

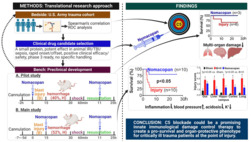
Abstract
Background and Purpose
Traumatic haemorrhage (TH) is the leading cause of potentially preventable deaths that occur during the prehospital phase of care. No effective pharmacological therapeutics are available for critical TH patients yet. Here, we identify terminal complement activation (TCA) as a therapeutic target in combat casualties and evaluate the efficacy of a TCA inhibitor (nomacopan) on organ damage and survival in vivo.
Experimental Approach
Complement activation products and cytokines were analysed in plasma from 54 combat casualties. The correlations between activated complement pathway(s) and the clinical outcomes in trauma patients were assessed. Nomacopan was administered to rats subjected to lethal TH (blast injury and haemorrhagic shock). Effects of nomacopan on TH were determined using survival rate, organ damage, physiological parameters, and laboratory profiles.
Key Results
Early TCA was associated with systemic inflammatory responses and clinical outcomes in this trauma cohort. Lethal TH in the untreated rats induced early TCA that correlated with the severity of tissue damage and mortality. The addition of nomacopan to a damage‐control resuscitation (DCR) protocol significantly inhibited TCA, decreased local and systemic inflammatory responses, improved haemodynamics and metabolism, attenuated tissue and organ damage, and increased survival.
Conclusion and Implications
Previous findings of our and other groups revealed that early TCA represents a rational therapeutic target for trauma patients. Nomacopan as a pro‐survival and organ‐protective drug, could emerge as a promising adjunct to DCR that may significantly reduce the morbidity and mortality in severe TH patients while awaiting transport to critical care facilities.
Keywords: complement, mortality, organ failure, prehospital care, traumatic haemorrhage
Abbreviations
- CH50
complement haemolytic 50% activity
- DCR
damage control resuscitation
- ETBV
estimated total blood volume
- HMGB1
high mobility group box 1
- ISS
injury severity score
- MAP
mean arterial pressure
- MODS
multi‐organ dysfunction syndrome
- SIRS
systemic inflammatory response syndrome
- TBI
traumatic brain injury
- TCA
terminal complement activation
- TH
traumatic haemorrhage
What is already known
Traumatic haemorrhage is the leading cause of potentially preventable deaths during the prehospital care phase.
Terminal complement activation has the potential to trigger inflammation and multi‐organ dysfunction syndrome.
What does this study add
Treatment with nomacopan attenuates organ damage and increases survival.
Nomacopan functions as a pro‐survival and organ‐protective drug.
What is the clinical significance
Terminal complement activation represents a rational therapeutic target for trauma patients.
Nomacopan is a promising drug to create a pro‐survival and organ‐protective phenotype in trauma patients.
1. INTRODUCTION
Traumatic haemorrhage (TH) is a major cause of potentially preventable death on the battlefield as well as in the civilian world. Blast injury was the predominant wounding mechanism during recent conflicts, accounting for 70%–80% of military casualties in Iraq and Afghanistan (Belmont et al., 2012; Ritenour et al., 2010). The pathophysiology of blast‐induced injury is distinctive and appears more complex than most other forms of trauma (Cernak, 2010).
TH involves tissue injury, ischemia, and subsequent reperfusion. Ischemia/reperfusion injury and damaged tissue activate a multifaceted network of plasma cascades (complement, coagulation, kinin, and fibrinolytic systems) that play a major role in the systemic inflammatory response syndrome (SIRS) and the compensatory anti‐inflammatory response syndrome. SIRS and anti‐inflammatory response syndrome ultimately lead to injury‐related multi‐organ dysfunction syndrome (MODS) (Duehrkop & Rieben, 2014). MODS represents a major cause of late mortality following severe trauma (Cole et al., 2020; Gentile et al., 2012; Huber‐Lang et al., 2018). The underlying immunological disturbance is complex, and early activation of the complement cascade plays a crucial role.
Complement activation appears to fuel a vicious cycle of inflammatory damage that exacerbates MODS pathology (Valparaiso et al., 2015). Growing evidence from our studies (Campbell et al., 2016; Dalle Lucca et al., 2011, 2013; Dalle Lucca, Chavko, et al., 2012; Dalle Lucca, Li, et al., 2012; Li et al., 2013; Li et al., 2018; Li, Zhao, et al., 2019; Yang et al., 2019) and those of others (Burk et al., 2012; Ganter et al., 2007; Huber‐Lang et al., 2018; Karasu et al., 2019; Lord et al., 2014; Rittirsch et al., 2012) illustrates that excessive activation of the complement system represents a key mechanism regulating the development of inflammation‐mediated MODS after TH. Pronounced early complement activation was identified in civilians sustaining major trauma (Burk et al., 2012). Elevated concentrations of C3a, C5a, and sC5b‐9 were correlated with injury severity and were associated with increased incidence of MODS and mortality (Burk et al., 2012; Ganter et al., 2007). Similarly, our recent findings in civilians with trauma or burn injury have revealed robustly elevated plasma levels of C3a; the terminal complement activation (TCA) products C5a/sC5b‐9; protein Bb, a product of the complement alternative pathway; and protein C4d, a derivative of the complement classical/lectin pathways (Li, Zhao, et al., 2019). Genetic and pharmacological manipulation of complement levels and complement activation in murine models of ischemia/reperfusion injury, traumatic brain injury (TBI), and TH demonstrate beneficial effects on survival, neuroprotection, inflammation, and tissue damage (Alawieh et al., 2015; Barrett et al., 2018; Danobeitia et al., 2017; Karasu et al., 2019; Leinhase et al., 2006). Our previous studies have demonstrated the beneficial effects of pharmacological inhibition of complement activity on increasing survival, improving haemodynamics, reducing fluid requirements, attenuating organ damage, and modulating systemic and local inflammatory responses in rats and pigs in short‐term studies (<6 h) after TH (Campbell et al., 2016; Dalle Lucca et al., 2011, 2013; Dalle Lucca, Li, et al., 2012).
The latter studies used either a C1 esterase inhibitor (which inhibits the classical and lectin pathways of complement C3 and C5 activation and the contact system that generates bradykinin), or a decay accelerating factor (which inhibits the classical, lectin and alternative pathways of complement C3 and C5 activation). There is debate about the best point at which to inhibit complement activation to improve outcomes in trauma. For example, should one inhibit all three complement pathways or only one and, further, should one interrupt the formation of all the main effectors (C3a, C5a and C5b‐9), only C5a and C5b‐9, or only C5a? Because secondary infection is a major risk after blast injury and other forms of trauma, some investigators believe that inhibition of complement activation at the C3 level may be less desirable than TCA‐specific blockade, because C3 opsonization has a significant antimicrobial function, and C3 and its activation products may have roles in tissue recovery (Huber‐Lang et al., 2020; Keshari et al., 2017). The relative importance of each of the three complement activation pathways in trauma is not yet clear and each may have a different degree of importance in different types of injury.
In the current study, we show that the terminal complement is the predominant activated complement pathway early after trauma in a cohort of combat casualties, most of whom had blast‐induced TBI. Based on these clinical findings, we then hypothesized that blocking C5a and C5b‐9, the TCA products, may reduce organ tissue damage and increase survival. To evaluate this hypothesis, we tested the effect of C5 inhibition using the clinical‐stage complement C5 inhibitor nomacopan in a rat model of TH that replicates the immunological responses seen in injured patients.
2. METHODS
2.1. Overview of study design
This study was designed (1) to determine the role of TCA‐induced morbidity and mortality in military trauma patients with two major mechanisms of injury (69% blast injury and 26% gunshot wounds) and (2) to test the efficacy of a dual specific inhibitor of C5 and LTB4 , nomacopan, against blast injury/haemorrhage‐induced organ damage, and to evaluate whether C5 inhibition provides a survival benefit in a military relevant preclinical rat model (Figure 1). The primary endpoints were survival and organ damage. Prior to performance of the main study, a pilot study was conducted (Figure 1). The main study was designed to generate groups of equal size. However, this was impossible due to the differential mortality rates between drug‐treated and vehicle‐treated groups. None of the animals were excluded from the study. Power calculations were performed on data obtained from the pilot study of complement haemolytic 50% activity (CH50) (Figure S3E) using the PROC POWER and PROC GLMPOWER of the SAS program (SAS Institute, Cary, NC). Using these data (drug‐treated vs. vehicle‐treated animals: 89% vs. 27% CH50 at 1 h after injury or repeated measures over the time period), we calculated a minimum size of 10 rats per treatment group to have a power >85% from both the t‐test for two mean difference at 1‐hour after injury (62% and pooled SD = 43.8%) and the repeated measure ANOVA for longitudinal data (pooled SD = 41.0%) with an expectation of 95% confidence intervals. After recovery (5–7 days) from surgical cannulation and prior to trauma, the animals were randomly assigned to one of two experimental groups (Figure 1). No data were excluded. Drug and vehicle administration was non‐blinded; no bias was applied during husbandry or tissue harvesting. A randomized blinded code for histological sections was used.
FIGURE 1.
