

Abstract
Sulfoxides and sulfinamides represent versatile sulfur functional groups found in ligands, chiral auxiliaries, and bioactive molecules. Canonical two‐component syntheses, however, rely on substrates with a preinstalled C−S bond and impede efficient and modular access to these sulfur motifs. Herein is presented the application of an easily prepared, bench‐stable sulfoxide reagent for one‐pot, three‐component syntheses of sulfoxides and sulfinamides. The sulfoxide reagent donates the SO unit upon the reaction with a Grignard reagent (RMgX) as a sulfenate anion (RSO−). While subsequent trapping reactions of this key intermediate with carbon electrophiles provide sulfoxides, a range of tertiary, secondary, and primary sulfinamides can be prepared by substitution reactions with electrophilic amines. The syntheses of sulfinamide analogs of amide‐ and sulfonamide‐containing drugs illustrate the utility of the method for the rapid preparation of medicinally relevant molecules.
Keywords: Grignard Reaction, One-Pot Synthesis, Sulfinamides, Sulfoxides, Synthetic Methods
Upon Grignard reactions, a simple sulfoxide reagent allows the formation of sulfenate anions that are subsequently transformed into sulfoxides and sulfinamides. This one‐pot, three‐component method does not require substrates with preinstalled sulfur functional groups.

Sulfoxides and sulfinamides are versatile SIV motifs used as ligands in transition metal catalysis, [1] chiral auxiliaries, [2] and precursors to SVI functional groups such as sulfoximines [3] and sulfonimidamides. [4] Synthetic methods for the preparation of sulfoxides and sulfinamides are typically based on substrates with preinstalled sulfur functional groups. The known substrates required for sulfinamide syntheses, for example, include malodorous thiols, [5] disulfides,[ 5a , 6 ] sulfinate esters, [7] metal sulfinates, [8] and others, [9] all of which have limited commercial availability (Figure 1). In addition, these protocols suffer from limited structural modularity, as only one of the two substituents can be introduced in a critical C−S or N−S bond‐forming step. The ideal approach for rapid access to a variety of sulfoxide and sulfinamide derivatives would be a three‐component coupling wherein two substituents are attached to the central SO unit in a one‐step (or one‐pot) operation. In this context, Willis and co‐workers disclosed the one‐pot, three‐component syntheses of sulfoxides [10] and sulfinamides [11] employing a sulfur dioxide surrogate DABSO (DABCO‐bis(sulfur dioxide)) as a source of the SO unit. The intermediate sulfinates (RSO2 −) serve as a diverging point to distinct product classes. Inspired by these notable examples, my strategy was based on the use of sulfenates (RSO−), [12] which have been well‐documented as key intermediates for sulfoxide syntheses. [13] Although sulfenates can be formally accessible via the addition of organometallic reagents to sulfur monoxide, this reaction is, in practice, inapplicable, as sulfur monoxide is an unstable gas under ambient conditions. [14] Moreover, the known precursors of sulfenate anions are equipped with a preinstalled carbon‐sulfur bond[ 13 , 15 ] and are inappropriate for realizing a three‐component coupling platform. Accordingly, a central question for my approach was finding a suitable “SO”‐donating reagent.
Figure 1.

Key precedents and this work.
Herein is presented the development of one‐pot, three‐component syntheses of sulfoxides and sulfinamides exploiting benzyl 2‐pyridyl sulfoxide (1 a) and its chlorine‐substituted variant 1 b as bench‐stable sulfur monoxide equivalents. The strategic use of these sulfoxides was based on a seminal report by Oae and co‐workers on the intramolecular C−C bond‐forming coupling of 1 a mediated by phenylmagnesium bromide (2). [16] This reaction was proposed to proceed via the formation of hypervalent sulfur intermediates, sulfuranes, followed by ligand coupling to form 2‐benzylpyridine (3) and benzenesulfenate (4). While the team reported the trapping of sulfenate 4 with methyl iodide, the yield of the resulting sulfoxide was not determined. [17] Another relevant contribution by the Stockman group revealed that similar intramolecular coupling reactions can also be promoted by methylmagnesium bromide and tert‐butylmagnesium chloride where methyl‐ and tert‐butylsulfenates should be the corresponding byproducts. [18]
These precedents constituted an entry point of my investigation. Using sulfoxide 1 a and phenylmagnesium bromide (2) as test substrates, sulfenate 4 was trapped with tert‐butyl bromoacetate (5). This reaction indeed formed sulfoxide 6; optimization of reaction conditions revealed that the sulfenate anion formation at −20 °C in THF proved optimal (see Supporting Information), and sulfoxide 6 was isolated in 77 % yield (Table 1). During optimization, sulfoxide 1 b, a Cl‐substituted variant of 1 a, turned out another viable reagent. [19]
Table 1.
