

Abstract
Many patients with heart failure have an iron‐deficient state, which can limit erythropoiesis in erythroid precursors and ATP production in cardiomyocytes. Yet, treatment with sodium–glucose cotransporter 2 (SGLT2) inhibitors produces consistent increases in haemoglobin and haematocrit, even in patients who are iron‐deficient before treatment, and this effect remains unattenuated throughout treatment even though SGLT2 inhibitors further aggravate biomarkers of iron deficiency. Heart failure is often accompanied by systemic inflammation, which activates hepcidin, thus impairing the duodenal absorption of iron and the release of iron from macrophages and hepatocytes, leading to a decline in circulating iron. Inflammation and oxidative stress also promote the synthesis of ferritin and suppress ferritinophagy, thus impairing the release of intracellular iron stores and leading to the depletion of bioreactive cytosolic Fe2+. By alleviating inflammation and oxidative stress, SGLT2 inhibitors down‐regulate hepcidin, upregulate transferrin receptor protein 1 and reduce ferritin; the net result is to increase the levels of cytosolic Fe2+ available to mitochondria, thus enabling the synthesis of heme (in erythroid precursors) and ATP (in cardiomyocytes). The finding that SGLT2 inhibitors can induce erythrocytosis without iron supplementation suggests that the abnormalities in iron diagnostic tests in patients with mild‐to‐moderate heart failure are likely to be functional, rather than absolute, that is, they are related to inflammation‐mediated trapping of iron by hepcidin and ferritin, which is reversed by treatment with SGLT2 inhibitors. An increase in bioreactive cytosolic Fe2+ is also likely to augment mitochondrial production of ATP in cardiomyocytes, thus retarding the progression of heart failure. These effects on iron metabolism are consistent with (i) proteomics analyses of placebo‐controlled trials, which have shown that biomarkers of iron homeostasis represent the most consistent effect of SGLT2 inhibitors; and (ii) statistical mediation analyses, which have reported striking parallelism of the effect of SGLT2 inhibitors to promote erythrocytosis and reduce heart failure events.
Keywords: SGLT2 inhibitors, Iron, Hepcidin, Ferritin, Transferrin receptor protein, Soluble transferrin receptor, Heart failure, Haemoglobin, Hematocrit, Ferritinophagy, Ferroptosis
One of the most striking features of treatment with sodium–glucose cotransporter 2 (SGLT2) inhibitors in heart failure is their effect to increase haemoglobin and haematocrit. The increase in red blood cell production produced by these drugs occurs within the first month of therapy and is triggered by an increase in erythropoietin. 1 , 2 The degree of erythrocytosis precedes, predicts and closely parallels the effect of SGLT2 inhibitors to reduce the risk of heart failure events in large‐scale cardiovascular outcome trials. 3 , 4 , 5 Patients who develop striking erythrocytosis while receiving an SGLT2 inhibitor experience the most marked reduction in heart failure hospitalizations.
The enigma of erythrocytosis during sodium–glucose cotransporter 2 inhibition
The erythrocytosis seen with SGLT2 inhibitors is not seen with other drugs used for the treatment of heart failure, and it is particularly impressive, given the fact that ≈50% of patients with heart failure and a reduced ejection fraction fulfill current criteria for an absolute or functional iron deficiency, manifested as (i) a low serum ferritin level (<100 μg/L), or (ii) low transferrin saturation (<20%) together with a serum ferritin level of <300 μg/L. 6 , 7 Iron deficiency indicates an impaired ability to deliver iron to erythroid precursors for adequate erythropoiesis or to cardiomyocytes to sustain optimal mitochondrial ATP production. Iron deficiency may result from (i) an absolute deficiency of iron, reflected by the absence of iron in the main storage depot in the liver or bone marrow and is typically due to poor dietary intake or gastrointestinal blood loss; or (ii) defective gastrointestinal absorption or impaired release of iron from hepatocytes and from macrophages involved in the recycling of senescent red blood cells, typically related to chronic inflammation. The relative importance of these two distinct aetiologies in patients with heart failure has been controversial, 8 since most studies have evaluated small highly selected cohorts and relied on diverse criteria for defining iron deficiency.
Many iron‐deficient patients with heart failure are anaemic, suggesting that most are not able to deliver sufficient iron to support adequate red blood cell production, regardless of whether the deficiency is absolute or functional. Yet, SGLT2 inhibitors produce an erythrocytosis even when patients are deemed to be iron‐deficient before treatment, and the magnitude of the increase in red blood cell mass following SGLT2 inhibition is not impaired as compared with patients who are deemed to be iron‐replete. 9 Even the presence of anaemia prior to treatment does not attenuate the magnitude of the increase in haemoglobin following treatment with SGLT2 inhibitors in patients with heart failure. 10
These findings are surprising since the depletion of systemic iron stores typically blunts the response to erythropoietin mimetics, thus necessitating intravenous iron supplementation to minimize the development of hyporesponsiveness or resistance to these drugs. 11 , 12 Furthermore, treatment of patients with heart failure with SGLT2 inhibitors typically aggravates conventional metrics of iron deficiency, as these drugs act to lower both serum ferritin and transferrin saturation. In a large‐scale trial, SGLT2 inhibition increased the risk of patients fulfilling conventional criteria for iron deficiency by 70% after 12 months of therapy. 10 Yet, despite the presence of iron deficiency before treatment and the apparent exacerbation of iron deficiency during treatment, the erythrocytosis produced by these drugs is sustained and does not become attenuated over time.
Why does iron deficiency not blunt responsiveness to the increased synthesis of erythropoietin produced by SGLT2 inhibitors? Similarly, why does the apparent exacerbation of iron deficiency during SGLT2 inhibition not negate the beneficial effects of these drugs on the myocardium to reduce the risk of major heart failure events?
Regulation of iron homeostasis in heme‐synthesizing cells
Iron enters the bloodstream when dietary iron is absorbed from the gastrointestinal tract or when it is released from hepatocytes involved in iron storage or from macrophages that are engaged in the recycling of senescent red blood cells (Figure 1 ). These points of entry are inhibited by hepcidin, 13 and suppression of hepcidin promotes the ingress of iron into the circulation, where it is bound to and carried by transferrin as ferric ion (Fe3+).
Figure 1.
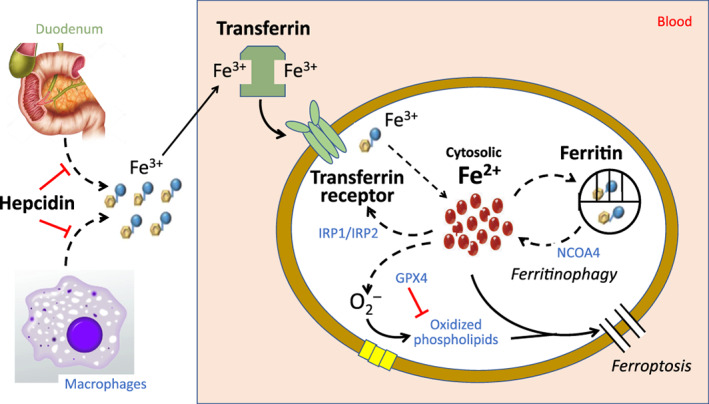
Mechanisms of iron homeostasis in erythroid precursors and cardiomyocytes. Hepcidin blocks the absorption of iron from the duodenum and the release of iron from macrophages and hepatocytes. Iron is bound to transferrin as ferric ion (Fe3+) and is internalized when transferrin docks with the transferrin receptor (transferrin receptor protein 1). Iron is released into the cytosolic pool in its reactive form (Fe2+), where it is available to be utilized by mitochondria in the synthesis of heme and iron–sulfur clusters. Cytosolic levels of Fe2+ are maintained in a tight range by the coordinated actions of iron regulatory proteins 1 and 2 (IRP1 and IRP2) and ferritin. Iron regulatory proteins stimulate TfR1 if cytosolic Fe2+ is low. Conversely, if Fe2+ increases, Fe2+ is sequestered as Fe3+ in a ferritin cage, which releases iron back into the cytosol by nuclear receptor coactivator 4 (NCOA4)‐mediated ferritinophagy. If cytosolic level of highly reactive Fe2+ increases excessively, the resulting production of reactive oxygen species oxidizes membrane‐bound phospholipids to promote ferroptosis; this oxidation and the resulting ferroptosis is prevented by glutathione peroxidase 4 (GPX4).