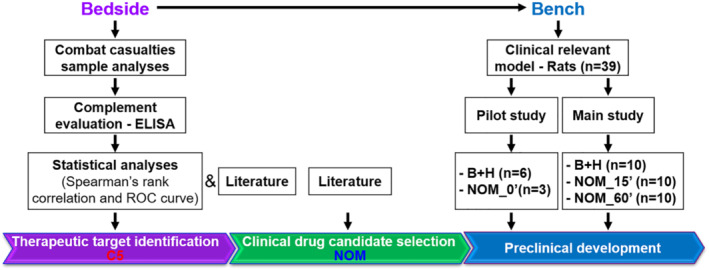
Workflow of translational study design. Blood plasma from 54 casualties on admission, 8 and 24 h after admission to a hospital and 10 civilian volunteers was used for the analysis of the complement activation. On the basis of the products of complement activation in the casualties' plasma, complement component C5 was identified as a reasonable therapeutic target. Prophylactic and therapeutic effects of nomacopan, an inhibitor of C5, were tested in rat models of blast (B) and haemorrhage (H) injury. Abbreviation: NOM, nomacopan
2.2. Clinical study
This study was conducted under a protocol reviewed and approved by the US Army Medical Research and Development Command Institutional Review Board and in accordance with the approved protocol (Protocol #MNC1‐07021). The study in trauma patients was designed to identify the clinical significance of early complement activation in casualties admitted to a US Army Combat Support Hospital (Role 3) in Baghdad, Iraq over a one‐year period (2007–2008, Figure 1). Foreign nationals, prisoners, enemy combatants, children and any patient undergoing therapeutic anticoagulation were excluded. Citrated plasma was collected from trauma patients after admission to the emergency department (n = 54), and if available, 8 h (n = 23) and 24 h (n = 9) later (Table 1). At these later time points, samples were collected after patients had received appropriate clinical care, including surgery and resuscitation. On admission (45–60 min after injury), the clinical and demographic characteristics of the patients were recorded, including base excess, MAP, blood product transfusion units, and SIRS score during the first 24 h. Injury severity was coded using the Abbreviated Injury Scale 2005‐Military system (Champion et al., 2010). Most casualties (n = 45) suffered a traumatic brain injury from explosions.
TABLE 1.
Demographics and characteristics of trauma patients and healthy controls
| Injured service‐members (n = 54) | Healthy controls (n = 10) | |
|---|---|---|
| Age in years, median (IQR) | 25 (22–30) | 36 (28–41) |
| Gender, n (%) | ||
| Male | 53 (98) | 6 (60) |
| Female | 1 (2) | 4 (40) |
| Combat theatre | ||
| OIF, n | 54 | N/A |
| Mortality, n (%) | 5 (9) | N/A |
| Injury severity score, median (IQR) | 16 (9–24) | N/A |
| Mechanism of injury, n (%) | ||
| Blast | 37 (69) | N/A |
| GSW | 14 (26) | N/A |
| Burn | 2 (4) | N/A |
| MVA | 1 (2) | N/A |
| Traumatic brain injury, n (%) | ||
| Yes | 45 (83) | N/A |
| No | 4 (7) | N/A |
| Unknown | 5 (9) | N/A |
| Glasgow coma scale, median (IQR) | 15 (14–15) | N/A |
Abbreviations: IQR, interquartile range; OIF, Operation Iraqi Freedom; GSW, gunshot wound; MVA, motor vehicle accidents. Data are presented as the numbers of patients (with percentages), unless otherwise indicated.
Blood collected at the hospital, was processed according to standard clinical practice (Lusczek et al., 2017) and the resultant plasma was frozen and transported to the US Army Institute of Surgical Research (USAISR) as described (Lusczek et al., 2017) and stored at −80°C until analysis in 2013. Ten healthy volunteers were enrolled at the authors' laboratory as reference controls. Volunteers were 18 years or older with no significant medical conditions. Blood samples were drawn once for analysis of selected complement components, coagulation parameters, and the levels of cytokines.
2.3. Animal study
All animal care and experimental protocols complied with the Animal Welfare Act, the corresponding Animal Welfare regulations and the principles of the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee approved all research conducted in this study. The facility where this research was conducted is fully accredited by the AAALAC. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley, Stanford et al., 2020).
2.3.1. Surgical procedures and injury model in rats
Specific‐pathogen‐free adult male Sprague–Dawley rats (10–12 weeks old) weighing 350–475 g, were purchased from Charles River Laboratories (Wilmington, MA). Under anaesthesia, the carotid artery and jugular vein were cannulated in all rats. The cannulated animals underwent with (the main study) or without (pilot study) a recovery period (5–7 days). The blast injury (B) was conducted as described previously (Chavko et al., 2009; Li et al., 2013; Yang et al., 2019). Briefly, rats were anaesthetised with ketamine/xylazine (60/5 mg·kg−1 body weight) via i.p. injection, and then placed on a rack holder, which was wheeled into the end of the expansion chamber of a compressed‐air‐driven shock tube (Applied Research Associates, Inc., Albuquerque, NM) (Yang et al., 2019). During the blast, the animals were immobilized to prevent movement upon impact and subsequent tertiary blast injury. Animals in prone positions with their heads turned to the blast wave were exposed to a single mild–moderate blast injury (Table S3). Fifteen minutes after blast exposure, animals were subjected to volume‐controlled haemorrhage over 15 min. The estimated total blood volume (ETBV) was calculated using the following formula: ETBV (ml) = weight in kg × 65 ml·kg−1. After haemorrhage, the animals were maintained for 30 min in the shock phase, then received double the shed blood volume of Plasma‐Lyte A. The animals were monitored under anaesthesia (1‐2.5% isoflurane) for 3 h after haemorrhagic shock (H), then returned to their cage and observed for up to 25 h.
Two doses of nomacopan were administered. The first dose (7.5 mg·kg−1, i.v.) was given either immediately before the blast exposure (NOM_0´), 15 min after the blast but prior to haemorrhage (NOM_15´), or 60 min post‐blast (at the end of shock but before fluid resuscitation, NOM_60´), The second dose (also 7.5 mg·kg−1) was given subcutaneously 11 h after blast injury. The dose of nomacopan via i.v. route of administration has previously demonstrated complete inhibition of serum haemolytic activity in rats with a half‐life of 8–12 h (28). Injured rats not treated with nomacopan received the same volume of saline by the same routes of administration at equivalent times.
In the pilot study, cannulated animals (non‐recovery) were allocated to three groups (Figures S3A and Table S3): (1) B + H (n = 6): a single blast injury and 40% haemorrhage with receiving an equal volume of normal saline, (2) NOM_0´ (n = 3): B + H animals treated with nomacopan, (3) Sham (n = 5): the animals underwent the same surgical cannulation, anaesthesia, and analgesia but without B + H. Statistical analysis was not performed for studies where each group size was n < 5.
For the main study, the recovered animals post‐cannulation were randomly assigned to four groups (Figures 1 and 5a and Table S3): (1) B + H (n = 10): a single blast injury and 52% haemorrhage (7 rats lost 50% of ETBV, and three animals lost 57% of ETBV) with receiving equal volume of normal saline, (2) NOM_15´ (n = 10, seven rats lost 50% of ETBV, and three animals lost 57% of ETBV), (3) NOM_60´ (n = 10, seven rats lost 50% of ETBV, and three animals lost 57% of ETBV), and (4) Sham (n = 10): the animals underwent all procedures except B + H and subsequently received an equal volume of normal saline. During the observation period, the mean arterial pressure was recorded by the BIOPAC data acquisition system (BIOPAC Systems, Inc., Goleta, CA). Blood samples were collected before the blast, at the end of haemorrhagic shock, then at 2, 4, 11, and 25 h after blast. Blood chemistry was analysed by i‐STAT (Abbott Laboratories), and PaO2/FiO2 ratio was based on collected i‐STAT data.
FIGURE 5.
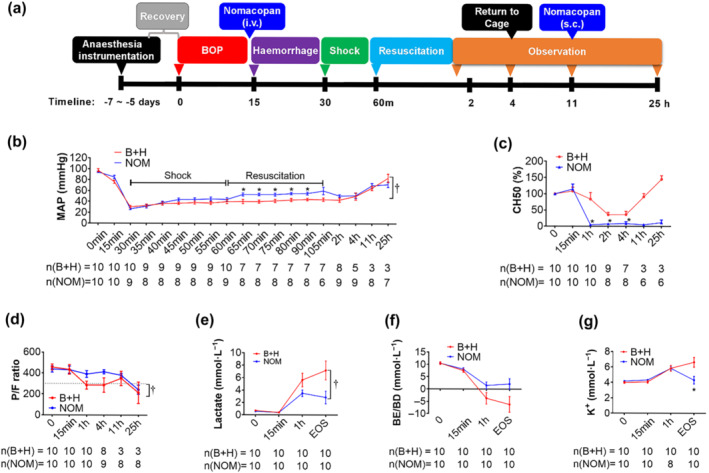
Effects of nomacopan treatment early after blast injury on MAP, CH50, and blood chemistry changes in a clinically relevant rat model of traumatic haemorrhagic shock. (a) Experimental design; (b) changes of MAP were monitored via the carotid artery using the BIOPAC system. During the shock and resuscitation period, the MAP was recorded every 5 min. Data are presented as mean ± SEM; (c) haemolytic TCA of sera was measured by CH50 and normalized to baseline level, which was a pre‐blast injury; the percentages of baseline are shown; (d) PaO2/FiO2 ratio (PFR) following injuries. The PFR was calculated at each time point based on the artery i‐STAT data. A PFR of less than 300 (dashed line) suggests acute respiratory distress syndrome (ARDS). E‐G, the blood chemistry of lactate, BE/BD, and K+ in injured and treated animals were measured by I‐stat and presented. Data shown are means ± SEM from groups of rats, sizes (n) as indicated. *P < 0.05, significant effect of NOM; unpaired t‐test with Welch's correction; †P < 0.05, significantly different as indicated; two‐way ANOVA. Abbreviations: BOP = blast overpressure; B + H = blast + haemorrhagic shock; NOM = nomacopan given to injured rats; MAP = mean arterial pressure; EOS = end of the study
2.4. Assays
2.4.1. Analysis of complement factors in human and rat plasma
Quantitative levels of complement factors in human plasma, including C3a (cat#A031), C5a (cat#A021), sC5b‐9 (cat#A020), Bb (cat#A027) and C4d (cat#A008) were measured by using commercial ELISA kits according to the manufacturer's instructions (Quidel, San Diego, CA). Rat plasma levels of complement C3 (cat#ab157731, Abcam, Cambridge, MA) and C1q (cat#HK211‐01, HycultBiotech, Plymouth Meeting, PA) were assessed using ELISA kits following the manufacturers' recommendations.
2.4.2. Analysis of human cytokines
Human cytokines in the plasma were analysed by Bio‐Plex® Pro Human Cytokine 27‐plex Assay (cat#M500KCF0Y, BIO‐RAD, Hercules, CA) according to the manufacturer's instructions.
