

Abstract
Chronic intraperitoneal injection of thioacetamide (TAA) in rats has been used as an animal model of human cirrhosis to study the effects of the disease on drug metabolism. However, TAA inhibits P450 enzymes directly and independently of cirrhosis. We investigated the effects of chronic cirrhosis in rats, induced by 10 weeks of intraperitoneal TAA, on the P450 enzymes after a 10‐day washout period to eliminate TAA. Liver histology and serum biomarkers of hepatic function confirmed cirrhosis in all animals. Microsomal total P450 content, P450 reductase activity and ethoxycoumarin O‐deethylase activity, a general marker of P450 activity, were significantly reduced by 30%–50% in cirrhotic animals. Additionally, the protein content and Michaelis–Menten kinetics of the activities of CYP2D, CYP2E1 and CYP3A were investigated. Whereas cirrhosis reduced the microsomal protein contents of CYP2D and CYP3A by 70% and 30%, respectively, the protein contents of CYP2E1 were not affected. However, the activities of all the tested isoenzymes were substantially lower in the cirrhotic livers. It is concluded that the TAA model of cirrhosis that incorporates a 10‐day washout period after intraperitoneal injection of the chemical to rats produces isoenzyme‐selective reductions in the P450 proteins or activities, which are independent of the direct inhibitory effects of TAA.
Keywords: animal model, CYP2D, CYP2E1, CYP3A, cytochrome P450, fibrosis, liver cirrhosis, Michaelis–Menten kinetics, thioacetamide
1. INTRODUCTION
Liver cirrhosis is a common disease worldwide, with a prevalence of >1 million patients in the United States alone. 1 Patients with liver cirrhosis usually receive several drugs both for the management of cirrhosis and also for other associated diseases and comorbidities. 2 , 3 However, liver cirrhosis significantly affects the drug metabolism and clearance in the liver through multiple mechanisms, including changes in the metabolic activity of the cytochrome P450 (P450) enzymes, liver blood flow, biliary excretion and protein binding of drugs. 4 , 5 , 6 Indeed, it has been reported that patients with liver cirrhosis experience a high frequency of adverse drug reactions and hospitalization. 7 Therefore, it is necessary to study the mechanisms and effects of cirrhosis‐induced changes in the hepatic clearance of drugs in order to improve drug therapy and dosage adjustments in these patients.
Several chemicals and surgical procedures have been used to generate hepatic cirrhosis models in experimental animals, including rodents. 8 , 9 , 10 , 11 , 12 , 13 , 14 Among these models, thioacetamide (TAA), an organosulfur chemical, has been widely used 8 , 9 , 10 , 15 because the histopathological changes in the TAA‐treated animals are similar to those observed in human cirrhosis. 9 To induce cirrhosis, TAA is usually administered orally, in drinking water, or injected intraperitoneally. Both models require chronic administration of TAA. However, the drinking water models usually require longer term administration of TAA (e.g., 6 months) 8 , 16 , 17 and generally result in less reproducible cirrhosis than the models utilizing intraperitoneal injection of the chemical. 8
In agreement with the reduced metabolic activity of P450 enzymes in human cirrhosis, previous studies using TAA‐induced cirrhosis in rodents have also shown decreases in the activities of some hepatic P450 enzymes in cirrhotic animals. 16 , 18 , 19 , 20 However, it has been reported 21 , 22 that the in vivo administration of TAA or its S‐oxide metabolite significantly decreases the liver P450 activities acutely (within 12 h), which is independent of the cirrhosis‐inducing effects of TAA. Further, another study 18 showed that the direct addition of TAA to the rat liver microsomes caused significant decreases in the activities of several P450 isoenzymes, which was NADPH dependent, suggesting a mechanism‐based inactivation of P450 by TAA metabolites. Collectively, these data indicate the need for a washout period for the TAA model to separate the direct mechanism‐based inhibition of P450 activities by TAA from the P450 inhibitory effects of the disease (cirrhosis) itself. Few studies 16 , 23 have reported the effects of TAA‐induced cirrhosis on the P450 enzymes after a washout period. However, these studies were carried out after oral administration of TAA in the drinking water, which requires longer treatment periods and is not as reproducible as the ip injection of the chemical to induce cirrhosis. Therefore, the purpose of the current study was to test the effects of experimental cirrhosis caused by the more reproducible ip injection of TAA in rats after a 10‐day washout period to eliminate the chemical and its direct effects on the P450 enzymes. We determined the Michaelis–Menten kinetics of major P450 enzymes in relation to their protein contents in the liver of cirrhotic animals and their control counterparts. We hypothesized that in the absence of mechanism‐based direct effects of TAA, the TAA‐induced cirrhosis in rats, similar to human cirrhosis, would result in reductions in protein contents and activities of P450 isoenzymes.
2. MATERIALS AND METHODS
2.1. Chemicals and reagents
TAA, cytochrome‐c, NADPH, 7‐ethoxycoumarin, 7‐hydroxycoumarin, chlorzoxazone, 6‐hydroxychlorzoxazone and 4‐hydroxymidazolam (4‐OH‐MDZ) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Dextromethorphan and midazolam (MDZ) were procured from the United States Pharmacopeial Convention (Rockville, MD, USA). Analytical metabolite standard 1′‐hydroxymidazolam (1′‐OH‐MDZ), dextrorphan tartrate and the stable isotopes dextrorphan‐d3 and 7‐hydroxycoumarin‐d5 were purchased from Cerilliant Corporation (Round Rock, TX, USA). The other stable isotopes 1′‐hydroxymidazolam‐d5 and 4‐hydroxymidazolam‐d5 methanoate were purchased from Toronto Research Chemicals (North York, ON, Canada). The stable isotope 6‐hydroxychlorzoxazone‐d2 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Primary antibodies raised in rabbits against CYP2D1 (ab22590; RRID:AB_447177), CYP2E1 (ab84598; RRID:AB_10673797) and calreticulin (ab92516; RRID:AB_10562796) were purchased from Abcam (Cambridge, MA, USA), while the primary antibody raised in mice against CYP3A (D2: sc271033; RRID:AB_10614144) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The secondary antibody HRP‐conjugated goat anti‐rabbit IgG H&L (ab6721; RRID:AB_955447) and HRP‐conjugated goat antimouse IgG H&L (ab6789; RRID:AB_955439) were purchased from Abcam. All the other chemicals and reagents were procured from commercially available sources.
2.2. Animals and experimental design
Male Sprague–Dawley rats, weighing around 150–180 g, were procured from Charles River Laboratories (Wilmington, MA, USA). The rats were housed in a temperature‐ and humidity‐controlled room under a 12‐h light–dark cycle with free access to food and water ad libitum. The animals were acclimated at the animal facility for a week before starting the experiments. The animals were randomly divided into two groups, with seven rats in each group. TAA, dissolved in saline, was injected intraperitoneally (200 mg/kg, 3 days a week for 10 weeks) to the TAA group to induce cirrhosis, while the control group received saline injections. At the end of TAA treatment, the animals were allowed a 10‐day washout period to remove residual TAA, followed by the collection of blood samples through cardiac puncture under isoflurane anaesthesia. After cardiac perfusion with cold saline, the liver samples were collected, snap‐frozen in liquid nitrogen and stored at −80°C.
The study was conducted in accordance with the Basic & Clinical Pharmacology & Toxicology policy for experimental and clinical studies. 24 Additionally, the Institutional Animal Care and Use Committee of Chapman University approved all the experimental animal procedures.
2.3. Serum biochemical and liver tissue histopathological analysis
The serum was separated from the blood samples collected from the saline‐ and TAA‐treated groups. The serum samples were utilized for the quantitation of the liver biomarkers albumin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin (TBil), alkaline phosphatase (ALP) and γ‐glutamyl transferase (GGT) by an automated blood chemical analyser.
For histological studies, sections of the livers from both saline‐ and TAA‐treated groups were taken and fixed in phosphate‐buffered 10% formaldehyde solution, followed by embedding in paraffin wax. Sections of the liver tissues (5‐μm thickness) were stained with haematoxylin–eosin (to assess necrotic inflammation) and Masson's trichrome (to assess fibrosis). They were examined for histopathological changes under the microscope, and their photomicrographs were captured. Histological grading for necro‐inflammatory activity and fibrosis staging was done according to the Knodell scoring system. 25 , 26 The necro‐inflammatory activity has a maximum score of 18, consisting of periportal bridging necrosis (0–10), intralobular degeneration and focal necrosis (0–4) and portal inflammation (0–4). The fibrosis score has a grade range of zero (no fibrosis) to 4 (cirrhosis). The grader was blinded to the treatment of samples.
