

Abstract
Background and purpose
Charcot–Marie–Tooth disease (CMT) is a heterogeneous group of disorders caused by mutations in at least 100 genes. However, approximately 60% of cases with axonal neuropathies (CMT2) still remain without a genetic diagnosis. We aimed at identifying novel disease genes responsible for CMT2.
Methods
We performed whole exome sequencing and targeted next generation sequencing panel analyses on a cohort of CMT2 families with evidence for autosomal recessive inheritance. We also performed functional studies to explore the pathogenetic role of selected variants.
Results
We identified rare, recessive variants in the MYO9B (myosin IX) gene in two families with CMT2. MYO9B has not yet been associated with a human disease. MYO9B is an unconventional single‐headed processive myosin motor protein with signaling properties, and, consistent with this, our results indicate that a variant occurring in the MYO9B motor domain impairs protein expression level and motor activity. Interestingly, a Myo9b‐null mouse has degenerating axons in sciatic nerves and optic nerves, indicating that MYO9B plays an essential role in both peripheral nervous system and central nervous system axons, respectively. The degeneration observed in the optic nerve prompted us to screen for MYO9B mutations in a cohort of patients with optic atrophy (OA). Consistent with this, we found compound heterozygous variants in one case with isolated OA.
Conclusions
Novel or very rare variants in MYO9B are associated with CMT2 and isolated OA.
Keywords: axonal neuropathy, Charcot–Marie–Tooth neuropathy, mutations, myosin
INTRODUCTION
Inherited peripheral neuropathies (IPNs) are a heterogeneous group of disorders that may be part of more complicated syndromes. Patients with IPNs present distal motor and/or sensory deficits that progress with age. When neuropathy is the predominant finding, then the IPN is usually classified as Charcot–Marie–Tooth disease (CMT), distal hereditary motor neuropathy (HMN; or distal spinal muscular atrophy), or hereditary sensory autonomic neuropathy (HSAN) [1, 2, 3]. When additional findings (such as encephalopathy, spasticity, ataxia, cranial nerve involvement, or myopathy) dominate the clinical phenotype, the disorder is usually given a different name [4]. Of particular interest here is the association of CMT and optic atrophy (OA) [5]. OA is found in some patients with CMT2A (which is caused by dominant MFN2 mutations) [6], and in all patients with OPA1 (caused by dominant OPA1 mutations), but 50%–60% of OA patients (isolated or combined with other neurological issues) lack a genetic diagnosis [5].
Since the advent of next generation sequencing (NGS), the number of novel genes and causative variants identified as responsible for IPNs has dramatically increased [7]. However, approximately 60% of cases of the axonal forms (including CMT2, HMN, and HSAN) remain genetically undiagnosed, due in part to a large number of unknown rare genetic causes [8]. By performing whole exome sequencing (WES) and NGS panel analyses on a cohort of HMN/CMT2 families with evidence of autosomal recessive inheritance, we recently reported novel or rare variants in known disease genes that were not previously associated with HMN or CMT2, thus further expanding the spectrum of clinical phenotypes that can be associated with a specific disease gene [9].
MYO9B is an unconventional single‐headed processive myosin motor protein with signaling properties. MYO9B is an actin filament plus end‐directed motor that converts chemical energy derived from adenosine triphosphate (ATP) hydrolysis into mechanical force (Figure 8a) [10, 11, 12]. The ATP‐bound state of myosin usually has a weak affinity for actin. In contrast, ATP‐bound MYO9B displays a high affinity for F‐actin due to the presence of a unique actin‐binding insertion in the head region. Following the motor head region, it encompasses four IQ motifs that have been shown to bind the Ca2+ sensor calmodulin. The binding of calmodulin generates a rigid lever arm whose length determines the step size of motor movements. At the C‐terminus, a RhoGAP domain was shown to switch off RhoA‐signaling by accelerating the guanosine triphosphate (GTP) hydrolysis by RhoA, switching it from the active/GTP‐bound state to the inactive/guanosine diphosphate‐bound state. Moreover, the RhoGAP domain has been found to interact with the cytosolic region of the single transmembrane receptor Robo1, and this interaction inhibits the guanosine triphosphatase (GTPase)‐activating protein (GAP) activity [13].
FIGURE 8.
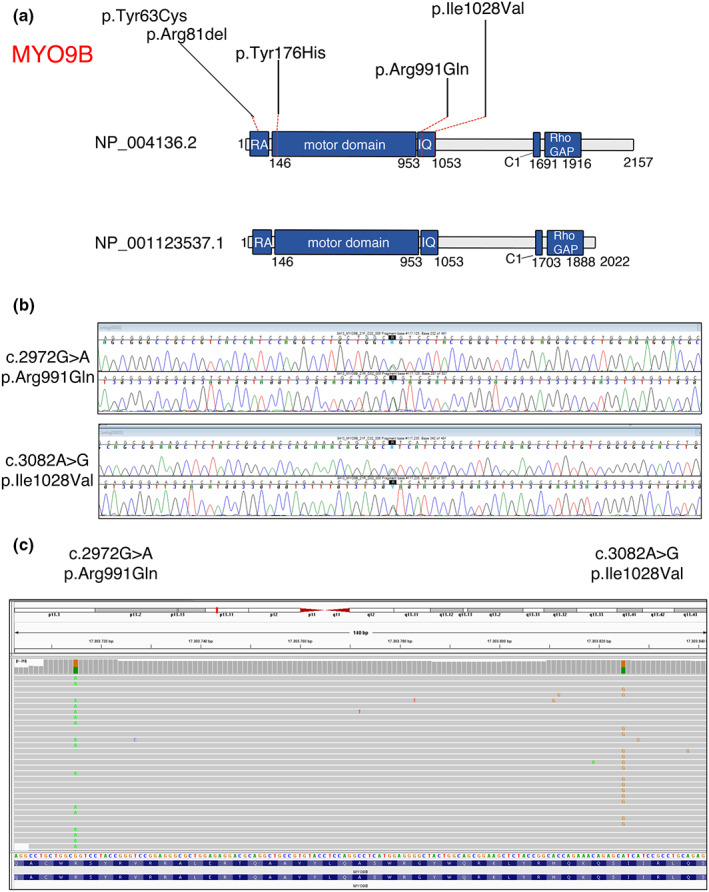
(a) Schematic representation of the predicted MYO9B protein isoforms, with the localization of the identified variants. RA is the Ras‐associated domain. The motor domain possesses adenosine triphosphatase activity; IQ motifs bind to calmodulin; C1 is a PKC‐like zinc‐binding domain; Rho GAP displays guanosine triphosphatase (GTPase)‐activating protein (GAP) activity for RhoA GTPase. (b) Electropherograms show the two variants identified in the MT8413 patient with optic atrophy, which are in trans, as indicated by reads alignment in c
Here, we report the identification of recessive variants in the MYO9B (myosin IX) gene in patients with CMT2 and isolated OA. The MYO9B gene has not been yet associated with a human disease [10, 11, 12, 14]. The five identified variants are located in MYO9B structural domains, thus having a predicted impact on protein function. One variant in the motor region alters protein expression level and localization at Rac1‐induced lamellipodia. Finally, consistent with the patient clinical presentation, Myo9b‐null mice have degenerating axons in their peripheral nerves and the optic nerve. In conclusion, here we report that novel or very rare variants in MYO9B are associated with CMT2 and OA.
MATERIALS AND METHODS
Patients
Patients gave informed consent to clinical and genetic studies according to respective national regulations and in agreement with the Declaration of Helsinki (64th World Medical Association General Assembly, Fortaleza, Brazil, October 2013). All data collected were anonymized. All subjects were interviewed for family and personal history and underwent a standard neurological examination, including evaluation for pyramidal or cerebellar signs, cranial nerve involvement, and the presence of bone abnormalities, joint contractures, and skin lesions, as well as standard clinical neurophysiology (nerve conduction and electromyography).
WES and NGS custom panel analysis
For Pedigree A, WES analysis and variant annotation were performed as recently reported [9]. For Pedigree B, WES was performed as previously reported and the generated NGS data of the two affected siblings were analyzed in the Genesis platform [15, 16]. Selected variants were confirmed by Sanger sequencing in all available family members. Droplet digital polymerase chain reaction (ddPCR) assay was performed as previously described [17].