2.4.3. Analysis of cytokines in the lung tissue from rats
Levels of several cytokines (IL‐1β, IL‐6, TNF‐α and KC/GRO) in homogenates of lung tissue were analysed with an electrochemical ELISA (cat#K15059G) using the MesoScale Discovery platform (Rockville, MD) (Li et al., 2018).
2.4.4. Protein assay
Levels of total protein in plasma were measured using a bicinchoninic acid protein assay kit (cat#23225, Pierce, Rockford, IL) according to the manufacturer's instructions.
2.4.5. Haemolytic complement activity assay
The complement haemolytic 50% activity (CH50) assay was performed to determine the function of the complement classical pathway as described previously (Li et al., 2013). Briefly, antibody‐sensitized chicken red blood cells (Colorado Serum Company, Denver, CO, cat#31151) were incubated for 30 min at 37°C with serial dilutions of rat serum samples in gelatin‐veronal buffer (GVB++ buffer, Complement Technology, Tyler, TX, cat#B100). After centrifugation, the supernatant was transferred to a new plate, and the absorbance of the supernatant was determined at 405 nm by SpectraMax microplate reader (Molecular Devices, San Jose, CA). The fold serum dilution inducing 50% of complement haemolytic activity was determined and presented as the CH50 value.
2.4.6. Levels of myeloperoxidase and high mobility group box 1 protein in plasma
Myeloperoxidase (MPO) levels in human/rat plasma were determined by quantitative sandwich enzyme‐linked immunosorbent assay (cat#HK324‐02 and cat#HK105‐02) using kits obtained from Hycult Biotech (Plymouth Meeting, PA). Rat plasma levels of the protein, high mobility group box 1 (HMGB1), were measured by ELISA (cat#ST51011, IBL‐International, Baldwin Park, CA) according to the manufacturer's instructions.
2.5. Histopathological evaluation
2.5.1. Immunohistochemical (IHC) staining in rat tissues
Lung tissue was processed for IHC staining as described previously (Li et al., 2018). Briefly, after 4% paraformaldehyde fixation for 24 h, the tissues were transferred to 20% sucrose (w/v) in PBS overnight at 4°C, followed by freezing in the Tissue‐Tek OCT mounting medium (Sakura, Japan). Frozen‐tissue sections were then cut at 5‐μm thickness with a cryostat and mounted onto glass slides. The slides were fixed in cold acetone or 4% paraformaldehyde for 20 min followed by permeabilization with 0.05% Triton X‐100 in PBS for a further 10 min. Next, sections were blocked by PBS/10% normal goat serum and incubated with primary antibodies, including mouse anti‐rat C5b‐9 (IgG1, clone#2A1, cat#HM3033, diluted at 1:20 in PBS/10% normal goat serum, Hycult Biotech, Plymouth Meeting, PA), chicken anti‐mouse C3/C3a (IgY, polyclonal, cat#ab48581, diluted at 1:100 in PBS/10% normal goat serum, Abcam, Cambridge, MA), rabbit anti‐mouse MPO (IgG, polyclonal, cat#ab9535, diluted at 1:100 in PBS/10% normal goat serum, Abcam, Cambridge, MA), and mouse anti‐rat ICAM‐1 (IgG1, clone#1A29, cat#ab171123, diluted at 1:100 in PBS/10% normal goat serum, Abcam, Cambridge, MA) overnight at 4°C. Following extensive washing, sections were incubated with secondary goat‐anti‐rabbit/mouse/chicken IgG‐labelled with Alexa Fluor 488‐conjugated antibodies (cat#150077/cat#150113, diluted at 1:400 in PBS/10% normal goat serum, Abcam, Cambridge, MA) or 594‐conjugated antibody (cat#ab150080/cat#ab150116, diluted at 1:400 in PBS/10% normal goat serum, Abcam, Cambridge, MA) for 1 h at room temperature. Subsequently, after washing, sections were mounted with ProLong Gold Antifade solution containing 4′,6‐diamidino‐2‐phenylindole (cat#P36931, Invitrogen, Carlsbad, CA) for staining the nuclear DNA and then visualized at a 200× magnification under a fluorescence microscope (Nikon Eclipse Ti). Experiments with negative controls were conducted by substituting the primary antibodies with corresponding immunoglobulin isotypes. The IHC procedures are used to comply with the BJP's recommendations and requirements (Alexander et al., 2018).
2.5.2. Quantification of IHC staining
The numbers of positively stained cells and the total numbers of cells in 20 randomly selected fields of a given section were determined based on particle size using ImageJ software (ImageJ 1.50b). Three to four animals in each group were analysed. The data were not subjected to statistical analysis owing to the small group size of n < 5.
2.6. Tissue pathological evaluation and semi‐quantitative scoring
Histological images of entire sections for each individual rat tissue were recorded with a 10× objective under a slide scanner (Axio Scan. Z1 v1.0, Zeiss, Germany), and representative images of each group were presented (magnification = 400× for lung, brain, and liver or 100× for jejunum). Semi‐quantitative scoring of lung, brain, liver, and jejunum tissues was performed in 30 randomly selected fields at a 400× or 100× magnification by a pathologist blinded to the treatment information, and the criteria for the evaluation of histological injury scores are as follows.
For the lung injury score, four parameters (alveolar fibrin oedema, alveolar haemorrhage, septal thickening, and intra‐alveolar inflammatory cells) were scored on each haematoxylin and eosin (H&E) stained slide based on (1) severity (0: absent; 1, 2, 3 and 4 for increasingly severe changes) and (2) the extent of injury (0: absent; 1: <25%; 2: 25%–50%; 3: 50%–75%; 4 > 75%). The total injury score for each slide was calculated as the sum of the severity plus the extent of injury (Dalle Lucca et al., 2013).
For the scoring brain injury score, we undertook the approach previously described (Li et al., 2013). Two parts of the brain tissue were scored, including the frontal cortex and hippocampus. Damage was assessed using five distinct morphological parameters: neuronal morphological changes (shrinkage of the cell body, pyknosis of the nucleus, disappearance of the nucleolus, and loss of Nissl substance, with intense eosinophilia of the cytoplasm), neuronal loss, cytotoxic oedema, vasogenic oedema, and inflammatory cell infiltration in the brain cortex and hippocampus. The changes were scored according to their extent (score 0, 1, 2, 3, and 4 for an extent of 0%, <25%, 25%–50%, 50%–75%, and 75%–100%, respectively) and the severity of the injury (score 0 = normal histology, score 1 = slight, 2 = mild, 3 = moderate, and 4 = severe alterations).
For the hepatic injury score, four parameters, including vascular congestion, hepatocyte death, degeneration, and inflammation were considered (Dalle Lucca et al., 2013), and these parameters were assayed for severity (score 0 for no change, score 1, 2, 3 and 4 for more severe changes) and for the extent of injury (0: absent; 1: <25%; 2: 25%–50%; 3: 50%–75%; 4: >75%). The injury score represents the sum of the extent and the severity of injury.
For the jejunum, each slide was scored according to the following scale: 0, normal villi; 1, villi with tip distortion; 2, villi lacking goblet cells and containing Guggenheim's spaces; 3, villi with patch disruption of the epithelial cells: 4, villi with exposed but intact lamina propria and epithelial cell sloughing; 5, villi in which the lamina propria was exuding; and 6, haemorrhaged or denuded villi.
2.7. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Statistical analysis was undertaken only when a minimum of n = 5 independent samples was acquired. Demographic data are presented as interquartile ranges (IQR) or percentages as appropriate. Other data are presented as mean ± SEM. To reduce unwanted sources of variation between the time points by dilution effects of the early fluid transfusion, complement and cytokine data obtained from trauma patients were normalized to the measured plasma protein levels. The animal experiments were randomized and data analysis was blinded. Group size is the number of independent values, and statistical analysis was performed using these independent values. Intergroup comparisons for complement factors and inflammatory mediators in patients or animals were assessed using Mann–Whitney U test, or unpaired t‐test with Welch's correction. Correlation analyses were analysed using Spearman's correlation test. For animal survival analysis, Kaplan–Meier plot and log‐rank test were performed. Two‐way ANOVA was performed to compare the animal groups on particular variables. Bonferroni post hoc tests were conducted only if F value in ANOVA achieved P < 0.05. The receiver operating characteristic curve (ROC) and area under the ROC survive (AUC) were performed using univariate logistic regression. The optimal cutoff values were obtained by ROC curves analysis with Youden index. P < 0.05 was considered significant. All data were included, and none were treated as outliers. All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA).
2.8. Materials
Nomacopan was supplied by Akari Therapeutics plc (London, UK). Ketamine was supplied by Zoetis Manufacturing and Research Spain, S.L. (Girona, Spain). xylazine by Akorn, Inc. (Lake Forest, IL, USA), and isoflurane by Baxter (Deerfield, IL, USA).
2.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander, Christopoulos et al., 2021; Alexander, Fabbro, et al., 2021; Alexander, Kelly, et al., 2021).
3. RESULTS
3.1. Patient demographics and clinical data
Patient demographics and clinical data are shown in Table 1. Most patients were male (98%) with a median (IQR) age of 25 (22–30) years, and an injury severity score of 16 (9–24). The predominant cause of injury was a blast (69%). Forty‐five of the 54 patients (83%) had TBI, of whom most sustained mild TBI (80%). During treatment in the hospital, seven patients (13%) used mechanical ventilation, and five patients (9%) died. All patients received operative care and intravenous resuscitation fluids. Ten healthy controls, six male and four female with a median (IRQ) age of 36 (28–41) years were sampled once, and served as controls. No clinical data were collected for healthy controls.
