

Abstract
Background
Antibody–drug conjugates (ADCs) have complex molecular structures and have been tested in numerous clinical trials. Therefore, understanding the mechanisms of their toxicity when applied in medical practice is of high importance.
Methods
In a systematic review and meta‐analysis of data gathered from different scientific databases (PubMed, Embase, Cochrane, and Web of Science) between January 1, 2000, and June 7, 2022, the authors applied a random‐effects model with logit transformation and evaluated the heterogeneity between studies using I2 statistics. The primary outcome was the incidence and 95% confidence interval (CI) for all‐grade and grade ≥3 treatment‐related adverse events and differences between different drugs, molecular structures, and cancer types.
Results
In total, 2511 records were identified that included 169 clinical trials involving 22,492 patients. The overall incidence of treatment‐related adverse events was 91.2% (95% CI, 90.7%–91.7%; I2 = 95.9%) for all‐grade adverse events and 46.1% (95% CI, 45.2%–47.0%; I2 = 96.3%) for grade ≥3 adverse events. The most common all‐grade adverse events were lymphopenia (53.0%; 95% CI, 48.7%–57.3%), nausea (44.1%; 95% CI, 43.2%–44.9%), neutropenia (43.7%; 95% CI, 42.6%–44.9%), blurred vision (40.5%; 95% CI, 37.4%–43.6%), and peripheral neuropathy (39.6%; 95% CI, 38.2%–41.1%); and the most common grade ≥3 adverse events were neutropenia (31.2%; 95% CI, 30.2%–32.3%), hypoesthesia (23.3%; 95% CI, 10.6%–35.9%), thrombocytopenia (22.6%; 95% CI, 21.3%–23.9%), febrile neutropenia (21.2%; 95% CI, 19.3%–23.1%), and lymphopenia (21.0%; 95% CI, 18.2%–23.7%).
Conclusions
Different ADCs appear to affect various treatment‐related adverse events and provide comprehensive data on treatment‐related adverse events for ADCs. The current results provide an important reference for clinicians and patients on how to care for toxicities from ADCs in clinical practice.
Lay summary
Unique anticancer drugs called antibody –drug conjugates (ADCs) have made significant progress in oncology in recent years because of their great success, and they are rapidly being used in the clinic as well as in hundreds of ongoing trials exploring their further use.
The occurrence of serious side effects (adverse events) related to the receipt of ADCs was studied using data from 169 clinical trials involving 22,492 patients to determine the treatment‐related causes of higher toxicity and adverse events in patients who receive ADCs, because these data are crucial for informing physicians how to safely treat patients using ADCs.
The results indicate that different ADCs appear to affect various adverse events related to their use, providing comprehensive data on these ADCs that provide an important reference for clinicians and patients on how to care for toxicities from ADCs in clinical practice.
Keywords: antibody–drug conjugates (ADCs), clinical trials, monoclonal antibody (MoAb), oncology, payload, treatment‐related adverse events
Short abstract
The overall incidence of treatment‐related adverse events was 91.2% for all‐grade adverse events and 46.1% for grade ≥3 adverse events, and the most common all‐grade and grade ≥3 adverse events were lymphopenia (53.0%) and neutropenia (31.2%), respectively. The current results provide an important reference for clinicians and patients on how to care for toxicity from antibody–drug conjugates in clinical practice.
INTRODUCTION
Novel potential anticancer drugs of antibody–drug conjugates (ADCs) have gathered recent attention. ADCs have made significant progress in oncology in recent years because of their great success, and they are rapidly being used in the clinic as well as in hundreds of ongoing trials exploring further indications. 1 , 2 , 3 , 4 ADCs have a complex molecular structure and consist of three main components, including: (1) a monoclonal antibody (MoAb) that has highly selective action targeting tumor‐associated antigens, (2) a potent cytotoxic small molecule (also known as the payload or warhead), and (3) a linker that connects the two substances. 1 , 2 , 3 , 4 The clinical efficacy and toxicity of ADCs are affected by each component. Data have established that ADCs as anticancer therapeutics have a unique mechanism of action, which includes the targeted delivery and release of its payload at the tumor site through MoAb antibody components, thus exerting simultaneous roles as both targeted therapy and chemotherapy. 2 , 5
Since 2000, several ADCs used as anticancer drugs have been approved by the US Food and Drug Administration. Some include gemtuzumab ozogamicin (Mylotarg; Pfizer Inc.), brentuximab vedotin (Adcetris; Seattle Genetics), inotuzumab ozogamicin (Besponsa; Pfizer Inc.), polatuzumab vedotin (Polivy; Genentech), trastuzumab emtansine (T‐DM1, Kadcyla; Genentech), trastuzumab deruxtecan (T‐DXd, Enhertu; Daiichi Sankyo Company Ltd.), sacituzumab govitecan (Trodelvy; Gilead), enfortumab vedotin (Padcev; Astellas Pharma US, Inc., and Seagen Inc.), and tisotoumab vedotin (Tivdak; Seagen Inc. Genmab). 4 , 6 , 7 , 8 , 9 The first four ADCs have been used to treat hematologic malignancies, and the last five have been used to treat solid tumors, including breast cancer, gastric cancer, urothelial cancer, and cervical cancer. 4 , 6 , 7 , 8 , 9 These ADCs circulate in vivo as the conjugated ADC, MoAb, and free payload. 9 The specific characteristics associated with each ADC structure affect the relative proportions of these three components, thereby determining the dose at which the nonconjugated payload can circulate freely and induce off‐target toxicity. 4 , 10 However, although ADCs are a kind of targeted chemotherapy, higher toxicity and adverse side effects have been reported. 4 Hematotoxicity, hepatotoxicity, and gastrointestinal reactions may be associated with the premature release of cytotoxic payloads into the blood. In addition, immune responses partially induced by antibodies to ADCs may cause secondary damage. 11
Given the advent of the era of ADCs, understanding their toxicologic characteristics is critical. To date, few authors have conducted a meta‐analysis on the clinical toxicity of ADCs, and those results indicate that ADCs with different vectors have different adverse reactions and that there are potential differences between ADCs of the same vector in solid and hematologic tumors. 12 Therefore, we believe it is very important to investigate the adverse reactions of different effective vectors of ADCs, to adopt standardized methods to summarize the incidence of different grades of adverse events exerted by different ADCs and their components, and to evaluate their toxicity profiles. Moreover, it is crucial to provide medical specialists with the knowledge to better assess the treatment risk and cope with the possible toxicity of these agents.
We performed a systematic review and meta‐analysis of treatment‐related adverse events based on ADC therapy. Our objective was to investigate the incidence and profile of different treatment‐related adverse events associated with ADCs in clinical trials. Potential differences in the incidence of adverse events among cancer types, drugs, and components were also summarized to provide a reference for clinicians and patients in practice. In particular, we compared the incidence of treatment‐related adverse events between different drugs and molecular structures in randomized controlled trials (RCTs). Because this study was based on previously conducted studies and did not include any new studies with human participants or animals performed by any of the authors, it did not require the approval of the Independent Ethics Committee.
MATERIALS AND METHODS
Search strategy and study selection
We conducted a systematic review and meta‐analysis based on the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses statement to identify published clinical trials of ADCs that reported treatment‐related adverse events. 13 We searched the PubMed, Cochrane, Embase, and Web of Science databases for articles published from January 1, 2000, to June 7, 2022. The key search terms were “antibody–drug conjugate,” “ADC,” “cancer,” and “clinical trial” (see Table S1). References from the searched reviews and articles also were checked manually to avoid missing relevant publications. The inclusion criteria for literature were as follows: (1) prospective clinical trials of cancer treatment published before June 7, 2022; (2) participants who received treatment with ADCs; (3) clinical trials that reported incidences and tabulated data of treatment‐related adverse events; and (4) English language was used in the reported documents. The exclusion criteria were: (1) abstracts of meetings; (2) older publications with incomplete adverse event data from the same study population; (3) a number of patients in the ADCs treatment group <10; and (4) reviews and systematic reviews, meta‐analyses, and cost‐effectiveness analyses.