Plasmids
The Tyr176His point mutation was introduced in the Myo9b cDNA by PCR using primers with the corresponding substitution and pBSK myr5 as a template [18]. The PCR construct was digested with XhoI and Pfl23II, and ligated into mcherry‐Myo9b Arg1695Met (R1695M; Addgene plasmid #134958) [19]. Plasmids encoding mCherry‐Myo9b (GAP−) and mCherry‐Myo9b Gly244Arg GAP− (G244R) were described previously [19].
Cell culture
Fibroblasts from CMT2 patients and age‐matched controls were established from skin biopsy samples and cultured at 5% O2 and 5% CO2 using Dulbecco modified Eagle medium and 10% fetal bovine serum supplemented with 2 mM sodium pyruvate. Mouse embryonic fibroblast NIH/3T3 cells were cultivated and transfected as described [19].
Rac1 photoactivation and fluorescence microscopy
Live cell imaging with PA‐Rac1 photoactivation was performed and analyzed as described by Hemkemeyer et al. [19].
Filopodia tip accumulation assay
To analyze the accumulation of Myo9b at filopodial tips, NIH/3T3 cells were seeded and transfected with different green fluorescent protein‐tagged MYO9B constructs in six‐well plates using Lipofectamin LTX. After 24 h, cells were detached using trypsin and seeded freshly on fibronectin‐precoated cover slips. Cells were allowed to attach and spread for 3 h before they were washed with phosphate‐buffered saline (PBS) and fixed with 4% paraformaldehyde for 15 min. Unreacted aldehydes were blocked with 0.1 M glycine, and cells were permeabilized using 0.1% Triton X‐100. To visualize filopodia, F‐actin was labeled with Texas Red‐phalloidin and cover slips were mounted with Mowiol. For each construct, filopodia of 30 transfected cells were counted and evaluated for MYO9B tip accumulation.
Antibodies
The following antibodies were used in this study: rabbit anti‐MYO9B (#12432‐1‐AP, Proteintech), mouse antivinculin (#05‐386, Millipore), rat anti‐myelin basic protein (MBP; #MAB386, Millipore), rat anti‐L1‐CAM (#MAB5272, Millipore), chicken anti‐NF‐M (#822701, Biolegend), goat anti‐ChAT (#AB144P, Millipore), and goat anti‐Brn3a (SC:8429, Santa Cruz Biotechnology).
For immunofluorescence, secondary antibodies included fluorescein (FITC)‐ and rhodamine (TRITC)‐conjugated donkey anti‐rat, chicken, or rabbit IgG (Jackson ImmunoResearch), and Alexa‐488 donkey anti‐Goat IgG (#A11055, Invitrogen).
For Western blotting, secondary antibodies included horseradish peroxidase‐conjugated goat antirabbit (#P0448, DAKO) and rabbit antimouse (#P0260, DAKO) immunoglobulins.
Immunohistochemistry
Sciatic nerves were collected, embedded in compound embedding medium for cryostat (OCT) (Killik, Bio‐Optica) and then snap‐frozen in liquid nitrogen. Spinal cords were collected and incubated overnight (O/N) in paraformaldehyde (PFA) 4% at room temperature (RT). The following day, samples were washed with PBS 1×, infiltrated in 30% sucrose O/N at RT, then embedded in OCT. Sciatic and optic nerve sections were cut and stained with rabbit anti‐MYO9B primary antibody O/N at 4°C. Spinal cord double immunostaining with MYO9B and ChAT was performed on 30‐μm‐thick sections. Samples were cut and left to air dry O/N, rehydrated in PBS 1× for 15 min, permeabilized in 50% ethanol (in PBS) for 30 min at RT, and then incubated with PBS‐Triton (PBS‐T) 0.3% for 1 h at RT. Slides were finally mounted using 4,6‐diamidino‐2‐phenylindole (DAPI) mounting medium (Vectashield), and then Z‐stacks were acquired with a confocal microscope SP5 (Leica).
Retina isolation and immunofluorescence of retinal ganglion cells
The eyes were dissected out and postfixed with 4% PFA for 60 min. Retinas were isolated under a dissecting microscope. Briefly, after making a circumferential cut of the cornea following the limbus, the cornea and lens were removed, and the retinas were visualized as a white surface covering the inside of the posterior eyecups. Eyecups were equilibrated in 30% sucrose O/N, embedded in OCT, and frozen in liquid nitrogen. Ten‐micrometer‐thick sections were postfixed with 4% PFA, permeabilized, and costained with Brn3a and MYO9B antibodies O/N at 4°C. Finally, sections were mounted with antifade mounting medium containing DAPI (H‐1800, Vector Laboratories).
Western blot analyses
Protein lysates from mouse tissues and human cells for Western blot analysis were prepared using a lysis buffer containing 2% sodium dodecyl sulfate (SDS), 25 mM Tris buffer pH 8.0, 95 mM NaCl, 10 mM EDTA, complete protease inhibitors (Roche), 1 mM β‐glycerophosphate (Sigma), and phosphatase inhibitor Cocktail 1 and 2 (Sigma). Protein quantification was performed using bicinchoninic acid assay (Pierce, Thermo Scientific). For Western blotting, SDS–polyacrylamide gel electrophoresis gels were transferred to polyvinylidene difluoride membranes (Millipore) at 4°C in 20% methanol blotting buffer. Filters were blocked in 5% dry milk in PBS–0.1% Tween 20 O/N at 4°C and immunoblotted with primary antibodies diluted in 3% dry milk in PBS–0.1% Tween. Horseradish peroxidase‐conjugated secondary antibodies (DAKO) were used and immunoblots revealed by using ECL/ECL Prime developing systems and films for chemiluminescent detection (Amersham).
Mice and morphological analysis
The generation and characterization of Myo9b knockout (KO) mice have been already reported [14], and sciatic nerve analysis by light and electron microscopy was performed as described previously [20]. To perform morphometric analysis, digitalized images of cross sections were obtained from corresponding levels of sciatic nerves with a 100× objective and Leica DFC300F digital camera. Five images per animal were analyzed using QWin software (Leica Microsystems), and the g‐ratio was calculated as the ratio between the mean diameter of an axon (without myelin) and the mean diameter of the same axon including the myelin sheath.
All animal experiments were conducted according to the guidelines of the Italian national regulations and covered by experimental protocols reviewed by local institutional animal care and use committees.
Data
The data that support the findings of this study are available from the corresponding author upon request.
RESULTS
By performing WES analysis in 15 unrelated Italian families with HMN/CMT2 and evidence for autosomal recessive inheritance, we identified a novel homozygous variant in the MYO9B gene (c.526T>C; p.Tyr176His) in a family with two siblings affected with CMT2 (Figure 1a, Tables 1 and 2). The healthy parents were both heterozygous carriers. Patient II,1 (37‐year‐old woman) had normal birth and developmental milestones. During childhood, her parents noted bilateral pes cavus and a tendency to turn the right ankle. Over years, she developed progressive walking difficulties, and her neurological examination at age 12 years showed bilateral foot drop, mild weakness of hand intrinsic muscles, and marked weakness of anterior leg muscles. Her last evaluation (Table 1), performed at age 37 years, showed atrophy and severe weakness of distal muscles of upper (thenar > hypothenar muscles) and lower (dorsiflexor > plantar flexor muscles) limbs. Deep tendon reflexes (DTRs) were reduced at knee and absent at ankle. Cranial nerve examination was unremarkable. Sensory examination showed normal pinprick sensation and reduced vibration sense at ankles. Disability assessed using the Charcot–Marie–Tooth Neuropathy Score (CMTNSv2 [21]) was moderate (16/36).
FIGURE 1.