3.2. Early complement activation in casualties
To avoid misinterpretation of the measured complement components (C3a, C4d, C5a, sC5b‐9, and Bb) (Figures 2 and S1) by the dilution effects of early fluid infusion, data were normalized to total plasma protein levels (Burk et al., 2012). Levels of activated complement factors, including C5a, sC5b‐9, and C4d, were significantly higher in injured patients on admission compared with healthy controls. These factors remained elevated at 8 and 24 h after admission. The mean values for C5a, sC5b‐9, and C4d at 24 h increased by 3.2‐, 3.6‐, and 7.0‐fold, respectively, compared to controls (Figure 2a,b,d). There was a strong positive correlation between complement C5a and sC5b‐9 with Bb levels (Figure 2e,f) on admission. However, C3a was not significantly changed during the observation period (Figure S1A, B). Patients with blast injuries showed a peak in activated complement factors at 8 h after admission during 24 h observation period (Figure S1C–F). Patients with gunshot‐wound injuries generally had elevated levels of C5a, sC5b‐9, Bb, and C4d throughout the 24 h after admission (Figure S1C–F), although Bb levels clearly peaked at 8 h (Figure S1E). This early Bb elevation indicates that the alternative complement pathway, unlike the classical or mannose‐binding lectin complement pathways, contributed to early complement activation in these patients.
FIGURE 2.
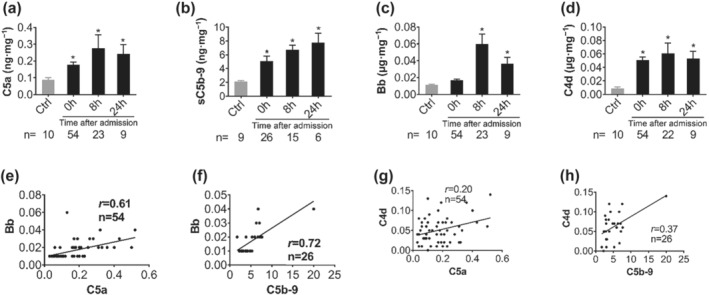
Early activation of complement terminal and alternative pathways after trauma in military casualties. Plasma levels of C5a (a), sC5b‐9 (b), Bb (c), and C4d (d) were measured to determine activation of terminal complement (C5a and sC5b9), alternative pathway (Bb), and classical and lectin pathways (C4d) by ELISA in healthy donors, and trauma patients on admission to hospital, 8 and 24 h after admission. The data are expressed as μg·mg−1 plasma protein, except for C5a and C5b‐9, which are shown as ng·mg−1 plasma protein. Data shown are means ± SEM, from the group sizes shown (n). *P < 0.05, significantly different from values in healthy subjects (Ctrl); Mann–Whitney U test). (e–h) Correlation of Bb and C4d with either C5a or sC5b‐9 in the injured patients at admission. Correlation analysis between complement factors bb or C4d and C5a or sC5b‐9 were performed using Spearman's rank correlation. Data are shown as individual values with the correlation coefficient (rs). Significant correlations (P < 0.05) are indicated by boldface type.
3.3. Early complement activation and early systemic inflammatory response in casualties
We measured the plasma levels of inflammatory mediators and determined the relationship between plasma concentrations of complement activation products (C3a, C5a, sC5b‐9, Bb, C4d) and inflammatory mediators at admission after trauma. We found that the levels of pro‐inflammatory mediators (TNF‐α, IFN‐γ, IL‐1β, IL‐6, IL‐8, CCL2 (MCP‐1), MPO, GM‐CSF) (Figure S2A–H), anti‐inflammatory cytokines, including IL‐4, IL‐10 and G‐CSF (Figure S2I, J, L), and regulatory cytokines such as IL‐12 and CCL4 (MIP‐1β) (Figure S2O, P) were significantly elevated on admission and at 8 h when compared with the healthy controls. Most of these mediators declined and returned to their baseline levels within 24 h. The ratios of the pro‐inflammatory factors (IL‐6, CCL2, or GM‐CSF) to IL‐10, a classical anti‐inflammatory cytokine, were significantly higher than in the controls (Figure S2S).
At admission, blood levels of IL‐6 and MPO strongly correlated with C5a, sC5b‐9, and Bb levels (Figure 3). CCL2 and CCL4 levels clearly correlated with the blood levels of C3a, C5a, sC5b‐9, and fragment Bb but not with C4d (Figure 3 and Table S1). Complement protein C4d, a degradation product of the classical/lectin pathways negatively correlated with the pro‐inflammatory cytokines IL‐1β, IL‐17, and TNF‐α (Figure 3j–l).
FIGURE 3.
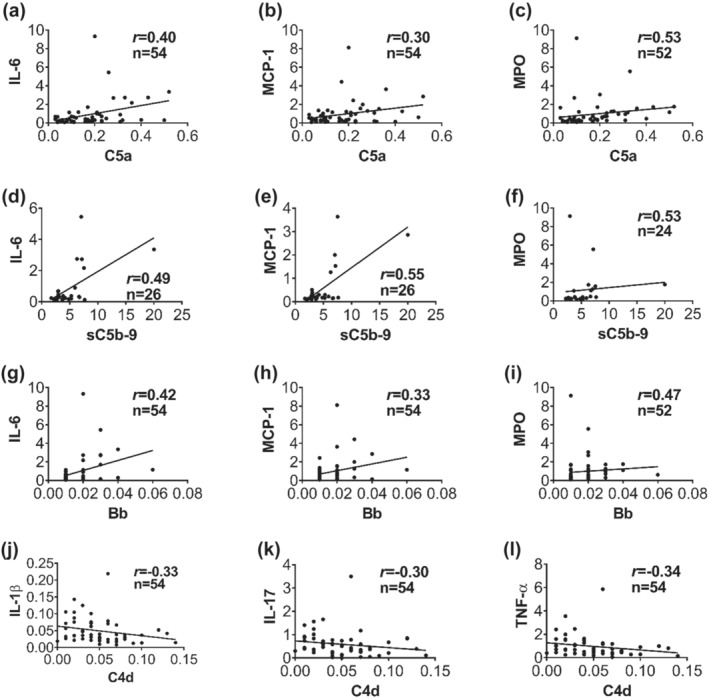
Activation of complement terminal and alternative pathways is related to systemic inflammatory response after trauma in battlefield casualties. Inflammatory factors and cytokines were measured by ELISA and by Bio‐Plex kits, respectively. Positive correlation between plasma concentrations of C5a in the trauma patients and IL‐6 (a), MCP‐1 (b), and MPO (c) in the blood plasma of the patients on admission. Positive correlation of plasma levels of C5b‐9 on admission with IL‐6 (d), MCP‐1 (e), and MPO (f) in the injured patients on admission. Plasma concentration of bb on admission positively correlated with IL‐6 (g), MCP‐1 (h), and MPO (i), whereas the plasma levels of C4d on admission inversely correlated with IL‐1β (j), IL‐17 (k), and TNF‐α (l). Correlation analysis between complement factors (C5a, C5b‐9, bb and C4d) and inflammatory factors/cytokines were performed by using Spearman's rank correlation. Data are shown as individual values with the correlation coefficient (rs). Significant correlations (P < 0.05) are indicated by boldface type.
3.4. Clinical outcomes and early complement activation in casualties
There was a positive correlation between admission plasma levels of C5a/sC5b‐9 (Figure 4a–h) or Bb (Figure 4i–l) and ISS, international normalized ratio (INR), severity of TBI, and crystalloid resuscitation requirements. Plasma levels of C5a, C3a, and Bb at admission were also positively correlated with increased RBC and FFP blood transfusion requirements (Table S2). However, we found an inverse correlation between plasma concentrations of C4d at admission and blood transfusion requirements or TBI (Table S2). No significant correlation was found between C4d plasma levels at admission and ISS and crystalloid resuscitation requirements (Table S2). We next tested the accuracy of admission plasma concentrations of C5a and Bb for distinguishing TBI patients from non‐TBI patients, or distinguishing ventilated patients from non‐ventilated patients using ROC analysis (Figure 4m–o,q). Admission plasma levels of C5a ≥ 0.185 ng·mg−1 plasma protein (AUC = 0.73, specificity = 68%, sensitivity = 71%) (Figure 4m,q) and Bb ≥ 0.015 μg·mg−1 plasma protein (AUC = 0.69, specificity = 65%, sensitivity = 71%) (Figure 4n,q) were associated with the presence of TBI. Admission plasma C5a ≥ 0.195 ng·mg−1 plasma protein was a fair predictor of mechanical ventilation requirements (AUC = 0.75, specificity = 71%, sensitivity = 86%, Figure 4o,q). However, patients who survived had higher levels of C4d than patients who did not survive, and admission plasma C4d ≥ 0.025 μg·mg−1 plasma protein was a fair predictor of mortality (AUC = 0.83, specificity = 78%, sensitivity = 80%) (Figure 4p,q). These data suggest that inhibition of complement terminal pathway or alternative pathway, but not classical/lectin pathways, may be the optimal therapeutic approach to improve clinical outcomes after TH.
FIGURE 4.
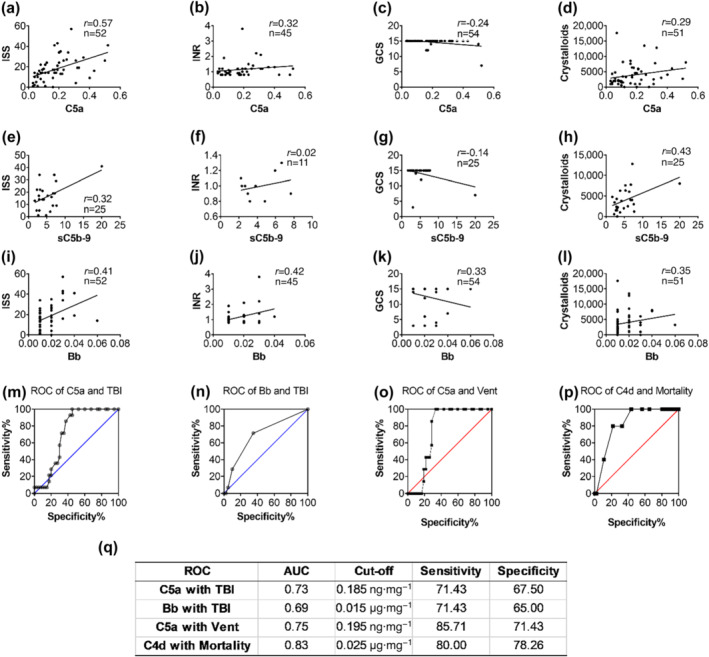
Complement activation correlated with clinical outcomes in trauma patients on admission. (a–d) Correlation of C5a plasma levels with clinical scores, infused fluid and INR, a standard coagulation test; (e–h) correlation of C5b‐9 with clinical scores, infused fluid and INR; (i–l) correlation of fragment bb with clinical scores, infused fluid and INR; (m, n) the receiver‐operator characteristic curve (ROC) analysis tested diagnostic ability of C5a and fragment bb in identifying traumatic brain injury (TBI); the cut‐off value, specificity, and sensitivity are shown (q); (o) patients on mechanical ventilation had significantly higher plasma levels of C5a in comparison to those not‐ventilated; (p) ROC analysis showed that the C4d plasma levels had a strong predictive value for the survival; the cut‐off value, specificity and sensitivity are presented (q). Data are shown as individual values with the correlation coefficient (rs). Significant correlations (P < 0.05) are indicated by boldface type.
