

Summary
The factor H (FH) protein family is emerging as a complex network of proteins controlling the fate of the complement alternative pathway (AP) and dictating susceptibility to a wide range of diseases including infectious, inflammatory, autoimmune, and degenerative diseases and cancer. Composed, in man, of seven highly related proteins, FH, factor H‐like 1, and 5 factor H‐related proteins, some of the FH family proteins are devoted to down‐regulating the AP, while others exert an opposite function by promoting AP activation. Recent findings have provided insights into the molecular mechanisms defining their biological roles and their pathogenicity, illustrating the relevance that the balance between the regulators and the activators within this protein family has in defining the outcome of complement activation on cell surfaces. In this review we will discuss the emerging roles of the factor H protein family, their impact in the complement cascade, and their involvement in the pathogenesis of complement‐mediated diseases associated with the AP dysregulation.
Keywords: alternative pathway, complement, Factor H, FHL‐1, FHRs
1. INTRODUCTION
The complement system is a major component of innate immunity with primary functions in protecting against infections, shaping adaptative immune responses and maintaining host homeostasis through the removal of immune complexes as well as cellular debris and apoptotic cells. 1 It has the intrinsic capacity to distinguish between host cells, pathogens and altered host cells, and exerts an exquisite control to limit its activation to the sites of interest (e.g., pathogens and apoptotic cells) while inhibiting bystander damage to the host. In contrast to the classical (CP) or lectin (LP) pathways, the alternative pathway (AP) does not require any specific surface recognition molecule for its activation. Instead, initiation of the AP occurs spontaneously by the hydrolysis of C3 to C3(H2O) in the fluid phase, which in turn may result in activation and generation of C3b molecules that bind to any cell surface in the vicinity. 2 If these C3b molecules are not inactivated, they may form AP C3 convertases (C3bBb) that will activate new C3 molecules into C3a and C3b. Thus, a remarkable characteristic of the AP is that it can rapidly amplify the activation of additional C3 molecules. Importantly, this amplification can account for up to 80%–90% of total complement activation, even when initially triggered by the classical or lectin pathways. 3 , 4 Therefore, a precise control of AP initiation and its amplification loop is of great importance to prevent complement from damaging host tissues. An imbalance due to either an excessive activation or a deficient regulation can result in a wide range of pathological conditions. 5 , 6 , 7
Factor H (FH) is the main regulator of the AP both in the fluid phase and on surfaces. 8 It has the potential to distinguish host cell surfaces from pathogens by the recognition of carbohydrates such as sialic acid‐containing glycosaminoglycans that are specifically present on host cell surfaces, where it will rapidly inhibit any accidental deposition of C3b preventing further complement activation and damage of host tissues. 9 FH belongs to a protein family that, in man, includes 6 other protein members, a FH‐like protein (FHL‐1) and 5 FH‐related proteins (FHRs; FHR‐1‐5). 10 , 11 , 12 , 13 , 14 While some family members are dedicated to inhibiting AP activation (i.e., FH and FHL‐1), others (i.e., the FHRs) can promote AP activation, and thus opposite complement regulatory functions are displayed within the family. 15 These apparent contradictory functions provide this set of proteins with the capacity to fine‐tune AP activation, which serves to modulate AP activation in different surface contexts. The relevance of the FH protein family in determining disease susceptibility is clearly illustrated by the multiple associations of common and rare variations in the genes encoding the FH/FHR1‐5 with a wide range of diseases including cancer, infectious, autoimmune, renal, ocular, and neurodegenerative diseases. 16 , 17 The characterization of diverse disease‐associated genetic variants has been crucial to our current understanding of the physiological role of the FH protein family and the pathogenic mechanisms by which alterations in these proteins lead to dysregulation of AP and disease. Recent findings also suggest physiological roles beyond the complement system, which illustrates the functional complexity of this protein family in diverse biological processes.
Here, we will discuss some of the recent findings in genetics, structural biology and functional aspects of the FH protein family that have shed light on the biological role of this family and its association with disease. Other important findings such as the non‐canonical functions of FH and the FHRs are beyond the scope of this review, but they have been recently discussed by Jozsi et al. 18
2. EVOLUTIONARY ORIGIN OF THE CFH‐CFHR1‐5 LOCI
The genetic architecture of the genes encoding the human FH protein family and its chromosomic location within the regulators of complement activation (RCA) gene cluster in chromosome 1q32 is well‐described. 8 , 19 , 20 , 21 Spanning almost 360 kbp, the FH gene family consists of 6 different genes positioned in tandem in the following order: CFH‐CFHR3‐CFHR1‐CFHR4‐CFHR2‐CFHR5 (Figure 1a). FH and FHL‐1 are alternative splice products of CFH, while the FHR1‐5 proteins are encoded by their respective CFHR1‐5 genes. Although the evolutionary origin of this gene family is not completely understood, phylogenetic studies point CFH as the most ancestral gene of the family. This is based on the observation that sand bass, the most divergent species from humans that is found to contain a regulator of complement activation, expresses a protein (i.e., SBP1) that shares structural and functional similarities with FH. 22 Subsequently, up to 12 incomplete segmental duplications of CFH with a few protein‐coding exons are believed to have given rise to the rest of the CFHR1‐5, providing a mechanism of rapid generation of partial or new functions to the FHRs. 23 Consistent with the idea of CFH being the most ancestral gene of the family and a scaffold for the generation of subsequent CFHRs, CFH is conserved in many different species. Interestingly, the presence of the FHRs, but not FHL‐1, 24 is also conserved, although the number of proteins and their similarities with FH vary from species to species. Despite these differences, it is noteworthy that a common denominator seems to be preserved in all species, and it is the fact that the conservation between FH and the FHRs lies within the surface recognition domains of FH and not within the regulatory domains of the molecule, as it was demonstrated for divergent species such as sand bass, mice, and humans. 8 , 22 , 25 These observations suggest that although the composition of the FH family members may have evolved differently, the functionality of the proteins within the family is conserved in all species.
FIGURE 1.
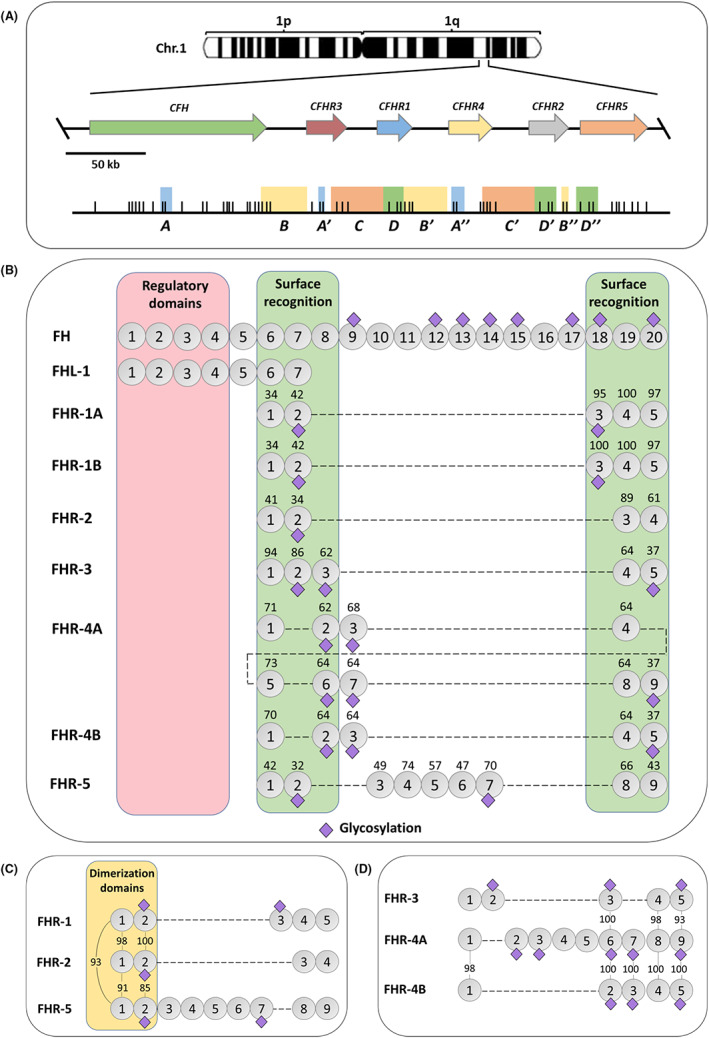
The FH family. (A) Genomic organization of CFH‐CFHR1‐5 within chromosome 1q32. Each gene is represented by an arrow. Large genomic duplications are depicted by colored boxes underneath. Vertical lines represent the position of exons in each gene. (B) Schematic representation of the different proteins composing the FH protein family. SCR domains are represented by circles and potential glycosylation sites (purple rhombus) are indicated for each protein. The proteins are aligned according to the conservation with FH, and the numbers above the SCRs of the FHRs indicate the percentage of amino acids that are identical to the corresponding amino acids in FH. FH and FHL‐1 are identical in their sequences, except for the last 4 amino acids (SFLT) in FHL‐1 SCR7 (grey square) that are not present in FH. FH N‐terminal SCR1‐4 domains are involved in complement regulatory activities (red box), while SCR6‐8 and the C‐terminal SCR18‐20 are domains involved in surface recognition (green boxes). Notably, the FHRs share a varying degree of conservation with the FH surface recognition domains, but none of the FHRs have homologous SCRs to the FH regulatory domains. In this panel, the two common allelic variants of FHR‐1, A and B, are depicted. (C) Alignment of FHR1, FHR‐2 and FHR‐5 shows high sequence similarity in their N‐terminal SCR1‐2 domains as illustrated by the numbers denoting the percentage of identical amino acids between proteins. SCR domains 1 and 2 in FHR‐1, −2 and − 5 contain a shared dimerization motif. (D) Alignment of FHR‐3 and FHR‐4 illustrating high amino acid sequence similarity in their C‐terminal domains
The reasons why the different types of segmental duplications have been fixed during evolution are not completely understood. One hypothesis relies on the interaction between the host and microbes, where the presence of specific FHRs along with FH may provide an advantage to the host to fight infections. As will be discussed below, some pathogens recruit FH to their surface to evade complement activation. In the presence of the FHRs, the binding of FH by microbes may be competed by the FHRs, thus becoming a mechanism to compensate the pathogen´s strategy to evade complement attack. Importantly, the presence of segmental duplications within the CFH‐CFHR1‐5 gene family also has a major impact on the generation of genetic variability at these loci. The high sequence similarity between large genomic regions makes the region genetically unstable and prone to gene conversion and non‐homologous recombination events that result in a source of common and rare genetic variations, many of which associate with susceptibility to complex diseases that will be described in the following sections.
3. THE HUMAN FACTOR H PROTEIN FAMILY: STRUCTURE AND FUNCTION
At the protein level, the human FH family consists of seven highly related members named FH, FH‐like 1 (FHL‐1) and FH‐related proteins −1, −2, −3, −4 and − 5 (FHR‐1‐5) (Figure 1b). All family members are exclusively composed of repetitive units named short consensus repeats (SCR) of ~60 amino acids, also known as sushi domains or control complement protein domains (CCPs), which are arranged in the form of “beads on a string”. These proteins are mainly produced by the liver, most of them are glycosylated and circulate in human plasma in concentrations that vary substantially between the different members of the family. Although there is still no consensus on the absolute plasma concentrations of each of the factor H family proteins, FH is one of the most abundant proteins followed by FHR‐1. 26 Extrahepatic expression of some of them has also been reported by various cell types, which may serve as an additional mechanism to increase their local concentration.
Because of the various genomic duplication events that gave rise to this gene family, the encoded proteins share a varying degree of similarity with each other (Figure 1). As mentioned above, the main conservation between FH and the FHRs lies within the surface recognition domains in FH (i.e., SCR6‐8 and the C‐terminal SCR18‐20), which enables these proteins to bind to similar ligands on surfaces, such as heparin or C3b. Importantly, the FHRs do not have homologous SCRs to the FH domains that mediate the complement regulatory activities (i.e., N‐terminal SCR1‐4). A variable degree of similarity is also observed between the different FHRs. A remarkable feature of FHR‐1, −2 and − 5 is that they are very similar in their N‐terminal SCR1‐2 domains, which contain a dimerization motif and, hence, these proteins circulate in plasma as either homo‐ or heterodimers, and even higher order complexes have been described with native proteins (Figure 1c and Figure 2). 27 , 28 This dimerization status confers avidity to the molecules and increases the interaction with surface‐bound ligands, as has been demonstrated for the different C3 fragments C3b, iC3b and C3dg. 27 , 28 , 29 Moreover, the formation of heterodimers increases the repertoire of molecules within the family, although the functional consequences of the heterodimeric species compared with the homodimeric species have not yet been investigated. FHR‐3 and FHR‐4, instead, are highly similar in their C‐terminal SCR domains (93%–100% amino acid sequence similarity) (Figure 1d). These structural similarities and differences amongst the FH protein family members explain the binding to specific or shared ligands (Figure 3) and define their functional activities. While FH and FHL‐1 display complement inhibiting activities, the FHRs may have a dual role by either promoting or inhibiting complement activation, although the physiological relevance of the latter is still controversial (Figure 4). Hence, this complex family of proteins provides a versatile means to both inhibit and promote AP activation, properties that are context‐dependent as will be discussed throughout the review.
FIGURE 2.