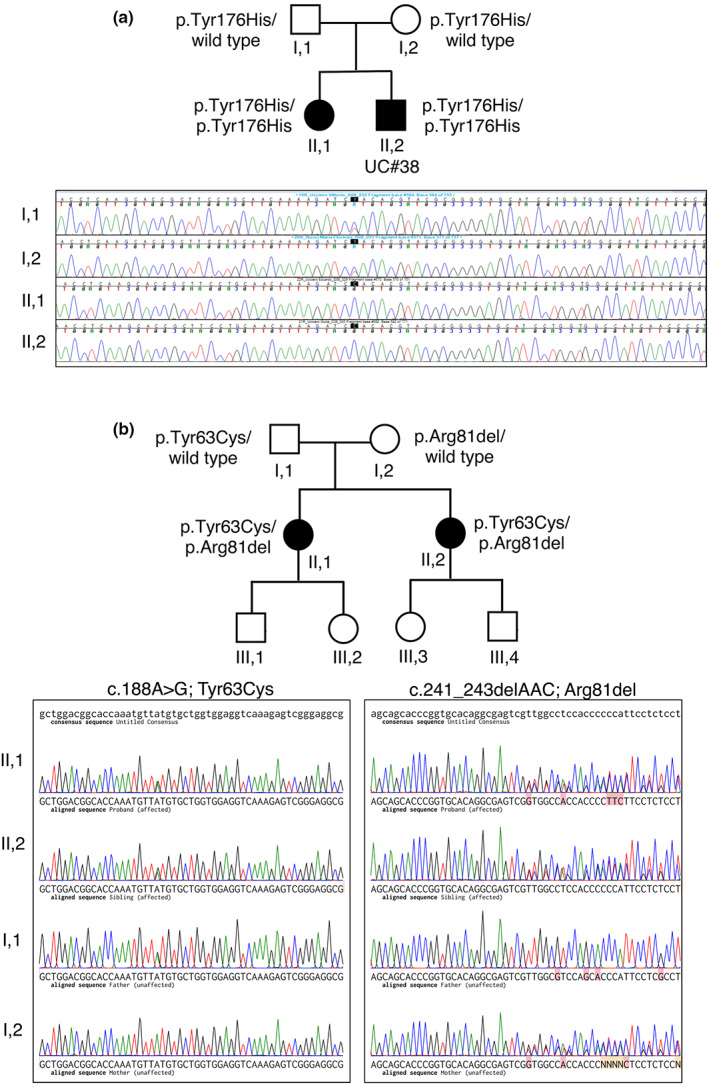
Identified variants in the MYO9B gene in siblings with Charcot–Marie–Tooth disease type 2 (CMT2) neuropathy. (a) Pedigree of the index Family A with autosomal recessive inheritance of CMT2. The p.Tyr176His variant was detected in the two alleles of II,1 and II,2 (UC#38) affected siblings and in heterozygosity in the healthy parents (I,1 and I,2), as illustrated by the electropherograms, where the c.526T>C change is shown. (b) Pedigree of Family B with autosomal recessive inheritance of CMT2. The two affected sisters II,1 and II,2 are compound heterozygous for the p.Tyr63Cys and p.Arg81del that were inherited in trans, as shown by electropherograms
TABLE 1.
Clinical and electrophysiological features of CMT2 patients
| Sex | Pedigree A, II,1 | Pedigree A, II,2 [UC#38] | Pedigree B, II,1 | Pedigree B, II,2 |
|---|---|---|---|---|
| Female | Male | Female | Female | |
| Age and type of onset | Childhood; walking difficulties | Adolescence; walking difficulties | Balance issues as child, trouble walking as teen | Late teens |
| Clinical features | ||||
| Age at last neurological examination, years | 37 | 36 | 52 | 50 |
| Gait | Bilateral foot drop | Bilateral foot drop | Bilateral foot drop | Bilateral foot drop |
| On heels | Impossible | Impossible | Impossible | Impossible |
| On toes | Impossible | Possible | Impossible | Impossible |
| Strength | ||||
| Upper limbs | Hand, thenar > hypothenar | Hand, thenar > hypothenar |
WE 4+, FE 4 Intrinsic hand muscles 2 or less Ankle dorsi‐ and plantarflexion 2/5 or less |
Triceps 4+, WE 4, FE 4‐, Intrinsic hand muscles 3/5 or less Hamstring 4, ankle dorsi‐ and plantarflexion 2 or less |
| Lower limbs | Ankle dorsiflexor > plantar flexor | Ankle dorsiflexor | ||
| Pinprick sensation | ||||
| Upper limbs | Normal | Normal | Normal | Normal |
| Lower limbs | Normal | Normal | Normal | Reduced to knees |
| Vibration sensation a | ||||
| Upper limbs | Normal | Normal | Not done | Not done |
| Lower limbs | Reduced (ankle) | Reduced (knee) | 0 knees | 3R 0L knee |
| Joint sense position | ||||
| Upper limbs | Normal | Normal | Not done | Not done |
| Lower limbs | Normal | Normal | Not done | Not done |
| Deep tendon reflexes | ||||
| Upper limbs | Normal | Normal | Normal | Normal |
| Lower limbs | Reduced (knee)/absent (ankle) | Reduced (knee)/absent (ankle) | 0 ankles, 1 knees, 3 biceps | 0 ankles, 1 knees, 1 biceps |
| Other features | Pes cavus | None | ||
| Nerve conduction study | ||||
| Median nerve | Age 36 | Age 37 | Age 55 | Age 51 |
| SAP, μV | 1.9 (antidromic) | NR (antidromic) | NR (orthodromic) | NR |
| SNCV, m/s | 32.6 | NR | NR | NR |
| DML, ms | NA | 4 | 6.4 | NR |
| dCMAP, mV | NR | 2.7 | 0.1 | NR |
| MNCV, m/s | NA | 38 | 24 | NR |
| Ulnar nerve | ||||
| SAP, μV | ND | NR (antidromic) | NR (orthodromic) | NR |
| SNCV, m/s | ND | NA | NR | NR |
| DML, ms | 3.3 | 3.5 | 3.9 | 4.6 |
| dCMAP, mV | 2.2 | 4.4 | 2.8 | 2.9 |
| MNCV, m/s | 46 | 39 | 33 | 32 |
| Tibial nerve | ||||
| DML, ms | 6.9 | ND | ND | ND |
| dCMAP, mV | 0.1 | |||
| MNCV, m/s | 35 | |||
| Peroneal nerve | ||||
| DML, ms | NA | 6.6 | ND | ND |
| dCMAP, mV | NR | 1.3 | ||
| MNCV, m/s | NA | 37 | ||
| Sural nerve | ||||
| SAP, μV | NR | NR | ND | ND |
| SNCV, m/s | NA | NA | ||
| Radial nerve | ||||
| SAP, μV | NR (antidromic) | NR (antidromic) | 3.1 (antidromic) | NR |
| SNCV, m/s | NA | NA | 46 | NR |
| CMTNS | 16 | 17 | 20 | 31 |
Abbreviations: CMT, Charcot–Marie–Tooth disease; CMT2, CMT type 2; CMTNS, Charcot–Marie–Tooth Neuropathy Score; dCMAP, distal compound muscle action potential; DML, distal motor latency; FE, finger extensor; L, left; MNCV, motor nerve conduction velocity; NA, not applicable; ND, not determined; NR, no response; R, right; SAP, sensory action potential; SNCV, sensory nerve conduction velocity; WE, wrist extensor.
Rydell–Seiffer tuning fork.
TABLE 2.
Variants identified in MYO9B
| Patient ID | cDNA | Protein | NFE AF | Overall AF | Hom AF | SIFT | PolyPhen | CADD | GERP |
|---|---|---|---|---|---|---|---|---|---|
| Pedigree A: II,1 and II,2 | c.526T>C | p.Tyr176His | 0 | 0 | 0 | DL | PD | 26.9 | 5.62 |
| Pedigree B: II,1 and II,2 | c.188A>G | p.Tyr63Cys | 0 | 0.0000041 | 0 | DL | PD | 14.54 | 4.93 |
| Pedigree B: II,1 and II,2 | c.241_243del | p.Arg81del | 0 | 0 | 0 | N/A | N/A | N/A | 4.93 |
| MT8413 | c.2972G>A | p.Arg991Gln | 0.0001 | 0.00016 | 0 | DL | PD | 27.9 | 4.88 |
| MT8413 | c.3082A>G | p.Ile1028Val | 0 | 0 | 0 | TL | B | 12.28 | 0.534 |
Abbreviations: AF, allele frequency; B, benign; CADD, Combined Annotation Dependent Depletion; DL, deleterious; GERP, Genomic Evolutionary Rate Profiling; Hom, homozygous count in GnomAD database; N/A, not available; NFE, Non‐Finnish European; PD, probably damaging; SIFT, Sorting Intolerant from Tolerant; TL, tolerated.