2.4. Preparation of microsomes
Rat liver microsomes were prepared using the differential centrifugation technique. Frozen liver samples were weighed and homogenized (1:10, w/v) in ice‐cold buffer (250‐mM Mannitol, 0.1‐mM EDTA, 5‐mM HEPES, pH 7.4). The homogenate was spun at 600 g at 4°C for 5 min to separate the large debris and nuclei. The supernatant was collected and centrifuged at 10 300 g at 4°C for 10 min to remove mitochondria. The supernatant (cytosol and microsomes) was collected and centrifuged at 110 000 g at 4°C for 70 min. The resulting microsomal pellet was washed and resuspended in 2 ml of storage buffer (250‐mM Mannitol, 0.1‐mM EDTA, 5‐mM HEPES, 20% glycerol, 0.1‐mM dithiothreitol, 22‐μM butylated hydroxytoluene and 0.1‐mM phenylmethylsulfonylfluoride; pH 7.4) and preserved at −80°C for further experiments. The total protein concentrations were estimated using the Bradford method.
2.5. Western blot analysis
The protein expressions of the major rat cytochrome P450 isoenzymes in the liver microsomal fractions were determined by Western blot analysis. In brief, the liver microsomal samples (0.5‐ to 5‐μg protein/lane) were separated by SDS‐polyacrylamide gel electrophoresis on a precast Bio‐Rad (Hercules, CA, USA) 12% MP TGX gels at a constant voltage (200 V) for 30 min. Proteins were transferred with the Bio‐Rad Trans‐Blot Turbo Transfer System onto 0.45‐μm polyvinylidene fluoride (PVDF) membranes. The PVDF membranes were blocked with 5% BSA (for CYP2D or CYP2E1) or ultra‐blocking reagent (for CYP3A) for 1 h at room temperature. The blots were then incubated (4°C) overnight with primary antibodies diluted in 5% BSA in wash buffer for CYP2D (1:10 000) or CYP2E1 (1:10 000). CYP3A blots were incubated for 1 h with the diluted (1:400) primary antibody. After washing, the blots were incubated with HRP‐conjugated goat anti‐rabbit (CYP2D [1:5000] and CYP2E1 [1:50000]) or HRP‐conjugated goat antimouse (CYP3A [1:2500]) secondary antibodies for 1 h at room temperature and washed before visualization of bands with the Bio‐Rad Chemi Doc Imager system.
After imaging for CYP2D, CYP2E1 and CYP3A, the membranes were stripped to detect calreticulin (endoplasmic reticulum marker) as a loading control. For stripping, the membranes were washed with wash buffer and incubated with Restore™ Western blot stripping buffer (ThermoFisher Scientific, Waltham, MA, USA) for 15 min at room temperature with gentle shaking. After washing, membranes were blocked with 5% BSA and probed with an anti‐calreticulin antibody (1:1000) at 4°C overnight. Subsequently, the membranes were incubated with the HRP‐conjugated goat anti‐rabbit (1:5000) secondary antibody for 1 h at room temperature, and the chemiluminescence was detected using a Bio‐Rad imager.
2.6. P450 reductase activity and total P450
Microsomal P450 reductase activity was quantitated using established methods, based on the reduction of cytochrome c. 27 Total P450 content of the liver microsomes was estimated based on the carbon monoxide difference spectrum method of Omura and Sato 28 and expressed as nanomoles per milligram of protein.
2.7. P450 enzymatic activities
Enzymatic activities of P450 isoenzymes were characterized based on the following reactions: ethoxycoumarin O‐deethylation (ECOD) activity as a general marker of P450 activity, 29 dextromethorphan O‐demethylation for CYP2D, 30 chlorzoxazone 6‐hydroxylation for CYP2E1 30 and midazolam 1′‐hydroxylation and 4‐hydroxylation for CYP3A. 31 For each probe drug, preliminary experiments were performed to determine whether metabolite formation rates were linear with the incubation time and microsomal protein concentrations. For all the assays, the percentage of metabolite conversion was less than 10% of substrate added.
2.7.1. ECOD activity
The assay for ECOD activity was carried out based on the formation of 7‐hydroxycoumarin, in a 0.1‐M phosphate buffer (pH 7.4), at a protein concentration of 0.05 mg/ml using the substrate (7‐ethoxycoumarin) at two concentrations of 50 and 500 μM. The reaction mixtures (100 μl) contained buffer, liver microsomes and the substrate. After 5 min of preincubation at 37°C, the reaction was initiated by the addition of 1‐mM NADPH. After 10 min of incubation at 37°C, reactions were terminated by the addition of ice‐cold acetonitrile (300 μl), containing 400‐nM internal standard (7‐hydroxycoumarin‐d5). Samples were centrifuged, and the supernatant was used for the LC‐MS/MS analysis.
2.7.2. Dextromethorphan O‐demethylase activity
The assay for dextromethorphan O‐demethylase activity was carried out based on the formation of dextrorphan, in a 100‐mM Tris–HCl buffer (pH 7.4), at a protein concentration of 0.05 mg/ml, using the substrate (dextromethorphan) at different concentrations (0.25, 0.5, 2.5, 5, 10, 50, 100, 200 and 500 μM). The reaction mixtures (25 μl) contained buffer, liver microsomes and the substrate. After 5 min of preincubation at 37°C, the reaction was initiated by the addition of 1‐mM NADPH. After 5 min of incubation at 37°C, reactions were terminated by the addition of ice‐cold acetonitrile (75 μl), containing 200‐nM internal standard (dextrorphan‐d3). Samples were centrifuged, and the supernatant was used for the LC‐MS/MS analysis.
2.7.3. Chlorzoxazone 6‐hydroxylase activity
The assay for chlorzoxazone‐6‐hydroxylation activity was carried out based on the formation of 6‐hydroxychlorzoxazone in a 100‐mM Tris–HCl buffer (pH 7.4), at a protein concentration of 0.4 mg/ml, using the substrate (chlorzoxazone) at various concentrations (0.5, 1, 5, 10, 50, 100, 250, 500 and 1000 μM). The reaction mixtures (50 μl) contained buffer, liver microsomes and the substrate. After 5 min of preincubation at 37°C, the reaction was initiated by the addition of 1‐mM NADPH. After 5 min of incubation at 37°C, reactions were terminated by the addition of ice‐cold acetonitrile (150 μl), containing 20‐nM internal standard (6‐hydroxy chlorzoxazone‐d2). Samples were centrifuged, and the supernatant was subjected to the LC‐MS/MS analysis.
2.7.4. Midazolam hydroxylation activity
The assay for midazolam hydroxylation activity was carried out based on the formation of 1′‐OH‐MDZ and 4‐OH‐MDZ, in a 100‐mM Tris–HCl buffer (pH 7.4), at a protein concentration of 0.025 mg/ml, using the substrate (midazolam) at different concentration (0.5, 2, 5, 10, 20, 50, 100 and 200 μM). The reaction mixtures (50 μl) contained buffer, liver microsomes and the substrate. After 5 min of preincubation at 37°C, the reaction was initiated by the addition of 1‐mM NADPH. After 10 min of incubation at 37°C, reactions were terminated by the addition of ice‐cold acetonitrile (150 μl) containing 20‐nM internal standards (1′‐OH‐MDZ‐d5 and 4‐OH‐MDZ‐d5). Samples were centrifuged, and the supernatant was subjected to the LC‐MS/MS analysis.
2.8. Analytical methods
The metabolites 7‐hydroxycoumarin, dextrorphan, 6‐hydroxychlorzoxazone and 1′‐ and 4‐hydroxymidazolam and their respective deuterated internal standards were analysed by UPLC‐tandem mass spectroscopy. The UPLC‐MS/MS system consisted of a Bruker EVOQ triple quadrupole mass spectrometer coupled to the Bruker Advance UPLC system equipped with an integrated column oven, degasser and a CTC PAL autosampler. The system was controlled, and the data were acquired and quantified by the Bruker MSWS‐8 software.