Transport of iron into heme‐producing cells
To exert its effects in the heart and bone marrow, iron must enter cells involved in the synthesis of heme and iron–sulfur clusters. This transport is achieved when transferrin attaches to transferrin receptor protein 1 (TfR1), forming a complex that is internalized by endocytosis (Figure 1 ). Movement of Fe3+ out of the endosome and its reduction by a ferrireductase leads to the release of ferrous iron (Fe2+) into the cytosol, which is the highly reactive form of weakly‐bound iron that is utilized by mitochondria in the synthesis of heme and iron–sulfur clusters, 14 which are essential to the production of haemoglobin (in erythrocytes) or to the generation of ATP (in cardiomyocytes). It has been estimated that this bioreactive pool of cytosolic iron represents 5–10% of the cellular iron content. 15
Because Fe2+ is highly reactive, cytosolic levels of Fe2+ are tightly controlled to ensure an adequate supply for heme synthesis, while minimizing deleterious excesses. A decrease in cytosolic Fe2+ is sensed by iron regulatory proteins (IRP1 and IRP2), which promote the expression of TfR1, 16 thus facilitating the entry of iron into cells (Figure 1 ). In addition, in states of cardiomyocyte iron depletion, hepcidin is selectively upregulated within cardiomyocytes to retard the egress of iron through ferroportin. 16 , 17 The coordinated actions of TfR1, IRP1/IRP2 and hepcidin in the heart act to preserve the levels of cytosolic iron in the face of systemic iron deficiency, potentially explaining why cardiac iron stores are not depleted even when iron is absent in the bone marrow. 18 , 19 , 20 Accordingly, experimental cardiac‐specific deletion of TfR1, IRP1/IRP2 or hepcidin leads to cytosolic iron deficiency and attenuation of iron‐dependent heme‐driven synthesis of ATP, thus impairing the heart's response to stress and leading to cardiomyopathy. 17 , 21 , 22
Intracellular sequestration of iron in heme‐producing cells
Homeostatic mechanisms in heme‐producing cells ensure that intracellular concentrations of highly reactive cytosolic iron do not rise to unhealthy levels. If the level of cytosolic Fe2+ increases beyond the capacity for mitochondrial utilization, Fe2+ can interact with oxidized lipids (especially in states of glutathione depletion) to promote an iron‐dependent apoptosis‐independent form of programmed cell death known as ferroptosis. Ferroptosis is prevented by iron chelation and by cellular mechanisms (e.g. glutathione peroxidase 4) that control lipid peroxidation. 23 Ferroptosis contributes to the development of cardiomyopathy due to both ischaemic and non‐ischaemic causes, even in clinical states that are not characterized by systemic or myocardial iron overload. 24 , 25
To prevent excess levels of bioreactive cytosolic Fe2+, cells involved in heme synthesis utilize intracellular ferritin as a nanocage, which sequesters excessive iron in a non‐reactive ferric form (Fe3+) for storage and subsequent release when needed. 26 The level of ferritin responds to deleterious changes in cytosolic Fe2+; as cytosolic Fe2+ levels increase and lead to oxidative stress, ferritin is synthesized to enhance the degree of intracellular iron sequestration. 27 Accordingly, the cardiac‐specific loss of ferritin leads to cytosolic iron overload, ferroptosis and cardiomyopathy. 28 Conversely, if cytosolic Fe2+ declines to dysfunctional levels, ferritin is degraded, releasing iron from sequestration. The primary mechanism that mediates the degradation of ferritin is ferritinophagy, a lysosomal‐dependent process that depends on nuclear receptor coactivator 4 (NCOA4), an autophagic cargo receptor that targets and directs ferritin for destruction (Figure 1 ). 29 If ferritinophagy is excessive, the resulting rise in cytosolic Fe2+ results in cardiomyocyte death and heart failure. 30 Conversely, if ferritinophagy is deficient, iron is not released from the ferritin nanocage in erythroid precursors, leading to anaemia, despite elevated tissue iron levels. 31 Excessive cellular iron uptake leads to enhanced degradation of NCOA4, suppression of ferritinophagy and augmented iron sequestration. 32
Assessment of iron deficiency in patients without heart failure
In states of absolute iron deficiency due to poor dietary intake or gastrointestinal blood loss, a decrease in hepcidin leads to the enhanced duodenal absorption of iron and the augmented release of iron from macrophages and hepatocytes. The increase in circulating iron is prioritized for transport into heme‐producing cells by an increased expression of TfR1. At the same time, a decline in ferritin enables the release of iron into the cytosol from intracellular stores. Therefore, the combination of a decrease in hepcidin, an increase in TfR1 (or its secreted form, i.e. soluble transferrin receptor [sTfR]) and a decline in ferritin is typically seen when cytosolic Fe2+ is depleted due to absolute iron deficiency. 33 , 34 , 35 However, it should be noted that changes in these iron homeostasis proteins do not measure cytosolic Fe2+ 14 , 15 instead, they assess the cellular responses that are evoked during iron deficiency to restore cytosolic Fe2+. In clinical practice, physicians have assumed that circulating levels of these proteins are closely correlated with their intracellular concentrations in heme‐producing cells, although this assumption may not be valid. Nevertheless, the calculation of a ratio of sTfR to serum ferritin has been proposed as a superior diagnostic test for the identification of patients with iron deficiency, 34 , 36 , 37 based on the premise that the ratio reflects cellular efforts to enhance the entry of Fe2+ into cytosol from both extracellular and intracellular sources.
Historically, the diagnostic accuracy of blood tests used for the identification of iron‐deficient states has been based on the evaluation of patients with anaemia due to absolute iron deficiency, typically established by the absence of iron stores in the bone marrow. However, the evaluation of iron staining in the bone marrow is challenging, since the aspirates are often inadequate and the assessment is subjective. 38 Many patients with anaemia and no detectable bone marrow iron are not iron‐deficient, 39 , 40 possibly because iron staining in bone marrow does not reflect iron stores in the liver, the most important storage depot for systemic iron. In light of these difficulties, serum ferritin has been considered to be the most accurate means of assessing systemic iron stores in patients without heart failure. In general, serum ferritin levels <12–40 μg/L are considered to be diagnostic of absolute iron deficiency, even in elderly patients with anaemia. 41 , 42 The requirement of an increase in sTfR (accompanying the low ferritin) further increases the likelihood that systemic iron stores are truly depleted. 43
Changes in iron homeostasis proteins in patients with heart failure
In heart failure, the assessment of iron deficiency is complicated by the fact that such patients often have a systemic inflammatory state, 44 which enhances the production of hepcidin in the liver (Figure 2 ). 45 Circulating levels of hepcidin are increased in patients with heart failure 9 , 46 , 47 in proportion to the activation of proinflammatory pathways, 48 , 49 , 50 , 51 and the increase retards the entry of iron from the gastrointestinal tract and from macrophages, leading to a state of functional iron deficiency (akin to the anaemia of chronic disease 49 , 52 , 53 ), which is resistant to oral iron supplementation, but can be responsive to treatment with intravenous iron. 54 Patients with heart failure can suffer from either absolute iron depletion or the functional iron deficiency of chronic inflammation, or both. 8 , 18 , 44 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 The relative importance of the two mechanisms in clinical practice has been difficult to discern from the available studies, most of which were small and evaluated highly selected patients. Furthermore, it is possible that the mechanisms leading to iron deficiency may shift as heart failure advances. 46 , 47
Figure 2.