Her 36‐year‐old brother (II,2, also UC#38) had normal birth and developmental milestones. During his teens, he developed walking difficulties and muscle cramps in the lower limbs at rest and after exercise. The first neurological examination at 22 years showed mild weakness of ankle dorsiflexor muscles with difficulties in standing on heels and calf muscle hypertrophy. The last examination (Table 1), at age 36 years, showed severe wasting and weakness of hand intrinsic muscles (thenar > hypothenar muscles) and ankle dorsiflexor muscles with bilateral foot drop. DTRs were reduced at knee and absent at ankle. Cranial nerve examination was unremarkable. Sensory examination showed normal pinprick and reduced vibration sense at knees. Clinical disability was moderate (CMTNSv2 = 17/36).
To look for additional variants, we interrogated the Inherited Neuropathy Consortium database (1790 CMT2 and 161 distal HMN families), and identified two sisters with CMT2 who shared compound heterozygous mutations (p.Tyr63Cys and p.Asn81del) in MYO9B (Table 2). These variants occur in the RA (Ras association) domain, which does not bind Ras; its function remains to be clarified [12]. Segregation analysis showed that the mutations were inherited in trans from their parents (Figure 1b); each sister had two unaffected children. The oldest sister was first evaluated at age 49 years. She reported trouble with balance for as long as she could remember, and was told that she walked funny when she was 12 years old. She was sufficiently athletic as a teenager, but by age 18 years she could not wear heels. She developed foot drops in her 20s, and was diagnosed with CMT during her second pregnancy. Her hands were insidiously involved, and she has developed clawing of her fingers around age 50 years. At her last examination at age 55 years, she had high arches, hammer toes, and severe wasting and weakness of hand intrinsic muscles and lower legs. Pinprick sensation was normal in the legs, and vibration sensation was absent at the knees. DTRs were absent at ankles and present elsewhere. Clinical disability on the CMTNSv2 was 11 (out of 16).
The youngest sister was first evaluated at age 44 years. She had developed motor and sensory dysfunction in her legs in her late teens, and had used ankle–foot orthoses since her 20s. She subsequently developed hand weakness, and later swelling and pain in her feet while standing. She had hammer toes that were fixed surgically in 2003 and 2005. She had hemifacial microsomia and congenital hearing loss on the left. Her last examination (Table 1), at age 50 years, showed high arches, severe wasting and weakness of hand intrinsic muscles and lower legs, mild weakness in the triceps (4+/5), and moderate weakness in the hamstrings (4/5). DTRs were absent at ankles and present elsewhere. Sensory examination showed pinprick sensation was reduced in the knees, and there was severely reduced vibration sense in the knees. In addition to her left‐sided hearing loss, cranial nerve examination was unremarkable, including a funduscopic examination. Clinical disability on the CMTNSv2 was 15 (out out 16).
Electrophysiological examination in the four affected patients shows a sensory–motor polyneuropathy with similar features (Table 1). The amplitudes of compound muscle action potentials and sensory action potentials were markedly reduced or absent, and most of the motor and sensory nerve conduction velocities showed “intermediate” slowing. Needle electromyography showed severe, chronic denervation that was most pronounced in distal muscles and nearly as severe in proximal muscles. Patient II,2 in Family A underwent visual evoked potential testing that was normal. Patients II,1 and II,2 in Family B had retinal scans that were normal in Il,1 and showed thinning of the nerve fiber lay in the left eye, consistent with optic nerve damage, in II,2.
Functional analysis of the MYO9B variants
We investigated the impact of the p.Tyr176His change, which occurs in the MYO9B motor region, the most studied domain at the functional level. To this aim, first we investigated MYO9B protein expression levels in fibroblasts of Patient II,2 (UC#38), who was homozygous for p.Tyr176His. By performing Western blot analysis, we observed that MYO9B expression levels were significantly decreased in UC#38 patient fibroblasts as compared to age‐matched controls (Figure 2a,a′), consistent with previous reports that artificial mutations in MYO9B motor domain decrease protein stability and expression [22].
FIGURE 2.
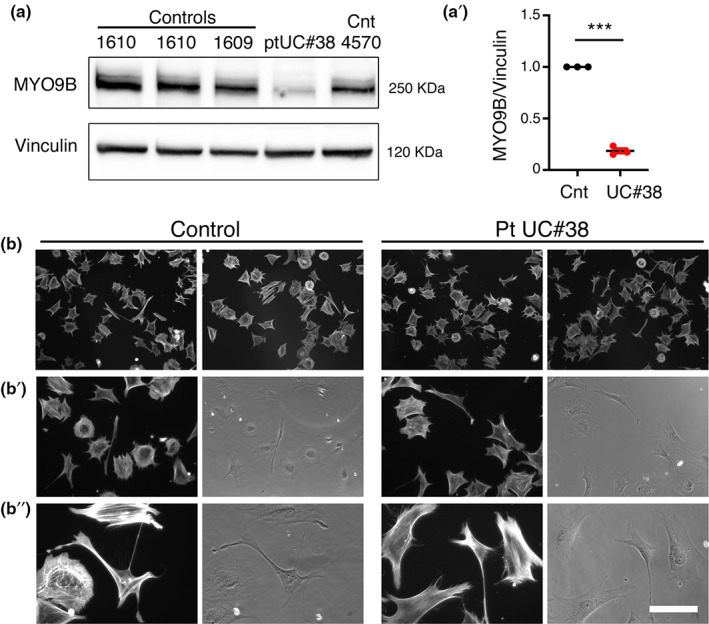
Functional analysis of p.Tyr176His variant of MYO9B. (a) Western blot analysis shows reduced MYO9B protein levels in UC#38 patient fibroblasts as compared to age‐matched controls (Cnt), as quantified (a′) from three independent experiments (one sample t‐test, t = 34, df = 2, ***p = 0.0009). (b, b″) Cell morphology is similar in UC#38 patient and control fibroblasts, as monitored 1 h after plating on a fibronectin substrate. Bar is 22.43 μm in b, 12.91 μm in b′, and 6.34 μm in b″
MYO9B is both a motor and signaling protein, and also possesses Rho GAP activity for RhoA GTPase [11, 12, 14]. Consistent with this, Myo9b KO mouse macrophages display a round morphology and decreased motility as a consequence of increased levels of GTP‐bound, active RhoA [14]. Thus, following the hypothesis that the p.Tyr176His variant in the motor domain may alter MYO9B localization and signaling activity, we monitored the morphology of UC#38 patient fibroblasts during adhesion, a process that relies on actomyosin cytoskeleton remodeling (Figure 2b,b″). However, the morphology of UC#38 patient fibroblasts was similar to controls, suggesting that, at least in these cells in these experimental conditions, the p.Tyr176His change does not alter the RhoA GAP activity of MYO9B.
To investigate the impact of the p.Tyr176His variant on MYO9B motor activity, we explored the ability of overexpressed MYO9B carrying p.Tyr176His to localize at regions of actin polymerization that depend on its motor activity (Figure 3) [19, 22]. It is known that the RhoGAP activity of overexpressed Myo9b leads to considerable morphological changes, whereas Myo9bGAP− (which has the R1695M mutation in the GAP domain that abrogates its RhoGAP activity) shows a dynamic subcellular distribution indistinguishable from the wild‐type Myo9B. Myo9bGAP− is enriched in extending lamellipodia, membrane ruffles, and occasionally at filopodial tips, all regions of active F‐actin polymerization [19, 22]. Thus, NIH/3T3 cells were cotransfected with photoactivatable PA‐Rac1, which induces actin polymerization and membrane protrusion upon photoactivation, and mCherry‐Myo9bR1695M (which abrogates RhoGAP activity and is henceforth termed GAP−), mCherry‐Myo9bG244R GAP− (G244R is predicted to abolish nucleotide binding), or mCherry‐Myo9bY176H GAP−. Cells were analyzed by live cell fluorescence microscopy. The GAP− mutant (mCherry‐GAPMyo9bR1695M) accumulates at the leading edge of photoactivatable Rac1‐induced protrusion, whereas the mCherry‐Myo9bG244R GAP− motor mutant and the mCherry‐Myo9bY176H GAP− mutant did not (Figure 3a–c). In a second approach, we transfected cells to express mCherry‐Myo9bGAP−, mCherry‐Myo9bG244R GAP−, and mCherry‐Myo9bY176H GAP−, and colabeled for F‐actin with FITC‐conjugated phalloidin. Both mCherry‐Myo9bG244R GAP− and mCherry‐Myo9bY176H GAP− failed to accumulate at filopodial tips as compared to mCherry‐Myo9bGAP− (Figure 3d,e). Together, these results indicate that the p.Tyr176His variant results in loss of function, as it negatively impacts MYO9B expression and motor activity.
