

Abstract
Efzofitimod is a first-in-class biologic immunomodulator based on a naturally occurring splice variant of histidyl-tRNA synthetase (HARS) that binds to neuropilin-2 (NRP2). Preclinical data found high expression of NRP2 in sarcoidosis granulomas. Treatment with efzofitimod reduced the granulomatous inflammation induced by P. acnes in an animal model of sarcoidosis. An ascending dose study of efzofitimod in sarcoidosis with chronic symptomatic pulmonary disease found that treatment with efzofitimod was associated with improved quality of life with a trend towards reduced glucocorticoid use and stable to improved pulmonary function. These studies have led to a large Phase 3 trial of efzofitimod in symptomatic pulmonary sarcoidosis.
Keywords: sarcoidosis, NRP-2, treatment, mechanism of action
Introduction
Efzofitimod (also known as ATYR1923 or KRP-R120) is a novel Fc fusion protein in development for the treatment of interstitial lung disease (ILD), a group of immune mediated fibrotic lung conditions including sarcoidosis. Efzofitimod is comprised of an active moiety, a human 59 amino-acid protein, fused to the Fc region of human IgG1. The amino acid sequence of the active moiety of efzofitimod corresponds identically to the extracellularly active N-terminal domain (amino acids 2 to 60) of histidyl-tRNA synthetase (HARS) (1) (Figure 1).
Figure 1.
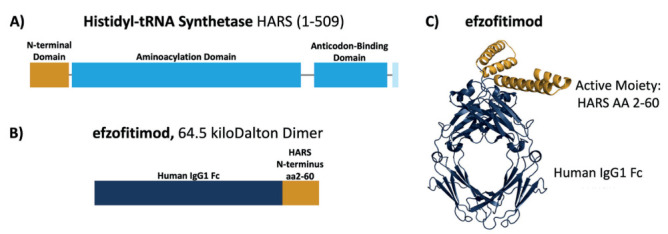
Domain organization of primary sequences of histidyl-tRNA synthetase (HARS) and efzofitimod. A schematic view of the domain organization of HARS (A) and efzofitimod (B). The ribbon structure of efzofitimod depicting the active moiety (HARS aa2-60) is shown in (C). Abbreviations: AA = amino acid; Fc = fragment crystallizable; HARS = histidyl-tRNA synthetase; IgG1 = immunoglobulin 1 (1).
HARS is one of a family of several aminoacyl tRNA synthetases (Figure 2 left panel)(2,3). The tRNA synthetases catalyze the esterification of a tRNA to its cognate (according to the genetic code) amino acid. Synthetases help to ensure accurate translation of the genetic code by using both highly accurate cognate substrate recognition and stringent proofreading of noncognate products. This canonical (intracellular) function is enabled by the presence of the anti-codon binding domain and the aminoacylation domain in each tRNA synthetase. As shown in figure 2 (center panel), fragments and splice variants of tRNA synthetases are also available extracellularly (4). This extracellular presence of tRNA synthetases is the focus of current research at aTyr.
Figure 2.
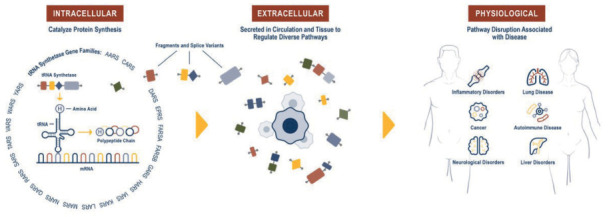
The intracellular (canonical) function (left panel), extracellular (novel) functions (center panel) and the possible physiological role for the tRNA synthetases (right panel) (4).
As with other tRNA synthetases, the gene for HARS also gives rise to a number of splice variants, and though most of these have lost their catalytic activity, they all retain the N-terminal domain (HARS amino acids 2-60) (5). This N-terminal domain, which is non-essential for the enzyme’s protein synthesis activity that is required in all living organisms, was appended to HARS during the evolutionary development of multicellular organisms and retained with high sequence identity across mammalian species. It is not found in lower organisms. One splice variant (SV9 – HARS amino acids 1 - 60), which encodes only the N-terminal domain of the protein, is enriched in human lung tissue (aTyr, data on file). Expression of this HARS splice variant is increased following inflammatory cytokine stimulation (IFN-γ and TNF-α, two key players in the initiation of lung inflammation and fibrosis) followed by subsequent secretion. This indicates that it is being regulated in response to local inflammation (aTyr, data on file). Furthermore, HARS, specifically the N-terminal domain, is targeted by autoantibodies in a rare autoimmune disorder, anti-Jo-1 syndrome. This syndrome is characterized by extensive activation and migration of immune cells into lung and muscle and is classically associated with the triad of ILD, myositis, and arthritis (3,6). HARS circulates in healthy individuals, but it is largely undetectable in the serum of anti-Jo-1-positive antisynthetase syndrome patients (3,7). It is hypothesized that the sequestration of HARS may play a causal role through disruption of its homeostatic immune-regulatory effects (8). The role of the N-terminal domain of HARS in inflammation and its association with ILD as part of the anti-synthetase syndrome led to its evaluation as a drug candidate.