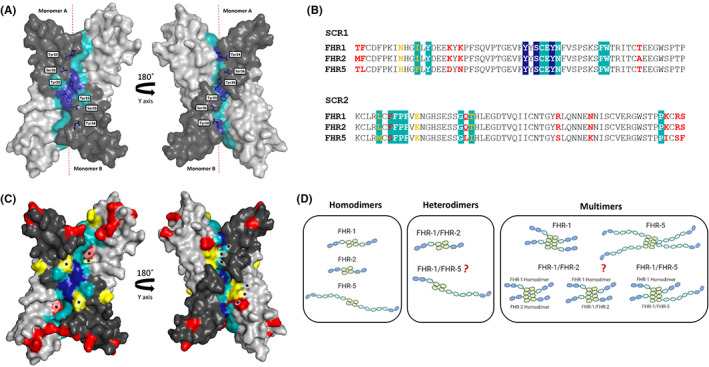
Structural features of a conserved dimerization motif in the SCR1‐2 domains of FHR‐1, −2 and – 5. (A) X‐ray crystal structure of recombinant SCR‐1 and SCR‐2 of FHR‐1 (FHR‐11–2, PDB 3zd2) demonstrates the formation of a head‐to‐tail dimer between two copies of FHR‐11–2. SCR1 domains are depicted in dark gray and SCR2 domains in light gray. Residues that play key roles in stabilizing the assembly (Tyr34, Ser36, and Tyr39) are indicated in dark blue and other interface residues are indicated in light blue. (B) Alignment of the amino acid sequences of SCR‐1 and SCR2 of FHR‐1, −2 and − 5. As above, dark blue and light blue boxes indicate the key amino acids in stabilizing the complex and other interface residues, respectively. Residues indicated in red are non‐conservative variations between the three proteins, whilst the yellow ones indicate conservative variations. (C) Mapping of the sequence variations between FHR‐1, −2 and − 5 in the crystal structure of FHR‐11–2. As above, residues depicted in red are non‐conservative variations and the yellow residues are conservative variations. Variations in the interface residues are indicated by an asterisk (*). Figures in a and c were drawn using program PyMol, www.pymol.org. (D) Schematic illustration of the potential homodimers, heterodimers and multimers of FHR‐1, −2 and − 5 that can be found in plasma. While homodimeric species of these proteins and the heterodimer between FHR‐1 and FHR‐2 are generally accepted, there is some controversy on whether FHR‐5 can form any heterodimer or whether these proteins form higher order complexes and further studies will be needed to clarify this issue 28 , 55
FIGURE 3.
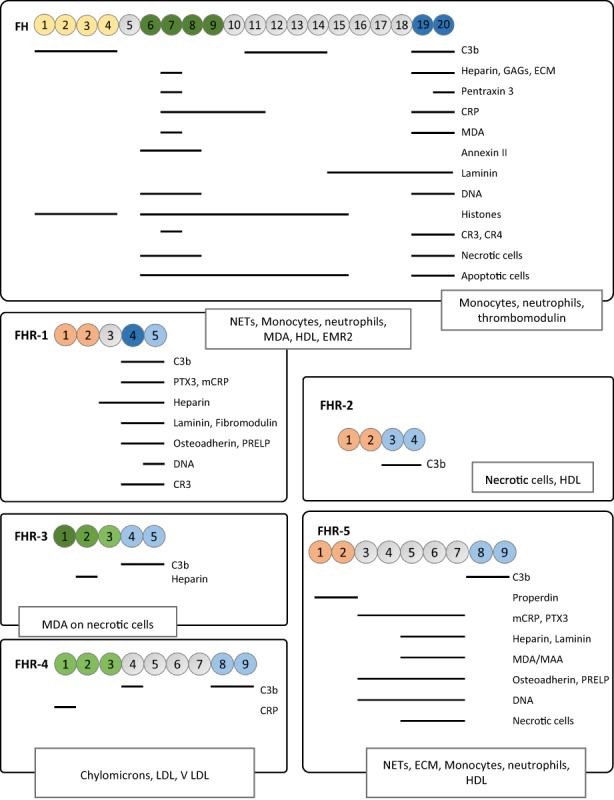
Host ligands identified for the FH protein family. For those ligands for which the binding site within the molecules is known, the SCRs involved in the binding are indicated by horizontal lines. In the cases where the binding sites are unknown, the ligands are framed in boxes. Abbreviations are as follows: CRP, C‐reactive protein; PTX3, Pentraxin III; ECM, components of the extracellular matrix; EMR2, adhesion G protein‐coupled receptor E2; GAGs, glycosaminoglycans; HDL, high density lipoprotein; LDL, low density lipoprotein; VLDL, very low‐density lipoprotein; MAA, Malondialdehyde‐acetaldehyde adducts; MDA, malondialdehyde; NETs, Neutrophil Extracellular Traps; PRELP, leucine‐rich repeat protein
FIGURE 4.
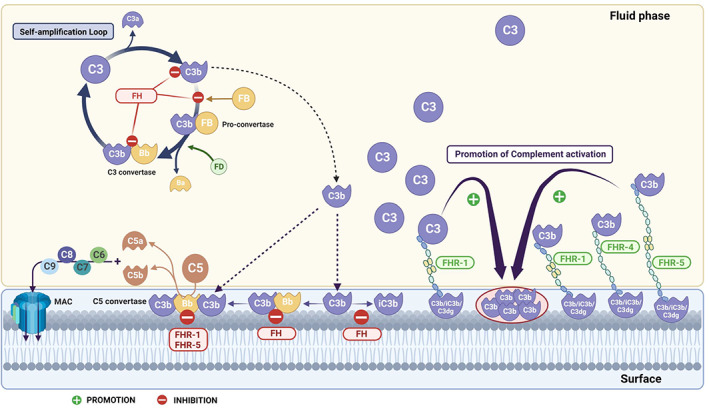
Schematic representation of complement alternative pathway activation in the fluid phase and on cell surfaces and its regulation by the FH family proteins. In the absence of complement regulators, activation of the alternative pathway (AP) results in the formation of C3 convertases (C3bBb) that cleave the C3 molecule, creating a self‐amplification loop and thus amplifying complement activation in fluid phase and on surfaces. On surfaces, AP amplification leads to the generation of C5 convertases (C3bBbC3b), which, in turn, cleaves C5 into C5a and C5b triggering inflammation and allowing MAC formation, respectively. Regulation of the AP amplification loop and the deposition of C3b on the surface is key for controlling homeostasis. FH is the main regulator of the AP both in the fluid phase and on cell surfaces. It acts as a cofactor for factor I proteolytic cleavage of C3b, accelerates the dissociation of the C3/C5 convertases and competes with factor B for C3b binding. While FH displays AP inhibiting activities, the FHRs may display both inhibiting and promoting AP activities. FHR‐1 and FHR‐5 have been shown to act as inhibitors of the C5 convertases, although the physiological relevance of this activity is not known. Conversely, surface‐bound FHRs such as FHR‐1, FHR‐4 and FHR‐5 have been shown to bind C3b from the fluid phase and to serve as a platform for the assembly of the C3 convertase (C3bBb) promoting AP activation. The binding of the FHRs to surfaces can be mediated through deposited‐C3 activated fragments (C3b, iC3b, C3dg, C3g). Additionally, surface‐bound FHR‐1 can recruit native C3 to the surface, increasing the local concentration of C3 and thus favoring AP activation towards the surface. Abbreviations for complement proteins depicted in the figure are: FB, factor B; FH, factor H; FHR‐1 and FHR‐5, factor H‐related protein 1 and 5; MAC, membrane attack complex. Figure created with BioRender.com
3.1. Factor H
FH is the largest protein of the family by far. It is composed by 20 SCRs and contains 8 potential N‐glycosylation sites throughout the molecule resulting in a glycoprotein of 155 kDa (Figure 1b). FH is an abundant plasma protein that can be found at concentrations ranging from 116–562 μg/mL in the population. 30 Although FH is constitutively expressed by the liver, many other cell types have been reported to express FH such as peripheral blood lymphocytes, fibroblasts, endothelial and epithelial cells, myoblasts, glia cells and retinal pigment cells. FH interacts with a broad spectrum of ligands in the host that may be present either in body fluids or on surfaces and, for many of them, the FH domains involved in the interaction have been described (Figure 3). FH also interacts with many other ligands present in pathogens, also interact with FH and have recently been reviewed by Moore et al. 31 FH is the main inhibitor of complement AP activation both in fluid‐phase and on surfaces. This FH regulatory activity is mediated through its interaction with C3b, acting as a cofactor for the proteolytic inactivation of C3b by factor I, competing with the binding of factor B to C3b and accelerating the decay of the AP C3 convertase (C3bBb) (Figure 4). Although the FH N‐terminal SCR1‐4 domains are responsible for such regulatory functions, it has been shown that this activity is enhanced by the adjacent SCR5‐7. 32 Importantly, the inhibition of C3b in the fluid phase only requires these N‐terminal domains, whereas inactivation of C3b deposited on surfaces depends on additional contacts that FH establishes with ligands present on the surface. As mentioned before, the FH regions that interact with ligands on surfaces are mainly SCR6‐7 and SCR19‐20, which interact, for example, with polyanions such as sialic acid‐containing glycosaminoglycans and heparan sulfates, increasing the affinity of FH for surface‐bound C3b and enhancing its regulatory capacity. Notably, the interaction of FH SCR19‐20 with 2,3′‐sialic acid moieties is critical for the discrimination of self and non‐self surfaces by FH, as will be discussed in detail in the following sections.
3.2. FHL‐1
Given that FHL‐1 is the result of an alternative splicing product of CFH, the protein is almost identical to FH as it contains the seven N‐terminal SCR domains of FH. The only difference lies in the last four amino acids at the C‐terminus of FHL‐1 (SLTF), which are encoded by exon 10 of CFH, an exon that is not included in the transcript encoding FH (Figure 1b). FHL‐1 is a non‐glycosylated protein of a molecular weight (MW) of 49 kDa and is estimated to circulate in plasma at a concentration about 10 times lower than that of FH. 26 , 33 Like FH, FHL‐1 displays AP inhibitory functions such as cofactor and decay accelerating activities, and it is a matter of debate whether these regulatory activities are comparable or not to FH, as contradictory results are found in the literature suggesting that both proteins may behave differently depending on the context. 34 Due to the low concentration of FHL‐1 in circulation, it is assumed that the main systemic AP regulator is FH. Another major difference between FHL‐1 and FH is that FHL‐1 cannot discriminate between self and non‐self surfaces because it cannot interact with the host sialic acids, due to the lack of the FH surface recognition domains SCR19‐20. However, FHL‐1 displays different surface specificities that are mediated by its SCR6‐7 domains and the unique 4 amino acids at the C‐terminus, which have been reported to influence the binding of FHL‐1 to pentraxin 3 and CRP. 35 These structural and functional differences between FHL‐1 and FH, together with the fact that both proteins display distinct expression patterns in some tissues despite having the same promoter, suggest that each protein is specialized in controlling AP activation at different sites. In fact, of major relevance is the role of FHL‐1 in the Bruch´s membrane in the eye, as FHL‐1, but not FH, is highly expressed in retinal pigment epithelium and is able to diffuse from the choroid through the Bruch´s membrane. Considering that the Bruch´s membrane is the site where AP dysregulation and tissue damage occur in age‐related macular degeneration, understanding the biological role of FHL‐1 is of key importance. A detailed description of the functions of FHL‐1 and its implication in pathology has been recently provided by Mannes et al. 34
3.3. FHR‐1
Amongst the FHRs, FHR‐1 is one of the best characterized molecules. FHR‐1 is composed of 5 SCR domains and circulates in two glycosylated forms in human plasma with an apparent MW of 37 kDa (FHR‐1α) or 43 kDa (FHR‐1β), depending on the glycosylation pattern. The estimated mean concentration of FHR‐1 in plasma is 122 μg/mL. This concentration is highly determined by the presence of a common genetic variant that results in the deletion of the two consecutive genes CFHR3 and CFHR1 (ΔCFHR3‐CFHR1). 36 As stated above, FHR‐1 is the FHR with the highest sequence similarity to the C‐terminal domains in FH that are crucial for recognizing and protecting host surfaces. Two FHR‐1 allelic variants, encoding an acidic (FHR‐1*A) and a basic (FHR‐1*B) molecule, that differ in the amino acid sequence of SCR3 have been described 37 (Figure 1b). It is noteworthy that SCR3 of FHR‐1*B is identical to FH SCR18, while FHR‐1*A differs in 3 amino acids (95% similarity). Importantly, only a couple of amino acids distinguish SCR5 of FHR‐1 from SCR20 of FH, L290 and A296 in FHR‐1 and S1191 and V1196 in FH. However, these amino acid differences have a major impact in the functionalities of these proteins. Recent structural and functional studies have shown that while FH can bind to sialic acid, FHR‐1 loses this ability, and therefore, there is a differential recognition of cell surfaces by these proteins that will determine where their regulatory activities will be directed. 38 These findings have been crucial to understand the physiological role of the FH protein family and their molecular mechanism leading to disease, as will be discussed in the following sections.
The physiological role of FHR‐1 has been controversial as both complement inhibitory and promotion activities have been described. Unlike FH, FHR‐1 does not display inhibitory activities at the level of C3b or the C3 convertase, instead FHR‐1 inhibits the C5 convertase and the assembly of the membrane attack complex. 39 However, other studies could not replicate such findings. 27 , 40 Importantly, other reports have shown that FHR‐1 promotes AP activation on cell surfaces rather than inhibiting it. Evidences supporting this include in vitro experiments showing that surface‐bound FHR‐1 binds to C3b and serves as a platform for the assembly of the AP C3 convertase, and hemolytic assays where the addition of FHR‐1 increases the cell lysis through the AP. 28 , 29 , 41 Additional structural and functional findings have recently described the interaction between FHR‐1 and native C3. This unexpected finding suggests that surface‐bound FHR‐1 promotes AP activation through the recruitment of native C3 from the fluid phase (or C3b), increasing the local concentration of C3 that may then be activated either spontaneously or through proteases, and hence, enriching of substrates to form the AP C3 convertase 42 (Figure 4).