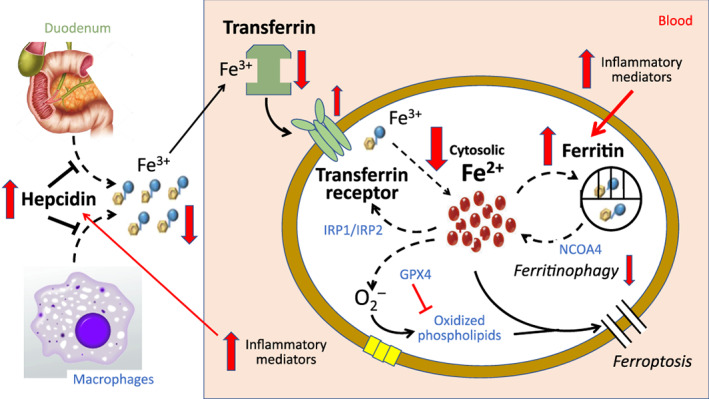
Mechanisms of functional iron deficiency in patients with heart failure. Red arrows indicate the shifts in iron homeostasis in patients with heart failure. Heart failure is often accompanied by increased systemic inflammation, which activates hepcidin, thus impairing the absorption of iron from the duodenum and the release of iron from macrophages and hepatocytes and leading to a decline in iron levels in circulating blood. Inflammation and oxidative stress also promote the synthesis of ferritin and suppress ferritinophagy, thus impairing the release of intracellular iron stores into the cytosol and leading to the depletion of bioreactive cytosolic Fe2+. Transferrin receptor protein 1 may be down‐regulated in the failing heart, but it is upregulated when cytosolic levels of Fe2+ are threatened. GPX4, glutathione peroxidase 4; IRP1 and IRP2, iron regulatory proteins 1 and 2; NCOA4, nuclear receptor coactivator 4.
In addition to their effects of hepcidin, activation of oxidative stress and proinflammatory pathways in heart failure can promote the synthesis of ferritin, independent of the level of cytosolic iron. 57 , 58 Additionally, the suppression of sirtuin‐1 (SIRT1) in heart failure 59 can impair autophagic flux, thus limiting the ability of ferritinophagy to replete cytosolic Fe2+ levels. 60 As a result, the expected relationship between cytosolic Fe2+ and ferritin levels is disrupted, leading to ferritin levels that are disproportionately higher than the level of cytosolic Fe2+, and undermining their ability to identify patients with iron depletion. Accordingly, the threshold for serum ferritin used for the diagnosis of iron‐deficiency anaemia is shifted to a substantially higher level in patients with heart failure (to 300–600 ng/ml), and it is coupled with the requirement for a transferrin saturation < 20%. However, the adequacy of this criterion adjustment for serum ferritin has been questioned. 7 , 61 In one study, patients with heart failure and an absolute iron deficiency often had normal serum levels of ferritin, 55 and in other reports, myocardial (but not serum) levels of ferritin reflected the presence of cardiac iron deficiency in experimental and clinical heart failure. 20 , 62 Furthermore, inflammation and nutritional status can influence transferrin levels, thus complicating the interpretation of transferrin saturation in heart failure. 63 , 64 , 65 , 66
Given the limitations of both hepcidin and ferritin in states of chronic inflammation, the measurement of sTfR has been advocated as an additional diagnostic tool. Serum levels of sTfR are increased in both patients with an absolute or functional iron deficiency, 43 , 67 since the receptor is upregulated in response to a decrease in cytosolic iron, regardless of cause. However, the failing heart often shows decreased expression of TfR1, 68 , 69 perhaps the result of neurohormonal activation, 70 , 71 which may contribute to an intracellular iron deficiency in cardiomyocytes. However, it is possible that this down‐regulation of TfR1 is modified into an upregulation of TfR1 when myocardial iron stores are depleted. 72 , 73
The conventional definition of iron deficiency in patients with heart failure – based on the combination of serum ferritin and transferrin saturation – does not distinguish between absolute and functional iron deficiency. From the perspective of the failing heart or endangered erythropoiesis, the distinction may be unimportant, since both absolute and functional iron deficiency would be expected to deprive erythroid precursors and the myocardium of the iron required to maintain haemoglobin synthesis and ATP production. Patients who fulfill criteria for either absolute or functional iron deficiency have greater impairment in cardiac ATP production than those who are iron‐replete. 74 It has been assumed that iron deficiency in the heart would be reflected in a depletion of myocardial iron stores, which can be assessed by myocardial biopsy or by non‐invasive imaging. 75 Myocardial iron is characteristically reduced in heart failure, 19 , 20 often more so in patients with accompanying anaemia. However, myocardial staining for iron does not reflect the disordered regulation of bioreactive cytosolic Fe2+ that is caused by changes in iron handling proteins in the failing heart. 18 In fact, an iron deficiency state identified by low serum or myocardial ferritin can be accompanied by normal myocardial iron stores. 7 , 20 Instead, cardiac iron deficiency may be most reliably identified by an increase in the myocardial expression of TfR1 and a decrease in myocardial ferritin, rather than semi‐quantitative assessments of myocardial iron content. 18 , 20 , 62 , 72 The ratio of TfR1 to ferritin in cardiomyocytes would reflect cellular efforts to enhance the entry of ferrous iron into cytosol from both extracellular and intracellular sources.
In light of the influence of inflammation on circulating levels of conventionally‐measured iron homeostatic proteins in heart failure, the available evidence suggests that the iron deficiency state in most patients with heart failure is functional, rather than absolute; a similar judgment has been reached by other investigators. 76 This conclusion is supported by observations that (i) hepcidin and ferritin are increased in patients with heart failure until the disease becomes advanced; 9 , 47 and (ii) iron‐deficient patients with heart failure show clinical benefits in response to intravenously (but not orally) administered iron supplements, 77 , 78 , 79 , 80 presumably related to hepcidin‐mediated inhibition of duodenal iron absorption. A similar discordance in the responsiveness to intravenous vs oral iron therapy has been observed when patients with chronic kidney disease and functional iron deficiency are treated with erythropoietin‐stimulating agents. 81 , 82
Effect of sodium–glucose cotransporter 2 inhibitors on iron homeostasis in patients with heart failure
Clinical studies of patients receiving SGLT2 inhibitors have shown that treatment with these drugs produces meaningful decreases in hepcidin, transferrin saturation and ferritin, while increasing sTfR (Figure 3 ). 2 , 10 , 68 , 69 , 70 This pattern has been noted consistently in placebo‐controlled trials of patients who receive these drugs for diabetes or heart failure.
Figure 3.