The chromatographic separation of 7‐hydroxycoumarin, dextrorphan or 6‐hydroxychlorzoxazone and their respective deuterated internal standards was achieved using a 5‐cm‐long Phenomenex Kinetex 1.7 μm C18 (100 A, 50 × 2.1 mm) column, connected to a Phenomenex C18 Security Guard ULTRA (2.1 mm) pre‐column maintained at 40°C. 1′‐Hydroxymidazolam, 4′‐hydroxymidazolam and their deuterated internal standards were separated on a 10‐cm‐long Phenomenex Kinetex 1.7 μm C18 (100 A, 100 × 2.1 mm) column.
All the processed samples (2 μl) were injected onto the column and eluted at a flow rate of 0.2 ml/min under gradient conditions consisting of solvent A (5‐mM ammonium formate:formic acid 100:0.05) and solvent B (acetonitrile:methanol:formic acid, 95:5:0.05). The mass spectrometer was equipped with an electrospray ionization source heated at 300°C. The common parameters like cone, probe and nebulizer gas flow were set at 20, 40 and 50 psi, respectively. Quantitation was achieved by MS/MS detection in positive ion mode set at 3000‐V ion spray voltage for dextrorphan, 1′‐hydroxymidazolam and 4‐hydroxymidazolam, while for 7‐hydroxycoumarin and 6‐hydroxychlorzoxazone, the quantitation was achieved in negative ion mode where the ion spray voltage was set at −3000 V. The detailed MS conditions, collision energy and the respective retention times are summarized in Table 1. Additionally, the linear (r 2 ≥ 0.99) range of calibration curves and the lower limit of quantitation (LLOQ) of the analytes are presented in Table 2.
TABLE 1.
Mass spectrometric conditions for the metabolites generated by the P450 substrates and their respective deuterated internal standards
| Analyte | Precursor (Q1) | Product (Q3) | Collision energy (V) | Retention time (min) |
|---|---|---|---|---|
| 7‐Hydroxycoumarin | 160.9 | 132.9 | 15.0 | 3.12 |
| 7‐Hydroxycoumarin‐d5 | 166.0 | 137.9 | 17.0 | 3.11 |
| Dextrorphan | 258 | 156.9 | 39.0 | 2.52 |
| Dextrorphan‐d3 | 261 | 156.9 | 39.0 | 2.52 |
| 6‐Hydroxychloroxazone | 184 | 120.0 | 16.0 | 2.81 |
| 6‐Hydroxychloroxazone‐d2 | 186 | 121.8 | 16.0 | 2.81 |
| 1′‐Hydroxymidazolam | 342 | 202.9 | 26.0 | 5.05 |
| 1′‐Hydroxymidazolam‐d5 | 346.9 | 207.9 | 26.0 | 5.05 |
| 4‐Hydroxymidazolam | 342 | 234.2 | 35.0 | 4.65 |
| 4‐Hydroxymidazolam‐d5 | 346.9 | 234.9 | 23.0 | 4.65 |
TABLE 2.
The linear (r 2 ≥ 0.99) range of calibration curves and the lower limit of quantitation (LLOQ) of the studied metabolites
| Metabolite | Calibration range (nM) | LLOQ (nM) |
|---|---|---|
| 7‐Hydroxycoumarin | 10–1000 | 10 |
| Dextrorphan | 10–2000 | 10 |
| 6‐Hydroxychlorzoxazone | 5–7500 | 5 |
| 1′‐Hydroxymidazolam | 2.5–500 | 2.5 |
| 4‐Hydroxymidazolam | 2.5–500 | 2.5 |
2.9. Data analysis
Data analysis was conducted using GraphPad Prism software (La Jolla, CA, USA). For the estimation of the Michaelis–Menten kinetic parameters, the formation rates of metabolites were plotted against the substrate concentrations. Subsequently, the data were fitted to the following two equations for a one‐ or two‐enzyme kinetic model using nonlinear regression analysis:
where V MAX and K M refer to the maximum velocity and Michaelis–Menten constant, respectively; C refers to the substrate concentration and numbers 1 and 2 denote the enzymes 1 and 2 in a two‐enzyme model. The fit of the data to the two models for each of the metabolites was compared using the sum‐of‐squares F test, which selects the simpler model (single‐enzyme model) unless the p value is less than 0.05.
A comparison of the liver histopathological scores between the TAA and saline groups was conducted using the non‐parametric Mann–Whitney test. All the other comparisons between the two groups were conducted using an unpaired, two‐tailed t‐test. Differences were regarded as statistically significant when p was <0.05. The data are presented as mean ± SE.
3. RESULTS
3.1. Biochemical and histopathological parameters
The serum biomarkers of hepatic functions are shown in Figure 1. Compared with the saline group, the TAA‐treated animals showed significant increases in the serum levels of ALP, GGT, TBil and bile acids and a significant decrease in the serum albumin levels, indicating the presence of cirrhosis and hepatocyte damage in the TAA group. However, no significant changes were observed in the serum levels of ALT, AST and creatinine as a result of cirrhosis after a 10‐day TAA washout period (Figure 1).
FIGURE 1.
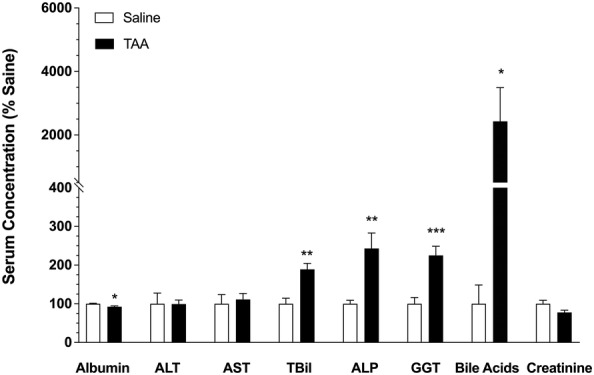
The effects of TAA‐induced cirrhosis on the serum biomarkers of hepatic function in rats. Animals were treated with ip injections of 200‐mg/kg TAA (n = 7) or saline (n = 7) three times a week for 10 weeks, followed by a 10‐day washout period. The columns and bars indicate the mean and SE values, respectively. *, p < 0.05; **, p < 0.01; ***, p < 0.001; two‐tailed unpaired t‐test, compared with the saline group. Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ‐glutamyl transferase; TBil, total bilirubin
Representative photomicrographs of liver sections of saline and TAA animals stained with Masson's trichrome and haematoxylin–eosin (H&E) are depicted in Figure 2. Additionally, the average scores for fibrosis (Masson's trichrome) and necro‐inflammation (H&E) for the two groups of animals are presented in this figure. As demonstrated (Figure 2), all the TAA livers had the highest score (4) for fibrosis without any fibrosis for the saline livers. Further, the necro‐inflammatory score in the TAA group (7.7 ± 0.7) was significantly (p < 0.001) higher than that in the saline animals (1.7 ± 0.4), confirming the presence of cirrhosis in the TAA animals.
FIGURE 2.
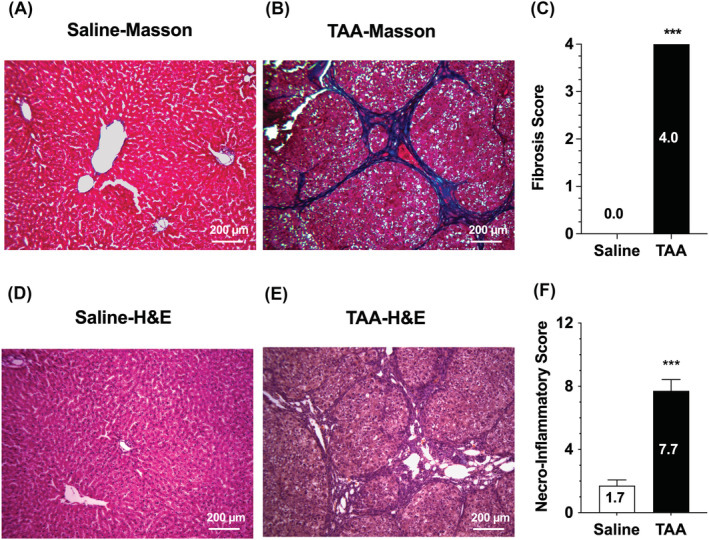
Representative photomicrographs of liver sections stained with Masson's trichrome (A and B) and haematoxylin–eosin (H&E) (D and E) along with fibrosis (C) and necro‐inflammatory (F) scores in rat livers. Animals were treated with ip injections of 200‐mg/kg TAA (n = 7) or saline (n = 7) three times a week for 10 weeks, followed by a 10‐day washout period. The columns and bars indicate the mean and SE values, respectively. ***, p < 0.001; Mann–Whitney test, compared with the saline group
3.2. Total CYP450 and CPR activity
The total 450 content and cytochrome P450 reductase (CPR) activity in the liver microsomes are presented in Figure 3. TAA‐induced cirrhosis caused significant reductions in both total P450 content (~50%, p < 0.01) and the CPR activity (~30%, p < 0.05) in the liver microsomes.
