

Abstract
Background
PARP (poly(ADP‐ribose) polymerase) inhibitors (PARPi) are now standard of care in metastatic castrate‐resistant prostate cancer (mCRPC) patients with select mutations in DNA damage repair (DDR) pathways, but patients with ATM‐ and BRCA2 mutations may respond differently to PARPi. We hypothesized that differences may also exist in response to taxanes, which may inform treatment sequencing decisions.
Methods
mCRPC patients (N = 158) with deleterious ATM or BRCA2 mutations who received taxanes, PARPi, or both were retrospectively identified from 11 US academic centers. Demographic, treatment, and survival data were collected. Kaplan−Meier analyses were performed and Cox hazard ratios (HR) were calculated for progression‐free survival (PFS) as well as overall survival (OS), from time of first taxane or PARPi therapy.
Results
Fifty‐eight patients with ATM mutations and 100 with BRCA2 mutations were identified. Fourty‐four (76%) patients with ATM mutations received taxane only or taxane before PARPi, while 14 (24%) received PARPi only or PARPi before taxane. Patients with ATM mutations had longer PFS when taxane was given first versus PARPi given first (HR: 0.74 [95% confidence interval [CI]: 0.37−1.50]; p = 0.40). Similarly, OS was longer in patients with ATM mutations who received taxane first (HR: 0.56 [CI: 0.20−1.54]; p = 0.26). Among patients with BRCA2 mutations, 51 (51%) received taxane first and 49 (49%) received PARPi first. In contrast, patients with BRCA2 mutations had longer PFS when PARPi was given first versus taxane given first (HR: 0.85 [CI: 0.54−1.35]; p = 0.49). Similarly, OS was longer in patients with BRCA2 mutations who received PARPi first (HR: 0.75 [CI: 0.41−1.37]; p = 0.35).
Conclusions
Our retrospective data suggest differential response between ATM and BRCA2 mutated prostate cancers in terms of response to PARPi and to taxane chemotherapy. When considering the sequence of PARPi versus taxane chemotherapy for mCRPC with DDR mutations, ATM, and BRCA2 mutation status may be helpful in guiding choice of initial therapy.
Keywords: ATM, BRCA2, DNA repair, mCRPC, overall survival, progression‐free survival
1. INTRODUCTION
Germline and somatic mutations in DNA damage repair (DDR) pathway genes are emerging therapeutic targets in advanced prostate cancer. 1 , 2 , 3 , 4 , 5 In particular, optimizing treatment for patients with mutations in the DDR genes (especially BRCA1, BRCA2, and ATM) is a key area of research, as these mutations portend aggressive clinical courses. 6 , 7 , 8 , 9 , 10 , 11 Poly(ADP‐ribose) polymerase (PARP) inhibitors have revolutionized cancer therapy for patients with BRCA2 mutations 5 , 12 , 13 , 14 , 15 ; however, the utility of PARP inhibitors in patients with ATM mutations is less clear. Further, optimal sequencing of PARP inhibitors relative to taxane chemotherapy is undefined.
ATM is mutated in approximately 5% of all cancers, in malignancies as diverse as mantle cell lymphoma to lung cancer. 16 Early gene panel assays that identified ATM as a successful treatment target for PARP inhibitors required a complete loss of function in the gene. 17 In the real world, the mutational landscape of ATM is vast and the clinical phenotype of many of these mutations are yet unknown. 16
Treatment outcomes in metastatic castration‐resistant prostate cancer (mCRPC) patients with ATM mutations is an area of active investigation. In the TOPARP‐B trial, complete loss of the ATM protein by immunohistochemistry was associated with notable improved clinical response to olaparib, but the overall response of patients with ATM mutations to olaparib remained significantly lower than those observed in patients with BRCA mutations. 18 A large multicenter retrospective analysis of mCRPC patients with BRCA2 and ATM mutations revealed that patients with ATM mutations had longer time to next treatment with first‐line enzalutamide, similar times with taxanes, and shorter times with PARP inhibitors compared to patients with BRCA mutations, respectively. 19 Cell culture studies have demonstrated limited PARP inhibitor response, even when the prostate cancer cells are fully ATM‐deficient. 20 This limited response parallels outcomes in patients with only ATM mutations in PROFOUND 21 (olaparib) and TRITON2 22 (rucaparib).
As there remains ambiguity regarding optimal treatment, we investigated the optimal sequencing of PARP inhibitors relative to conventional chemotherapy (taxanes) in ATM‐mutated mCRPC via a large multicenter retrospective review. In light of the limited response to PARP inhibitors reported in previous studies, we hypothesized that using taxanes first (before PARP inhibition) may yield better survival than using PARP inhibitors first in mCRPC patients with ATM mutations, and vice versa in patients with BRCA2 mutations. We assembled a retrospective cohort of 158 mCRPC patients with ATM or BRCA2 mutations to answer this question. For perspective, we analyzed 58 patients with ATM mutations, relative to the number of patients with ATM mutations in the prospective trials PROFOUND (86), TRITON2 (49), and TOPARP‐B (21).