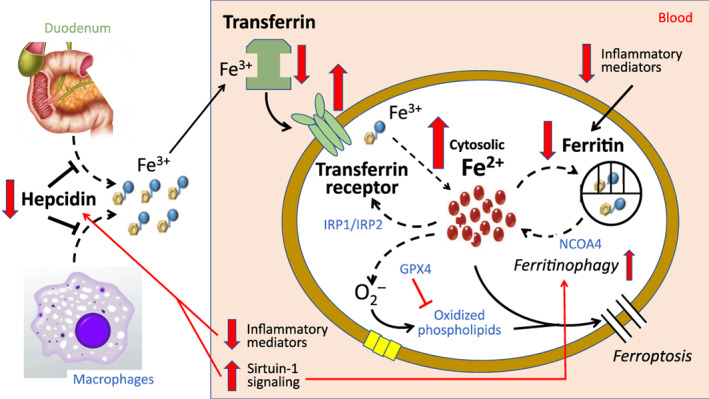
Alleviation of functional iron deficiency in patients with heart failure during treatment with sodium–glucose cotransporter 2 inhibitors (SGLT2). Red arrows indicate the shifts in iron homeostasis in patients with heart failure treated with SGLT2 inhibitors. Due to their action to upregulate sirtuin‐1 and to suppress inflammatory mediators, SGLT2 inhibitors down‐regulate hepcidin, thus enhancing the absorption of iron from the duodenum and the release of iron from macrophages and hepatocytes. Concomitant increased expression of transferrin receptor protein 1 – often measured as soluble transferrin receptor in circulating blood – augments the transit of iron into erythroid precursors and cardiomyocytes. Suppression of inflammation and upregulation of sirtuin‐1 also mutes the synthesis of ferritin and promotes ferritinophagy; thus, SGLT2 inhibitors promote the release of Fe2+ into the cytosol from intracellular stores. The net effect of these pathways is to alleviate the deficit of bioreactive Fe2+, thus enabling the synthesis of heme (in erythroid precursors) and ATP (in cardiomyocytes). GPX4, glutathione peroxidase 4; IRP1 and IRP2, iron regulatory proteins 1 and 2; NCOA4, nuclear receptor coactivator 4.
Changes in iron homeostasis biomarkers in placebo‐controlled trials
In a placebo‐controlled trial of 52 patients with type 2 diabetes studied after 12 weeks of treatment with dapagliflozin, Ghanim et al. 83 reported a 24% decrease in hepcidin (together with a 70% increase in erythroferrone, a hepcidin suppressor), a 22% decrease in transferrin saturation and a 32% decline in ferritin (all measured in plasma), while noting a 59% increase in TfR1 in mononuclear cells. In a placebo‐controlled trial of 44 patients with type 2 diabetes studied after 12 weeks with empagliflozin, Thiele et al. 84 reported a 29% decrease in transferrin saturation and a 33% decline in ferritin in circulating blood. In a placebo‐controlled trial of >2200 patients with heart failure and a reduced ejection fraction studied after 12 months of treatment with dapagliflozin, Docherty et al. 9 noted meaningful decreases in hepcidin and ferritin (with modest decreases in transferrin saturation) together with increases in sTfR. In all studies, the effects of SGLT2 inhibition were significantly different from placebo.
In a proteomics analysis of 72 patients with type 2 diabetes reported by Ferrannini et al., 85 SGLT2 inhibition for 4 weeks led to increases in sTfR and decreases in ferritin and neogenin (a positive regulator of hepcidin), all with false discovery rates <1%. Interestingly, the effect on ferritin was exceptionally consistent, with a false discovery rate p‐value of 6.7 × 10−8. Of >3700 proteins examined, 43 were differentially‐expressed as a result of SGLT2 inhibition, and of these, 5 were involved in iron metabolism. In a proteomics analysis of >1100 patients with heart failure reported by Zannad et al., 2 SGLT2 inhibition for 12 weeks led to increases in TfR1 and erythropoietin, both with false discovery rates <1%. TfR1 was the protein that was most consistently changed by empagliflozin among >1200 proteins that were measured, with a false discovery rate p‐value of 2.5 × 10−12. Increases in erythropoietin with SGLT2 inhibitors have been reported in other placebo‐controlled trials. 1
Mechanistic implications of changes in iron homeostasis during sodium–glucose cotransporter 2 inhibition
Increases in erythropoietin will not yield meaningful increases in red blood cell production if a deficiency in cytosolic levels of bioreactive Fe2+ impairs the synthesis of haemoglobin. It is therefore interesting that SGLT2 inhibition leads to changes in iron regulatory proteins that ensure that cytosolic levels of Fe2+ are supported (Figure 3 ). The decreases in hepcidin seen following SGLT2 inhibition should increase the gastrointestinal absorption of iron and the release of iron from macrophages and hepatocytes. SGLT2 inhibition produces a small decrease in transferrin saturation, presumably because iron is rapidly transported out of the bloodstream and into cells as a consequence of the upregulation of TfR1/sTfR. The augmented transport of iron into erythroid precursors and cardiomyocytes would increase cytosolic levels of bioreactive Fe2+, as long as iron is not sequestered within the ferritin nanocage. It is therefore noteworthy that SGLT2 inhibitors produce a decrease in serum ferritin; if this decline reflects a decline in ferritin in erythroid precursors and cardiomyocytes, such an action would ensure an increase in cytosolic levels of Fe2+.
How do SGLT2 inhibitors cause increases in sTfR/TfR1 and erythropoietin and decreases in hepcidin and ferritin? SGLT2 inhibitors promote a state of starvation mimicry, which is characterized by upregulation of nutrient deprivation signalling, especially SIRT1. 59 SIRT1 is responsible for the increased gluconeogenesis and ketogenesis that follows the loss of calories in the urine seen following inhibition of SGLT2. 86 , 87 Upregulation of SIRT1 increases signalling through hypoxia‐inducible factor‐2α (HIF‐2α), 88 the primary positive regulator of erythropoietin synthesis. At the same time, activation of SIRT1 also leads to the suppression of hepcidin synthesis in both the liver and macrophages. 89 Upregulation of SIRT1 also increases the activation of PGC‐1α, which can upregulate the expression of TfR1. 90 Finally, since the principal mechanism by which intracellular ferritin declines is related to ferritinophagy, it is noteworthy that SIRT1 can promote ferritinophagy either by a direct effect to promote autophagic flux 59 , 60 or through stimulation of HIF‐2α‐mediated increases in the expression of NCOA4, 91 thus promoting ferritinophagy. Additionally, the well‐established action of SGLT2 inhibitors to mute inflammation in the heart and bone marrow 92 , 93 , 94 (also the result of enhanced SIRT1 signalling 93 , 94 , 95 ) might contribute to the observed decline in both hepcidin and ferritin. Finally, increased erythropoietin activity upregulates TfR1 and reduces transferrin saturation, ferritin and hepcidin. 96 , 97 , 98 Importantly, if cytosolic Fe2+ levels were to rise excessively, SIRT1 can prevent the deleterious consequences of iron overload by its action to suppress ferroptosis. 99 , 100
Therefore, through their effect to enhance nutrient deprivation signalling and suppress inflammation, SGLT2 inhibitors act to increase in bioreactive cytosolic Fe2+ together with an increase in erythropoietin, thus stimulating erythroid precursors and haemoglobin synthesis to produce an increase in red blood cell mass. Similar changes in the heart would promote the synthesis of heme‐containing proteins in iron‐deficient cardiomyocytes, which are essential for the synthesis of ATP in the energy‐starved failing heart. Upregulation of TfR1 also promotes autophagic clearance of damaged mitochondria, which is essential for both erythroid maturation and cardioprotection. 101 Accordingly, an increase in bioreactive cytosolic Fe2+ may explain the ability of SGLT2 inhibitors to simultaneously increase ATP production within the heart and to promote erythrocytosis in the bone marrow. 102 This parallelism may explain why erythrocytosis closely parallels and predicts the cardioprotective effect of these drugs. In fact, an increase in haemoglobin is the most important statistical mediator of the ability of empagliflozin, canagliflozin and ertugliflozin to reduce the risk of major heart failure events in large‐scale cardiovascular outcome trials. 3 , 4 , 5