FIGURE 3.
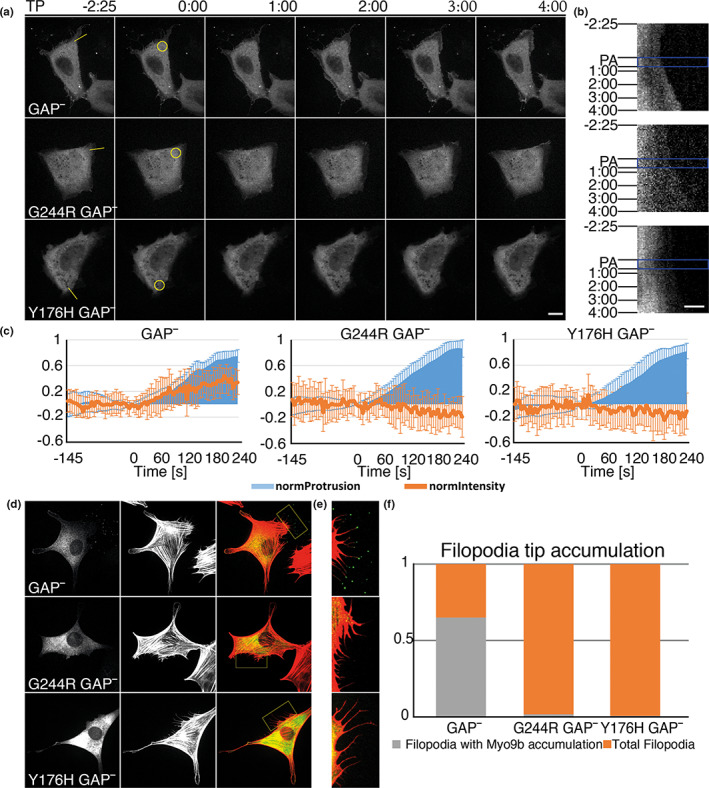
The p.Tyr176His variant in the motor domain of MYO9B mimics a Myo9b variant with defective motor activity, as it is not recruited to the tip of filopodia or to the leading edge of Rac1‐induced lamellipodia. (a, b) NIH/3 T3 cells coexpressing photoactivatable PA‐Rac1 and either mCherry‐Myo9bR1695M (GAP−), which contains a point mutation known to abrogate RhoGAP activity of Myo9b, mCherry‐Myo9bG244R GAP− (G244R is predicted to abolish nucleotide binding and thus disrupt motor function), or mCherry‐Myo9bY176H GAP−, the MYO9B variant identified in the index family (Figure 1a), and analyzed by live cell fluorescence microscopy. Time‐lapse movies were recorded with a frame rate of 5 s on a spinning disc microscope to monitor the distribution of the indicated Myo9b constructs. PA‐Rac1 was activated after 30 frames (145 s) in the indicated region of interest (yellow circle in a, zero time point image) by irradiation with 405 nm light using a FRAP (fluorescence recovery after photobleaching) module. Photoactivation included short pulses of light every 5 s for eight cycles, and the distribution of mCherry‐Myo9b constructs was monitored until several minutes afterward. (a) Single images are shown from characteristic time‐lapse movies. Time stamp indicates the time relative to photoactivation onset. The cell forms a protrusion in response to the photoactivation that persists for several minutes after photoactivation. GAP− (Myo9bR1695M) accumulates at the leading edge of this protrusion, whereas the Myo9bG244R GAP− motor mutant and the Myo9bY176H GAP− mutant did not. Scale bar is 15 μm. (b) Kymographs of the corresponding movies recorded along the yellow line indicated in the first images of a. PA (blue rectangle) indicates the photoactivation phase. Time points of the single images shown in a are indicated to the left. (c) For each Myo9b construct, the averaged normalized protrusion (light blue) fluorescence intensities at the leading edge (brown lines) were calculated from kymographs of 10 independent photoactivation experiments. Error bars represent SDs. (d–f) NIH3T3 cells were transfected with the indicated Myo9b construct (labeled with enhanced green fluorescent protein) and F‐actin was labeled in fixed cells with fluorescent phalloidin (red). (d) Images of the transfected construct (left row) and F‐actin (middle row) are shown as well as the merged images (right row). The boxes in the merged images are enlarged in e. Note the lack of MYO9B at the filopodial tips for Myo9bG244R GAP− and Myo9bY176H GAP− mutants as compared to the GAP− (Myo9bR1695M) mutant. (f) The accumulation of the indicated Myo9b construct at filopodia was quantified. GAP−, fluorescent protein (FP)‐Myo9bR1695M (GAP−) which lacks RhoGAP activity; G244R GAP−, FP‐Myo9bG244R GAP− motor mutant, which also lacks RhoGAP activity; Y176H GAP−, FP‐Myo9bY176H GAP− identified in the index family, which also lacks RhoGAP activity. In a–c, FP corresponds to mCherry and in d–f to enhanced green fluorescent protein. TP, Time points
Myo9b is expressed in both the peripheral nervous system and central nervous system
The MYO9B gene is predicted to be transcribed in different mRNA isoforms that differ at the 5′‐end for an additional exon 1 and at the 3′‐end for an intron retention between exons 39 and 40 (Figure 4a). The longest mRNA isoform is translated into a protein of 2157 amino acids in length, whereas a shorter mRNA isoform is predicted to generate a protein of 2022 amino acids. In the shorter isoform, intron 39 is transcribed, producing a premature stop codon and a truncated MYO9B protein lacking 135 amino acids at the C‐terminus. To explore expression of these predicted isoforms, we performed ddPCR analysis on blood and fibroblasts from three controls. We were able to amplify only the shorter isoform containing intron 39, suggesting that this isoform and the corresponding 2022 amino acid MYO9B protein are physiologically relevant (Figure 4b).
FIGURE 4.
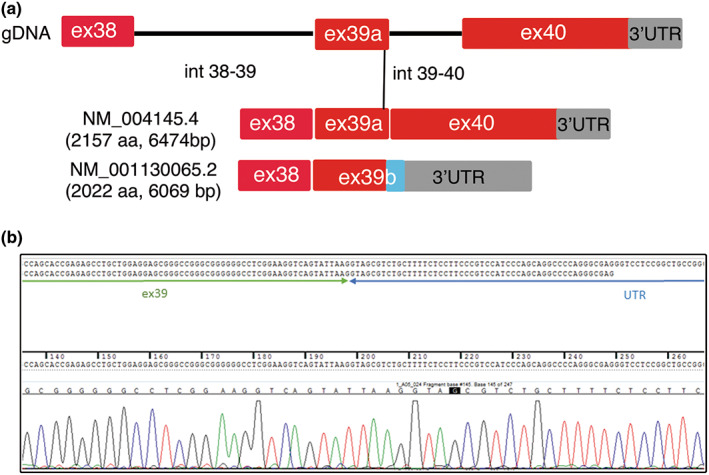
Predicted mRNA isoforms of MYO9B. (a) Schematic representation of the genomic organization of exons 38–40 (gDNA) transcribed into a longer mRNA isoform of 6474‐bp open reading frame and a shorter isoform, which derives from retention of intron 39–40. Coding region of exon 39b includes 11 bp of intron 39–40, which introduces a stop codon and a premature truncation of the MYO9B protein lacking 135 amino acids (aa) at the C‐terminus. (b) Sequencing analysis (electropherograms) of reverse transcriptase polymerase chain reaction amplification of exons 38–40 from control blood and fibroblasts confirmed the presence of the short MYO9B mRNA isoform. UTR, untranslated region
We then investigated MYO9B expression in the nervous system. First, by performing Western blot analysis, we detected expression of Myo9b in isolated rat Schwann cell lysates as well as in mouse sciatic nerve and spinal cord (Figure 5a). To explore Myo9b localization in these tissues, we performed immunohistochemistry on sciatic nerve sections at different time points of postnatal development. MYO9B was expressed in both myelin‐forming (MBP‐positive fibers, Figure 5b) and non‐myelin‐forming Schwann cells (L1‐CAM‐positive cells, Figure 5d) in the nerve, as well as in axons (neurofilament medium‐positive, Figure 5c). In myelin‐forming Schwann cells, Myo9b is detected in cytoplasmic regions surrounding the nucleus (Figure 5b,c) and in the abaxonal Schwann cytoplasm, the outermost layer of Schwann cell membrane (Figure 5c). In the spinal cord, MYO9B was localized in astrocytic processes in the white matter, and diffusely in the gray matter (Figure 6a,a′), but not in ChAT‐positive motor neurons. Consistent with findings in the sciatic nerve, MYO9B was detected in both Schwann cells and axons in ventral roots (Figure 6b,b′).
