than one individual: DYNC1H1 (n = 3), ALG11 (n = 2), CHD2 (n = 2), FRRS1L (n = 2), NEXMIF (n = 2), PACS2(n = 2), SCN8A (n = 2), and WDR45 (n = 2) (Fig 3). Additional genetic details are provided in Table S3.
FIGURE 2.
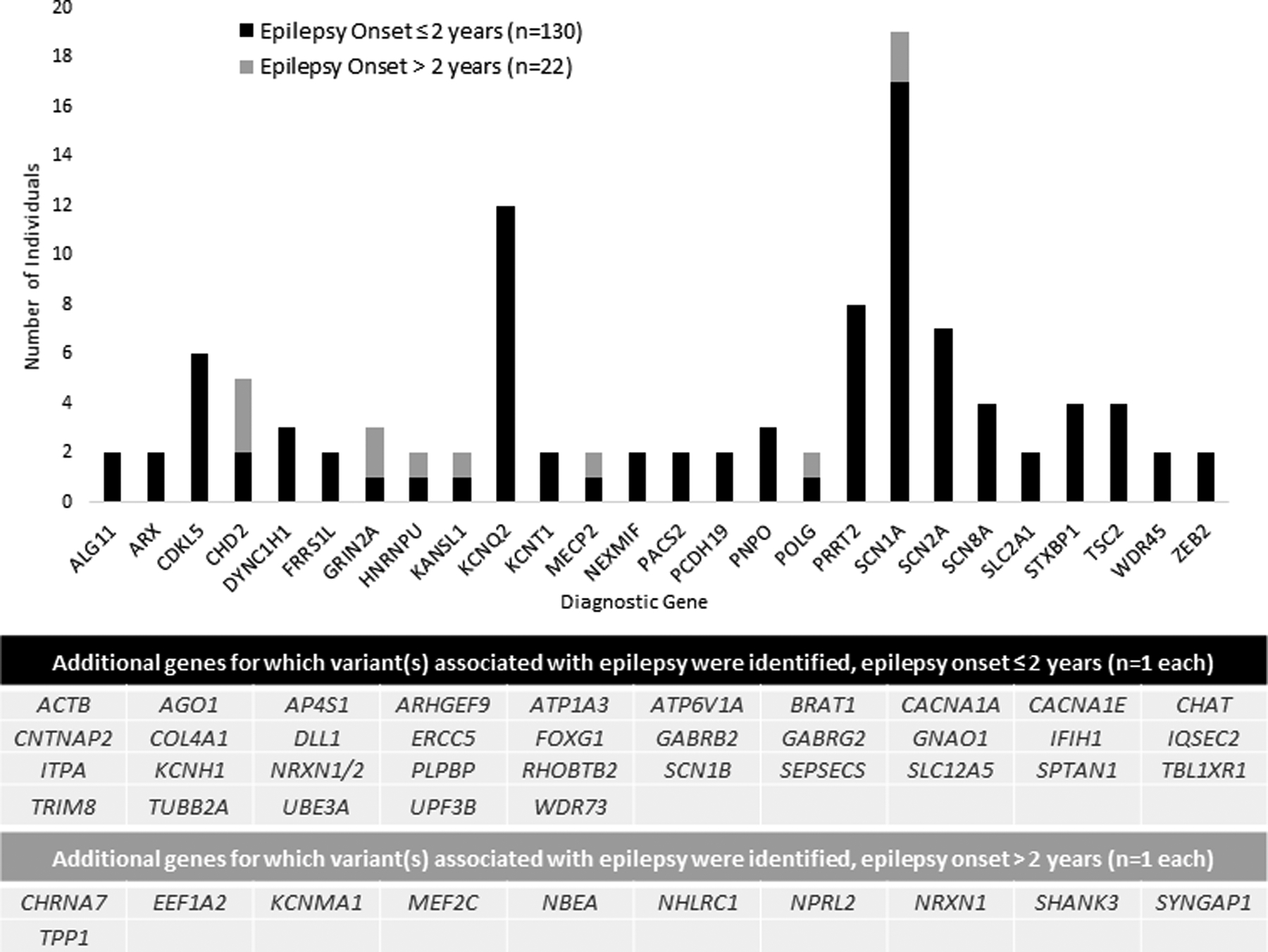
Implicated genes for epilepsy diagnosis in a clinical cohort of 602 individuals with infantile- or childhood-onset epilepsy from Boston Children’s Hospital. Genes with variants identified on panel or exome thought to clinically explain epilepsy for 152 individuals, 130 with epilepsy onset less than or equal to two years of age. The most common genes for which causative variants were identified included SCN1A (n = 19), KCNQ2 (n = 12), PRRT2 (n = 8), SCN2A (n = 7), CDKL5 (n = 6), CHD2 (n = 5), TSC2 (n = 4) STXBP1 (n = 4), SCN8A (n = 4) CHD2 and SCN8A, which were later added to panels, were identi fied on both panels and exomes in our series.