3.4. FHR‐2
FHR‐2, with only 4 SCR domains, is the smallest protein of the family, and can be found in plasma as a non‐glycosylated form (24 kDa) or as a glycosylated form (29 kDa). 43 While the N‐terminal domains SCR1 and SCR2 of FHR‐2 share low similarity to SCR 6 (41%) and SCR7 (34%) of FH, a higher similarity is observed between the C‐terminal domains SCR3 and SCR4 of FHR‐2 and the FH domains SCR19 and SCR20 (89% and 61% similarity, respectively). However, important structural differences were noted when comparing the crystal structures of SCR3‐4 of FHR‐2 and SCR19‐20 of FH, which indicated that while the C3b interface is conserved in FHR‐2, the GAG binding site is altered or lost. 27 These observations agree with the fact that FHR‐2 binds to C3b but not to heparin (Figure 3). The biological role of FHR‐2 is poorly understood and somewhat controversial. On the one hand, Eberhard et al. showed that FHR‐2 is a complement regulator that inhibits the activity of the AP C3 convertase and prevents MAC formation. 44 However, Xiao et al. described a C3G‐associated hybrid mutant protein consisting of SCR1‐2 of FHR‐5 and the full length of FHR‐2 that caused complement overactivation, which suggests that FHR‐2 rather promotes AP activation. 45 In addition to binding C3b, FHR‐2 binds to necrotic cells and has been found in association with high density lipoproteins, but the functional consequences of these interactions are not known. 46
3.5. FHR‐3
FHR‐3 consists of 5 SCR domains and circulates in plasma in various glycosylated forms ranging from 35 to 56 kDa. The N‐terminal SCR1 and SCR2 domains of FHR‐3 share high amino acid similarity with SCR6 (91%) and SCR7 (85%) of FH, and SCR3 and SCR4 of FHR‐3 have a considerable homology with SCR8 (62%) and SCR19 (64%) of FH. These similarities with FH may explain why FHR‐3 also binds to C3b and heparin (Figure 3). Remarkably, the C‐terminal SCR3‐5 domains of FHR‐3 and the respective conserved domains in FHR‐4A (SCR2, −4, −6, −8 and − 9) and FHR‐4B (SCR2, −4 and − 5) are almost identical (Figure 1c). The plasma concentration of FHR‐3 is highly dependent on the presence of common genetic variants such as the ΔCFHR3‐CFHR1 and the FHR‐3*A/B isoforms, the latter being distinct allelic variants defined by 4 SNPs in CFHR3 that are associated with low (FHR‐3*A) or with high (FHR‐3*B) protein levels. For instance, the median concentration of FHR‐3 in individuals with two copies of the gene is 0.68 μg/mL, while in individuals with one copy of the gene it is 0.36 μg/mL. 47 From the functional point of view, weak cofactor activity and an enhancement of the FH cofactor activity were originally reported, 48 and a marginal inhibitory effect was observed in hemolytic assays using FH depleted serum. 49 However, the physiological relevance of these experiments is questioned as rather high and non‐physiological concentrations were used.
3.6. FHR‐4
FHR‐4 is the only FHR for which two splice variants have been reported, giving rise to a long protein (86 kDa) composed of 9 SCR domains and a short protein (42 kDa) composed of 5 SCR domains, named FHR‐4A and FHR‐4B, respectively. 50 Because the gene encoding the protein (i.e., CFHR4) has most likely originated by a partial internal duplication, the N‐terminal domains SCR1‐4 are almost identical to the C‐terminal SCR5‐8. Of note, all SCR domains of FHR‐4B are contained in FHR‐4A and the amino acid sequence is identical between the two proteins except for SCR1 that shows a 98% similarity (Figure 1d). These extremely high similarities between the two proteins make it challenging to determine the specific concentration of each protein in plasma. Pouw et al. developed a battery of monoclonal antibodies that allowed the discrimination between FHR‐4A and FHR‐4B and estimated the concentration of FHR‐4A to be 2.55 ± 1.46 μg/mL, while FHR‐4B could not be detected. Although the authors cannot rule out whether FHR‐4B is expressed in plasma, it seems clear that the predominant circulating form is FHR‐4A. 51 Not many host ligands have been reported to bind to FHR‐4 so far (Figure 3). Like the rest of the FHRs, FHR‐4 can bind to C3b, consistent with the complement regulatory activities that have been reported. By binding C3b, FHR‐4 also serves as a platform for the assembly of the convertase C3bBb, thus promoting AP activation. In addition, FHR‐4 has been shown to play a role in the opsonization of necrotic cells through the binding and recruitment of CRP. 52 , 53 , 54
3.7. FHR‐5
FHR‐5 is a glycosylated plasma protein of 65 kDa and is composed of 9 SCR domains. As indicated above, FHR‐1, FHR‐2 and FHR‐5 show high amino acid identity in their SCR‐1 and SCR‐2 and share a dimerization motif in these SCRs. It is worth noting that FHR‐5 is the least conserved protein of the three, which suggest that FHR‐5 is less prone to form heterodimers with FHR‐1 and FHR‐2 and may explain why some studies could not detect these heterodimers. 55 In contrast to the other FHRs, FHR‐5 contains SCR domains (i.e., SCR3‐7) that share a certain degree of sequence identity to the central SCR10‐14 domains of FH (Figure 1b). The C‐terminal SCR8‐9 domains of FHR‐5 share 64% and 43% amino acid sequence similarity with SCR19‐20 of FH, respectively. Almost 20 y ago, FHR‐5 was estimated to circulate in plasma at a concentration of 3–6 μg/mL, 56 and such concentrations have recently been confirmed by different groups. 55 , 57 Like FH and FHR‐1, many host ligands have been identified for FHR‐5, several of which are shared between the three proteins such as C3b, heparin, CRP, PTX3 and components of the extracellular matrix. Notably, the central domains in FHR‐5 (SCR3‐7) mediate many of the identified interactions with host ligands (Figure 3). FHR‐5 was first discovered as a component that co‐localizes with C3 in glomerular deposits of immune complex‐mediated diseases, the first observation suggesting that a FHR protein might be involved in pathology. 58 , 59 The first functional studies described complement regulatory activities for FHR‐5 such as FI cofactor activity and C3 convertase decay accelerating activity. However, these activities are not considered physiologically relevant as rather high and non‐physiological concentrations were required. 56 Instead, various reports have shown that FHR‐5 rather enhances the promotion of AP activation on cell surfaces. 27 , 60 , 61 Like FHR‐1 and FHR‐4, surface‐bound FHR‐5 serves as a platform for the assembly of the AP C3 convertase. In addition, FHR‐5 may compete with FH binding to certain surface ligands and inhibit FH regulatory activities. 60
4. THE FACTOR H PROTEIN FAMILY IN DISEASE
The association of the FH protein family with disease dates back more than 40 y ago, when the first complete FH deficiency was described in a patient with atypical hemolytic uremic syndrome (aHUS). 62 Since then, numerous studies have resulted in the identification of many susceptibility factors implicating the FH protein family in a wide range of diseases. These genetic variants include both common and rare variations in CFH/CFHR1‐5 that may affect either the function of the proteins or may alter the expression levels of the molecules. Today, all members of the FH family have been associated with pathology in one way or another and, on many occasions, the same CFH/CFHR1‐5 genes are associated with different diseases. Interestingly, specific alterations within a gene are commonly found in one disease but not in others, illustrating the existence of genotype–phenotype correlations. Understanding the molecular mechanisms underlying these associations is crucial and often represents the bottleneck to drawing definitive conclusions about the involvement of the FH/FHRs in disease pathogenesis. In this section, we will describe the most informative CFH‐CFHR1‐5 genetic variants associated with disease, with a particular focus on the ones associated with the complement‐mediated renal diseases aHUS, C3 glomerulopathy (C3G) and IgA nephropathy (IgAN), as they are amongst the best characterized, and have shed light into the biological role of the FH protein family and its pathogenic mechanisms leading to disease.
4.1. Rare genetic variants in CFH‐CFHR1‐5 associated with disease
As mentioned above, complete FH deficiencies were the first alterations described in the FH protein family associated with pathology and were identified in families with high degree of consanguinity. 62 , 63 As expected, the lack of this regulator in plasma causes uncontrolled activation of the AP, which results in a secondary deficiency of C3. Although the complete deficiency of FH is rare, this extreme phenotype is probably the reason why these cases could be detected by conventional biochemical and immunological studies at a time when genetic testing was underdeveloped. In 1998, a landmark work by Warwiker et al. reported the first molecular evidence of the involvement of FH with disease. On the one hand, a 4‐base pair deletion in CFH exon 1 causing FH haploinsufficiency was identified in a sporadic case of aHUS and, on the other hand, a missense mutation in SCR20 (R1197G) was identified in a familial case of aHUS. 64 In subsequent years, a significant number of CFH mutations associated with various pathological conditions such as aHUS, C3G and IgAN, were identified. 17 , 20 For instance, missense mutations in the C‐terminal region of FH that result in the expression of a FH protein that cannot control complement activation on surfaces, while maintaining its regulatory activity in fluid phase, are prototypical of aHUS. 12 , 64 , 65 , 66 Consistently, carriers of this type of mutations are not hypocomplementemic. In contrast, CFH mutations resulting in either null alleles or missense mutations affecting the N‐terminal regulatory domains of the molecule impair all regulatory activities of FH and lead to the consumption of plasma C3. Despite complete FH deficiency being firstly described in cases of aHUS, the presence of homozygous or compound heterozygous FH mutations leading to massive C3 consumption in plasma are mainly associated with dense deposit disease (DDD), a form of C3G. Interestingly, partial deficiencies of FH are associated with various diseases including aHUS, C3G, IgAN, systemic lupus erythematosus (SLE) and age‐related macular degeneration (AMD), suggesting that additional factors would be responsible for determining the disease phenotype. 67
The distinct association of specific FH variants with specific pathological situations represented an important step forward in the understanding of the pathogenic mechanisms leading to disease. In the case of aHUS, it was postulated that the combination of an active complement system and a defective protection of cell surfaces by FH is required to cause the glomerular endothelium damage that is characteristic of the disease, a situation that is clearly different to the massive complement activation leading to C3G. 8 This hypothesis was demonstrated in animal models a few years later. As in humans, FH deficiencies in animals lead to uncontrolled AP activation and C3 consumption. In pigs and mice, the complete deficiency of plasma FH leads to renal failure due to the development of progressive glomerulonephritis, which resembles type II membranoproliferative glomerulonephritis (MPGNII) in humans, 68 , 69 now renamed DDD. Interestingly, the expression of a mouse FH truncated protein lacking the C‐terminal SCR16‐20 domains (FHΔ16‐20) in FH‐knockout mice resulted in the phenotype switch from MPGNII to aHUS. As expected, the truncated FHΔ16‐20 protein efficiently restores C3 levels in plasma but lacks the capacity to regulate complement on cell surfaces, clearly demonstrating that aHUS results from a defective protection of cell surfaces by FH in the presence of an active complement system. 70 Consistent with this pathogenic mechanism, loss‐of‐function mutations in MCP, a membrane regulator of the AP, or gain‐of function mutations in C3 and CFB that affect complement activation on surfaces, have also been specifically associated with aHUS. 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78
As mentioned before, the CFH‐CFHR1‐5 genomic region is prone to gene conversion or non‐homologous recombination events that are an additional source of genetic variability. As a result, loss or gain of whole genes or parts of genes have been identified, as well as the generation of hybrid genes between different members of the CFH gene family. The most abundant genomic rearrangement involving the CFHR1‐5 is the deletion of CFHR3 and CFHR1 (ΔCFHR3‐CFHR1), a common polymorphism that will be described in the next section. Rarer events such as hybrid genes between CFH and CFHR1 or CFHR3 that result in the exchange of the C‐terminal domains of FH by those in the corresponding FHR proteins (i.e., FH::FHR‐1 and FH::FHR‐3) are specifically associated with aHUS. The functional characterization of such hybrid proteins demonstrates their impaired ability to control complement activation on cell surfaces, resulting in the same functional defect as the missense mutations localized at the C‐terminus of FH. Similarly, the exchange of the C‐terminal SCR5 domain in FHR‐1 by FH SCR20 results in a FH competitor that interferes specifically with the regulatory activity of FH on cell surfaces. 40 , 79 These findings demonstrate that the C‐terminal SCR domains of FH and the FHRs have different surface recognition properties despite the high homology between them and that they are not exchangeable.
Genomic rearrangements involving exclusively the CFHR1‐5, but not CFH, are specifically associated with C3 glomerulonephritis (C3GN), a type of C3G not normally associated with hypocomplementemia. Among them, rearrangements leading to the duplication of the dimerization domain of FHR‐1, FHR‐2 and FHR‐5 have been very informative for a better understanding of the pathophysiological role of the FHRs. The first mutation of this type was identified in CFHR5 thanks to the implementation of copy number variation analysis that allowed the identification of an internal duplication of exons 2 and 3 that was missed by Sanger sequencing. 80 This mutation is endemic in Cyprus and more than 100 patients so far have been reported, demonstrating an autosomal dominant mode of inheritance. 81 In contrast to the low disease penetrance observed for other complement genetic variants, C3GN penetrance in carriers of this CFHR5 internal duplication is strikingly high (>90%), supporting a crucial role for this mutant protein in causing complement dysregulation. Recently, an elegant animal model of spontaneous C3GN was developed by introducing a transgene that encodes a FHR‐5 protein with a duplicated dimerization domain in a mouse that was previously humanized for the CFH locus, thus demonstrating the pathogenicity of the mutant protein. 82 In the following years, similar rearrangements leading to internal duplications of the dimerization domains (e.g., Dup.CFHR1 (exons 2 to 5) and Dup. CFHR1 (exons 2 and 3)) or to the generation of hybrid genes (e.g., CFHR‐2::CFHR‐5, CFHR‐5::CFHR2 and CFHR‐1::CFHR‐5 hybrids) with similar features were identified. 28 , 29 , 45 , 83 , 84 Notably, disease penetrance is always high in carriers of these types of mutations.
