

Abstract
Objective
To assess the efficacy and safety of deucravacitinib, an oral, selective, allosteric inhibitor of TYK2, in a phase II trial in adult patients with active systemic lupus erythematosus (SLE).
Methods
Adults with active SLE were enrolled from 162 sites in 17 countries. Patients (n = 363) were randomized 1:1:1:1 to receive deucravacitinib 3 mg twice daily, 6 mg twice daily, 12 mg once daily, or placebo. The primary end point was SLE Responder Index 4 (SRI‐4) response at week 32. Secondary outcomes assessed at week 48 included SRI‐4, British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) response, Cutaneous Lupus Erythematosus Disease Area and Severity Index 50 (CLASI‐50), Lupus Low Disease Activity State (LLDAS), and improvements in active (swollen plus tender), swollen, and tender joint counts.
Results
At week 32, the percentage of patients achieving SRI‐4 response was 34% with placebo compared to 58% with deucravacitinib 3 mg twice daily (odds ratio [OR] 2.8 [95% confidence interval (95% CI) 1.5, 5.1]; P < 0.001 versus placebo), 50% with 6 mg twice daily (OR 1.9 [95% CI 1.0, 3.4]; P = 0.02 versus placebo), and 45% with 12 mg once daily (OR 1.6 [95% CI 0.8, 2.9]; nominal P = 0.08 versus placebo). Response rates were higher with deucravacitinib treatment for BICLA, CLASI‐50, LLDAS, and joint counts compared to placebo. Rates of adverse events were similar across groups, except higher rates of infections and cutaneous events, including rash and acne, with deucravacitinib treatment. Rates of serious adverse events were comparable, with no deaths, opportunistic infections, tuberculosis infections, major adverse cardiovascular events, or thrombotic events reported.
Conclusion
Deucravacitinib treatment elicited higher response rates for SRI‐4 and other end points compared with placebo, with an acceptable safety profile, in adult patients with active SLE.

INTRODUCTION
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by the presence of antinuclear autoantibodies and diverse clinical manifestations (1). Many patients with SLE do not reach therapeutic goals (2, 3), and uncontrolled SLE leads to end‐organ damage and an increased risk of premature death (1, 3, 4). Furthermore, current therapies are associated with undesirable side effects (1, 5, 6). Despite expanding treatment options, including targeted therapies (7, 8, 9), an unmet need remains for novel therapies that effectively control symptoms and modify the disease course.
TYK2 is an intracellular kinase that mediates signaling of key cytokines involved in the pathogenesis of SLE, including type I interferons (IFNs) and interleukin‐10 (IL‐10), IL‐12, and IL‐23 (10, 11, 12). A biologic agent targeting the type I IFN receptor has been approved in SLE (7, 13). Deucravacitinib is an oral, selective, allosteric inhibitor of TYK2 that binds the regulatory domain and locks the enzyme in an inactive state (10), distinguishing it from inhibitors of JAK1, JAK2, and/or JAK3 that bind the highly conserved active domains. Previous studies in human cellular assays have demonstrated that deucravacitinib is a potent inhibitor of TYK2 activation and downstream signaling mediated by type I IFN, IL‐10, IL‐12, and IL‐23 (10). Deucravacitinib is approved by the US Food and Drug Administration for the treatment of adults with moderate‐to‐severe plaque psoriasis who are candidates for systemic therapy or phototherapy (14). The approval was based on 2 pivotal phase III trials in patients with plaque psoriasis that demonstrated robust efficacy and an acceptable safety and tolerability profile (15, 16). Deucravacitinib was also efficacious in a phase II trial in psoriatic arthritis (17) with no reported opportunistic infections or laboratory abnormalities characteristic of treatment with JAK1, JAK2, and/or JAK3 inhibitors.
Based on the mechanism of action of deucravacitinib, this phase II trial (PAISLEY) was designed to evaluate the efficacy and safety of deucravacitinib in adult patients with active SLE. Dose selection was based on ability to suppress type I IFN– and IL‐23–mediated pathways in earlier clinical trials (18, 19), and 3 dosages were chosen for assessment of efficacy and safety.
PATIENTS AND METHODS
Trial design and participants
This 48‐week, multicenter, randomized, double‐blind, placebo‐controlled, phase II trial enrolled patients at 162 sites in 17 countries (ClinicalTrials.gov identifier NCT03252587; EudraCT database no. 2017‐001203‐79). The trial included patients 18–75 years of age, inclusive, at the time of screening. The eligibility criteria used for this trial were as follows: 1) met the Systemic Lupus International Collaborating Clinics classification criteria for SLE (20); 2) had at least 1 positive test for antinuclear antibodies, anti–double‐stranded DNA antibodies, or anti‐Sm antibodies, as determined by a central laboratory; 3) had a SLE Disease Activity Index 2000 (SLEDAI‐2K; a 24‐item weighted score of lupus activity) (21, 22) score ≥6; and 4) had at least 1 2004 British Isles Lupus Assessment Group (BILAG) grade A (very active disease) or at least 2 BILAG grade B (moderate disease activity) manifestations from the musculoskeletal or mucocutaneous domains (23). Patients were required to be receiving background therapy with at least 1 antimalarial or immunosuppressant drug and were permitted to be receiving background glucocorticoid therapy with up to 30 mg/day prednisolone or equivalent (see Supplementary Methods available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/art.42391). The trial excluded patients with drug‐induced lupus erythematosus; active, severe lupus nephritis; active neuropsychiatric SLE; a history of herpes zoster, herpes simplex, or influenza infection within 12 weeks before randomization; or a history of disseminated or complicated herpes zoster infection.
Eligible patients were randomized 1:1:1:1 to receive oral deucravacitinib (3 mg twice daily, 6 mg twice daily, or 12 mg once daily) or placebo for 48 weeks (Supplementary Figure 1, available at https://onlinelibrary.wiley.com/doi/art.42391). Randomization was stratified by screening background glucocorticoid dosage (≥10 mg/day versus <10 mg/day), SLEDAI‐2K score (≥10 versus <10), and geographic region (North America, Latin America, Japan, rest of world).
For patients receiving >7.5 mg/day prednisone or equivalent, glucocorticoid tapering to ≤7.5 mg/day was required from week 8 to week 20, and doses then had to remain stable from week 20 to week 32. Further tapering was optional from week 32 to week 40, with a stable dose required from week 40 to week 48.
Ethics approval and trial oversight
This trial was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines for Good Clinical Practice, and local regulations. An institutional review board or independent ethics committee at each site approved the protocol, the consent form, and any other written information provided to patients. All patients provided written informed consent before trial entry. An independent data monitoring committee assessed safety and efficacy data throughout the trial. The trial was sponsored by Bristol Myers Squibb, which designed the trial; all authors vouch for the completeness and accuracy of the data and analyses, and reporting of adverse events.
