

Abstract
Background
We aimed to develop a single step method for the production of human platelet lysate (hPL). The method must result in high hPL yields, be closed system and avoid heparin use.
Study Design and Methods
The method aimed at using glass beads and calcium. An optimal concentration of calcium and glass beads was determined by serial dilution. This was translated to a novel method and compared to known methods: freeze‐thawing and high calcium. Quality outcome measures were transmittance, fibrinogen and growth factor content, and cell doubling time.
Results
An optimal concentration of 5 mM Ca2+ and 0.2 g/ml glass beads resulted in hPL with yields of 92% ± 1% (n = 50) independent of source material (apheresis or buffy coat‐derived). The transmittance was highest (56% ± 9%) compared to known methods (<39%). The fibrinogen concentration (7.0 ± 1.1 μg/ml) was well below the threshold, avoiding the need for heparin. Growth factor content was similar across hPL production methods. The cell doubling time of adipose derived stem cells was 25 ± 1 h and not different across methods. Batch consistency was determined across six batches of hPL (each n = 25 constituting concentrates) and was <11% for all parameters including cell doubling time. Calcium precipitation formed after 4 days of culturing stem cells in media with hPL prepared by the high (15 mM) Ca2+ method, but not with hPL prepared by glass bead method.
Discussion
The novel method transforms platelet concentrates to hPL with little hands‐on time. The method results in high yield, is closed system, without heparin and non‐inferior to published methods.
Keywords: component processing, human platelet lysate, method development
1. INTRODUCTION
Platelets carry many different (bio)molecules that serve diverse purposes in human physiology. Besides factors with known roles in hemostasis and coagulation also cytokines, chemokines, and growth factors are found within the platelet's confinements. These contribute to inflammation and infection control, and support wound healing. 1 This implies that platelets and their cargo may have applications in regenerative medicine.
In the 1970s and 1980s, human platelet lysate (hPL) was indeed shown to support in vitro cell culture. 2 , 3 , 4 , 5 This concept was rediscovered in the early 2000s when hPL was suggested as a xeno‐free replacement for fetal bovine serum (FBS). 6 , 7 The latter has been standard as a supplement in eukaryotic cell culture media for over 50 years of tissue research. 8 The advent of cells and tissues as direct clinical application for treating disease in humans meant that FBS faced increased regulatory scrutiny. Risks of zoonotic infection, risks of patient immunization against bovine antigens, and some ethical concerns 9 , 10 , 11 have been raised.
In the past decade, the European Medicines Agency (EMA) has released guidelines to minimize the use of FBS in cell therapy. 12 , 13 , 14 A potential alternative therefore is hPL, in particular for those cell types that divide slowly or not at all without complex growth factor supplements. 15 Blood establishments manage the platelet inventory for transfusion to the best of their abilities but nonetheless still face losses to expiry between 0.1% and 24.5%. 16 While no longer considered suited for transfusion, these concentrates can serve for hPL preparation. Despite the opportunity to reappraise this leftover donor material, hPL has not replaced FBS. The latter is still intensively used in cell culture research and clinical trials of cellular therapy. 17 The reasons for this have not been mapped, but user's conservatism may play a role as well as genuine concerns over compatibility in specific applications, concerns over (in)sufficient supply, price, and hPL standardization.
A number of hPL production methods exist and each has its particular up‐ and downside making it difficult to appoint a unified method suited to process all platelet concentrates across countries and continents. Repeated freeze‐thawing or sonication for instance does not remove fibrinogen, thus requiring the addition of heparin to basal media so to avoid fibrin formation during cell growth. Using (human) thrombin to generate hPL solves this issue but comes at cost and leaves behind a serine protease in the final product. 18 Alternatively, hPL may be produced by raising ionized calcium (Ca2+) concentrations in platelet concentrates, resulting in similar growth factor levels as freeze‐thawing. 19 This hPL will inevitably contain high Ca2+ concentrations which can sometimes interfere with cellular biology. 20 In addition, many blood establishments prepare concentrates in platelet additive solutions (PAS) that often contain millimolar concentrations of phosphate anions. High Ca2+ will thus form insoluble phosphate complexes leading to turbid salt precipitations that are detrimental to cells. Finally and perhaps foremost, the ideal hPL production method is easily blended onto the blood establishment's production chain at low cost without compromising high clinical grade standards.
In this paper, we propose a one‐step method that exploits the catalytic effect of glass on coagulation allowing to significantly lower Ca2+ levels required to obtain full platelet activation. Our method overcomes the problems described above and is compatible with closed system manufacturing. It does not require centrifugation nor filtration and can easily be implemented in a blood establishment's routine, thereby contributing to standardization and reduce intra‐ but also inter‐manufacturer variability.
2. MATERIALS AND METHODS
2.1. Platelet concentrate preparation
Platelet concentrates (PC) were prepared by apheresis or by the buffy coat method as described. 21 , 22 The buffy coat method manually pools six ABO blood group matched buffy coats. This pool was supplemented with 280 ml of a platelet additive solution, PAS‐E (Macopharma, Mouvaux, France). This bag was centrifuged at 542 g for 450 s at 22°C. The buoyant platelet suspension was taken off by automated separation (Macopress Smart, Macopharma) while passing over a WBC reduction filter. The entire process of pooling, centrifugation, and leukoreduction was performed with a CompoStop set (Fresenius Kabi, Bad Homburg, Germany). All PC were treated with the amotosalen and ultraviolet light pathogen inactivation method (Cerus Corporation, Concord, CA). The PC were stored in a temperature controlled environment (22°C) with continuous agitation. All donations were from voluntary non‐remunerated donors who gave informed consent for all experiments performed on the use of the donation or of fractions of the donation for scientific research purposes. Experiments adhered to legislation and were within the goals and activities of the issuing biobank (BB190034) that were approved by an Institutional Review Board (UZ/KU Leuven, ethical committee with approval code S62549).