Having shown that complement was activated in combat casualties and that the alternative pathway activation and terminal activation pathway were associated with inflammatory mediators that may drive organ damage, we investigated the effect of inhibiting terminal complement activation in a rat model of TH.
3.5. Pilot study results
We first carried out a pilot study with the following goals: (1) test the effect of surgical cannulation on complement activation; (2) evaluate blast overpressure intensity; (3) measure TH‐induced complement activation; (4) determine an effective therapeutic regimen; and (5) assess the efficacy of the therapeutic regimen on organ damage and survival. Our preliminary data showed that surgical cannulation alone triggered activation of the complement cascade, reducing functional classical pathway complement activity by >50% (Figure S3B and Table S3); and that TH further rapidly activated the complement system (Figure S3B–D). Complement activation peaked about 1–2 h after blast injury (or after the shock phase in haemorrhaged rats) and then gradually recovered. In injured rats, low serum levels of C1q (a subunit of the C1 enzyme complex) (Figure S3C) and of C3 (Figure S3D) also indicated that early consumption of complement factors was driving the decrease in CH50. Pretreatment with nomacopan prior to blast (Figure S3E), increased the base excess (Figure S3F), MAP (Figure S3G), and survival (Figure S3H). It also improved organ‐damage scores on histology including those of lung, brain, and liver (Figure S3I). We then decided to address the problem of surgery‐induced complement activation by introducing a recovery period between surgery and the injury.
3.6. Nomacopan improved haemodynamics, respiration, and blood chemistry
We used the data generated from the pilot study to formulate the design of the main study in which we (1) allowed the animals to recover full complement activity by adding a post‐surgical recovery period of 5–7 days before the study; (2) increased haemorrhage volume (from 40% to 52%); and (3) adjusted the treatment window to administer the study drug after the blast but before haemorrhage to be consistent with early battlefield use (Figure 5a).
In this main study (Figure 5a and Table S3), the MAP in the injured/untreated animals (B + H) was 97.27 (±2.93) mmHg at baseline (Figure 5b). MAP decreased to 30.35 (±2.55) mmHg at the end of haemorrhage. In the injured/treated (NOM) group, the MAP baseline was 93.96 (±1.19) mmHg and decreased to 26.57 (±1.71) mmHg at the end of haemorrhage. However, injured/treated animals showed significantly increased MAP in the first 30 min of the resuscitation phase when compared with injured/untreated rats. CH50 in the blood of injured rats started to decrease 15 min after blast injury due to complement consumption, reached its lowest level 1 h after shock, and then returned to baseline level 11 h after blast injury (Figure 5c). CH50 was even higher than 100% in injured/untreated rats at the end of the observation period. In the injured/treated rats, CH50 was significantly lower than in the injured/untreated group and almost completely inhibited at the end of haemorrhagic shock, approximately 45 min after the drug administration. In the injured/treated group, CH50 remained at a very low level until the end of the experiment.
Nomacopan treatment also improved blood chemistry (Table S4). The pH value in injured/untreated animals changed from a baseline of 7.44 (±0.02) to 7.07 (±0.37) at 4 h after the blast, while the pH in injured/treated animals had almost returned to the baseline level at that point (7.41 ± 0.16 to 7.44 ± 0.03). The PaO2/FiO2 ratio in the injured/treated rats was clearly higher at the end of the shock period and throughout the first 4 h when compared with injured/untreated rats (Figure 5d). There was a significant difference in the lactate level (Figure 5e), and a late, although not significant, difference in the base excess (Figure 5f). Injured/treated animals had significantly lower potassium levels at the end of the observation period in comparison to injured/untreated rats (Figure 5g).
3.7. Nomacopan reduced systemic and local inflammatory responses
Next, we measured inflammatory cytokines in the plasma. The level of HMGB1 protein increased after TH, reaching a peak 4 h after injury. Nomacopan treatment significantly reduced HMGB1 levels at 2 h (Figure 6a) after the blast. MPO gradually increased after TH, and nomacopan treatment significantly slowed this increase (Figure 6b).
FIGURE 6.
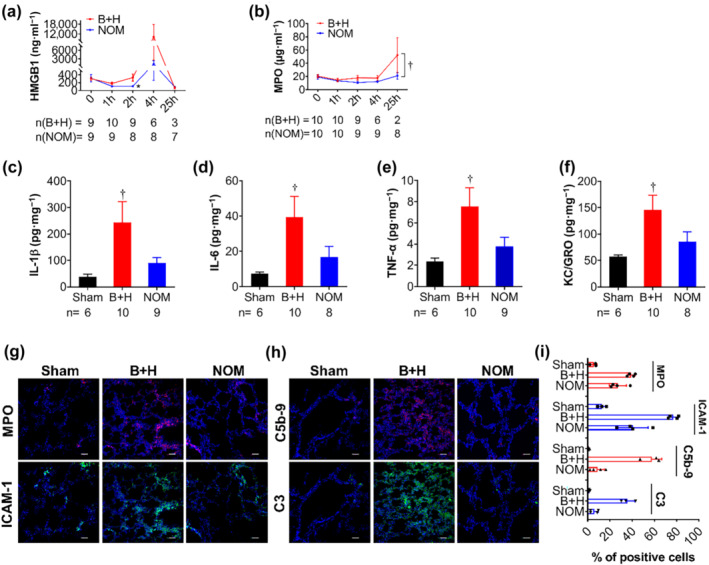
Effect of nomacopan treatment early after injury on systemic and local inflammatory response in rats after blast injury and haemorrhage. (a, b) Inflammatory mediators HMGB1and MPO, were measured by ELISA. Data shown are means ± SEM from groups of rats, sizes (n) as indicated. *P < 0.05, significantly different from B+H at this time point; unpaired t‐test with Welch's correction; †P < 0.05, group data significantly difference in group data: two‐way ANOVA. (c–f) Cytokines/chemokines were measured in the lung homogenates. Data shown are means ± SEM from groups of rats, sizes (n) as indicated. †P < 0.05, significantly different from sham; Mann–Whitney U test. (g) Lung tissue at necropsy or at the end of study were collected, fixed by PFA, and stained by IHC. The representative images of immunostaining of MPO (red) and ICAM1 (green) are shown; (h) antibodies for detecting C5b‐9 (red) and C3 (green) were used to detect complement deposition in the lung tissue. Representative images (g, h) are shown (original magnification, 200×), and semi‐quantitative analysis (i) of the positively stained cells to total cells are presented. Data are shown as individual values with means +SEM; n = 3‐4 animals per group. Scale bar = 50 μm. Abbreviations: B + H = blast + haemorrhagic shock; NOM = nomacopan treated rats; MPO = myeloperoxidase
In lung homogenates, we found significantly increased levels of IL‐1β, IL‐6, TNF‐α, and KC/GRO in rats subjected to TH compared to the sham group (Figure 6c–f). These tissue cytokine levels trended lower with nomacopan treatment, but the reduction was not significant.
Immunohistochemical (IHC) images demonstrated that the MPO level was increased in the lung tissue after TH. However, after treatment with nomacopan, MPO expression was reduced (Figure 6g,i). ICAM‐1, a marker of endothelial injury, was expressed in the lung tissue of injured/untreated rats. Treatment with nomacopan reduced ICAM‐1 expression in the lung tissue (Figure 6g,i). It also reduced C5b‐9 and complement C3 deposition in lung tissue (Figure 6h,i). These findings indicate that treatment with nomacopan reduced both systemic and local inflammatory responses.
3.8. Effect of nomacopan on MODS and survival
Histological evaluation revealed that nomacopan treatment reduced the severity of MODS following TH. After TH, extensive cellular inflammatory infiltrates were present in the pulmonary tissue; the majority of these were neutrophils and macrophages. Lung oedema was severe after TH but nomacopan treatment alleviated this phenomenon (Figure 7a). Typical neuronal apoptosis, neuronal loss, and neuronal degeneration were seen in the cerebral cortex and hippocampus, which were significantly reduced by nomacopan treatment (Figure 7a). TH induced hepatic tissue damage characterized by hepatic cell apoptosis or necrosis and severe thrombosis. Nomacopan treatment alleviated this damage (Figure 7a). TH also induced intestinal mucosal injury. Photomicrographs of jejunal tissue show that TH denuded the villi to the level of the lamina propria with inflammatory cell infiltration while nomacopan clearly helped to preserve villus structure and improved the recovery of jejunal mucosa (Figure 7a). Semi‐quantitative scoring of injury severity on histology further validated these observations (Figure 7b).
FIGURE 7.
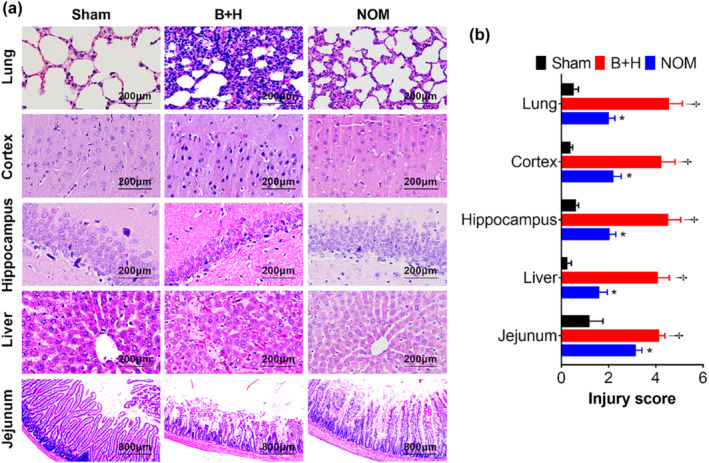
Effect of nomacopan treatment on histological changes in rats after blast injury and haemorrhage. (a, b) Representative H&E photomicrographs of organs collected at necropsy (a), and organ injury scored based on the criteria described in Section 2 (b). The data are presented as means ± SEM. †P < 0.05, significantly different from sham; *P < 0.05, significantly different from B + H; Mann–Whitney U test).