Scope of Grignard reagents in the one‐pot preparation of sulfoxides.[a]
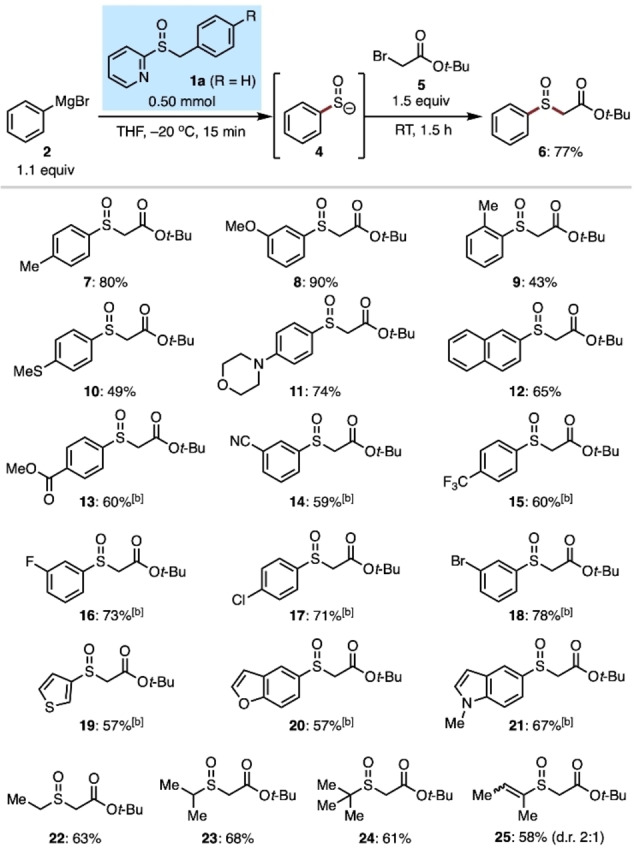
[a] Isolated yields are shown. [b] Sulfoxide reagent 1 b (R=Cl) was used in place of 1 a, and the formation of the sulfenate anions was performed at 0 °C. Additional scope of Grignard reagents is shown in Supporting Information.
With optimized conditions in hand, a critical question was whether sulfoxides 1 a and 1 b can be broadly applicable reagents for the generation of sulfenate anions upon Grignard reactions. To address this point, the scope of Grignard reagents was evaluated (Table 1). Commercially available Grignard reagents were used as supplied. Other Grignard reagents were prepared either by the magnesium insertion into aryl bromides in THF or the halogen/magnesium exchange of aryl bromides (or aryl iodides) with the Turbo Grignard reagent (i‐PrMgCl⋅LiCl). [20] Phenyl, naphthyl, and aryl Grignard reagents with electron‐donating substituents with varied steric character provided the desired sulfoxides (6–12) under the optimized conditions. Note that sulfoxide 10 cannot be easily prepared by traditional thioether oxidation methods. Aryl Grignard reagents with electron‐withdrawing groups and halogens, all of which were prepared by the Turbo Grignard method, proved viable substrates to give functionalized sulfoxides (13–18). In these cases, the generation of sulfenate anions was performed at 0 °C, as the incomplete consumption of reagent 1 b was observed at −20 °C. The formation of heteroaryl, alkyl, and alkenyl sulfenates was also successful using the corresponding Grignard reagents and allowed the synthesis of sulfoxides 19–25.
The scope of carbon electrophiles for trapping benzenesulfenate (4) was briefly examined. The results in Table 2 demonstrate that even in the presence of the reagent‐derived byproduct, 2‐benzylpyridine (3), a range of sulfoxides can be synthesized from 4 employing activated alkyl bromides [13a] with oxidation‐sensitive functional groups, primary and secondary alkyl iodides, [21] and hypervalent iodine reagents [22] to deliver sulfoxides 26–33. Importantly, this sulfoxide synthesis can be performed on multi‐gram scale (see Supporting Information).