FIGURE 3.
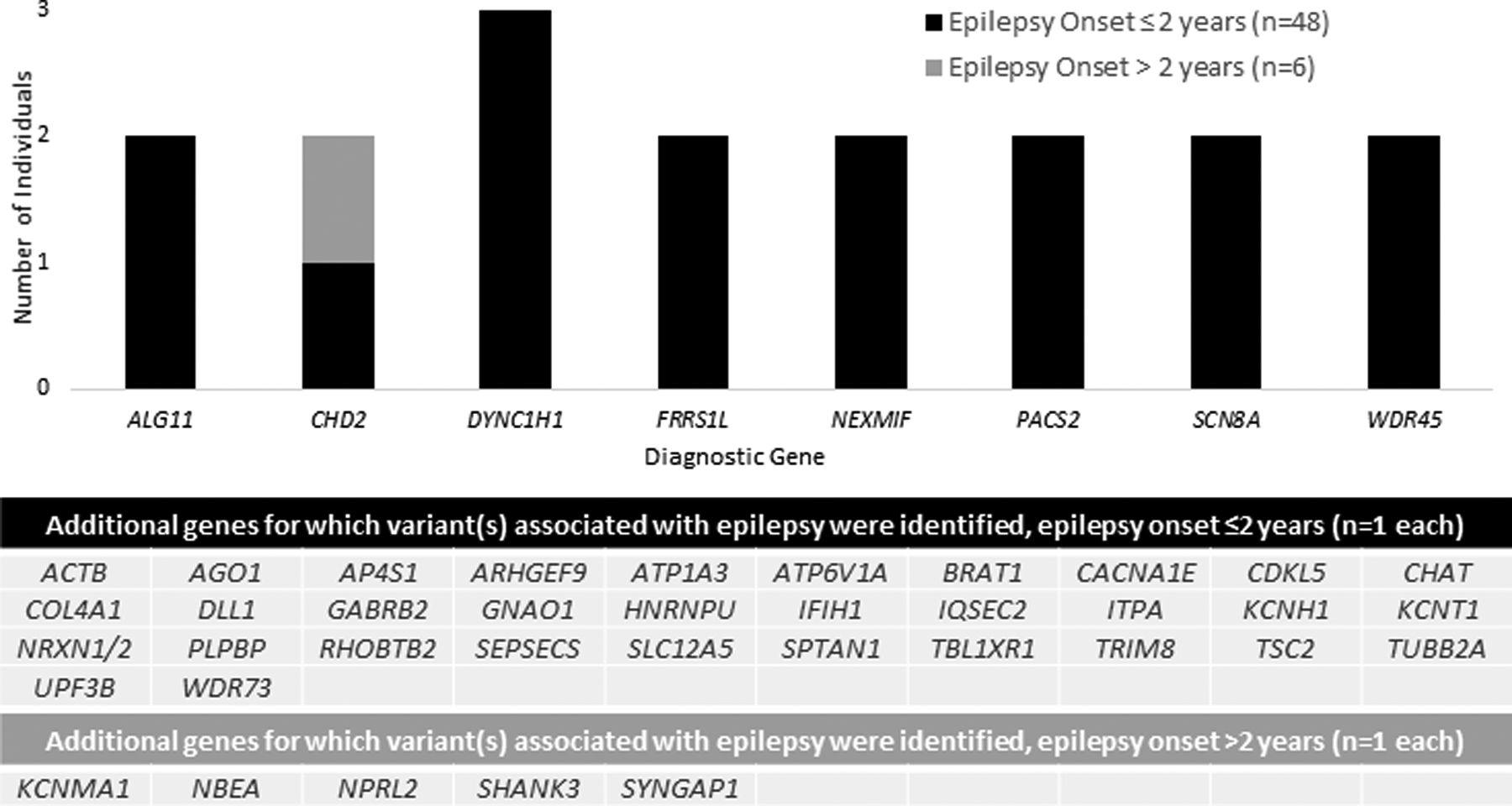
Implicated genes for epilepsy diagnosis in a clinical cohort of 183 individuals with infantile- or childhood-onset epilepsy from Boston Children’s Hospital and initial negative epilepsy gene panel testing. Genes with causative variants identified on clinical or research exome, following a nondiagnostic panel, in 54 individuals with epilepsy, 48 with age of epilepsy onset less than or equal to two years. The genes for which causative variants were identified in more than one individual included DYNC1H1 (n = 3), ALG11(n = 2), CHD2 (n = 2), FRRS1L (n = 2), NEXMIF (n = 2), PACS2 (n = 2), SCN8A (n = 2), and WDR45 (n = 2).