Trial end points
The primary end point, evaluated at week 32, was the proportion of patients meeting the SLE Responder Index 4 (SRI‐4), defined as meeting all of the following criteria: a ≥4‐point reduction from baseline in SLEDAI‐2K score, no new disease activity measured by a new BILAG grade A or >1 BILAG grade B score, and no worsening (an increase of <0.3 points from baseline) in the physician global assessment of disease activity (24).
Secondary efficacy end points, evaluated at week 48, were the proportion of patients achieving the following 5 end points: 1) SRI‐4 response; 2) BILAG‐based Composite Lupus Assessment (BICLA) response (improvement in all organ systems with BILAG grade A or B at baseline, no new BILAG A, ≤1 new BILAG B, no increase from baseline in SLEDAI‐2K score, no increase ≥0.3 points from baseline in physician global assessment score, and no discontinuation of trial medication or use of restricted medications beyond the protocol‐allowed threshold) (25); 3) Lupus Low Disease Activity State (LLDAS) response (defined as SLEDAI‐2K score ≤4, with no activity in major organ systems and no hemolytic anemia or gastrointestinal activity as assessed by BILAG; no new or worsening individual BILAG parameters; physician global assessment score ≤1; glucocorticoid dose ≤7.5 mg/day; and stable background doses of immunosuppressive drugs and approved biologics) (26); 4) Cutaneous Lupus Erythematosus Disease Area and Severity Index 50 (CLASI‐50) response (decrease of ≥50% from baseline CLASI activity score, a measure of skin disease activity with a score range of 0 to 70) (27) in patients with a baseline CLASI activity score ≥10; and 5) change from baseline in the 40‐joint count for tender plus swollen (active), swollen, and tender joints.
In a protocol amendment prior to trial data unblinding, the time point for assessing secondary end points was changed from week 32 to week 48 to evaluate the durability of response, and these end points were moved into the multiplicity‐controlled hierarchy.
Prespecified exploratory end points analyzed at week 48 included the percent of patients achieving Joint Count‐50 (a decrease in active joint count of ≥50% from baseline in patients with ≥6 active joints at baseline) (28); time to SRI‐4 response; and change from baseline in mean complement (C3, C4) and anti–double‐stranded DNA levels. Change from baseline in type I IFN gene signature in patients’ whole blood was determined by a central laboratory (DxTerity) using an analytically validated 5‐gene (MX1, HERC5, IFIT1, RSAD2, EIF2AK2) chemical ligation–dependent probe amplification–based test (29).
Safety assessments included adverse event reporting, physical examination, vital sign measurements, electrocardiography, and clinical laboratory parameter evaluations, and were conducted periodically throughout the trial as described in the trial protocol.
Statistical analysis
A sample size of 90 patients per treatment group provided 84% power to detect a treatment difference of 20% in SRI‐4 response rates between the deucravacitinib twice daily treatment groups and placebo, assuming a 30% response rate in the placebo group. Due to unequal allocation of alpha (Supplementary Figure 2, available at https://onlinelibrary.wiley.com/doi/art.42391) to the different dosage groups, this same sample size provided 66% power to detect a treatment difference of 20% in SRI‐4 response rates between the deucravacitinib 12 mg once daily treatment group and placebo. The power determination by a chi‐square test was based on the assumption of a placebo response rate of 30%, as mentioned above.
All analyses were conducted in the modified intention‐to‐treat population, which included all patients who were randomized and received at least 1 dose of study treatment. The primary efficacy analysis used a logistic regression model with treatment group (4 levels) and stratification factors (background glucocorticoid dosage, SLEDAI‐2K score, geographic region) as covariates, and nonresponder imputation for missing values or subjects who took prespecified prohibited medications. There were several rules for nonresponder imputation: 1) patients who were lost to follow‐up or discontinued the study early were considered nonresponders from that point forward; 2) patients who were taking prohibited medications were imputed as nonresponders, with prohibited medications defined as glucocorticoid dosages ≥7.5 mg/day prednisone or equivalent after week 20, glucocorticoid burst/rescue (increase in dose) above the protocol‐defined maximum dosage of 40 mg/day prednisone equivalent, or >1 glucocorticoid burst or any glucocorticoid burst for treatment of SLE after week 8; and 3) any other increase in background medications (see Supplementary Methods, https://onlinelibrary.wiley.com/doi/art.42391).
The odds ratios (ORs) between each deucravacitinib treatment group and placebo, and differences between each deucravacitinib dose group, were estimated along with their corresponding 95% confidence intervals (95% CIs). Similar analyses were performed for the binary secondary and exploratory efficacy end points. Continuous secondary end points were analyzed using a mixed model for repeated measures. The adjusted mean changes and corresponding 95% CIs were calculated for the difference between each treatment group and placebo at each specified visit.
The trial had an overall probability of a one‐sided Type I error (alpha value) of 0.05. To control the overall Type I error rate, testing of the primary and secondary end points for the 3 treatment groups versus placebo was split into 2 branches, 1 branch for the deucravacitinib 12 mg once daily dosage group versus placebo and 1 branch for the 2 twice daily dosage groups versus placebo, with the 6 mg twice daily group at the top of the branch (Supplementary Figure 2, https://onlinelibrary.wiley.com/doi/art.42391). The Type I error rate was allocated unequally between the dosage groups (one‐sided α = 0.01 for the 12 mg once daily treatment group; one‐sided α = 0.04 for the 2 twice daily treatment groups). Testing was done in a fixed sequence order, as follows: SRI‐4 response at week 32, followed by SRI‐4 response, BICLA response, LLDAS response, CLASI‐50 response, and 40‐joint count for active (tender plus swollen), then swollen, then tender joints at week 48. Testing continued until a test resulted in a nonsignificant P value, at which point no further testing was possible; however, in this case, nominal P values were provided.
RESULTS
Patients
From September 2017 through October 2021, 772 patients were screened, and 363 patients meeting eligibility criteria were randomized to receive treatment (placebo, n = 90; deucravacitinib 3 mg twice daily, n = 91; deucravacitinib 6 mg twice daily, n = 93; deucravacitinib 12 mg once daily, n = 89) (Supplementary Figure 3, available at https://onlinelibrary.wiley.com/doi/art.42391). Among randomized patients, 275 (75.8%) completed 48 weeks of treatment. The most common reasons for discontinuation across the treatment groups were adverse events and patient withdrawal (Supplementary Figure 3). The adverse event classes most commonly leading to discontinuation included gastrointestinal disorders and skin and cutaneous disorders, with the incidence balanced across treatment groups (Supplementary Table 1, https://onlinelibrary.wiley.com/doi/art.42391). Reasons for patient withdrawal showed no clear pattern, with the most common reasons being relocation and personal problems.