The N-terminal domain itself has a short half-life in serum, thus it was fused with the Fc portion of human IGg1 to extend its half-life (about 10 days in humans). In solution, the efzofitimod molecule forms a homodimer, similar to other Fc fusion proteins.
Preclinical Pharmacology
Neuropilin-2 (NRP2) (9) was identified as the sole binding partner for efzofitimod through screening via a cell microarray system in which over 4,500 cell surface proteins are represented (Retrogenix Cell Microarray, Charles River). This screening approach identified two NRP2 isoforms (Neuropilin 2A and 2B) as the only convincing and specific binding partners of efzofitimod. Notably, efzofitimod is selective in that it does not exhibit binding to the closely related neuropilin-1 (NRP1) receptor. NRP2 is a cell surface receptor that is present on multiple immune cell types. NRP2 expression is often upregulated upon inflammatory insult, immune cell differentiation or stimulation. Growing evidence indicates that NRP2 plays an important role in myeloid cell biology influencing their activation and recruitment to inflammatory sites. For instance, NRP2 expression on alveolar macrophages has been shown to regulate airway inflammatory responses to inhaled lipopolysaccharide in mice (10). Blinded sample studies analyzing sarcoidosis granulomas from the skin and lung using in situ hybridization (ISH) and immunohistochemistry (IHC) methods demonstrated significantly increased NRP2 expression on macrophage populations within the granulomas (Figure 3A-C) (11). Studies have demonstrated that the target receptor of efzofitimod, NRP2, is expressed primarily on immune cells of myeloid lineage, such as monocytes, macrophages, and dendritic cells, and has been detected in lymphoid cells, such as T cells, albeit at lower levels than that of monocytes and macrophages (Figure 4) (12). NRP2 is particularly enriched on circulating monocytes from sarcoidosis patients (Figure 5).
Figure 3.
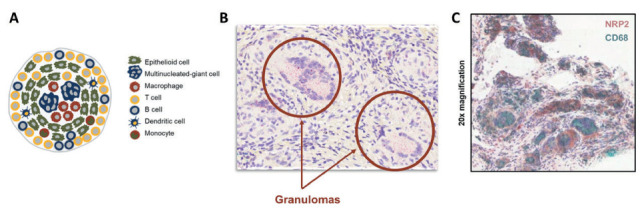
(A) Schematic of key immune cells that form sarcoidosis granulomas. (B) NRP2 mRNA expression measure by in situ hybridization (ISH). (C) NRP2 in both skin and lung tissue samples from sarcoidosis patients is highly expressed throughout granulomas but not in the surrounding tissue. Lung tissue from a sarcoidosis patient was co-stained for NRP2 and the CD68 macrophage marker. Granulomas in this sample were composed primarily of CD68+ macrophages that express NRP2 (11).
Figure 4.
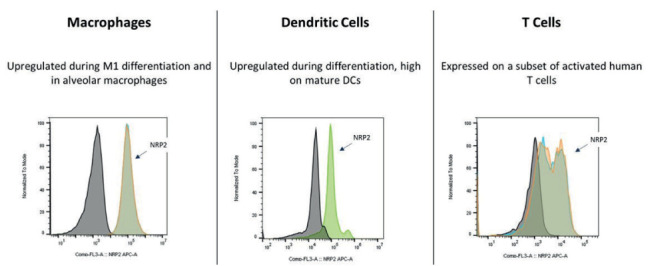
NRP2 expression on primary human cells: activated macrophages, differentiated dendritic cells, and T cells (12).
Figure 5.