Although this is largely a nonmalformation-related epilepsy cohort, in the case of variants in genes known to be associated with brain malformations, including DYNC1H1, FOXG1, KANSL1, TSC2, TUBB2A, and ZEB2, the patterns of malformation identified were consistent with expectations for the genetic diagnoses. In two individuals with DYNC1H1 variants, a malformation was identified only on detailed review after genetic diagnosis (one after statistical analysis, recognized only following research review for this study). There were also malformations identified in individuals with nonmalformation-related genetic diagnoses, including SCN1A and AGO1 diagnoses with cortical dysplasia, SCN8A diagnosis with mild inferior vermian hypoplasia, GRIN2A diagnosis with prominent thalamic adhesion, and CDKL5 diagnosis with a cortical heterotopia.
Yield of diagnosis by epilepsy gene panel and/or ES by predefined phenotypic features included the following: ASD (if age greater than or equal to three years) 22.9%, ID (if age greater than or equal to five years) 25.9%, GDD (if age greater than or equal to two years) 31.7%, neurotypical development (if age greater than or equal to two years) 8.8%, and DEE 30% (Tables 3 and S2). Comparing the cohort of individuals with ES after a nondiagnostic panel (n = 183) with those without ES after a negative panel (n = 310), age of epilepsy onset was less than or equal to two years in 69.4% and 57.1%, respectively; ASD in 31.4% and 18.6%, respectively; GDD in 69.4% and 50.9%, respectively; abnormal muscle tone (hypotonia, spasticity, dystonia, or mixed patterns) in 58.5% and 37.7%, respectively; and cortical visual impairment in 23.2% and 11.9%, respectively.
Using multivariate regression analysis, positive or negative predictors of genetic diagnosis by epilepsy gene panel, ES, or a combination included age of epilepsy onset less than or equal to two years (odds ratio [OR], 3.82; 95% confidence interval [CI], 2.19,6.66), malformation of brain development (OR, 0.27; CI 0.15, 0.51), focal motor seizures (OR, 2.29; CI, 1.43, 3.65), and DD (OR, 2.64; CI, 1.47, 4.73) (Tables 5 and S2). Controlling for malformation of brain development, seizure types, and DD, the odds of a diagnostic test result were 3.82 times higher (CI, 2.19, 6.66) in individuals with epilepsy onset less than or equal to two years compared with individuals with later epilepsy onset.
Table 5.
Predictors of genetic diagnosis by panel or exome. Multivariate logistic regression analysis for phenotypic predictors of A) diagnostic epilepsy gene panel or exome sequencing and B) diagnostic compared to non-diagnostic exome sequencing after a non-diagnostic epilepsy gene panel. Variables with p-value of <0.25 on univariate testing were initially included in the model, then backwards selection was used to reach the final model with variables having p <0.05 and age of seizure onset ≤ 2 years as a required variable.