Data extraction
Two researchers conducted the literature search, study selection, data extraction, and assessment of each article’s quality (Y.Z. and K.L.), and another senior investigator of the team (H.Z.) reviewed differences and resolved conflicts by consensus. Predetermined information data tables were prepared (see Tables S2 and S3). Summary estimates of incidence (number of events vs. total number of patients) were extracted from published reports for analysis. We extracted primary data, including authors’ names, year of publication, clinical trials name and phase, type of cancer, name and type of ADC, the number of patients treated with ADCs, and the number of patients who had adverse events. In addition, the numbers of adverse events in the ADC treatment group and the control group were extracted from RCTs. Data were extracted for all‐grade (according to Common Terminology Criteria for Adverse Events [CTCAE] grades 1–5) and grade 3 or 4 (CTCAE grade 3–4) adverse events, treatment‐related deaths (CTCAE grade 5), and treatment‐related adverse events leading to treatment discontinuation. Adverse event terms were coded according to the Medical Dictionary of Regulatory Activity.
Statistical analysis
The incidence of adverse events was calculated by division, using the number of events as the numerator and the total number of patients as the denominator. We summarized and analyzed the final incidence and 95% confidence intervals (CIs) using a random‐effects model with Logit transformation. In addition to pooling the overall incidence of all‐grade adverse events, we also analyzed the incidence of grade ≥3 adverse events. In addition, we performed subgroup analyses according to drug types, payload types, target agents, and cancer types. Importantly, we also analyzed the overall incidence and toxicity profiles of treatment‐related deaths and treatment‐related adverse events that led to treatment discontinuation, including the subgroup analyses, by drug type, payload type, etc. All models were fitted using the restrictive maximum‐likelihood estimation method.
The risk of adverse events in the ADC treatment group was indirectly compared using the chemotherapy control group as the intermediary and the odds ratio (OR) with the 95% CI as the effective value using the frequentist method weighted by generic inverse variance. We applied the random‐effects model to the pooled analysis of the OR. All p values <.05 were considered statistically significant. In addition, an analysis of variance (ANOVA) or the nonparametric test was used to compare differences in incidence between different cancer types, payload types, and target agents. When the p value of the homogeneity test of variance was <.05, the ANOVA was adopted. Otherwise, we used the nonparametric test. When the p value of the ANOVA or the nonparametric test was <.05, we conducted a post‐hoc analysis. In the ANOVA, we used the least significant difference method for post‐hoc analysis. In the nonparametric test, we used the Kruskal–Wallis test. In the post‐hoc analysis, when the p value adjusted by using the Bonferroni correction method was <.05, we considered that there was a difference in the incidence of adverse events between the two groups.
The I2 statistic was used to evaluate heterogeneity between studies, and univariate regression analysis was used to explore the effect of study‐level factors on heterogeneity. 14 A funnel plot and the Egger test were used to assess publication bias. 15 These two results may overstate the aggregate evidence in the meta‐analysis. 16 , 17 We used the trim‐and‐fill method to draw the modified funnel plot as a secondary evaluation of publication bias, and the visual asymmetry of the modified funnel plot was used to indicate publication bias. 18 The Cochrane Bias of Risk tool was used to assess the risk of bias in included studies. 19 The statistical software programs STATA (version 17; StataCorp), SPSS (version 28.0.1.1; IBM Corporation), and R (version 4.2.1 [metafor and metan packages]; R Foundation for Statistical Computing) were used for all analyses.
RESULTS
Characteristics of patients in included trials
The literature search and systematic review identified 2511 records. After intensive screening, we identified 169 eligible clinical studies involving 22,492 patients, of whom 17,005 were selected for quantitative analysis (Figure 1; see Table S1). In total, 115 clinical trials involving 12,943 patients were analyzed to determine the overall incidence of treatment‐related adverse events, and 97 trials involving 10,495 patients were analyzed to determine the profile of treatment‐related adverse events. In addition, 20 randomized controlled trials involving 7164 patients were included in the analysis comparing treatment‐related adverse events between different ADCs, and 16 trials involving 2250 patients were included in the profile of treatment‐related adverse events with combination regimens. Because most of the RCTs included were open‐label, all trials had a low risk of bias, except for blinding bias (see Table S4). The ADCs used included trastuzumab emtansine (n = 17), brentuximab vedotin (n = 15), trastuzumab deruxtecan (n = 12), rovalpituzumab tesirine (n = 9), sacituzumab govitecan (n = 8), glembatumumab vedotin (n = 7), inotuzumab ozogamicin (n = 5), depatuxizumab mafodotin (n = 5), lorvotuzumab mertansine (n = 4), enfortumab vedotin (n = 4), telisotuzumab vedotin (n = 3), pinatuzumab vedotin (n = 3), mirvetuximab soravtansine (n = 3), lifastuzumab vedotin (n = 3), camidanlumab tesirine (n = 3), loncastuximab tesirine (n = 3), coltuximab ravtansine (n = 3), TAK‐264 (n = 3), anetumab ravtansine (n = 2), RC48‐ADC (n = 2), and SGN‐CD70A (n = 2). Each specific ADC agent had a corresponding payload and target class. The trials that involved payload classes included MMAE (n = 63), emtansine (DM1; n = 24), pyrrolobenzodiazepine (PBD; n = 22), topoisomerase I inhibitor (DXd; n = 13), ravtansin (DM4; n = 13), MMAF (n = 11), irinotecan derivatives (SN‐38; n = 10), calicheamicin (n = 8), and auristatin‐0101 (n = 2). The target agents included human epidermal growth factor receptor 2 (HER2; n = 35), CD30 (n = 16), delta‐like protein 3 (n = 10), human trophoblast cell‐surface antigen 2 (Trop‐2; n = 9), CD79b (n = 8), CD22 (n = 8), C19 (n = 7), glycoprotein NMB (n = 7), epidermal growth factor receptor (n = 7), nectin cell adhesion molecule 4 (n = 4), CD56 (n = 4), folate receptor alpha (n = 4), mesothelin (n = 4), guanylyl cyclase C (n = 3), solute carrier family 34 member 2 (n = 3), CD70 (n = 3), cellular‐mesenchymal epithelial transition factor (n = 3), tissue factor (n = 2), ectonucleotide pyrophosphatase family member 3 (n = 2), prostate‐specific membrane antigen (n = 2), mucin 16 (n = 2), and CD33 (n = 2). In addition, the clinical trials involved the treatment of hematologic malignant neoplasms (n = 41) breast cancer (n = 27), lung cancer (n = 21), urinary cancer (n = 12), gynecologic cancer (n = 9), melanoma (n = 6), gastrointestinal cancer (n = 9), glioma (n = 4), melanoma (n = 3), other cancers (n = 4), and mixed cancer types (n = 28). Those with smaller sample sizes are not listed (see Table S2).
FIGURE 1.