Demographic and baseline disease characteristics were similar across groups, with 12.1% of enrolled patients of Asian race and 9.1% of patients of Black race. The trial population had active disease at baseline with groups being balanced (Table 1). At baseline, 80.4% of patients were taking glucocorticoids, and 32.2% of patients were taking a combination of antimalarials, immunosuppressants, and glucocorticoids. The distribution of concomitant immunosuppressant medications taken by each group was balanced at baseline (Supplementary Table 2, https://onlinelibrary.wiley.com/doi/art.42391). Moreover, at baseline, 47 (12.9%) of 363 randomized patients either had antiphospholipid syndrome (n = 10) or were positive for antiphospholipid antibodies (n = 37). Of those 47 patients, 24 (51.1%) were receiving anticoagulation and/or antithrombotic therapy (Supplementary Table 3, https://onlinelibrary.wiley.com/doi/art.42391).
Table 1.
Baseline demographic and clinical characteristics of patients in the deucravacitinib treatment groups and placebo group*
| Deucravacitinib 3 mg twice daily (n = 91) | Deucravacitinib 6 mg twice daily (n = 93) | Deucravacitinib 12 mg once daily (n = 89) | Placebo (n = 90) | Total (n = 363) | |
|---|---|---|---|---|---|
| Age, mean ± SD years | 40.2 ± 11.9 | 40.9 ± 12.5 | 39.0 ± 10.6 | 40.1 ± 13.1 | 40.1 ± 12.0 |
| Body mass index, mean ± SD kg/m2 | 26.5 ± 6.7 | 26.1 ± 6.9 | 27.1 ± 6.9 | 27.5 ± 6.7 | 26.8 ± 6.8 |
| Female | 85 (93.4) | 88 (94.6) | 81 (91.0) | 80 (88.9) | 334 (92.0) |
| Race/ethnicity | |||||
| American Indian or Alaska Native | 3 (3.3) | 5 (5.4) | 2 (2.2) | 4 (4.4) | 14 (3.9) |
| Asian | 9 (9.9) | 15 (16.1) | 10 (11.2) | 10 (11.1) | 44 (12.1) |
| Black or African American | 10 (11.0) | 8 (8.6) | 9 (10.1) | 6 (6.7) | 33 (9.1) |
| Other | 7 (7.7) | 10 (10.8) | 11 (12.4) | 10 (11.1) | 38 (10.5) |
| White | 62 (68.1) | 55 (59.1) | 57 (64.0) | 60 (66.7) | 234 (64.5) |
| Hispanic or Latino | 31 (34.1) | 29 (31.2) | 36 (40.4) | 31 (34.4) | 127 (35.0) |
| Geographic region | |||||
| Latin America† | 26 (28.6) | 29 (31.2) | 26 (29.2) | 27 (30.0) | 108 (29.8) |
| North America | 23 (25.3) | 22 (23.7) | 21 (23.6) | 21 (23.3) | 87 (24.0) |
| Japan | 5 (5.5) | 5 (5.4) | 5 (5.6) | 4 (4.4) | 19 (5.2) |
| Rest of world | 37 (40.7) | 37 (39.8) | 37 (41.6) | 38 (42.2) | 149 (41.0) |
| Years since initial diagnosis | |||||
| <3 years | 24 (26.4) | 27 (29.0) | 32 (36.0) | 26 (28.9) | 109 (30.0) |
| 3 to 6 years | 13 (14.3) | 19 (20.4) | 13 (14.6) | 16 (17.8) | 61 (16.8) |
| >6 years | 54 (59.3) | 47 (50.5) | 44 (49.4) | 48 (53.3) | 193 (53.2) |
| Baseline treatment for SLE | |||||
| Glucocorticoid use | 74 (81.3) | 73 (78.5) | 71 (79.8) | 74 (82.2) | 292 (80.4) |
| ≥10 mg/day prednisone or equivalent | 45 (49.5) | 46 (49.5) | 43 (48.3) | 47 (52.2) | 181 (49.9) |
| Antimalarial agent | 81 (89.0) | 84 (90.3) | 75 (84.3) | 75 (83.3) | 315 (86.8) |
| Immunosuppressant agent‡ | 53 (58.2) | 43 (46.2) | 46 (51.7) | 46 (51.1) | 188 (51.8) |
| Antimalarial, immunosuppressant, and glucocorticoid | 38 (41.8) | 26 (28.0) | 27 (30.3) | 26 (28.9) | 117 (32.2) |
| SLEDAI‐2K score, mean ± SD | 11.1 ± 3.2 | 10.8 ± 3.2 | 10.7 ± 3.0 | 10.8 ± 3.1 | 10.8 ± 3.1 |
| Overall BILAG‐2004 A/B grades | |||||
| ≥1 A grade | 51 (56.0) | 44 (47.3) | 51 (57.3) | 51 (56.7) | 197 (54.3) |
| No A grade or ≥2 B grades | 40 (44.0) | 46 (49.5) | 37 (41.6) | 39 (43.3) | 162 (44.6) |
| BILAG‐2004 Organ Domain, patients with A/B/C grades | |||||
| Musculoskeletal | 90 (98.9) | 93 (100.0) | 89 (100.0) | 88 (97.8) | 360 (99.2) |
| Mucocutaneous | 87 (95.6) | 92 (98.9) | 84 (94.4) | 87 (96.7) | 350 (96.4) |
| Hematologic | 54 (59.3) | 55 (59.1) | 50 (56.2) | 57 (63.3) | 216 (59.5) |
| Renal | 13 (14.3) | 10 (10.8) | 12 (13.5) | 7 (7.8) | 42 (11.6) |
| Constitutional | 8 (8.8) | 12 (12.9) | 6 (6.7) | 9 (10.0) | 35 (9.6) |
| Cardiorespiratory | 4 (4.4) | 2 (2.2) | 4 (4.5) | 5 (5.6) | 15 (4.1) |
| Ophthalmic | 0 (0.0) | 1 (1.1) | 0 (0.0) | 2 (2.2) | 3 (0.8) |
| Neuropsychiatric | 1 (1.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.3) |
| Gastrointestinal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| PGA score, mean ± SD | 1.80 ± 0.3 | 1.84 ± 0.4 | 1.86 ± 0.4 | 1.82 ± 0.4 | 1.83 ± 0.4 |
| CLASI‐A score, mean ± SD | 8.6 ± 7.6 | 8.2 ± 6.5 | 8.4 ± 5.8 | 8.0 ± 5.1 | 8.3 ± 6.3 |
| Forty‐joint count§ | |||||
| Active (swollen + tender) joints, mean ± SD | 8.6 ± 4.8 | 8.8 ± 6.1 | 9.4 ± 5.7 | 9.2 ± 6.0 | 9.0 ± 5.7 |
| Tender joints, mean ± SD | 13.5 ± 7.6 | 14.8 ± 8.9 | 13.5 ± 7.8 | 14.1 ± 9.2 | 14.0 ± 8.4 |
| Swollen joints, mean ± SD | 9.0 ± 4.9 | 9.4 ± 6.2 | 9.9 ± 5.8 | 9.4 ± 6.1 | 9.4 ± 5.7 |
Except where indicated otherwise, values are the number (%) of patients. SLE = systemic lupus erythematosus; SLEDAI‐2K = Systemic Lupus Erythematosus Disease Activity Index 2000; BILAG‐2004 = British Isles Lupus Assessment Group 2004 index; PGA = physician global assessment of disease activity; CLASI‐A = Cutaneous Lupus Erythematosus Disease Area and Severity Index activity score.