Unravelling the pathogenic mechanism of FHR mutations has been a matter of great complexity due to the uncertainties about the biological role of the FHRs. Our current understanding is that disease‐associated FHR mutations are gain‐of‐function mutations that compromise the regulatory activity of FH causing dysregulation of the alternative pathway, although the underlying molecular mechanism depends on the type of mutation, as will be discussed in detail in section 5.
4.2. Common genetic variants in CFH‐CFHR1‐5 associated with disease
Like the rare genetic variations, common variants spanning the CFH‐CFHR1‐5 region have also been associated with disease demonstrating specific genotype–phenotype correlations. Apart from the deletion ΔCFHR3‐CFHR1, these variants are mostly single nucleotide polymorphisms (SNPs) that can affect the function of the proteins or their expression levels, although with a minor effect compared with the mutations. Notably, many SNPs are localized in CFH but, because of the high linkage disequilibrium across this genomic region, this genetic variability can be simplified into a few haplotype blocks (e.g., haplotypes H1‐H4), some of which extend from CFH to the CFHR1‐5. 85 , 86 One of the best described SNPs is the Y402H variant in FH and its association with increased susceptibility to the development of AMD. 87 , 88 , 89 , 90 This variant defines haplotype H1 and lies within SCR7, a region of FH important for surface recognition that is involved in the interaction with various ligands such as heparin, GAGs, CRP, and MDA. 8 , 91 , 92 , 93 The presence of H at position 402 has been shown to reduce the affinity for the indicated ligands compared to the Y residue, suggesting that the recruitment of FH to the Bruch´s membrane in the eye is less efficient. Another well‐characterized common variant in FH is V62I. In contrast to Y402H, FH variant V62I (that defines the CFH‐H2 haplotype) encodes for a more potent FH regulator and confers protection not only for AMD but also for aHUS and C3G. 70 , 94
Other common variants in CFH‐CFHR1‐5 are known to affect the plasma levels of the FH protein family. One of the most studied ones is the genomic rearrangement leading to the deletion of CFHR3 and CFHR1 (ΔCFHR3‐CFHR1). This is a common polymorphism of varying allele frequency (2%–50%) depending on the ethnic group, 95 that has been associated with several diseases with opposite effects. The ΔCFHR3‐CFHR1 is associated with protection against AMD, IgAN and C3G, while it is a risk factor for SLE. 29 , 96 , 97 , 98 Notably, the presence of the deletion is not a risk factor for aHUS, although in homozygosis it is highly associated with the appearance of anti‐factor H antibodies that are pathogenic because they are directed against the C‐terminus of the molecule and block the capacity of FH to regulate complement on cell surfaces. 99 , 100 This differential association of the deletion with disease illustrates the complexity of the impact that the FHR‐1 and FHR‐3 deficiency can have depending on the pathological context.
SNPs across the CFH‐CFHR1‐5 genes have also been associated with elevated or reduced plasma levels of various members of the FH protein family. Recently, a SNP in CFHR4 (rs6685931) has been associated with elevated FHR‐4 plasma levels, higher evidence of systemic complement activation and increased risk for AMD. 86 , 101 On the other hand, the extended haplotype CFH‐H3‐CFHR3*B‐CFHR1*B that associates with low FH levels, high FHR‐3 levels, and the allelic variant FHR‐1*B (more similar to FH), is a risk factor for aHUS. Interestingly, this extended haplotype associates with lower susceptibility to meningococcal disease. A possible explanation for this association is the fact that Neisseria meningitidis recruits FH to its surface via a FH‐binding protein (FHBP) to evade the complement attack. As demonstrated by Caesar et al., FHBP also binds FHR‐3, suggesting that the increased plasma FHR‐3/FH observed in carriers of the CFH‐H3‐CFHR3*B‐CFHR1*B would favor the competition between FHR‐3 and FH for FHBP binding, impairing FH surface regulation and thus avoiding complement evasion. 102
Altogether these data illustrate that rare and common genetic variants in the CFH‐CFHR1‐5 determine the functionality of FH and the FHRs and thus the capacity to activate/regulate the alternative pathway. The inheritance of genetic variants resulting in elevated ratios of FHRs/FH plasma levels may compromise a proper regulation of the alternative pathway activation by FH and, hence, is a risk factor for diseases associated with AP dysregulation, while being a protective factor in the context of infectious diseases. Conversely, reduced FHRs/FH ratios increase the susceptibility for the development of SLE. It is noteworthy that although common variants are not pathogenic per se, they modulate the disease susceptibility and contribute to increase disease penetrance in carriers of pathogenic mutations. For detailed information on rare and common genetic variants in CFH‐CFHR1‐5 and its association with disease see review by Rodriguez de Córdoba. 76
4.3. Quantitative variations of the FH protein family
It is well‐known that the plasma levels of complement proteins may have a wide range of variation in the population, and this is also the case for the FH family proteins. As can be deduced from the preceding section, the plasma levels of FH/FHL‐1 and the FHRs are partially determined by genetics, but environmental factors or pathological conditions themselves may also play a role, as will be discussed here. In the case of FH, a family‐based study including 358 individuals concluded that 62% of the plasma FH variation is genetically determined and three different genomic regions (1q32, 2p2‐24 and 15q22‐24) responsible for regulating FH expression were identified. 30 In 1q32, common genetic variations in the CFH‐CFHR1‐5 loci (e.g., haplotypes CFH‐H3 and CFH‐H4) are most likely responsible for such regulation. In addition to genetics, many other situations have been shown to modulate FH levels. FH plasma levels increased with age, while they are reduced in smokers. 30 Certain cell stress conditions such as hypoxia and metal‐sulfate‐induced stress, as well as infections, lead to down‐regulation of FH expression. 103 , 104 , 105 Importantly, it is also well known that FH plasma levels are consumed upon complement activation and that there is a positive correlation between FH levels and the plasma levels of other complement components of the alternative pathway such as C3 or FB. 106
In the case of the FHRs, in‐depth genetic studies to examine the heritability of the plasmatic variation of the proteins and to identify quantitative trait loci responsible for that are missing. However, as described earlier, certain common genetic variants such as the ΔCFHR3‐CFHR1 are known to have a profound effect in defining the plasma levels of FHR‐1 and FHR‐3. An SNP in CFHR2 (c.C420T, p.C140C, rs4085749) that has a high allele frequency in the population (e.g., 0.23% in non‐Finnish Europeans according to gnomAD) is known to introduce a new donor splice site and predicted to result in a near‐null allele. 107 Alike FH, the plasma levels of FHR‐1, FHR4A and FHR‐5 appear to increase with age, as the levels of these proteins in healthy children were slightly lower compared to the levels in adults. However, such differences were not observed for FHR‐2 and FHR‐3, although it cannot be excluded that the same situation would apply after correcting for genetic factors. 108 In contrast to FH, the levels of some FHRs have been shown to increase during a pathological process that involved complement activation. The first report suggesting such elevation described that, relative to serum, FHRs were enriched in the middle ear fluid (MEE) of patients with otitis media with effusion, a common disease in childhood that is characterized by chronic inflammation. 109 Interestingly, MEE specimens were shown to strongly activate complement AP in normal human serum, suggesting that the elevation of FHRs in relation to FH interfere with the regulatory activity of FH. A more detailed description of the elevation of plasma FHR levels during disease was provided by two independent groups many years later, 36 , 57 demonstrating the elevation of plasma FHR‐1 levels in patients with IgAN despite normal FH levels. Notably, FHR‐1 levels in the patients were elevated compared with controls independently of the ΔCFHR3‐CFHR1 genotype, and the levels were even higher in patients with disease progression compared with non‐progressors. Of note, such increase of FHR‐1 was also observed in a non‐complement‐related autosomal dominant polycystic kidney disease (ADPKD), suggesting that renal function impairment elevates FHR‐1 plasma levels. 36 Similarly, FHR‐5 plasma levels were also significantly elevated in IgAN, but whether this is due to genetic or environmental factors is unknown. 57 In contrast, reduced plasma FHR‐5 levels compared with controls have been observed in different cohorts of C3G and in immune complex‐mediated membranoproliferative glomerulonephritis. 110 , 111 One of these studies also showed that the reduced FHR‐5 levels were independent of the presence of CFHR5 null alleles, and that the levels correlated positively with the levels of C3, C4, FH and the activity of the AP and the CP, suggesting that FHR‐5 is consumed upon complement activation. 110 A study by Pouw et al. reported increased FHR‐3 plasma levels in aHUS compared with controls independently of the genetic factors known to determine FHR‐3 plasma levels (i.e., ΔCFHR3‐CFHR1 and FHR‐3*A/B isoforms), supporting that additional factors may modulate the protein levels. It is tempting to speculate that, similarly to FHR‐1 levels, the renal function impairment in aHUS patients elevates plasma FHR‐3 levels, but proper studies will be needed to address it. Moreover, infections have also been shown to alter the plasma levels of the FHRs. In HUS‐associated with S. pneumoniae (SP‐HUS), FHR‐5 plasma levels were increased in patient samples at disease onset compared to samples in remission, 112 which suggests that increasing FHR‐5 levels is a host defense mechanism to fight infection.
5. HISTORICAL PERSPECTIVE OF THE ROLE OF THE FACTOR H PROTEIN FAMILY IN PHYSIOLOGICAL AND PATHOLOGICAL CONDITIONS
Since the first description of FH as a complement regulator back in 1976, 113 , 114 , 115 tremendous pieces of work during the following decades provided detailed insight into the molecular mechanisms of its complement inhibiting activities becoming, by far, the best described molecule of the family. Based on the high degree of similarity between FH and the FHRs, it was initially postulated that the FHRs could also act as complement regulators. Functional studies, however, have failed to demonstrate that the FHRs, at physiological concentrations, have the same regulatory activities as FH. 15 , 27 , 41 , 60 Yet, complement regulatory activities have been shown for FHR‐2 at the level of the C3 convertase and the formation of the MAC, and at the level of the C5 convertase in the case of FHR‐1 and FHR‐5. However, the relevance of these complement regulatory activities is questioned since these proteins do not seem to prevent complement‐mediated lysis of sheep erythrocytes in hemolytic assays, and whether they exert different regulatory activities depending on the context remains to be demonstrated.
The recognition that FH complement regulatory activities lie within the first four SCR domains (SCR1‐4) and SCR6‐7 and SCR19‐20 of FH are surface recognition domains, was key to postulate that the FHRs do not regulate complement like FH but, instead, they may interfere with the binding of FH to its ligands on cell surfaces. This idea came to the forefront with the discovery of the dimeric status of FHR‐1, FHR‐2 and FHR‐5, and the recognition that this structural property confers avidity to interact with ligands on a surface. 27 , 28 Consistently, it was demonstrated that the presence of FHR‐1, FHR‐2 and FHR‐5 increased the cell lysis in hemolytic assays that were dependent on the regulatory activity of FH. 27 Because FHR‐1, FHR‐2 and FHR‐5, like FH, can bind to C3b deposited on a surface, it was then proposed that these proteins promote complement activation by competing with FH for surface‐bound C3b, preventing FH regulation and allowing further complement activation. 27 , 28 This activity of the FHRs was coined as FH deregulation activity, and it was greatly affected by the ability of the FHRs to dimerize. 27 In this context, it was then interpreted that the C3G‐associated FHR mutant proteins with duplicated dimerization domain would result in an enhanced deregulation activity, as these mutant proteins form multimers that increase the avidity for its ligands and thus may present an increased capacity to compete with FH for C3b binding. 27 , 28 In this regard, competition between FH and the FHRs for the binding of other ligands has also been described. For instance, FHR‐5 can strongly compete the binding of FH to pentraxin 3, C‐reactive protein, extracellular matrix and malondialdehyde‐acetaldehyde epitopes, 60 , 116 and FHR‐1 can also compete CRP, although to a lower extent. 41 Altogether, these data illustrate that the modulation of the regulatory activity of FH by the FHRs may be different dependent on the ligands that are present on cell surfaces.
Further mechanistic insights reveal additional complement functions for the FHRs. It has been shown that surface‐bound FHR‐1, FHR‐4 and FHR‐5 bind C3b and serve as a platform for the assembly of an active C3 convertase (C3bBb), thereby enhancing AP activation. 41 , 52 , 60 Importantly, Merinero et al. have recently discovered that surface‐bound FHR‐1 binds native C3 just as well as C3b. 38 This unexpected FHR‐1‐C3 interaction becomes very relevant considering that the concentration of C3 in plasma largely exceeds that of C3b and, thus, the expected preferential ligand of FHR‐1 would be C3. By interacting with native C3, surface‐bound FHR‐1 is expected to increase the local concentration of C3 molecules towards the proximity of the surface. This C3 might then end up being activated spontaneously or through proteases, increasing the local deposition of C3b‐like molecules and hence promoting AP complement activation 38 (Figure 4). An additional mechanism for promoting AP activation was also reported for FHR‐5, as it was shown to recruit properdin via its SCR1 and 2 to a surface and to activate the AP. 117 Altogether, these observations led to a change of paradigm regarding the biological role of the FHRs, shifting from the original idea of the FHRs being complement regulators like FH, to consider these proteins as deregulators or promoters of AP activation. This new functional dimension of the FHRs illustrates the complexity of the FH protein family, where the intricate interaction between family members having opposite regulatory functions will influence the outcome of the AP activation/regulation on surfaces. In this context, the balance between FH and the FHRs and the presence of ligands specific for the different FH family members will have a major impact on the local activity of the AP.