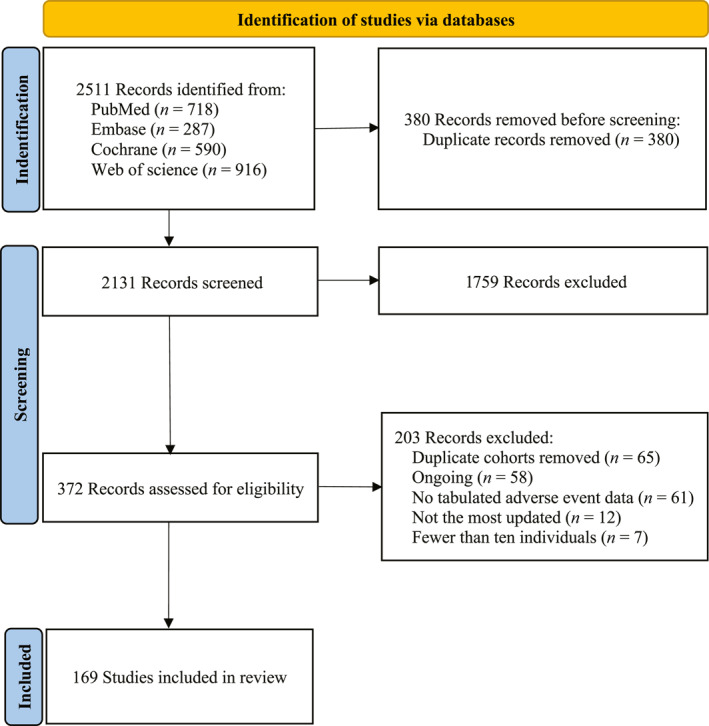
Study selection process.
Overall incidence of treatment‐related adverse events
In our meta‐analysis of the overall incidence of adverse events, we investigated 107 studies that reported all‐grade treatment‐related adverse events and 96 studies that reported grade ≥3 treatment‐related adverse events. The incidence of treatment‐related adverse events was 91.2% (95% CI, 90.7%–91.7%; I2 = 95.5%) for all‐grade of adverse events and 46.1% (95% CI, 45.2%–47.0%; I2 = 96.3%) for grade ≥3 adverse events. We pooled an overview of the overall adverse event rate for each ADC based on the drug agent. In the chemotherapy combinations, the ADCs that most commonly caused all‐grade adverse events were trastuzumab deruxtecan (98.0%; 95% CI, 97.3%–98.8%), polatuzumab vedotin (97.7%; 95% CI, 96.3%–99.0%), sacituzumab govitecan (97.1%; 95% CI, 96.0%–98.2%), patritumab deruxtecan (96.3%; 95% CI, 92.2%–100.4%), tisotumab vedotin (96.0%; 95% CI, 93.5%–98.4%), and trastuzumab emtansine (95.7%; 95% CI, 94.6%–96.7%). The ADCs that most commonly caused grade ≥3 adverse events were brentuximab vedotin (72.9%; 95% CI, 69.9%–75.9%), inotuzumab ozogamicin (68.0%; 95% CI, 63.1%–72.9%), sacituzumab govitecan (61.1%; 95% CI, 57.6%–64.6%), polatuzumab vedotin (60.7%; 95% CI, 56.1%–65.3%), belantamab mafodotin (56.8%; 95% CI, 46.9%–66.8%), and glembatumumab vedotin (56.6%; 95% CI, 51.6%–61.6%; Figures 2 and 3).
FIGURE 2.
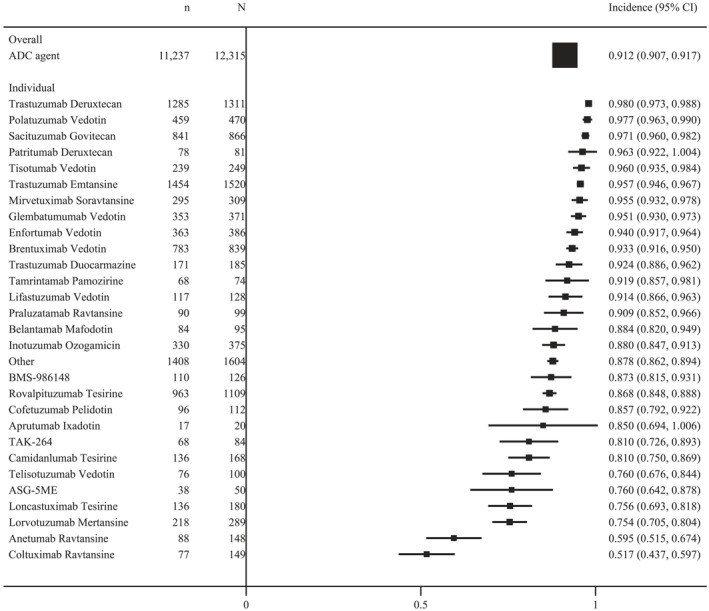
The overall incidence of all‐grade treatment‐related adverse events in ADC regimens. ADC indicates antibody–drug conjugate.
FIGURE 3.
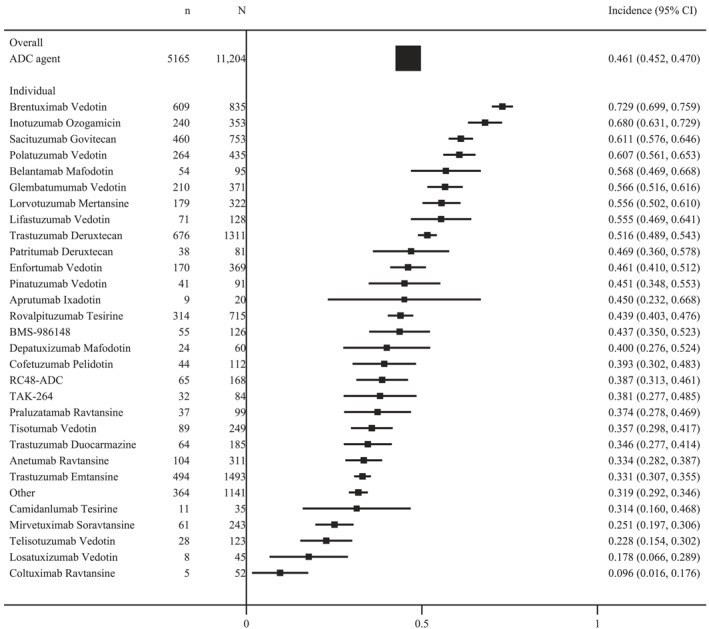
The overall incidence of grade ≥3 treatment‐related adverse events in ADC regimens. ADC indicates antibody–drug conjugate.
We documented more than 600 different types of adverse events from 169 trials. To reflect the most clinically relevant adverse events, we mainly included >20% of all‐grade adverse events and 10% of grade ≥3 adverse events. The most common all‐grade adverse events were lymphopenia (53.0%; 95% CI, 48.7%–57.3%) nausea (44.1%; 95% CI, 43.2%–44.9%), neutropenia (43.7%; 95% CI, 42.6%–44.9%), blurred vision (40.5%; 95% CI, 37.4%–43.6%), and peripheral neuropathy (39.6%; 95% CI, 38.2%–41.1%); and the most common grade ≥3 adverse events were neutropenia (31.2%; 95% CI, 30.2%–32.3%), hypoesthesia (23.3%; 95% CI, 10.6%–35.9%), thrombocytopenia (22.6%; 95% CI, 21.3%–23.9%), febrile neutropenia (21.2%; 95% CI, 19.3%–23.1), and lymphopenia (21.0%; 95% CI, 18.2%–23.7%; Figures 4 and 5).
FIGURE 4.
