

Abstract
The thioredoxin (TRX) system is an important contributor to cellular redox balance and regulates cell growth, apoptosis, gene expression, and antioxidant defense in nearly all living cells. Oxidative stress, the imbalance between reactive oxygen species (ROS) and antioxidants, can lead to cell death and tissue damage, thereby contributing to aging and to the development of several diseases, including cardiovascular and allergic diseases, diabetes, and neurological disorders. Targeting its activity is also considered as a promising strategy in the treatment of cancer. Over the past years, immunologists have established an essential function of TRX for activation, proliferation, and responses in T cells, B cells, and macrophages. Upon activation, immune cells rearrange their redox system and activate the TRX pathway to promote proliferation through sustainment of nucleotide biosynthesis, and to support inflammatory responses in myeloid cells by allowing NF‐κB and NLRP3 inflammasome responses. Consequently, targeting the TRX system may therapeutically be exploited to inhibit immune responses in inflammatory conditions. In this review, we summarize recent insights revealing key roles of the TRX pathway in immune cells in health and disease, and lessons learnt for cancer therapy.
Keywords: cancer, cellular redox, immunoregulation, ROS, TRX system
The thioredoxin (TRX) system is a critical contributor to cellular redox homeostasis in all living cells. Here, we review the key roles of TRX for activation, proliferation, and responses in T cells, B cells, and myeloid cells. Further, we conceptualize how this knowledge may be therapeutically exploited for cancer treatment.
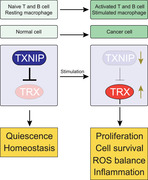
Introduction
Reactive oxygen species (ROS) are highly reactive molecules that contain oxygen, such as hydrogen peroxide and superoxide among others. In addition to exogenous sources (e.g. pollutants, radiation, etc), ROS can also arise endogenously by mitochondrial respiratory chain and by the action of NADPH oxidase (NOX) [1]. Despite their essential role in killing pathogens and regulating cell proliferation and differentiation, exacerbated ROS can induce damage to DNA, proteins, and lipids, thereby contributing to aging and cell death [1, 2, 3]. Aerobic organisms are equipped with a sophisticated network of antioxidant proteins that maintain reduction‐oxidation (redox) homeostasis [4, 5, 6]. Tilting of this redox balance towards oxidation, which is known as “oxidative stress”, has been associated with multiple pathologies, such as cardiovascular diseases [7], cancer [8], diabetes [9], asthma [10], neurological disorders [11], and aging [1].
The thioredoxin (TRX) system is one of the major cellular antioxidant pathways that control redox homeostasis. This system comprises NADPH, TRX reductase (TRXR, encoded by TXNRD), TRX (encoded by TXN) itself, and the negative regulator TRX‐interacting protein (TXNIP, also known as vitamin D3 upregulated protein 1; encoded by the TXNIP gene). TXNIP binds to reduced TRX via intermolecular disulfide interactions and blocks its activity. TRXR is a selenoenzyme that has the unique capacity to utilize reducing equivalents from NADPH generated by the pentose phosphate pathway (PPP) to keep TRX in its reduced state (Fig. 1A). Three different TRXR isoforms have been described in mammals, which differ in their subcellular localization and tissue‐specific expression: TRXR1 is found in the cytoplasm, TRXR2 is localized in mitochondria, and TRXR3 is specifically expressed in testis. Similarly, the two TRX isoforms TRX1 and TRX2 are localized in the cytoplasm and mitochondria, respectively. This specific expression pattern of the components of the TRX system builds up a robust antioxidant system that ubiquitously maintains redox homeostasis [4, 5, 12].
Figure 1.
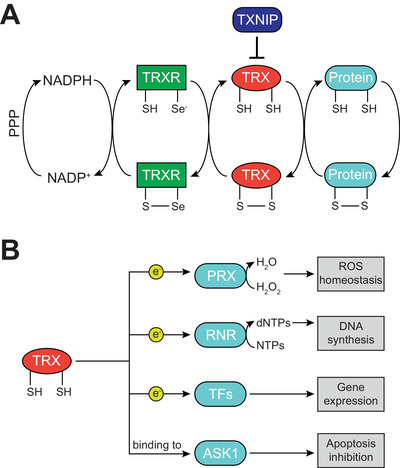
The components of the TRX system and its cellular functions. (A) The pentose phosphate pathway (PPP) produces NADPH, which acts as the most upstream electron donor of the thioredoxin (TRX) system. NADPH keeps TRX reductase (TRXR) in its reduced state, which in turn provides reducing equivalents to TRX. Reduced TRX can ultimately donate electrons to several cellular proteins. The activity of the TRX system is regulated by TRX‐interacting protein (TXNIP), which binds to TRX and inhibits its function (S, sulfur atom in cysteine; Se, selenium atom in selenocysteine). (B) Cellular functions of reduced TRX. By donation of electrons (e−), TRX supports the functionality of cellular peroxiredoxins (PRX), which scavenge hydrogen peroxide (H2O2); of ribonucleotide reductase (RNR), which generates 2’‐deoxyribonucleotides (dNTPs) for DNA synthesis; and of numerous transcription factors (TFs), thus regulating gene expression. Furthermore, by binding to ASK1 and inhibiting its activity, reduced TRX prevents apoptosis.
The TRX system regulates several processes, including gene expression, antioxidant response, apoptosis, and cell proliferation [13]. For instance, TRX modulates the activity of various transcription factors such as NF‐κB, REF1, and HIF1α [14, 15, 16]; it donates reducing equivalents to the antioxidant enzymes peroxiredoxins (PRX) and methionine sulfoxide reductases [17, 18]; it sustains nucleotide biosynthesis by ribonucleotide reductase (RNR) [19]; and it regulates apoptosis by suppressing ASK1 activity [20] (Fig. 1B).
The glutathione (GSH)/glutaredoxin (GRX) system, similarly to the TRX system, utilizes electrons from NADPH and shuttles them to glutathione reductase (GSR), which regenerates reduced GSH from it oxidized form (GSSG). In addition to directly maintain redox balance [21, 22], GSH can in turn provide reducing equivalents to glutaredoxins (GRX) [23]. TRX and GRX shuttle electrons to numerous shared substrates and therefore possess overlapping bioactivities [23]. To which extent, however, TRX and GRX can compensate for each other in vivo is poorly understood and may vary in distinct cell types and environmental conditions.
Over the past decade, by studying conditional knockouts of critical components of these redox systems, immunologists have established their cell‐autonomous roles in development and function of lymphocytes and myeloid cells. These studies implicate how a dysregulated control of the cellular redox state may contribute to immune dysfunction and disease [4]. In this Review, we summarize recent insights revealing the role of the cytosolic TRX system in immune cells and discuss potential therapeutic strategies in pathological conditions.