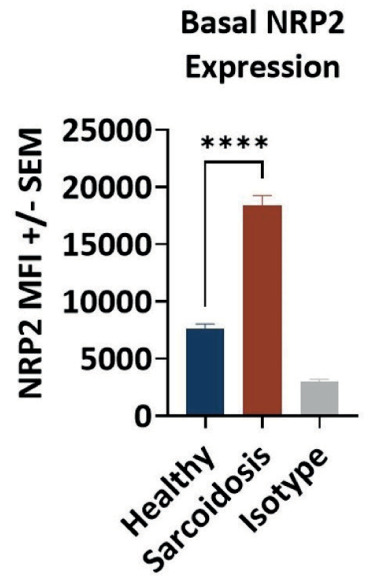
NRP2 expression is enriched on monocytes from sarcoidosis patients. Peripheral blood mononuclear cells from healthy donors (N=3) and sarcoidosis patients (N=5) were evaluated for CD14 and NRP2 expression by flow cytometry. NRP2 expression was determined by median fluorescence intensity (MFI) of NRP2 on CD14+
Following an inflammatory insult, there is a strong upregulation of NRP2 in inflammatory cells. When efzofitimod was introduced into mouse models of acute lung inflammation (ALI), recruitment of immune cells to the inflamed lungs was significantly inhibited, affecting cells of myeloid lineage, including primarily alveolar macrophages (Figure 6) (13). Thus, subsequent research has focused on elucidating the potential for efzofitimod to modulate the inflammatory response through myeloid cells. These findings indicate that efzofitimod could be a novel therapeutic approach to inflammation associated fibrotic diseases such as pulmonary sarcoidosis.
Figure 6.
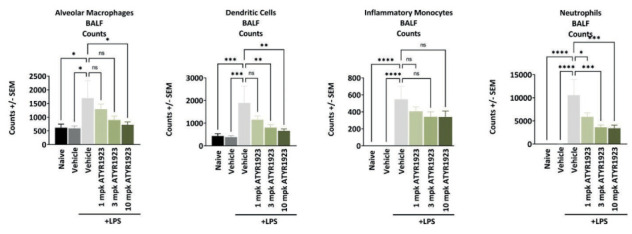
Reduction of immune cell infiltrates in bronchoalveolar lavage (BAL) and lung tissue from mice treated with efzofitimod in an Lipopolysaccharide (LPS) acute lung inflammation (ALI) model. BAL samples obtained by lavage using PBS and lung tissue homogenate obtained by enzymatic and mechanical tissue processing. Statistical analyses were performed using one-way ANOVA with Dunnett’s post-hoc test (alpha 0.05) (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001) (13).
A model of pulmonary granulomatosis generated by intratracheal administration of Propionibacterium acnes ( P. acnes) was used to assess potential therapeutic activity of efzofitimod in pulmonary granulomatous inflammation. P. acnes is hypothesized to be involved in the etiology of sarcoidosis (14,15) and the murine model resembles some clinical features of sarcoidosis such as pulmonary inflammation and formation of granulomas. Several key inflammatory and pro-fibrotic cytokines were reduced following efzofitimod treatment (16). Animals dosed with efzofitimod at 3 mg/kg had significantly reduced levels of IFN-γ, IL-6, and MCP-1/CCL2, when compared with the matched vehicle IV control (Figure 7). In addition, the therapeutic activity of efzofitimod was assessed in a mouse model of chronic hypersensitivity pneumonitis (CHP). An experimental model of CHP was induced by repeated intranasal administration of Saccharopolyspora rectivirgula (S. rectivirgula). Histological analysis (Day 22 when the study was terminated) revealed a significant reduction of the bronchus-associated lymphoid tissue area in efzofitimod-treated animals as compared with matched vehicle control. Furthermore, animals dosed with efzofitimod at 3 mg/kg had significantly reduced levels of IFN γ, IL-6, IP-10/CXCL10, and MCP-1/CCL2 when compared with matched vehicle IV control (Figure 8). Reduction of inflammatory biomarkers in these key mouse models of interstitial lung disease provides evidence of the ability for efzofitimod to reduce the inflammatory response in the lung in response to different stimuli.
Figure 7.
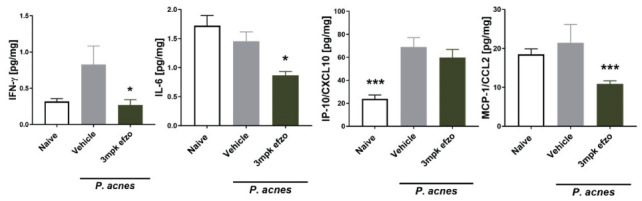
Treatment with efzofitimod lowers several fibrosis-associated and Inflammatory proteins in murine model for experimental sarcoidosis induced by P. acnes. Lung homogenates were subjected to bead-based multiplex ELISAs (Luminex platform). One-Way ANOVA with Dunn’s multiple comparisons test – efzofitimod buffer IV control (All data are shown as mean ± SEM. * p < 0.05; *** p < 0.001) (11).
Figure 8.