| Phenotypic predictor variable | Odds ratio (95% confidence interval) for relationship with diagnostic panel or exome | P-value, Wald test |
|---|---|---|
| A. Multivariate logistic regression model for predictors of diagnostic epilepsy gene panel or exome, combined | ||
| Age of epilepsy onset ≤ 2 years | 3.82 (2.19, 6.66) | <0.0001 |
| Malformation of brain development (Y/N) | 0.27 (0.15, 0.51) | <0.0001 |
| Focal non-motor | 1.15 (0.73, 1.83) | 0.55 |
| Developmental delay, global or in one area | 2.64 (1.46, 4.73) | 0.0012 |
| B. Multivariate logistic regression model for predictors of diagnostic exome after a negative epilepsy gene panel | ||
| Age of epilepsy onset ≤ 2 | 2.23 (0.81, 6.19) | 0.12 (required in the model) |
| Tone | 3.02 (1.20, 7.59) | 0.002 |
| GDD | 4.45 (1.14, 17.31) | 0.03 |
All individuals with diagnostic ES after a negative epilepsy panel had DD in at least one area, so GDD was used for statistical modeling. Using multivariate regression analysis, features predictive of genetic diagnosis by ES after a negative epilepsy panel were abnormal muscle tone (OR, 3.02; CI, 1.20, 7.59) and GDD (OR, 4.45; CI, 1.14, 17.31) (Table 5 and S2). Age of epilepsy onset was required in the model but was below significance in the final model (P = 0.12; OR, 2.23; CI, 0.81, 6.19). Controlling for age of epilepsy onset less than or equal to two years and GDD, the odds of diagnostic ES after a negative panel were 3.02 times higher (CI 1.20, 7.59) in individuals with abnormal muscle tone compared with individuals with normal muscle tone. There is evidence of colinearity of age of epilepsy onset less than or equal to two years with GDD and abnormal muscle tone. If GDD is removed from the model, age of epilepsy onset less than or equal to two years is a significant predictor of diagnostic ES (OR, 3.21; CI, 1.22, 8.44), P. = 0.02, and the P value for tone decreases to 0.0003 (Table 5).
Genetic diagnosis had a direct impact on medical management for 72.4% of individuals with a diagnostic result (110 of 152), including 33.6% (51 of 152) with impact in more than one category. No impact on medical management was documented for 22.3% of individuals (34 of 152), including for four individuals who were deceased before the availability of genetic results. Median length of follow-up after genetic diagnosis was 1.7 years (IQR 0.3, 3.8). Impact on medical management was unknown in eight individuals who were not followed after diagnosis (5.2%). According to specific medical management categories, 45.4% of individuals were found to have impact on treatment (69 of 152), including 35.5% on choice of ASMs, 6.6% on use of disease-specific vitamin or metabolic treatments (including ketogenic diet), 3.3% on pathway-driven off-label use of medications, and 9.9% on discussion of disease or gene-specific clinical trials between family and clinicians or investigational new drug use. Examples are provided in Table 4 and detailed for all participants in Table S4.
We documented impact on care coordination in 48.0% of individuals (73 of 152), including surveillance for other disease-associated features (including non-neurological features), additional disease-specific diagnostic testing, specialist referrals, and referrals to disease-specific clinics (e.g., tuberous sclerosis complex, CDKL5 deficiency disorder) (Tables 4 and S5). Although we did not systematically collect information on outcomes of testing and specialist evaluation as some individuals were not followed at BCH, there were instances in which referrals led to identification of disease-associated features (e.g., TRIM8 variant led to nephrology referral and diagnosis of nephrotic syndrome and focal segmental glomerulosclerosis). In 27.6% of individuals, we documented counseling related to change in prognosis or life expectancy, such as discussion of degenerative diseases (e.g., NHLRC1-related Lafora disease), risk for early mortality or sudden unexpected death in epilepsy (e.g., BRAT1, SCN1A), and optimistic long-term outcomes (e.g., PRRT2). Although most participants lacked a diagnosis before genetic testing, 1.3% of individuals had a correction of diagnosis, replacing a nonspecific diagnosis of mitochondrial disorder with a precise genetic diagnosis (GNAO1, CACNA1A). For 73.0% of individuals (n = 111), there exists a gene-specific family organization related to their diagnosis. Of those with a genetic diagnosis and data available on medical management impact (n = 144), impact was documented in 55% of those with epilepsy onset greater than two years (12 of 22), compared with 80% of those with onset at less than or equal to two (98 of 122) (chi-square test, P = 0.0088). Medical management was impacted in 73.5% of those with a developmental disorder (DD, ASD, and/or ID) (97 of 131) and all 13 individuals with neurotypical development (Fisher exact test, P = 0.0386).