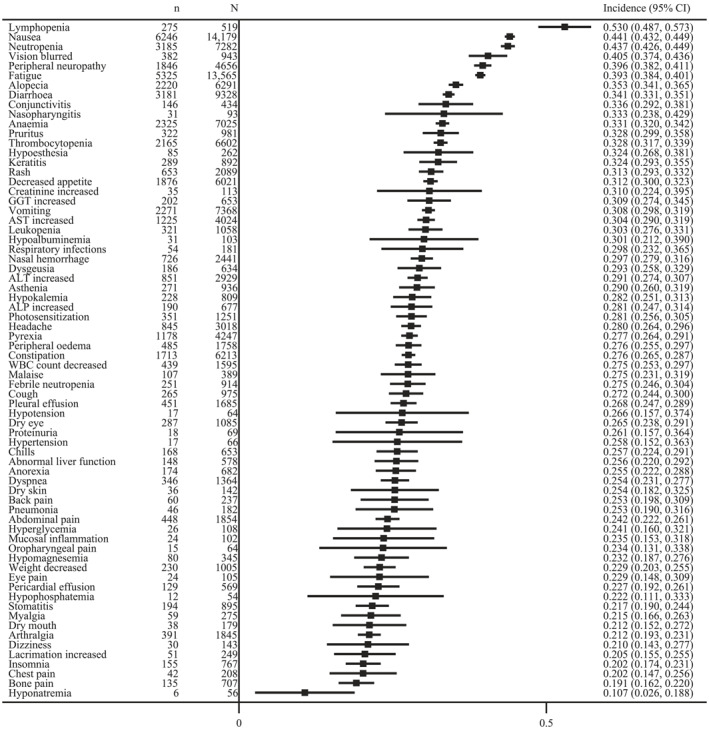
The incidence of most common all‐grade adverse events. ALP indicates alkaline phosphatase; ALT, aspartate aminotransaminase; AST, alanine aminotransferase; GGT, gamma‐glutamyl transpeptidase; WBC, white blood cell.
FIGURE 5.
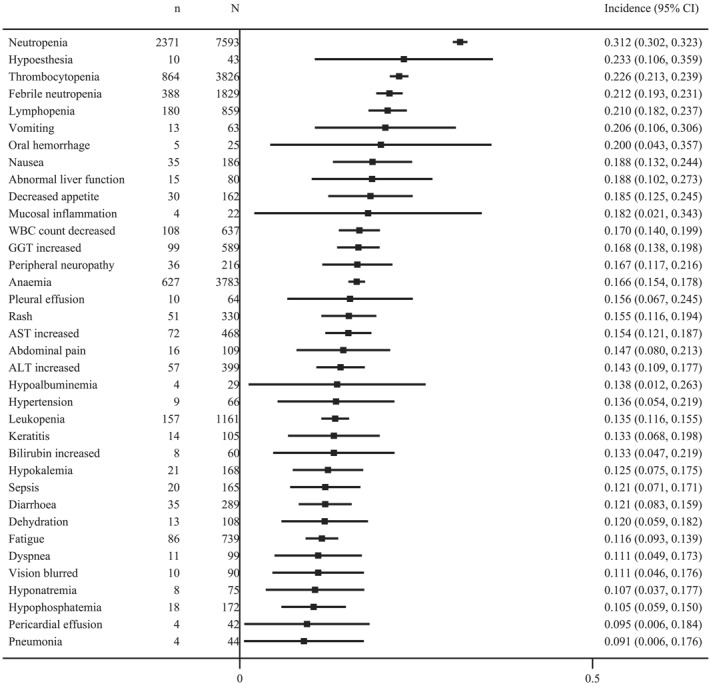
The incidence of most common grade ≥3 adverse events. ALT indicates aspartate aminotransaminase; AST, alanine aminotransferase; GGT, gamma‐glutamyl transpeptidase; WBC, white blood cell.
Incidence of treatment‐related adverse events leading to discontinuation
Of 169 studies, 154 (91.1%) reported treatment‐related adverse events leading to treatment discontinuation. Among the 16,126 patients involved, there were 2132 treatment‐related discontinuations. The overall incidence of treatment‐related discontinuation was 13.2% (95% CI, 12.7%–13.7%; I2 = 86.4%). The ADCs that most commonly caused treatment‐related adverse events leading to treatment discontinuation were BMS‐936561 (42.3%; 95% CI, 23.3%–61.3%), SC‐002 (37.1%; 95% CI, 21.1%–53.2%), and DCDS0780A (35.0%; 95% CI, 22.9%–47.1%; see Figure S1). The payload agents that most commonly caused treatment‐related adverse events leading to treatment discontinuation were MMAW (25.0%; 95% CI, 6.0%–44.0%), MSTP2109A (22.1%; 95% CI, 12.8%–31.3%), and seco‐DUBA (20.5%; 95% CI, 14.7%–26.4%; see Figure S2). The target agents that most commonly caused treatment‐related adverse events leading to treatment discontinuation were CD25 (66.6%; 95% CI, 61.4%–71.7%), CD70 (30.6%; 95% CI, 22.1%–39.2%), and CD33 (26.0%; 95% CI, 18.4%–33.5%; see Figure S2). There were 132 types of adverse events reported in the studies, and 1030 events were not reported in detail. After excluding unknown adverse events, we identified the most common causes of treatment‐related adverse events leading to treatment discontinuation (n = 1113) as peripheral sensory neuropathy (n = 161; 14.5%), thrombocytopenia (n = 125; 11.2%), and peripheral neuropathy (n = 122; 11.0%; see Table S5).
Incidence of treatment‐related deaths
Of the 169 studies, 153 (90.5%) reported treatment‐related adverse events leading to death. Among the 15,340 patients involved, 204 treatment‐related deaths occurred. The overall incidence of treatment‐related deaths was 1.3% (95% CI, 1.1%–1.5%; I2 = 68.2%). The ADCs that most commonly caused treatment‐related deaths were SGN‐CD70A (13.2%; 95% CI, 2.4%–23.9%), DMUC5754A (6.5%; 95% CI, 1.0%–12.0%), and pinatuzumab vedotin (5.8%; 95% CI, 2.1%–9.5%; see Figure S3). There were 59 types of adverse events leading to death reported in the study, and 22 were not reported in detail. After excluding unknown adverse events, we identified the most common causes of treatment‐related death (n = 169) as pneumonitis (n = 21; 12.4%), pneumonia (n = 17; 10.1%), sepsis (n = 13; 7.7%), and respiratory failure (n = 11; 6.5%; Table 1).
TABLE 1.
Spectrum of treatment‐related deaths
| Cause of death a | ADC therapy: No. (%), N = 169 |
|---|---|
| Pulmonary | |
| Pneumonitis | 21 (12.4) |
| Pneumonia | 17 (10.1) |
| Respiratory failure | 11 (6.5) |
| Interstitial lung disease | 5 (3.0) |
| Pulmonary infection | 4 (2.4) |
| Acute respiratory distress syndrome | 3 (4.1) |
| Aspiration pneumonia | 2 (1.2) |
| Bronchopneumonia | 1 (0.6) |
| Bronchopulmonary hemorrhage | 1 (0.6) |
| Hemoptysis | 1 (0.6) |
| Pneumothorax | 1 (0.6) |
| Atypical pneumonia | 1 (0.6) |
| Pulmonary edema | 1 (0.6) |
| Pulmonary cavitation | 1 (0.6) |
| Pneumonitis chemical | 1 (0.6) |
| Pleural effusion | 1 (0.6) |
| Pulmonary arrest | 1 (0.6) |
| Other | 1 (0.6) |
| Infectious | |
| Sepsis | 13 (7.7) |
| Septic shock | 8 (4.7) |
| Neutropenic sepsis | 3 (4.1) |
| Clostridium difficile sepsis | 1 (0.6) |
| Other | 1 (0.6) |
| Hematologic | |
| Neutropenia | 8 (4.7) |
| Febrile neutropenia | 5 (3.0) |
| Disseminated intravascular coagulation | 1 (0.6) |
| Neurologic | |
| Encephalopathy | 2 (1.2) |
| Hepatic encephalopathy | 2 (1.2) |
| Progressive polyneuropathy | 1 (0.6) |
| Subarachnoid hemorrhage | 1 (0.6) |
| Metabolic encephalopathy | 1 (0.6) |
| Cardiovascular | |
| Myocardial infarction | 2 (1.2) |
| Cardiac arrest | 2 (1.2) |
| Ventricular fibrillation | 1 (0.6) |
| Heart attack | 1 (0.6) |
| Cardiac death | 1 (0.6) |
| Renal | |
| Acute kidney injury | 4 (2.4) |
| Renal failure | 2 (1.2) |
| Renal dysfunction | 1 (0.6) |
| Urinary tract obstruction | 1 (0.6) |
| Gastrointestinal | |
| Intestinal perforation | 2 (1.2) |
| Intracranial hemorrhage | 2 (1.2) |
| Ascites | 2 (1.2) |
| Hepatic dysfunction | 2 (1.2) |
| Ischemic colitis | 1 (0.6) |
| Pancreatitis | 1 (0.6) |
| Drug‐induced liver injury | 1 (0.6) |
| Hepatic failure | 1 (0.6) |
| IPEX | 1 (0.6) |
| Other | |
| VOD/SOS | 6 (3.6) |
| Multiorgan failure | 4 (2.4) |
| Multiple organ dysfunction syndrome | 3 (4.1) |
| Diabetic ketoacidosis | 1 (0.6) |
| Generalized edema | 1 (0.6) |
| Sudden death | 1 (0.6) |
| Device related infection | 1 (0.6) |
| General physical health deterioration | 1 (0.6) |
| Disseminated intravascular coagulation | 1 (0.6) |
| Metabolic acidosis | 1 (0.6) |
Abbreviations: IPEX, immune‐dysregulation polyendocrinopathy enteropathy X‐linked; VOS/SOS, veno‐occlusive disease/sinusoidal obstruction syndrome.