Immunoregulation by TRX1 and TRXR1
TRX1‐TRXR1 system: An accelerator of T cell proliferation
T cells undergo massive proliferation during thymic development and upon antigen encounter in secondary lymphoid organs. T cell activation is accompanied by the upregulation of cellular redox regulators, including the components of the TRX system TRX1 and TRXR1 [24, 25] (Fig. 2A). Genetic deletion of Txnrd1 in mice, which encodes the key enzyme that recycles oxidized TRX1 into its reduced form, results in impaired expansion of activated T cells during viral infections and other immune responses [25]. Mechanistically, the TRX1 system donates reducing equivalents to RNR, which ultimately reduces ribonucleotides into the corresponding 2’‐deoxyribonucleotides (dNTPs) during DNA biosynthesis at the last step of the PPP [25] (Fig. 2B and C). Although the GSH system can also provide reducing power to RNR and compensate for the absence of TRX1 in other cell types, GRX1 expression is extremely low and not upregulated upon T cell activation [25] (Fig. 2B). Of note, despite the antioxidant function of TRX1, Txnrd1‐deficient T cells do not display increased levels of ROS [25]. Consistent with the key function of the TRX1 system in T cells, a recent in vivo CRISPR‐Cas9 mutagenesis screen identified Txnrd1 as a positive regulator of the antitumor response of CD8+ T cells [26], and another report showed that overexpression of Trx1 improves the antitumor function of T cells [27]. By contrast, the TRX1‐TRXR1 system is largely dispensable for homeostatic maintenance of naïve T cells [25], likely due to their modest requirement of dNTP biosynthesis through the PPP [28].
Figure 2.
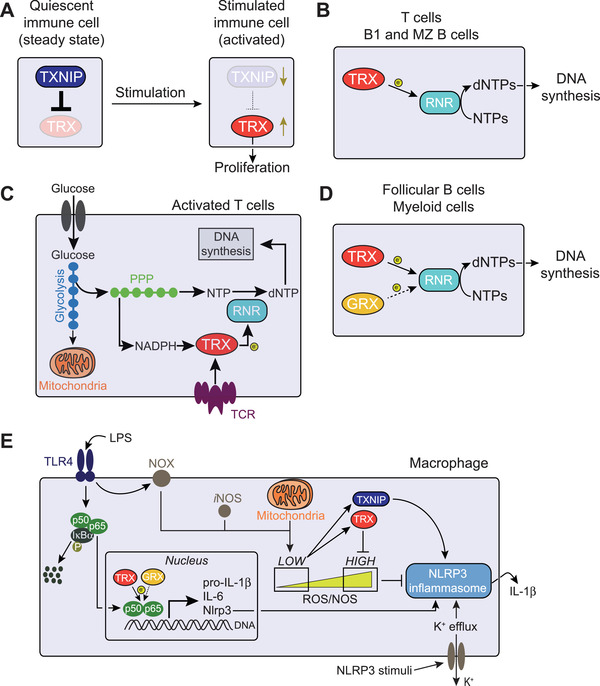
Immunoregulation by the TRX system. (A) Stimulated immune cells activate the thioredoxin (TRX) system by both increasing its transcriptional expression and by repressing its inhibitor thioredoxin‐interacting protein (TXNIP), thus promoting proliferation. (B) In T cells, B1, and marginal zone (MZ) B cells, the TRX system is the only pathway supporting ribonucleotide reductase (RNR)‐mediated reduction (electron [e−] donation) of ribonucleotides (NTPs) into the corresponding 2’‐deoxyribonucleotides (dNTPs) for DNA synthesis. (C) T cell stimulation reprogram cellular metabolism towards increased glycolysis and pentose phosphate pathway (PPP), which generates NADPH and building blocks for DNA synthesis. Concomitantly, upregulated TRX shuttles electrons from NADPH to RNR for dNTP production. (D) In follicular B cells and myeloid cells, both the TRX and the glutaredoxin (GRX) system can sustain DNA biosynthesis, although GRX‐dependent reaction is less efficient (dotted arrow). (E) TRX system‐dependent redox regulation of NLRP3 inflammasome activation. Lipopolysaccharide (LPS) binding to Toll‐like receptor 4 (TLR4) leads to phosphorylation (P) and consequent proteolysis of the NF‐κB inhibitor IκB‐α. This results in the release and translocation of NF‐κB p50 and p65 to the nucleus for transcription of genes encoding pro‐inflammatory cytokines (e.g. pro‐IL‐1β and IL‐6) and NLRP3 itself. TRX, and partially also GRX, positively regulates the binding of NF‐κB to target DNA, promoting transcriptional activity of target genes. NADPH oxidase (NOX) and mitochondrial metabolism generate reactive oxygen species (ROS), and nitric oxide (NO) synthase (iNOS) produces NO. Low ROS levels releases TRX from TXNIP‐mediated inhibition, which then leads to NLRP3 inflammasome activation and IL‐1β secretion. This is achieved by both TRX scavenging excessive ROS that would otherwise prevent inflammasome formation and by TXNIP potentially interacting with NLRP3 and enhancing IL‐1β production.
Naïve T cells are quiescent and predominantly rely on oxidative phosphorylation (OXPHOS) for energy generation. By contrast, activated T cells are metabolically more active and utilize the PPP and aerobic glycolysis, a process that converts glucose into lactate despite sufficient oxygen supply, to meet their bioenergetic demands, including the synthesis of nucleotides, fatty acids and amino acids [25, 28]. Metabolic changes and the TRX1 pathway are tightly interconnected in activated T cells: Shift toward increased PPP upon antigen encounter allows the generation of NADPH, which fuels the TRX1 system that ultimately sustains the PPP by donating reducing power to RNR for nucleotide biosynthesis in activated T cells [25] (Fig. 2C).
TRX1‐TRXR1 system in B cells and compensatory mechanisms
B cell activation also results in increased mitochondrial activity and glycolysis to fuel the PPP [29]. However, deletion of Txnrd1 in B cells does not affect development, homeostasis, germinal center reactions, and antibody responses in mice [30], implying that other compensatory pathways sustain the PPP by providing reducing power to RNR. GRX1 is highly expressed in B cells and indeed fuels dNTP production with reducing equivalents, which may allow proliferation of Txnrd1‐deficient B cells in the bone marrow and in germinal centers [30]. However, GRX1 is unable to fully sustain rapid expansion, and thus Txnrd1‐deficient B cells display a partial proliferative delay that may arise from either a less efficient donation of reducing power to RNR or from the time window required to shift from TRX to GRX utilization [30] (Fig. 2D).
In addition to the TRX system, B cell responses can also relinquish on the ROS scavenging enzymes glutathione peroxidase 1 (GPX1) and GPX4, PRX1‐PRX4, and NF‐E2‐related factor 2 (NRF2) [31, 32], indicating that B cells have a more robust and flexible redox system compared to T cells. B cells express NADPH oxidases and show electron leakage from mitochondrial complex I and III upon cell stimulation leading to ROS; Moreover, proper immunoglobulin folding plasma cells also results in ROS cascade from endoplasmic reticulum [31, 33, 34]. Thus, exposure of B cells to high levels of ROS may have evolutionary selected development of redundant pathways allowing to deal with oxidative stress.