2. METHODS
2.1. Study population
A retrospective chart review of mCRPC patients across 11 academic centers in the United States was conducted from June to August 2021. Patients were included if any deleterious somatic or germline ATM or BRCA2 mutation was detected on available clinical‐grade genetic sequencing performed by individual study sites. Deleterious mutations were defined as genetic changes that led to predicted protein truncation or loss (frameshift, nonsense or splicing mutations, or homozygous deletions) or missense mutations that were classified as deleterious by the sequencing platform used. Patients were excluded from final analysis if they harbored a mutation in both BRCA2 and ATM. In addition to the presence of a deleterious ATM or BRCA2 mutation, patient must have also received a taxane (docetaxel or cabazitaxel) and/or a PARP inhibitor (any type) for the diagnosis of castration‐resistant prostate cancer. Prior therapy with abiraterone and/or enzalutamide was permitted. Patients were additionally excluded from final analysis if they received ≤21 days of therapy.
2.2. Study outcomes
Demographic, staging, treatment, and genomic characteristics were collected from all patients. This included age, Gleason sum, histology, presence of M1 disease at initial diagnosis, site of metastases, prior enzalutamide and abiraterone exposure, duration of taxane and PARP inhibitor therapy, progression (prostate‐specific antigen [PSA] or radiologic/clinical), and vital status. Genomic data included mutation mechanism, mutation origin (germline or somatic), and zygosity status (monoallelic vs. biallelic). The presence of concurrent genomic alterations in BRCA1, CDK12, CHD1, FOXA1, FOXO1, MED12, MYC, PIK3CA, PTEN, RB1, SPOP, and TP53 were also collected. Statistical analysis between cooccurrence of BRCA2, ATM, PTEN, RB1, and TP53 alterations was performed.
The primary study outcome was progression‐free survival (PFS) on first taxane or PARP inhibitor therapy. This PFS outcome was a composite outcome combining both PSA progression and investigator‐assessed radiographic or clinical progression. Disease progression was defined as PSA progression (≥25% increase in PSA from baseline or nadir) or investigator‐assessed radiographic or clinical progression. In patients with both PSA progression and investigator‐assessed radiographic/clinical progression, the earlier date was denoted as the date of disease progression. The secondary study outcome was overall survival (OS), from the time of first taxane or PARP inhibitor therapy until death from any cause.
2.3. Statistical analysis
Univariate analyses were performed for demographic, staging, treatment, and genomic characteristics using Pearson's χ 2 or Fisher's exact tests and Wilcoxon rank‐sum test for categorical and continuous variables, respectively. Kaplan−Meier survival analysis was performed for the primary and secondary time‐to‐event outcomes. Multivariable Cox proportional‐hazards modeling with backward stepwise selection was employed to assess the contribution of possible confounders on the primary and secondary outcomes. All analyses were performed using SAS version 9.4. Institutional review board approval was obtained at the local level at each participating site.
3. RESULTS
3.1. Study population
The patient selection schema is shown in Figure 1. The final patient population comprised 158 patients, with 100 (63%) and 58 (37%) patients having deleterious BRCA2 and ATM mutations, respectively. Among patients with BRCA2 mutations, 51 (51%) received a taxane only or taxane before PARP inhibitor treatment, while 49 (49%) received a PARP inhibitor only or PARP inhibitor before taxane treatment. Among patients with ATM mutations, 44 (76%) received a taxane only or taxane before a PARP inhibitor, while 14 (24%) received a PARP inhibitor only or PARP inhibitor before a taxane.
Figure 1.
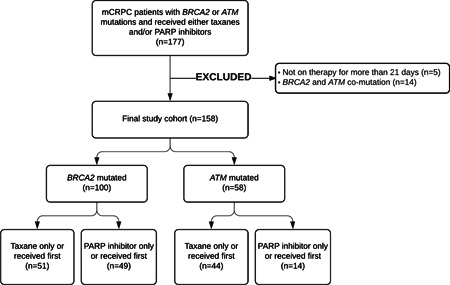
Patient selection schematic. mCRPC, metastatic castrate‐resistant prostate cancer; PARP, poly(ADP‐ribose) polymerase.