Discussion
Overall, as genetic testing practices evolved from 2012 to 2019, we identified genetic diagnoses in 152 (25.3%) of 602 individuals with early-onset epilepsy of whom over 70% had direct impact on medical management. Expanding on prior reports establishing yield of genetic diagnosis in epilepsy with limited information on direct medical impact, this study emphasizes direct clinical utility.9 Furthermore, although medical impact was higher in those with epilepsy onset under two years, impact was found in over 50% of individuals with epilepsy onset after age two years and in all 13 individuals with neurotypical development.
We observed age of onset less than two years, DD, and focal motor seizures to be phenotypic features associated with higher odds of genetic diagnosis. Furthermore, we demonstrate meaningful additional yield of clinical ES after a negative epilepsy gene panel (39.5%), especially in the setting of GDD and abnormal muscle tone. The yield of diagnosis by panel or ES in our study was similar to the yield reported in prior literature that included broad cohorts of individuals with epilepsy.9,34 Genes for which causative variants were most commonly identified in our cohort closely matched the genes most commonly implicated in epilepsy in other series of next-generation sequencing.34,35 Genes for which variants were identified on ES after a negative panel in this cohort similarly included genes coding for proteins and pathways known to be linked to epilepsy and brain malformations and consistent with prior literature, although in many cases not typically included in epilepsy panels (e.g., congenital disorders of glycosylation).30,34
In recent years, clinical genetic testing in neurology has been shifting from panel testing to an exome-first approach at some larger academic centers with adequate genetic counseling resources, as supported by literature for neurodevelopmental disorders and by a cost-effectiveness study in epilepsy genetics.36,37 Findings on panel testing that would not have been expected to be identified by ES were seen in the eight of 602 individuals in our cohort (1.3%) with copy number variants identified by epilepsy panel. Between 2012 and 2019, Copy nuber variant analysis was not routinely performed on exomes, but it is now an emerging practice. Thus, given the significant additional yield of ES over epilepsy gene panel, improvement of certainty in variant classification with a trio ES approach, the ability to reanalyze data over time, and the falling costs of ES, we suggest ES as an appropriate first-line test in the diagnosis of unexplained epilepsy. If ES is performed and not revealing of a definite etiology, we suggest consideration of exon-level copy number variant assessment.
This is the first study to demonstrate the direct medical impact of genetic diagnosis in epilepsy in a clinical cohort, providing evidence of consequent tailored care in more than 70% of individuals, including impact on treatment, care coordination, prognosis, and correction of diagnosis. Although prior studies have reported the impact of ES on medical management in broader pediatric populations with a variety of diagnoses, as well as theoretical impact of genetic diagnoses on medical management in epilepsy,19–21,34,36,38–42 these reports did not evaluate individualized medical management impact as we have done in this current large clinical series. Over one-third of participants had documented impact on ASM choice, and we demonstrate increasing impact of pathway-driven treatments and experimental therapies with improved precision diagnosis. Results are consistent with impacts of the highest-yield genes on recommended treatment approaches38–42 as well as novel approaches to precision treatment and identification of new genes in treatable pathways.12–18,43 Reported percentages may be an underestimate, because some individuals were seen only for a consult at BCH but were followed elsewhere. Furthermore, lack of documented medical impact in 30 individuals (excluding four participants who passed away before genetic results) does not preclude the possibility of future impact for these recently reported genetic diagnoses when more information becomes available.
In addition to the documented impact on medical management, families routinely received genetic counseling consultations, which include discussion of recurrence risk, reproductive planning, cascade testing of at-risk family members, and referral to gene-specific family advocacy organizations. For those with a genetic diagnosis without an existing family advocacy organization, our Epilepsy Genetics Program informs families about the Rare Epilepsy Network (REN), Syndromes Without A Name (SWAN), Genome-Connect, and applicable social media groups and utilizes GeneMatcher.
Although prior reports have shown a correlation between age of epilepsy onset, presence of comorbid neurodevelopmental features, and diagnostic yield,5,16,30–32 these features have not been previously studied in relation to medical impact. Our results show that although early age of epilepsy onset (less than or equal to two years) may be associated with an increased likelihood of medical impact, impact was nevertheless noted in >50% of those with onset greater than two years. Impact was significant both in those with developmental disorders and neurotypical development.