aUnknown were not included (22 cases).
Subgroup analysis of the incidence of treatment‐related adverse events
We also pooled 15 different payload agents from 115 clinical trials. The payload agents that most commonly caused all‐grade adverse events were DXd (97.9%; 95% CI, 97.2%–98.7%), SN‐38 (97.2%; 95% CI, 96.1%–98.3%), AS269 (97.1%; 95% CI, 93.1%–101.1%), DM1 (95.5%; 95% CI, 94.7%–96.3%), and eribulin (95.5%; 95% CI, 86.8%–104.2%). The payload agents that most commonly caused grade ≥3 adverse events were MMAW (90.0%; 95% CI, 71.4%–108.6%), calicheamicin (61.3%; 95% CI, 56.7%–65.8%), SN‐38 (61.1%; 95% CI, 57.6%–64.6%), MMAE (53.4%; 95% CI, 51.7%–55.1%), and DXd (50.6%; 95% CI,48.0%–53.2%; see Figure S4). We pooled 38 different target agents from 115 trials. The target agents that most commonly caused all‐grade adverse events were Trop‐2 (97.2%; 95% CI, 96.1%–98.3%), HER2 (96.9%; 95% CI, 96.4%–97.5%), and CD79b (96.8%; 95% CI, 95.3%–98.3%); and the target agents that most commonly caused grade ≥3 adverse events were CD30 (72.9%; 95% CI, 69.9%–75.9%), CD22 (63.3%; 95% CI, 58.8%–67.8%), and CD79b (60.7%; 95% CI, 56.1%–65.3%; see Figure S5).
To focus on the adverse events most associated with payload and target agents, we mainly included >20% of all‐grade adverse events and 10% of grade ≥3 adverse events. Therefore, we selected molecular structure‐related adverse events in a population sample of ≥200. In the MMAE‐based ADCs, the most common all‐grade adverse events were decreased white blood cell count (53.6%; 95% CI, 46.0%–61.1%) and neutropenia (45.3%; 95% CI, 43.1%–47.5%), and the most common grade ≥3 adverse events were neutropenia (37.0%; 95% CI, 34.9%–39.2%) and thrombocytopenia (28.8%; 95% CI, 16.5%–41.2%). In the DM1‐based ADCs, the most common all‐grade adverse events were liver function test abnormalities (72.7%; 95% CI, 46.4%–99.0%) and peripheral neuropathy (56.3%; 95% CI, 48.2%–64.5%), and the most common grade ≥3 adverse events were neutropenia (29.9%; 95% CI, 25.1%–34.8%) and lymphopenia (27.3%; 95% CI, 19.7%–24.6%). In the anti‐HER2–based ADCs, the most common all‐grade adverse events were liver function test abnormalities (72.7%; 95% CI, 46.4%–99.0%) and increased gamma‐glutamyl transferase (48.3%; 95% CI, 30.1%–66.5%), and the most common grade ≥3 adverse events were thrombocytopenia (23.2%; 95% CI, 25.1%–34.8%) and hypoesthesia (23.3%; 95% CI, 10.6%–35.9%). In the anti–delta‐like protein 3 MoAb–based ADCs, the most common all‐grade adverse events were thrombocytopenia (46.4%; 95% CI, 41.6%–51.1%) and increased aspartate transaminase (41.4%; 95% CI, 23.5%–59.3%); and the most common grade ≥3 adverse events were neutropenia (13.8%; 95% CI, 1.2%–26.3%), increased aspartate transaminase (13.8%; 95% CI, 1.2%–26.3%), and hypoalbuminemia (13.8%; 95% CI, 1.2%–26.3%). Other molecular structure‐related adverse events are listed in Tables S6 and S7 and in Figures S6 and S7.
Based on the type of cancer treated in the clinical trials, 115 clinical trials were divided into 12 groups: breast and lung cancers, lymphoma, leukemia, urinary cancer, gynecologic cancer, melanoma, gastrointestinal cancer, glioma, melanoma, other cancers, and mixed cancer types. The highest mean incidence of all‐grade adverse events was observed in breast cancer (97.2%; 95% CI, 96.7%–97.7%), followed by urinary cancer (93.9%; 95% CI, 92.0%–95.7%), and lymphoma (91.0%; 95% CI, 89.9%–92.3%). The highest mean incidence of grade ≥3 adverse events was observed in lymphoma (65.4%; 95% CI, 63.0%–67.7%), followed by leukemia (62.4%; 95% CI, 56.3%–68.5%), and lung cancer (51.8%; 95% CI, 49.0%–54.5%; see Figure S8).
Statistical analysis demonstrated that the mean incidence of grade ≥3 adverse events was similar across cancer types (p = .0004), payload types (p = .032), and target agents (p = .006). However, except for breast cancer, there was no significant difference in the overall incidence of treatment‐related adverse events for the remaining cancer types. There was no significant difference in the overall incidence of grade ≥3 treatment‐related adverse events between different payload types and target agents (see Figures S4, S5, and S8).
Indirect comparison of treatment‐related adverse events
We focused on 20 RCTs to evaluate the correlation between 12 types of ADCs, 8 payload categories, and 10 target agents and the mean incidence of treatment‐related adverse events. Among the specified drugs, sacituzumab govitecan had a higher mean incidence of all‐grade adverse events (OR, 10.94; 95% CI, 4.59–26.04; p < .0001), and lorvotuzumab mertansine had a higher mean incidence of grade ≥3 adverse events (OR, 3.20; 95% CI, 1.32–7.77; p = .0101) compared with the other ADCs. We observed that, among all payload categories, DXd‐based ADCs had a higher mean incidence of all‐grade adverse events (OR, 27.76; 95% CI, 12.00–63.78; p < .0001), whereas SN‐38‐based ADCs had a higher mean incidence of grade ≥3 adverse events (OR, 2.05; 95% CI, 1.42–2.95; p = .0001) compared with the other payload categories. Among the ADC targets, HER2‐expressing ADCs had a higher mean incidence of all‐grade adverse events (OR, 7.53; 95% CI, 5.92–9.58; p < .0001), and CD30‐expressing ADCs had a higher mean incidence of grade ≥3 adverse events (OR, 2.74; 95% CI, 2.18–3.44; p < .0001) compared with the other target agents (see Table S8).