Differential requirement of the TRX1‐TRXR1 system in B1 and MZ B cells
B cells are generally subdivided into two major subsets which differ in ontogeny, homeostasis, and functionality: B2 and B1 B cells. B2 cells, which are often referred to as “classical” B cells, develop in the bone marrow, produce high‐affinity antibodies against foreign antigens and generate immunological memory in secondary lymphoid organs. While B2 cells encompass both follicular and marginal zone (MZ) B cells, the aforementioned features of B2 cells exclusively apply to follicular B cells. MZ B cells functionally differ from follicular B cells and, together with B1 cells, are often referred to as innate‐like lymphocytes because of their capacity to rapidly respond to blood‐borne antigens and to initiate low‐affinity antibody responses [35, 36]. In contrast to follicular B cells, the redox system of innate‐like B cells, including both B1 and MZ B cells, is weaker as it has been presented for T cells in the previous chapter. Txnrd1‐deficient MZ B cells and B1 cells display impaired development, homeostatic maintenance, and antibody responses, since they are unable to engage GRX1 as a compensatory pathway to sustain dNTP production [30] (Fig. 2B). This redox similarity between T cells and innate‐like B cells also further applies to their strict requirement of lipid peroxide‐scavenging enzyme glutathione peroxidase‐4 (GPX4) [32] and of the redox‐sensitive organelle “peroxisomes” [37].
Inflammatory role of the TRX1‐TRXR1 pathway in dendritic cells and macrophages
In contrast to T cells, the TRX1‐TRXR1 pathway is largely dispensable for the development and maintenance of tissue and blood monocytes, dendritic cells, neutrophils, eosinophils, and macrophages in mice due to their flexibility in shifting toward the compensatory GSH/GRX pathway to sustain thiol‐based reactions [38]. Despite being dispensable during homeostatic maintenance, the GSH/GRX pathway is unable to fully compensate for the absence of the TRX1 system in a situation of emergency myelopoiesis driven by endotoxin when proliferation of myeloid precursors bursts [38] (Fig. 2D). As described above, this has also been observed for follicular B cells, in which the TRX1 system is dispensable for homeostasis but strictly required during rapid expansion in the germinal centers upon infection.
In the inflammatory phase, M1 macrophages rearrange their metabolism to support production of pro‐inflammatory cytokines, lipid mediators, and ROS to kill invading pathogens. A shift toward the PPP generates NADPH, the key electron donor that promotes ROS and nitric oxide (NO) production by NOX enzymes and NO synthase (iNOS), respectively [39]. In the resolution phase, however, NADPH donates reducing equivalents to the TRX1‐TRXR1 pathway, which in turn scavenges excessive ROS and helps preventing excessive tissue damage after pathogen clearance (Fig. 2E). Consistently, deletion of Txnrd1 results in ROS overproduction upon TLR stimulation [38]. How exactly NADPH can both promote and inhibit ROS production remains unknown, but it may be explained by highly dynamic metabolic alterations during an immune response, paralleling functional transitions [4, 40].
The transcription factor NF‐κB is a key mediator of inflammatory responses that induces the expression of several pro‐inflammatory cytokines and chemokines after its translocation into the nucleus [41]. Based on the numerous target genes under control, NF‐κB activation is tightly regulated both in the cytoplasm and in the nucleus by distinct mechanisms. TRX1‐dependent redox regulation of NF‐κB has been previously proposed, although its role in inflammation remains somewhat controversial when considering that both activating and inhibitory roles have been reported. Indeed, while generation of cellular ROS result in NF‐κB activation and nuclear translocation, oxidized NF‐κB displays impaired DNA‐binding activity. Similarly, TRX1 has a dual role: While preventing NF‐κB activation by blocking the dissociation and degradation of the IκB‐α inhibitor, TRX1 oxidation promotes NF‐κB binding to the target DNA by the reduction of cysteine 62 in the p50 subunit of NF‐κB [42, 43, 44]. Indeed, TLR activation of Txnrd1‐deficient bone‐marrow‐derived dendritic cells (BMDCs) results in impaired DNA‐binding capacity of the p65 subunit of NF‐κB in the nucleus rather than cytoplasmic activation and nuclear translocation [38]. Enhanced NF‐kB DNA binding and transcriptional activity may be achieved via structural modifications, such as the reduction of disulfide bonds, of NF‐κB itself or other components of the transcriptional complex [38] (Fig. 2E). By contrast, bone‐marrow‐derived macrophages (BMDMs) express high levels of GRX proteins, which take over the function of TRX1 when Txnrd1 is genetically deleted, keep nuclear NF‐κB p65 reduced, and allow normal transcription of pro‐inflammatory target genes [38]. Dendritic cells, tissue‐resident macrophages, and other myeloid cell types express variable levels of GRX proteins in vivo. It remains to be investigated which ones rely strictly on the TRX1‐TRXR1 system similar to BMDCs, and which ones are more flexible and can use both the TRX1 and GRX pathways similar to BMDM.
Inflammasomes are large cytosolic proteins that trigger IL‐1β processing and release [45]. IL‐1β is produced as an inactive precursor (pro‐IL‐1β) after exposure to pathogens or danger signals, and requires a second signal leading to inflammasome assembly to be processed into its active form by caspase‐1 [45]. Several mechanisms have been reported to describe NLRP3 inflammasome activation. In addition to potassium (K+) efflux, which is a common event that is associated with several NLRP3 stimuli, ROS production has been also proposed to promote inflammasome activation [46]. In particular, mitochondrial ROS, oxidized mitochondrial DNA, and compounds that induce mitochondrial outer membrane permeabilization lead to IL‐1β production and secretion [47, 48, 49, 50]. Paradoxically, despite this NLRP3‐promoting function of ROS, a defect of the TRX1 system prevents IL‐1β processing by the NLRP3 inflammasome due to impaired scavenging of excessive ROS (such as hydrogen peroxide) that may oxidize and damage other inflammasome components [38, 42] (Fig. 2E). NLRP3 itself and caspase‐1 can be oxidized and damaged by exacerbated ROS and NO levels [38, 51–53].
Taken together, TRX1 promotes the production of pro‐inflammatory cytokines (e.g., IL‐12, IL‐6, IL‐1β, and TNF‐α) by both enhancing NF‐κB binding to target DNA in the nucleus and allowing proper NLRP3 inflammasome formation in the cytoplasm.
TXNIP, the inhibitor of the TRX1‐TRXR1 pathway
TXNIP as brake of lymphocyte proliferation
TXNIP binds to TRX1 and negatively regulates the activity of the TRX1 system [54]. T cell activation results in rapid TXNIP downregulation, release of TRX1 from inhibition, and donation of electrons to RNR for dNTP production [25, 55] (Fig. 2A). The transcription factor c‐Myc mediates TXNIP repression and thereby contributes to the activation of the TRX1 system upon TCR triggering, in addition to promoting T cell metabolic reprogramming [25]. Consequently, absence of TXNIP results in increased proliferation of effector T cells and germinal center B cell in immune responses [55].