There are limitations inherent to our retrospective study design, including inability to systematically collect outcomes of surveillance testing and referrals. EEG patterns and developmental diagnoses were reported as documented in medical records. Because DEE was only defined during the course of this study and not consistently documented in medical records, we defined a triad of features that are suggestive of this diagnosis in this particular population with epilepsy of suspected genetic etiology. Our sample size for evaluating predictors of genetic diagnosis by ES after a negative panel was low, and additional predictors may have reached significance with higher power. Phenotypic features such as ID and ASD were not identified as significant predictors of genetic diagnosis, possibly related to limited sample size; requirement for age at last follow-up after five years and three years, respectively; and confounding with other significant predictors. Age of epilepsy onset under two years was not a significant predictor of diagnostic ES after negative gene panel likely due to confounding with abnormal tone and GDD. We suspect that focal motor seizures were a significant predictor of diagnosis because it is a common seizure type in neonatal-onset epilepsy. There was clinical selection bias in those who went on to ES for individuals with a broader neurodevelopmental disorder, and the yield of ES for the entire cohort may have been lower. Yield of research exomes was lower than for clinical exomes, likely explained by ongoing data analysis and selection bias due to inclusion of individuals with benign epilepsy syndromes and others with prior negative clinical genetic testing.
This study was conducted during a time period characterized by a shifting landscape of genetic testing in epilepsy. Although this study included multiple types of epilepsy panels with gene lists that varied in size and over time, the yield did not substantially increase over the time of the study and the highest yield was seen in the panel types sent in individuals with infantile-onset epilepsy. Testing in the earlier years, however, was likely influenced by selection bias, with more severely affected individuals with epilepsy sequenced, compared with more recent years, when genetic testing in epilepsy has been applied to a broader group of patients with epilepsy at BCH. We used data primarily from one diagnostic laboratory due to institutional contracting with our hospital. We do not have reason to believe results would be significantly different with different diagnostic laboratories, but it may have been higher with use of larger panels. Last, the majority of individuals in our cohort were white and non-Hispanic. Similar evaluations in other race and ethnic groups would be beneficial.
By including individuals with an epilepsy gene panel rather than ES only or brain malformation panels, our cohort focused on those with nonmalformation-related epilepsy as the predominant phenotype. Brain malformations were a negative predictor of genetic diagnosis in our series, which can be explained by the fact that epilepsy panels are targeted to nonmalformation-related epilepsy. Malformations identified were consistent with genetic diagnosis or, rarely, found nonspecifically in association with nonmalformation-related epilepsy genes such as SCN1A as has been reported.38,44,45
In conclusion, our study demonstrates substantial impact of genetic diagnosis on medical management in individuals with epilepsy. The likelihood of a diagnostic genetic test was highest in individuals with DD, abnormal muscle tone, and focal motor seizures. Medical impact is relevant for both individuals with neuro-typical development and with developmental disorders, and regardless of age of seizure onset. This study supports the inclusion of genetic testing, ES in most clinical scenarios, as part of the standard evaluation for individuals with unexplained epilepsy as a means of achieving diagnostic precision and potentially informing clinical management.
Supplementary Material
Acknowledgments
We thank Shaye Moore and Elizabeth Jarvis for editing and formatting support. We thank GeneDx for their support of this research effort. This study was supported by the National Institute of Neurologic Disorders and Stroke (K23 NS107646-05, PI Olson) and the Children’s Rare Disease Cohort Initiative at Boston Children’s Hospital with administrative core support through Boston Children’s Hospital IDDRC (1U54HD090255). T.B. was supported by Ponzio Family Chair in Neurology Research/Children’s Hospital Colorado Foundation. A.P. was supported by the Boston Children’s Hospital Translational Research Program.