Implications for understanding the pathogenesis and consequences of iron deficiency in heart failure
In patients with heart failure, the effect of SGLT2 inhibitors to increase TfR1 and decrease hepcidin and ferritin would be expected to increase the level of bioreactive cytosolic Fe2+ in cells involved in the production of heme and iron–sulfur clusters. This increase in cytosolic iron – coupled with an increase in erythropoietin – can explain the effect of SGLT2 inhibitors to promote erythrocytosis even in patients who are deemed to be iron‐deficient before treatment. 9 In fact, SGLT2 inhibitors induced significant erythropoiesis even in iron‐deficient patients who were anaemic prior to treatment 10 ; that is, before SGLT2 inhibition, cytosolic levels of Fe2+ in these patients were apparently insufficient to support adequate red blood cell production. Patients who have heart failure and anaemia are often poorly responsive to erythropoietin‐stimulating agents if they are iron‐deficient despite the use of oral supplements. 103 The finding that SGLT2 inhibitors can rapidly correct anaemia without iron supplementation 10 indicates that iron deficiency in these patients (who had mild‐to‐moderate heart failure) was likely to be functional, rather than absolute, that is, it was related to inflammation‐mediated increases in hepcidin and ferritin, which were reversed by treatment with SGLT2 inhibitors. Such a conclusion is consistent with previous studies. 7 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 Reports that patients with heart failure suffer from an absolute iron deficiency have been based on a small sample size, 55 , 56 , 63 the lack of confirmed bone marrow iron depletion, 53 , 55 or the evaluation of people with advanced disease. 47
Importantly, an increase in bioreactive cytosolic Fe2+ is likely to be relevant in augmenting mitochondrial production of ATP in cardiomyocytes, 22 , 73 and thus, retarding the progression of heart failure. It is therefore noteworthy that the effect of SGLT2 inhibitors to reduce heart failure hospitalizations was particularly marked in patients who were most iron‐deficient prior to therapy (i.e. serum ferritin <100 ng/ml) 9 and who might be expected to be particularly responsive to interventions that would increase cytosolic Fe2+. Analogously, patients with heart failure who have decreased transferrin saturations (i.e. <20%) were most likely to experience an improvement in cardiac performance and a decrease in heart failure hospitalizations following intravenous iron supplementation, 77 , 78 although this finding remains uncertain. 79 Nevertheless, the parallelism of these observations suggests that a SIRT1‐mediated improvement in iron homeostasis may mediate many of the benefits of SGLT2 inhibitors in heart failure. This hypothesis is consistent with the findings of both statistical mediation analyses and proteomics analyses of placebo‐controlled trials, which have highlighted the effects of these drugs on erythropoiesis and its regulators as prominent features of their pharmacological profile and therapeutic benefits. 2 , 3 , 4 , 5 , 6 , 85
It should be noted that SGLT2 inhibitors exert their parallel benefits on red blood cell production and heart failure events even though treatment with these drugs produces decreases in transferrin saturation and serum ferritin and increases in sTfR. This pattern of changes would typically lead physicians to conclude that patients were becoming more (rather than less) iron‐deficient during treatment with these drugs. In fact, after 12 months of treatment, patients receiving dapagliflozin in the DAPA‐HF trial were far more likely to fulfill diagnostic criteria of an iron‐deficiency state than patients receiving placebo, even though patients treated with the SGLT2 inhibitor continued to demonstrate a robust erythrocytic response and the magnitude of erythrocytosis was not attenuated in those with a diagnosis of iron deficiency prior to treatment. 9 These intriguing observations raise important questions about our current reliance on the measurement of circulating levels of iron homeostatic proteins to reach conclusions about the level of bioreactive cytosolic Fe2+ within erythrocyte precursor cells or cardiomyocytes when patients are being treated with SGLT2 inhibitors.
Conflict of interest: none declared.
References
- 1. Mazer CD, Hare GMT, Connelly PW, Gilbert RE, Shehata N, Quan A, et al. Effect of empagliflozin on erythropoietin levels, iron stores, and red blood cell morphology in patients with type 2 diabetes mellitus and coronary artery disease. Circulation. 2020;141:704–7. [DOI] [PubMed] [Google Scholar]
- 2. Zannad F, Ferreira JP, Butler J, Filippatos G, Januzzi JL, Sumin M, et al. Effect of empagliflozin on circulating proteomics in heart failure: mechanistic insights from the EMPEROR Program. Eur Heart J. 2022. 10.1093/eurheartj/ehac495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Segar MW, Kolkailah AA, Frederich R, Pong A, Cannon CP, Cosentino F, et al. Mediators of ertugliflozin effects on heart failure and kidney outcomes among patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2022;24:1829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li J, Woodward M, Perkovic V, Figtree GA, Heerspink HJL, Mahaffey KW, et al. Mediators of the effects of canagliflozin on heart failure in patients with type 2 diabetes. JACC Heart Fail. 2020;8:57–66. [DOI] [PubMed] [Google Scholar]
- 5. Fitchett D, Inzucchi SE, Zinman B, Wanner C, Schumacher M, Schmoor C, et al. Mediators of the improvement in heart failure outcomes with empagliflozin in the EMPA‐REG OUTCOME trial. ESC Heart Fail. 2021;8:4517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Klip IT, Comin‐Colet J, Voors AA, Ponikowski P, Enjuanes C, Banasiak W, et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J. 2013;165:575–82. [DOI] [PubMed] [Google Scholar]
- 7. Masini G, Graham FJ, Pellicori P, Cleland JGF, Cuthbert JJ, Kazmi S, et al. Criteria for iron deficiency in patients with heart failure. J Am Coll Cardiol. 2022;79:341–51. [DOI] [PubMed] [Google Scholar]
- 8. Anand IS, Gupta P. Anemia and iron deficiency in heart failure. Circulation. 2018;138:80–98. [DOI] [PubMed] [Google Scholar]
- 9. Docherty KF, Welsh P, Verma S, De Boer RA, O'Meara E, Bengtsson O, et al. Iron deficiency in heart failure and effect of dapagliflozin: findings from DAPA‐HF. Circulation. 2022;146:980–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Docherty KF, Curtain JP, Anand IS, Bengtsson O, Inzucchi SE, Køber L, et al. Effect of dapagliflozin on anaemia in DAPA‐HF. Eur J Heart Fail. 2021;23:617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ogawa T, Mitta K. Erythropoiesis‐stimulating agent hyporesponsiveness in end‐stage renal disease patients. Contrib Nephrol. 2015;185:76–86. [DOI] [PubMed] [Google Scholar]