Table 2.
Scope of carbon electrophiles for in situ trapping of benzenesulfenate (4).[a]

[a] Isolated yields are shown. For detailed conditions for each electrophile, see Supporting Information.
Next, an effort was made to devise a practical, one‐pot method to prepare sulfinamides. While a considerable number of transformations from sulfenates into sulfoxides are known in the literature, sulfinamide synthesis via sulfenate has only been described in Dai and Zhang's work. [23] Their copper‐catalyzed method, however, requires hydroxylamine derivatives whose preparation needs additional steps and is only applicable to the synthesis of tertiary sulfinamide. With this in mind, I wondered whether sulfenates undergo simple substitution reactions with electrophilic amines without the need for a transition metal catalyst and custom‐made aminating agents. I focused on N‐chloroamines, readily prepared in situ from amines and chlorinating agents, inspired by Willis's sulfonamide synthesis through the substitution between sulfinates and N‐chloroamines. [24]
Fortunately, N‐chloroamines can be generated in situ from commercially available amines and N‐chlorosuccinimide (NCS), and readily react with sulfenate anions to give sulfinamides. Importantly, this protocol applied to both secondary and primary amines as coupling partners (Table 3). Both cyclic and acyclic secondary amines including medicinally relevant motifs (e.g., morpholine, piperazine, and pyrrolidine) are viable substrates to afford tertiary sulfinamides (34–48). Concerning the scope of primary amines, benzyl, phenethyl, cyclopropyl, cyclobutyl, cyclohexyl, and even sterically hindered adamantyl amines provided the desired secondary sulfinamides (49–60). Other amines including aniline and amines with further functionalization handles are also within the scope (61–64). It is worth noting that despite the relatively simple structures, all of the tertiary and secondary sulfinamides presented in Table 3 were synthesized and characterized for the first time.
Table 3.
Scope of one‐pot, three‐component syntheses of sulfinamides.[a,b]

[a] Tertiary and secondary sulfinamides: sulfoxide 1 a (0.50 mmol, 1.0 equiv), Grignard reagent (1.1 equiv), THF, −20 °C, 15 min, then amine (3.0 equiv), NCS (1.1 equiv), room temperature 1 h. Isolated yields are shown. [b] Primary sulfinamides: sulfoxide 1 a (1.2 equiv), Grignard reagent (1.3 equiv), THF, −20 °C, 15 min, then DPPH (0.50 mmol, 1.0 equiv), room temperature, 1 h. Isolated yields are shown. [c] Sulfoxide reagent 1 b was used in place of 1 a, and the formation of the sulfenate anions was performed at 0 °C.
The preparation of primary sulfinamides required a modified protocol. Initial efforts to prepare sulfinamide 65 using ammonia (as a solution in THF) and NCS were unsuccessful. I then tested commercially available electrophilic aminating agents, namely, hydroxylamine O‐sulfonic acid, O‐(2,4‐dinitrophenyl)hydroxylamine, and O‐(diphenylphosphinyl)hydroxylamine (DPPH). Among these, DPPH gave the highest yield of 65 (77 % yield). The modified procedure worked well for other sulfenates including functionalized aryl, heteroaryl, and alkyl sulfenates to prepare a collection of primary sulfinamides (66–80).
Although sulfinamides have been employed as amide bioisosteres, [25] their widespread applications in a medicinal chemistry context have yet to be seen possibly because this structural unit was difficult to access by traditional synthetic methods. To illustrate the utility of the presented method, three sulfinamide analogs of known drug molecules were synthesized (Scheme 1). First, the sulfenate anion from the reaction of sulfoxide reagent 1 a and 3,4,5‐trimethoxyphenylmagnesium bromide (81) underwent the substitution with in situ generated N‐chloromorpholine to give 82 in 90 % yield, which is a sulfinamide analog of a sedative, Trimetozine. [26] In a similar substitution reaction with N‐chloroazocane, the same sulfenate anion was transformed into 83, a sulfinamide analog of an antidepressant, Trocimine. [27] The last example is the synthesis of a sulfinamide analog of a sulfonamide drug used to treat gout and hyperuricemia, Probenecid. [28] The Grignard reagent with an ester functional group 84 reacted with sulfoxide reagent 1 b, and the resulting sulfenate coupled with dipropylamine in the presence of NCS to furnish 85. The conversion of the methyl ester to the carboxylic acid effected by potassium trimethylsilanolate (KOTMS) provided 86. These syntheses demonstrate the potential use of the developed method to rapidly construct medicinally relevant molecules.