Klein Geltink et al. proposed that CD28 signaling (signal 2) leads to the downregulation of TXNIP, which then translates into the engagement of mitochondrial fatty acid oxidation [56]. Muri et al., however, found that TCR triggering alone (anti‐CD3; signal 1) is sufficient to potently inhibit TXNIP expression in CD8+ T cells, and that CD28 engagement has only a minor contribution at weak TCR engagement (low concentration of anti‐CD3) [55]. This discrepancy may arise from the fact that Klein Geltink et al. only focused on the minor difference between anti‐CD3 alone and anti‐CD3/anti‐CD28 without a comparison to the expression of TXNIP in naïve T cells, thus missing that signal 1 is the main contributor to TXNIP inhibition.
Controversial role of TXNIP in inflammasome activation
TXNIP has originally been suggested as a redox‐sensitive ligand of NLRP3 that links ROS production to NLRP3 inflammasome activation in macrophages [57]. A physical interaction of TXNIP and NLRP3 has also been proposed in pancreatic islets, leading to enhanced IL‐1β production in type 2 diabetes [57, 58]. However, the requirement of TXNIP in NLRP3 inflammasome activation has been questioned in other studies. Txnip‐deficient macrophages display indeed normal NLRP3 activation and IL‐1β production both in vitro and in vivo [38, 59]. In conclusion, despite the controversial role of TXNIP in inflammasome activation, the TRX1‐TRXR1 pathway is key in the production of IL‐1β (Fig. 2E).
The TRX system in cancer and therapeutic approaches
Roles of TRX and TRXR in cancer and therapeutic interventions
Cancer is a complex and heterogeneous disease ranking among the top 10 leading causes of death worldwide. Cancer cells switch metabolism from oxidative phosphorylation to glycolysis (Warburg effect), which fuels the synthesis of anabolic molecules required for accelerated cell proliferation. In addition to metabolic reprogramming, exacerbated ROS production, mainly driven by aberrant electron flow in the mitochondrial electron transport chain and NADPH oxidases, is a hallmark of cancer cells [60]. Rapid cancer cell proliferation induces ROS and oxidation of redox‐sensitive signaling pathways (i.e., PI3K/Akt/mTOR and MAPK) and transcription factors (such as NF‐kB, HIF‐1α, p53) promote cell growth and a pro‐tumorigenic state. However, oxidative stress also induces DNA damage, genome instability, and toxic lipid oxidation resulting in senescence, apoptotic, necrotic, or ferroptotic cell death. To counteract oxidative stress and prevent cytotoxicity, cancer cells adapt to elevated ROS levels by upregulation of antioxidant pathways [61, 62, 63]. It is therefore not surprising that TRX and TRXR levels are increased in numerous types of solid tumors (i.e. lung [64], colorectal [65], pancreatic [66], gastric [67], breast [68, 69], and prostate [70]) as well as hematopoietic malignancies and correlate with poor prognosis [71, 72, 73].
Upregulation of the TRX1 system has been associated with each of the six cancer hallmarks proposed by Hanahan and Weinberg [74]. Beyond scavenging ROS, TRX is a driver of tumor development, propagation, metastasis, angiogenesis, resistance to chemotherapies [73, 75, 76] by regulating transcription factors (such as p53, NF‐κB, and AP1, HIF‐1α), signaling pathways (including p38 MAPK, ASK‐1, JNK, PTEN), and synthesis of DNA building (i.e. RNR) that are all involved in regulation of cell growth and death. Moreover, extracellular activity of TRX can also promote cell growth [73, 75, 76]. Consequently, inhibiting TRX/TRXR activity is a promising tumor therapy.
Several chemotherapeutic drugs have been developed to target the TRX pathway and act mainly by inducing oxidative stress and apoptosis in tumor cells, as reviewed elsewhere [61, 62, 77]. For instance, the compound 1‐methylpropyl 2‐imidazolyl disulfide (PX‐12) inhibits TRX activity through binding to the cysteine 73 residue of TRX. PX‐12 has previously entered clinical trials [62] and has been shown to possess potent antitumor activity against different types of tumors, including lung cancer [78], acute myeloid leukemia [79], and lung colorectal cancer [80] among others. Furthermore, the gold‐based compound auranofin, which is FDA‐approved for the treatment of rheumatoid arthritis [81], targets primarily TRXR (Fig. 3). Based on its capacity to induce apoptosis, auranofin has been repurposed to treat cancer, such as ovarian cancer [82], breast cancer [83], multiple myeloma [84], and chronic lymphocytic leukemia [85, 86]. Despite the availability of numerous drugs that target the TRX system, and some promising effects in preclinical reports, only few compounds went to clinical trials, and they do not inhibit TRX/TRXR specifically. Novel strategies for development of better and more specific drugs have been summarized in excellent reviews recently [61, 62, 77].
Figure 3.
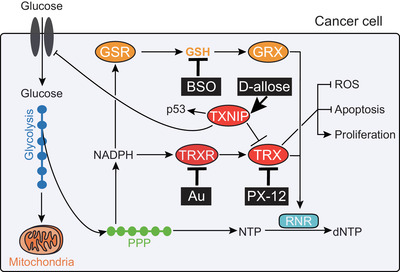
Targeting the TRX system for cancer therapy. Cancer cells adapt to elevated ROS by upregulating cellular antioxidants, including thioredoxin (TRX) and TRX reductase (TRXR), and by downregulating their inhibitor TRX‐interacting protein (TXNIP). The chemotherapeutic drugs auranofin (Au) and PX‐12, which respectively inhibit TRXR and TRX, lead to oxidative stress, reduced 2’‐deoxyribonucleotide (dNTP) biosynthesis and cell death of cancer cells. D‐Allose inhibits cancer cell survival and proliferation by boosting TXNIP expression, which results in the induction of the tumor suppressor p53 and in the inhibition of TRX and of glucose transporter 1 (GLUT1)‐dependent glucose import. Of note, while blockade of TRX or TRXR alone may be sufficient to inhibit T cell leukemia and lymphomas, additional inhibition of the glutathione (GSH)/glutaredoxin (GRX) pathway by the drug buthionine sulfoximine (BSO) may be required to treat B cell malignancies and other tumors due to their redundant functions (e.g. donation of electrons to ribonucleotide reductase [RNR]). Abbreviations: GSR, glutathione reductase; PPP, pentose phosphate pathway; black rectangular boxes indicate chemotherapeutic drugs.
Dual targeting of the TRX and GSH/GRX system
As both the TRX and the GSH/GRX systems regulate cellular redox homeostasis in cancer cells, the two pathways are functionally overlapping and absence or inefficiency of one system can be compensated by the other. Consistently, genetic deletion and pharmacological inhibition of both the TRX and GSH/GRX pathways in vitro and in vivo synergistically induce cancer cell death [87, 88]. However, the efficiency of the therapeutic interference with the two redox systems depends on the type of tumor. Although inhibition of the TRX pathway alone could be exploited to inhibit T cell leukemia and lymphomas based on their exclusive utilization of the TRX pathway for proliferation [25], blockade of both the TRX and GSH/GRX systems may be needed to treat B cell lymphoma, myeloid tumors, sarcoma, mammary tumors and possibly others due to redundancy of the two systems [30, 38, 87–89] (Fig. 3). As an additional level of complexity, other cellular antioxidants, including PRX, GPX, and other components of the GSH pathway, are also upregulated in tumor cells and promote tumorigenesis [90, 91, 92]. Development of drugs that target the catalytically active selenocysteine group present in TRX and GPX, but not the thiol group of cysteine residues present in many proteins and enzymes that are vital for the function of non‐tumor cells will facilitate reaching several goals at once.