3.2. Patient characteristics
Patient demographic, staging, and treatment data are shown in Table 1. The median age of all patients at the time of first taxane or PARP inhibitor treatment was 67 years (interquartile range 62−72 years). There was a significant difference in age at receipt of first taxane or PARP inhibitor therapy between the patients with BRCA2 and ATM mutations (median age 66 years for the BRCA2‐mutated group and 69 years for the ATM‐mutated group, p < 0.01). There was also a significant difference between bone metastases, present in 79% and 91% of patients with BRCA2 and ATM mutations, respectively (p = 0.04). There were no significant differences between the BRCA2‐mutated and ATM‐mutated groups with respect to Gleason sum at diagnosis, M1 disease at diagnosis, presence of metastases (nodal, liver, or lung), prior enzalutamide therapy, or prior abiraterone therapy. Among patients with BRCA2 mutations, 21 (21%) received a taxane only, 29 (29%) received a PARP inhibitor only, 30 (30%) received a taxane first then a PARP inhibitor, and 20 (20%) received a PARP inhibitor first then a taxane. Among patients with ATM mutations, 30 (52%) received a taxane only, 9 (16%) received a PARP inhibitor only, 14 (24%) received a taxane first then a PARP inhibitor, and 5 (9%) received a PARP inhibitor first then a taxane. This overall treatment pattern is significantly different between patients with BRCA2 and ATM mutations (p < 0.001).
Table 1.
Demographic, staging, and treatment characteristics of study population
| Characteristic | Overall | BRCA2‐mutated patients | ATM‐mutated patients | p Value |
|---|---|---|---|---|
| N | 158 | 100 | 58 | |
| Median age at first taxane or PARP inhibitor therapy, years (interquartile range) | 72 (62−72) | 66 (60−71) | 69 (63−74) | <0.01 |
| Gleason sum at diagnosis (%) | 0.34 | |||
| 8−10 | 107 (75%) | 69 (78%) | 38 (70%) | |
| 6−7 | 36 (25%) | 20 (22%) | 16 (30%) | |
| Unknown | 15 | 11 | 4 | |
| M1 disease at initial diagnosis (%) | 0.37 | |||
| Yes | 70 (44%) | 47 (47%) | 23 (40%) | |
| No | 88 (56%) | 53 (53%) | 35 (60%) | |
| Presence of bone metastases | 0.04 | |||
| Yes | 132 (84%) | 79 (79%) | 53 (91%) | |
| No | 26 (16%) | 21 (21%) | 5 (9%) | |
| Presence of nodal metastases | 0.25 | |||
| Yes | 99 (63%) | 66 (66%) | 33 (57%) | |
| No | 59 (37%) | 34 (34%) | 25 (43%) | |
| Presence of liver metastases | 0.32 | |||
| Yes | 35 (22%) | 25 (25%) | 10 (17%) | |
| No | 123 (78%) | 75 (75%) | 48 (83%) | |
| Presence of lung metastases | 0.43 | |||
| Yes | 35 (22%) | 20 (20%) | 15 (26%) | |
| No | 123 (78%) | 80 (80%) | 43 (74%) | |
| Prior enzalutamide therapy | 0.68 | |||
| Yes | 96 (61%) | 62 (62%) | 34 (59%) | |
| No | 62 (39%) | 39 (38%) | 24 (41%) | |
| Prior abiraterone therapy | 0.61 | |||
| Yes | 126 (80%) | 81 (81%) | 45 (78%) | |
| No | 32 (20%) | 19 (19%) | 13 (22%) | |
| Taxane and PARP inhibitor treatment pattern | <0.001 | |||
| Taxane only | 51 (32%) | 21 (21%) | 30 (52%) | |
| PARP inhibitor only | 38 (24%) | 29 (29%) | 9 (16%) | |
| Taxane first, then PARP inhibitor | 44 (28%) | 30 (30%) | 14 (24%) | |
| PARP inhibitor first, then taxane | 25 (16%) | 20 (20%) | 5 (9%) | |
Abbreviation: PARP, poly(ADP‐ribose) polymerase.
Mutation characteristics are shown in Table 2. Tissue samples were obtained from primary tumor (41%), metastatic tissue (36%), circulating tumor DNA (10%), or in some cases by germline‐only testing (13%). The mechanisms of BRCA2 mutation included homozygous deletions (55%), frameshift mutations (31%), missense mutations (6%), and nonsense mutations (8%); while the mechanisms for ATM mutations included deletions (28%), frameshift mutations (28%), missense mutations (22%), nonsense mutations (17%), and splice site mutations (5%). In the overall patient population, 50 (32%) had germline mutations, and 105 (68%) had somatic mutations. A total of 31 (20%) of patients had confirmed biallelic mutations. There was a significant difference in concurrent RB1 alteration between BRCA2‐mutated and ATM‐mutated patients (35% vs. 17%, p = 0.04); there were no differences in cooccurrence in TP53 and PTEN alterations between the two cohorts.
Table 2.