- 12. Gidaro A, Delitala AP, Berzuini A, Soloski MJ, Manca P, Castro D, et al. Ferric carboxymaltose and erythropoiesis‐stimulating agent treatment reduces the rate of blood transfusion in refractory anemia. J Clin Med. 2022;11:4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Afsar RE, Kanbay M, Ibis A, Afsar B. In‐depth review: is hepcidin a marker for the heart and the kidney? Mol Cell Biochem. 2021;476:3365–81. [DOI] [PubMed] [Google Scholar]
- 14. Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A. 2010;107:10775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krijt M, Jirkovska A, Kabickova T, Melenovsky V, Petrak J, Vyoral D. Detection and quantitation of iron in ferritin, transferrin and labile iron pool (LIP) in cardiomyocytes using 55Fe and storage phosphorimaging. Biochim Biophys Acta Gen Subj. 2018;1862:2895–901. [DOI] [PubMed] [Google Scholar]
- 16. Kozłowska B, Sochanowicz B, Kraj L, Palusińska M, Kołsut P, Szymański Ł, et al. Clinical and molecular aspects of iron metabolism in failing myocytes. Life (Basel). 2022;12:1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lakhal‐Littleton S, Wolna M, Chung YJ, Christian HC, Heather LC, Brescia M, et al. An essential cell‐autonomous role for hepcidin in cardiac iron homeostasis. Elife. 2016;5:e19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petrak J, Havlenova T, Krijt M, Behounek M, Franekova J, Cervenka L, et al. Myocardial iron homeostasis and hepcidin expression in a rat model of heart failure at different levels of dietary iron intake. Biochim Biophys Acta Gen Subj. 2019;1863:703–13. [DOI] [PubMed] [Google Scholar]
- 19. Leszek P, Sochanowicz B, Brzóska K, Kraj L, Kuśmierczyk M, Śmigielski W, et al. Accurate noninvasive assessment of myocardial iron load in advanced heart failure patients. Dis Markers. 2020;2020:8885189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leszek P, Sochanowicz B, Szperl M, Kolsut P, Brzóska K, Piotrowski W, et al. Myocardial iron homeostasis in advanced chronic heart failure patients. Int J Cardiol. 2012;159:47–52. [DOI] [PubMed] [Google Scholar]
- 21. Xu W, Barrientos T, Mao L, Rockman HA, Sauve AA, Andrews NC. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep. 2015;13:533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haddad S, Wang Y, Galy B, Korf‐Klingebiel M, Hirsch V, Baru AM, et al. Iron‐regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. Eur Heart J. 2017;38:362–72. [DOI] [PubMed] [Google Scholar]
- 23. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. 2022. 10.1038/s41569-022-00735-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li D, Pi W, Sun Z, Liu X, Jiang J. Ferroptosis and its role in cardiomyopathy. Biomed Pharmacother. 2022;153:113279. [DOI] [PubMed] [Google Scholar]
- 25. Komai K, Kawasaki NK, Higa JK, Matsui T. The role of ferroptosis in adverse left ventricular remodeling following acute myocardial infarction. Cell. 2022;11:1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Behera RK, Torres R, Tosha T, Bradley JM, Goulding CW, Theil EC. Fe2+ substrate transport through ferritin protein cage ion channels influences enzyme activity and biomineralization. J Biol Inorg Chem. 2015;20:957–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Corna G, Santambrogio P, Minotti G, Cairo G. Doxorubicin paradoxically protects cardiomyocytes against iron‐mediated toxicity: role of reactive oxygen species and ferritin. J Biol Chem. 2004;279:13738–45. [DOI] [PubMed] [Google Scholar]
- 28. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11‐mediated ferroptosis. Circ Res. 2020;127:486–501. [DOI] [PubMed] [Google Scholar]
- 29. Qin Y, Qiao Y, Wang D, Tang C, Yan G. Ferritinophagy and ferroptosis in cardiovascular disease: mechanisms and potential applications. Biomed Pharmacother. 2021;141:111872. [DOI] [PubMed] [Google Scholar]
- 30. Ito J, Omiya S, Rusu MC, Ueda H, Murakawa T, Tanada Y, et al. Iron derived from autophagy‐mediated ferritin degradation induces cardiomyocyte death and heart failure in mice. Elife. 2021;10:e62174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santana‐Codina N, Gableske S, Quiles del Rey M, Małachowska B, Jedrychowski MP, Biancur DE, et al. NCOA4 maintains murine erythropoiesis via cell autonomous and non‐autonomous mechanisms. Haematologica. 2019;104:1342–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryu MS, Duck KA, Philpott CC. Ferritin iron regulators, PCBP1 and NCOA4, respond to cellular iron status in developing red cells. Blood Cells Mol Dis. 2018;69:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ambroszkiewicz J, Klemarczyk W, Mazur J, Gajewska J, Rowicka G, Strucińska M, et al. Serum hepcidin and soluble transferrin receptor in the assessment of iron metabolism in children on a vegetarian diet. Biol Trace Elem Res. 2017;180:182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin DH, Kim HS, Park MJ, Suh IB, Shin KS. Utility of access soluble transferrin receptor (sTfR) and sTfR/log ferritin index in diagnosing iron deficiency anemia. Ann Clin Lab Sci. 2015;45:396–402. [PubMed] [Google Scholar]
- 35. Jonker FA, Boele van Hensbroek M, Leenstra T, Vet RJ, Brabin BJ, Maseko N, et al. Conventional and novel peripheral blood iron markers compared against bone marrow in Malawian children. J Clin Pathol. 2014;67:717–23. [DOI] [PubMed] [Google Scholar]
- 36. Punnonem K, Irjala K, Rajamäki A. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood. 1997;89:1052–7. [PubMed] [Google Scholar]
- 37. Rimon E, Levy S, Sapir A, Gelzer G, Peled R, Ergas D, et al. Diagnosis of iron deficiency anemia in the elderly by transferrin receptor‐ferritin index. Arch Intern Med. 2002;162:445–9. [DOI] [PubMed] [Google Scholar]
- 38. Thomas DW, Hinchliffe RF, Briggs C, Macdougall IC, Littlewood T, Cavill I. British Committee for Standards in Haematology. Guideline for the laboratory diagnosis of functional iron deficiency. Br J Haematol. 2013;161:639–48. [DOI] [PubMed] [Google Scholar]
- 39. Ganti AK, Moazzam N, Laroia S, Tendulkar K, Potti A, Mehdi SA. Predictive value of absent bone marrow iron stores in the clinical diagnosis of iron deficiency anemia. In Vivo. 2003;17:389–92. [PubMed] [Google Scholar]
- 40. Barron BA, Hoyer JD, Tefferi A. A bone marrow report of absent stainable iron is not diagnostic of iron deficiency. Ann Hematol. 2001;80:166–9. [DOI] [PubMed] [Google Scholar]
- 41. Thompson WG, Meola T, Lipkin M Jr, Freedman ML. Red cell distribution width, mean corpuscular volume, and transferrin saturation in the diagnosis of iron deficiency. Arch Intern Med. 1988;148:2128–30. [PubMed] [Google Scholar]
- 42. Guyatt GH, Patterson C, Ali M, Singer J, Levine M, Turpie I, et al. Diagnosis of iron‐deficiency anemia in the elderly. Am J Med. 1990;88:205–9. [DOI] [PubMed] [Google Scholar]
- 43. Means RT Jr, Allen J, Sears DA, Schuster S. Serum soluble transferrin receptor and the prediction of marrow aspirate iron results in a heterogeneous group of patients. J Clin Lab Haematol. 1999;21:161–7. [DOI] [PubMed] [Google Scholar]
- 44. Perticone M, Zito R, Miceli S, Pinto A, Suraci E, Greco M, et al. Immunity, inflammation and heart failure: their role on cardiac function and iron status. Front Immunol. 2019;10:2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute‐phase protein. Blood. 2003;101:2461–3. [DOI] [PubMed] [Google Scholar]