Scheme 1.

Syntheses of sulfinamide analogs of amide‐ and sulfonamide‐containing drugs.
Like any other reaction, the strategy presented herein is not without limitations. The first is the formation of 2‐benzylpyridine (3) as a byproduct. This non‐volatile molecule is relatively polar which occasionally required careful separation from the desired sulfoxides and sulfinamides using column chromatography. Second, sulfoxide/magnesium exchange was observed as a minor side reaction. [29] For example, the reaction between sulfoxide 1 a and phenylmagnesium bromide (2) under the condition in Table 1, followed by column chromatography yielded 2‐benzylpyridine (3) in 87 % yield as a mixture containing diphenyl sulfoxide (3 % yield), which was likely formed by sequential sulfoxide/magnesium exchange reactions between 1 a and 2. A similar exchange reaction was also observed as a minor pathway when ethylmagnesium bromide was employed. Lastly, among substrates tested to date, 2‐cyanophenyl, 2‐methoxycarbonylphenyl, 2‐thienyl, and allyl Grignard reagents failed to deliver the expected sulfenates. In the case of allylmagnesium bromide (87), sulfoxide 88 and 2‐allylpyridine (89) were isolated in 76 % and 69 % yields, respectively (Scheme 2). These products could form either a direct S N Ar reaction between 1 a and 87 (path a) or the facile intramolecular coupling between the allyl and 2‐pyridyl groups via a sulfurane intermediate 90 (path b). [30] The new reagent design of a sulfur monoxide equivalent should aim to overcome these limitations.
Scheme 2.

Possible scenarios for the formation of sulfoxide 88 and 2‐allylpyridine (89).
In summary, a versatile method for one‐pot, three‐component syntheses of sulfoxides and sulfinamides was developed employing easily prepared, bench‐stable sulfoxide reagents. A broad range of Grignard reagents can be used to generate the corresponding sulfenates as key intermediates to access sulfoxides. The sulfenates also undergo substitution reactions with nitrogen electrophiles, thus allowing the preparation of tertiary, secondary, and primary sulfinamides. The rapid preparation of medicinally relevant molecules is demonstrated through the syntheses of sulfinamide analogs of amide‐ and sulfonamide‐containing drugs. The modular approach described herein will find applications such as the array synthesis of sulfoxides and sulfinamides to accelerate the drug discovery process and preparation of SIV building blocks for chiral ligand/auxiliary synthesis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by the Walter Benjamin Program (500656103) of the Deutsche Forschungsgemeinschaft. The author thanks Prof. Oliver Trapp (Ludwig‐Maximilians‐Universität München) and Prof. Peter R. Schreiner (Justus‐Liebig‐Universität Gießen) for their generous support. Open Access funding enabled and organized by Projekt DEAL.
F. Saito, Angew. Chem. Int. Ed. 2022, 61, e202213872; Angew. Chem. 2022, 134, e202213872.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐ql75g).
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1.
- 1a. Trost B. M., Rao M., Angew. Chem. Int. Ed. 2015, 54, 5026–5043; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5112–5130; [Google Scholar]
- 1b. Otocka S., Kwiatkowska M., Madalińska L., Kiełbasiński P., Chem. Rev. 2017, 117, 4147–4181. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Robak M. T., Herbage M. A., Ellman J. A., Chem. Rev. 2010, 110, 3600–3740; [DOI] [PubMed] [Google Scholar]
- 2b. Salom-Roig X., Bauder C., Synthesis 2020, 52, 964–978. [Google Scholar]
- 3.