The role of the FHRs as promoters of the AP activation immediately raised relevant questions that could not be easily explained and required further mechanistic understanding. For instance, how is AP activation controlled by the FH protein family to avoid complement‐mediated damage on host surfaces? In this regard, it is of particular interest to know what prevents FHR‐1, which has almost identical C‐terminal domains to FH and is present at a similar molar concentration, from competing with the binding of FH to host cell surfaces? As mentioned before, the protection of host cells by FH relies on its C‐terminal domains SCR19‐20. The concerted interaction of these domains with the TED domain of deposited‐C3b and the sialic acid‐containing glycans on host cell surfaces, results in a strong interaction and in an efficient regulation by FH. 118 Recently, Martín‐Merinero et al. reported that, despite the high homology between FHR‐1 and FH, the two amino acid differences in SCR20 between these two proteins are sufficient to alter the way host cell surfaces are recognized by each protein. In contrast to FH, the presence of leucine at position 290 and an alanine at position 296 in FHR‐1 (amino acids 1191 and 1197 in FH), resulted in an increased interaction with the TED domain in C3 but in a loss of sialic acid binding capacity. 38 Importantly, the lack of sialic acid binding capacity in FHR‐1 is likely the reason why this protein does not compete with FH for the binding to surface‐bound C3b in host cell surfaces and, hence, does not deregulate complement. These ideas would be consistent with the fact that the aHUS‐associated mutations that exchange the C‐terminal of FH for that of FHR‐1, result in a mutant protein that can no longer target host cell surfaces efficiently and do not regulate complement activation properly. 119 Conversely, aHUS‐associated FHR‐1 mutations at the C‐terminal of the molecule (i.e., FHR‐1L290S,A296V and FHR‐1L290V) become pathogenic because they acquire the capacity to bind sialic acid‐containing glycans. This new property increases the avidity of the molecules for surface‐bound C3‐activated fragments in host cell surfaces, favoring the C3b‐binding competition with FH even when there is low level of C3b deposition. 38 , 79
These recent structural and functional insights have important implications for FHR‐1 pathophysiology. First, it implies that de‐regulation activity is limited to the aHUS‐associated FHR‐1 mutant proteins that acquire the ability to interact with sialic acids and not to wild‐type FHR‐1 (Figure 5). Second, the promotion of AP activation by surface‐bound FHR‐1 molecules is mainly due to the attraction of native C3 to the surface. This new framework prompted a re‐evaluation of other contexts where FHRs were thought to become pathogenic because of an enhancement of the de‐regulation activity, like the C3G‐associated FHR mutant proteins with duplicated dimerization domain. 27 , 28 , 45 , 80 , 83 , 84 , 117 , 120 In a recent publication, we reported the structural and functional characterization of a novel C3G‐associated FHR‐1 mutant with duplicate dimerization domain (SCR1 and SCR2). 29 As expected, this mutant protein formed higher order complexes beyond a dimer and displayed enhanced avidity to interact with all types of C3 molecules, including the native form and the activated fragments. Knowing the key role of sialic acids for the physiological recognition and regulation of FH on host cell surfaces, it was fundamental to assess if the FHR‐1 protein with a duplicated dimerization domain competes with the binding of FH to surface‐bound C3b in the presence of sialic acid. Interestingly, when a C3b‐binding competition assay is performed on sheep red blood cells containing sialic acids, neither the wild‐type FHR‐1 nor the FHR‐1 mutant protein with duplicated domain competes with FH for the binding to C3b. Only the aHUS‐associated FHR‐1 mutations with capacity to bind sialic acids can do so in such scenario. 29 Altogether, these experiments provide the formal proof that the de‐regulation activity is the pathogenic mechanism of FHR‐1 mutations leading to aHUS, but it is not the molecular mechanism that explains how the FHR mutant proteins with duplicated dimerization domains lead to complement dysregulation in C3G.
FIGURE 5.
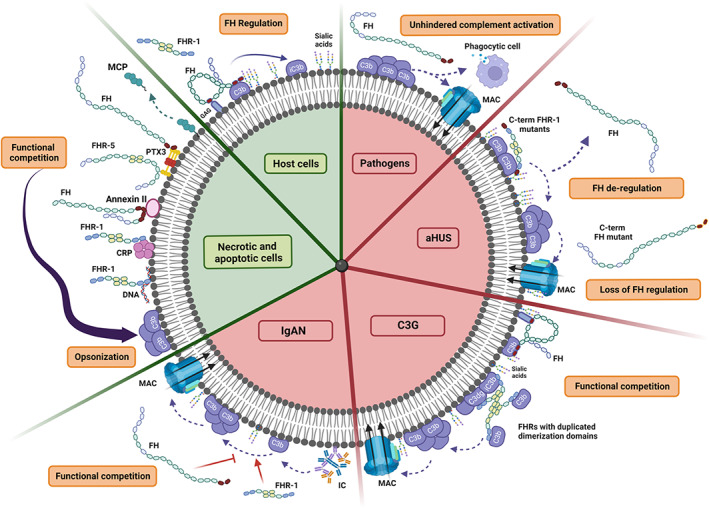
Regulation of AP activation on cell surfaces by the FH protein family under physiological and pathological conditions. The figure depicts different physiological cell surfaces such as a normal host cell and an apoptotic/necrotic cell, as well as different pathological scenarios including infections (pathogen) and the complement‐mediated renal diseases associated with AP dysregulation including atypical hemolytic uremic syndrome (aHUS), C3 glomerulopathy (C3G) and IgA nephropathy (IgAN). On host cells, any accidental deposition of C3b is rapidly inhibited by FH, as the regulatory activity of FH is very efficient in the presence of sialic acids and is not interfered by the FHRs (they can neither deregulate FH nor promote AP activation). Apoptotic and necrotic cells require an efficient and silent removal by opsonophagocytosis without causing excessive inflammation and tissue damage. The exposure of certain molecules on its surface such as pentraxins (CRP and PTX3) and DNA, which are shared ligands between FH and FHR‐1 and FHR‐5, allows a FH/FHR competition that may be beneficial for the correct opsonization of the cells. In contrast to host surfaces, pathogens lack 2,3´sialic acid‐containing glycosaminoglycans and, hence, FH does not regulate C3b molecules that may deposit on their surface. Thus, AP activation gets amplified, and unhindered complement activation occurs to eliminate the infection. In aHUS, genetic alterations in the CFH/CFHR1‐5 cause AP dysregulation on glomerular endothelial cells. Prototypical aHUS‐associated mutations include missense mutations at the C‐terminal of either FH or FHR‐1, which results in a FH that cannot regulate efficiently surface‐bound C3b or in an FHR‐1 protein that acquires the ability to bind to sialic acids and thus, can out‐compete FH for the binding of C3b preventing FH regulation (deregulation activity). IgAN and C3G conditions reflect scenarios where host cell surfaces or host surfaces such as the glomerular basement membrane become altered because of the deposition of immune complexes or C3 activated fragments (C3b, iC3b, C3dg, C3d), which are the perfect substrates where FHRs would bind promoting AP activation. The pathogenic mechanism in these cases probably implies a functional competition between FH and the FHRs, where the regulatory activity of FH is overwhelmed by the promotion of AP activation by the FHRs. Figure created with BioRender.com
As mentioned before, FHR proteins bind to C3 activated fragments deposited on a cell surface (i.e., mainly iC3b and C3dg) and promote complement activation. 38 , 41 , 60 In contrast to the aHUS‐associated FHR‐1 mutants that would strongly bind to surfaces with low‐density deposition of C3 fragments, the binding of wild‐type FHRs would require a higher deposition of such fragments, which means that their binding is mainly restricted to heavily C3‐opsonized surfaces. In the case of FHR‐1 with duplicated domains, Marquez‐Tirado et al. demonstrated that the multimerization of the protein increases the avidity to C3 activated fragments and, consequently, required less density of C3 deposition to target a surface compared with wild‐type FHR‐1. 29 Once these proteins are bound to a surface, they will all promote complement activation by recruiting native C3 (or C3b) molecules towards the surface, which may result in an increased generation and deposition of local C3b. This promotion of complement activation is counterbalanced by the regulatory activity of FH and, thus, a functional competition between the FHRs and FH is established. If the regulatory activity of FH is overwhelmed by the promotion of complement activation by FHRs, then complement dysregulation will occur on that surface. In this context, the C3G‐associated FHR mutant proteins are more likely to break the balance towards excessive complement activation (Figure 5).
6. DEFINING THE FATE OF AP ACTIVATION BY THE FH PROTEIN FAMILY
The control of AP activation by the factor H protein family is context‐dependent and several factors are expected to play a role in defining the outcome. In the first place, a clear distinction between AP activation in the fluid phase and on cell surfaces must be made. FH is the main regulator of the AP in the fluid phase, as evidenced by the massive C3 consumption occurring in complete FH deficiencies. This regulatory activity of FH in the fluid phase is, hence, crucial for maintaining host homeostasis and this is probably the reason why none of the FHRs have evolved to interfere with it (i.e., FH N‐terminal SCR domains are not conserved in any FHR). In line with this observation, the lack of the FHR proteins does not seem to have any impact on fluid phase AP activity, as evidenced by the common deficiency of FHR‐1 and FHR‐3 in the population, or by the complete removal of FHRs in a mouse model. 82 , 95 Conversely, FHRs display different degrees of similarity with the FH surface recognition domains and share surface ligands with FH, indicating that the functional activities of the FHRs will most likely be manifested on surfaces.
The regulation of AP activation at cell surfaces is a complex process and different outcomes may be expected depending on the context and type of cell surface. Normal host cell surfaces require a strict regulation of complement activation to avoid any complement‐mediated damage. However, full‐blown of complement activation on the surface of a pathogen would be ideal to fight infection. On the other hand, maintenance of host homeostasis would also require the proper removal of apoptotic and necrotic cells, and for that a certain degree of complement activation would be necessary. Additionally, pathological conditions, genetic and environmental factors or just a normal aging process may alter normal host surfaces impairing a correct regulation of complement activation.
Unlike the classical and lectin pathways, whose activation at surfaces is generally an active process that involves the binding of specific recognition molecules (e.g., C1q, MBLs, etc…), the activation of the AP on surfaces is a passive process driven by the non‐specific binding of C3b molecules that are generated in the proximity of the surface. Hence, whether AP activation progress or not depends on the control by complement regulators such as FH and the membrane bound proteins MCP, DAF and CR1. Importantly, the regulation of AP activation on non‐cell surfaces like the glomerular basement membrane or the Bruch´s membrane, which lack the membrane complement regulators, relies on the control by FH.
The FH protein family has evolved to regulate AP activation on surfaces in a very sophisticated way, exerting either a strict inhibition or potentiating its activation when needed. The existence of several FHRs, in addition to FH and FHL‐1, each one with different binding properties and the possibility of combining some of them by heterodimerization, gives rise to a large repertoire of molecules with different functional capabilities, providing an exquisite mean to modulate AP activity. Considering that FH and the FHRs exert opposite complement functions on surfaces, the ratio between these proteins in an individual would determine the capacity to activate/regulate the AP on surfaces. Many factors are expected to influence this balance: (1) the protein level of each member, (2) the functionality of the proteins, (3) the surface context (e.g., type of surface, ligands present at the surface). In the following sections, a few scenarios are discussed.