FIGURE 5.
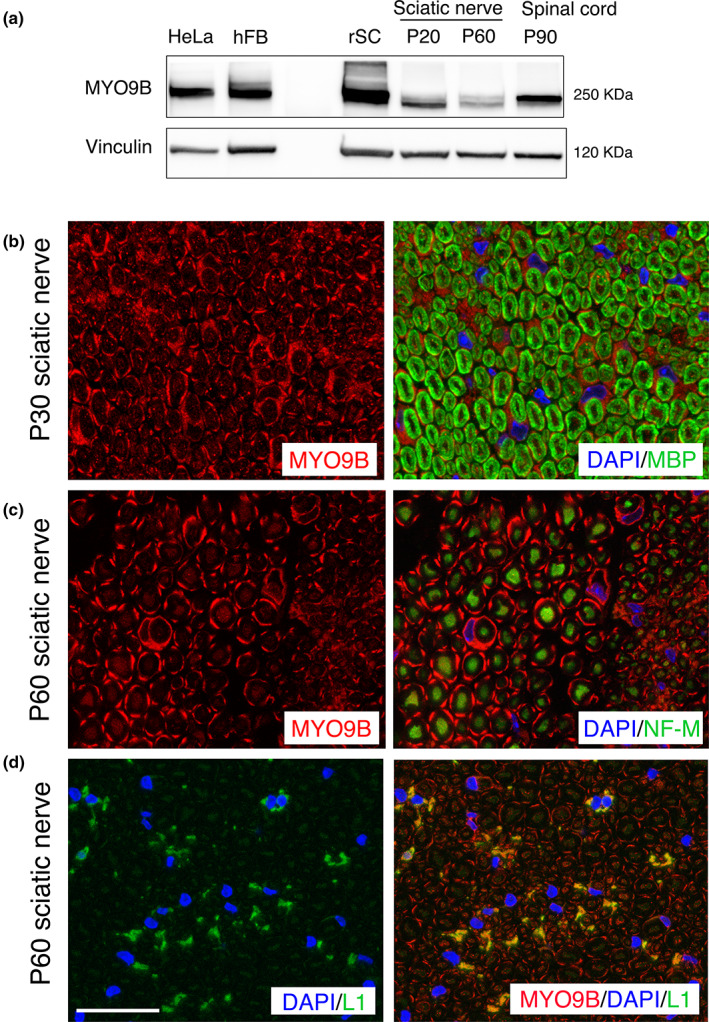
Myo9b expression analysis in the peripheral nervous system. (a) Western blot analysis shows that Myo9b is expressed in isolated rat Schwann cell lysate (rSC), as well as in mouse sciatic nerve and spinal cord lysates. HeLa cells and human fibroblasts (hFB) were used as controls. (b–d) Images of mouse sciatic nerve labeled for the indicated antigens: Myo9B, myelin basic protein (MBP; which stains compact myelin), neurofilament medium (NF‐M; which labels axons), and L1‐CAM (which labels non‐myelin‐forming Schwann cells). Myo9b is expressed in the cytoplasm of myelin‐forming Schwann cells at P30 and at P60, localized to the abaxonal Schwann cell cytoplasm, and to a lesser degree in axons at P60. Myo9b staining is also detected in non‐myelin‐forming Schwann cells (d). 4,6‐Diamidino‐2‐phenylindole (DAPI) marks Schwann cell nuclei. Bar is 13.31 μm for d and 12.28 μm for b and c
FIGURE 6.
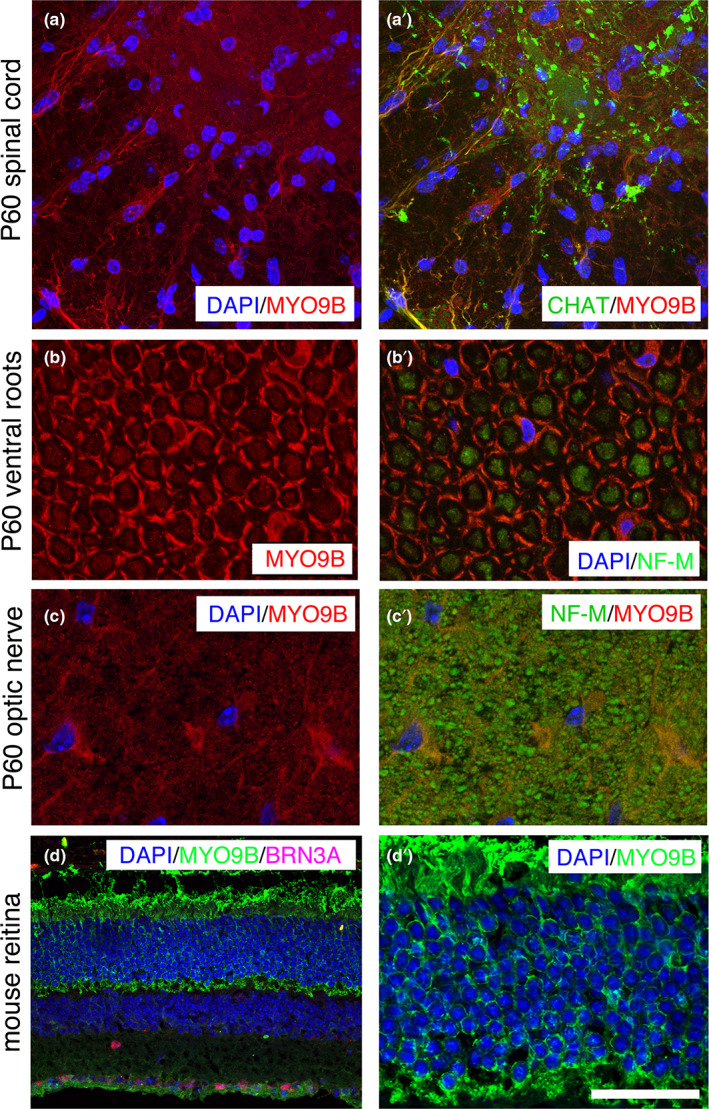
Myo9b expression analysis in the central nervous system. Images show mouse tissues labeled for the indicated antigens: Myo9B, choline acetyltranserase (CHAT; which labels motor neurons), neurofilament medium (NF‐M; which labels axons), and BRN3A (Pou4f1, which labels retinal ganglion cells), and counterstained with DAPI. (a, a′) In the ventral region of the mouse spinal cord at P60, Myo9b in found in astrocytic processes and diffusely in the gray matter, but does not colocalize with CHAT‐positive motor neurons. (b, b′) In ventral roots, Myo9b is localized to the abaxonal Schwann cell cytoplasm, and also to axons. (c, c′) Staining of optic nerves in transverse section shows Myo9b localization in glial cells but not axons. (d, d′) Staining of mouse retina shows Myo9b expression in retinal ganglion cells (BRN3A‐positive) and in photoreceptors. Bar is 9.32 μm in d′; 19.56 μm in a and a′, 8.18 μm in b‐c′, and 31.55 μm in d. DAPI, 4,6‐diamidino‐2‐phenylindole
Myo9b/ MYO9B is associated with optic atrophy in mouse and human
To further explore the function of MYO9B, we analyzed the sciatic nerve, spinal cord, and optic nerve from Myo9b KO mice. Except for a few degenerated myelinated axons and clusters of regenerated axons in sciatic nerves of this mutant at 12 months (Figure 7a), the KO nerves looked normal, including a morphometric analysis of myelin thickness (Figure 7b) and axonal diameters (Figure 7c). Ultrastructural analysis of optic nerves revealed loss of myelinated fibers, and occasional enlarged axons that contained accumulations of organelles (Figure 7d,e). To further explore this finding, we extended MYO9B expression analysis to the optic nerve and retina. Interestingly, we observed Myo9b expression in processes and perinuclear cytoplasm of glial cells in the optic nerve (Figure 6c,c′). Myo9b was expressed in the photoreceptors and retinal ganglion cells (Figure 6d,d′).