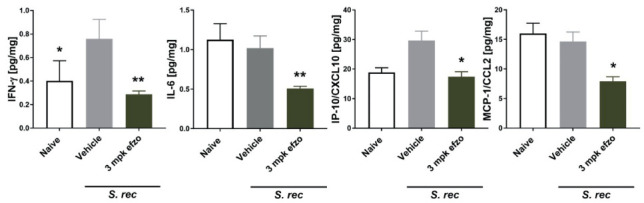
Treatment with efzofitimod lowers several fibrosis-associated and inflammatory proteins in murine model for chronic hypersensitivity pneumonitis induced by S. rectivirgula (S. rec). Lung homogenates were subjected to bead-based multiplex ELISAs (Luminex platform). One-Way ANOVA with Dunn’s multiple comparisons test with efzofitimod buffer IV as control (All data are shown as mean ± SEM. * p < 0.05; ** p < 0.01) (11).
In addition to demonstrating efficacy in the P. Acnes and S. rectivirgula models, efzofitimod has also demonstrated a significant reduction in histological measures of fibrosis and inflammation in other models of ILD, including the bleomycin model of lung fibrosis (data on file).
Clinical Studies
Sarcoidosis is a granulomatous disease characterized by granulomatous inflammation (17). The clinical outcome is variable, with up to a third of patients witnessing spontaneous resolution without therapy and a third of patients progressing to chronic disease (18). Of those with chronic disease, fibrosis can occur in the lung and other parts of the body. Overall, less than ten percent of sarcoidosis patients die from the disease. Mortality attributed to sarcoidosis is usually associated with advanced fibrosis of the lung or the presence of pulmonary hypertension (19) or cardiac disease.
As opposed to other progressive pulmonary fibrotic conditions, advanced pulmonary sarcoidosis has evidence of continued inflammation as well as progressive fibrosis. Patients with fibrotic pulmonary sarcoidosis often have positive positron emission tomography (PET) scans, indicating ongoing inflammation (20). Additionally explants of lungs from sarcoidosis patients undergoing lung transplant often show active granulomas (21).
The observation of increased neuropilin-2 in the granulomatous model suggest that efzofitimod may be effective treatment against advanced pulmonary sarcoidosis (Figure 9). Figure 10 shows the granulomatous response and targets for treatment in sarcoidosis (adapted from Obi et al (22)). Several of the currently available treatments for sarcoidosis are targeted for specific proteins (Figure 10A), such as monoclonal antibodies binding tumor necrosis factor (anti-TNF). Other therapies are more diffuse anti-inflammatory treatments, such as glucocorticoids and methotrexate (Figure 10B). Both strategies have been successful. However, given the multiple pathways of the inflammatory response within the granuloma, to date anti-inflammatory therapies have been more widely used.
Figure 9.
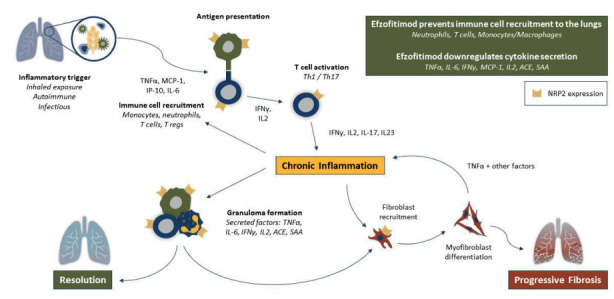
Proposed effect of efzofitimod on various inflammatory cells involved in the initiation and perpetuation of sarcoidosis.
Figure 10.