Role of TXNIP in cancer
TXNIP acts as a tumor suppressor that is commonly silenced in various human cancers, including breast cancer [93] and hepatocellular carcinoma [94] among others, and exploited by the tumor cells to maintain redox homeostasis [61]. Moreover, in a TRX‐independent manner, TXNIP negatively regulates glucose metabolism by inhibiting the expression of glucose transporter 1 (GLUT1) in cancer cells [95], and it can act as tumor suppressor by directly regulating the induction of p53 [96]. d‐Allose, a sugar that induces expression of TXNIP, exhibits antitumor effects in several cancers [61] (Fig. 3). In this regard, a recent genetic screen identified TXNIP to possess key metabolic gatekeeper functions in B cell acute lymphoblastic cells and proposed that pharmacological TXNIP agonists, such as d‐allose, synergize with glucocorticoids and thus represent a potential therapeutic strategy [97].
In summary, the different components of the TRX system, namely TRX, TRXR, and TXNIP, can all be targeted by chemotherapeutic drugs and offer opportunities for cancer therapy.
Concluding remarks
Recent reports unraveled an essential role of the TRX system for the expansion of effector and memory T cells, and macrophage inflammatory responses. B cells are more flexible and can use both the GSH/GRX and TRX systems for germinal center and antibody responses. Cancer cells upregulate the TRX system to allow rapid growth, alike activated T cells, and avoid damage and death due to oxidative stress. Thus, interference with redox homeostasis is considered the Achilles’ heel of cancer cells and targeting the components of the TRX and GRX systems as a promising therapeutical strategy. The main challenge will be to selectively target the TRX system in tumor cells that rely solely on TRX and to target both the TRX and GSH/GRX systems in tumors with redundant antioxidant activity. At the same time, antitumor immunity and successful immune therapy relies on intact TRX function. Further research is required to define the specific functions of the TRX pathway in CD4+ T cell subsets (such as TH1/TH2/TH17 cells and T regulatory (Treg) cells), natural killer (NK) cells, and in myeloid cell populations including neutrophils, conventional DCs (cDC1/cDC2), tissue‐resident macrophages, and inflammatory versus alternatively activated macrophages. Moreover, it would be of general interest to investigate the expression and function of the TRX system in exhausted T cells (Tex). Since they are known to have reduced proliferative potential and accumulated ROS, Tex cells may potentially display a high TXNIP/TRX ratio. A better understanding may be exploited for therapeutic interventions in cancer and other inflammatory diseases.
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Author contribution
The authors contributed equally to all aspects of the article.
Acknowledgments
We appreciate financial support by research grants from Swiss National Science Foundation (SNF 310030B_182829) and ETH Zürich (ETH 23‐16‐2).
Contributor Information
Jonathan Muri, Email: Jonathan.Muri@irb.usi.ch.
Manfred Kopf, Email: Manfred.Kopf@ethz.ch.
Data availability statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
- 1. Finkel, T. and Holbrook, N. J. , Oxidants, oxidative stress and the biology of ageing. Nature 2000. 408: 239–247. [DOI] [PubMed] [Google Scholar]
- 2. Franchina, D. G. , Dostert, C. and Brenner, D. , Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol. 2018. 39: 489–502. [DOI] [PubMed] [Google Scholar]
- 3. Sena, L. A. and Chandel, N. S. , Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012. 48: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muri, J. and Kopf, M. , Redox regulation of immunometabolism. Nat. Rev. Immunol. 2020. [DOI] [PubMed] [Google Scholar]
- 5. Arner, E. S. , Focus on mammalian thioredoxin reductases–important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009. 1790: 495–526. [DOI] [PubMed] [Google Scholar]
- 6. Kalinina, E. V. , Chernov, N. N. and Novichkova, M. D. , Role of glutathione, glutathione transferase, and glutaredoxin in regulation of redox‐dependent processes. Biochemistry (Mosc) 2014. 79: 1562–1583. [DOI] [PubMed] [Google Scholar]
- 7. Ceriello, A. and Motz, E. , Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler. Thromb. Vasc. Biol. 2004. 24: 816–823. [DOI] [PubMed] [Google Scholar]
- 8. Toyokuni, S. , Okamoto, K. , Yodoi, J. and Hiai, H. , Persistent oxidative stress in cancer. FEBS Lett. 1995. 358: 1–3. [DOI] [PubMed] [Google Scholar]
- 9. Brownlee, M. , Biochemistry and molecular cell biology of diabetic complications. Nature 2001. 414: 813–820. [DOI] [PubMed] [Google Scholar]
- 10. Andreadis, A. A. , Hazen, S. L. , Comhair, S. A. and Erzurum, S. C. , Oxidative and nitrosative events in asthma. Free Radic. Biol. Med. 2003. 35: 213–225. [DOI] [PubMed] [Google Scholar]
- 11. Lyras, L. , Cairns, N. J. , Jenner, A. , Jenner, P. and Halliwell, B. , An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J. Neurochem. 1997. 68: 2061–2069. [DOI] [PubMed] [Google Scholar]
- 12. Mustacich, D. and Powis, G. , Thioredoxin reductase. Biochem. J. 2000. 346: 1–8. [PMC free article] [PubMed] [Google Scholar]
- 13. Arner, E. S. and Holmgren, A. , Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000. 267: 6102–6109. [DOI] [PubMed] [Google Scholar]
- 14. Hayashi, T. , Ueno, Y. and Okamoto, T. , Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J. Biol. Chem. 1993. 268: 11380–11388. [PubMed] [Google Scholar]
- 15. Ueno, M. , Masutani, H. , Arai, R. J. , Yamauchi, A. , Hirota, K. , Sakai, T. , Inamoto, T. et al., Thioredoxin‐dependent redox regulation of p53‐mediated p21 activation. J. Biol. Chem. 1999. 274: 35809–35815. [DOI] [PubMed] [Google Scholar]
- 16. Welsh, S. J. , Bellamy, W. T. , Briehl, M. M. and Powis, G. , The redox protein thioredoxin‐1 (Trx‐1) increases hypoxia‐inducible factor 1alpha protein expression: Trx‐1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002. 62: 5089–5095. [PubMed] [Google Scholar]
- 17. Rhee, S. G. , Chae, H. Z. and Kim, K. , Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005. 38: 1543–1552. [DOI] [PubMed] [Google Scholar]