6.1. FH/FHR regulation on host surfaces
Host cell surfaces are covered by the glycocalyx, a carbohydrate meshwork of negatively charged glycoproteins, proteoglycans, glycosamioglycans and associated plasma proteins that contributes to many biological processes. 121 The content of these glycans vary among species, tissues, or cell types and changes throughout physiological and pathological conditions. FH, as the master regulator of the AP on surfaces, discriminates host versus non‐host by recognizing host‐specific cellular markers such as polyanionic and sialylated glycans. Through the distinct surface binding sites at SCR6‐7 and SCR19‐20, FH provides versatile recognition of these host cell markers, with each of these regions involved in the binding of specific ligands and defining the regulatory activity of FH on surfaces. As mentioned before, the binding of FH to sialic acid‐containing glycosaminoglycans is of major importance for the protection of host surfaces from AP activation. Notably, amongst the different sialic acid species, FH recognizes exclusively the species with α2‐3´ linkages, which allows to discriminate self from non‐self. The recognition of this sialic acid by the C‐terminal SCR20 increases the affinity of FH to surface‐bound C3b 10‐fold, allowing an efficient regulation of any accidental deposition of C3b. 9 , 122 , 123 To preserve the important function of FH in protecting host surfaces, the FHRs have evolved in a way that they can be tolerated by the host. One of the most illustrative examples is FHR‐1. As described previously, SCR5 of FHR‐1 is almost identical to FH SCR20, but the only two amino acid differences between these proteins are enough to avoid the binding of FHR‐1 to sialic acid and, hence, any competition of the binding of FH to surface‐bound C3b is prevented. 38 This differential recognition of host markers between FH and FHR‐1 is particularly critical, as illustrated by the association of FH and FHR‐1 mutant proteins that either lose the ability to bind to sialic acid or acquire the ability to bind to them, respectively, and the development of aHUS, as described in the previous section and shown in Figure 5. Consistent with this, loss of sialic acids from cell surfaces following Streptoccocus pneumoniae infection due to neuraminidase secretion, prevents the efficient binding and regulation by FH and leads to the development of aHUS, illustrating the critical role of sialic acids in protecting host cell surfaces from complement attack. 124
Despite the C‐terminus of FH being critical for the protection of host cell surfaces, the involvement of the surface recognition domains SCR6‐7 seem to play a key role on certain host surfaces such as the Bruch´s membrane in the eye. 125 For instance, the common variant Y402H in FH SCR7 has been shown to influence the binding to various ligands such as heparan sulfate, MDA, CRP, and chondroadherin, from whom the differential binding to heparan sulfate seems critical to prevent excessive complement activation and is particularly relevant in the pathogenesis of AMD. 126 , 127
6.2. FH/FHR regulation on pathogens
In contrast to self surfaces, non‐self surfaces such as pathogens, lack sialic acids species with α2‐3´ linkages and, hence, FH does not regulate C3b molecules that may deposit on their surface. This initial deposition of C3b is then rapidly amplified allowing unimpeded AP activation, which will hopefully lead to the elimination of the infection by formation of the membrane attack complex or by opsonophagocytosis 1 (Figure 5). Although the initial tagging of C3b is normally considered a non‐specific process, the reactivity of the thioester moiety to specific carbohydrates may lead to the preferential opsonization of foreign particles and, hence, may be interpreted as a first mechanism of pattern recognition. 128 As recently reviewed by Moore et al., one of the several strategies of pathogens to evade complement attack is the recruitment of FH to their surface to inhibit AP activation. 31 Interestingly, this is one of the possible arguments to explain the origin and conservation of the various FHRs throughout evolution. From an evolutionary perspective, where host and pathogens are in constant co‐evolution, the host, to counterbalance the hijacking of FH by certain pathogens, has developed a battery of FHRs that compete the FH binding to the surface of the pathogens, thus restoring complement attack. In support of this hypothesis is the description in the literature of several interactions between pathogens and all the FHRs members. 31 Illustrative examples of the competition of the binding of FH to the surface of a pathogen by an FHR include the competition between FH and FHR‐3 for the binding of the FH‐binding protein present in Neisseria meningitides, and the competition between FH and FHR‐1 for the binding of Plasmodium falciparum. 102 , 129 As mentioned in previous sections, the levels of some FHRs (e.g., FHR‐1 and FHR‐5) have been shown to be elevated upon infection, suggesting that this is an additional mechanism of host defense to counteract the binding of FH to the pathogen surface to fight the infection more efficiently. Interestingly, a computational model of the complement system´s attack on pathogens suggests that the FHRs will favor a rapid response of the host to cover the pathogen surface with complement, leading to the opsonization of the cells and to MAC formation within 1 min. 130 Furthermore, according to this mathematical modelling, pathogens are rapidly coated by complement components in the bloodstream, although MAC only covers <<1.0% of the total pathogen surface. Hence, it is predicted that if a pathogen enters the bloodstream, phagocytosis, following opsonization, may serve as the primary mechanism of eliminating pathogens and, in this context, the FHRs may be crucial.
6.3. FH/FHR regulation on apoptotic and necrotic cells (or altered host surfaces)
In addition to combating infections, one of the canonical functions of the complement system is to maintain host homeostasis by removing waste material such as apoptotic and necrotic cells. 131 Dying cells are a source of self‐antigens, that if not properly removed, may lead to inadequate immune responses and generation of autoantibodies. During apoptosis, cells are efficiently and silently eliminated through a complex mechanism that requires the concerted action of the complement activation and terminal pathways to ensure adequate opsonization of cells to allow phagocytic uptake, while controlling excessive inflammation and damage by the MAC. 132 , 133 The role of the CP activation has been proven crucial in this task, as illustrated by the strong association between C1q, C2, C4 or C3 deficiencies and systemic lupus erythematosus (SLE), a chronic autoimmune disease characterized by an impaired removal of apoptotic cells by macrophages. 134 In addition, both common and rare gene variants in the CFH‐CFH1‐5 have also been associated with susceptibility to develop SLE, supporting the involvement of the factor H protein family in the silent clearance of dying cells.
A relevant question is how the complement system recognizes that a particular cell needs to be cleared. During cell death, several alterations occur in the cell membranes that are detected by complement‐related pattern recognition molecules triggering the activation of complement pathways to opsonize the surface. For instance, apoptotic cells expose DNA and annexin II, and recruit C‐reactive protein (CRP) and pentraxin‐3 (PTX3) to their surface. CRP promotes the binding of C1q, initiating CP activation. MBL and ficolins bind to DNA on late apoptotic cells leading to the LP activation and facilitating phagocytosis. 135 Importantly, several factors also influence AP activity. The content of sialic acids on cell surfaces gets decreased during aging and senescence, as was initially reported in human erythrocytes. 136 Notably, α2‐3´ and α2‐6´ sialic acids were shown to be decreased on the surface of apoptotic cells. 137 Additionally, the membrane regulator MCP is rapidly detached from the surfaces of apoptotic cells. 138 However, this apparent loss of AP regulators is compensated by other mechanisms to ensure control of the amplification loop. For instance, FH was reported to bind to apoptotic cell surfaces independently of GAGs and sialic acid 139 and to display cofactor activity. In addition to recognizing polyanions, FH also binds to DNA, annexin II, histones, CRP and PTX3, which serve as ligands to recruit FH to the surface of apoptotic cells, limiting the amplification of the AP and, thus, preventing the formation of the C5 convertase, and subsequent damage by lysis and inflammation 139 , 140 , 141 (Figure 5).
It is not completely clear what the role of the FHRs might be in the context of dying cell removal, although recent in vitro studies point to the contribution of these proteins in facilitating opsonization and clearance of the cells. The binding of FHR‐1, FHR‐2 and FHR‐5 to both apoptotic and necrotic cells, but not to living cells, has been reported. 61 , 117 Like FH, it has also been shown that FHR‐1 and FHR‐5 bind to DNA and the pentraxins mCRP and PTX3. Importantly, both FHR‐5 and FHR‐1 compete with FH binding to DNA preventing the regulatory activity of FH. In addition, FHR‐1 and FHR‐5 can either bind to surface‐bound CRP and PTX3 or can recruit these proteins to the surface, resulting in the enhancement of complement activation by both the CP and AP and contributing to opsonization. 61 Interestingly, in vitro models have shown that FHR‐1 and FHR‐5 can regulate at the level of C5 convertases. 39 , 142 Although the relevance of these regulatory functions under physiological conditions is unknown, it is tempting to speculate that the FHRs have the potential to promote C3 activation while shutting down C5 activation, thus specifically enhancing opsonization of the surfaces to which they bind (Figure 5). The beneficial role of the FHRs in this process is supported by the genetic association of the deletion of CFHR3‐CFHR1 with increased susceptibility to develop SLE, which suggests that either FHR‐1 or FHR‐3 or both contribute to the efficient removal of the cells.
6.4. FH/FHR regulation on altered host surfaces in pathological conditions
Host cell surfaces may also be altered by other factors without implying “eat me signals” and the removal of the cells. These alterations may be caused by pathological processes themselves that lead to dysregulation of the complement AP due to an imbalance of the factor H protein family. Prototypic examples are IgAN and C3G. IgAN is the most common cause of primary glomerulonephritis and chronic renal damage worldwide. The pathogenesis of IgAN is complex and a multi‐hit model that includes the formation of galactose‐deficient IgA, and the binding of anti‐glycan antibody to form circulating immune complexes that deposit in the mesangium and induce renal damage has been proposed. The presence of C3 colocalizing with both circulating immune complexes and IgA deposits within the glomerulus, pointed to the role of complement in the disease pathogenesis. More than a decade ago, genome wide association studies reported the association of the ΔCFHR3‐CFHR1 with strong protection for the development of IgAN, supporting the involvement of AP dysregulation in the disease pathogenesis and highlighting the relevance of the factor H protein family. 96 Consistent with these genetic findings, patients presented abnormally elevated plasma levels of FHR‐1 and increased FHR‐1:FH ratios compared with controls. 36 , 57 Notably, the highest FHR‐1 and FHR‐1:FH ratios were associated with disease progression. Importantly, it was also shown that the increased FHR‐1:FH ratios could be a consequence of low factor H levels, some of which are due to the presence of FH null alleles in heterozygosity. 36 In addition to FHR‐1, plasma levels of FHR‐5 were also reported to be significantly increased in IgAN patients compared to controls and appear as an independent risk factor for disease progression. 57 , 143 Although the exact molecular mechanisms are not completely understood, it is tempting to speculate that the initial deposition of C3‐containing immune complexes in the mesangium may then serve as a surface ligand for the binding of the FHRs, which will then contribute to the local promotion of AP activation overcoming FH regulation and exacerbating kidney damage (Figure 5). In agreement with this hypothesis, the ΔCFHR3‐CFHR1 was found associated with lower mesangial deposition of C3, lower levels of complement activation split product C3a and reduced kidney injury. 143 , 144 , 145
C3G is a rare and heterogeneous entity that encompasses several glomerular diseases characterized by the predominant deposition of glomerular C3 without significant immunoglobulin deposition. The pathogenic mechanisms leading to dysregulation of the AP in C3G are diverse, as both massive systemic complement activation and normal complement levels have been observed in patients. While significant systemic complement activation is normally associated with the presence of null FH alleles or autoantibodies (e.g., C3 nephritic factor and anti‐factor H antibodies), normal complement levels are generally observed in patients with alterations at the level of the FHRs, supporting the concept that the FHRs mainly impact complement activation locally. A relevant question is what determines the binding of the FHRs in the glomerular microenvironment? As previously mentioned, the FHRs bind to many different ligands such as GAGs, C3 fragments, pentraxins, MDA epitopes and ECM proteins, amongst others. The presence or accumulation of such ligands within the glomerular microenvironment may then set the scene for the local recruitment of the FHRs. Notably, the binding of the FHRs to C3‐opsonized surfaces is particularly critical in C3G, as illustrated by the advantage of the C3G‐associated FHR mutants with duplicated dimerization domains in targeting those surfaces and in promoting complement activation. 29 Therefore, situations that lead to a certain degree of opsonization within the glomerulus or that increase the presence of other FHR ligands on surfaces, would trigger the binding of the FHRs and favor local AP dysregulation. One of such situations is infection, a known trigger for both C3GN and IgAN, but other inflammatory conditions may have similar effects.
7. CONCLUSIONS
Over the last decades, seminal contributions in the field of the factor H protein family have provided insight into their biological role and their association with disease. There is now compelling evidence that this protein family should be viewed as a master regulator of the AP with the potential to modulate AP activation in different ways depending on the context. The interplay between FH, FHL‐1 and the different FHRs provides a sophisticated network of inhibitors and promoters, which are crucial for fine‐tuning AP activation on cell surfaces. While FH and FHL‐1 exert complement inhibitory functions, the FHRs promote AP activation on cell surfaces through various mechanisms, including FH‐dependent and FH‐independent means. Recent structural and functional studies have provided mechanistic insights and illustrated that the proposed FH deregulation activity of the FHRs is restricted to FHR proteins with the capacity to bind to sialic acids. That is the case of the C‐terminal FHR‐1 mutant proteins and is the molecular mechanism by which these mutant proteins cause complement dysregulation and lead to the development of aHUS. Furthermore, it is well established that surface‐bound FHRs can promote AP activation independently of FH by the recruitment of native C3 or C3b to the surface. This previously unrecognized FHR activity represents a paradigm shift in our understanding on how the AP is regulated, as it implies that AP activation can be directed and potentiated to specific surfaces. Importantly, the outcome of AP activation will be determined by the balance between FH and the FHRs on that surface. Disruption of this balance because of genetic or environmental factors is the underlying cause of many diseases associated with AP dysregulation and supports the FH protein family as potential therapeutic target for future interventions.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We would like to thank Cristina Sobrino Fernández‐Miranda for her assistance in preparing the figures and editing of the manuscript. EGdJ is supported by the Spanish “Ministerio de Ciencia Innovación y Universidades” (RTI2018‐095955‐B‐100), the Autonomous Region of Madrid (P2022/BMD‐7278), and the European Union´s Horizon 2020 Research and Innovation Programe (N° 899163). LLC is supported by the European Union´s Horizon 2020 Research and Innovation Programe (N° 899163). BMT is supported by the Spanish “Instituto de Salud Carlos III” (ISCIII) and the European Regional Development Fund from the European Union grant PI19/01624.