Baseline mutation characteristics of study population
| Characteristic | Overall | BRCA2‐mutated patients | ATM‐mutated patients | p Value |
|---|---|---|---|---|
| N | 158 | 100 | 58 | |
| Sample source | 0.21 | |||
| Primary tissue | 60 (41%) | 42 (46%) | 18 (33%) | |
| Metastatic tissue | 52 (36%) | 30 (33%) | 22 (41%) | |
| Circulating tumor DNA | 14 (10%) | 6 (7%) | 8 (15%) | |
| Germline‐only testing | 19 (13%) | 13 (14%) | 6 (11%) | |
| Unknown | 13 | 9 | 4 | |
| Mechanism of mutation | <0.001 | |||
| Homozygous deletion | 69 (45%) | 53 (55%) | 16 (28%) | |
| Frameshift | 46 (30%) | 30 (31%) | 16 (28%) | |
| Missense | 19 (12%) | 6 (6%) | 13 (22%) | |
| Nonsense | 18 (12%) | 8 (8%) | 10 (17%) | |
| Splicing | 3 (2%) | 0 (0%) | 3 (5%) | |
| Unknown | 3 | 3 | 0 | |
| Origin of mutation | 0.09 | |||
| Germline | 50 (32%) | 36 (37%) | 14 (24%) | |
| Somatic | 105 (68%) | 61 (63%) | 44 (76%) | |
| Unknown | 3 | 3 | 0 | |
| Allelic status of mutation | 0.49 | |||
| Bialleic | 31 (20%) | 21 (22%) | 10 (17%) | |
| Monoalleic or unconfirmed | 123 (80%) | 75 (78%) | 48 (83%) | |
| Unknown | 4 | 4 | 0 | |
| TP53 comutation (missing data = 35) | 0.70 | |||
| No | 80 (65%) | 49 (64%) | 31 (67%) | |
| Yes | 43 (35%) | 28 (36%) | 15 (33%) | |
| PTEN comutation (missing data = 35) | 0.85 | |||
| No | 76 (62%) | 47 (61%) | 29 (63%) | |
| Yes | 47 (38%) | 30 (39%) | 17 (37%) | |
| RB1 comutation (missing data = 35) | 0.04 | |||
| No | 88 (72%) | 50 (65%) | 38 (83%) | |
| Yes | 35 (28%) | 27 (35%) | 8 (17%) | |
The pattern of cooccurring mutations in 14 preselected prostate cancer‐relevant genes is shown in Figure 2. Statistical analyses between the most commonly expressed genes other than ATM and BRCA2 (PTEN, RB1, and TP53) demonstrate significant coalteration of RB1 in PTEN mutated patients (47% vs. 17% in PTEN nonmutated patients, p < 0.001).
Figure 2.
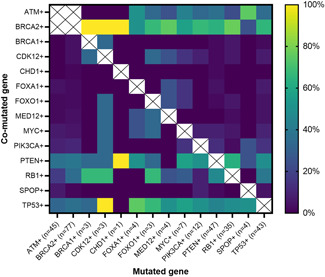
Heatmap of cooccurring alterations. The X‐axis indicates the primary mutated gene (n indicates the number of patients), and the Y‐axis indicates the second comutated gene. The colored squares demonstrate the percentage of patients with a genetic mutation shown in the X‐axis, who also have a concurrent mutation denoted by the Y‐axis. Patients with both BRCA2 and ATM mutations were specifically excluded. [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Study outcomes
The primary outcome of PFS on first taxane or PARP inhibitor therapy by ATM or BRCA2 mutation status is shown in Figure 3. Patients with ATM mutations who received a taxane first had numerically longer median PFS compared to those who received a PARP inhibitor first (6.2 vs. 3.3 months, hazard ratio [HR] with 95% confidence interval [CI] 0.74 [0.37−1.50]; p = 0.40). In contrast, patients with BRCA2 mutations who received a PARP inhibitor first had numerically longer median PFS compared to those who received a taxane first (11.2 vs. 7.2 months, HR: 0.85 (0.54−1.35); p = 0.49). These differences were not statistically significant.
Figure 3.