- 18. Boschi‐Muller, S. and Branlant, G. , Methionine sulfoxide reductase: chemistry, substrate binding, recycling process and oxidase activity. Bioorg. Chem. 2014. 57: 222–230. [DOI] [PubMed] [Google Scholar]
- 19. Holmgren, A. and Sengupta, R. , The use of thiols by ribonucleotide reductase. Free Radic. Biol. Med. 2010. 49: 1617–1628. [DOI] [PubMed] [Google Scholar]
- 20. Matsuzawa, A. and Ichijo, H. , Redox control of cell fate by MAP kinase: physiological roles of ASK1‐MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008. 1780: 1325–1336. [DOI] [PubMed] [Google Scholar]
- 21. Mak, T. W. , Grusdat, M. , Duncan, G. S. , Dostert, C. , Nonnenmacher, Y. , Cox, M. , Binsfeld, C. et al., Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017. 46: 675–689. [DOI] [PubMed] [Google Scholar]
- 22. Franchina, D. G. , Kurniawan, H. , Grusdat, M. , Binsfeld, C. , Guerra, L. , Bonetti, L. , Soriano‐Baguet, L. et al., Glutathione‐dependent redox balance characterizes the distinct metabolic properties of follicular and marginal zone B cells. Nat. Commun. 2022. 13: 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lillig, C. H. and Holmgren, A. , Thioredoxin and related molecules–from biology to health and disease. Antioxid. Redox. Signal. 2007. 9: 25–47. [DOI] [PubMed] [Google Scholar]
- 24. Levring, T. B. , Kongsbak, M. , Rode, A. K. , Woetmann, A. , Odum, N. , Bonefeld, C. M. and Geisler, C. , Human CD4+ T cells require exogenous cystine for glutathione and DNA synthesis. Oncotarget 2015. 6: 21853–21864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muri, J. , Heer, S. , Matsushita, M. , Pohlmeier, L. , Tortola, L. , Fuhrer, T. , Conrad, M. et al., The thioredoxin‐1 system is essential for fueling DNA synthesis during T‐cell metabolic reprogramming and proliferation. Nat. Commun. 2018. 9: 1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wei, J. , Long, L. , Zheng, W. , Dhungana, Y. , Lim, S. A. , Guy, C. , Wang, Y. et al., Targeting REGNASE‐1 programs long‐lived effector T cells for cancer therapy. Nature 2019. 576: 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chakraborty, P. , Chatterjee, S. , Kesarwani, P. , Thyagarajan, K. , Iamsawat, S. , Dalheim, A. , Nguyen, H. et al., Thioredoxin‐1 improves the immunometabolic phenotype of antitumor T cells. J. Biol. Chem. 2019. 294: 9198–9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang, R. , Dillon, C. P. , Shi, L. Z. , Milasta, S. , Carter, R. , Finkelstein, D. , McCormick, L. L. et al., The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011. 35: 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akkaya, M. , Traba, J. , Roesler, A. S. , Miozzo, P. , Akkaya, B. , Theall, B. P. , Sohn, H. et al., Second signals rescue B cells from activation‐induced mitochondrial dysfunction and death. Nat. Immunol. 2018. 19: 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muri, J. , Thut, H. , Heer, S. , Krueger, C. C. , Bornkamm, G. W. , Bachmann, M. F. and Kopf, M. , The thioredoxin‐1 and glutathione/glutaredoxin‐1 systems redundantly fuel murine B‐cell development and responses. Eur. J. Immunol. 2019. [DOI] [PubMed] [Google Scholar]
- 31. Bertolotti, M. , Yim, S. H. , Garcia‐Manteiga, J. M. , Masciarelli, S. , Kim, Y. J. , Kang, M. H. , Iuchi, Y. et al., B‐ to plasma‐cell terminal differentiation entails oxidative stress and profound reshaping of the antioxidant responses. Antioxid. Redox. Signal. 2010. 13: 1133–1144. [DOI] [PubMed] [Google Scholar]
- 32. Muri, J. , Thut, H. , Bornkamm, G. W. and Kopf, M. , B1 and marginal zone B cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 2019. 29: 2731–2744.e2734. [DOI] [PubMed] [Google Scholar]
- 33. Aronov, M. and Tirosh, B. , Metabolic control of plasma cell differentiation‐ what we know and what we don't know. J. Clin. Immunol. 2016. 36: 12–17. [DOI] [PubMed] [Google Scholar]
- 34. Vene, R. , Delfino, L. , Castellani, P. , Balza, E. , Bertolotti, M. , Sitia, R. and Rubartelli, A. , Redox remodeling allows and controls B‐cell activation and differentiation. Antioxid. Redox. Signal. 2010. 13: 1145–1155. [DOI] [PubMed] [Google Scholar]
- 35. Baumgarth, N. , The double life of a B‐1 cell: self‐reactivity selects for protective effector functions. Nat. Rev. Immunol. 2011. 11: 34–46. [DOI] [PubMed] [Google Scholar]
- 36. Pillai, S. and Cariappa, A. , The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 2009. 9: 767–777. [DOI] [PubMed] [Google Scholar]
- 37. Muri, J. , Corak, B. , Matsushita, M. , Baes, M. and Kopf, M. , Peroxisomes are critical for the development and maintenance of B1 and marginal zone B cells but dispensable for follicular B cells and T cells. J. Immunol. 2022. 208: 839–850. [DOI] [PubMed] [Google Scholar]
- 38. Muri, J. , Thut, H. , Feng, Q. and Kopf, M. , Thioredoxin‐1 distinctly promotes NF‐kappaB target DNA binding and NLRP3 inflammasome activation independently of Txnip. Elife 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jha, A. K. , Huang, S. C. , Sergushichev, A. , Lampropoulou, V. , Ivanova, Y. , Loginicheva, E. , Chmielewski, K. et al., Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015. 42: 419–430. [DOI] [PubMed] [Google Scholar]
- 40. Seim, G. L. and Fan, J. , A matter of time: temporal structure and functional relevance of macrophage metabolic rewiring. Trends Endocrinol. Metab. 2022. 33: 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu, T. , Zhang, L. , Joo, D. and Sun, S. C. , NF‐kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Isakov, E. , Weisman‐Shomer, P. and Benhar, M. , Suppression of the pro‐inflammatory NLRP3/interleukin‐1beta pathway in macrophages by the thioredoxin reductase inhibitor auranofin. Biochim. Biophys. Acta 2014. 1840: 3153–3161. [DOI] [PubMed] [Google Scholar]
- 43. Kabe, Y. , Ando, K. , Hirao, S. , Yoshida, M. and Handa, H. , Redox regulation of NF‐kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox. Signal. 2005. 7: 395–403. [DOI] [PubMed] [Google Scholar]
- 44. Morgan, M. J. and Liu, Z. G. , Crosstalk of reactive oxygen species and NF‐kappaB signaling. Cell Res. 2011. 21: 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martinon, F. , Petrilli, V. , Mayor, A. , Tardivel, A. and Tschopp, J. , Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 2006. 440: 237–241. [DOI] [PubMed] [Google Scholar]