2.2. Preparation of hPL
For determination of the optimal glass bead content and Ca2+ concentration, paired experiments were performed in conical 50 ml tubes (Greiner Bio‐One, Kremsmünster, Austria). Based on these results, hPL was next prepared in prototype bags using PC between 6 and 10 days post donation. The individual PC were transferred to a single bag containing 0.2 g/ml autoclaved glass beads of 3 mm in diameter (Sigmund Lindner, Warmensteinach, Germany) and 1% (vol/vol) of a sterile calcium chloride solution in water (Bio‐world, Dublin, OH) reaching a final average concentration of 5 mM Ca2+, assuming a mean PC volume of 350 ml. An overview of this single‐step method is shown in Figure S1. After mixing PC were rested for 3 h at 22°C to complete coagulation and retraction. Time to onset of retraction was the time lapsed from PC transfer to the bag until the coagulum visually detached from the bag's periphery. After retraction, the bags were stored at −80°C for at least 24 h. Thawing was at 22°C for 16 h after which the hPL was separated from the coagulum by welding and transferring to an empty transfusion bag. For paired experiments, PC were pooled and split and subsequently used to produce hPL according to either the above mentioned single‐step method or one of the following two methods: (1) hPL produced with the freeze‐thaw method was performed as described. 7 , 23 , 24 , 25 , 26 , 27 , 28 Briefly, a PC was frozen and thawed three times at −80°C for at least 24 h and 22°C for 16 h. The resulting product was centrifuged at 4500 g for 10 min to remove cell debris. (2) hPL produced with the high calcium method was performed by transferring the PC to a single bag containing a concentrated, sterile calcium chloride solution in water reaching a final 15 mM Ca2+ after transfer. For comparison of batches, 25 individual hPL products were pooled per batch regardless of blood type but with distinction of source product (apheresis or buffy coat‐derived).
2.3. Biophysical and biochemical assays
Transmittance was calculated using the formula T = 10(2‐A680nm), where T is transmittance and A680nm the absorbance of undiluted hPL measured at a wavelength of 680 nm in a spectrophotometer (NanoDrop OneC, Thermo Fisher Scientific, Waltham, MA). The system was calibrated using 0.2 μm filtered distilled water as a blank. Yield of hPL was the fraction of (hPL) volume retrieved from the original PC volume, given as percent. Concentrations of Ca2+ were measured in a point‐of‐care blood gas analyzer (RAPIDPoint 500, Siemens, Munich, Germany). pH was determined by glass electrode (pH1000L, VWR, Radnor, PA). Phosphate concentration was determined with a colorimetric assay kit following the manufacturer's instructions (Abcam, Cambridge, UK).
2.4. Growth factor concentration
Concentrations of Epidermal Growth Factor (EGF), Fibroblast Growth Factor (FGF), Insulin‐like Growth Factor (IGF)‐I, Platelet‐Derived Growth Factor (PDGF)‐AB, Transforming Growth Factor Beta 1 (TGF‐ß1), and Vascular Endothelial Growth Factor (VEGF) were measured using a quantitative sandwich‐type Enzyme‐Linked Immuno Sorbent Assay (ELISA) kit (R&D Systems, Minneapolis, MN) according to the kit's manual. In brief, samples were serially diluted in calibrator diluent and added to a 96‐well microtiter plate coated with antibodies specific for the antigens listed. For IGF‐I analysis, samples were first diluted in an acidic dissociation solution and a blue dyed buffer. For TGF‐ß1 analysis, the sample was first activated by addition of 1.0 M HCl and neutralized with 1.2 M NaOH and 0.5 M 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES). Detection was with a polyclonal anti‐human antibody specific for the antigens listed and conjugated with horseradish peroxidase. Chromogenic substrate was tetramethylbenzidine. Resulting optical densities at 450 nm were measured using a multiwell plate reader (Infinite F200 Pro).
2.5. Fibrinogen and total protein
Fibrinogen was quantified using ELISA (Molecular Innovations, Novi, MI) according to the provider's instructions. Samples were added to the pre‐coated 96‐well microtiter plate and incubated at 22°C for 30 min on a horizontal orbital microplate shaker. Polyclonal anti‐human fibrinogen primary biotinylated antibody was added and incubated at 22°C for 30 min. Detection of bound antibody was with horseradish peroxidase‐conjugated streptavidin. Detection was as described for the growth factors ELISA's. Total protein in samples was determined with a colorimetric bicinchoninic acid assay (Thermo Fisher Scientific) according to the kit's manual.
2.6. Cell expansion and detection of calcium complex precipitation
Complete culture media were prepared by supplementation of 1% (vol/vol) penicillin–streptomycin (100 U/ml, Thermo Fisher Scientific) and 10% (vol/vol) hPL or Fetal Bovine Serum (FBS, Thermo Fisher Scientific) to DMEM (Thermo Fisher Scientific, reference 11885084) low in glucose, with pyruvate. Culture media containing hPL obtained from freeze‐thawing PCs was supplemented with heparin (2 U/ml final, Zen‐Bio, Durham, NC). Prior to use all complete media preparations were filtered by vacuum filtertop 0.2 μm flasks (Rapid‐Flow, Thermo Fisher Scientific). Human adipose derived mesenchymal stem cells (hADSCs, Lonza, Basel, Switzerland) were thawed at 37°C and washed with culture medium using centrifugation at 300 g for 5 min. Efficacy was tested in three different clones. Cells were cultured in flasks (Greiner Bio‐One) at 37°C and 5% partial CO2 pressure. Culture medium was changed every 2–3 days until the cells reached 80%–90% confluence at which point the cultures were split using trypsin‐ethylenediaminetetraacetic acid (EDTA, Thermo Fisher Scientific). Calcium complex precipitation was identified using Alizarin Red S staining. Medium was removed from the culture flasks and 2% (wt/vol) Alizarin Red S (Sigma‐Aldrich) staining solution in dH2O was added. After a brief incubation of 10 min at room temperature, the Alizarin Red S staining solution was discarded and the flask was thoroughly washed with distilled water.
2.7. Cell count and cell doubling time
After expansion, hADSCs were diluted in trypan blue (0.16% (wt/vol), Carl Roth, Karlsruhe, Germany) and counted using a Malassez counting chamber (Brand, Wertheim, Germany). The cells were seeded at 3000 cells/cm2 in culture flasks and placed in culture conditions. Medium was changed after 24 h. After a total incubation of 96 h, all cells were trypsinized and counted. Cell doubling time (CDT) was calculated according to following formula where t is the time from seeding to detachment in hours, N 0 is the amount of cells at seeding, and N 1 is the amount of cells after 96 h expansion: . 29
2.8. Statistical analysis
All statistical analyses were performed using Prism Version 9.4.1 (GraphPad Software Inc.). A repeated measures one‐way ANOVA for comparison of hPL production methods was used. A Sidak's correction for multiple comparison was included. Null hypotheses were rejected when p < .05.