Study heterogeneity
The heterogeneity between studies was statistically quantified using a metaregression model, and the sources of heterogeneity were investigated, including the ADC agent, the payload type, the target agent, and the cancer type. The factors listed are significant sources of heterogeneity (p < .05; see Table S9). There was no obvious asymmetry between the classical funnel plot and the revised funnel plot, indicating that there was no evidence of significant publication bias (if the revised funnel plot showed asymmetry using the scissor‐supplement method, it would indicate publication bias), and the Egger test confirmed that there was no publication bias at all caused by reporting the incidence of grade ≥3 adverse events (p > .05; see Table S9 and Figures S9 and S10).
DISCUSSION
The relatively high incidence of adverse events and their main causes need to be assessed very thoroughly because these data are crucial for determining the safety and efficacy of ADC‐associated medication approaches. Therefore, the detailed study of all treatment‐related adverse events in clinical trials is crucial. This information represents a significant orientation for medical practitioners. A global review of the incidence of adverse events from ADCs provides opportunities and breakthroughs for the wide application of ADCs to clinical practice, supplements the management guidelines for ADC‐induced adverse events, and provides information for clinical practice guidelines.
In the current literature review, we performed a meta‐analysis of 169 studies that examined the incidence and profile of treatment‐related adverse events in ADC therapy. To the best of our knowledge, this systematic review is the largest and the most complete to date. There are previous reports that focus on certain treatment‐related adverse events like neutropenia, thrombocytopenia, leukopenia, hepatic toxicity, peripheral neuropathy, and ocular toxicity, but they analyzed a limited number of literature sources. 12 Moreover, those studies on ADC treatment‐related adverse events mainly reported on the types of payload molecular structures, with less attention to the payload types themselves. 12
Frequently, the dose‐limiting toxicities for most ADCs in medical practice currently appear to be off‐target. 10 This is because the payloads are small molecules, which exploit the typical mechanisms of action of standard chemotherapeutics. Therefore, once released from the MoAbs, the payloads may drive toxicity. 4 , 9 , 10 Furthermore, the influence of the expression level of the molecular target and its biologic distribution of the target also causes relevant clinical toxicity. 20 , 21 For example, efficient and toxic payloads may be the preferred choice for tumor‐specific targets (clean targets); however, conversely, they could be bad payloads for targets that present in healthy tissues. 12 The reason for this is the difficulty in mitigating the toxicity inevitably associated with the target. 12 Therefore, when discussing clinical safety, it is of great importance to pay close attention to the payloads and molecular targets and their interactions with MoAbs and connectors. This may provide a more useful idea of the complete map of ADC toxicity drivers.
The current meta‐analysis demonstrated that the toxicity characteristics of ADC‐associated therapy were significant across the incidence of treatment‐related adverse events for all‐grade (91.2%) and grade ≥3 (46.1%) events. Among the studied ADCs, those that caused side effects for all‐grade and grade ≥3 events were trastuzumab deruxtecan and brentuximab vedotin, respectively. These common adverse events included hematologic events (neuropathy, thrombocytopenia, febrile neutropenia, lymphopenia, anemia, etc.), gastrointestinal effects, ocular toxicities, peripheral neuropathy, fatigue, and hepatic dysfunction. However, some of the adverse events were significant and were associated only with the ADC agents. Close monitoring and early recognition of associated symptoms and signs may help in their proper management and diagnosis. In the case of trastuzumab emtansine and inotuzumab ozogamicin, thrombocytopenia arose as the most recurrent adverse events. 22 , 23 The key reason was that DM1 and MMAF ADCs caused the apoptosis of megakaryocyte progenitors. 24 , 25 For patients receiving brentuximab vedotin, neutropenia presented as the predominant adverse event, which was largely attributed to the disruption of microtubule function caused by MMAE during bone marrow mitosis. 20 , 26 , 27 Therefore, we advise clinicians to take exceptional care with routine blood tests during treatment with MMAE or MMAF ADCs. ABT‐414 and SGN‐75 most commonly caused ocular toxicities, which seemed to be related to the accumulation of DM4 or MMAF ADCs within cells. 20 , 28 , 29 Therefore, steroid eye drops may be used in the event of ocular toxicity caused by these medications. Notably, with brentuximab vedotin, peripheral neuropathy was also the most frequent adverse event, which seemed to occur because of disruption of the interphase microtubule function. 20 , 26 , 27 , 30 It is particularly associated with MMAE ADCs. 20 For peripheral neuropathy events, we have needed to choose neurotrophic drugs for symptomatic treatment in clinical practice. For patients receiving bivatuzumab mertansine and cantuzumab mertansine therapy, hepatic dysfunction events presented as the predominant adverse events of DM1‐nased ADCs by mannose receptors on the surface of hepatic sinusoidal cells. 31 , 32 , 33 Therefore, we recommend that, during ADC‐associated treatment, special attention should be paid to liver enzyme levels by the appropriate use of drugs that lower the liver enzymes and protect them. Furthermore, we also need to pay attention to the adverse events caused by ADC target types. For example, anti‐HER2 ADCs cause pulmonary toxicities and cardiotoxicity, 34 , 35 , 36 , 37 anti‐CD33 ADCs induce neutropenia, 38 antiepidermal growth factor receptor ADCs may be particularly susceptible to ocular and skin toxicities, 20 , 39 and anti‐CD44v6 ADCs can also induce very severe skin toxicities. 40 Obviously, to better understand the adverse events driven by ADCs, it is essential that we include the target information of different drugs, payload types, and target agents in the drug safety models.
Our results also suggest that the ADC regimens continued to cause fatal toxic events, with treatment‐related deaths occurring in 1.3% of all treated patients (204 of 15,340). In all ADC categories, 5 of 38 patients (13.2%) receiving SGN‐CD70A experienced treatment‐related deaths. Respiratory diseases (pneumonitis, 12.4% incidence) were the most common cause of treatment‐related death in patients treated with ADCs. In addition, infection‐related (sepsis, 7.7%) and blood‐related (neutropenia, 4.7%) diseases were the most likely causes of fatally related adverse events. ADC regimens also resulted in a higher incidence of serious toxic events leading to treatment cessation, with treatment‐related discontinuation occurring in 13.2% of all treated patients (2132 of 16,126). Among all ADC categories, 11 of 26 patients (42.6%) receiving BMS‐936561 experienced treatment‐related treatment discontinuation. Neurologic diseases (peripheral sensory neuropathy, 14.5% incidence) were the most common cause of treatment‐related death in patients treated with ADCs. In addition, infection‐related (sepsis, 11.2%) and blood‐related (thrombocytopenia, 11.2%) diseases were the most likely causes of adverse events leading to treatment discontinuation. Because treatment‐related deaths and events leading to treatment discontinuation were important factors for physicians and patients to consider when using innovative drugs, it is crucial to improve clinicians’ awareness and vigilance of routine screening and early treatment.