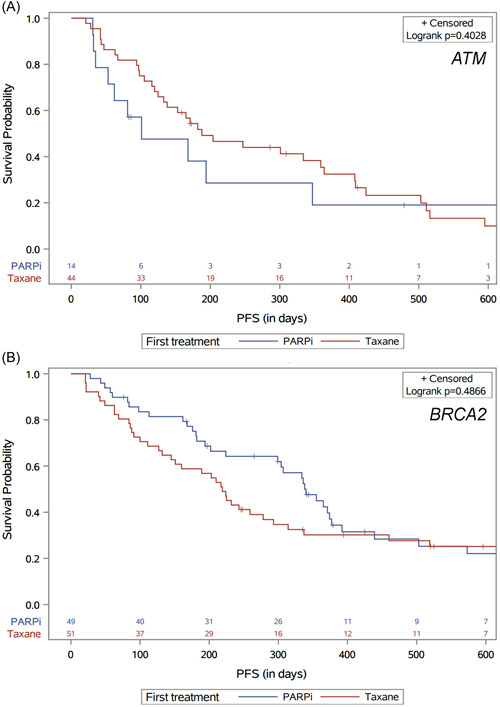
Progression‐free survival (PFS) by first taxane or PARP inhibitor therapy, in (A) patients with ATM mutations and (B) patients with BRCA2 mutations.PARP, poly(ADP‐ribose) polymerase. [Color figure can be viewed at wileyonlinelibrary.com]
The secondary outcome of OS from the time of first taxane or PARP inhibitor therapy to death, by ATM or BRCA2 mutation status, is shown in Figure 4. Patients with ATM mutations who received taxanes first had numerically longer median OS compared to those who received PARP inhibitors first (38.1 vs. 33.0 months, HR: 0.56 (0.20−1.54); p = 0.26). In contrast, patients with BRCA2 mutations who received PARP inhibitors first had numerically longer median OS compared to those who received taxanes first (36.6 vs. 32.8 months, HR: 0.75 (0.41−1.37); p = 0.35). Again, these differences did not reach statistical significance.
Figure 4.
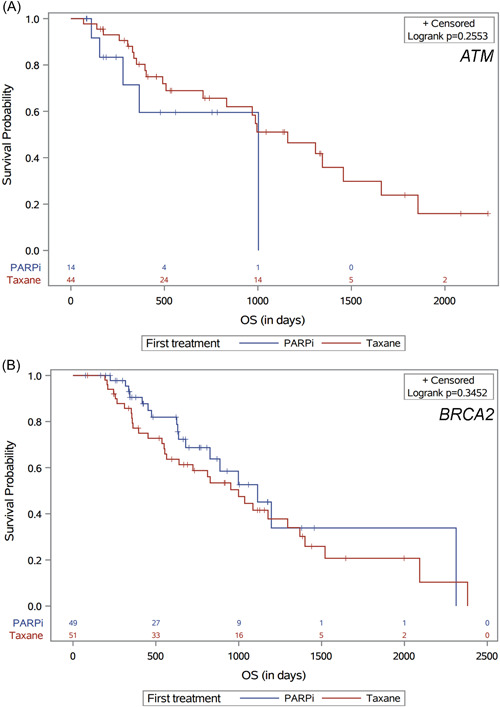
Overall survival (OS) by first taxane or PARP inhibitor therapy, in (A) patients with ATM mutations and (B) patients with BRCA2 mutations.PARP, poly(ADP‐ribose) polymerase. [Color figure can be viewed at wileyonlinelibrary.com]
Multivariable Cox proportional‐hazards modeling using backward stepwise selection to evaluate the impact of possible factors, including choice of first therapy (taxane vs. PARP inhibitor), age, Gleason score, presence of M1 disease at initiation diagnosis, presence of metastases (nodal, liver, or lung), and prior enzalutamide or abiraterone exposure on patients with ATM or BRCA2 mutations did not demonstrate a significant association of any factor with PFS or OS in our model.
4. DISCUSSION
In our multicenter retrospective chart review investigating the optimal sequencing of PARP inhibitors and taxanes in mCRPC patients with ATM or BRCA2 mutations, we found that patients with ATM mutations demonstrated a trend toward longer PFS and OS when taxane was given first rather than PARP inhibitors. The reverse was true for patients with BRCA2 mutations: that PARP inhibitors demonstrated numerically longer PFS and OS when given first over taxanes. However, none of these survival analyses were statistically significant.
The following factors may have contributed to the observed PFS and OS results. First, previous studies 17 , 18 have demonstrated that complete loss of ATM is associated with improved response to PARP inhibitors. As 28% of patients with ATM mutations in our cohort had homozygous deletions in ATM, these patients may have demonstrated a better response to PARP inhibitors compared to other ATM‐mutated patients. It is important to note, however, that other types of ATM‐mutations observed in our cohort may have also led to a loss‐of‐function phenotype, increasing the number of patients in this group. Second, there may have been other unmeasured differences in baseline demographics, clinical characteristics, or previous prostate cancer therapy between the cohorts that affected survival. However, our baseline demographics and clinical characteristics are similar to ATM and BRCA2 patient cohorts in other mCRPC studies. 19 , 23 Third, our cohorts may not have contained sufficient patient numbers to detect a significant difference in outcomes given the unknown relative hazard of PARP inhibitor and taxane therapy in patients with ATM or BRCA2 mutations, and thus our analysis was likely underpowered to interrogate differential treatment sequences.