FIGURE 3.
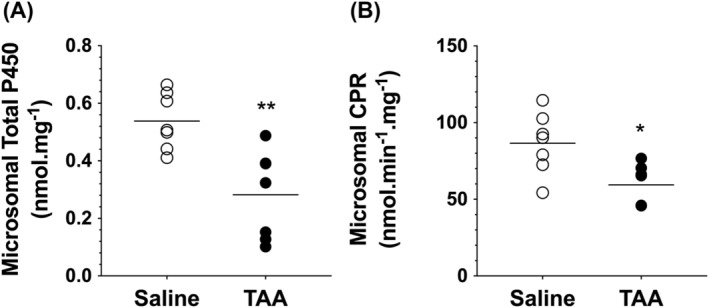
Microsomal total cytochrome P450 (A) and cytochrome P450 reductase (CPR) (B) in rat livers. Animals were treated with ip injections of 200‐mg/kg TAA (n = 7) or saline (n = 7) three times a week for 10 weeks, followed by a 10‐day washout period. The symbols and horizontal lines indicate the individual and mean values, respectively. *, p < 0.05; **, p < 0.01; two‐tailed unpaired t‐test, compared with the saline group
3.3. Western blot analysis of P450 isoenzymes
The protein contents of CYP2D, CYP2E1 and CYP3A in the liver microsomal preparations are shown in Figure 4. For all the isoenzymes, calreticulin was used as the loading control. The TAA treatment showed significant reductions in the protein contents of CYP2D (~70%, p < 0.001; Figure 4A) and CYP3A (~30%, p < 0.05; Figure 4C) enzymes as compared with those of the saline control group. However, there were no significant changes in the protein levels of CYP2E1 as a result of TAA‐induced cirrhosis (Figure 4B).
FIGURE 4.
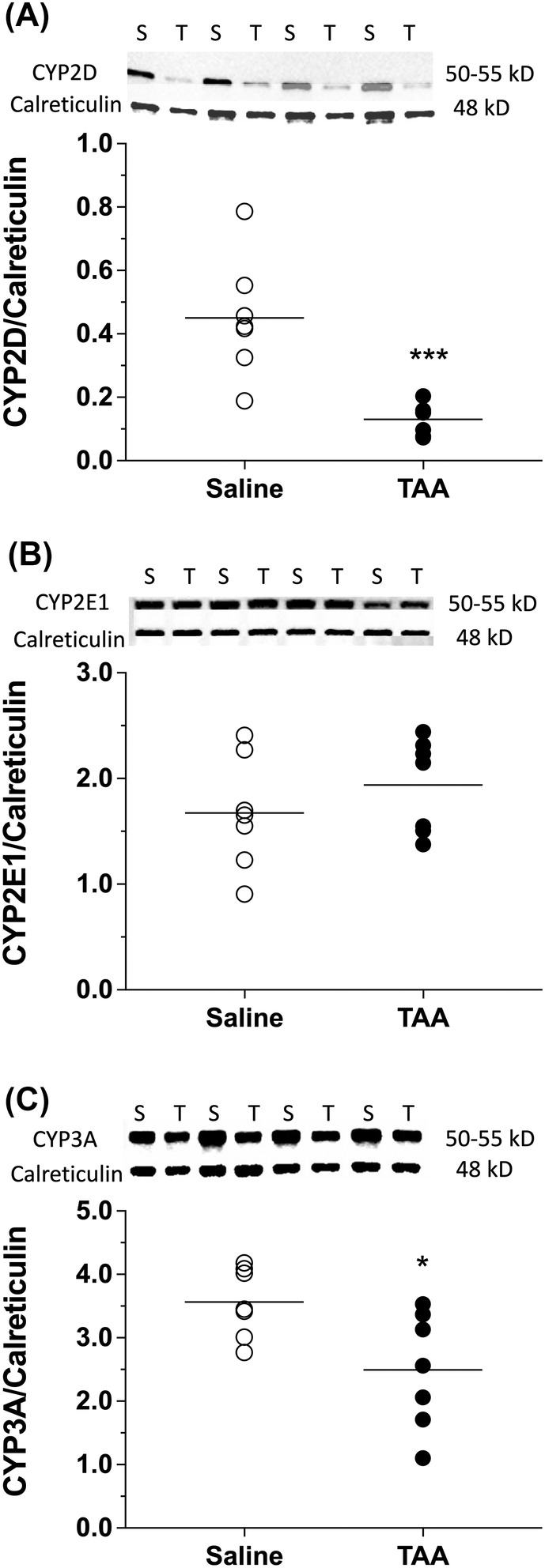
Western blot images and densitometric data for microsomal content of CYP2D (A), CYP2E1 (B) and CYP3A (C) along with their respective loading control calreticulin in TAA (T)‐ or saline (S)‐treated animals. Animals were treated with ip injections of 200‐mg/kg TAA (n = 7) or saline (n = 7) three times a week for 10 weeks, followed by a 10‐day washout period. For brevity, the Western blot images are shown for four livers in each group. The symbols and horizontal lines indicate the individual and mean values, respectively. *, p < 0.05; ***, p < 0.001; two‐tailed unpaired t‐test, compared with the saline group
3.4. General and isoenzyme‐specific activity of P450
The ECOD activities, as a general marker of P450 activity, at substrate concentrations of 50 and 500 μM are presented in Figure 5. Compared with the saline‐treated animals, the ECOD activities of the liver microsomes of TAA‐treated animals were significantly reduced by ~40%–45% (p < 0.05) at the tested substrate concentrations of 50 and 500 μM (Figure 5).
FIGURE 5.
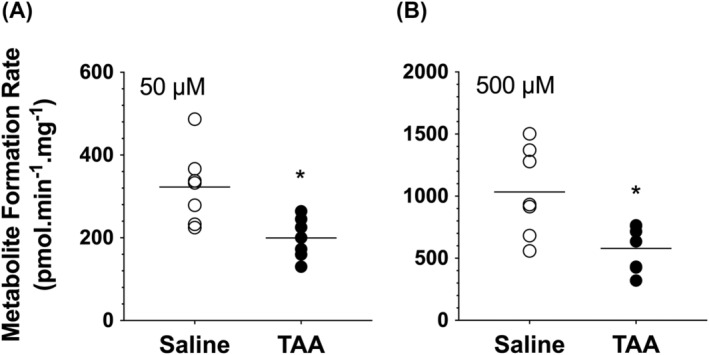
Ethoxycoumarin O‐deethylation (ECOD) activities at substrate concentrations of 50 (A) or 500 (B) μM in rat livers. Animals were treated with ip injections of 200‐mg/kg TAA (n = 7) or saline (n = 7) three times a week for 10 weeks, followed by a 10‐day washout period. The symbols and horizontal lines indicate the individual and mean values, respectively. *, p < 0.05; two‐tailed unpaired t‐test, compared with the saline group
The Michaelis–Menten plots of dextromethorphan O‐demethylation, chlorzoxazone 6‐hydroxylation and midazolam 1′‐ and 4‐hydroxylation as markers of CYP2D, CYP2E1 and CYP3A, respectively, in the saline‐ and TAA‐treated animals are presented in Figure 6. Additionally, their respective Michaelis–Menten kinetic parameters are listed in Table 3. The incubation conditions, including microsomal protein levels and incubation time, were optimized to ensure the linear production of metabolites for each substrate. The data for dextromethorphan O‐demethylation (CYP2D) was best fitted by a two‐enzyme model, indicating the involvement of at least two enzymes in the metabolism of dextromethorphan in the rat livers (Figure 6A). However, the best fit for the chlorzoxazone 6‐hydroxylation (CYP2E1) (Figure 6B) and midazolam 1′‐ (Figure 6C) and 4‐ (Figure 6D) hydroxylation was a single‐enzyme model.
FIGURE 6.