In the subgroup analysis based on cancer type, ADC therapy was associated with a higher risk of all‐grade adverse events in breast cancer compared with lymphoma, leukemia, and mixed tumors. For the subgroup analysis based on payload type, ADC treatment with DXd‐based ADCs was associated with a higher risk of all‐grade adverse events compared with PBD‐based ADCs. For the subgroup analysis based on target type, anti‐HER2 ADCs were associated with a greater danger of all‐grade adverse events compared with anti‐CD19 ADCs. There was no significant difference in the risk of grade ≥3 adverse events for the subgroups studied. We further investigated whether specific adverse events were associated with specific payload and target types. All‐grade neurotoxicities and ocular toxicities were associated most with MMAE‐based ADCs; whereas constitutional effects, gastrointestinal effects, hematotoxicities, and hematotoxicity were linked with SN‐38‐based ADCs. DM4‐based ADCs were linked with abnormalities in hepatorenal responses and skin reactions, whereas pulmonary adverse events were linked with PBD‐based ADCs. Grade ≥3 gastrointestinal effects, hematotoxicities, hepatorenal responses, ocular toxicities, and pulmonary effects were associated most with MMAE‐based ADCs; and neurotoxicities and constitutional effects were associated with DM1‐based ADCs. All‐grade ocular toxicities and skin reactions were associated most with antifolate receptor alpha MoAb ADCs, pulmonary effects were associated most with anti‐HER2 ADCs, gastrointestinal effects were associated most with anti‐Trop‐2 ADCs, neurotoxicities were associated most with anti‐CD56 ADCs, hematotoxicities and hepatorenal responses were associated most with anti‐CD19 ADCs, and constitutional effects were associated most with anti‐CD22 ADCs. Grade ≥3 neurotoxicities and hepatorenal responses were associated most with anti‐HER2 ADCs, hematotoxicities and constitutional effects were associated most with anti‐CD30 ADCs, and gastrointestinal effects were associated most with anti‐CD22 ADCs. In addition, we also observed that most ADCs (any payload type and any target ADC) produce some degree of hematotoxicities (all‐grade and grade ≥3 adverse events); thus we pay attention to the examination of patients’ routine blood screenings in clinical practice, and those results also provide guidance for symptomatic treatment.
In this meta‐analysis, we conducted indirect comparisons of different drugs, payload types, and target drugs based on RCTs to determine whether the differences were statistically significant, which included much smaller samples but provided the most unbiased results. Sacituzumab govitecan had a higher mean incidence of all‐grade adverse events, and lorvotuzumab mertansine had a higher mean incidence of grade ≥3 adverse events compared with other ADCs. DXd‐based ADCs had a higher mean incidence of all‐grade adverse events, and SN‐38–based ADCs had a higher mean incidence of grade ≥3 adverse events compared with other payload categories. HER2‐expressing ADCs had a higher mean incidence of all‐grade adverse events, and CD30‐expressing ADCs had a higher mean incidence of grade ≥3 adverse events compared with other target agents. The main reason for the significant differences in these results was the variety of ADCs and the diversity of molecular structures. We recommend the drugs included in the RCTs, and we recommend AGS‐16C3F, MMAF‐based ADCs, and ectonucleotide pyrophosphatase family member 3–expressing ADCs. Therefore, in future clinical work, we should choose a treatment plan with a balance of efficacy and safety.
Our study has several limitations. First, the Medical Dictionary of Regulatory Activity was used to record adverse events in clinical trials, but there was overlap in the pooled adverse events in different clinical trials. For example, patients who had increased alanine transaminase and aspartate transaminase levels may have been recorded with elevated liver enzymes or rigid dysfunctions, and keratitis and blurred vision may be directly recorded as ocular toxicities. We suggest establishing more standardized and practical guidelines for recording adverse events. Second, all of our current analyses were based on published clinical trials; however, we observed a small study effect when single‐drug, payload, and target studies had a higher incidence of adverse events and a wider CI. These errors may be caused by the small sample sizes, and the results which should be interpreted with caution. Third, the current study was also affected by any bias or error by the original study investigator, and the results applied only to the group of patients eligible for the included trial. In future work, we need to conduct large, real‐world studies with large populations to verify our results. Fourth, we did not perform computational analyses or classical continuity correction for zero cells and corresponding sample sizes, which may overestimate the incidence. Fifth, we used an indirect comparison method based on RCTs to investigate the differences in ADCs and the adverse events caused by their molecular structures. We assumed that there were no differences in patient characteristics among studies, so there was potential uncertainty about accuracy. However, head‐to‐head comparisons were not conducted, and these results should be interpreted with caution. Finally, the linker, as the driving factor, is the key to ADC toxicity, but we did not perform a separate subgroup analysis for payload linkers. As more and more ADCs with various combinations of payload and linker are used in clinical practice, the key driver of ADC toxicity needs to be explored in the future.
CONCLUSIONS
In the absence of consistency in safety data reporting on oncology drugs, this meta‐analysis summarizes the incidence of all common treatment‐related adverse events for ADC therapy and details the treatment‐related causes of treatment cessation and death. As the published clinical data on ADCs increase in quantity and quality, the drivers of ADC toxicity need to be further investigated. Payload‐related toxicity and molecular target‐related toxicity have different toxicity characteristics. We can also select patient groups for optimal ADC dose and management based on drug and payload types, molecular target expression and distribution, and cancer type by subgroup analysis. This global overview of ADC adverse events serves as a reference for clinicians and may guide clinical practice.
AUTHOR CONTRIBUTIONS
Hong Zhu: Conceptualization, methodology, software, resources, data curation, writing–original draft, writing–review and editing, supervision, project administration, and funding acquisition. Youwen Zhu: Conceptualization, methodology, validation, formal analysis, investigation, writing–original draft, writing–review and editing, and visualization. Kun Liu: Conceptualization, methodology, validation, formal analysis, investigation, writing–original draft, and writing–review and editing. Kailing Wang: Formal analysis, investigation, writing–original draft, and writing–review & editing.
CONFLICTS OF INTEREST
The authors made no disclosures.
Supporting information
Supporting Information 1
ACKNOWLEDGMENTS
This work was partly supported by the Clinical Researh Project of Xiangya Hospital (grant number, 2016L06 to H.Z.). We thank Hong Zhu for providing us with the analysis tools and funding.
Zhu Y, Liu K, Wang K, Zhu H. Treatment‐related adverse events of antibody–drug conjugates in clinical trials: a systematic review and meta‐analysis. Cancer. 2023;129(2):283‐295. doi: 10.1002/cncr.34507
The first two authors contributed equally to this article.