Although the PFS and OS differences did not reach statistical significance, our study provides insight into the current treatment landscape of mCRPC patients with ATM or BRCA2 mutations. First, the observed numeric advantage in PFS and OS of upfront taxanes compared to upfront PARP inhibitors in ATM‐mutated mCRPC should be confirmed in larger datasets and prospective studies, such as TRITON3. Since mCRPC patients with ATM mutations also appear to be less sensitive to platinum chemotherapy, 19 , 24 ascertaining an efficacious treatment agent in this population is especially important. Second, 49% of patients with BRCA2 mutations in our cohort received PARP inhibitors as the first line of mCRPC therapy, compared to only 25% of patients with ATM mutations, showing the increasing uptake of upfront PARP inhibitor therapy in mCRPC patients with BRCA2 mutations.
There were several limitations to our study, including the retrospective nature and the possible heterogenous phenotypes in our ATM‐mutated cohort. Second, composite PFS was defined as the earliest of three possible indicators of disease progression (biochemical progression, radiological progression, investigator‐determined clinical progression) in this study which may have affected the PFS analysis. Although using a defined biochemical progression would be preferable, limitations in retrospective clinical data across multiple institutions made standardizing this data challenging. Third, retrospective sequencing studies in the metastatic population require controlling for previous treatment courses that may have affected tumor biology at the time of receipt of the treatments of interest. To address this, we did account for prior abiraterone and enzalutamide use in our analysis. Despite these limitations, we were able to collate the largest retrospective ATM‐ and BRCA2‐mutated mCRPC cohort in the literature to our knowledge that contains detailed diagnostic and treatment data with respect to sequencing of taxane and PARP inhibitor agents in these patients. 19 , 23
5. CONCLUSION
Our retrospective multicenter analysis of mCRPC patients with ATM or BRCA2 mutations demonstrates a numerically increased PFS and OS when taxanes are given upfront in patients with ATM mutations, and vice versa with PARP inhibitors in patients with BRCA2 mutations. These differences in clinical outcomes, while not statistically significant, support increasing genomic profiling uptake and the use of tailored optimal sequencing of therapies for mCRPC patients with specific classes of DDR mutations. We hope that these hypothesis‐generating results inspire additional clinical consortia to confirm or refute these findings in larger genetically‐defined mCRPC populations.
CONFLICTS OF INTEREST
Emily Nizialek: Consulting or Advisory Role: AstraZeneca. Jacob E. Berchuck: Honoraria: Digital Science Press. Consulting or Advisory Role: Genome Medical, VetOncoDx. Equity: Genome Medical, VetOncoDx, Cityblock Health (spouse). Employment: Cityblock Health (spouse). Travel, Accommodations, Expenses: Digital Science Press. Patents: Institutional patent filed on methods to detect neuroendocrine prostate cancer through tissue‐informed cell‐free DNA methylation analysis. Pedro C. Barata: Consultant (Institutional): Astellas; Eisai; AVEO Oncology, Janssen, EMD Serono; Dendreon; Pfizer, Seattle Genetics, BMS, Bayer, Guardant Health. Contracted Research (Institutional): AstraZeneca, Merck, AVEO Oncology. Research Grant (Institutional): Blue Earth Diagnostics. Speaker's Bureau (Institutional): Bayer, Caris, Myovant. Rahul R. Aggarwal: Honoraria: Clovis Oncology. Consulting or Advisory Role: Advanced Accelerator Applications; Alessa Therapeutics; Alessa Therapeutics; Amgen; AstraZeneca; Axiom Biotechnologies; Clovis Oncology; Dendreon; Jubilant Pharmaceuticals; Merck; Pfizer. Research Funding: Abbvie (Inst); Amgen (Inst); AstraZeneca (Inst); BioXCel therapeutics (Inst); Cancer Targeted Technology (Inst); Janssen (Inst); Merck (Inst); Novartis (Inst); Xynomic Pharma (Inst); Zenith Epigenetics (Inst). Rana R. McKay: Consulting or Advisory Role: Aveo, Astellas, Medivation, AstraZeneca, Bayer, Bristol