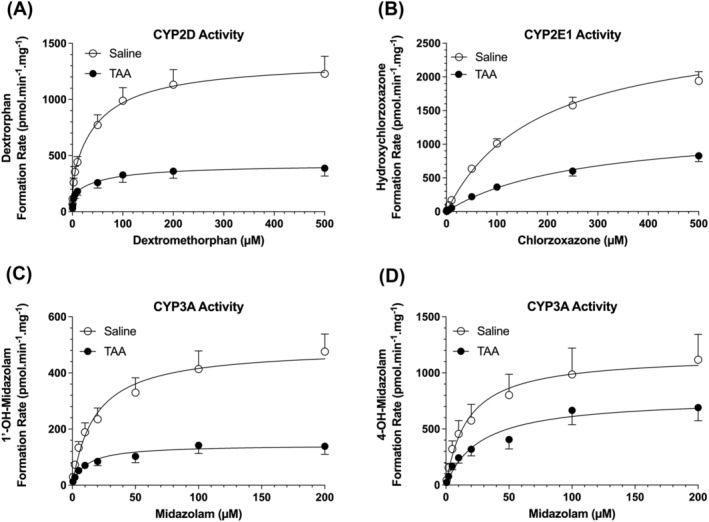
Michaelis–Menten plots of CYP2D‐mediated dextromethorphan O‐demethylation (A), CYP2E1‐mediated chlorzoxazone hydroxylation (B) and CYP3A‐mediated midazolam 1′‐ (C) and 4‐ (D) hydroxylation in rat liver microsomes. Animals were treated with ip injections of 200‐mg/kg TAA (n = 7) or saline (n = 7) three times a week for 10 weeks, followed by a 10‐day washout period. The symbols and error bars represent the mean and SE values, respectively, and the lines represent the nonlinear regression fit of the experimental data.
TABLE 3.
Apparent K M and V MAX values (mean ± SE, n = 7) for dextromethorphan O‐demethylation, chlorzoxazone 6‐hydroxylation and midazolam 1′‐ and 4‐hydroxylation in rat liver microsomal preparations of saline‐ and TAA‐treated groups
| Saline group | TAA group | |||
|---|---|---|---|---|
| Enzyme/substrate | V MAX pmol/(min.mg) | K M μM | V MAX pmol/(min.mg) | K M (μM) |
| CYP2D/dextromethorphan | ||||
| High affinity | 308 ± 63 | 1.08 ± 0.30 | 151 ± 29* | 1.04 ± 0.09 |
| Low affinity | 1040 ± 105 | 75.5 ± 25.2 | 274 ± 47**** | 72.6 ± 18.8 |
| CYP2E1/chlorzoxazone | 2630 ± 168 | 190 ± 20 | 1270 ± 80**** | 337 ± 82 |
| CYP3A/midazolam | ||||
| 1′‐Hydroxylation | 494 ± 64 | 20.4 ± 2.5 | 145 ± 30*** | 15.2 ± 3.5 |
| 4‐Hydroxylation | 1190 ± 235 | 25.0 ± 5.8 | 792 ± 128 | 32.5 ± 7.3 |
Note: Two‐tailed unpaired t‐test, compared with the saline group.
p < 0.05.
p < 0.001.
p < 0.0001.
Except for the 4‐hydroxylation of MDZ, cirrhosis resulted in significant reductions in the V MAX values for all the tested microsomal activities (Figure 6 and Table 3). However, the K M values of the metabolism of the probe substrates were not significantly affected by TAA‐induced cirrhosis (Table 3). The microsomal high‐ and low‐capacity V MAX values for CYP2D demethylase activity were reduced by 51% (p < 0.05) and 74% (p < 0.0001), respectively, in the TAA group (Table 3). For CYP2E1, cirrhosis caused a 52% reduction in the V MAX value (p < 0.0001) for the chlorzoxazone hydroxylation activity. The V MAX of microsomal CYP3A hydroxylase activity, resulting in 1′‐hydroxylation of midazolam, was reduced by 71% (p < 0.001). However, a 33% reduction in the V MAX of midazolam 4‐hydroxylation activity did not reach statistical significance (p = 0.1672) (Table 3).
4. DISCUSSION
Compared with the other chemically induced cirrhosis models, the TAA model of liver cirrhosis in rodents is one of the preferred experimental models of human cirrhosis because of its reproducibility, low mortality and reduced toxicity. 9 However, different protocols with regard to the route of administration (oral in drinking water or intraperitoneal injection), dose/dosing frequency and length of treatment have been reported in the literature. Our chronic cirrhosis model in rats, which was based on the intraperitoneal injection of 200‐mg/kg TAA three times a week for 10 weeks, followed by a 10‐day washout period, resulted in substantial necro‐inflammation and fibrosis of the liver (Figure 2) and significant increases in the plasma levels of the liver injury markers ALP, GGT and TBil (Figure 1). Additionally, the plasma levels of bile acids were increased by 24 times in the TAA group (Figure 1), suggesting the presence of severe cholestasis. These results are consistent with the previous reports after intraperitoneal injection of TAA. 10 , 18 , 20 , 32 , 33 For example, similar to our high necro‐inflammatory (cirrhosis) and fibrosis scores in the TAA group (Figure 2), gross cirrhosis and a fibrosis score of 3.98 were reported in rats receiving ip injection of 300‐mg/kg TAA, thrice a week for 10 weeks, after a 1‐week washout period. 32 Similarly, an average fibrosis score of 3.5 was reported in rats 3 weeks after the cessation of ip injections of 200‐mg/kg TAA every other day for 10 weeks. 10 The histological findings in our study (Figure 2) and these literature data 10 , 32 indicate that the TAA‐induced cirrhosis using these protocols is not reversible after 1–3 weeks of washout.
Despite large increases in the plasma levels of ALP, GGT, TBil and bile acids in the TAA group, the plasma levels of ALT and AST were similar in the saline‐ and TAA‐treated groups in our study (Figure 1). Although some studies have reported higher plasma AST and ALT levels in the TAA‐induced cirrhotic rats, 18 , 19 , 33 others 9 , 20 have shown no significant changes in these markers in the TAA model. This discrepancy may be related to the length of TAA treatment. For example, in a time course study of the effects of repeated ip administration of TAA to rats, Wallace et al. 9 showed that while the plasma levels of AST and ALT increased sharply during the first 2 weeks of treatment, they returned to normal levels at Week 4 and remained normal for up to 12 weeks of TAA treatment. However, the portal pressure, collagen content and fibrosis‐associated genes peaked at Week 8 and remained high up to 12 weeks of treatment. 9 The lack of changes in the AST/ALT levels in our 10‐week TAA model is in agreement with the latter report and consistent with the reports that AST and ALT levels are more sensitive to acute liver injury and are often close to the normal range in patients with cirrhosis. 34
Although cirrhosis induced in rats surgically by bile duct ligation or chemically by carbon tetrachloride causes substantial (~60%) reductions in the serum albumin concentrations, 35 the chronic administration of TAA in our model caused only ~10% decline in the serum albumin levels (Figure 1). The smaller reduction in the albumin concentrations in our animal model is closer to the 10%–20% reductions in the serum albumin concentrations observed in moderate to severe cirrhosis in human beings. 4 Further, our results are in agreement with previous studies after chronic administration of TAA to rats, 8 , 33 which showed small or no reductions in the serum albumin levels. Nevertheless, collectively, our biochemical (Figure 1) and histological (Figure 2) findings clearly show the presence of liver cirrhosis in our model, which also has a 10‐day washout period.
To test the effects of liver cirrhosis in our rat model on the overall content and activities of P450, we first measured the microsomal total P450 contents and 7‐ECOD activity that is a measure of activities of multiple P450 enzymes in CYP1, CYP2 and CYP3 families. 29 The 50% and 40%–45% reductions in the total P450 (Figure 3) and ECOD activity (Figure 5), respectively, in our TAA group after 10 days of TAA washout indicate that the observed effects of P450 are due to cirrhosis and not the direct effects of the chemical.
Previous in vitro and in vivo studies have shown that TAA and its metabolite TAA S‐oxide reduce the P450 content and activity using mechanisms that are independent of cirrhosis. 18 , 21 , 22 Hunter et al. 21 reported that the in vivo administration of TAA or its S‐oxide metabolite to rats caused a relatively rapid decrease in the P450 activities, which reached its maximum effect at 24 h. The effect was dose dependent for both compounds and more pronounced and rapid for the metabolite than the parent drug. 21 In a follow‐up study, Matsuura et al. 22 reported that a relatively rapid decrease in the P450 content and activity by the in vivo administration of both TAA or its S‐oxide metabolite was associated with a rapid induction of heme oxygenase and inhibition of 5‐aminolevulinic acid synthetase, which is the rate‐limiting enzyme in heme synthesis. Therefore, the authors suggested that the rapid effects of in vivo administration of TAA or its S‐oxide metabolites on the P450 enzymes in rats are most likely due to increased degradation and reduced synthesis of heme by both compounds. They also showed in a time course study that after injection of a single ip dose of 150 mg/kg of TAA to rats, P450 concentration and activity declined relatively rapidly, reaching a maximum effect at 32 h and returning to near control values by 96 h. 22 Therefore, in our studies, we chose a 10‐day washout period to separate the effects of the disease (cirrhosis) on the P450 concentration and activity from the other potential effects of the chemical itself.