Patients in the Latin America region were in Argentina, Brazil, Colombia, or Mexico.
Immunosuppressants could include azathioprine, 6‐mercaptopurine, methotrexate, leflunomide, or mycophenolate mofetil.
For details on joints included in the 40‐joint count, see protocol in the Supplementary Methods, available at https://onlinelibrary.wiley.com/doi/art.42391.
Efficacy
The trial met its primary end point. A significantly higher percentage of patients achieved an SRI‐4 response at week 32 with deucravacitinib 3 mg twice daily compared with placebo (58.2% versus 34.4%; adjusted OR 2.8 [95% CI 1.5, 5.1]; P < 0.001 versus placebo) and 6 mg twice daily (49.5% versus 34.4%; OR 1.9 [95% CI 1.0, 3.4]; P = 0.02 versus placebo) (Figure 1 and Supplementary Table 4, https://onlinelibrary.wiley.com/doi/art.42391). The deucravacitinib 12 mg once daily group demonstrated a higher SRI‐4 response rate versus placebo (44.9% versus 34.4%; OR 1.6 [95% CI 0.8, 2.9]; nominal P = 0.08 versus placebo). The between‐group adjusted differences from placebo in SRI‐4 response rate were as follows: deucravacitinib 3 mg twice daily, 23.8% (95% CI 8.5, 37.7); deucravacitinib 6 mg twice daily, 15.0% (95% CI –0.0, 29.2); and deucravacitinib 12 mg once daily, 10.5% (95% CI –4.5, 24.9). The median time to onset of SRI‐4 response was 85 days (95% CI 85, 113 days) with deucravacitinib 3 mg twice daily, 92 days (95% CI 85, 138) with deucravacitinib 6 mg twice daily, and 111 days (95% CI 85, 115) with deucravacitinib 12 mg once daily versus 116 days (95% CI 112, 144) with placebo.
Figure 1.
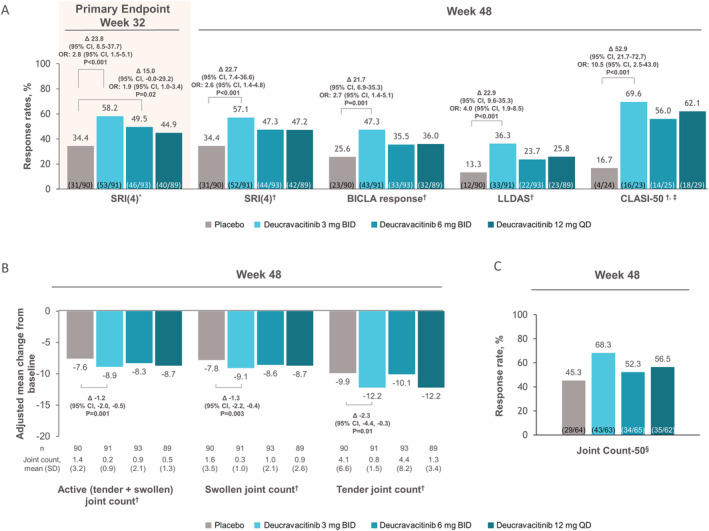
Key efficacy end points of patient groups receiving deucravacitinib 3 mg twice daily (BID), deucravacitinib 6 mg twice daily, deucravacitinib 12 mg once daily (QD), or placebo for treatment of systemic lupus erythematosus. Nonresponder imputation was used to impute missing data for all end points except change from baseline in joint count. A, Patient response rates using the Systemic Lupus Erythematosus Responder Index 4 (SRI‐4) at week 32, and SRI‐4, British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA), Lupus Low Disease Activity State (LLDAS), and Cutaneous Lupus Erythematosus Disease Area and Severity Index 50 (CLASI‐50) at week 48. B, Adjusted mean change from baseline in the number of active (swollen plus tender), swollen, and tender joint counts at week 48. The adjusted mean change in each joint count was calculated using the mixed model for repeated measures approach. For analysis visits where the nonresponder imputation criteria were met, the observed values were set to missing and the overall cohort response modeled. C, Patient response rates using Joint Count‐50 at week 48 assessed in patients with ≥6 active (both swollen and tender) joints at baseline. In A and B, multiplicity‐controlled primary and secondary end points, between‐group differences (Δ), and odds ratios (ORs) for treatment groups versus placebo, along with their respective 95% confidence intervals (95% CIs), are shown. P values were not calculated for exploratory end points. * = primary end point; † = multiplicity‐adjusted secondary end point; ‡ = in patients with a baseline CLASI activity score ≥10. § = exploratory non–multiplicity‐controlled end point.
All multiplicity‐controlled secondary efficacy end points, assessed at week 48, were more frequently achieved in patients treated with deucravacitinib 3 mg twice daily versus placebo. Secondary efficacy end point results in the deucravacitinib 6 mg twice daily and 12 mg once daily dosage groups were higher compared with placebo; however, the results did not achieve statistical significance (Figure 1 and Supplementary Table 4, https://onlinelibrary.wiley.com/doi/art.42391). Significant differences were observed in 2 composite disease measures between the 3 mg twice daily group versus placebo: SRI‐4 response (57.1% versus 34.4%, respectively; P < 0.001 versus placebo) and BICLA response (47.3% versus 25.6%, respectively; P = 0.001 versus placebo) (Figure 1 and Supplementary Table 4). Attainment of the treat‐to‐target end point LLDAS also favored deucravacitinib 3 mg twice daily, with a very low placebo response rate for this stringent end point (36.3% versus 13.3%, respectively; P < 0.001 versus placebo). Organ‐specific end points for skin were significantly improved with deucravacitinib 3 mg twice daily versus placebo (CLASI‐50 response, 69.6% versus 16.7%, respectively; P < 0.001 versus placebo) and for arthritis (mean change from baseline in active joint count, –8.9 versus –7.6, respectively; P = 0.001 versus placebo) (Figure 1 and Supplementary Table 4, https://onlinelibrary.wiley.com/doi/art.42391).
Among exploratory end points, the number of patients achieving a Joint Count‐50 response was greater with deucravacitinib treatment (68.3% at 3 mg twice daily, 52.3% at 6 mg twice daily, and 56.5% at 12 mg once daily) compared with placebo (45.3%) (Figure 1 and Supplementary Table 4). Over 48 weeks of treatment, levels of anti–double‐stranded DNA antibodies decreased, and in patients with low levels of complement C3 (<0.9 gm/liter) or C4 (<0.1 gm/liter) at baseline, levels of C3 and C4 increased over follow‐up (Figure 2). In addition, all dosages of deucravacitinib, but not placebo, were associated with reduced IFN‐regulated gene expression through 44 weeks of treatment, with suppression evident as early as week 4 (Figure 3).