3. RESULTS
3.1. Optimal Ca2+ and glass bead content for high hPL yield
To determine the optimal combination of Ca2+ and glass content, serial dilutions of both constituents were prepared in conical test tubes before adding platelet concentrate (Figure S2). As a primary outcome measure the moment of clot retraction was determined (Figure 1A). At 15 mM Ca2+ platelet suspensions fully retracted within 90 min independent of glass beads. Glass beads alone or the addition of 4 mM or 5 mM Ca2+ alone could not initiate coagulation or retraction within 5 h of observation. The combination of both glass and these low Ca2+ concentrations however always caused coagulation and retraction within that time frame. This catalytic effect was furthermore dose dependent because more glass beads caused faster clot retraction. The combination of any glass quantity and 5 mM Ca2+ was always more efficient than any glass quantity and 4 mM Ca2+. Subsequently, hPL yield was highest when glass was combined with 5 mM Ca2+ compared to with 4 mM Ca2+ (Figure 1B).
FIGURE 1.
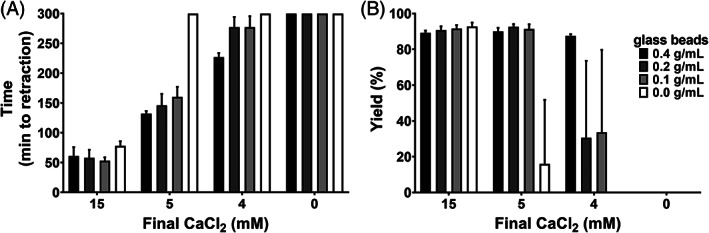
Optimal CaCl2 concentration and glass contents for coagulum retraction. (A) the time (minutes, min) lapsed from addition of CaCl2 and glass until the start of coagulum retraction was determined. Onset of retraction was confirmed or refuted by visual inspection every 5 min for a maximum follow‐up of 5 h. (B) the resulting hPL was harvested and the yield (%) was calculated by dividing the volume of harvested hPL by the original PC volume. Data in both panels are shown as mean with SD (n = 5). Gray shading of bars in both panels corresponds to glass content, given in the legend as inset.
3.2. Comparison of three hPL production methods
The optimal experimental conditions of 5 mM Ca2+ and 0.2 g/ml of glass were next investigated in bags, so to recreate blood banking operations (Figure 2A and Video S1). Individual platelet concentrates of either apheresis or buffy coat origin all coagulated and retracted within 120 min (n = 50). The hPL yield was 92% ± 1% independent of apheresis or buffy coat origin (Figure 2B). The time lapse video depicts the dynamic process of coagulation, platelet activation, and clot retraction in and around the glass fraction.
FIGURE 2.
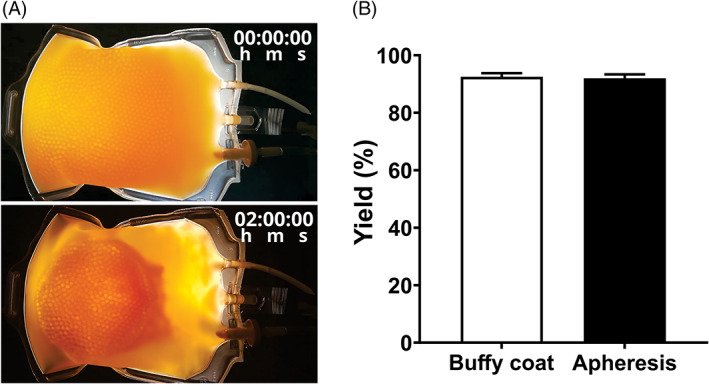
Visual representation of retraction and yielded hPL. (A) Selected timeframes of the supplementary video representing a PC treated with the glass bead method before (top) and after (bottom) clot retraction. (B) hPL yield (%) was determined by dividing the volume of harvested hPL by the original PC volume of either buffy coat‐derived PC or apheresis PC. Data are shown as mean with SD (n = 50).
We hypothesized that the glass beads act as a scaffold for retaining particulate debris. To test this, we compared light transmittance of hPL prepared by three different methods: (i) repeated freeze‐thawing, (ii) 15 mM Ca2+ without glass, or (iii) the low calcium method of 5 mM Ca2+ and 0.2 g/ml glass. Light transmittance was twofold higher (56% ± 9%) in hPL prepared by Ca2+ with glass compared to Ca2+ without glass (27% ± 7%, n = 4) (Figure 3A). These data show minimal turbidity when 0.2 g/ml glass is combined with 5 mM Ca2+. In addition, rest fibrinogen was <10 μg/ml in hPL prepared by either Ca2+‐based method while the freeze‐thaw method retained high fibrinogen concentrations >3 mg/ml (Figure 3B). As a marker for degranulation, PDGF‐AB concentration was determined by ELISA and was not different between preparations (Figure 3C). Assuming that PDGF‐AB release is maximal using freeze‐thawing, this method can be used as a reference, indicating that PDGF‐AB recovery in hPL prepared by 15 mM Ca2+ without glass is 93% and in hPL prepared by 5 mM Ca2+ with 0.2 g/ml glass is 100%. The cell doubling time of hADSC was not different between the hPL preparations at 10% (vol/vol) in DMEM (Figure 3D). For this experiment a single clone of primary hADSC was used.
FIGURE 3.