It may be argued that the direct effects of TAA on the degradation and synthesis of heme after multiple doses of TAA, used in our study, may last longer than that reported after a single dose of TAA (<96 h). 22 Although this possibility cannot be ruled out, given the very short in vivo half‐lives of TAA and its S‐oxide metabolite in rats (1–2 h), 36 it is less likely that the cirrhosis‐independent effects of TAA last longer than our washout period (i.e., 10 days).
In addition to its effects on heme synthesis and degradation, it has been reported that TAA exerts a mechanism‐based inhibitory effect on P450 enzymes. 18 Direct in vitro addition of TAA at different concentrations to rat liver microsomes inhibited the activities of several P450 isoenzymes in a concentration, time and NADPH‐dependent manner, suggesting a mechanism‐based inhibitory activity for TAA. These studies clearly indicate the need for a washout period in the TAA‐induced cirrhosis model to remove the drug and its metabolites and their mechanism‐based inhibitory effects on P450 enzymes in order to study the effects of liver cirrhosis on drug metabolism.
Studies in human beings with liver cirrhosis have shown that the disease‐induced reductions in the P450 activities are isoenzyme selective. 37 , 38 , 39 Similarly, the results of our study in the TAA model indicate that the contents (Figure 4) and activities (Figure 6 and Table 3) of the individual isoenzymes were selectively affected by the disease. For example, a 50% reduction in the microsomal total P450 (Figure 3A) was associated with 70% and 30% reductions in the contents of CYP2D (Figure 4A) and CYP3A (Figure 4C), respectively, whereas the microsomal contents of CYP2E1 remained unaffected (Figure 4B). Although the TAA‐induced reductions in the activities of CYP2D and CYP3A were in agreement with the reductions in their protein contents, the activities of CYP2E1 were also significantly reduced in the TAA group despite no changes in its protein contents (Figures 4 and 6 and Table 3). This discrepancy may be explained by a significant cirrhosis‐induced reduction in the CPR activity (Figure 3B), which is the rate‐limiting step in the P450‐mediated enzymatic reactions. A previous study 40 has also reported 20% and 40% reductions in the CPR activity per milligrams of liver tissue in human livers with mild and moderate cirrhosis, respectively. Our results in the TAA model in rats, which showed a 30% reduction in the CPR activity (Figure 3B), are in agreement with this report 40 in human cirrhotic livers.
The decrease in the CYP2E1 activity (Figure 6B) despite no change in its protein contents (Figure 4B) in cirrhotic animals may also be related to the post‐translational modifications of this isoenzyme in cirrhotic animals. For example, it has been reported 41 that whereas the CYP2E1 mRNA and protein contents remain unaffected in rats from 8 to 18 months of age, the activity of CYP2E1, measured by chlorzoxazone hydroxylation, significantly decreases during the same period. This observation was selective for CYP2E1 because the CYP3A activity did not change during the same time period, an observation that was consistent with no change in its mRNA or protein content. The authors attributed the discrepancy in the protein–activity relationship of CYP2E1 to its protein modifications by age‐related oxidative stress, such as reductions in reduced glutathione and increases in thiobarbituric acid reactive substances, selectively affecting CYP2E1 and not CYP3A. 41 Cirrhosis induced by TAA is also known to produce oxidative stress. 12 Therefore, similar oxidative stress‐induced post‐translational modifications of CYP2E1 protein may have also contributed to the reduction in the activity of this enzyme in our cirrhotic animals in the absence of any changes in its protein.
The reductions in the P450 enzyme content and activities observed in our study after chronic ip administration of TAA to rats (Figures 3, 4, 5, 6 and Table 3) are in general agreement with the previous reports that used ip injection of TAA to induce liver cirrhosis in rats. 18 , 19 , 20 Nakajimal et al. 19 reported that after 8 weeks of ip injection of TAA (200 mg/kg, three times a week), the microsomal total P450, the protein contents of several P450 isoenzymes and the metabolism of nicotine to cotinine declined significantly. Similarly, Xie et al. 18 reported significant reductions in the protein contents of several P450 isoenzymes 6 weeks after ip injection of 200‐mg/kg TAA twice a week. However, both studies 18 , 19 also found a >50% decline in the microsomal CYP2E1 protein, which contrasts with our finding of no change in the microsomal protein content of this isoenzyme in the TAA group (Figure 4B). This discrepancy may be related to the fact that, as opposed to our 10‐day washout period, these studies 18 , 19 were conducted in microsomes prepared soon after the last injection of TAA.
Among P450 isoenzymes, CYP2E1 is unique because its catalytic cycling in the absence of substrates and inhibitors generates a substantial amount of reactive oxygen species, resulting in its own degradation. Therefore, CYP2E1 is the most unstable microsomal P450 enzyme with a short half‐life of 4–7 h. 42 However, substrates and inhibitors of CYP2E1 increase the half‐life of the enzyme several times by inhibiting its degradation. 42 TAA and its S‐oxide metabolite are reportedly CYP2E1 substrates, which are converted by CYP2E1 to the highly reactive S,S‐dioxide metabolite that causes hepatic damage. Indeed, a previous study 43 reported that by binding to the active site of CYP2E1, TAA reduces the degradation of CYP2E1 in hepatocytes. Therefore, the lack of change in the protein concentrations of CYP2E1 (Figure 4B), compared with the TAA‐induced reductions in the microsomal total P450 (Figure 3A), CYP2D (Figure 4A) and CYP3A (Figure 4C) proteins, might, at least in part, be due to the stabilizing effects of TAA on CYP2E1. Further, the catalytic cycling of CYP2E1, resulting in its fast degradation, is dependent on the activity of NADPH‐dependent CPR. 42 Therefore, a 30% reduction in the CPR activity in our cirrhosis model (Figure 3B) may also have contributed selectively to the lack of reduction of CYP2E1 protein in our model.
The in vitro studies using liver microsomes from TAA‐induced liver cirrhosis conducted here and in previous reports 18 , 19 only address the effects of cirrhosis on the metabolic activity of P450 enzymes. However, the metabolism of drugs by the liver after their in vivo administration is affected by other parameters, such as hepatic blood flow and degree of protein binding of drugs, in addition to the metabolic activity of P450. 44 It is known that liver cirrhosis causes intra‐ and extra‐hepatic portosystemic shunting and reduction in the hepatic blood flow. 6 Additionally, liver cirrhosis causes a reduction in the synthesis of albumin, which is responsible for the binding of most drugs in plasma. 6 The cirrhosis‐associated changes in the metabolic activity of P450 enzymes, hepatic blood flow and degree of protein binding of drugs can have varying and complex effects on the hepatic clearance and bioavailability of drugs, depending on the extent of hepatic extraction ratio of drug and its route of administration. 6 Although some studies have shown the effects of cirrhosis induced by ip injection of TAA on the in vivo metabolism of drugs, 20 future studies could shed more light on the suitability of the model to investigate the effects of liver cirrhosis on the in vivo pharmacokinetics of drugs.
The cirrhosis‐induced declines in the total P450 content (Figure 3A) and protein (Figure 4) and activities (Figure 6 and Table 3) of CYP2D, CYP2E1 and CYP3A observed in our TAA model in rats are consistent with the effects of cirrhosis on the total P450 and these isoenzymes reported before in human beings. 38 , 45 , 46 , 47 , 48 For instance, the 50% reduction in total P450 content in our experimental model (Figure 3A) is similar to the extent of reduction in the total P450 contents in patients with severe liver cirrhosis. 47 , 48 Additionally, similar to our results with CYP3A (Figures 4 and 6 and Table 3), previous studies 45 , 47 reported significant reductions in the in vitro protein content and activity of CYP3A4 in patients with liver cirrhosis. Further, in agreement with our in vitro activity results (Figure 6), the in vivo metabolism of the CYP3A4 substrate midazolam, 46 CYP2E1 substrate chlorzoxazone 38 and CYP2D6 substrate debrisoquine 38 were significantly lower in patients with liver cirrhosis. However, as noted before, the in vivo results may be affected by other parameters, such as cirrhosis‐induced changes in the liver blood flow and/or degree of protein binding of the drugs, in addition to reductions in the activity of isoenzymes. Additionally, different isoforms of the same P450 subfamily may be affected in rats versus human beings (e.g., CYP2D1 in rats versus CYP2D6 in human beings).