Figure 2.
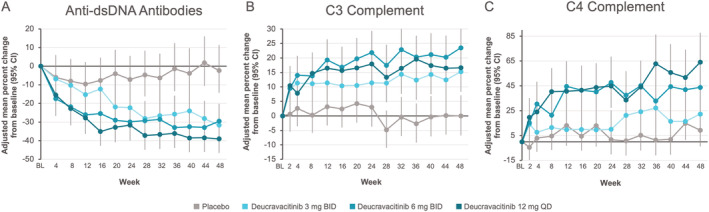
Adjusted mean percentage change from baseline (BL) over 48‐week follow‐up in levels of anti–double‐stranded DNA (anti‐dsDNA) antibodies in patients with detectable anti‐dsDNA at baseline (A), C3 complement in patients with C3 <0.9 gm/liter at baseline (B), and C4 complement in patients with C4 <0.1 gm/liter at baseline (C) for patient groups receiving deucravacitinib 3 mg twice daily (BID), deucravacitinib 6 mg twice daily, deucravacitinib 12 mg once daily (QD), or placebo for treatment of systemic lupus erythematosus. In A–C, vertical bars indicate 95% confidence intervals (95% CIs). Imputation was done using a mixed‐effects model. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42391/abstract.
Figure 3.
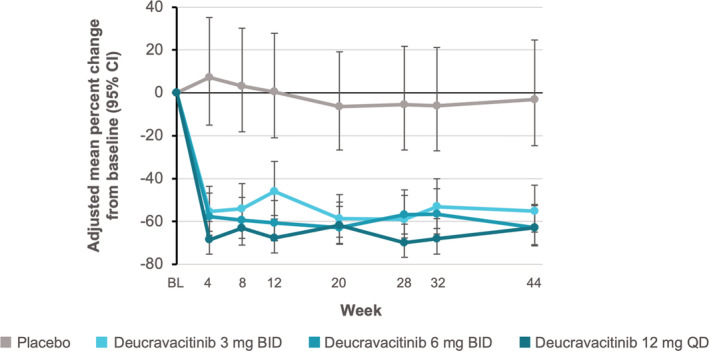
Adjusted mean percentage change in interferon gene signature from baseline (BL) over 44‐week follow‐up in patient groups receiving deucravacitinib 3 mg twice daily (BID), deucravacitinib 6 mg twice daily, deucravacitinib 12 mg once daily (QD), or placebo for treatment of systemic lupus erythematosus. Vertical bars indicate 95% confidence intervals (95% CIs). Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42391/abstract.
Safety
A similar frequency of adverse events was observed in patients across deucravacitinib treatment groups (93.4% [85 of 91 patients] at 3 mg twice daily, 87.1% [81 of 93 patients] at 6 mg twice daily, and 84.3% [75 of 89 patients] at 12 mg once daily) and in patients receiving placebo (87.8% [79 of 90 patients]). Adverse events occurring in at least 5% of patients in any group are listed in Table 2.
Table 2.
Summary of adverse events in patients in the deucravacitinib treatment groups and placebo group over the course of weeks 0 to 48*
| Deucravacitinib 3 mg twice daily (n = 91) | Deucravacitinib 6 mg twice daily (n = 93) | Deucravacitinib 12 mg once daily (n = 89) | Placebo (n = 90) | |
|---|---|---|---|---|
| Deaths | 0 | 0 | 0 | 0 |
| Adverse events | 85 (93.4) | 81 (87.1) | 75 (84.3) | 79 (87.8) |
| Serious adverse events | 7 (7.7) | 8 (8.6) | 7 (7.9) | 11 (12.2) |
| Serious infections/infestations | 1 (1.1) † | 2 (2.2) ‡ | 1 (1.1) § | 1 (1.1) ¶ |
| Adverse events leading to treatment discontinuation | 8 (8.8) | 6 (6.5) | 11 (12.4) | 3 (3.3) |
| Adverse events occurring in at least 5% of patients# | ||||
| Upper respiratory tract infection | 13 (14.3) | 18 (19.4) | 8 (9.0) | 8 (8.9) |
| Nasopharyngitis | 8 (8.8) | 13 (14.0) | 8 (9.0) | 11 (12.2) |
| Urinary tract infection | 10 (11.0) | 6 (6.5) | 7 (7.9) | 3 (3.3) |
| Cystitis | 5 (5.5) | 1 (1.1) | 2 (2.2) | 0 |
| Pharyngitis | 7 (7.7) | 5 (5.4) | 2 (2.2) | 2 (2.2) |
| Oral herpes | 4 (4.4) | 4 (4.3) | 5 (5.6) | 0 |
| COVID‐19 | 3 (3.3) | 5 (5.4) | 3 (3.4) | 3 (3.3) |
| Sinusitis | 4 (4.4) | 5 (5.4) | 0 | 2 (2.2) |
| Bronchitis | 3 (3.3) | 5 (5.4) | 0 | 6 (6.7) |
| Rhinorrhea | 1 (1.1) | 0 | 1 (1.1) | 5 (5.6) |
| Diarrhea | 4 (4.4) | 8 (8.6) | 3 (3.4) | 5 (5.6) |
| Nausea | 6 (6.6) | 5 (5.4) | 4 (4.5) | 8 (8.9) |
| Vomiting | 3 (3.3) | 4 (4.3) | 1 (1.1) | 6 (6.7) |
| Acne | 3 (3.3) | 8 (8.6) | 7 (7.9) | 4 (4.4) |
| Rash | 2 (2.2) | 3 (3.2) | 7 (7.9) | 0 |
| Headache | 7 (7.7) | 8 (8.6) | 11 (12.4) | 15 (16.7) |
| Back pain | 1 (1.1) | 8 (8.6) | 2 (2.2) | 6 (6.7) |
| Arthralgia | 5 (5.5) | 1 (1.1) | 0 | 1 (1.1) |
| Hypertension | 4 (4.4) | 3 (3.2) | 6 (6.7) | 3 (3.3) |
Values are the number (%) of patients who experienced an event.
In the deucravacitinib 3 mg twice daily group, 1 patient had serious chronic pyelonephritis.
In the deucravacitinib 6 mg twice daily group, 1 patient had serious COVID‐19, and 1 patient had serious herpes zoster.
In the deucravacitinib 12 mg once daily group, 1 patient had serious urinary tract infection.
In the placebo group, 1 patient had serious COVID‐19 pneumonia.
Adverse events are grouped by system organ class.