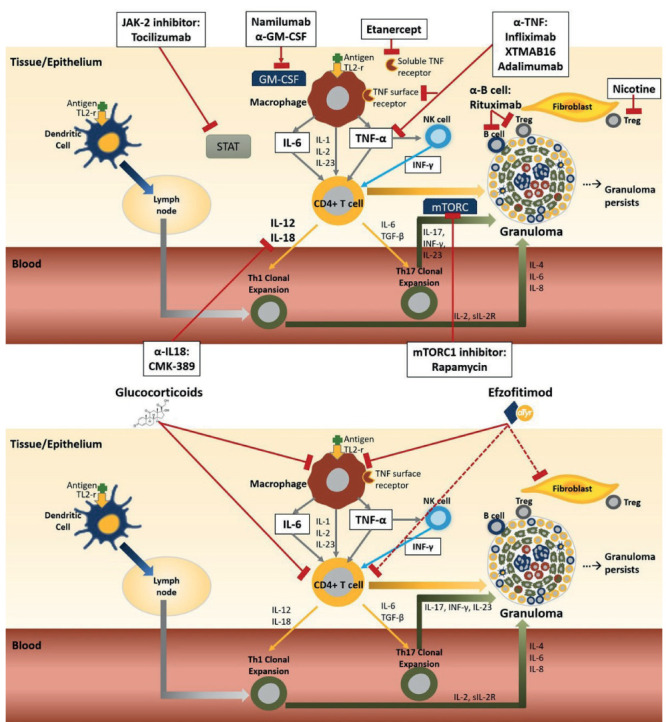
Inhaled antigen comes into contact with cells at the epithelial layer. Activation of both macrophages and dendritic cells occurs through the toll-like receptor-2 (TLR-2). The dendritic cells transport the antigen across the epithelium to the lymph node, where it is processed leading to differentiation and clonal expression of T helper cells (Th1 and Th17). The antigen also stimulates macrophages on the surface of the epithelium and leads to the release of tumor necrosis factor alpha (TNF-α) and several other pro-inflammatory cytokines including IL-1, IL-2. IL-6 and IL-23. TNF-α crosses epithelial layer where it activates tissue macrophages and natural killer (NK) cells. Activated NK releases interferon gamma (IFN-γ) which act in concert with TNF-α to upregulate the inflammatory response by attracting CD4+ cells, monocytes and Tregs to the site of inflammation. TNF-α along with the other pro-inflammatory cytokines also activates CD4+ cells and causes them to further differentiate down the Th1 and Th17 effector pathways. The activated macrophages and clonal Th1/Th17 cells form the core of the granuloma. Other cells in the granuloma include T regulatory cells (Treg) and B cells. The Th17 pathway has been more implicated in chronic disease as has impaired Treg cell function and persistent accumulation of serum amyloid A within the granuloma. Biologic therapies for sarcoidosis specifically targeted for one or two cytokines or cell types (Figure 10A). Targets of therapy for two broad based anti-inflammatory therapies for sarcoidosis: glucocorticoids and efzofitimod (Figure 10B). Dashed lines indicate potential targets. Adapted from Obi et al 32.
Glucocorticoids, especially prednisone, remain the cornerstone of treatment for symptomatic sarcoidosis (18). Many patients have received prednisone for years. The toxicity of prolonged glucocorticoid use has been well recognized in many diseases, including sarcoidosis (23,24). Treatment strategies which are steroid sparing have been developed. In particular, methotrexate, azathioprine, leflunomide, and mycophenolate have become standard second line treatments to reduce prednisone dosage. However, most of the data comes from retrospective studies (18).
It has been recommended that treatment of pulmonary sarcoidosis follow evidence-based guidelines. These recently published guidelines emphasized the need for more treatment options for treating advanced, chronic pulmonary sarcoidosis. In pursuit of a new treatment for pulmonary sarcoidosis, a 24-week placebo-controlled ascending dose study of efzofitimod was performed (NCT03824392). The study was designed to evaluate safety and prove preliminary efficacy (25). Evaluation of therapy included steroid sparing, quality of life, and impact on lung function. Efzofitimod was well tolerated at all three doses studied (1, 3, and 5 mg/kg). Significant improvement in quality of life compared to placebo was seen at the higher doses.
Steroid sparing remains an important target for second- and third-line therapy in chronic pulmonary and extra-pulmonary disease. The efzofitimod study included a specific protocol for steroid withdrawal. Those treated with either 3 or 5 mg/kg efzofitimod had a higher rate of steroid withdrawal compared to placebo.
In addition to steroid sparing, improved health related quality of life (HRQoL) with stable to improved lung function are the major goals of treatment for symptomatic pulmonary sarcoidosis. Table 1 provides details of placebo-controlled trials in symptomatic pulmonary sarcoidosis in which prednisone dose reduction was studied. Some of these studies employed a prednisone tapering schedule. In all studies, the dose of prednisone was reduced for both the active and placebo arms of the study. Three studies, including efzofitimod, reported a higher rate of steroid sparing with active drug versus placebo. However, only the efzofitimod study was associated with significant improvement in some patient reported quality of life measures. Neither the methotrexate nor pentoxifylline studies showed any tendency for improved forced vital capacity (FVC), while there was a trend to improvement in FVC in the efzofitimod trial.
Table 1.