FIGURE 7.
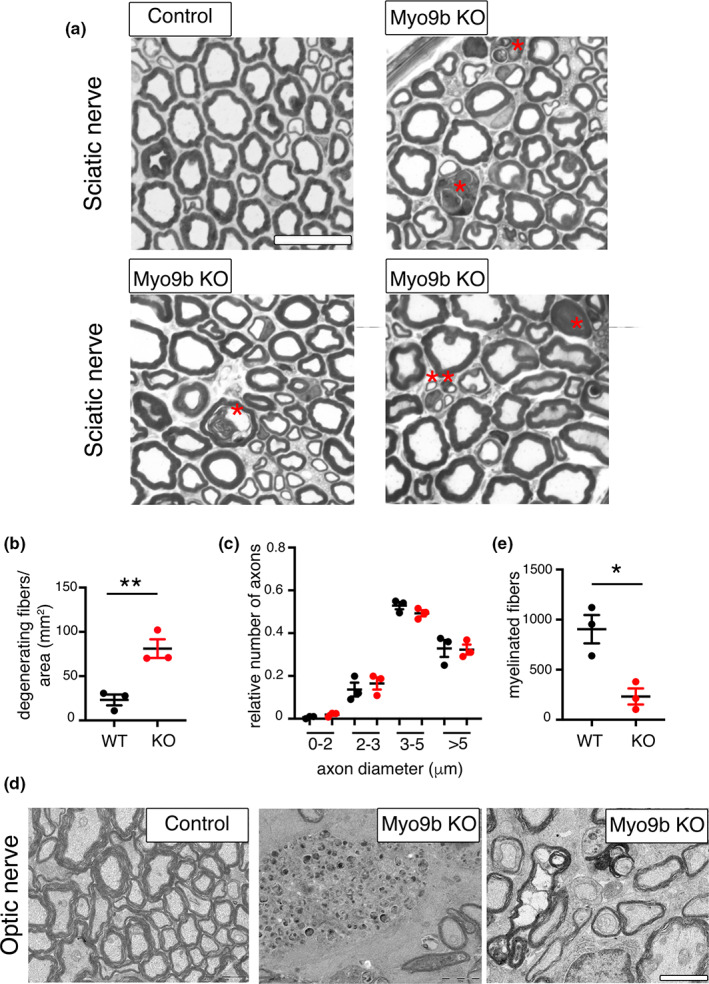
Morphological analysis of Myo9b knockout (KO) mice. (a) Semithin section analysis of sciatic nerves from Myo9b KO mice at 12 months of age (n = 3) shows degenerating myelinated axons (red asterisks) as quantified in (b), two‐tailed unpaired t‐test, t = 4.759, df = 4, p = 0.0089. The double asterisks indicate a cluster of regenerating axons. (c) Loss of MYO9B does not alter myelin thickness and axonal diameter distribution in these nerves; the g‐ratios of wild‐type (WT) nerves (0.660 ± 0.003 for 1035 fibers, n = 3 animals) and KO nerves (0.658 ± 0.004 for 983 fibers analyzed, n = 3 animals) were not significantly different (p > 0.9999; two‐tailed Mann–Whitney t‐test). (d) Ultrastructural analysis of optic nerves shows a decreased density of myelinated axons, as compared in two‐tailed unpaired t‐test, t = 4.134, df = 4, p = 0.0145 (n = 3 per genotype; e), as well as axons with abnormal accumulations of organelles. Bar is 2.65 μm in a and 2 μm in d
We thus sought to assess whether mutations in MYO9B could also be associated with OA in humans. We performed NGS custom panel analysis of 72 patients (17–60 years of age) presenting with OA of unknown origin as main clinical manifestation, but some also with a neuropathy or myopathy. Toxic cause such as smoke and alcohol abuse were excluded, and vitamin B12 and folic acid levels were normal in all patients. The patients were negative for Leber hereditary optic neuropathy (LHON), common mitochondrial DNA mutations, and genes associated with OA such as OPA1, OPA3, AFG3L2, WFS1, ACO2, CISD2, SPG7, RTN4iP, TMEM126A, DNM1L, and YMEL1L. We identified two additional variants, c.2972G>A; p.Arg991Gln and c.3082A>G; p.Ile1028Val, in a 28‐year‐old woman with isolated OA (MT8413, Figure 8a,b and Table 2), both located in the MYO9B IQ motifs, which bind calmodulin and regulate protein step size (Figure 8a).
The patient had a normal developmental history, a slight bilateral myopia (corrected with lenses), and bilateral visual pallor on ophthalmological examination. The peripapillary retinal nerve fiber layer was slightly reduced at the OCT evaluation. No other neurological signs or symptoms were identified. NGS analysis showed that these variants were in trans (Figure 8c).
DISCUSSION
We identified five novel variants in the MYO9B gene in two independent families with CMT2 and in one case with isolated optic atrophy. All variants identified were located in structural domains of MYO9B (Figure 8a). We showed that the p.Tyr176His, which lies in the well‐characterized motor domain, had reduced expression in patient fibroblasts, as well as that Myo9b constructs carrying this mutation abrogated motor activity, and did not accumulate at Rac1‐induced lamellipodia. There was an increased number of degenerating myelinated axons in Myo9b KO sciatic and optic nerves.
The four patients with CMT had a similar phenotype: disease onset at younger than 20 years, with slow progression to a moderate to severe phenotype in adulthood. Motor strength and function, as well as vibration, were severely affected, demonstrating a loss of large, myelinated axons. Clinical neurophysiology showed reduced motor and sensory amplitudes, with “intermediate” slowing of motor conduction velocities in the arms. This degree of slowing is found in some neuropathies that are considered “demyelinating,” such as CMTX1 [23], but also in some neuropathies that are probably “axonal,” such as recessive GDAP1 mutations [24]. There was preserved pinprick sensation in three of the patients, indicating that nociceptive axons were relatively spared. The Myo9b KO provides some insight into this issue, as there was some axonal loss but no features of de/remyelination.
The reduced expression of p.Tyr176His mutation is consistent with previous findings that artificial mutations that inactivate the Myo9b motor domain decrease protein MYO9B levels. In addition, this mutant did not accumulate at Rac1‐induced lamellipodia, demonstrating that its motor function is impaired. This tyrosine residue is highly conserved in myosins, and an analogous mutation (p.Tyr115His) in beta‐cardiac myosin II (MYH7) causes a dominantly inherited cardiomyopathy [25]. Cytoskeleton remodeling is a fundamental process not only in migrating cells but also for synaptic maintenance in neurons [26]. Interestingly, Myo9b has been found to inhibit RhoA signaling and modulate dendritic morphogenesis in cortical neurons [27]. Overactivated RhoA signaling in neurons has been reported in several neurodegenerative disorders. This is a direct consequence of gene mutations or, indirectly, a result of imbalance between Rac1 and RhoA, which have opposite functions on cytoskeletal remodeling [28]. Two other forms of demyelinating CMT have also been associated with a gain of function of RhoA GTPase activity. Mutations in ARHGEF10, a RhoA regulator, result in RhoA activation and low nerve conduction velocities [29]. Loss of function of INF2, another RhoA regulator, results in RhoA activation and CMT [30].