Myers Squibb, Calithera Biosciences, Caris, Dendreon, Exelixis, Janssen, Merck, Myovant, Novartis, Pfizer, Sanofi, Sorrento Therapeutics, Tempus, and Vividion Therapeutics. R. R. M. receives research funding from Tempus, Bayer. Neerja Agarwal: Consulting or Advisory Role: Astellas, Astra Zeneca, Aveo, Bayer, Bristol Myers Squibb, Calithera, Clovis, Eisai, Eli Lilly, EMD Serono, Exelixis, Foundation Medicine, Genentech, Gilead, Janssen, Merck, MEI Pharma, Nektar, Novartis, Pfizer, Pharmacyclics, and Seattle Genetics. Funding to Institution: Astellas, Astra Zeneca, Bavarian Nordic, Bayer, Bristol Myers Squibb, Calithera, Celldex, Clovis, Eisai, Eli Lilly, EMD Serono, Exelixis, Genentech, Gilead, Glaxo Smith Kline, Immunomedics, Janssen, Medivation, Merck, Nektar, New Link Genetics, Novartis, Pfizer, Prometheus, Rexahn, Roche, Sanofi, Seattle Genetics, Takeda, and Tracon. Alan H. Bryce: Funding to Institution: Janssen, AstraZeneca, Gilead. Consulting: Merck, Bayer. Honoraria: Elsevier, Fallon Medica, Horizon CME, PRIME Education, MJH Life Sciences. Patents: Therapeutic Targeting of Cancer Patients wiht NRG1 Rearrangements 15/735,289. Oliver Sartor: Consulting: Advanced Accelerator Applications (AAA), Astellas, AstraZeneca, Bayer, Blue Earth Diagnostics, Inc., Bavarian Nordic, Bristol Myers Squibb, Clarity Pharmaceuticals, Clovis, Constellation, Dendreon, EMD Serono, Fusion, Isotopen Technologien Meunchen, Janssen, Myovant, Myriad, Noria Therapeutics, Inc., Novartis, Noxopharm, Progenics, POINT Biopharma, Pfizer, Sanofi, Tenebio, Telix, Theragnostics. Grant/Research Support: Advanced Accelerator Applications, Amgen, AstraZeneca, Bayer, Constellation, Endocyte, Invitae, Janssen, Lantheus, Merck, Progenics, Tenebio. Heather H. Cheng: Research Funds to Institution: Clovis Oncology, Color Genomics, Janssen, Medivation, Phosplatin, Sanofi; Consultant: AstraZeneca; Royalties: UpToDate. Nabil Adra: Consulting or Advisory Role: Astellas Pharma; Aveo; Bristol Myers Squibb Foundation; Bristol Myers Squibb Foundation; Exelixis; Merck; Sanofi. Research Funding: Exelixis (Inst); Genentech (Inst); Merck (Inst). Cora N. Sternberg: Funding, consultancy, advisory: Astellas Pharma/AstraZeneca/Bayer/Genzyme/Gilead/Medscape/Janssen; Merck/MSD/Pfizer/Roche/Impact Pharma/BMS/Sanofi‐Genzyme/UroToday/CCO Clinical, GLC; NCI. Mary‐Ellen Taplin: Consulting or Advisory Role: Janssen, Arcus Bioscience, Roviant, Arvinas, Blue Earth, Clovis, AstraZeneca, Epizyme, Research To Practice, Pfizer, UpToDate. Ajjai S. Alva: Consulting or Advisory Role: AstraZeneca; BMS; Merck; Pfizer. Research Funding: Arcus Biosciences (Inst); Astellas Pharma (Inst); AstraZeneca (Inst); Bayer (Inst); Bristol‐Myers Squibb (Inst); Celgene (Inst); Genentech (Inst); Harpoon Therapeutics (Inst); Janssen (Inst); Merck Sharp & Dohme (Inst); Mirati Therapeutics (Inst); Progenics (Inst); Progenics (Inst); Prometheus (Inst); Roche (Inst). Travel, Accommodations, Expenses: BMS; Merck. Emmanuel S. Antonarakis: Consulting or Advisory Role: Janssen, Astellas, Sanofi, Dendreon, Bayer, BMS, Amgen, ESSA, Constellation, Blue Earth, Exact Sciences, Invitae, Curium, Pfizer, Merck, AstraZeneca, Clovis, Eli Lilly. Grant/Research Support: Janssen, J&J, Sanofi, BMS, Pfizer, AstraZeneca, Novartis, Curium, Constellation, ESSA, Celgene, Merck, Bayer, Clovis. Inventions/patents: Coinventor of AR‐V7 technology licensed to Qiagen. The remaining authors declare no conflict of interest.
ACKNOWLEDGMENTS
This study was funded by National Cancer Institute T32‐CA‐236621 (to Christopher Su) and PNW SPORE:P50 CA097186, CCSG:P30 CA015704 & T32CA009515 (to Alexandra Sokolova).
Su CT, Nizialek E, Berchuck JE, et al. Differential responses to taxanes and PARP inhibitors in ATM‐ versus BRCA2‐mutated metastatic castrate‐resistant prostate cancer. The Prostate. 2023;83:227‐236. 10.1002/pros.24454
DATA AVAILABILITY STATEMENT
Deidentified data that support the findings of this study may be available upon reasonable request from the corresponding author. The data set is not publicly available due to privacy concerns related to protected health information.