Potential conflicts of interest:
Isabel Haviland, Carolyn Daniels, Caitlin Greene, Jacqueline Drew, Jamie Love-Nichols, Lacey Smith, Bo Zhang, and Beth Sheidley: Nothing to report. Lindsay Swanson: Lindsay Swanson receives research support from the International Foundation for CDKL5 Research. Tim A. Benke: Dr. Benke conducts clinical trials sponsored by Rett Syndrome Research Trust, Neuren, Acadia, Ovid, and Marinus. He has received consulting fees from RettSyndrome.org, AveXis, Ovid, Takeda, and Marinus. Annapurna Poduri: Dr Poduri is a member of the SAB for TevardBio and for SYNGAP Research Fund (both without personal compensation). Heather Olson: Dr Olson received consulting fees from Takeda Pharmaceuticals and Zogenix regarding clinical trial design, Ovid Therapeutics regarding clinical trial results, Marinus Pharmaceuticals regarding CDKL5 Deficiency Disorder, and has done consulting for the FOXG1 Research Foundation.
Footnotes
Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.pediatrneurol.2022.10.006.
References
- 1.Eltze CM, Chong WK, Cox T, et al. A population-based study of newly diagnosed epilepsy in infants. Epilepsia. 2013;54:437–445. [DOI] [PubMed] [Google Scholar]
- 2.Wirrell EC, Grossardt BR, Wong-Kisiel LCL, Nickels KC. Incidence and classification of new-onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: a population-based study. Epilepsy Res. 2011;95:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg AT, Coryell J, Saneto RP, et al. Early-life epilepsies and the emerging role of genetic testing. JAMA Pediatr. 2017;171:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mercimek-Mahmutoglu S, Patel J, Cordeiro D, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56:707–716. [DOI] [PubMed] [Google Scholar]
- 5.Shellhaas RA, Wusthoff CJ, Tsuchida TN, et al. Profile of neonatal epilepsies: characteristics of a prospective US cohort. Neurology. 2017;89:893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rochtus A, Olson HE, Smith L, et al. Genetic diagnoses in epilepsy: the impact of dynamic exome analysis in a pediatric cohort. Epilepsia. 2020;61:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Gotway G, Pascual JM, Park JY. Diagnostic yield of clinical next-generation sequencing panels for epilepsy. JAMA Neurol. 2014;71:650–651. [DOI] [PubMed] [Google Scholar]
- 8.Allen AS, Berkovic SF, Cossette P, et al. , Epi4K Consortium, Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheidley BR, Malinowski J, Bergner AL, et al. Genetic testing for the epilepsies: a systematic review. Epilepsia. 2022;63:375–387. [DOI] [PubMed] [Google Scholar]
- 10.Epi4K consortium, Epilepsy Phenome/Genome Project. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16:135–143. [DOI] [PubMed] [Google Scholar]
- 11.Kjeldsen MJ, Corey LA, Christensen K, Friis ML. Epileptic seizures and syndromes in twins: the importance of genetic factors. Epilepsy Res. 2003;55:137–146. [DOI] [PubMed] [Google Scholar]
- 12.Axeen EJT, Olson HE. Neonatal epilepsy genetics. Semin Fetal Neonatal Med. 2018;23:197–203. [DOI] [PubMed] [Google Scholar]
- 13.Striano P, Minassian BA. From genetic testing to precision medicine in epilepsy. Neurotherapeutics. 2020;17:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perucca P, Perucca E. Identifying mutations in epilepsy genes: impact on treatment selection. Epilepsy Res. 2019;152:18–30. [DOI] [PubMed] [Google Scholar]
- 15.Symonds JD, Zuberi SM, Stewart K, et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019;142:2303–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson HE, Kelly M, LaCoursiere CM, et al. Genetics and genotype-phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol. 2017;81:419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amador A, Bostick CD, Olson H, et al. Modelling and treating GRIN2A developmental and epileptic encephalopathy in mice. Brain. 2020;143:2039–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Millichap JJ, Park KL, Tsuchida T, et al. KCNQ2 encephalopathy: features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. 2016;2:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iglesias A, Anyane-Yeboa K, Wynn J, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med. 2014;16:922–931. [DOI] [PubMed] [Google Scholar]
- 20.Matias M, Wusik K, Neilson D, Zhang X, Valencia CA, Collins K. Comparison of medical management and genetic counseling options pre- and post-whole exome sequencing for patients with positive and negative results. J Genet Couns. 2019;28:182–193. [DOI] [PubMed] [Google Scholar]