Clinical trials for Reducing Glucocorticoids in Symptomatic Sarcoidosis
| Drug studied | Number of patients Drug/Placebo | Background therapy | Change in FVC % predicted | Prednisone Taper | Change in Quality of Life | Comments |
|---|---|---|---|---|---|---|
| Methotrexate 27 | 9 Methotrexate; 6 Placebo |
Prednisone | 11% | Significant prednisone tapering |
Not reported | No significant change in FVC absolute value. Excluded from analysis 7 patients treated started on methotrexate and withdrew from study |
| Fluticasone 28 | 10 Fluticasone; 12 Placebo |
Prednisone | No significant change | No significant difference | No difference in SF-36 | Less cough reported with fluticasone. No objective measurement reported. |
| Pentoxifylline 29 | 12 Pentoxifylline; 13 Placebo |
Prednisone | No significant difference | yes | Not reported | Significant steroid sparing only at 8 and 10 months; Significantly less flares with prednisone tapering; Did not report FVC results |
| Golimumab 30 | 42 Golimumab; 44 Placebo |
Prednisone plus other agents | -0.87% | no change in prednisone | No significant changes | Same placebo patients for golimumab and ustekinumab trial |
| Ustekinumab 30 | 46 Ustekinumab; 44 Placebo | Prednisone plus other agents | -2.17% | no change in prednisone | No significant changes | Same placebo patients for golimumab and ustekinumab trial |
| Efzofitimod 31 | 27 Efzofitimod; 12 Placebo; |
Prednisone plus other agents | 2.81 % at 3 mg/kg; 3.30 at 5 mg/kg | Dose response effect seen for prednisone tapering | Statistically significant (p value <0.05) improvement in KSQ Lung, KSQ GH and FAS |
Underpowered to demonstrate significant changes in FVC |
In the higher dose arm of efzofitimod (5 mg/kg), there was statistically significant improvement (p value <0.05) in the HRQoL, mainly Fatigue Assessment Scale (FAS), King’s Sarcoidosis Questionnaire (KSQ) Lung (L) and KSQ General Health (GH). Figure 11 shows the changes in FAS, KSQ-L, and KSQ-GH. The minimal clinically important difference (MCID) for FAS and KSQ-L is 4 points while that for KSQ GH is 8 points (26). In the higher dose arm (5 mg/kg), significant improvement in these HRQoL measures was reported well above previously determined MCID.
Figure 11.
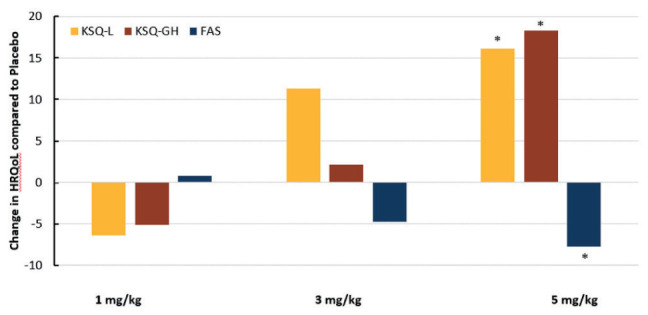
The change in quality-of-life instruments Kings Sarcoidosis Questionnaire Lung (KSQ-L), Kings Sarcoidosis Questionnaire General Health (KSQ-GH), and Fatigue Assessment Score (FAS) after 24 weeks for three treatment regimens versus placebo. The minimal clinically important difference (MCID) for FAS and KSQ-L is 4 points while that for KSQ GH is 8 points. There was significant improvement for KSQ-L and KSQ-GH (as indicated by higher score) and significant reduction in fatigue as shown by a lower FAS. For KSQ-L and KSQ-GH, an increase in score means improvement, while for FAS, a decrease in score means improvement (* p-value <0.05) (31).
Changes in pulmonary function included analysis of changes in the forced vital capacity (FVC). The difference in least square means (from a mixed effects model adjusted for baseline value) for FVC % predicted at 24 weeks when compared to that of placebo was -0.08% for the 1 mg/kg group. For the higher dosages, there was a dose dependent trend for improvement at 3 mg/kg (2.81%) and 5 mg/kg (3.30%) when compared to that of placebo.
These encouraging results have led to an ongoing Phase 3 trial of efzofitimod for symptomatic pulmonary sarcoidosis. Patients who have persistent dyspnea despite glucocorticoid therapy will be randomized to either active drug or placebo in a double blind study. Prednisone will be withdrawn per protocol. The trial will be evaluating steroid sparing and improvement of quality of life while evaluating stability to improvement of lung function during the prednisone withdrawal. In addition, further information regarding safety of the drug will be obtained.
Conclusion
Efzofitimod is a first in class biologic with significant anti-inflammatory properties. Sarcoidosis is a diffuse granulomatous disease in which chronic inflammation leads to significant morbidity and some mortality. Preclinical data found that efzofitimod reduced inflammation and key biomarkers for fibrosis in sarcoidosis animal models. An ascending dose study provided preliminary evidence for safety. Additionally, patients receiving active drug versus placebo had significant improvement in sarcoidosis specific health related quality of life instruments. This was associated with a trend towards steroid sparing and improved lung function. A larger study adequately powered to address all three outcomes of therapy is underway (NCT05415137).