The most common adverse events observed in at least 10% of patients treated with deucravacitinib were upper respiratory tract infection, nasopharyngitis, headache, and urinary tract infection. The majority of events occurring with deucravacitinib treatment were mild‐to‐moderate as assessed by investigators, and most were considered unrelated to treatment. The incidence of serious adverse events was comparable across deucravacitinib treatment groups (7.7% [7 of 91 patients] at 3 mg twice daily, 8.6% [8 of 93 patients] at 6 mg twice daily, and 7.9% [7 of 89 patients] at 12 mg once daily) and placebo (12.2% [11 of 90 patients]) (Table 2). Adverse events resulting in treatment discontinuation occurred in 8.8% of patients (8 of 91) in the deucravacitinib 3 mg twice daily group, 6.5% of patients (6 of 93) in the 6 mg twice daily group, 12.4% of patients (11 of 89) in the 12 mg once daily group, and 3.3% of patients (3 of 90) in the placebo group. Solid organ malignancies occurred in 1 patient in the deucravacitinib 3 mg twice daily group (breast carcinoma at day 150) and 1 patient in the 12 mg once daily group (vaginal squamous cell carcinoma at day 136); 1 case of nonmelanoma skin cancer occurred in 1 patient in the placebo group (basal cell carcinoma at day 168) (Supplementary Table 1, https://onlinelibrary.wiley.com/doi/art.42391). No deaths, systemic opportunistic infections, tuberculosis infections, hematologic malignancies, major cardiovascular events, or thromboembolic events occurred in any treatment group.
Among protocol‐specified adverse events of interest, no differences in incidence of herpes zoster infections, including herpes zoster, herpes ophthalmicus, and genital herpes zoster, were observed with deucravacitinib treatment. Herpes zoster infections occurred in 3.3% of patients (3 of 91) treated with deucravacitinib 3 mg twice daily, 3.2% of patients (3 of 93) treated with 6 mg twice daily, 2.2% of patients (2 of 89) treated with 12 mg once daily, and 4.4% of patients (4 of 90) receiving placebo. All cases of herpes zoster infection were localized with none being disseminated. Oral herpes simplex was observed at a higher frequency with deucravacitinib treatment, with lesions seen in 4.4% of patients (4 of 91) treated with deucravacitinib 3 mg twice daily, 4.3% of patients (4 of 93) treated with 6 mg twice daily, 5.6% of patients (5 of 89) treated with 12 mg once daily, and 0 patients receiving placebo. The incidence of influenza infection was comparable across treatment groups and occurred in 3.3% of patients (3 of 91) treated with deucravacitinib 3 mg twice daily, 1.1% of patients (1 of 93) treated with 6 mg twice daily, 3.4% of patients (3 of 89) treated with 12 mg once daily, and 1.1% of patients (1 of 90) receiving placebo. COVID‐19 was reported in 3.3% of patients (3 of 91) treated with deucravacitinib 3 mg twice daily, 5.4% of patients (5 of 93) treated with 6 mg twice daily, 3.4% of patients (3 of 89) treated with 12 mg once daily, and 3.3% of patients (3 of 90) receiving placebo (Table 2).
Cutaneous adverse events were observed more frequently with deucravacitinib treatment, occurring in 16.5% of patients (15 of 91) treated with deucravacitinib 3 mg twice daily, 34.4% of patients (32 of 93) treated with 6 mg twice daily, 33.7% of patients (30 of 89) treated with 12 mg once daily, and 13.3% of patients (12 of 90) receiving placebo. The most common cutaneous adverse events were acne and rash (Table 2). Cutaneous adverse events leading to discontinuation occurred in 0 patients treated with deucravacitinib 3 mg twice daily, 3.2% of patients (3 of 93) treated with 6 mg twice daily, 4.5% of patients (4 of 89) treated with 12 mg once daily, and 2.2% of patients (2 of 90) receiving placebo (Supplementary Table 1, https://onlinelibrary.wiley.com/doi/art.42391).
No meaningful abnormalities were observed over time in mean levels of hematology and laboratory parameters (Figure 4).
Figure 4.
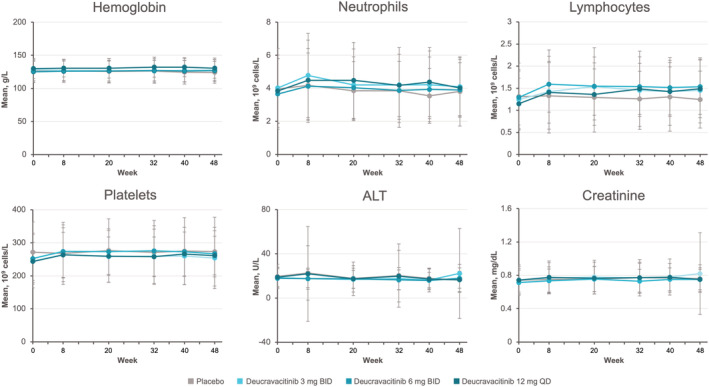
Mean change in laboratory parameters over 48‐week follow‐up in patient groups receiving deucravacitinib 3 mg twice daily (BID), deucravacitinib 6 mg twice daily, deucravacitinib 12 mg once daily (QD), or placebo for treatment of systemic lupus erythematosus. Vertical bars represent standard deviations. ALT = alanine aminotransferase. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42391/abstract.
DISCUSSION
Deucravacitinib is a first‐in‐class oral, selective, allosteric inhibitor of the intracellular signaling kinase TYK2. This kinase transduces signals of multiple cytokines, including type I IFNs, IL‐10, IL‐12, and IL‐23, which are implicated in the pathogenesis of SLE (11, 12). Deucravacitinib previously demonstrated robust efficacy with acceptable tolerability in trials in plaque psoriasis and psoriatic arthritis (15, 16, 17, 30). The concept that deucravacitinib could be effective and safe for SLE was suggested by the profile of cytokines that TYK2 could inhibit, and the persisting unmet need for improved therapies for SLE justified testing this concept.
In this trial, significantly higher proportions of patients treated with deucravacitinib 3 mg twice daily or 6 mg twice daily met the primary outcome of increased SRI‐4 response versus placebo at week 32. In addition, patients treated with deucravacitinib 3 mg twice daily met all secondary end points, including SRI‐4, BICLA, LLDAS, and CLASI‐50 response, and reduction in active joint count at week 48. As BICLA response requires improvement of all active domains and worsening of none, and as LLDAS response from a population with highly active baseline disease requires resolution of multiple domains, these results together suggest a broad set of effects on SLE clinical manifestations beyond skin and joint involvement. Alongside the lack of new safety signals, these findings suggest the potential for deucravacitinib to be beneficial in the management of SLE.