Lucientes‐Continente L, Márquez‐Tirado B, Goicoechea de Jorge E. The Factor H protein family: The switchers of the complement alternative pathway. Immunol Rev. 2023;313:25‐45. doi: 10.1111/imr.13166
This article is part of a series of reviews covering The Alternative Pathway or Amplification Loop of Complement appearing in Volume 313 of Immunological Reviews.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN. The tick‐over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb). Mol Immunol. 2008;45(8):2370‐2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harboe M, Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. 2008;12(4):1074‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138(3):439‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. 2012;217(11):1034‐1046. [DOI] [PubMed] [Google Scholar]
- 6. Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190(8):3831‐3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12(7):383‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodriguez de Cordoba S, Esparza‐Gordillo J, Goicoechea de Jorge E, Lopez‐Trascasa M, Sanchez‐Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41(4):355‐367. [DOI] [PubMed] [Google Scholar]
- 9. Fearon DT. Regulation by membrane sialic acid of beta1H‐dependent decay‐dissociation of amplification C3 convertase of the alternative complement pathway. Proc Natl Acad Sci U S A. 1978;75(4):1971‐1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Díaz‐Guillén MA, Rodríguez de Córdoba S, Heine‐Suñer D. A radiation hybrid map of complement factor H and factor H‐related genes. Immunogenetics. 1999;49(6):549‐552. [DOI] [PubMed] [Google Scholar]
- 11. McRae JL, Murphy BE, Eyre HJ, Sutherland GR, Crawford J, Cowan PJ. Location and structure of the human FHR‐5 gene. Genetica. 2002;114(2):157‐161. [DOI] [PubMed] [Google Scholar]
- 12. Perez‐Caballero D, Gonzalez‐Rubio C, Gallardo ME, et al. Clustering of Missense Mutations in the C‐Terminal Region of Factor H in Atypical Hemolytic Uremic Syndrome. Am J Hum Genet. 2001;68(2):478‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zipfel PF, Skerka C. Complement factor H and related proteins: an expanding family of complement‐regulatory proteins? Immunol Today. 1994;15(3):121‐126. [DOI] [PubMed] [Google Scholar]
- 14. Zipfel PF, Skerka C. FHL‐1/reconectin: a human complement and immune regulator with cell‐adhesive function. Immunol Today. 1999;20(3):135‐140. [DOI] [PubMed] [Google Scholar]
- 15. Jozsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodriguez de Cordoba S. Factor H‐related proteins determine complement‐activating surfaces. Trends Immunol. 2015;36(6):374‐384. [DOI] [PubMed] [Google Scholar]
- 16. Garred P, Tenner AJ, Mollnes TE. Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics. Pharmacol Rev. 2021;73(2):792‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goicoechea de Jorge E, Lopez Lera A, Bayarri‐Olmos R, Yebenes H, Lopez‐Trascasa M, Rodriguez de Cordoba S. Common and rare genetic variants of complement components in human disease. Mol Immunol. 2018;102:42‐57. [DOI] [PubMed] [Google Scholar]
- 18. Jozsi M, Schneider AE, Karpati E, Sandor N. Complement factor H family proteins in their non‐canonical role as modulators of cellular functions. Semin Cell Dev Biol. 2019;85:122‐131. [DOI] [PubMed] [Google Scholar]
- 19. Rodriguez de Cordoba S, Diaz‐Guillen MA, Heine‐Suñer D. An integrated map of the human regulator of complement activation (RCA) gene cluster on 1q32. Mol Immunol. 1999;36:803‐808. [DOI] [PubMed] [Google Scholar]
- 20. Rodríguez de Córdoba S, Goicoechea de Jorge E. Translational mini‐review series on complement factor H: Genetics and disease associations of the human complement factor H. Clin Exp Immunol. 2008;151:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez de Cordoba S, Lublin DM, Rubinstein P, Atkinson JP. Human genes for three complement components that regulate the activation of C3 are tightly linked. J Exp Med. 1985;161(5):1189‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krushkal J, Bat O, Gigli I. Evolutionary relationships among proteins encoded by the regulator of complement activation gene cluster. Mol Biol Evol. 2000;17(11):1718‐1730. [DOI] [PubMed] [Google Scholar]
- 23. Cantsilieris S, Nelson BJ, Huddleston J, et al. Recurrent structural variation, clustered sites of selection, and disease risk for the complement factor H (CFH) gene family. Proc Natl Acad Sci U S A. 2018;115(19):E4433‐E4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vik DP, Keeney JB, Munoz‐Canoves P, Chaplin DD, Tack BF. Structure of the murine complement factor H gene. J Biol Chem. 1988;263(32):16720‐16724. [PubMed] [Google Scholar]
- 25. Antonioli AH, White J, Crawford F, et al. Modulation of the Alternative Pathway of Complement by Murine Factor H‐Related Proteins. J Immunol. 2018;200(1):316‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poppelaars F, Goicoechea de Jorge E, Jongerius I, et al. A Family Affair: Addressing the Challenges of Factor H and the Related Proteins. Front Immunol. 2021;12:660194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goicoechea de Jorge E, Caesar JJ, Malik TH, et al. Dimerization of complement factor H‐related proteins modulates complement activation in vivo. Proc Natl Acad Sci U S A. 2013;110(12):4685‐4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tortajada A, Yebenes H, Abarrategui‐Garrido C, et al. C3 glomerulopathy‐associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest. 2013;123(6):2434‐2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marquez‐Tirado B, Gutierrez‐Tenorio J, Tortajada A, et al. Factor H‐Related Protein 1 Drives Disease Susceptibility and Prognosis in C3 Glomerulopathy. J Am Soc Nephrol. 2022;33:1137‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esparza‐Gordillo J, Soria JM, Buil A, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics. 2004;56(2):77‐82. [DOI] [PubMed] [Google Scholar]
- 31. Moore SR, Menon SS, Cortes C, Ferreira VP. Hijacking Factor H for Complement Immune Evasion. Front Immunol. 2021;12:602277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dopler A, Guntau L, Harder MJ, et al. Self versus Nonself Discrimination by the Soluble Complement Regulators Factor H and FHL‐1. J Immunol. 2019;202(7):2082‐2094. [DOI] [PubMed] [Google Scholar]
- 33. Friese MA, Hellwage J, Jokiranta TS, et al. FHL‐1/reconectin and factor H: two human complement regulators which are encoded by the same gene are differently expressed and regulated. Mol Immunol. 1999;36(13–14):809‐818. [DOI] [PubMed] [Google Scholar]
- 34. Mannes M, Dopler A, Huber‐Lang M, Schmidt CQ. Tuning the Functionality by Splicing: Factor H and Its Alternative Splice Variant FHL‐1 Share a Gene but Not All Functions. Front Immunol. 2020;11:596415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Swinkels M, Zhang JH, Tilakaratna V, et al. C‐reactive protein and pentraxin‐3 binding of factor H‐like protein 1 differs from complement factor H: implications for retinal inflammation. Sci Rep. 2018;8(1):1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tortajada A, Gutierrez E, Goicoechea de Jorge E, et al. Elevated factor H‐related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int. 2017;92(4):953‐963. [DOI] [PubMed] [Google Scholar]
- 37. Abarrategui‐Garrido C, Martinez‐Barricarte R, Lopez‐Trascasa M, de Cordoba SR, Sanchez‐Corral P. Characterization of complement factor H‐related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. 2009;114(19):4261‐4271. [DOI] [PubMed] [Google Scholar]
- 38. Martin Merinero H, Subias M, Pereda A, et al. Molecular bases for the association of FHR‐1 with atypical hemolytic uremic syndrome and other diseases. Blood. 2021;137(25):3484‐3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heinen S, Hartmann A, Lauer N, et al. Factor H‐related protein 1 (CFHR‐1) inhibits complement C5 convertase activity and terminal complex formation. Blood. 2009;114(12):2439‐2447. [DOI] [PubMed] [Google Scholar]
- 40. Valoti E, Alberti M, Tortajada A, et al. A novel atypical hemolytic uremic syndrome‐associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H‐dependent complement regulation. J Am Soc Nephrol. 2015;26(1):209‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Csincsi AI, Szabo Z, Banlaki Z, et al. FHR‐1 Binds to C‐Reactive Protein and Enhances Rather than Inhibits Complement Activation. J Immunol. 2017;199(1):292‐303. [DOI] [PubMed] [Google Scholar]
- 42. Martin Merinero H, Zhang Y, Arjona E, et al. Functional characterization of 105 factor H variants associated with aHUS: lessons for variant classification. Blood. 2021;138(22):2185‐2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skerka C, Timmann C, Horstmann RD, Zipfel PF. Two additional human serum proteins structurally related to complement factor H. Evidence for a family of factor H‐related genes. J Immunol. 1992;148(10):3313‐3318. [PubMed] [Google Scholar]
- 44. Eberhardt HU, Buhlmann D, Hortschansky P, et al. Human factor H‐related protein 2 (CFHR2) regulates complement activation. PLoS One. 2013;8(11):e78617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiao X, Ghossein C, Tortajada A, et al. Familial C3 glomerulonephritis caused by a novel CFHR5‐CFHR2 fusion gene. Mol Immunol. 2016;77:89‐96. [DOI] [PubMed] [Google Scholar]
- 46. Park CT, Wright SD. Plasma lipopolysaccharide‐binding protein is found associated with a particle containing apolipoprotein A‐I, phospholipid, and factor H‐related proteins. J Biol Chem. 1996;271(30):18054‐18060. [DOI] [PubMed] [Google Scholar]
- 47. Pouw RB, Gomez Delgado I, Lopez Lera A, et al. High Complement Factor H‐Related (FHR)‐3 Levels Are Associated With the Atypical Hemolytic‐Uremic Syndrome‐Risk Allele CFHR3*B. Front Immunol. 2018;9:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hellwage J, Jokiranta TS, Koistinen V, Vaarala O, Meri S, Zipfel PF. Functional properties of complement factor H‐related proteins FHR‐3 and FHR‐4: binding to the C3d region of C3b and differential regulation by heparin. FEBS Lett. 1999;462(3):345‐352. [DOI] [PubMed] [Google Scholar]
- 49. Fritsche LG, Lauer N, Hartmann A, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age‐related macular degeneration (AMD). Hum Mol Genet. 2010;19(23):4694‐4704. [DOI] [PubMed] [Google Scholar]
- 50. Jozsi M, Richter H, Loschmann I, et al. FHR‐4A: a new factor H‐related protein is encoded by the human FHR‐4 gene. Eur J Hum Genet. 2005;13(3):321‐329. [DOI] [PubMed] [Google Scholar]
- 51. Pouw RB, Brouwer MC, van Beek AE, Jozsi M, Wouters D, Kuijpers TW. Complement Factor H‐Related Protein 4A Is the Dominant Circulating Splice Variant of CFHR4. Front Immunol. 2018;9:729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hebecker M, Jozsi M. Factor H‐related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem. 2012;287(23):19528‐19536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hebecker M, Okemefuna AI, Perkins SJ, Mihlan M, Huber‐Lang M, Jozsi M. Molecular basis of C‐reactive protein binding and modulation of complement activation by factor H‐related protein 4. Mol Immunol. 2010;47(6):1347‐1355. [DOI] [PubMed] [Google Scholar]
- 54. Mihlan M, Hebecker M, Dahse HM, et al. Human complement factor H‐related protein 4 binds and recruits native pentameric C‐reactive protein to necrotic cells. Mol Immunol. 2009;46(3):335‐344. [DOI] [PubMed] [Google Scholar]
- 55. van Beek AE, Pouw RB, Brouwer MC, et al. Factor H‐Related (FHR)‐1 and FHR‐2 Form Homo‐ and Heterodimers, while FHR‐5 Circulates Only As Homodimer in Human Plasma. Front Immunol. 2017;8:1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McRae JL, Duthy TG, Griggs KM, et al. Human Factor H‐Related Protein 5 Has Cofactor Activity, Inhibits C3 Convertase Activity, Binds Heparin and C‐Reactive Protein, and Associates with Lipoprotein. J Immunol. 2005;174(10):6250‐6256. [DOI] [PubMed] [Google Scholar]
- 57. Medjeral‐Thomas NR, Lomax‐Browne HJ, Beckwith H, et al. Circulating complement factor H‐related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017;92(4):942‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McRae JL, Cowan PJ, Power DA, et al. Human Factor H‐related Protein 5 (FHR‐5). A new complement‐associated protein. J Biol Chem. 2001;276(9):6747‐6754. [DOI] [PubMed] [Google Scholar]
- 59. Murphy B, Georgiou T, Machet D, Hill P, McRae J. Factor H‐related protein‐5: a novel component of human glomerular immune deposits. Am J Kidney Dis. 2002;39(1):24‐27. [DOI] [PubMed] [Google Scholar]
- 60. Csincsi AI, Kopp A, Zoldi M, et al. Factor H‐related protein 5 interacts with pentraxin 3 and the extracellular matrix and modulates complement activation. J Immunol. 2015;194(10):4963‐4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Karpati E, Papp A, Schneider AE, et al. Interaction of the Factor H Family Proteins FHR‐1 and FHR‐5 With DNA and Dead Cells: Implications for the Regulation of Complement Activation and Opsonization. Front Immunol. 2020;11:1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thompson RA, Winterborn MH. Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin Exp Immunol. 1981;46(1):110‐119. [PMC free article] [PubMed] [Google Scholar]
- 63. Levy M, Halbwachs‐Mecarelli MC, Guber G, et al. H deficiency in two brothers with atypical dense intramembraneous deposit disease. Kidney Int. 1986;30:949‐956. [DOI] [PubMed] [Google Scholar]
- 64. Warwicker P, Goodship THJ, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53(4):836‐844. [DOI] [PubMed] [Google Scholar]
- 65. Caprioli J, Castelletti F, Bucchioni S, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C‐257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12(24):3385‐3395. [DOI] [PubMed] [Google Scholar]
- 66. Neumann HPH, Salzmann M, Bohnert‐Iwan B, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry‐based study of German speaking countries. J Med Genet. 2003;40(9):676‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de Cordoba SR. Complement genetics and susceptibility to inflammatory disease. Lessons from genotype‐phenotype correlations. Immunobiology. 2016;221(6):709‐714. [DOI] [PubMed] [Google Scholar]
- 68. Høgåsen K, Jansen JH, Mollnes TE, Hovdenes J, Harboe M. Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiency. J Clin Invest. 1995;95(3):1054‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pickering MC, Cook HT, Warren J, et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31(4):424‐428. [DOI] [PubMed] [Google Scholar]
- 70. Pickering MC, Goicoechea de Jorge E, Martinez‐Barricarte R, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204(6):1249‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267‐1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Goicoechea de Jorge E, Harris CL, Esparza‐Gordillo J, et al. Gain‐of‐function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2007;104(1):240‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martinez‐Barricarte R, Heurich M, Valdes‐Canedo F, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120(10):3702‐3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Noris M, Brioschi S, Caprioli J, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. 2003;362(9395):1542‐1547. [DOI] [PubMed] [Google Scholar]