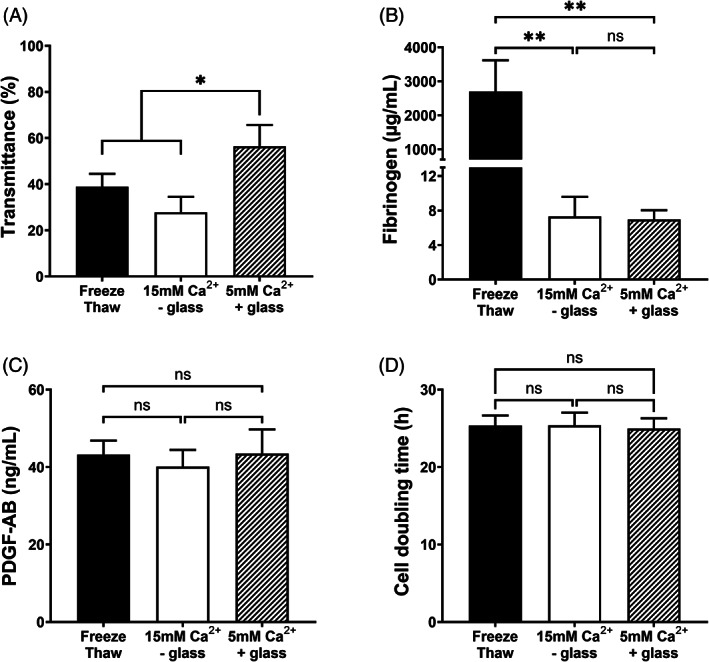
Physical and biochemical properties of hPL per method. The freeze‐thawing (black bars) and high calcium (15 mM Ca2+ without glass, white bars) methods were compared with the 5 mM Ca2+ with glass method (shaded bars) for (A) transmittance, (B) fibrinogen concentration, (C) PDGF‐AB concentration, and (D) hADSC doubling time (h). Data are shown as mean with SD (n = 4), *p < .05 and **p < .01.
3.3. Calcium complex precipitation in complete medium and its influence on cell culture
In a paired experiment, hADSCs were split and cultured in DMEM supplemented with 10% (vol/vol) hPL prepared by repeated freeze‐thawing, by addition of 15 mM Ca2+ without glass or by addition of 5 mM Ca2+ with 0.2 g/ml glass. Supplementation with FBS was used as control condition (Figure 4). Qualitative analysis by bright field microscopy showed particulate precipitates on day 4 in cultures prepared with hPL by 15 mM Ca2+ without glass. Filtration (0.2 μm) of media could not prevent calcium complex precipitate formation. Although cells were present in conditions with precipitate these could not be harvested using trypsin. Alizarin Red S staining was performed to identify the precipitate as calcium deposits. Furthermore, precipitates were easily dissolved within seconds upon incubation with EDTA 50 mM (pH 7.4) suggesting involvement of Ca2+ cations (Figure S3). In addition, we studied cell doubling time of three different hADSC clones in hPL prepared from the same source product but by different preparation methods (Figure 5). We found that some clones expanded slower in medium containing hPL prepared by addition of 15 mM Ca2+ without glass compared to the other two hPL production methods.
FIGURE 4.
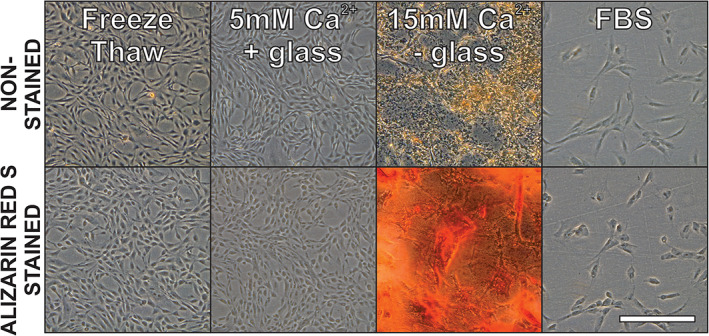
Calcium complexes precipitate during hADSC culture. Representative pictures of hADSCs culture in DMEM with 10% (vol/vol) hPL prepared by three different methods are shown. Top row depicts the cells after 4 days of adherent culture but before Alizarin Red S staining. The bottom row depicts the same flasks but after alizarin red S staining. Data were retrieved by bright field microscopy on day 4 post seeding at 50x total magnification. Scale bar = 500 μm.
FIGURE 5.
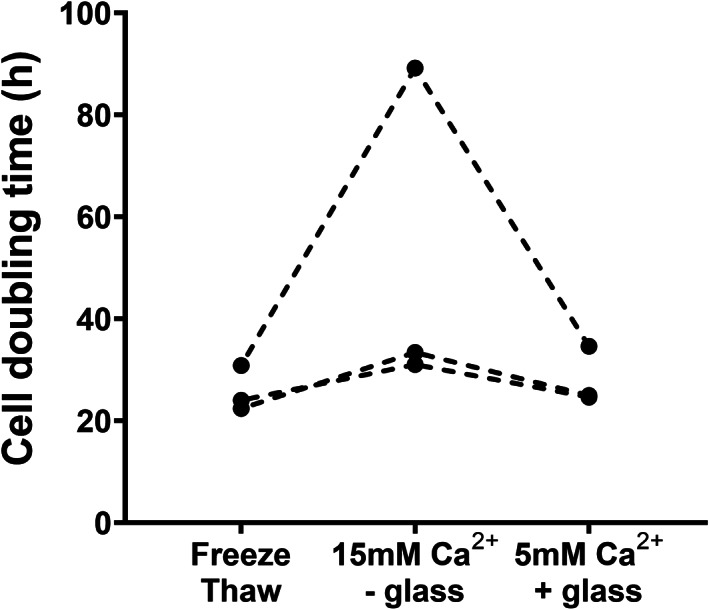
Cell doubling time of three hADSC clones in hPL. Three different hADSC clones were cultured with hPL prepared by different methods but from the same platelet concentrate as starting material. Cells were detached after 4 days of culture and counted in a hemocytometer. Only viable cells were counted using trypan blue staining. Individual data are shown and paired clones are connected with dotted lines.
Chemical analysis demonstrated a fourfold higher (p < .0001) calcium concentration in hPL prepared by 15 mM Ca2+ without glass compared to hPL prepared by 5 mM Ca2+ with 0.2 g/ml glass (Figure 6A). In addition, significantly lower pH (p < .01) was found in hPL by 15 mM Ca2+ without glass compared to hPL by 5 mM Ca2+ and 0.2 g/ml glass (Figure 6B). Phosphate levels were around 20 mM in both hPL preparations and not significantly different (Figure 6C).
FIGURE 6.
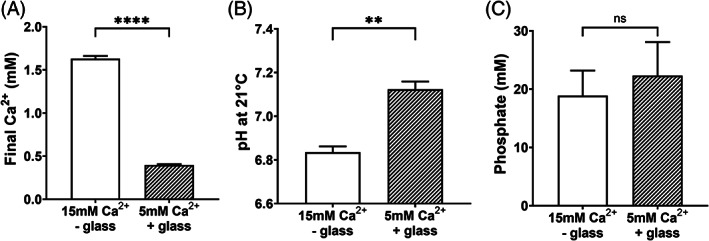
Comparison of two hPL methods by chemical composition. The high calcium (15 mM Ca2+ without glass, white bars) method was compared with the 5 mM Ca2+ with glass method (shaded bars) for (A) final Ca2+ concentrations, (B) pH at 21°C, and (C) phosphate concentrations. Data are shown as mean with SD (n = 4), **p < .01 and ****p < .0001.