- 21.Valencia CA, Husami A, Holle J, et al. Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr. 2015;3:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the international league against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:522–530. [DOI] [PubMed] [Google Scholar]
- 24.Rockowitz S, LeCompte N, Carmack M, et al. Children’s rare disease cohorts: an integrative research and clinical genomics initiative. NPJ Genom Med. 2020;5:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association For Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63: 1349–1397. [DOI] [PubMed] [Google Scholar]
- 27.Specchio N, Curatolo P. Developmental and epileptic encephalopathies: what we do and do not know. Brain. 2021;144:32–43. [DOI] [PubMed] [Google Scholar]
- 28.Trinka E, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus–report of the ILAE task force on classification of status epilepticus. Epilepsia. 2015;56:1515–1523. [DOI] [PubMed] [Google Scholar]
- 29.Sakki HEA, Dale NJ, Sargent J, Perez-Roche T, Bowman R. Is there consensus in defining childhood cerebral visual impairment? A systematic review of terminology and definitions. Br J Ophthalmol. 2018;102:424–432. [DOI] [PubMed] [Google Scholar]
- 30.Stefanski A, Calle-López Y, Leu C, Pérez-Palma E, Pestana-Knight E, Lal D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: a systematic review and meta-analysis. Epilepsia. 2021;62: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trump N, McTague A, Brittain H, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Kong W, Gao Y, et al. Gene mutation analysis in 253 Chinese children with unexplained epilepsy and intellectual/developmental disabilities. PLoS One. 2015;10, e0141782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drew J, Greene C, Daniels C, et al. Yield and Clinical Predictors of Diagnosis with Exome Sequencing after a Non-diagnostic Epilepsy Panel in Pediatric Patients with Epilepsy. Washington: Poster Presented at American Epilepsy Society Annual Meeting, Seattle; 2020. [Google Scholar]
- 34.Symonds JD, McTague A. Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur J Paediatr Neurol. 2020;24:15–23. [DOI] [PubMed] [Google Scholar]
- 35.Lindy AS, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59:1062–1071. [DOI] [PubMed] [Google Scholar]
- 36.Srivastava S, Love-Nichols JA, Dies KA, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sánchez Fernández I, Loddenkemper T, Gaínza-Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: a meta-analysis and cost-effectiveness study. Neurology. 2019;92:e418–e428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knupp KG, Wirrell EC. Treatment strategies for dravet syndrome. CNS Drugs. 2018;32:335–350. [DOI] [PubMed] [Google Scholar]
- 39.Howell KB, McMahon JM, Carvill GL, et al. SCN2A encephalopathy: a major cause of epilepsy of infancy with migrating focal seizures. Neurology. 2015;85: 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolff M, Johannesen KM, Hedrich UBS, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140:1316–1336. [DOI] [PubMed] [Google Scholar]
- 41.Pisano T, Numis AL, Heavin SB, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. 2015;56:685–691. [DOI] [PubMed] [Google Scholar]
- 42.Lim Z, Wong K, Olson HE, Bergin AM, Downs J, Leonard H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: experience of >100 patients. Epilepsia. 2017;58:1415–1422. [DOI] [PubMed] [Google Scholar]
- 43.Olson HE, Kelly ML, Rosen-Sheidley B, Pearl P, Poduri A. Precision Treatment of KCNQ2 Encephalopathy with Ezogabine. In: Poster Presented at American Epilepsy Society Annual Meeting, Washington, DC. 2017. [Google Scholar]
- 44.Guerrini R, Striano P, Catarino C, Sisodiya SM. Neuroimaging and neuropathology of Dravet syndrome. Epilepsia. 2011;52:30–34. [DOI] [PubMed] [Google Scholar]
- 45.Barba C, Parrini E, Coras R, et al. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia. 2014;55:1009–1019. [DOI] [PubMed] [Google Scholar]
- 46.Rochtus AM, Trowbridge S, Goldstein RD, et al. Mutations in NRXN1 and NRXN2 in a patient with early-onset epileptic encephalopathy and respiratory depression. Cold Spring Harb Mol Case Stud. 2019;5, a003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgess R, Wang S, McTague A, et al. The genetic landscape of epilepsy of infancy with migrating focal seizures. Ann Neurol. 2019;86:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srivastava S, Olson HE, Cohen JS, et al. BRAT1 mutations present with a spectrum of clinical severity. Am J Med Genet A. 2016;170:2265–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.