Multiple clinical trials of novel agents for treating SLE have failed to achieve their primary and/or secondary end points (13, 31, 32). It has been postulated that such failures may be due to factors such as disease heterogeneity or lack of efficacy of a drug, but also due to well‐characterized issues with study design and end points (33). In this trial, higher rates of response with deucravacitinib 3 mg twice daily treatment versus placebo were observed across the primary end point and all key multiplicity‐adjusted secondary end points, including 2 different composite end points, SRI‐4 and BICLA response. Such consistent findings are unusual for trials in SLE, and they differentiate these results from those of other studies where these end points were discordant (34). Results favoring deucravacitinib were also seen in attainment of LLDAS, a treat‐to‐target end point that has been associated with protection from morbidity, mortality, and loss of quality of life (2, 26, 35, 36). The placebo response rate was very low with this stringent outcome. Finally, significantly higher rates of response with deucravacitinib 3 mg twice daily treatment versus placebo were observed for both skin‐ and joint‐specific end points, supporting the potential benefit of deucravacitinib across a range of SLE clinical manifestations. Improvements in serologic markers of lupus activity (anti–double‐stranded DNA, C3, C4) and reduction of type I IFN signals at all dosages were also observed.
TYK2 does not transduce signals of endocrine or hematopoietic factors, suggesting a different safety profile from that of JAK inhibitors (10). Safety and tolerability of deucravacitinib were similar to previous experiences with this agent in plaque psoriasis and psoriatic arthritis (15, 16, 17, 30), and there were no changes in laboratory parameters typically observed with JAK inhibitors (37). While serious infections were infrequent across treatment groups, some nonserious infections (e.g., upper respiratory tract infection, urinary tract infection) occurred more frequently with deucravacitinib, without increases in infections such as herpes zoster, influenza, or SARS–COV‐2. Dose‐related increases in cutaneous adverse events, distinct from lupus‐related skin findings, were consistent with those seen in trials with deucravacitinib in other diseases. Most of the skin findings were mild‐to‐moderate, with few leading to discontinuations. Adverse events resulting in treatment discontinuation were higher with deucravacitinib versus placebo and highest in the deucravacitinib 12 mg once daily group, but no specific pattern was observed.
No added benefit in efficacy was observed with the higher deucravacitinib dosages of 6 mg twice daily and 12 mg once daily. The 3 deucravacitinib dosages were selected based on earlier trials that assessed their ability to suppress type I IFN– and IL‐23–mediated pathways (18, 19). It should be noted that 3 mg twice daily and 6 mg twice daily are different total daily doses (6 mg versus 12 mg, respectively) while 6 mg twice daily and 12 mg once daily are the same total daily dose (12 mg) with different dosing regimens. Thus, this trial investigated a dose response (3 mg twice daily versus 6 mg twice daily) as well as the pharmacokinetic parameters (e.g., Cavg versus Ctrough) affecting responses (6 mg twice daily versus 12 mg once daily).
Although treatment with all 3 dosages resulted in greater proportions of responders than placebo at week 32 and week 48, the 3 mg twice daily dosage showed the greatest difference versus placebo across all end points. However, there was less statistical power to detect a difference with the 12 mg once daily dose due to unequal allocation of alpha, and the trial was not designed or powered to formally compare doses of deucravacitinib. Nevertheless, the results in the 3 mg twice daily dosage group were the most consistent in this study, and higher doses seem unlikely to improve outcomes. These findings suggest that suppression of pathways operative in active SLE was sufficient at the deucravacitinib 3 mg twice daily dosage. There was minimal if any additional impact of higher doses on IFN gene signature. A trend toward greater improvements in anti–double‐stranded DNA and complement levels with higher doses was not accompanied by increased clinical benefit at those doses. Taken together, these observations may suggest a saturation of responses at lower exposures with a plateau effect in the inhibition of pathologic pathways, and/or optimized effects on disease‐controlling pathways at the 3 mg twice daily dosage. Further evaluations of biomarkers, including cytokine levels and their relationship to informative patient subsets and clinical responses, and exposure–response relationships are ongoing.
The generalizability of the results in this trial is limited by participation being restricted to patients with moderate‐to‐severe disease despite conventional therapy, exclusion of patients with active severe renal or neuropsychiatric disease, and a relatively low proportion of Black patients relative to the incidence of SLE in this population. The strengths of this trial include the double‐blind, placebo‐controlled design over 48 weeks, a relatively large sample size for a phase II trial in SLE, the effect sizes in key responses, and consistency across outcomes.
In conclusion, in this phase II trial, deucravacitinib, an oral, selective, allosteric TYK2 inhibitor, was superior to placebo in reducing SLE disease activity across multiple measures, with an acceptable safety profile. These results support the potential benefits of TYK2 inhibition in SLE. The potential of deucravacitinib to be an efficacious agent in this disease will be explored in larger phase III trials.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Morand had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Pike, Catlett, Banerjee, Singhal.
Acquisition of data
Pike, Hobar, Delev, Shah, Wegman, Catlett.
Analysis and interpretation of data
Morand, Pike, Merrill, van Vollenhoven, Werth, Hobar, Delev, Shah, Sharkey, Wegman, Catlett, Banerjee, Singhal.
ROLE OF THE STUDY SPONSOR
Bristol Myers Squibb designed the trial, participated in the collection, analysis, and interpretation of the data along with external authors, and paid for professional writing assistance. There were confidentiality agreements between the authors and the sponsor. All authors had full access to the trial data, participated in the interpretation of the data and development of the draft, approved the final draft before submission, and vouch for adherence of the trial to the protocol, completeness and accuracy of the data and analyses, and reporting of adverse events. Publication of this article was not contingent upon approval by Bristol Myers Squibb. Medical writing and editorial assistance were provided by Julianne Hatfield, PhD, of Peloton Advantage, LLC, an OPEN Health company, and funded by Bristol Myers Squibb.
ADDITIONAL DISCLOSURES
Author Brian Sharkey was an employee of Bristol Myers Squibb at the time the study was conducted.
Supporting information
Disclosure Form
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
We would like to thank the patients enrolled in the trial as well as the clinical trial teams. We acknowledge Christina Crater, MD, who was employed by Bristol Myers Squibb at the time the study was conducted, for contributions to study conduct.
Listen to the podcast Here
A video abstract of this article can be found at: https://players.brightcove.net/3806881048001/default_default/index.html?videoId=6315678068112
ClinicalTrials.gov identifier: NCT03252587. EudraCT database no. 2017‐001203‐79.
Sponsored by Bristol Myers Squibb.
For the Bristol Myers Squibb policy on data sharing and information about requesting access to clinical study data, see https://www.bms.com/researchers‐and‐partners/independent‐research/data‐sharing‐request‐process.html.
A graphical abstract and author disclosures are available online at http://onlinelibrary.wiley.com/doi/10.1002/art.42391.