DATA AVAILABILITY STATEMENT
All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Thomas A, Teicher BA, Hassan R. Antibody‐drug conjugates for cancer therapy. Lancet Oncol. 2016;17(6):e254‐e262. doi: 10.1016/s1470-2045(16)30030-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chau CH, Steeg PS, Figg WD. Antibody‐drug conjugates for cancer. Lancet. 2019;394(10200):793‐804. doi: 10.1016/s0140-6736(19)31774-x [DOI] [PubMed] [Google Scholar]
- 3. Hafeez U, Parakh S, Gan HK, Scott AM. Antibody‐drug conjugates for cancer therapy. Molecules. 2020;25(20):4764. doi: 10.3390/molecules25204764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tarantino P, Carmagnani Pestana R, Corti C, et al. Antibody‐drug conjugates: smart chemotherapy delivery across tumor histologies. CA Cancer J Clin. 2022;72(2):165‐182. doi: 10.3322/caac.21705 [DOI] [PubMed] [Google Scholar]
- 5. Sigorski D, Roanowski P, Izycka‐Swieszewska E, Wiktorska K. Antibody‐drug conjugates in uro‐oncology. Target Oncol. 2022;17(3):203‐221. doi: 10.1007/s11523-022-00872-3 [DOI] [PubMed] [Google Scholar]
- 6. Baron J, Wang ES. Gemtuzumab ozogamicin for the treatment of acute myeloid leukemia. Expet Rev Clin Pharmacol. 2018;11(6):549‐559. doi: 10.1080/17512433.2018.1478725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richardson NC, Kasamon YL, Chen H, et al. FDA approval summary: brentuximab vedotin in first‐line treatment of peripheral T‐cell lymphoma. Oncologist. 2019;24(5):e180‐e187. doi: 10.1634/theoncologist.2019-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amiri‐Kordestani L, Blumenthal GM, Xu QC, et al. FDA approval: ado‐trastuzumab emtansine for the treatment of patients with HER2‐positive metastatic breast cancer. Clin Cancer Res. 2014;20(17):4436‐4441. doi: 10.1158/1078-0432.ccr-14-0012 [DOI] [PubMed] [Google Scholar]
- 9. Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody‐drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327‐344. doi: 10.1038/s41571-021-00470-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody‐drug conjugates. Nat Rev Drug Discov. 2017;16(5):315‐337. doi: 10.1038/nrd.2016.268 [DOI] [PubMed] [Google Scholar]
- 11. Mecklenburg L. A brief introduction to antibody‐drug conjugates for toxicologic pathologists. Toxicol Pathol. 2018;46(7):746‐752. doi: 10.1177/0192623318803059 [DOI] [PubMed] [Google Scholar]
- 12. Masters JC, Nickens DJ, Xuan D, Shazer RL, Amantea M. Clinical toxicity of antibody drug conjugates: a meta‐analysis of payloads. Invest New Drugs. 2018;36(1):121‐135. doi: 10.1007/s10637-017-0520-6 [DOI] [PubMed] [Google Scholar]
- 13. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi: 10.1371/journal.pmed.1000097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003;327(7414):557‐560. doi: 10.1136/bmj.327.7414.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629‐634. doi: 10.1136/bmj.315.7109.629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L. Comparison of two methods to detect publication bias in meta‐analysis. JAMA. 2006;295(6):676‐680. doi: 10.1001/jama.295.6.676 [DOI] [PubMed] [Google Scholar]
- 17. Macaskill P, Walter SD, Irwig L. A comparison of methods to detect publication bias in meta‐analysis. Stat Med. 2001;20(4):641‐654. doi: 10.1002/sim.698 [DOI] [PubMed] [Google Scholar]
- 18. Duval S, Tweedie R. Trim and fill: a simple funnel‐plot‐based method of testing and adjusting for publication bias in meta‐analysis. Biometrics. 2000;56(2):455‐463. doi: 10.1111/j.0006-341x.2000.00455.x [DOI] [PubMed] [Google Scholar]
- 19. Cumpston M, Li T, Page MJ, et al. Updated guidance for trusted systematic reviews: a new edition of the Cochrane Handbook for Systematic Reviews of Interventions. Cochrane Database Syst Rev. 2019;10:ED000142. doi: 10.1002/14651858.ED000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody‐drug conjugates. MAbs. 2016;8(4):659‐671. doi: 10.1080/19420862.2016.1156829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bouchard H, Viskov C, Garcia‐Echeverria C. Antibody‐drug conjugates—a new wave of cancer drugs. Bioorg Med Chem Lett. 2014;24(23):5357‐5363. doi: 10.1016/j.bmcl.2014.10.021 [DOI] [PubMed] [Google Scholar]
- 22. Advani A, Coiffier B, Czuczman MS, et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B‐cell non‐Hodgkin's lymphoma: results of a phase I study. J Clin Oncol. 2010;28(12):2085‐2093. doi: 10.1200/jco.2009.25.1900 [DOI] [PubMed] [Google Scholar]
- 23. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33‐positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19(13):3244‐3254. doi: 10.1200/jco.2001.19.13.3244 [DOI] [PubMed] [Google Scholar]
- 24. Zeuner A, Signore M, Martinetti D, Bartucci M, Peschle C, De Maria R. Chemotherapy‐induced thrombocytopenia derives from the selective death of megakaryocyte progenitors and can be rescued by stem cell factor. Cancer Res. 2007;67(10):4767‐4773. doi: 10.1158/0008-5472.can-06-4303 [DOI] [PubMed] [Google Scholar]
- 25. Genentech Inc . Kadcyla prescribing information. Genentech Inc; 2015.
- 26. Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN‐35) for relapsed CD30‐positive lymphomas. N Engl J Med. 2010;363(19):1812‐1821. doi: 10.1056/nejmoa1002965 [DOI] [PubMed] [Google Scholar]
- 27. Seattle Genetics . Adcetris (brentuximab vedotin) prescribing information. Seattle Genetics; 2015. [Google Scholar]
- 28. Reardon DA, Lassman AB, van den Bent M, et al. Efficacy and safety results of ABT‐414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro Oncol. 2017;19(7):965‐975. doi: 10.1093/neuonc/now257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tannir NM, Forero‐Torres A, Ramchandren R, et al. Phase I dose‐escalation study of SGN‐75 in patients with CD70‐positive relapsed/refractory non‐Hodgkin lymphoma or metastatic renal cell carcinoma. Invest New Drugs. 2014;32(6):1246‐1257. doi: 10.1007/s10637-014-0151-0 [DOI] [PubMed] [Google Scholar]
- 30. Vaklavas C, Forero‐Torres A. Safety and efficacy of brentuximab vedotin in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Ther Adv Hematol. 2012;3(4):209‐225. doi: 10.1177/2040620712443076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rupp U, Schoendorf‐Holland E, Eichbaum M, et al. Safety and pharmacokinetics of bivatuzumab mertansine in patients with CD44v6‐positive metastatic breast cancer: final results of a phase I study. Anti Cancer Drugs. 2007;18(4):477‐485. doi: 10.1097/cad.0b013e32801403f4 [DOI] [PubMed] [Google Scholar]
- 32. Tolcher AW, Ochoa L, Hammond LA, et al. Cantuzumab mertansine, a maytansinoid immunoconjugate directed to the CanAg antigen: a phase I, pharmacokinetic, and biologic correlative study. J Clin Oncol. 2003;21(2):211‐222. doi: 10.1200/jco.2003.05.137 [DOI] [PubMed] [Google Scholar]
- 33. Gorovits B, Krinos‐Fiorotti C. Proposed mechanism of off‐target toxicity for antibody‐drug conjugates driven by mannose receptor uptake. Cancer Immunol Immunother. 2013;62(2):217‐223. doi: 10.1007/s00262-012-1369-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med. 2012;367(19):1783‐1791. doi: 10.1056/nejmoa1209124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Modi S, Saura C, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2‐positive breast cancer. N Engl J Med. 2020;382(7):610‐621. doi: 10.1056/nejmoa1914510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Banerji U, van Herpen CML, Saura C, et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2‐expressing breast cancer: a phase 1 dose‐escalation and dose‐expansion study. Lancet Oncol. 2019;20(8):1124‐1135. doi: 10.1016/s1470-2045(19)30328-6 [DOI] [PubMed] [Google Scholar]
- 37. Cote GM, Sawyer DB, Chabner BA. ERBB2 inhibition and heart failure. N Engl J Med. 2012;367(22):2150‐2153. doi: 10.1056/nejmcibr1203156 [DOI] [PubMed] [Google Scholar]
- 38. Sievers EL, Appelbaum FR, Spielberger RT, et al. Selective ablation of acute myeloid leukemia using antibody‐targeted chemotherapy: a phase I study of an anti‐CD33 calicheamicin immunoconjugate. Blood. 1999;93(11):3678‐3684. doi: 10.1182/blood.v93.11.3678.411k24_3678_3684 [DOI] [PubMed] [Google Scholar]
- 39. Renouf DJ, Velazquez‐Martin JP, Simpson R, Siu LL, Bedard PL. Ocular toxicity of targeted therapies. J Clin Oncol. 2012;30(26):3277‐3286. doi: 10.1200/jco.2011.41.5851 [DOI] [PubMed] [Google Scholar]
- 40. Tijink BM, Buter J, de Bree R, et al. A phase I dose escalation study with anti‐CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res. 2006;12(20 pt 1):6064‐6072. doi: 10.1158/1078-0432.ccr-06-0910 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information 1
Data Availability Statement
All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.