3.4. Growth factor content and batch variation
Three independent batches of pooled hPL were prepared next. Hereto, 25 individual hPL products were mixed. One batch was derived from buffy coat PC and two from apheresis PC, the former consisting of 150 donors and the latter two of 25 donors. Growth factor content in every batch is presented in Table 1. To determine batch consistency on general quality outcome measures, six additional batches of pooled hPL were prepared (Table 2). Three batches were derived from buffy coat PC and three from apheresis PC. The coefficient of variation was always below 11% for the parameters tested: volume yield, calcium levels, transmittance, pH, fibrinogen concentration, total protein concentration, PDGF‐AB concentration, and cell doubling time and this was independent of the nature of the source product.
TABLE 1.
Growth factor content in three hPL batches prepared by pooling of 25 individual hPL solutions prepared by the 5 mM Ca2+ with glass bead method
| EGF | FGF | IGF‐I | PDGF‐AB | TGF‐ß1 | VEGF | |
|---|---|---|---|---|---|---|
| (ng/ml) | (ng/ml) | (ng/ml) | (ng/ml) | (ng/ml) | (ng/ml) | |
| Batch 1 | 2.52 | 0.144 | 44.25 | 37.42 | 119.9 | 0.66 |
| Batch 2 | 2.64 | 0.138 | 44.63 | 40.78 | 141.0 | 0.73 |
| Batch 3 | 2.72 | 0.148 | 44.18 | 39.13 | 128.9 | 0.65 |
| Mean | 2.63 | 0.143 | 44.35 | 39.11 | 129.9 | 0.68 |
| ±SD | 0.09 | 0.004 | 0.20 | 1.37 | 8.6 | 0.03 |
Note: The concentration of a selection of growth factors in the final hPL batch is presented. The bottom rows present means with SD (n = 3).
TABLE 2.
Batch consistency in six independent hPL batches
| Yield (%) | Transmittance (%) | pH (at 21°C) | Calcium (mM) | Fibrinogen (μg/ml) | Total protein (g/L) | PDGF‐AB (ng/ml) | Cell doubling time (h) | ||
|---|---|---|---|---|---|---|---|---|---|
| Buffy coat | Batch 1 | 84.6 | 50.5 | 6.990 | 0.34 | 5.0 | 21.7 | 35.7 | 30.7 |
| Batch 2 | 87.0 | 51.2 | 7.043 | 0.41 | 4.9 | 20.4 | 40.9 | 32.8 | |
| Batch 3 | 88.0 | 53.8 | 7.040 | 0.40 | 4.5 | 23.1 | 42.1 | 35.6 | |
| Mean | 86.5 | 51.8 | 7.024 | 0.38 | 4.8 | 21.7 | 39.6 | 33.0 | |
| SD | 1.4 | 1.4 | 0.024 | 0.03 | 0.2 | 1.1 | 2.8 | 2.0 | |
| CV (%) | 1.6 | 2.8 | 0.3 | 8.1 | 4.2 | 5.0 | 7.0 | 6.1 | |
| Apheresis | Batch 1 | 87.8 | 65.6 | 7.042 | 0.48 | 5.1 | 24.3 | 40.4 | 33.4 |
| Batch 2 | 87.0 | 60.3 | 6.977 | 0.43 | 5.2 | 21.1 | 35.9 | 38.0 | |
| Batch 3 | 90.0 | 60.6 | 7.029 | 0.46 | 4.6 | 20.1 | 35.6 | 35.0 | |
| Mean | 88.3 | 62.1 | 7.016 | 0.46 | 5.0 | 21.8 | 37.3 | 35.5 | |
| SD | 1.3 | 2.5 | 0.028 | 0.02 | 0.3 | 1.8 | 2.2 | 1.9 | |
| CV (%) | 1.4 | 4.0 | 0.4 | 4.5 | 5.4 | 8.1 | 5.9 | 5.3 | |
| All batches | Mean | 87.4 | 57.0 | 7.020 | 0.42 | 4.9 | 21.8 | 38.4 | 34.2 |
| SD | 1.6 | 5.5 | 0.027 | 0.05 | 0.3 | 1.5 | 2.8 | 2.3 | |
| CV (%) | 1.8 | 9.7 | 0.4 | 10.7 | 5.2 | 6.7 | 7.2 | 6.8 | |
Abbreviations: CV, coefficient of variation; SD, standard deviation.
4. DISCUSSION
Cell and tissue culture is a pivotal laboratory practice for both basic and applied (clinical) research. For many cell types, chemically defined media have been developed avoiding the need for undefined crude proteinaceous growth factor additives like FBS. However, some cell types cannot yet cost‐effectively expand without it. One important example is the mesenchymal stem cell family that is a primary source for many promising cell therapies under investigation worldwide. 30
Excess PC are currently discarded as medical waste in blood institutions worldwide 16 , 31 despite being a precious source of human derived growth factors to supplement cell and tissue culture media. These products are an opportunity for blood establishments and for their donors to diversify product portfolio's and donate for medical needs beyond transfusion respectively. Because PC are however not manufactured in a uniform manner across blood establishments, hPL production would benefit from a simple one‐step procedure that can be emulated on the blood component production systems of blood institutions. 32 , 33 With this in mind, we aimed to resolve four critical factors targeting the development of a disposable that allows quick and standardized production of hPL; (1) safeguard closed system manufacturing, (2) reduce hands‐on time, (3) reduce hardware investments (e.g., filtration systems, pumps, centrifuges, additional freezers) and (4) avoid biomolecular additives (e.g., heparin, thrombin).
Glass beads and CaCl2 solutions are stable and can thus be (steam) sterilized inside a custom built container. Equipped with compatible tubing a PC can be connected using sterile welding. This allows transfer in a closed system followed by platelet activation, coagulation, and retraction over a maximal time course of 3 h without further manipulation. The resulting product is then stored at <−20°C until used for pooling.