- 3a. Moragas T., Liffey R. M., Regentová D., Ward J.-P. S., Dutton J., Lewis W., Churcher I., Walton L., Souto J. A., Stockman R. A., Angew. Chem. Int. Ed. 2016, 55, 10047–10051; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10201–10205; [Google Scholar]
- 3b. Zenzola M., Doran R., Degennaro L., Luisi R., Bull J. A., Angew. Chem. Int. Ed. 2016, 55, 7203–7207; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7319–7323; [Google Scholar]
- 3c. Yu H., Li Z., Bolm C., Angew. Chem. Int. Ed. 2018, 57, 324–327; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 330–333; [Google Scholar]
- 3d. Aota Y., Kano T., Maruoka K., J. Am. Chem. Soc. 2019, 141, 19263–19268. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Izzo F., Schäfer M., Stockman R., Lücking U., Chem. Eur. J. 2017, 23, 15189–15193; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Richards-Taylor C. S., Martínez-Lamenca C., Leenaerts J. E., Trabanco A. A., Oehlrich D., J. Org. Chem. 2017, 82, 9898–9904; [DOI] [PubMed] [Google Scholar]
- 4c. Greed S., Briggs E. L., Idiris F. I. M., White A. J. P., Lücking U., Bull J. A., Chem. Eur. J. 2020, 26, 12533–12538; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Terhorst S., Jansen T., Langletz T., Bolm C., Org. Lett. 2022, 24, 4109–4113. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Taniguchi N., Eur. J. Org. Chem. 2016, 2157–2162; [Google Scholar]
- 5b. Chatterjee S., Makai S., Morandi B., Angew. Chem. Int. Ed. 2021, 60, 758–765; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 769–776. [Google Scholar]
- 6. Cogan D. A., Liu G., Kim K., Backes B. J., Ellman J. A., J. Am. Chem. Soc. 1998, 120, 8011–8019. [Google Scholar]
- 7.
- 7a. Davis F. A., Zhang Y., Andemichael Y., Fang T., Fanelli D. L., Zhang H., J. Org. Chem. 1999, 64, 1403–1406; [Google Scholar]
- 7b. García Ruano J. L., Parra A., Yuste F., Mastranzo V. M., Synthesis 2008, 2008, 311–319. [Google Scholar]
- 8.
- 8a. Billard T., Greiner A., Langlois B. R., Tetrahedron 1999, 55, 7243–7250; [Google Scholar]
- 8b. Zhu R.-H., Shi X.-X., Tetrahedron: Asymmetry 2011, 22, 387–393. [Google Scholar]
- 9.
- 9a. Harmata M., Zheng P., Huang C., Gomes M. G., Ying W., Ranyanil K.-O., Balan G., Calkins N. L., J. Org. Chem. 2007, 72, 683–685; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Wei J., Sun Z., Org. Lett. 2015, 17, 5396–5399; [DOI] [PubMed] [Google Scholar]
- 9c. Wang Q., Tang X.-Y., Shi M., Angew. Chem. Int. Ed. 2016, 55, 10811–10815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10969–10973; [Google Scholar]
- 9d. Yu H., Li Z., Bolm C., Angew. Chem. Int. Ed. 2018, 57, 15602–15605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15828–15831; [Google Scholar]
- 9e. Wen D., Zheng Q., Wang C., Tu T., Org. Lett. 2021, 23, 3718–3723. [DOI] [PubMed] [Google Scholar]
- 10. Lenstra D. C., Vedovato V., Ferrer Flegeau E., Maydom J., Willis M. C., Org. Lett. 2016, 18, 2086–2089. [DOI] [PubMed] [Google Scholar]
- 11. Lo P. K. T., Oliver G. A., Willis M. C., J. Org. Chem. 2020, 85, 5753–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. O'Donnell J. S., Schwan A. L., J. Sulfur Chem. 2004, 25, 183–211; [Google Scholar]
- 12b. Riddell A. B., Smith M. R. A., Schwan A. L., J. Sulfur Chem. 2022, 43, 540–592. [Google Scholar]
- 13.
- 13a. Refvik M. D., Froese R. D. J., Goddard J. D., Pham H. H., Pippert M. F., Schwan A. L., J. Am. Chem. Soc. 1995, 117, 184–192; [Google Scholar]
- 13b. Caupène C., Boudou C., Perrio S., Metzner P., J. Org. Chem. 2005, 70, 2812–2815; [DOI] [PubMed] [Google Scholar]
- 13c. Maitro G., Vogel S., Sadaoui M., Prestat G., Madec D., Poli G., Org. Lett. 2007, 9, 5493–5496; [DOI] [PubMed] [Google Scholar]
- 13d. Gelat F., Gaumont A.-C., Perrio S., J. Sulfur Chem. 2013, 34, 596–605; [Google Scholar]
- 13e. Zong L., Ban X., Kee C. W., Tan C.-H., Angew. Chem. Int. Ed. 2014, 53, 11849–11853; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12043–12047; [Google Scholar]
- 13f. Jia T., Zhang M., Jiang H., Wang C. Y., Walsh P. J., J. Am. Chem. Soc. 2015, 137, 13887–13893; [DOI] [PubMed] [Google Scholar]
- 13g. Wang L., Chen M., Zhang P., Li W., Zhang J., J. Am. Chem. Soc. 2018, 140, 3467–3473; [DOI] [PubMed] [Google Scholar]
- 13h. Schwan A. L., Michalski M. M., Findlay J. P., Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 692–697. [Google Scholar]
- 14. Herron J. T., Huie R. E., Chem. Phys. Lett. 1980, 76, 322–324. [Google Scholar]
- 15.