- 46. Tschopp, J. and Schroder, K. , NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010. 10: 210–215. [DOI] [PubMed] [Google Scholar]
- 47. Zhou, R. , Yazdi, A. S. , Menu, P. and Tschopp, J. , A role for mitochondria in NLRP3 inflammasome activation. Nature 2011. 469: 221–225. [DOI] [PubMed] [Google Scholar]
- 48. Shimada, K. , Crother, T. R. , Karlin, J. , Dagvadorj, J. , Chiba, N. , Chen, S. , Ramanujan, V. K. et al., Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012. 36: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Muri, J. , Feng, Q. , Wolleb, H. , Shamshiev, A. , Ebner, C. , Tortola, L. , Broz, P. et al., Cyclopentenone prostaglandins and structurally related oxidized lipid species instigate and share distinct pro‐ and anti‐inflammatory pathways. Cell Rep. 2020. 30: 4399–4417.e4397. [DOI] [PubMed] [Google Scholar]
- 50. Muri, J. , Wolleb, H. , Broz, P. , Carreira, E. M. and Kopf, M. , Electrophilic Nrf2 activators and itaconate inhibit inflammation at low dose and promote IL‐1beta production and inflammatory apoptosis at high dose. Redox. Biol. 2020. 36: 101647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meissner, F. , Molawi, K. and Zychlinsky, A. , Superoxide dismutase 1 regulates caspase‐1 and endotoxic shock. Nat. Immunol. 2008. 9: 866–872. [DOI] [PubMed] [Google Scholar]
- 52. Kim, Y. M. , Talanian, R. V. , Li, J. and Billiar, T. R. , Nitric oxide prevents IL‐1beta and IFN‐gamma‐inducing factor (IL‐18) release from macrophages by inhibiting caspase‐1 (IL‐1beta‐converting enzyme). J. Immunol. 1998. 161: 4122–4128. [PubMed] [Google Scholar]
- 53. Mishra, B. B. , Rathinam, V. A. , Martens, G. W. , Martinot, A. J. , Kornfeld, H. , Fitzgerald, K. A. and Sassetti, C. M. , Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome‐dependent processing of IL‐1beta. Nat. Immunol. 2013. 14: 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hwang, J. , Suh, H. W. , Jeon, Y. H. , Hwang, E. , Nguyen, L. T. , Yeom, J. , Lee, S. G. et al., The structural basis for the negative regulation of thioredoxin by thioredoxin‐interacting protein. Nat. Commun. 2014. 5: 2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Muri, J. , Thut, H. and Kopf, M. , The thioredoxin‐1 inhibitor Txnip restrains effector T‐cell and germinal center B‐cell expansion. Eur. J. Immunol. 2020. [DOI] [PubMed] [Google Scholar]
- 56. Klein Geltink, R. I. , O'Sullivan, D. , Corrado, M. , Bremser, A. , Buck, M. D. , Buescher, J. M. , Firat, E. et al., Mitochondrial priming by CD28. Cell 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou, R. , Tardivel, A. , Thorens, B. , Choi, I. and Tschopp, J. , Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010. 11: 136–140. [DOI] [PubMed] [Google Scholar]
- 58. Oslowski, C. M. , Hara, T. , O'Sullivan‐Murphy, B. , Kanekura, K. , Lu, S. , Hara, M. , Ishigaki, S. et al., Thioredoxin‐interacting protein mediates ER stress‐induced beta cell death through initiation of the inflammasome. Cell Metab. 2012. 16: 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Masters, S. L. , Dunne, A. , Subramanian, S. L. , Hull, R. L. , Tannahill, G. M. , Sharp, F. A. , Becker, C. et al., Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL‐1beta in type 2 diabetes. Nat. Immunol. 2010. 11: 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Raimondi, V. , Ciccarese, F. and Ciminale, V. , Oncogenic pathways and the electron transport chain: a dangeROS liaison. Br. J. Cancer 2020. 122: 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jia, J. J. , Geng, W. S. , Wang, Z. Q. , Chen, L. and Zeng, X. S. , The role of thioredoxin system in cancer: strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019. 84: 453–470. [DOI] [PubMed] [Google Scholar]
- 62. Jovanovic, M. , Podolski‐Renic, A. , Krasavin, M. and Pesic, M. , The role of the thioredoxin detoxification system in cancer progression and resistance. Front. Mol. Biosci. 2022. 9: 883297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Franchina, D. G. , Grusdat, M. and Brenner, D. , B‐cell metabolic remodeling and cancer. Trends Cancer 2018. 4: 138–150. [DOI] [PubMed] [Google Scholar]
- 64. Gasdaska, P. Y. , Oblong, J. E. , Cotgreave, I. A. and Powis, G. , The predicted amino acid sequence of human thioredoxin is identical to that of the autocrine growth factor human adult T‐cell derived factor (ADF): thioredoxin mRNA is elevated in some human tumors. Biochim. Biophys. Acta 1994. 1218: 292–296. [DOI] [PubMed] [Google Scholar]
- 65. Raffel, J. , Bhattacharyya, A. K. , Gallegos, A. , Cui, H. , Einspahr, J. G. , Alberts, D. S. and Powis, G. , Increased expression of thioredoxin‐1 in human colorectal cancer is associated with decreased patient survival. J. Lab. Clin. Med. 2003. 142: 46–51. [DOI] [PubMed] [Google Scholar]
- 66. Nakamura, H. , Bai, J. , Nishinaka, Y. , Ueda, S. , Sasada, T. , Ohshio, G. , Imamura, M. et al., Expression of thioredoxin and glutaredoxin, redox‐regulating proteins, in pancreatic cancer. Cancer Detect. Prev. 2000. 24: 53–60. [PubMed] [Google Scholar]
- 67. Lim, J. Y. , Yoon, S. O. , Hong, S. W. , Kim, J. W. , Choi, S. H. and Cho, J. Y. , Thioredoxin and thioredoxin‐interacting protein as prognostic markers for gastric cancer recurrence. World J. Gastroenterol. 2012. 18: 5581–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Park, B. J. , Cha, M. K. and Kim, I. H. , Thioredoxin 1 as a serum marker for breast cancer and its use in combination with CEA or CA15‐3 for improving the sensitivity of breast cancer diagnoses. BMC Res. Notes 2014. 7: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bhatia, M. , McGrath, K. L. , Di Trapani, G. , Charoentong, P. , Shah, F. , King, M. M. , Clarke, F. M. , et al. The thioredoxin system in breast cancer cell invasion and migration. Redox. Biol. 2016. 8: 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Samaranayake, G. J. , Troccoli, C. I. , Huynh, M. , Lyles, R. D. Z. , Kage, K. , Win, A. , Lakshmanan, V. et al., Thioredoxin‐1 protects against androgen receptor‐induced redox vulnerability in castration‐resistant prostate cancer. Nat. Commun. 2017. 8: 1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li, C. , Thompson, M. A. , Tamayo, A. T. , Zuo, Z. , Lee, J. , Vega, F. , Ford, R. J. , et al. Over‐expression of Thioredoxin‐1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B‐cell lymphoma. Oncotarget 2012. 3: 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shao, L. , Diccianni, M. B. , Tanaka, T. , Gribi, R. , Yu, A. L. , Pullen, J. D. , Camitta, B. M. , et al. Thioredoxin expression in primary T‐cell acute lymphoblastic leukemia and its therapeutic implication. Cancer Res. 2001. 61: 7333–7338. [PubMed] [Google Scholar]