We tested the efficacy of two treatment windows of nomacopan in the rats. Early treatment with nomacopan 15 min after the blast but immediately before haemorrhage significantly improved survival by 50% (Figure 8), whereas later treatment with nomacopan during the resuscitation phase immediately after TH (NOM 60 min) did not improve survival compared to the vehicle control (Figure S4B). Nomacopan's beneficial effect on survival depended on its administration time, which resulted in early complement inhibition (Figure S4A, B).
FIGURE 8.
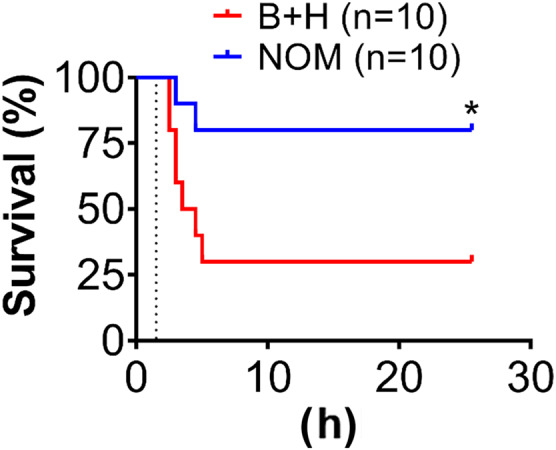
Effect of nomacopan on injury‐induced mortality. All rats were subjected to blast and severe haemorrhage (B + H), treated with vehicle control (saline) or nomacopan (NOM), and monitored for survival up to 25 h after blast injury. Survival distribution of these two groups was determined by using the log‐rank Mantel‐Cox test. *P < 0.05, significantly different from B + H.
4. DISCUSSION
Haemorrhage and/or TBI are the leading cause of death in the prehospital phase of care (Alam, 2017), with most deaths occuring within a few hours after trauma despite recent advances in trauma care (Alam, 2017; Brohi et al., 2019; CRASH‐2 trial collaborators et al., 2010). Approximately 90% of battlefield casualties die in the prehospital environment, with 85% of these prehospital deaths due to bleeding, and of those about 25% are deemed potentially survivable (Alam, 2017; Eastridge et al., 2012). Furthermore, although patients with TH may survive the initial injury by means of haemorrhage control and resuscitation, they may still succumb to a complex series of inflammatory events ending in multi‐organ failure that is the major contributor to late mortality (Arbore & Kemper, 2016; De Blasio et al., 2019; Hepburn et al., 2007; Lord et al., 2014; Mannucci & Levi, 2007). Thus, the current primary goal of early care is to keep patients alive long enough and to maintain organ functions, while awaiting transport to higher echelons of care for definitive treatment.
In this study, using a translational medicine approach, we identified a complement therapeutic target (C5) in a cohort of military casualties, selected a clinical drug candidate (nomacopan) with desirable properties (a bifunctional anti‐inflammatory protein, thermostability, easy‐to‐transport/store/reconstitution, amenable‐to‐manufacture in single‐use dual chamber autopen, and multiple routes of administration) that make it suitable for battlefield/prehospital use, and evaluated its efficacy in a clinically relevant animal model of blast injury and haemorrhagic shock.
We first investigated the relationship between complement activation, cytokine production, and clinical variables in casualties. The study revealed that several complement factors (C5a, sC5b‐9, Bb, and C4d) were significantly increased on admission to the emergency department of a Combat Support Hospital, suggesting that injury induces rapid complement activation in patients. This is consistent with previous findings that complement was activated in animal models of trauma (Dalle Lucca, Chavko, et al., 2012; Li et al., 2013) and in both civilian burn (Li, Zhao, et al., 2019) and non‐burn trauma patients (Burk et al., 2012; Ganter et al., 2007). We observed that the activation of the terminal complement and the alternative pathways was associated with ISS, TBI, coagulopathy, the requirements of crystalloid fluid resuscitation and blood transfusion, and the need of mechanical ventilation. Other studies showed that activation of these pathways in civilian trauma patients was directly related to clinical outcomes, including acute lung injury, renal injury, TBI, and mortality (Bellander et al., 2011; Burk et al., 2012; Ganter et al., 2007). These observations are consistent with our previous publications demonstrating that complement blockade by decay‐accelerating factor or C1 inhibitor mitigated organ damage and increased survival of rats and pigs subjected to blast injury and haemorrhagic shock (Campbell et al., 2016; Dalle Lucca et al., 2011, 2013; Dalle Lucca, Li, et al., 2012; Li et al., 2013; Lu et al., 2011). Demonstration that mice deficient in the gene for factor B had significantly decreased neuronal cell death after experimental brain injury (Leinhase et al., 2006) also indicates the involvement of the alternative pathway in the body's response to trauma. Further highlighting the importance of complement, De Blasio et al. (De Blasio et al., 2019) reported that complement system activation in human brain contusions depends on the lectin pathway and is likely on amplification via the alternative pathway. Increased levels of MASP‐2, a driving enzyme of lectin pathway activation, were associated with TBI severity. Moreover, complement activation is elicited not only by the traumatic event itself, but also by secondary insults after trauma (Bellander et al., 2011) and therapeutic interventions such as extracorporeal life support, mechanical ventilation, damage‐control surgery, instrumentation, volume resuscitation, and blood transfusion (Al‐Fares et al., 2019; Huber‐Lang et al., 2020; Nilsson et al., 2007). Immunomodulatory approaches aim to rebalance trauma‐induced complement activation and thus may change trauma management procedures to improve outcomes.
Huber‐Lang et al reported that thrombin functions as a C5 convertase and can independently activate the complement terminal pathway in C3‐null mice (Huber‐Lang et al., 2006). Unlike TCA in this study, circulating C3a was not significantly altered in the military casualties during the observation period when compared with other published findings in multiply injured patients with a similar ISS range (Ganter et al., 2007), indicating that extrinsic/common pathway‐induced TCA is the major pathway of complement activation after trauma (Ganter et al., 2007; Hoth et al., 2014), or the difference in storage time may affect the observed differences.
Complement is a critical component of innate immunity and a major initiator of the inflammatory reaction. In this study, we found that increased complement factors C3a, C5a, sC5b‐9, and Bb correlated with the levels of many cytokines, suggesting an interplay between the complement system and other immune systems. The complement system also functions as a bridge between innate and adaptive immunities (Chakraborty et al., 2018; Ricklin & Lambris, 2007). Specifically, complement's role in the regulation of the inflammatory response may be through activation of innate immune cells (neutrophils, monocytes, mast cells, macrophages and dendritic cells) and adaptive immune cells (Th1, Th2, Th17, natural Treg, and B cells) via a synergistic interaction between C3a/C5a‐C3a receptor/C5a receptor‐MAPK‐NF‐κ‐NLRP3 inflammasome and HMGB1‐TLR4‐MAPK‐NF‐κ‐NLRP3 inflammasome in response to trauma (Arbore & Kemper, 2016; Yang et al., 2020). Indeed, our data showed that increased plasma levels of C5a, sC5b‐9, and Bb were positively correlated with blood levels of MPO, IL‐6, CCL2, and CCL4, and with INR. The positive correlation between activated complement products (C5a, sC5b‐9, Bb) and INR in military casualties indicates that coagulation pathways may contribute to early complement activation, which is aligned with the previous findings (Amara et al., 2010; Kenawy et al., 2015). Furthermore, increased concentrations of MPO and IL‐1β in plasma, and prolonged PT/increased INR, were associated with clinical variables like blood transfusion requirements, SIRS and hypotension after trauma. These findings suggest that an interplay between these signalling pathways may synergistically increase morbidity and mortality after trauma.
Previous studies reported that C4d, C5b‐9, and C3d deposited on the surface of RBC, the deposition of C4d and C5b‐9 decreased faster over course of a 3‐day study in subjects with ISS < 9, and the complement deposition contributed to organ damage in trauma (Muroya et al., 2014; Satyam et al., 2020). In alignment with these studies, our data demonstrated that admission plasma levels of C5a, C3a, and Bb positively correlated with increased RBC transfusion requirements, ISS and TBI, suggesting that complement decoration on the RBC surface may, at least partly, play a role in organ damage and increased RBC transfusion requirements after trauma. Further studies on the relationship between complement deposition on RBC and clinical outcomes should be more informative. Typically, C4d is increased after trauma in animals and patients, as reported earlier (Ganter et al., 2007). It is interesting to note that the C4d level at admission was significantly higher in surviving trauma patients, compared with levels in those who died. The C4d level inversely correlated with many clinical variables including blood transfusion requirements and cytokine storm, suggesting that classical and/or lectin complement pathway activation may play a role in protecting against damage to the host. The negative correlation between C4d and cytokines may be due to inhibition of cytokine secretion in monocytes and macrophages via C4d‐Ig‐like transcript interaction (Hofer et al., 2016). Indeed, genetic manipulation of complement levels and activation in murine models demonstrated that the classical and/or lectin complement pathways play a crucial role in clearance of damaged cells and host defence against infection (Gullstrand et al., 2009; Held et al., 2008).