Among the P450 probes used in this study, dextromethorphan O‐demethylation, which is commonly used as an indication of CYP2D activity, was best described by a two‐enzyme system (Table 3). However, the intrinsic clearance (V MAX/K M ) of the high‐affinity enzyme, which is likely related to CYP2D, was >20 times higher than that for the low‐affinity component (Table 3). This observation is in complete agreement with a previous study, 49 which reported a two‐enzyme system for dextromethorphan O‐demethylation in the Sprague–Dawley rat liver microsomes. Similar to our findings, the high‐affinity component of dextromethorphan O‐demethylation in that study constituted 98% of the total intrinsic clearance of the pathway.
In conclusion, a TAA model of liver cirrhosis in rats using chronic ip injection of the chemical for 10 weeks, followed by a 10‐day washout period, caused substantial fibrosis and cirrhosis in the liver of these animals. Similar to human cirrhosis, the levels of total P450 and the protein levels or activities of several P450 enzymes were significantly reduced in this model. The model may be suitable for investigations of the mechanisms of the effects of liver cirrhosis on the in vivo pharmacokinetics of drugs used in this patient population.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
This study was partially funded by Chapman University School of Pharmacy and a Postdoctoral Research Fellowship Award from the American Liver Foundation to Barent DuBois.
Chandrashekar DV, DuBois BN, Rashid M, Mehvar R. Effects of chronic cirrhosis induced by intraperitoneal thioacetamide injection on the protein content and Michaelis–Menten kinetics of cytochrome P450 enzymes in the rat liver microsomes. Basic Clin Pharmacol Toxicol. 2023;132(2):197‐210. doi: 10.1111/bcpt.13813
Devaraj Venkatapura Chandrashekar and Barent N. DuBois contributed equally to the study.
Funding information Chapman University School of Pharmacy, Grant/Award Number: School of Pharmacy; American Liver Foundation, Grant/Award Number: Postdoctoral Research Fellowship
REFERENCES
- 1. Tapper EB, Ufere NN, Huang DQ, Loomba R. Review article: current and emerging therapies for the management of cirrhosis and its complications. Aliment Pharmacol Ther. 2022;55(9):1099‐1115. doi: 10.1111/apt.16831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lucena MI, Andrade RJ, Tognoni G, Hidalgo R, De La Cuesta FS. Spanish collaborative study group on therapeutic management in liver disease. Multicenter hospital study on prescribing patterns for prophylaxis and treatment of complications of cirrhosis. Eur J Clin Pharmacol. 2002;58(6):435‐440. doi: 10.1007/s00228-002-0474-1 [DOI] [PubMed] [Google Scholar]
- 3. Lucena MI, Andrade RJ, Tognoni G, Hidalgo R, Sanchez de la Cuesta F. Spanish collaborative study group on therapeutic management of liver diseases. Drug use for non‐hepatic associated conditions in patients with liver cirrhosis. Eur J Clin Pharmacol. 2003;59(1):71‐76. doi: 10.1007/s00228-003-0586-2 [DOI] [PubMed] [Google Scholar]
- 4. Duthaler U, Bachmann F, Suenderhauf C, et al. Liver cirrhosis affects the pharmacokinetics of the six substrates of the basel phenotyping cocktail differently. Clin Pharmacokinet. 2022;61(7):1039‐1055. doi: 10.1007/s40262-022-01119-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weersink RA, Burger DM, Hayward KL, Taxis K, Drenth JPH, Borgsteede SD. Safe use of medication in patients with cirrhosis: pharmacokinetic and pharmacodynamic considerations. Expert Opin Drug Metab Toxicol. 2020;16(1):45‐57. doi: 10.1080/17425255.2020.1702022 [DOI] [PubMed] [Google Scholar]
- 6. Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147‐1161. doi: 10.1007/s00228-008-0553-z [DOI] [PubMed] [Google Scholar]
- 7. Franz CC, Hildbrand C, Born C, Egger S, Ratz Bravo AE, Krahenbuhl S. Dose adjustment in patients with liver cirrhosis: impact on adverse drug reactions and hospitalizations. Eur J Clin Pharmacol. 2013;69(8):1565‐1573. doi: 10.1007/s00228-013-1502-z [DOI] [PubMed] [Google Scholar]
- 8. Guerra RR, Trotta MR, Aloia TPA, Dagli MLZ, Hernandez‐Blazquez FJ. A novel chronic cirrhosis TAA‐induced model in rats. Braz J Vet Pathol. 2010;3:9‐16. [Google Scholar]
- 9. Wallace MC, Hamesch K, Lunova M, et al. Standard operating procedures in experimental liver research: thioacetamide model in mice and rats. Lab Anim. 2015;49(1_suppl):21‐29. doi: 10.1177/0023677215573040 [DOI] [PubMed] [Google Scholar]
- 10. Oe S, Fukunaka Y, Hirose T, Yamaoka Y, Tabata Y. A trial on regeneration therapy of rat liver cirrhosis by controlled release of hepatocyte growth factor. J Control Release. 2003;88(2):193‐200. doi: 10.1016/S0168-3659(02)00463-7 [DOI] [PubMed] [Google Scholar]
- 11. Liu Y, Meyer C, Xu C, et al. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol. 2013;304(5):G449‐G468. doi: 10.1152/ajpgi.00199.2012 [DOI] [PubMed] [Google Scholar]
- 12. Ingawale DK, Mandlik SK, Naik SR. Models of hepatotoxicity and the underlying cellular, biochemical and immunological mechanism(s): a critical discussion. Environ Toxicol Pharmacol. 2014;37(1):118‐133. doi: 10.1016/j.etap.2013.08.015 [DOI] [PubMed] [Google Scholar]
- 13. Faccioli LAP, Dias ML, Paranhos BA, Dos Santos Goldenberg RC. Liver cirrhosis: an overview of experimental models in rodents. Life Sci. 2022;301:120615. doi: 10.1016/j.lfs.2022.120615 [DOI] [PubMed] [Google Scholar]
- 14. Ghosh Dastidar S, Warner JB, Warner DR, McClain CJ, Kirpich IA. Rodent models of alcoholic liver disease: role of binge ethanol administration. Biomolecules. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laleman W, Vander Elst I, Zeegers M, et al. A stable model of cirrhotic portal hypertension in the rat: thioacetamide revisited. Eur J Clin Invest. 2006;36(4):242‐249. doi: 10.1111/j.1365-2362.2006.01620.x [DOI] [PubMed] [Google Scholar]
- 16. Muller D, Sommer M, Kretzschmar M, et al. Lipid peroxidation in thioacetamide‐induced macronodular rat liver cirrhosis. Arch Toxicol. 1991;65(3):199‐203. doi: 10.1007/BF02307309 [DOI] [PubMed] [Google Scholar]
- 17. Corbin IR, Minuk GY. Serial percutaneous liver biopsies in laboratory rats. Dig Dis Sci. 2003;48(10):1939‐1943. doi: 10.1023/A:1026228018643 [DOI] [PubMed] [Google Scholar]
- 18. Xie Y, Wang G, Wang H, et al. Cytochrome P450 dysregulations in thioacetamide‐induced liver cirrhosis in rats and the counteracting effects of hepatoprotective agents. Drug Metab Dispos. 2012;40(4):796‐802. doi: 10.1124/dmd.111.043539 [DOI] [PubMed] [Google Scholar]
- 19. Nakajima M, Iwata K, Yamamoto T, Funae Y, Yoshida T, Kuroiwa Y. Nicotine metabolism in liver microsomes from rats with acute hepatitis or cirrhosis. Drug Metab Dispos. 1998;26(1):36‐41. [PubMed] [Google Scholar]
- 20. Kaneko H, Otsuka Y, Katagiri M, et al. Reassessment of monoethylglycinexylidide as preoperative liver function test in a rat model of liver cirrhosis and man. Clin Exp Med. 2001;1(1):19‐26. doi: 10.1007/s10238-001-8005-4 [DOI] [PubMed] [Google Scholar]