- 15a. Singh S. P., O'Donnell J. S., Schwan A. L., Org. Biomol. Chem. 2010, 8, 1712–1717; [DOI] [PubMed] [Google Scholar]
- 15b. Bernoud E., Le Duc G., Bantreil X., Prestat G., Madec D., Poli G., Org. Lett. 2010, 12, 320–323; [DOI] [PubMed] [Google Scholar]
- 15c. Jia T., Bellomo A., Montel S., Zhang M., El Baina K., Zheng B., Walsh P. J., Angew. Chem. Int. Ed. 2014, 53, 260–264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 264–268; [Google Scholar]
- 15d. Gelat F., Lohier J. F., Gaumont A. C., Perrio S., Adv. Synth. Catal. 2015, 357, 2011–2016; [Google Scholar]
- 15e. Chang M.-Y., Cheng Y.-C., Chan C.-K., Tetrahedron 2016, 72, 4068–4075. [Google Scholar]
- 16. Oae S., Kawai T., Furukawa N., Tetrahedron Lett. 1984, 25, 69–72. [Google Scholar]
- 17. Oae S., Kawai T., Furukawa N., Iwasaki F., J. Chem. Soc. Perkin Trans. 2 1987, 405–411. [Google Scholar]
- 18. Dean W. M., Šiaučiulis M., Storr T. E., Lewis W., Stockman R. A., Angew. Chem. Int. Ed. 2016, 55, 10013–10016; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10167–10170. [Google Scholar]
- 19. Wakabayashi S., Ishida M., Takeda T., Oae S., Tetrahedron Lett. 1988, 29, 4441–4444. [Google Scholar]
- 20. Krasovskiy A., Knochel P., Angew. Chem. Int. Ed. 2004, 43, 3333–3336; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3396–3399. [Google Scholar]
- 21. Sandrinelli F., Perrio S., Beslin P., J. Org. Chem. 1997, 62, 8626–8627. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Yu H., Li Z., Bolm C., Org. Lett. 2018, 20, 7104–7106; [DOI] [PubMed] [Google Scholar]
- 22b. Amos S. G. E., Nicolai S., Gagnebin A., Le Vaillant F., Waser J., J. Org. Chem. 2019, 84, 3687–3701; [DOI] [PubMed] [Google Scholar]
- 22c. Wang L., Chen M., Zhang J., Org. Chem. Front. 2019, 6, 32–35. [Google Scholar]
- 23. Dai Q., Zhang J., Adv. Synth. Catal. 2018, 360, 1123–1127. [Google Scholar]
- 24. Deeming A. S., Russell C. J., Willis M. C., Angew. Chem. Int. Ed. 2015, 54, 1168–1171; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1184–1187. [Google Scholar]
- 25. Moree W. J., van der Marel G. A., Liskamp R. J., J. Org. Chem. 1995, 60, 5157–5169. [Google Scholar]
- 26. Borsy J., Ther. Hung. 1960, 8, 3–7. [PubMed] [Google Scholar]
- 27. Palosi E., Szporny L., Nádor K., J. Pharm. Sci. 1968, 57, 709–710. [DOI] [PubMed] [Google Scholar]
- 28. Mason R. M., Ann. Rheum. Dis. 1954, 13, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Ogawa S., Furukawa N., J. Org. Chem. 1991, 56, 5723–5726; [Google Scholar]
- 29b. Rauhut C. B., Melzig L., Knochel P., Org. Lett. 2008, 10, 3891–3894. [DOI] [PubMed] [Google Scholar]
- 30. Oae S., Takeda T., Wakabayashi S., Tetrahedron Lett. 1988, 29, 4445–4448. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.