Several studies have reported that the treatment with nomacopan improved morbidity and mortality in TBI (Fluiter et al., 2014), bacterial infectious disease, and sepsis models, particularly when used together with an anti‐CD14 inhibitor (Hellerud et al., 2017; Huber‐Lang et al., 2014; Keshari et al., 2017; Skjeflo et al., 2015), but no information has been published on the efficacy of nomacopan in TH. Data from the present study showed that early nomacopan treatment significantly increased survival in rats after TH. Nomacopan promptly abolished TCA within 45 min of drug administration, as measured by CH50. Nomacopan also improved haemodynamics and blood chemistry, and significantly reduced systemic and local inflammatory responses. In addition, nomacopan prevented MODS as substantially less tissue damage was observed in the lungs, brain, small intestine, and liver tissues. The survival of rats subjected to TH was significantly better in those receiving nomacopan than in rats receiving the saline control.
Complement activation is the early innate immune response activated soon after traumatic injury (Huber‐Lang et al., 2018). We have previously shown that the complement activation appears as early as 3 h after blast injury in rats (Dalle Lucca, Chavko, et al., 2012; Li et al., 2013) and by 3 h after haemorrhagic shock in swine (Dalle Lucca, Li, et al., 2012). Here, we found that the complement was rapidly activated even after surgical cannulation, with TCA, as measured by CH50, reduced by more than 50% immediately after surgery in the pilot study. To avoid the surgical cannulation interfering with our experimental goals, we allowed the animals to recover full complement activity over 5–7 days before exposing the animals to trauma and haemorrhagic shock.
In injured animals, we observed that the TCA as measured by CH50 was reduced by about 65% 1 h after haemorrhagic shock. This quick decrease of CH50 indicated a rapid complement activation after injury. In addition, complement deposition, including C5b‐9 and C3, was soon detected in the lung tissue. These findings suggest that both systemic and local complement activation occurred in the injured animals. Our rodent model of traumatic haemorrhagic shock closely mimics the clinical setting, as a typical early complementopathy is observed after trauma (Burk et al., 2012).
Traumatic injury, including blast and haemorrhage, triggers a complex cascade of post‐traumatic events that are related to inflammatory and immune responses. Excessive or maladaptive activation of inflammatory pathway is considered to contribute to MODS, and eventually death (Ciesla et al., 2005; Jaffer et al., 2010). We found increased TCA measured by CH50 (up to 150%) in injured but untreated rats at 25 h at the end of the observation period. We explain this phenomenon by referring to the systemic acute‐phase immune response that includes pro‐inflammatory cytokines and increased C3 (Gruys et al., 2005). Thus, this study correlates with previous reports, which also observed the elevation of multiple inflammatory factors and cytokines, such as MPO, NF‐κB, TNF‐α, IL‐1β, IL‐6, IL‐12, IL‐13 and IL‐18, both systemically and locally after blast and haemorrhagic shock in rats and swine (Dalle Lucca, Chavko, et al., 2012; Gill et al., 2017; Li et al., 2013; Li et al., 2018; Li, Yang, et al., 2019; Sillesen et al., 2014; Yang et al., 2019). Synchronous activation of pro‐inflammatory and anti‐inflammatory cytokines was also observed in battlefield trauma patients as presented in this study. The inflammatory response seen in battlefield trauma patients correlated with clinical variables. Modulation of trauma‐induced inflammation may therefore be important in improving clinical outcomes. In this study, we found that nomacopan significantly attenuated both systemic and local levels of HMGB and MPO and, therefore, systemic and local inflammatory responses.
Nomacopan may alter the inflammatory cytokine milieu in several ways. First, nomacopan inhibits TCA (C5a and C5b‐9) and consequently prevents infiltration of inflammatory cells via C5a, a critical neutrophil and monocyte chemotactic factor (Guo & Ward, 2005; Ward et al., 2016). Accordingly, we found that inflammatory cell infiltration in the lung, brain and liver was reduced after nomacopan treatment. Second, nomacopan may inhibit DAMP‐induced inflammatory responses. After trauma, tissue debris, ATP, mtDNA, potassium, haem, reactive oxygen species (ROS), F‐actin, HMGB1 and other DAMPs are released. These can induce a rapid inflammatory response via multiple signalling pathways, including TLR‐MyD88‐MAPK‐NF‐κB‐inflammasome and ATP‐P2X7‐inflammasome pathways (Bortolotti et al., 2018; Swanson et al., 2019; Yang et al., 2020), with complement functioning as an initial and amplifying mediator (Li et al., 2018; Ratajczak et al., 2018; Triantafilou et al., 2016; Yang et al., 2020). Indeed, systemic and local levels of inflammatory mediators (HMGB1, MPO, IL‐1β, IL‐6, TNF‐α, KC, ICAM‐1, C3, and sC5b‐9) were significantly reduced in our rodent trauma model after nomacopan treatment. Unexpectedly, decreased lung C3 deposition in nomacopan‐treated animals was noted, indicating that C5b‐9‐induced tissue damage and/or C5a‐C5aR‐mediated cellular infiltration/activation may have triggered complement activation, neo‐synthesis, and subsequent C3 deposition/expression (Hepburn et al., 2007).
We observed that nomacopan treatment clearly improved haemodynamic and blood chemistry variables. During the resuscitation period, nomacopan significantly increased the MAP in injured rats when compared to placebo. This finding is consistent with the previous report that administration of an anti‐C5 antibody reduced fluid requirements and improved responsiveness to fluid resuscitation in a rat model of haemorrhagic shock (Peckham et al., 2007). It is reported that complement activation products directly or indirectly alter vascular tone after trauma and haemorrhagic shock (Karasu et al., 2019). Thus, C5a induces a reversible decrease of the MAP and an increase in central venous pressure when used in vivo in rabbits (Lundberg et al., 1987). Nomacopan may reverse the C5b‐9‐induced damage to the endothelial barrier and reduce vascular leakage caused by blast and haemorrhage (Wang & Chen, 2016). Moreover, the significantly lower plasma level of potassium in nomacopan‐treated rats implies a C5b‐9‐mediated K+ efflux (Triantafilou et al., 2016). C5a‐C5aR‐induced electrolyte imbalance (Denk et al., 2017; Karasu et al., 2019), and/or tissue damage may have contributed, at least partly, to hyperkalemia‐induced early death after TH.
Because of unbound nomacopan's short half‐life, Ort et al. suggested daily subcutaneous injections to maintain complement inhibition (Ort et al., 2020). In an experimental setting, Barratt‐Due et al. applied a continuous infusion of the drug in order to inhibit newly synthesized C5 (Barratt‐Due et al., 2013). Our treatment with nomacopan was performed 15 min after blast injury and immediately before haemorrhage. When treatment was delayed until 60 min after TH, nomacopan showed no effect on survival at 25 h. This finding is aligned with a report that administration of nomacopan 15 min after TBI in mice reduced neuropathology and improved neurological performance, whereas delaying treatment to 30 min after TBI was not beneficial (Fluiter et al., 2014). These observations indicate that there is an optimal therapeutic window for timely and effective intervention after TH.
Our studies have certain limitations. The limited data set at 8 and 24 h might have been biased by several factors related to patient evacuation, early death, or recovery to the point that they were not available for further sampling, as well as hospital treatments that may negatively impact the profiles of complement and cytokines. Consequently, we focused on the data acquired on admission and performed correlation analysis only using these data. Additionally, compared with the rat model used in this study, we wished to evaluate patients with a higher risk of mortality. Further analyses from larger datasets with higher mortality rates and stratified injury severity are warranted. Another limitation of the current study is with a few new mechanisms because this current work was designed to focus two major objectives as described in the abstract and Figure 1. Further studies are needed to explore more extensively how C5 blockade creates organ‐protective and pro‐survival phenotypes in cases of TH. Blood samples from injured service members were obtained from 2007 to 2008, shipped frozen to the United States, and stored, whereas samples from the healthy controls were collected in 2013 and assayed the same day. The effects of storage time on the observed differences are unknown, but it is assumed that storage may reduce, rather than increase such levels (Huang et al., 2017; Wagner‐Golbs et al., 2019). This difference in storage times may account for some of the observed differences in C3a profiles between our and other published findings (Ganter et al., 2007). The unique setting of field samples in combat injury limits the ability to address this issue. Nevertheless, our majority of clinical data show that changes in complement, cytokines and coagulation after trauma are a dynamic process, and this point is relevant to both civilian and military critical care physicians.
Taken together, our clinical and preclinical findings demonstrate that early TCA represents a rational therapeutic target for TH, and that early treatment with nomacopan attenuates multi‐organ damage and increases survival in a rat TH model. Therefore, TCA‐targeted therapy could be a promising adjunct to the DCR solution to create a pro‐survival and organ‐protective phenotype for severely injured patients on the battlefield and in prehospital environments.
AUTHOR CONTRIBUTIONS
Conceptualization: YL, LCC, MAN, AEP. Methodology: ZY, YL, BJL, MAN, TDL, PRE, BL, JLB. Investigation: ZY, YL, BJL, MOS, TDL, PRE, BL, JLB. Visualization: YL, ZY. Funding acquisition: LCC, YL, AEP. Project administration: CDH, AEP. Supervision: LCC, YL. Writing – original draft: YZ, MOS, YL. Writing – review & editing: YZ, MOS, PRE, TDL, CDH, BJL, AEP, MAN, LCC, YL.
CONFLICTS OF INTEREST
MAN is an inventor of Coversin and an employee of Akari Therapeutics, which currently develops Coversin as a novel therapeutic agent. The remaining authors declare that the research was conducted in absence of any commercial or financial relationships that could be constructed as a potential conflict of interest. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defence.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Table S1. Correlations of complement and inflammatory mediators/cytokines/chemokines. Abbreviations: FGF basic, basic fibroblast growth factor; G‐CSF, granulocyte‐colony stimulating factor; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; MCP‐1, monocyte chemoattractant protein‐1; MPO, myeloperoxidase; MIP, macrophage inflammatory protein; PDGF‐bb, platelet‐derived growth factor‐BB; RANTES, regulated on activation, normal T cell expressed and secreted; VEGF, vascular endothelial growth factor. n/a, not applicable. The correlation analyses were performed by Spearman's rank correlation. A significant correlation (p < .05) is indicated by boldface type.