- 46. Klip IT, Voors AA, Swinkels DW, Bakker SJ, Kootstra‐Ros JE, Lam CS, et al. Serum ferritin and risk for new‐onset heart failure and cardiovascular events in the community. Eur J Heart Fail. 2017;19:348–56. [DOI] [PubMed] [Google Scholar]
- 47. Jankowska EA, Malyszko J, Ardehali H, Koc‐Zorawska E, Banasiak W, von Haehling S, et al. Iron status in patients with chronic heart failure. Eur Heart J. 2013;34:827–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martínez‐Ruiz A, Tornel‐Osorio PL, Sánchez‐Más J, Pérez‐Fornieles J, Vílchez JA, Martínez‐Hernández P, et al. Soluble TNFα receptor type I and hepcidin as determinants of development of anemia in the long‐term follow‐up of heart failure patients. Clin Biochem. 2012;45:1455–8. [DOI] [PubMed] [Google Scholar]
- 49. Enko D, Wagner H, Kriegshäuser G, Kimbacher C, Stolba R, Worf E, et al. Hepcidin‐25 vs. conventional clinical biomarkers in the diagnosis of functional iron deficiency. Eur J Haematol. 2015;95:507–13. [DOI] [PubMed] [Google Scholar]
- 50. Markousis‐Mavrogenis G, Tromp J, Ouwerkerk W, Devalaraja M, Anker SD, Cleland JG, et al. The clinical significance of interleukin‐6 in heart failure: results from the BIOSTAT‐CHF study. Eur J Heart Fail. 2019;21:965–73. [DOI] [PubMed] [Google Scholar]
- 51. Weinmann K, Werner J, Rottbauer W, Keßler M. Immunoadsorption for heart failure is associated with normalization of iron metabolism. Biomarkers. 2021;26:395–400. [DOI] [PubMed] [Google Scholar]
- 52. Bregman DB, Morris D, Koch TA, He A, Goodnough LT. Hepcidin levels predict non‐responsiveness to oral iron therapy in patients with iron deficiency anemia. Am J Hematol. 2013;88:97–101. [DOI] [PubMed] [Google Scholar]
- 53. Przybylowski P, Wasilewski G, Golabek K, Bachorzewska‐Gajewska H, Dobrzycki S, Koc‐Zorawska E, et al. Absolute and functional iron deficiency is a common finding in patients with heart failure and after heart transplantation. Transplant Proc. 2016;48:173–6. [DOI] [PubMed] [Google Scholar]
- 54. Opasich C, Cazzola M, Scelsi L, De Feo S, Bosimini E, Lagioia R, et al. Blunted erythropoietin production and defective iron supply for erythropoiesis as major causes of anaemia in patients with chronic heart failure. Eur Heart J. 2005;26:2232–7. [DOI] [PubMed] [Google Scholar]
- 55. Grote Beverborg N, Klip IT, Meijers WC, Voors AA, Vegter EL, van der Wal HH, et al. Definition of iron deficiency based on the gold standard of bone marrow iron staining in heart failure patients. Circ Heart Fail. 2018;11:e004519. [DOI] [PubMed] [Google Scholar]
- 56. Nanas JN, Matsouka C, Karageorgopoulos D, Leonti A, Tsolakis E, Drakos SG, et al. Etiology of anemia in patients with advanced heart failure. J Am Coll Cardiol. 2006;48:2485–9. [DOI] [PubMed] [Google Scholar]
- 57. Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Iron‐independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci U S A. 1991;88:4946–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bulvik B, Grinberg L, Eliashar R, Berenshtein E, Chevion MM. Iron, ferritin and proteins of the methionine‐centered redox cycle in young and old rat hearts. Mech Ageing Dev. 2009;130:139–44. [DOI] [PubMed] [Google Scholar]
- 59. Packer M. Role of deranged energy deprivation signaling in the pathogenesis of cardiac and renal disease in states of perceived nutrient overabundance. Circulation. 2020;141:2095–105. [DOI] [PubMed] [Google Scholar]
- 60. Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)‐JQ1 in cancer cells. Cell Death Dis. 2019;10:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cuthbert JJ, Ransome N, Clark AL. Re‐defining iron deficiency in patients with heart failure. Expert Rev Cardiovasc Ther. 2022;20:667–81. [DOI] [PubMed] [Google Scholar]
- 62. Paterek A, Kępska M, Sochanowicz B, Chajduk E, Kołodziejczyk J, Polkowska‐Motrenko H, et al. Beneficial effects of intravenous iron therapy in a rat model of heart failure with preserved systemic iron status but depleted intracellular cardiac stores. Sci Rep. 2018;8:15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chiari MM, Bagnoli R, De Luca PD, Monti M, Rampoldi E, Cunietti E. Influence of acute inflammation on iron and nutritional status indexes in older inpatients. J Am Geriatr Soc. 1995;43:767–71. [DOI] [PubMed] [Google Scholar]
- 64. Ikeda‐Taniguchi M, Takahashi K, Shishido K, Honda H. Total iron binding capacity is a predictor for muscle loss in maintenance hemodialysis patients. Clin Exp Nephrol. 2022;26:583–92. [DOI] [PubMed] [Google Scholar]
- 65. Bross R, Zitterkoph J, Pithia J, Benner D, Rambod M, Kovesdy CP, et al. Association of serum total iron‐binding capacity and its changes over time with nutritional and clinical outcomes in hemodialysis patients. Am J Nephrol. 2009;29:571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stoffel NU, El‐Mallah C, Herter‐Aeberli I, Bissani N, Wehbe N, Obeid O, et al. The effect of central obesity on inflammation, hepcidin, and iron metabolism in young women. Int J Obes (Lond). 2020;44:1291–300. [DOI] [PubMed] [Google Scholar]
- 67. Sierpinski R, Josiak K, Suchocki T, Wojtas‐Polc K, Mazur G, Butrym A, et al. High soluble transferrin receptor in patients with heart failure: a measure of iron deficiency and a strong predictor of mortality. Eur J Heart Fail. 2021;23:919–32. [DOI] [PubMed] [Google Scholar]
- 68. Kozłowska B, Sochanowicz B, Kraj L, Palusińska M, Kołsut P, Szymański Ł, et al. Expression of iron metabolism proteins in patients with chronic heart failure. J Clin Med. 2022;11:837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maeder MT, Khammy O, dos Remedios C, Kaye DM. Myocardial and systemic iron depletion in heart failure implications for anemia accompanying heart failure. J Am Coll Cardiol. 2011;58:474–80. [DOI] [PubMed] [Google Scholar]
- 70. Moliner P, Enjuanes C, Tajes M, Cainzos‐Achirica M, Lupón J, Garay A, et al. Association between norepinephrine levels and abnormal iron status in patients with chronic heart failure: is iron deficiency more than a comorbidity? J Am Heart Assoc. 2019;8:e010887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tajes M, Díez‐López C, Enjuanes C, Moliner P, Ferreiro JL, Garay A, et al. Neurohormonal activation induces intracellular iron deficiency and mitochondrial dysfunction in cardiac cells. Cell Biosci. 2021;11:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, et al. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail. 2017;19:522–30. [DOI] [PubMed] [Google Scholar]
- 73. Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, et al. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail. 2018;20:910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Papalia F, Jouhra F, Amin‐Youssef G, Shah AM, Charles‐Edwards G, Okonko DO. Cardiac energetics in patients with chronic heart failure and iron deficiency: an in‐vivo 31P magnetic resonance spectroscopy study. Eur J Heart Fail. 2022;24:716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang H, Jamieson KL, Grenier J, Nikhanj A, Tang Z, Wang F, et al. Myocardial iron deficiency and mitochondrial dysfunction in advanced heart failure in humans. J Am Heart Assoc. 2022;11:e022853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Savarese G, von Haehling S, Butler J, Cleland JG, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Eur Heart J. 2022. 10.1093/eurheartj/ehac569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Anker SD, Kirwan BA, van Veldhuisen DJ, Filippatos G, Comin‐Colet J, Ruschitzka F, et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron‐deficient heart failure patients: an individual patient data meta‐analysis. Eur J Heart Fail. 2018;20:125–33. [DOI] [PubMed] [Google Scholar]