- 75. Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2003;100(22):12966‐12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rodriguez de Cordoba S. Genetic variability shapes the alternative pathway complement activity and predisposition to complement‐related diseases. Immunol Rev. 2022:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119(18):4182‐4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Roumenina LT, Jablonski M, Hue C, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood. 2009;114(13):2837‐2845. [DOI] [PubMed] [Google Scholar]
- 79. Goicoechea de Jorge E, Tortajada A, Garcia SP, et al. Factor H Competitor Generated by Gene Conversion Events Associates with Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol. 2018;29(1):240‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gale DP, de Jorge EG, Cook HT, et al. Identification of a mutation in complement factor H‐related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376(9743):794‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Deltas C, Gale D, Cook T, Voskarides K, Athanasiou Y, Pierides A. C3 glomerulonephritis/CFHR5 nephropathy is an endemic disease in Cyprus: clinical and molecular findings in 21 families. Adv Exp Med Biol. 2013;735:189‐196. [DOI] [PubMed] [Google Scholar]
- 82. Malik TH, Gitterman DP, Lavin DP, et al. Gain‐of‐function factor H‐related 5 protein impairs glomerular complement regulation resulting in kidney damage. Proc Natl Acad Sci U S A. 2021;118(13):e2022722118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen Q, Wiesener M, Eberhardt HU, et al. Complement factor H‐related hybrid protein deregulates complement in dense deposit disease. J Clin Invest. 2014;124(1):145‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Togarsimalemath SK, Sethi SK, Duggal R, et al. A novel CFHR1‐CFHR5 hybrid leads to a familial dominant C3 glomerulopathy. Kidney Int. 2017;92(4):876‐887. [DOI] [PubMed] [Google Scholar]
- 85. Bernabeu‐Herrero ME, Jimenez‐Alcazar M, Anter J, et al. Complement factor H, FHR‐3 and FHR‐1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. 2015;67(2):276‐286. [DOI] [PubMed] [Google Scholar]
- 86. Cipriani V, Lores‐Motta L, He F, et al. Increased circulating levels of Factor H‐Related Protein 4 are strongly associated with age‐related macular degeneration. Nat Commun. 2020;11(1):778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement Factor H Polymorphism and Age‐Related Macular Degeneration. Science. 2005;308(5720):421‐424. [DOI] [PubMed] [Google Scholar]
- 88. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age‐related macular degeneration. Proc Natl Acad Sci U S A. 2005;102(20):7227‐7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Haines JL, Hauser MA, Schmidt S, et al. Complement Factor H Variant Increases the Risk of Age‐Related Macular Degeneration. Science. 2005;308(5720):419‐421. [DOI] [PubMed] [Google Scholar]
- 90. Klein RJ, Zeiss C, Chew EY, et al. Complement Factor H Polymorphism in Age‐Related Macular Degeneration. Science. 2005;308(5720):385‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL. Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol. 1996;157(12):5422‐5427. [PubMed] [Google Scholar]
- 92. Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S. Regulation of Complement Activation by C‐Reactive Protein: Targeting the Complement Inhibitory Activity of Factor H by an Interaction with Short Consensus Repeat Domains 7 and 8‐11. J Immunol. 1999;163(7):3957‐3962. [PubMed] [Google Scholar]
- 93. Weismann D, Hartvigsen K, Lauer N, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478(7367):76‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tortajada A, Montes T, Martinez‐Barricarte R, Morgan BP, Harris CL, de Cordoba SR. The disease‐protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum Mol Genet. 2009;18(18):3452‐3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Holmes LV, Strain L, Staniforth SJ, et al. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS One. 2013;8(4):e60352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gharavi AG, Kiryluk K, Choi M, et al. Genome‐wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hughes AE, Orr N, Esfandiary H, Diaz‐Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age‐related macular degeneration. Nat Genet. 2006;38(10):1173‐1177. [DOI] [PubMed] [Google Scholar]
- 98. Zhao J, Wu H, Khosravi M, et al. Association of genetic variants in complement factor h and factor h‐related genes with systemic lupus erythematosus susceptibility. PLoS Genet. 2011; 7(5):e1002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jozsi M, Strobel S, Dahse HM, et al. Anti factor H autoantibodies block C‐terminal recognition function of factor H in hemolytic uremic syndrome. Blood. 2007;110(5):1516‐1518. [DOI] [PubMed] [Google Scholar]
- 100. Józsi M, Strobel S, Zipfel SLH, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency and affect recognition functions. Mol Immunol. 2007;44(16):3024.17292472 [Google Scholar]
- 101. Lores‐Motta L, Paun CC, Corominas J, et al. Genome‐Wide Association Study Reveals Variants in CFH and CFHR4 Associated with Systemic Complement Activation: Implications in Age‐Related Macular Degeneration. Ophthalmology. 2018;125(7):1064‐1074. [DOI] [PubMed] [Google Scholar]
- 102. Caesar JJ, Lavender H, Ward PN, et al. Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility. Elife. 2014;3:e04008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Li JF, Dai XP, Zhang W, et al. Upregulation of microRNA‐146a by hepatitis B virus X protein contributes to hepatitis development by downregulating complement factor H. MBio. 2015;6(2):e02459‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Okroj M, Corrales L, Stokowska A, Pio R, Blom AM. Hypoxia increases susceptibility of non‐small cell lung cancer cells to complement attack. Cancer Immunol Immunother. 2009;58(11):1771‐1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pogue AI, Li YY, Cui JG, et al. Characterization of an NF‐kappaB‐regulated, miRNA‐146a‐mediated down‐regulation of complement factor H (CFH) in metal‐sulfate‐stressed human brain cells. J Inorg Biochem. 2009;103(11):1591‐1595. [DOI] [PubMed] [Google Scholar]
- 106. Chen SF, Wang FM, Li ZY, Yu F, Zhao MH, Chen M. Plasma complement factor H is associated with disease activity of patients with ANCA‐associated vasculitis. Arthritis Res Ther. 2015;17:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hughes AE, Bridgett S, Meng W, et al. Sequence and Expression of Complement Factor H Gene Cluster Variants and Their Roles in Age‐Related Macular Degeneration Risk. Invest Ophthalmol Vis Sci. 2016;57(6):2763‐2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. van Beek AE, Kamp A, Kruithof S, et al. Reference Intervals of Factor H and Factor H‐Related Proteins in Healthy Children. Front Immunol. 2018;9:1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Narkio‐Makela M, Hellwage J, Tahkokallio O, Meri S. Complement‐regulator factor H and related proteins in otitis media with effusion. Clin Immunol. 2001;100(1):118‐126. [DOI] [PubMed] [Google Scholar]
- 110. Garam N, Cserhalmi M, Prohaszka Z, et al. FHR‐5 Serum Levels and CFHR5 Genetic Variations in Patients With Immune Complex‐Mediated Membranoproliferative Glomerulonephritis and C3‐Glomerulopathy. Front Immunol. 2021;12:720183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Vernon KA, Goicoechea de Jorge E, Hall AE, et al. Acute presentation and persistent glomerulonephritis following streptococcal infection in a patient with heterozygous complement factor H‐related protein 5 deficiency. Am J Kidney Dis. 2012;60(1):121‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gomez Delgado I, Corvillo F, Nozal P, et al. Complement Genetic Variants and FH Desialylation in S. pneumoniae‐Haemolytic Uraemic Syndrome. Front Immunol. 2021;12:641656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U S A. 1976;73(9):3268‐3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Whaley K, Ruddy S. Modulation of the alternative complement pathways by beta 1 H globulin. J Exp Med. 1976;144(5):1147‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Whaley K, Ruddy S. Modulation of C3b hemolytic activity by a plasma protein distinct from C3b inactivator. Science. 1976;193(4257):1011‐1013. [DOI] [PubMed] [Google Scholar]
- 116. Rudnick RB, Chen Q, Stea ED, et al. FHR5 Binds to Laminins, Uses Separate C3b and Surface‐Binding Sites, and Activates Complement on Malondialdehyde‐Acetaldehyde Surfaces. J Immunol. 2018;200(7):2280‐2290. [DOI] [PubMed] [Google Scholar]
- 117. Chen Q, Manzke M, Hartmann A, et al. Complement Factor H‐Related 5‐Hybrid Proteins Anchor Properdin and Activate Complement at Self‐Surfaces. J Am Soc Nephrol. 2016;27(5):1413‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T. Structural basis for sialic acid‐mediated self‐recognition by complement factor H. Nat Chem Biol. 2015;11(1):77‐82. [DOI] [PubMed] [Google Scholar]
- 119. Venables JP, Strain L, Routledge D, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3(10):e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Malik TH, Lavin PJ, Goicoechea de Jorge E, et al. A hybrid CFHR3‐1 gene causes familial C3 glomerulopathy. J Am Soc Nephrol. 2012;23(7):1155‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nieuwdorp M, Meuwese MC, Vink H, Hoekstra JB, Kastelein JJ, Stroes ES. The endothelial glycocalyx: a potential barrier between health and vascular disease. Curr Opin Lipidol. 2005;16(5):507‐511. [DOI] [PubMed] [Google Scholar]
- 122. Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci U S A. 1990;87(10):3982‐3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Pangburn MK, Muller‐Eberhard HJ. Complement C3 convertase: cell surface restriction of beta1H control and generation of restriction on neuraminidase‐treated cells. Proc Natl Acad Sci U S A. 1978;75(5):2416‐2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Syed S, Hakala P, Singh AK, et al. Role of Pneumococcal NanA Neuraminidase Activity in Peripheral Blood. Front Cell Infect Microbiol. 2019;9:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Langford‐Smith A, Keenan TD, Clark SJ, Bishop PN, Day AJ. The role of complement in age‐related macular degeneration: heparan sulphate, a ZIP code for complement factor H? J Innate Immun. 2014;6(4):407‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Clark SJ, Higman VA, Mulloy B, et al. His‐384 Allotypic Variant of Factor H Associated with Age‐related Macular Degeneration Has Different Heparin Binding Properties from the Non‐disease‐associated Form. J Biol Chem. 2006;281(34):24713‐24720. [DOI] [PubMed] [Google Scholar]
- 127. Prosser BE, Johnson S, Roversi P, et al. Structural basis for complement factor H linked age‐related macular degeneration. J Exp Med. 2007;204(10):2277‐2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sahu A, Kozel TR, Pangburn MK. Specificity of the thioester‐containing reactive site of human C3 and its significance to complement activation. Biochem J. 1994;302(Pt 2):429‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Reiss T, Rosa TFA, Blaesius K, et al. Cutting Edge: FHR‐1 Binding Impairs Factor H‐Mediated Complement Evasion by the Malaria Parasite Plasmodium falciparum. J Immunol. 2018;201(12):3497‐3502. [DOI] [PubMed] [Google Scholar]
- 130. Zewde NT, Hsu RV, Morikis D, Palermo G. Systems Biology Modeling of the Complement System Under Immune Susceptible Pathogens. Front Phys. 2021;9:603704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Flierman R, Daha MR. The clearance of apoptotic cells by complement. Immunobiology. 2007;212(4–5):363‐370. [DOI] [PubMed] [Google Scholar]
- 132. Fishelson Z, Attali G, Mevorach D. Complement and apoptosis. Mol Immunol. 2001;38(2–3):207‐219. [DOI] [PubMed] [Google Scholar]
- 133. Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol. 2008;45(5):1199‐1207. [DOI] [PubMed] [Google Scholar]
- 134. Gullstrand B, Martensson U, Sturfelt G, Bengtsson AA, Truedsson L. Complement classical pathway components are all important in clearance of apoptotic and secondary necrotic cells. Clin Exp Immunol. 2009;156(2):303‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Jensen ML, Honore C, Hummelshoj T, Hansen BE, Madsen HO, Garred P. Ficolin‐2 recognizes DNA and participates in the clearance of dying host cells. Mol Immunol. 2007;44(5):856‐865. [DOI] [PubMed] [Google Scholar]
- 136. Lutz HU, Fehr J. Total sialic acid content of glycophorins during senescence of human red blood cells. J Biol Chem. 1979;254(22):11177‐11180. [PubMed] [Google Scholar]
- 137. Meesmann HM, Fehr EM, Kierschke S, et al. Decrease of sialic acid residues as an eat‐me signal on the surface of apoptotic lymphocytes. J Cell Sci. 2010;123(Pt 19):3347‐3356. [DOI] [PubMed] [Google Scholar]
- 138. Cole DS, Hughes TR, Gasque P, Morgan BP. Complement regulator loss on apoptotic neuronal cells causes increased complement activation and promotes both phagocytosis and cell lysis. Mol Immunol. 2006;43(12):1953‐1964. [DOI] [PubMed] [Google Scholar]
- 139. Leffler J, Herbert AP, Norstrom E, et al. Annexin‐II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J Biol Chem. 2010;285(6):3766‐3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gershov D, Kim S, Brot N, Elkon KB. C‐Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med. 2000;192(9):1353‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Martin M, Leffler J, Smolag KI, et al. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016;23(5):903‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zwarthoff SA, Berends ETM, Mol S, et al. Functional Characterization of Alternative and Classical Pathway C3/C5 Convertase Activity and Inhibition Using Purified Models. Front Immunol. 2018;9:1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zhu L, Guo WY, Shi SF, et al. Circulating complement factor H‐related protein 5 levels contribute to development and progression of IgA nephropathy. Kidney Int. 2018;94(1):150‐158. [DOI] [PubMed] [Google Scholar]
- 144. Jullien P, Laurent B, Claisse G, et al. Deletion Variants of CFHR1 and CFHR3 Associate with Mesangial Immune Deposits but Not with Progression of IgA Nephropathy. J Am Soc Nephrol. 2018;29(2):661‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Xie J, Kiryluk K, Li Y, et al. Fine Mapping Implicates a Deletion of CFHR1 and CFHR3 in Protection from IgA Nephropathy in Han Chinese. J Am Soc Nephrol. 2016;27(10):3187‐3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.