The method furthermore does not require centrifugation or filtration steps. Such steps are expensive and labor‐intensive in addition to the requirements of certain investment and overhead costs for centrifuges and pumps. Centrifugation and filtration also unavoidably cause lower yields as both techniques lead to dead volumes. In addition, the single step closed bag system can easily be implemented in the current blood banking routine because it is compatible with standard blood bank operations.
The original method for releasing platelet content and produce hPL is freeze‐thawing. 34 Although elegant and compatible with closed system manufacturing, this method does not remove fibrinogen leading to “gel formation” when added to common basal media. 35 Basal media contain up to 1.8 mM of Ca2+ causing spontaneous coagulation of freeze‐thaw hPL in culture flasks, hence “gel formation”. In addition, current platelet containers (e.g., PL1240 polyvinylchloride) have not been validated for repeated freeze‐thawing. To our knowledge this issue has not been specifically investigated, but the risk for material tear and sterility breach during cold processing exists and requires attention. In our proposed method, the PC is first transferred to a new bag by sterile welding. This cryocompatible bag contains an optimized content of glass beads and Ca2+ ions. Glass beads act as a catalyst for coagulation initiation, increasing the rate of coagulation near its surface thereby also entrapping both platelet debris and coagulum. Glass is an established catalyst for coagulation and has been used in experimental hPL production. 36 In platelet suspensions containing anticoagulant solution it was however never optimized to maximally reduce the required Ca2+ concentration.
The latter is important, in particular when platelets are kept in phosphate buffered conditions as in most contemporary platelet additive solutions. 37 Calcium phosphates have poor solubility at 37°C and at neutral pH. 38 Consequently, supplementation of hPL (prepared by high Ca2+) to common basal media can lead to calcium complex precipitation of insoluble particulate matter. This phenomenon progresses gradually during cell culture and is often mistakenly interpreted as bacterial contamination. In addition, such precipitation precludes normal microscopic assessment and prevents further manipulation, such as cell harvesting. Using glass as a catalyst, the threshold Ca2+ concentration for platelet activation and component coagulation can thus be lowered so to reduce or entirely prevent calcium complex precipitation without compromising hPL yield.
This hPL was more translucent than hPL prepared by other methods. It contained only trace fibrinogen, but high concentrations of growth factors. Final Ca2+ concentration and pH were at physiologic levels and this hPL remained free of animal derived components, including heparin. Moreover, the hPL was at least as effective for expansion of hADSCs as hPL produced by other methods. Although this study focused on hADSCs, many other sources of stromal cells have already been shown to expand well in hPL such as bone marrow‐derived, corneal stroma‐derived cells, and periosteum‐derived cells. 29 , 39 , 40 Additionally, other cells lines were previously demonstrated to benefit from hPL. 41 , 42 , 43 , 44 , 45 , 46 We anticipate that other cell types may equally benefit from hPL produced by our method.
In conclusion, our method is easy, quick, and efficient to produce high quality and highly standardized clinical grade hPL. By using glass beads to lower Ca2+ concentrations for platelet activation and hPL production, this method is also applicable to most PC worldwide with or without additive solution, collected by cytapheresis or from buffy coats, fresh or “expired”. This method is highly suited for standardization across blood establishments.
CONFLICT OF INTEREST
PV, VC, and HBF are authors on a pending patent concerning the technology discussed in the paper. All other authors declare no conflict of interest.
Supporting information
Figure S1. Single step hPL production. A PC is transferred to a sterile bag containing a CaCl2 in water solution (final ~5 mM Ca2+) and 0.2 g/ml. of glass beads. After gentle mixing, the bag is rested for 3 h at 22°C at which point coagulation and retraction is final. The remaining supernatant is a platelet releasate rich in growth factors. The bag with coagulum can be stored <−20°C until further use.
Figure S2. Finding the optimal ratio of Ca2+ and glass to consistently produce hPL. Four conditions are depicted. From left to right: (i) 15 mM Ca2+; (ii) 5 mM Ca2+; (iii) 0.2 g/ml glass; (iv) 5 mM Ca2+ + 0.2 g/ml glass. After 3 h of observation, coagulation and retraction was consistently found in the conditions with high (15 mM) Ca2+ and in the condition with low (5 mM) Ca2+ combined with glass. Low calcium or glass alone could not elicit a response.
Figure S3. Dissolution of calcium complex precipitation by EDTA. Representative pictures of hADSCs culture in DMEM with 10% (vol/vol) hPL prepared by 15 mM Ca2+ are shown. The left panel depicts the bottom of the tissue culture flask with hADSC after 4 days of adherent culture and the right panel depicts the same flask after addition of 50 mM EDTA (pH 7.4). Data were retrieved by bright field microscopy at 50× total magnification. Scale bar = 500 μm.
Video S1. Single step production of hPL. A PC was transferred to a sterile bag containing 5 mM Ca2+ and 0.2 g/ml of glass beads and filmed for three consecutive hours. The time‐lapse video was digitally increased 120‐fold and static parts (0–1 h real‐time) are not included.
ACKNOWLEDGMENTS
This work was supported by the Foundation for Scientific Research of the Belgian Red Cross‐Flanders. This work was supported in part by a Flanders Innovation and Entrepreneurship (VLAIO) grant with reference HBC.2017.0522. This study received funding through the Regenerative Medicine Crossing Borders REGMEDXB (http://www.regmedxb.com) program, powered by EWI Flanders.