- 78. Martens P, Dupont M, Dauw J, Nijst P, Herbots L, Dendale P, et al. The effect of intravenous ferric carboxymaltose on cardiac reverse remodelling following cardiac resynchronization therapy – the IRON‐CRT trial. Eur Heart J. 2021;42:4905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ponikowski P, Kirwan BA, Anker SD, McDonagh T, Dorobantu M, Drozdz J, et al.; AFFIRM‐AHF Investigators Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: a multicentre, double‐blind, randomised, controlled trial. Lancet. 2020;396:1895–904. [DOI] [PubMed] [Google Scholar]
- 80. Lewis GD, Malhotra R, Hernandez AF, McNulty SE, Smith A, Felker GM, et al.; NHLBI Heart Failure Clinical Research Network Effect of oral iron repletion on exercise capacity in patients with heart failure with reduced ejection fraction and iron deficiency: the IRONOUT HF randomized clinical trial. JAMA. 2017;317:1958–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cirillo L, Somma C, Allinovi M, Bagalà A, Ferro G, Di Marcantonio E, et al. Ferric carboxymaltose vs. ferrous sulfate for the treatment of anemia in advanced chronic kidney disease: an observational retrospective study and cost analysis. Sci Rep. 2021;11:7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Van Wyck DB, Roppolo M, Martinez CO, Mazey RM, McMurray S. A randomized, controlled trial comparing IV iron sucrose to oral iron in anemic patients with nondialysis‐dependent CKD. Kidney Int. 2005;68:2846–56. [DOI] [PubMed] [Google Scholar]
- 83. Ghanim H, Abuaysheh S, Hejna J, Green K, Batra M, Makdissi A, et al. Dapagliflozin suppresses hepcidin and increases erythropoiesis. J Clin Endocrinol Metab. 2020;105:dgaa057. [DOI] [PubMed] [Google Scholar]
- 84. Thiele K, Rau M, Hartmann NK, Möllmann J, Jankowski J, Böhm M, et al. Effects of empagliflozin on erythropoiesis in patients with type 2 diabetes: data from a randomized, placebo‐controlled study. Diabetes Obes Metab. 2021;23:2814–8. [DOI] [PubMed] [Google Scholar]
- 85. Ferrannini E, Murthy AC, Lee YH, Muscelli E, Weiss S, Ostroff RM, et al. Mechanisms of sodium‐glucose cotransporter 2 inhibition: insights from large‐scale proteomics. Diabetes Care. 2020;43:2183–9. [DOI] [PubMed] [Google Scholar]
- 86. Wallenius K, Kroon T, Hagstedt T, Löfgren L, Sörhede‐Winzell M, Boucher J, et al. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β‐oxidation, and induces ketosis. J Lipid Res. 2022;63:100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wolf P, Fellinger P, Pfleger L, Beiglböck H, Krumpolec P, Barbieri C, et al. Gluconeogenesis, but not glycogenolysis, contributes to the increase in endogenous glucose production by SGLT‐2 inhibition. Diabetes Care. 2021;44:541–8. [DOI] [PubMed] [Google Scholar]
- 88. Chen R, Xu M, Hogg RT, Li J, Little B, Gerard RD, et al. The acetylase/deacetylase couple CREB‐binding protein/sirtuin 1 controls hypoxia‐inducible factor 2 signaling. J Biol Chem. 2012;287:30800–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xin H, Wang M, Tang W, Shen Z, Miao L, Wu W, et al. Hydrogen sulfide attenuates inflammatory hepcidin by reducing IL‐6 secretion and promoting SIRT1‐mediated STAT3 deacetylation. Antioxid Redox Signal. 2016;24:70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Salazar G, Cullen A, Huang J, Zhao Y, Serino A, Hilenski L, et al. SQSTM1/p62 and PPARGC1A/PGC‐1alpha at the interface of autophagy and vascular senescence. Autophagy. 2020;16:1092–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li X, Lozovatsky L, Sukumaran A, Gonzalez L, Jain A, Liu D, et al. NCOA4 is regulated by HIF and mediates mobilization of murine hepatic iron stores after blood loss. Blood. 2020;136:2691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Elrakaybi A, Laubner K, Zhou Q, Hug MJ, Seufert J. Cardiovascular protection by SGLT2 inhibitors – do anti‐inflammatory mechanisms play a role? Mol Metab. 2022;64:101549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Day EA, Ford RJ, Lu JH, Lu R, Lundenberg L, Desjardins EM, et al. The SGLT2 inhibitor canagliflozin suppresses lipid synthesis and interleukin‐1 beta in ApoE deficient mice. Biochem J. 2020;477:2347–61. [DOI] [PubMed] [Google Scholar]
- 94. Wu L, Zhang G, Guo C, Zhao X, Shen D, Yang N. MiR‐128‐3p mediates TNF‐α‐induced inflammatory responses by regulating Sirt1 expression in bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. 2020;521:98–105. [DOI] [PubMed] [Google Scholar]
- 95. Ying Y, Jiang C, Zhang M, Jin J, Ge S, Wang X. Phloretin protects against cardiac damage and remodeling via restoring SIRT1 and anti‐inflammatory effects in the streptozotocin‐induced diabetic mouse model. Aging (Albany NY). 2019;11:2822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Berezovsky B, Báječný M, Frýdlová J, Gurieva I, Rogalsky DW, Přikryl P, et al. Effect of erythropoietin on the expression of murine transferrin receptor 2. Int J Mol Sci. 2021;22:8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Honda H, Kobayashi Y, Onuma S, Shibagaki K, Yuza T, Hirao K, et al. Associations among erythroferrone and biomarkers of erythropoiesis and iron metabolism, and treatment with long‐term erythropoiesis‐stimulating agents in patients on hemodialysis. PLoS One. 2016;11:e0151601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gammella E, Diaz V, Recalcati S, Buratti P, Samaja M, Dey S, et al. Erythropoietin's inhibiting impact on hepcidin expression occurs indirectly. Am J Physiol Regul Integr Comp Physiol. 2015;308:R330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li D, Liu X, Pi W, Zhang Y, Yu L, Xu C, et al. Fisetin attenuates doxorubicin‐induced cardiomyopathy in vivo and in vitro by inhibiting ferroptosis through SIRT1/Nrf2 signaling pathway activation. Front Pharmacol. 2022;12:808480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ma S, Sun L, Wu W, Wu J, Sun Z, Ren J. USP22 protects against myocardial ischemia‐reperfusion injury via the SIRT1‐p53/SLC7A11‐dependent inhibition of ferroptosis‐induced cardiomyocyte death. Front Physiol. 2020;11:551318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lin J, Duan J, Wang Q, Xu S, Zhou S, Yao K. Mitochondrial dynamics and mitophagy in cardiometabolic disease. Front Cardiovasc Med. 2022;9:917135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Thirunavukarasu S, Jex N, Chowdhary A, Hassan IU, Straw S, Craven TP, et al. Empagliflozin treatment is associated with improvements in cardiac energetics and function and reductions in myocardial cellular volume in patients with type 2 diabetes. Diabetes. 2021;70:2810–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. van der Meer P, Grote Beverborg N, Pfeffer MA, Olson K, Anand IS, Westenbrink BD, et al. Hyporesponsiveness to darbepoetin alfa in patients with heart failure and anemia in the RED‐HF study (Reduction of Events by Darbepoetin Alfa in Heart Failure): clinical and prognostic associations. Circ Heart Fail. 2018;11:e004431. [DOI] [PubMed] [Google Scholar]