Disclosures:
Dr. Baughman is consultant for the Ann Theodore Foundation Abbvie, Actelion, aTyr, Bellerophon, Kinevant, Mallinckrodt, Novartis, Novartis, Xentria and is on speaker’s bureau for Boehringer Ingelheim, Mallinckrodt, United Therapeutics. Dr. Niranjan has received payments from aTyr Pharma Inc. for consultancy. Dr. Walker, Dr. Burkart, Dr. Chong, Dr. Siefker, Dr. Paz, Dr. Sun, Dr. Nangle, and Dr. Shukla are employed and own stock in aTyr Pharma Inc. Dr. Förster and Dr. Muders have received research collaboration payments from aTyr Pharma. Dr. Lower is consultant for Xentria and is on speaker’s bureau for Gilead. Dr. Culver has received grants from Mallinckrodt Pharmaceuticals, Boehringer Ingelheim, the Foundation for Sarcoidosis Research (FSR), and the Ann Theodore Foundation; serves as a consultant for Roivant Sciences and Boehringer Ingelheim; serves as a member of the Adjudication committee for Pliant Therapeutics; serves as President of the World Association for Sarcoidosis and Other Granulomatous Disorders (WASOG).
References
- 1.Freist W, Verhey JF, Rahlmann A, et al. Histidyl-tRNA synthetase. Biol Chem. 1999;380(6):623–646. doi: 10.1515/BC.1999.079. [DOI] [PubMed] [Google Scholar]
- 2.Rajendran V, Kalita P, Shukla H, et al. Aminoacyl-tRNA synthetases: Structure, function, and drug discovery. Int J Biol Macromol. 2018;111:400–414. doi: 10.1016/j.ijbiomac.2017.12.157. doi: 10.1016/j.ijbiomac.2017.12.157. Epub;%2018 Jan 3.:400-414. [DOI] [PubMed] [Google Scholar]
- 3.Galindo-Feria AS, Notarnicola A, Lundberg IE, et al. Aminoacyl-tRNA Synthetases: On Anti-Synthetase Syndrome and Beyond. Front Immunol. 2022;13:866087. doi: 10.3389/fimmu.2022.866087. doi: 10.3389/fimmu.2022.866087. eCollection;%2022.:866087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubio Gomez MA, Ibba M. Aminoacyl-tRNA synthetases. RNA. 2020;26(8):910–936. doi: 10.1261/rna.071720.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lo WS, Gardiner E, Xu Z, et al. Human tRNA synthetase catalytic nulls with diverse functions. Science. 2014;345(6194):328–332. doi: 10.1126/science.1252943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R, Zhao Y, Qi F, et al. Analysis of the clinical features of antisynthetase syndrome: a retrospective cohort study in China. Clin Rheumatol. 2022:10-06404. doi: 10.1007/s10067-022-06404-8. [DOI] [PubMed] [Google Scholar]
- 7.Cavagna L, Nuño L, Scirè CA, et al. Clinical Spectrum Time Course in Anti Jo-1 Positive Antisynthetase Syndrome: Results From an International Retrospective Multicenter Study. Medicine (Baltimore) 2015;94(32):e1144. doi: 10.1097/MD.0000000000001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams RA, Fernandes-Cerqueira C, Notarnicola A, et al. Serum-circulating His-tRNA synthetase inhibits organ-targeted immune responses. Cell Mol Immunol. 2021;18(6):1463–1475. doi: 10.1038/s41423-019-0331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy S, Bag AK, Singh RK, et al. Multifaceted Role of Neuropilins in the Immune System: Potential Targets for Immunotherapy. Front Immunol. 2017;8:1228. doi: 10.3389/fimmu.2017.01228. doi: 10.3389/fimmu.2017.01228. eCollection;%2017.:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Immormino RM, Lauzier DC, Nakano H, et al. Neuropilin-2 regulates airway inflammatory responses to inhaled lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol. 2018;315(2):L202–L211. doi: 10.1152/ajplung.00067.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paz S, Julian MW, Siefker D, et al. Immunomodulatory protein ATYR1923 disrupts an in vitro model of sarcoid granuloma formation. 2021:OA3968. [Google Scholar]
- 12.Paz S, Chu D, Ferrer M, et al. Neuropilin-2, the Specific Binding Partner to ATYR1923, Is Expressed in Sarcoid Granulomas and Key Immune Cells. 2020:A3099. [Google Scholar]
- 13.Paz S, Polizzi C, Chu D, et al. ATYR 1923 reduces neutrophil infiltration in an acute lipopolysaccharide (LPS) lung injurty model. 2019 [Google Scholar]