We found that one variant in MYO9B is associated with isolated OA. Approximately 30%–50% of patients with primary OA have mutations in OPA1 or mitochondrial DNA (LHON) [5], and 50%–60% of OA cases remain without a genetic diagnosis. Whether MYO9B mutations cause OA needs confirmation.
AUTHOR CONTRIBUTIONS
Conception and design of the study: Alessandra Bolino. Acquisition and analysis of data: all authors. Alessandra Bolino wrote the paper with the contribution of Matt Danzi, Davide Pareyson, Steven S. Scherer, Fiore Manganelli, and Martin Bähler.
CONFLICT OF INTEREST
The authors have declared no conflict of interest for this article. S.S.S. has served on the scientific advisory board for Mitochondria in Motion and Disarm, and has consulted for Pfizer, Eli Lilly, Applied Therapeutics, Passage Bio, and Toray Industries.
ACKNOWLEDGMENTS
This work was supported by the Italian Ministry of Health (RF‐2011‐02347127), and CoFin Regione Lombardia (n. 96) to A.B. and S.C.P; by the Italian Ministry of Health (RRC) to D.P.; by the E‐Rare project GENOMIT and the “Cell Line and DNA Bank of Genetic Movement Disorders and Mitochondrial Diseases” of the Telethon Network of Genetic Biobanks (grant GTB12001J) to C.L. C.P., C.L., D.P., and S.C.P. are members of the European Reference Network for Rare Neuromuscular Diseases. C.P., D.P., S.Z., and S.S.S. are members of the Inherited Neuropathies Consortium, which is a part of the NIH Rare Diseases Clinical Research Network. S.S.S. was also supported by the Judy Seltzer Levenson Memorial Fund for CMT Research. He thanks Tanya Bardakjian and Pooja Patel for their contributions. We are grateful to Prof Giampietro Schiavo, UCL, London, for protocol and advice on how to perform ChAT staining of mouse spinal cord. We also thank Dr Maria Carla Panzeri (Advanced Light and Electron Microscopy BioImaging Center, San Raffaele Scientific Institute) for help with electron microscopy imaging and Dr Roberta Di Guardo for technical assistance. Open access funding provided by BIBLIOSAN.
Cipriani S, Guerrero‐Valero M, Tozza S, et al. Mutations in MYO9B are associated with Charcot–Marie–Tooth disease type 2 neuropathies and isolated optic atrophy. Eur J Neurol. 2023;30:511‐526. doi: 10.1111/ene.15601
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Laura M, Pipis M, Rossor AM, Reilly MM. Charcot‐Marie‐tooth disease and related disorders: an evolving landscape. Curr Opin Neurol. 2019;32(5):641‐650. [DOI] [PubMed] [Google Scholar]
- 2. Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies. Pract Neurol. 2015;15(3):187‐198. [DOI] [PubMed] [Google Scholar]
- 3. Rossor AM, Tomaselli PJ, Reilly MM. Recent advances in the genetic neuropathies. Curr Opin Neurol. 2016;29(5):537‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6‐14. [DOI] [PubMed] [Google Scholar]
- 5. Allen KF, Gaier ED, Wiggs JL. Genetics of primary inherited disorders of the optic nerve: clinical applications. Cold Spring Harb Perspect Med. 2015;5(7):a017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pipis M, Feely SME, Polke JM, et al. Natural history of Charcot‐Marie‐tooth disease type 2A: a large international multicentre study. Brain. 2020;143(12):3589‐3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pipis M, Rossor AM, Laura M, Reilly MM. Next‐generation sequencing in Charcot‐Marie‐tooth disease: opportunities and challenges. Nat Rev Neurol. 2019;15(11):644‐656. [DOI] [PubMed] [Google Scholar]
- 8. Fridman V, Bundy B, Reilly MM, et al. CMT subtypes and disease burden in patients enrolled in the INC natural history study (6601) from 2009‐2013. J Neurol Neurosurg Psychiat. 2015;86:873‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Previtali SC, Zhao E, Lazarevic D, et al. Expanding the spectrum of genes responsible for hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2019;90(10):1171‐1179. [DOI] [PubMed] [Google Scholar]
- 10. Inoue A, Saito J, Ikebe R, Ikebe M. Myosin IXb is a single‐headed minus‐end‐directed processive motor. Nat Cell Biol. 2002;4(4):302‐306. [DOI] [PubMed] [Google Scholar]
- 11. Bahler M, Elfrink K, Hanley PJ, Thelen S, Xu Y. Cellular functions of class IX myosins in epithelia and immune cells. Biochem Soc Trans. 2011;39(5):1166‐1168. [DOI] [PubMed] [Google Scholar]
- 12. Hanley PJ, Vollmer V, Bahler M. Class IX Myosins: motorized RhoGAP signaling molecules. Adv Exp Med Biol. 2020;1239:381‐389. [DOI] [PubMed] [Google Scholar]
- 13. Kong R, Yi F, Wen P, et al. Myo9b is a key player in SLIT/ROBO‐mediated lung tumor suppression. J Clin Invest. 2015;125(12):4407‐4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanley PJ, Xu Y, Kronlage M, et al. Motorized RhoGAP myosin IXb (Myo9b) controls cell shape and motility. Proc Natl Acad Sci USA. 2010;107(27):12145‐12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez M, Falk MJ, Gai X, Postrel R, Schule R, Zuchner S. Innovative genomic collaboration using the GENESIS (GEM.App) platform. Hum Mutat. 2015;36(10):950‐956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lassuthova P, Rebelo AP, Ravenscroft G, et al. Mutations in ATP1A1 cause dominant Charcot‐Marie‐tooth type 2. Am J Hum Genet. 2018;102(3):505‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hindson BJ, Ness KD, Masquelier DA, et al. High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604‐8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reinhard J, Scheel AA, Diekmann D, Hall A, Ruppert C, Bahler M. A novel type of myosin implicated in signalling by rho family GTPases. EMBO J. 1995;14(4):697‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hemkemeyer SA, Vollmer V, Schwarz V, et al. Local Myo9b RhoGAP activity regulates cell motility. J Biol Chem. 2020;296:100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolino A, Bolis A, Previtali SC, et al. Disruption of Mtmr2 produces CMT4B1‐like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol. 2004;167(4):711‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot‐Marie‐tooth disease. J Periph Nerv Syst. 2011;16:191‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van den Boom F, Dussmann H, Uhlenbrock K, Abouhamed M, Bahler M. The myosin IXb motor activity targets the myosin IXb RhoGAP domain as cargo to sites of Actin polymerization. Mol Biol Cell. 2007;18(4):1507‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Panosyan FB, Laura M, Rossor AM, et al. Cross‐sectional analysis of a large cohort of patients with X‐linked Charcot‐Marie‐tooth disease (CMTX1). Neurology. 2017;89:927‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sevilla T, Cuesta A, Chumillas MJ, et al. Clinical, electrophysiological and morphological findings of Charcot‐Marie‐tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain. 2003;126:2023‐2033. [DOI] [PubMed] [Google Scholar]
- 25. Van Driest SL, Jaeger MA, Ommen SR, et al. Comprehensive analysis of the beta‐myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44(3):602‐610. [DOI] [PubMed] [Google Scholar]
- 26. Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601‐635. [DOI] [PubMed] [Google Scholar]
- 27. Long H, Zhu X, Yang P, Gao Q, Chen Y, Ma L. Myo9b and RICS modulate dendritic morphology of cortical neurons. Cereb Cortex. 2013;23(1):71‐79. [DOI] [PubMed] [Google Scholar]
- 28. Stankiewicz TR, Linseman DA. Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci. 2014;8:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chaya T, Shibata S, Tokuhara Y, et al. Identification of a negative regulatory region for the exchange activity and characterization of T332I mutant of rho guanine nucleotide exchange factor 10 (ARHGEF10). J Biol Chem. 2011;286(34):29511‐29520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roos A, Weis J, Korinthenberg R, et al. Inverted formin 2‐related Charcot‐Marie‐tooth disease: extension of the mutational spectrum and pathological findings in Schwann cells and axons. J Peripher Nerv Syst. 2015;20(1):52‐59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.