Delabie W, De Bleser D, Vandewalle V, Vandekerckhove P, Compernolle V, Feys HB. Single step method for high yield human platelet lysate production. Transfusion. 2023;63(2):373–383. 10.1111/trf.17188
Funding information Agentschap Innoveren en Ondernemen, Grant/Award Number: HBC.2017.0522; Foundation for Scientific Research of the Belgian Red Cross‐Flanders; Regenerative Medicine Crossing Borders REGMEDXB, powered by EWI Flanders
REFERENCES
- 1. Burnouf T, Strunk D, Koh MB, Schallmoser K. Human platelet lysate: replacing fetal bovine serum as a gold standard for human cell propagation? Biomaterials. 2016;76:371–87. [DOI] [PubMed] [Google Scholar]
- 2. Kohler N, Lipton A. Platelets as a source of fibroblast growth‐promoting activity. Exp Cell Res. 1974;87:297–301. [DOI] [PubMed] [Google Scholar]
- 3. Westermark B, Wasteson A. A platelet factor stimulating human normal glial cells. Exp Cell Res. 1976;98:170–4. [DOI] [PubMed] [Google Scholar]
- 4. Choi YC, Morris GM, Sokoloff L. Effect of platelet lysate on growth and sulfated glycosaminoglycan synthesis in articular chondrocyte cultures. Arthritis Rheum. 1980;23:220–4. [DOI] [PubMed] [Google Scholar]
- 5. Hara Y, Steiner M, Baldini MG. Platelets as a source of growth‐promoting factor(s) for tumor cells. Cancer Res. 1980;40:1212–6. [PubMed] [Google Scholar]
- 6. Lucarelli E, Beccheroni A, Donati D, Sangiorgi L, Cenacchi A, Del Vento AM, et al. Platelet‐derived growth factors enhance proliferation of human stromal stem cells. Biomaterials. 2003;24:3095–100. [DOI] [PubMed] [Google Scholar]
- 7. Doucet C, Ernou I, Zhang Y, Llense JR, Begot L, Holy X, et al. Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell‐based therapy applications. J Cell Physiol. 2005;205:228–36. [DOI] [PubMed] [Google Scholar]
- 8. Puck TT, Cieciura SJ, Robinson A. Genetics of somatic mammalian cells. III. Long‐term cultivation of euploid cells from human and animal subjects. J Exp Med. 1958;108:945–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Erickson GA, Bolin SR, Landgraf JG. Viral contamination of fetal bovine serum used for tissue culture: risks and concerns. Dev Biol Stand. 1991;75:173–5. [PubMed] [Google Scholar]
- 10. Jochems CE, van der Valk JB, Stafleu FR, Baumans V. The use of fetal bovine serum: ethical or scientific problem? Altern Lab Anim. 2002;30:219–27. [DOI] [PubMed] [Google Scholar]
- 11. Gstraunthaler G, Lindl T, van der Valk J. A plea to reduce or replace fetal bovine serum in cell culture media. Cytotechnology. 2013;65:791–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. EMEA/CHMP . In: EMEA , editor. Guideline on human cell‐based medicinal products. Volume 7. London, UK: Westferry Circus, Canary Wharf; 2008. [Google Scholar]
- 13. EMA/CHMP/BWP . In: EMA , editor. Guideline on the use of bovine serum in the manufacture of human biological medicinal products. Volume 7. London (UK): Westferry Circus Canary Wharf; 2013. [Google Scholar]
- 14. EMA . Note for guidance on minimising the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinal products. Vol 7. London, UK: Westferry Circus Canary Wharf; 2011. [Google Scholar]
- 15. van der Valk J, Bieback K, Buta C, Cochrane B, Dirks WG, Fu J, et al. Fetal Bovine Serum (FBS): Past ‐ Present ‐ Future. ALTEX. 2018;35:99–118. [DOI] [PubMed] [Google Scholar]
- 16. Flint AWJ, McQuilten ZK, Irwin G, Rushford K, Haysom HE, Wood EM. Is platelet expiring out of date? A systematic review. Trans Med Rev. 2020;34:42–50. [DOI] [PubMed] [Google Scholar]
- 17. Karnieli O, Friedner OM, Allickson JG, Zhang N, Jung S, Fiorentini D, et al. A consensus introduction to serum replacements and serum‐free media for cellular therapies. Cytotherapy. 2017;19:155–69. [DOI] [PubMed] [Google Scholar]
- 18. Cheng CM, Meyer‐Massetti C, Kayser SR. A review of three stand‐alone topical thrombins for surgical hemostasis. Clin Ther. 2009;31:32–41. [DOI] [PubMed] [Google Scholar]
- 19. Mareschi K, Marini E, Niclot A, Barone M, Pinnetta G, Adamini A, et al. A new human platelet lysate for mesenchymal stem cell production compliant with good manufacturing practice conditions. Int J Mol Sci. 2022;23:3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gigout A, Jolicoeur M, Buschmann MD. Low calcium levels in serum‐free media maintain chondrocyte phenotype in monolayer culture and reduce chondrocyte aggregation in suspension culture. Osteoarthr Cartil. 2005;13:1012–24. [DOI] [PubMed] [Google Scholar]
- 21. Hardwick J. Blood processing. ISBT Sci Series. 2008;3:148–76. [Google Scholar]
- 22. Van Aelst B, Feys HB, Devloo R, Vanhoorelbeke K, Vandekerckhove P, Compernolle V. Riboflavin and amotosalen photochemical treatments of platelet concentrates reduce thrombus formation kinetics in vitro. Vox Sang. 2015;108:328–39. [DOI] [PubMed] [Google Scholar]
- 23. Schallmoser K, Bartmann C, Rohde E, Reinisch A, Kashofer K, Stadelmeyer E, et al. Human platelet lysate can replace fetal bovine serum for clinical‐scale expansion of functional mesenchymal stromal cells. Transfusion. 2007;47:1436–46. [DOI] [PubMed] [Google Scholar]
- 24. Fekete N, Gadelorge M, Furst D, Maurer C, Dausend J, Fleury‐Cappellesso S, et al. Platelet lysate from whole blood‐derived pooled platelet concentrates and apheresis‐derived platelet concentrates for the isolation and expansion of human bone marrow mesenchymal stromal cells: production process, content and identification of active components. Cytotherapy. 2012;14:540–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lohmann M, Walenda G, Hemeda H, Joussen S, Drescher W, Jockenhoevel S, et al. Donor age of human platelet lysate affects proliferation and differentiation of mesenchymal stem cells. PLoS One. 2012;7:e37839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kerkis I, Jonsdottir‐Buch SM, Lieder R, Sigurjonsson OE. Platelet lysates produced from expired platelet concentrates support growth and osteogenic differentiation of mesenchymal stem cells. PLoS One. 2013;8:e68984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kinzebach S, Dietz L, Kluter H, Thierse HJ, Bieback K. Functional and differential proteomic analyses to identify platelet derived factors affecting ex vivo expansion of mesenchymal stromal cells. BMC Cell Biol. 2013;14:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Castiglia S, Mareschi K, Labanca L, Lucania G, Leone M, Sanavio F, et al. Inactivated human platelet lysate with psoralen: a new perspective for mesenchymal stromal cell production in good manufacturing practice conditions. Cytotherapy. 2014;16:750–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matthyssen S, Ni Dhubhghaill S, Van Gerwen V, Zakaria N. Xeno‐free cultivation of mesenchymal stem cells from the corneal stroma. Invest Ophthalmol Vis Sci. 2017;58:2659–65. [DOI] [PubMed] [Google Scholar]