REFERENCES
- 1. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus [review]. Nat Rev Dis Primers 2016;2:16039. [DOI] [PubMed] [Google Scholar]
- 2. Piga M, Floris A, Cappellazzo G, et al. Failure to achieve lupus low disease activity state (LLDAS) six months after diagnosis is associated with early damage accrual in Caucasian patients with systemic lupus erythematosus. Arthritis Res Ther 2017;19:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kandane‐Rathnayake R, Louthrenoo W, Hoi A, et al. ‘Not at target’: prevalence and consequences of inadequate disease control in systemic lupus erythematosus‐a multinational observational cohort study. Arthritis Res Ther 2022;24:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bultink IE, de Vries F, van Vollenhoven RF, et al. Mortality, causes of death and influence of medication use in patients with systemic lupus erythematosus vs matched controls. Rheumatology (Oxford) 2021;60:207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruce IN, O'Keeffe AG, Farewell V, et al. Factors associated with damage accrual in patients with systemic lupus erythematosus: results from the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Ann Rheum Dis 2015;74:1706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Basta F, Fasola F, Triantafyllias K, et al. Systemic lupus erythematosus (SLE) therapy: the old and the new [review]. Rheumatol Ther 2020;7:433–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morand EF, Furie R, Tanaka Y, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med 2020;382:211–21. [DOI] [PubMed] [Google Scholar]
- 8. Furie R, Petri M, Zamani O, et al. A phase III, randomized, placebo‐controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum 2011;63:3918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stohl W, Schwarting A, Okada M, et al. Efficacy and safety of subcutaneous belimumab in systemic lupus erythematosus: a fifty‐two–week randomized, double‐blind, placebo‐controlled study. Arthritis Rheumatol 2017;69:1016–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burke JR, Cheng L, Gillooly KM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med 2019;11:eaaw1736. [DOI] [PubMed] [Google Scholar]
- 11. Bengtsson AA, Rönnblom L. Role of interferons in SLE [review]. Best Pract Res Clin Rheumatol 2017;31:415–28. [DOI] [PubMed] [Google Scholar]
- 12. Dai H, He F, Tsokos GC, et al. IL‐23 limits the production of IL‐2 and promotes autoimmunity in lupus. J Immunol 2017;199:903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Furie RA, Morand EF, Bruce IN, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP‐1): a randomised, controlled, phase 3 trial. Lancet Rheumatol 2019;1:e208–19. [DOI] [PubMed] [Google Scholar]
- 14. Sotyktu full prescribing information. US Food and Drug Administration. Revised September 2022. Accessed October 19, 2022. URL: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214958s000lbl.pdf.
- 15. Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52‐week, randomized, double‐blinded, placebo‐controlled phase 3 POETYK PSO‐1 trial. J Am Acad Dermatol 2022. E‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16. Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52‐week, randomized, double‐blinded, phase 3 POETYK PSO‐2 trial. J Am Acad Dermatol 2022. E‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 17. Mease PJ, Deodhar AA, van der Heijde D, et al. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann Rheum Dis 2022;81:815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Catlett I, Aras U, Liu Y, et al. A first‐in‐human, study of BMS‐986165, a selective, potent, allosteric small molecule inhibitor of tyrosine kinase 2 [abstract SAT0226]. Ann Rheum Dis 2017;76:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aras U, Bei D, He B, et al. Comparison of pharmacokinetics of BMS‐986165, a TYK2 inhibitor, in Japanese and non‐Japanese healthy subjects [poster PII‐010]. Presented at: Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics; March 2018; Orlando, Florida. [Google Scholar]
- 20. Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2002;29:288–91. [PubMed] [Google Scholar]
- 22. Parker B, Bruce IN. Clinical markers, metrics, indices, and clinical trials. In: Wallace DJ, Hahn BH, eds. Dubois' Lupus Erythematosus and Related Syndromes. 9th ed. London: Elsevier; 2019:614–30. [Google Scholar]
- 23. Isenberg DA, Rahman A, Allen E, et al. BILAG 2004. Development and initial validation of an updated version of the British Isles Lupus Assessment Group's disease activity index for patients with systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:902–6. [DOI] [PubMed] [Google Scholar]
- 24. Furie RA, Petri MA, Wallace DJ, et al. Novel evidence‐based systemic lupus erythematosus responder index. Arthritis Rheum 2009;61:1143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wallace DJ, Kalunian K, Petri MA, et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double‐blind, placebo‐controlled, multicentre study. Ann Rheum Dis 2014;73:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franklyn K, Lau CS, Navarra SV, et al. Definition and initial validation of a Lupus Low Disease Activity State (LLDAS). Ann Rheum Dis 2016;75:1615–21. [DOI] [PubMed] [Google Scholar]
- 27. Klein RS, Morganroth PA, Werth VP. Cutaneous lupus and the Cutaneous Lupus Erythematosus Disease Area and Severity Index instrument. Rheum Dis Clin North Am 2010;36:33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morand EF, Furie RA, Bruce IN, et al. Efficacy of anifrolumab across organ domains in patients with moderate‐to‐severe systemic lupus erythematosus: a post‐hoc analysis of pooled data from the TULIP‐1 and TULIP‐2 trials. Lancet Rheumatol 2022;4:e282–92. [DOI] [PubMed] [Google Scholar]
- 29. Kim CH, Abedi M, Liu Y, et al. A novel technology for multiplex gene expression analysis directly from whole blood samples stabilized at ambient temperature using an RNA‐stabilizing buffer. J Mol Diagn 2015;17:118–27. [DOI] [PubMed] [Google Scholar]
- 30. Papp K, Gordon K, Thaçi D, et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med 2018;379:1313–21. [DOI] [PubMed] [Google Scholar]
- 31. Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately‐to‐severely active systemic lupus erythematosus: the randomized, double‐blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 2010;62:222–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mahieu MA, Strand V, Simon LS, et al. A critical review of clinical trials in systemic lupus erythematosus [review]. Lupus 2016;25:1122–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Connelly K, Golder V, Kandane‐Rathnayake R, et al. Clinician‐reported outcome measures in lupus trials: a problem worth solving [review]. Lancet Rheumatol 2021;3:e595–603. [DOI] [PubMed] [Google Scholar]
- 34. Bruce IN, Furie RA, Morand EF, et al. Concordance and discordance in SLE clinical trial outcome measures: analysis of three anifrolumab phase 2/3 trials. Ann Rheum Dis 2022;81:962–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Golder V, Kandane‐Rathnayake R, Hoi AY, et al. Association of the lupus low disease activity state (LLDAS) with health‐related quality of life in a multinational prospective study. Arthritis Res Ther 2017;19:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharma C, Raymond W, Eilertsen G, et al. Association of achieving lupus low disease activity state fifty percent of the time with both reduced damage accrual and mortality in patients with systemic lupus erythematosus. Arthritis Care Res (Hoboken) 2020;72:447–51. [DOI] [PubMed] [Google Scholar]
- 37. Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease [review] [published correction appears in Nat Rev Rheumatol 2017;13:320]. Nat Rev Rheumatol 2017;13:234–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1 Supporting Information