- 73. Arner, E. S. and Holmgren, A. , The thioredoxin system in cancer. Semin. Cancer Biol. 2006. 16: 420–426. [DOI] [PubMed] [Google Scholar]
- 74. Hanahan, D. and Weinberg, R. A. , The hallmarks of cancer. Cell 2000. 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 75. Karlenius, T. C. and Tonissen, K. F. , Thioredoxin and cancer: a role for thioredoxin in all states of tumor oxygenation. Cancers (Basel) 2010. 2: 209–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Powis, G. and Kirkpatrick, D. L. , Thioredoxin signaling as a target for cancer therapy. Curr. Opin. Pharmacol. 2007. 7: 392–397. [DOI] [PubMed] [Google Scholar]
- 77. Zhang, J. , Li, X. , Han, X. , Liu, R. and Fang, J. , Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 2017. 38: 794–808. [DOI] [PubMed] [Google Scholar]
- 78. You, B. R. , Shin, H. R. and Park, W. H. , PX‐12 inhibits the growth of A549 lung cancer cells via G2/M phase arrest and ROS‐dependent apoptosis. Int. J. Oncol. 2014. 44: 301–308. [DOI] [PubMed] [Google Scholar]
- 79. Tan, Y. , Bi, L. , Zhang, P. , Wang, F. , Lin, F. , Ni, W. , Wu, J. , et al. Thioredoxin‐1 inhibitor PX‐12 induces human acute myeloid leukemia cell apoptosis and enhances the sensitivity of cells to arsenic trioxide. Int. J. Clin. Exp. Pathol. 2014. 7: 4765–4773. [PMC free article] [PubMed] [Google Scholar]
- 80. Wang, F. , Lin, F. , Zhang, P. , Ni, W. , Bi, L. , Wu, J. and Jiang, L. , Thioredoxin‐1 inhibitor, 1‐methylpropyl 2‐imidazolyl disulfide, inhibits the growth, migration and invasion of colorectal cancer cell lines. Oncol. Rep. 2015. 33: 967–973. [DOI] [PubMed] [Google Scholar]
- 81. Roder, C. and Thomson, M. J. , Auranofin: repurposing an old drug for a golden new age. Drugs R. D. 2015. 15: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Marzano, C. , Gandin, V. , Folda, A. , Scutari, G. , Bindoli, A. and Rigobello, M. P. , Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin‐resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007. 42: 872–881. [DOI] [PubMed] [Google Scholar]
- 83. Kim, N. H. , Park, H. J. , Oh, M. K. and Kim, I. S. , Antiproliferative effect of gold(I) compound auranofin through inhibition of STAT3 and telomerase activity in MDA‐MB 231 human breast cancer cells. BMB Rep. 2013. 46: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nakaya, A. , Sagawa, M. , Muto, A. , Uchida, H. , Ikeda, Y. and Kizaki, M. , The gold compound auranofin induces apoptosis of human multiple myeloma cells through both down‐regulation of STAT3 and inhibition of NF‐kappaB activity. Leuk. Res. 2011. 35: 243–249. [DOI] [PubMed] [Google Scholar]
- 85. Fiskus, W. , Saba, N. , Shen, M. , Ghias, M. , Liu, J. , Gupta, S. D. , Chauhan, L. et al., Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014. 74: 2520–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fidyt, K. , Pastorczak, A. , Goral, A. , Szczygiel, K. , Fendler, W. , Muchowicz, A. , Bartlomiejczyk, M. A. et al., Targeting the thioredoxin system as a novel strategy against B‐cell acute lymphoblastic leukemia. Mol. Oncol. 2019. 13: 1180–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Harris, I. S. , Treloar, A. E. , Inoue, S. , Sasaki, M. , Gorrini, C. , Lee, K. C. , Yung, K. Y. et al., Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015. 27: 211–222. [DOI] [PubMed] [Google Scholar]
- 88. Mandal, P. K. , Schneider, M. , Kolle, P. , Kuhlencordt, P. , Forster, H. , Beck, H. , Bornkamm, G. W. , et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res. 2010. 70: 9505–9514. [DOI] [PubMed] [Google Scholar]
- 89. Kiebala, M. , Skalska, J. , Casulo, C. , Brookes, P. S. , Peterson, D. R. , Hilchey, S. P. , Dai, Y. et al., Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: a novel synergistic therapeutic approach. Exp. Hematol. 2015. 43: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kinowaki, Y. , Kurata, M. , Ishibashi, S. , Ikeda, M. , Tatsuzawa, A. , Yamamoto, M. , Miura, O. et al., Glutathione peroxidase 4 overexpression inhibits ROS‐induced cell death in diffuse large B‐cell lymphoma. Lab. Invest. 2018. 98: 609–619. [DOI] [PubMed] [Google Scholar]
- 91. Dai, L. , Cao, Y. , Chen, Y. , Kaleeba, J. A. , Zabaleta, J. and Qin, Z. , Genomic analysis of xCT‐mediated regulatory network: Identification of novel targets against AIDS‐associated lymphoma. Oncotarget 2015. 6: 12710–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Trzeciecka, A. , Klossowski, S. , Bajor, M. , Zagozdzon, R. , Gaj, P. , Muchowicz, A. , Malinowska, A. et al., Dimeric peroxiredoxins are druggable targets in human Burkitt lymphoma. Oncotarget 2016. 7: 1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cadenas, C. , Franckenstein, D. , Schmidt, M. , Gehrmann, M. , Hermes, M. , Geppert, B. , Schormann, W. et al., Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010. 12: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kwon, H. J. , Won, Y. S. , Suh, H. W. , Jeon, J. H. , Shao, Y. , Yoon, S. R. , Chung, J. W. et al., Vitamin D3 upregulated protein 1 suppresses TNF‐alpha‐induced NF‐kappaB activation in hepatocarcinogenesis. J. Immunol. 2010. 185: 3980–3989. [DOI] [PubMed] [Google Scholar]
- 95. Wu, N. , Zheng, B. , Shaywitz, A. , Dagon, Y. , Tower, C. , Bellinger, G. , Shen, C. H. et al., AMPK‐dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013. 49: 1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jung, H. , Kim, M. J. , Kim, D. O. , Kim, W. S. , Yoon, S. J. , Park, Y. J. , Yoon, S. R. et al., TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 2013. 18: 75–85. [DOI] [PubMed] [Google Scholar]
- 97. Chan, L. N. , Chen, Z. , Braas, D. , Lee, J. W. , Xiao, G. , Geng, H. , Cosgun, K. N. et al., Metabolic gatekeeper function of B‐lymphoid transcription factors. Nature 2017. 542: 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.