- 21. Hunter AL, Holscher MA, Neal RA. Thioacetamide‐induced hepatic necrosis. I. Involvement of the mixed‐function oxidase enzyme system. J Pharmacol Exp Ther. 1977;200(2):439‐448. [PubMed] [Google Scholar]
- 22. Matsuura Y, Takizawa Y, Fukuda T, Yoshida T, Kuroiwa Y. Induction of heme oxygenase and inhibition of delta‐aminolevulinic acid synthetase of rat liver by thioacetamide and thioacetamide‐S‐oxide. J Pharmacobiodyn. 1983;6(5):340‐345. doi: 10.1248/bpb1978.6.340 [DOI] [PubMed] [Google Scholar]
- 23. Kraul H, Zimmermann T, Pocha C, Truckenbrodt J, Hoffmann A. Cytochrome P‐450‐dependent biotransformation in Uje:WIST rats with chronic liver injury induced by thioacetamide. Z Versuchstierkd. 1989;32(6):269‐273. [PubMed] [Google Scholar]
- 24. Tveden‐Nyborg P, Bergmann TK, Jessen N, Simonsen U, Lykkesfeldt J. BCPT policy for experimental and clinical studies. Basic Clin Pharmacol Toxicol. 2021;128(1):4‐8. doi: 10.1111/bcpt.13492 [DOI] [PubMed] [Google Scholar]
- 25. Knodell RG, Ishak KG, Black WC, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1(5):431‐435. doi: 10.1002/hep.1840010511 [DOI] [PubMed] [Google Scholar]
- 26. Brunt EM. Grading and staging the histopathological lesions of chronic hepatitis: the Knodell histology activity index and beyond. Hepatology. 2000;31(1):241‐246. doi: 10.1002/hep.510310136 [DOI] [PubMed] [Google Scholar]
- 27. Guengerich FP, Martin MV, Sohl CD, Cheng Q. Measurement of cytochrome P450 and NADPH‐cytochrome P450 reductase. Nat Protoc. 2009;4(9):1245‐1251. doi: 10.1038/nprot.2009.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Omura T, Sato R. The carbon monoxide‐binding pigment of the liver microsomes. Evidence for its hemoprotein nature. J Biol Chem. 1964;239(7):2370‐2378. doi: 10.1016/S0021-9258(20)82244-3 [DOI] [PubMed] [Google Scholar]
- 29. Waxman DJ, Chang TK. Use of 7‐ethoxycoumarin to monitor multiple enzymes in the human CYP1, CYP2, and CYP3 families. In: Phillips IR, Shephard EA, eds. Cytochrome P450 Protocols Methods Mol Biol. Totowa, NJ: Humana Press; 2006:153‐156. [DOI] [PubMed] [Google Scholar]
- 30. Walsky RL, Obach RS. Validated assays for human cytochrome P450 activities. Drug Metab Dispos. 2004;32(6):647‐660. doi: 10.1124/dmd.32.6.647 [DOI] [PubMed] [Google Scholar]
- 31. Gorski JC, Hall SD, Jones DR, VandenBranden M, Wrighton SA. Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem Pharmacol. 1994;47(9):1643‐1653. doi: 10.1016/0006-2952(94)90543-6 [DOI] [PubMed] [Google Scholar]
- 32. Low TY, Leow CK, Salto‐Tellez M, Chung MC. A proteomic analysis of thioacetamide‐induced hepatotoxicity and cirrhosis in rat livers. Proteomics. 2004;4(12):3960‐3974. doi: 10.1002/pmic.200400852 [DOI] [PubMed] [Google Scholar]
- 33. Hao H, Zhang L, Jiang S, et al. Thioacetamide intoxication triggers transcriptional up‐regulation but enzyme inactivation of UDP‐glucuronosyltransferases. Drug Metab Dispos. 2011;39(10):1815‐1822. doi: 10.1124/dmd.111.039172 [DOI] [PubMed] [Google Scholar]
- 34. Johnston DE. Special considerations in interpreting liver function tests. Am Fam Physician. 1999;59(8):2223‐2230. [PubMed] [Google Scholar]
- 35. Bastien MC, Leblond F, Pichette V, Villeneuve JP. Differential alteration of cytochrome P450 isoenzymes in two experimental models of cirrhosis. Can J Physiol Pharmacol. 2000;78(11):912‐919. doi: 10.1139/y00-066 [DOI] [PubMed] [Google Scholar]
- 36. Porter WR, Gudzinowicz MJ, Neal RA. Thioacetamide‐induced hepatic necrosis. II. Pharmacokinetics of thioacetamide and thioacetamide‐S‐oxide in the rat. J Pharmacol Exp Ther. 1979;208(3):386‐391. [PubMed] [Google Scholar]
- 37. Branch RA. Drugs in liver disease. Clin Pharmacol Ther. 1998;64(4):462‐465. doi: 10.1016/S0009-9236(98)90077-7 [DOI] [PubMed] [Google Scholar]
- 38. Frye RF, Zgheib NK, Matzke GR, et al. Liver disease selectively modulates cytochrome P450–mediated metabolism. Clin Pharmacol Ther. 2006;80(3):235‐245. doi: 10.1016/j.clpt.2006.05.006 [DOI] [PubMed] [Google Scholar]
- 39. El‐Khateeb E, Achour B, Al‐Majdoub ZM, Barber J, Rostami‐Hodjegan A. Non‐uniformity of changes in drug‐metabolizing enzymes and transporters in liver cirrhosis: implications for drug dosage adjustment. Mol Pharm. 2021;18(9):3563‐3577. doi: 10.1021/acs.molpharmaceut.1c00462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. El‐Khateeb E, Achour B, Scotcher D, et al. Scaling factors for clearance in adult liver cirrhosis. Drug Metab Dispos. 2020;48(12):1271‐1282. doi: 10.1124/dmd.120.000152 [DOI] [PubMed] [Google Scholar]
- 41. Wauthier V, Verbeeck RK, Buc CP. Age‐related changes in the protein and mRNA levels of CYP2E1 and CYP3A isoforms as well as in their hepatic activities in Wistar rats. What role for oxidative stress? Arch Toxicol. 2004;78(3):131‐138. doi: 10.1007/s00204-003-0526-z [DOI] [PubMed] [Google Scholar]
- 42. Zhukov A, Ingelman‐Sundberg M. Relationship between cytochrome P450 catalytic cycling and stability: fast degradation of ethanol‐inducible cytochrome P450 2E1 (CYP2E1) in hepatoma cells is abolished by inactivation of its electron donor NADPH‐cytochrome P450 reductase. Biochem J. 1999;340(Pt 2):453‐458. doi: 10.1042/bj3400453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hajovsky H, Hu G, Koen Y, et al. Metabolism and toxicity of thioacetamide and thioacetamide S‐oxide in rat hepatocytes. Chem Res Toxicol. 2012;25(9):1955‐1963. doi: 10.1021/tx3002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mehvar R. Clearance concepts: fundamentals and application to pharmacokinetic behavior of drugs. J Pharm Pharm Sci. 2018;21(1s):88s‐102s. doi: 10.18433/jpps29896 [DOI] [PubMed] [Google Scholar]
- 45. Yang LQ, Li SJ, Cao YF, et al. Different alterations of cytochrome P450 3A4 isoform and its gene expression in livers of patients with chronic liver diseases. World J Gastroenterol. 2003;9(2):359‐363. doi: 10.3748/wjg.v9.i2.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Albarmawi A, Czock D, Gauss A, et al. CYP3A activity in severe liver cirrhosis correlates with Child‐Pugh and model for end‐stage liver disease (MELD) scores. Br J Clin Pharmacol. 2014;77(1):160‐169. doi: 10.1111/bcp.12182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. George J, Murray M, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology. 1995;21(1):120‐128. [PubMed] [Google Scholar]
- 48. Farrell GC, Cooksley WG, Powell LW. Drug metabolism in liver disease: activity of hepatic microsomal metabolizing enzymes. Clin Pharmacol Ther. 1979;26(4):483‐492. doi: 10.1002/cpt1979264483 [DOI] [PubMed] [Google Scholar]
- 49. Kerry NL, Somogyi AA, Mikus G, Bochner F. Primary and secondary oxidative metabolism of dextromethorphan. In vitro studies with female Sprague‐Dawley and Dark Agouti rat liver microsomes. Biochem Pharmacol. 1993;45(4):833‐839. doi: 10.1016/0006-2952(93)90166-T [DOI] [PubMed] [Google Scholar]