- 30. Bieback K, Fernandez‐Munoz B, Pati S, Schafer R. Gaps in the knowledge of human platelet lysate as a cell culture supplement for cell therapy: a joint publication from the AABB and the International Society of Cell Therapy. Cytotherapy. 2019;21:911–24. [DOI] [PubMed] [Google Scholar]
- 31. Veihola M, Aroviita P, Linna M, Sintonen H, Kekomaki R. Variation of platelet production and discard rates in 17 blood centers representing 10 European countries from 2000 to 2002. Transfusion. 2006;46:991–5. [DOI] [PubMed] [Google Scholar]
- 32. Astori G, Amati E, Bambi F, Bernardi M, Chieregato K, Schafer R, et al. Platelet lysate as a substitute for animal serum for the ex‐vivo expansion of mesenchymal stem/stromal cells: present and future. Stem Cell Res Ther. 2016;7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henschler R, Gabriel C, Schallmoser K, Burnouf T, Koh MBC. Human platelet lysate current standards and future developments. Transfusion. 2019;59:1407–13. [DOI] [PubMed] [Google Scholar]
- 34. Strunk D, Lozano M, Marks DC, Loh YS, Gstraunthaler G, Schennach H, et al. International forum on GMP‐grade human platelet lysate for cell propagation. Vox Sang. 2018;113:e1–e25. [DOI] [PubMed] [Google Scholar]
- 35. McKee TJ, Komarova SV. Is it time to reinvent basic cell culture medium? Am J Physiol Cell Physiol. 2017;312:C624–C6. [DOI] [PubMed] [Google Scholar]
- 36. Kao YC, Bailey A, Samminger B, Tanimoto J, Burnouf T. Removal process of prion and parvovirus from human platelet lysates used as clinical‐grade supplement for ex vivo cell expansion. Cytotherapy. 2016;18:911–24. [DOI] [PubMed] [Google Scholar]
- 37. Ringwald J, Zimmermann R, Eckstein R. The new generation of platelet additive solution for storage at 22 degrees C: development and current experience. Transfus Med Rev. 2006;20:158–64. [DOI] [PubMed] [Google Scholar]
- 38. Mekmene O, Quillard S, Rouillon T, Bouler J‐M, Piot M, Gaucheron F. Effects of pH and Ca/P molar ratio on the quantity and crystalline structure of calcium phosphates obtained from aqueous solutions. Dairy Sci Technol. 2009;89:301–16. [Google Scholar]
- 39. Schallmoser K, Rohde E, Reinisch A, Bartmann C, Thaler D, Drexler C, et al. Rapid large‐scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Eng Part C Methods. 2008;14:185–96. [DOI] [PubMed] [Google Scholar]
- 40. Gupta P, Hall GN, Geris L, Luyten FP, Papantoniou I. Human platelet lysate improves bone forming potential of human progenitor cells expanded in microcarrier‐based dynamic culture. Stem Cells Transl Med. 2019;8:810–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ranzato E, Mazzucco L, Patrone M, Burlando B. Platelet lysate promotes in vitro wound scratch closure of human dermal fibroblasts: different roles of cell calcium, P38, ERK and PI3K/AKT. J Cell Mol Med. 2009;13:2030–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reinisch A, Hofmann NA, Obenauf AC, Kashofer K, Rohde E, Schallmoser K, et al. Humanized large‐scale expanded endothelial colony‐forming cells function in vitro and in vivo. Blood. 2009;113:6716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fazzina R, Iudicone P, Mariotti A, Fioravanti D, Procoli A, Cicchetti E, et al. Culture of human cell lines by a pathogen‐inactivated human platelet lysate. Cytotechnology. 2016;68:1185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tasev D, van Wijhe MH, Weijers EM, van Hinsbergh VW, Koolwijk P. Long‐term expansion in platelet lysate increases growth of peripheral blood‐derived endothelial‐Colony forming cells and their growth factor‐induced sprouting capacity. PLoS One. 2015;10:e0129935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Siegel G, Fleck E, Elser S, Hermanutz‐Klein U, Waidmann M, Northoff H, et al. Manufacture of endothelial colony‐forming progenitor cells from steady‐state peripheral blood leukapheresis using pooled human platelet lysate. Transfusion. 2018;58:1132–42. [DOI] [PubMed] [Google Scholar]
- 46. Thieme D, Reuland L, Lindl T, Kruse F, Fuchsluger T. Optimized human platelet lysate as novel basis for a serum‐, xeno‐, and additive‐free corneal endothelial cell and tissue culture. J Tissue Eng Regen Med. 2018;12:557–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Single step hPL production. A PC is transferred to a sterile bag containing a CaCl2 in water solution (final ~5 mM Ca2+) and 0.2 g/ml. of glass beads. After gentle mixing, the bag is rested for 3 h at 22°C at which point coagulation and retraction is final. The remaining supernatant is a platelet releasate rich in growth factors. The bag with coagulum can be stored <−20°C until further use.
Figure S2. Finding the optimal ratio of Ca2+ and glass to consistently produce hPL. Four conditions are depicted. From left to right: (i) 15 mM Ca2+; (ii) 5 mM Ca2+; (iii) 0.2 g/ml glass; (iv) 5 mM Ca2+ + 0.2 g/ml glass. After 3 h of observation, coagulation and retraction was consistently found in the conditions with high (15 mM) Ca2+ and in the condition with low (5 mM) Ca2+ combined with glass. Low calcium or glass alone could not elicit a response.
Figure S3. Dissolution of calcium complex precipitation by EDTA. Representative pictures of hADSCs culture in DMEM with 10% (vol/vol) hPL prepared by 15 mM Ca2+ are shown. The left panel depicts the bottom of the tissue culture flask with hADSC after 4 days of adherent culture and the right panel depicts the same flask after addition of 50 mM EDTA (pH 7.4). Data were retrieved by bright field microscopy at 50× total magnification. Scale bar = 500 μm.
Video S1. Single step production of hPL. A PC was transferred to a sterile bag containing 5 mM Ca2+ and 0.2 g/ml of glass beads and filmed for three consecutive hours. The time‐lapse video was digitally increased 120‐fold and static parts (0–1 h real‐time) are not included.