REFERENCES
- 1. Hussain M, Daignault‐Newton S, Twardowski PW, et al. Targeting androgen receptor and DNA repair in metastatic castration‐resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2018;36(1):991‐999. 10.1200/JCO.2017.75.7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA‐repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443‐453. 10.1056/NEJMoa1603144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robinson DR, Wu YM, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548(7667):297‐303. 10.1038/nature23306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mateo J, Carreira S, Sandhu S, et al. DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697‐1708. 10.1056/NEJMoa1506859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Castro E, Romero‐Laorden N, Del Pozo A, et al. PROREPAIR‐B: a prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration‐resistant prostate cancer. J Clin Oncol. 2019;37(6):490‐503. 10.1200/JCO.18.00358 [DOI] [PubMed] [Google Scholar]
- 6. Nguyen‐Dumont T, Dowty JG, MacInnis RJ, et al. Rare germline pathogenic variants identified by multigene panel testing and the risk of aggressive prostate cancer. Cancers. 2021;13(7):1495. 10.3390/cancers13071495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Warner E, Herberts C, Fu S, et al. BRCA2, ATM, and CDK12 defects differentially shape prostate tumor driver genomics and clinical aggression. Clin Cancer Res. 2021;27(6):1650‐1662. 10.1158/1078-0432.CCR-20-3708 [DOI] [PubMed] [Google Scholar]
- 8. Castro E, Goh C, Olmos D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31(14):1748‐1757. 10.1200/JCO.2012.43.1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Darst BF, Dadaev T, Saunders E, et al. Germline sequencing DNA repair genes in 5545 men with aggressive and nonaggressive prostate cancer. JNCI: J Nat Cancer Inst. 2021;113(5):616‐625. 10.1093/jnci/djaa132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Na R, Zheng SL, Han M, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71(5):740‐747. 10.1016/j.eururo.2016.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dall'Era MA, McPherson JD, Gao AC, DeVere White RW, Gregg JP, Lara PN. Germline and somatic DNA repair gene alterations in prostate cancer. Cancer. 2020;126(13):2980‐2985. 10.1002/cncr.32908 [DOI] [PubMed] [Google Scholar]
- 12. Annala M, Struss WJ, Warner EW, et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair‐deficient prostate cancer. Eur Urol. 2017;72(1):34‐42. 10.1016/j.eururo.2017.02.023 [DOI] [PubMed] [Google Scholar]
- 13. Marshall CH, Sokolova AO, McNatty AL, et al. Differential response to olaparib treatment among men with metastatic castration‐resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol. 2019;76(4):452‐458. 10.1016/j.eururo.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clarke N, Wiechno P, Alekseev B, et al. Olaparib combined with abiraterone in patients with metastatic castration‐resistant prostate cancer: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Oncol. 2018;19(7):975‐986. 10.1016/S1470-2045(18)30365-6 [DOI] [PubMed] [Google Scholar]
- 15. Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration‐resistant prostate cancer with DNA repair gene aberrations (TOPARP‐B): a multicentre, open‐label, randomised, phase 2 trial. Lancet Oncol. 2020;21(1):162‐174. 10.1016/S1470-2045(19)30684-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jette NR, Kumar M, Radhamani S, et al. ATM‐deficient cancers provide new opportunities for precision oncology. Cancers. 2020;12(3):687. 10.3390/cancers12030687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP‐ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109‐8115. 10.1158/0008-5472.CAN-06-0140 [DOI] [PubMed] [Google Scholar]
- 18. Carreira S, Porta N, Arce‐Gallego S, et al. Biomarkers associating with PARP inhibitor benefit in prostate cancer in the TOPARP‐B trial. Cancer Dis. 2021;11(11):2812‐2827. 10.1158/2159-8290.CD-21-0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwon DH, Chou J, Yip SM, et al. Differential treatment outcomes in BRCA1/2‐, CDK12‐, and ATM‐mutated metastatic castration‐resistant prostate. Cancer. 2021;127(12):1965‐1973. 10.1002/cncr.33487 [DOI] [PubMed] [Google Scholar]
- 20. Rafiei S, Fitzpatrick K, Liu D, et al. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res. 2020;80(11):2094‐2100. 10.1158/0008-5472.CAN-19-3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hussain M, Mateo J, Fizazi K, et al. Survival with olaparib in metastatic castration‐resistant prostate cancer. N Engl J Med. 2020;383(24):2345‐2357. 10.1056/NEJMoa2022485 [DOI] [PubMed] [Google Scholar]
- 22. Abida W, Campbell D, Patnaik A, et al. Non‐BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration‐resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res. 2020;26(11):2487‐2496. 10.1158/1078-0432.CCR-20-0394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sokolova AO, Marshall CH, Lozano R, et al. Efficacy of systemic therapies in men with metastatic castration resistant prostate cancer harboring germline ATM versus BRCA2 mutations. Prostate. 2021;81(16):1382‐1389. 10.1002/pros.24236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mota JM, Barnett E, Nauseef JT, et al. Platinum‐based chemotherapy in metastatic prostate cancer with DNA repair gene alterations. JCO Precision Oncol. 2020;4:355‐366. 10.1200/po.19.00346 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data that support the findings of this study may be available upon reasonable request from the corresponding author. The data set is not publicly available due to privacy concerns related to protected health information.