- 14.Kishi J, Nishioka Y, Kuwahara T, et al. Blockade of Th1 chemokine receptors ameliorates pulmonary granulomatosis in mice. Eur Respir J. 2011;38(2):415–424. doi: 10.1183/09031936.00070610. [DOI] [PubMed] [Google Scholar]
- 15.Eishi Y, Suga M, Ishige I, et al. Quantitative analysis of mycobacterial and propionibacterial DNA in lymph nodes of Japanese and European patients with sarcoidosis. J Clin Microbiol. 2002;40(1):198–204. doi: 10.1128/JCM.40.1.198-204.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burkart C, Seikkula L, Eide L, et al. ATYR1923 Modulates the Inflammatory Response in Experimental Models of Interstitial Lung Disease. 2019:A2421. [Google Scholar]
- 17.Crouser ED, Maier LA, Wilson KC, et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;201(8):e26–e51. doi: 10.1164/rccm.202002-0251ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. 2021;58(6):2004079–2020. doi: 10.1183/13993003.04079-2020. [DOI] [PubMed] [Google Scholar]
- 19.Kirkil G, Lower EE, Baughman RP. Predictors of Mortality in Pulmonary Sarcoidosis. Chest. 2018;153(1):105–113. doi: 10.1016/j.chest.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Mostard RL, Voo S, van Kroonenburgh MJ, et al. Inflammatory activity assessment by F18 FDG-PET/CT in persistent symptomatic sarcoidosis. Respir Med. 2011;105(12):1917–1924. doi: 10.1016/j.rmed.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Xu L, Kligerman S, Burke A. End-stage sarcoid lung disease is distinct from usual interstitial pneumonia. Am J Surg Pathol. 2013;37(4):593–600. doi: 10.1097/PAS.0b013e3182785a2d. [DOI] [PubMed] [Google Scholar]
- 22.Obi ON, Lower EE, Baughman RP. Biologic and advanced immunomodulating therapeutic options for sarcoidosis: a clinical update. Expert Rev Clin Pharmacol. 2021;14(2):179–210. doi: 10.1080/17512433.2021.1878024. [DOI] [PubMed] [Google Scholar]
- 23.Khan NA, Donatelli CV, Tonelli AR, et al. Toxicity risk from glucocorticoids in sarcoidosis patients. Respir Med. 2017;132:9–14. doi: 10.1016/j.rmed.2017.09.003. doi: 10.1016/j.rmed.2017.09.003. Epub;%2017 Sep 8.:9-14. [DOI] [PubMed] [Google Scholar]
- 24.Judson MA, Chaudhry H, Louis A, et al. The effect of corticosteroids on quality of life in a sarcoidosis clinic: the results of a propensity analysis. Respir Med. 2015;109(4):526–531. doi: 10.1016/j.rmed.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Culver DA, Aryal S, Barney J, et al. Efzofitimod for the treatment of pulmonary sarcoidosis. Chest. 2022;(22):10. doi: 10.1016/j.chest.2022.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baughman RP, Judson MA, Beaumont JL, et al. Evaluating the Minimal Clinically Important Difference of the King’s Sarcoidosis Questionnaire in a Multicenter Prospective Study. Ann Am Thorac Soc. 2021;18(3):477–485. doi: 10.1513/AnnalsATS.202006-607OC. [DOI] [PubMed] [Google Scholar]
- 27.Baughman RP, Winget DB, Lower EE. Prospective, randomized, double blind trial of methotrexate as a steroid sparing agent for sarcoidosis: analysis of first 25 patients. Am J Respir Crit Care Med. 1997;155:A944. [Google Scholar]
- 28.Baughman RP, Iannuzzi MC, Lower EE, et al. Use of fluticasone for acute symptomatic pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19:198–204. [PubMed] [Google Scholar]
- 29.Park MK, Fontana JR, Babaali H, et al. Steroid sparing effects of pentoxifylline in pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2009;26:121–131. [PMC free article] [PubMed] [Google Scholar]
- 30.Judson MA, Baughman RP, Costabel U, et al. Safety and efficacy of ustekinumab or golimumab in patients with chronic sarcoidosis. Eur Respir J. 2014;44:1296–1307. doi: 10.1183/09031936.00000914. [DOI] [PubMed] [Google Scholar]
- 31.Culver DA, Aryal S, Barney J, et al. Efzofitimod for the treatment of pulmonary sarcoidosis. Chest. 2022;(22):10. doi: 10.1016/j.chest.2022.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obi ON, Saketkoo LA, Russell AM, et al. Sarcoidosis: Updates on therapeutic drug trials and novel treatment approaches. Front Med (Lausanne) 2022;9:991783. doi: 10.3389/fmed.2022.991783. doi: 10.3389/fmed.2022.991783. eCollection;%2022.:991783. [DOI] [PMC free article] [PubMed] [Google Scholar]