Table S2. Correlations between complement/inflammatory cytokines/chemokines and clinical outcomes. Abbreviations: MPO, myeloperoxidase; FGF basic, basic fibroblast growth factor; G‐CSF, granulocyte‐colony stimulating factor; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor. MCP‐1, monocyte chemoattractant protein‐1 (also known as CCL2); MIP, macrophage inflammatory protein (also known as CCL3); PDGF‐bb, platelet‐derived growth factor‐BB; RANTES, regulated on activation, normal T cell expressed and Secreted (also known as CCL5); VEGF, vascular endothelial growth factor. n/a, not applicable. The correlation analyses were performed by Spearman's rank correlation. A significant correlation (p < .05) is indicated by boldface type.
Table S3. Blast wave parameters from pilot and main (treatment) studies. Legend: B + H group = blast + haemorrhage; NOM group = nomacopan i.v. + blast + haemorrhage + nomacopan s.c.; P0 (peak pressure) in kPa (the kilopascal, a unit of pressure); t + [the positive‐pressure phase duration in milliseconds (ms)]; I [impulse (kPa‐ms)].
Table S4. Blood chemistry changes in control and nomacopan treatment (NOM_15′) groups. Legend: Data are expressed as mean ±SD; statistical analyses were performed by Mann–Whitney U test; * = p < .05 vs. the vehicle (saline); # = p < .05 vs. baseline (0 hours).
Figure S1. Systemic activation of the complement pathways in battlefield trauma. A‐F, plasma levels of complement factors (C3a, C5a, sC5b‐9, Bb, and C4d) were measured by ELISA in healthy donors and trauma patients at admission to the hospital (0 h), and at 8 and 24 hours after admission. A, plasma levels of C3a for all admitted patients; B‐F, plasma levels of complement components in patients with two major mechanisms of injury ‐ blast or gunshot wounds. The data are expressed as nanograms (C3a, C5a, C5b‐9) or microgram (Bb, C4d) per milligram of total plasma proteins and presented as mean ± SEM, * = p < .05, ** = p < .01, *** = p < .001 vs. Healthy.
Figure S2. Systemic inflammatory response to trauma. Inflammatory factors and cytokines were measured by ELISA and by Bio‐Plex Kits, respectively. Pro‐inflammatory factors/cytokines (A‐H), anti‐inflammatory cytokines (I‐L), and regulatory cytokines (M‐R) from healthy donors (n = 10) and trauma patients on admission (n = 54), and at 8 (n = 23) and 24 hours (n = 9) after admission were presented. The data are expressed as nanogram per milligram plasma protein and presented as mean ± SEM, * = p < .05, ** = p < .01, *** = p < .001 vs. Healthy. Pro‐inflammatory versus anti‐inflammatory response (S). The ratio of systemic inflammatory factors (TNF‐α, IFN‐γ, IL‐1β, IL‐6, IL‐8, MCP‐1, MPO, and GM‐CSF) to IL‐10 on admission is given. C, Healthy controls (n = 10); P, trauma patients (n = 54). * = p < .05 vs. respective control (Unpaired t‐ test with Welch's correction).
Figure S3. Preventive effects of nomacopan treatment on the acidosis, hemodynamics, tissue damage, and survival after blast injury and haemorrhage in rats (Pilot study). (A) Experimental design. Anaesthetised male rats were subjected to a moderate blast overpressure (BOP = 111.65 ± 2 kPa, t+ = 3.16 ± 0.03 ms, impulse = 143 ± 2.26 kPa‐ms) and a controlled haemorrhage (40% blood volume). After 30 min of shock, animals were resuscitated with Plasma‐Lyte A (2 × shed blood volume). Animals were randomized to three study arms: nomacopan (15 mg/kg, n = 3), B + H (saline, n = 6) and Sham (no injury, n = 4). The first dose of nomacopan (7.5 mg/kg, i.v.) and a repeated dose of nomacopan (7.5 mg/kg, s.c.) were given immediately before blast injury and at 11 hours after blast injury, respectively. Blood pressure was monitored and recorded with the BIOPAC MP160 Data Acquisition and Analysis Systems via the carotid arterial catheter. Blood and tissue samples were collected for blood complement/chemistry analysis and histopathological evaluation, respectively. (B‐D) Bar graphs showing serum CH50, and plasma concentrations of C1q and C3, respectively. The data were presented as mean ± SEM, * = p < .05, ** = p < .01, *** = p < .001 vs. Sham, † = p < .05, †† = p < .01 vs. baseline (0 hour; by Mann–Whitney U test). (E‐G) Bar graphs displaying the effect of nomacopan on CH50, BE, and MAP, respectively. The data were presented as mean ± SEM. (H) Effect of nomacopan treatment on survival. (I) Representative H & E images show the effect of nomacopan on histological changes of the organs. Labels: B + H = blast overpressure (BOP) + haemorrhage; NOM = nomacopan i.v. + BOP + haemorrhage + nomacopan s.c.; MAP = mean arterial pressure; BE = base excess/base deficit.
Figure S4. Complement hemolytic activity and nomacopan effect on survival dependent on its administration time. Experimental groups: All animals were subjected to blast and haemorrhage, and treated with vehicle (saline, B + H group) or nomacopan; B + H = blast + haemorrhage; NOM = blast + nomacopan i.v. + haemorrhage + nomacopan s.c. in resuscitation phase; NOM‐Late = nomacopan i.v., with the first dose was infused at the end of hemorrhagic shock, immediately before fluid resuscitation + nopmacopan s.c. with the second dose was given in the resuscitation phase; i.v. = intravenous; s.c. = subcutaneous. A, the CH50 test data throughout the observation period; B, the survival distribution for three experimental groups was compared using the log‐rank Mantel‐Cox test; * = p < .05. NOM, nomacopan.
ACKNOWLEDGEMENTS
We thank Akari Therapeutics Plc for kindly providing the drug nomacopan (coversin). We gratefully acknowledge the contributions of Dr. Michael A. Dubick for constructive input into experimental design. We also thank Celina A. Valdez, Amber M. Woodson, Olawale A. Aderemi, Pedro A. Garcia, and Stacy T. Bernetskie for excellent technical assistance. This research was funded by the DoD US Army Medical Research & Development Command (C_038_2014 to YL) and the DoD US Army Medical Research & Development Command FY15 Broad Agency Announcement (BA150310 to LCC and YL).
Yang, Z. , Nunn, M. A. , Le, T. D. , Simovic, M. O. , Edsall, P. R. , Liu, B. , Barr, J. L. , Lund, B. J. , Hill‐Pryor, C. D. , Pusateri, A. E. , Cancio, L. C. , & Li, Y. (2023). Immunopathology of terminal complement activation and complement C5 blockade creating a pro‐survival and organ‐protective phenotype in trauma. British Journal of Pharmacology, 180(4), 422–440. 10.1111/bph.15970
Funding information DoD US Army Medical Research & Development Command, Grant/Award Numbers: BA150310, C_038_2014
Contributor Information
Leopoldo C. Cancio, Email: leopoldo.c.cancio.civ@health.mil.
Yansong Li, Email: ysli916@gmail.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alam, H. B. (2017). Trauma care: Finding a better way. PLoS Medicine, 14(7), e1002350. 10.1371/journal.pmed.1002350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh, A. , Elvington, A. , & Tomlinson, S. (2015). Complement in the homeostatic and ischemic brain. Frontiers in Immunology, 6, 417. 10.3389/fimmu.2015.00417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Abbracchio, M. P. , Alexander, W. , Al‐hosaini, K. , Bäck, M. , Barnes, N. M. , Bathgate, R. , … Ye, R. D. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: G protein‐coupled receptors. British Journal of Pharmacology, 178(S1), S27–S156. 10.1111/bph.15538 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Beuve, A. , Brouckaert, P. , Bryant, C. , Burnett, J. C. , Farndale, R. W. , Friebe, A. , Garthwaite, J. , … Waldman, S. A. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Catalytic receptors. British Journal of Pharmacology, 178(S1), S264–S312. 10.1111/bph.15541 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Buneman, O. P. , Cidlowski, J. A. , Christopoulos, A. , Davenport, A. P. , Fabbro, D. , Spedding, M. , Striessnig, J. , Davies, J. A. , Ahlers‐Dannen, K. E. , … Zolghadri, Y. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Other Protein Targets. British Journal of Pharmacology, 178(S1), S1–S26. 10.1111/bph.15537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , Cirino, G. , Docherty, J. R. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Mangum, J. , Wonnacott, S. , & Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Fares, A. , Pettenuzzo, T. , & Del Sorbo, L. (2019). Extracorporeal life support and systemic inflammation. Intensive Care Medicine Experimental, 7(Suppl 1), 46. 10.1186/s40635-019-0249-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara, U. , Flierl, M. A. , Rittirsch, D. , Klos, A. , Chen, H. , Acker, B. , Bruckner, U. B. , Nilsson, B. , Gebhard, F. , Lambris, J. D. , & Huber‐Lang, M. (2010). Molecular intercommunication between the complement and coagulation systems. Journal of Immunology, 185(9), 5628–5636. 10.4049/jimmunol.0903678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbore, G. , & Kemper, C. (2016). A novel “complement‐metabolism‐inflammasome axis” as a key regulator of immune cell effector function. European Journal of Immunology, 46(7), 1563–1573. 10.1002/eji.201546131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt‐Due, A. , Thorgersen, E. B. , Egge, K. , Pischke, S. , Sokolov, A. , Hellerud, B. C. , Lindstad, J. K. , Pharo, A. , Bongoni, A. K. , Rieben, R. , Nunn, M. , Scott, H. , & Mollnes, T. E. (2013). Combined inhibition of complement C5 and CD14 markedly attenuates inflammation, thrombogenicity, and hemodynamic changes in porcine sepsis. Journal of Immunology, 191(2), 819–827. 10.4049/jimmunol.1201909 [DOI] [PubMed] [Google Scholar]
- Barrett, C. D. , Hsu, A. T. , Ellson, C. D. , Miyazawa, B. Y. , Kong, Y. W. , Greenwood, J. D. , Dhara, S. , Neal, M. D. , Sperry, J. L. , Park, M. S. , Cohen, M. J. , Zuckerbraun, B. S. , & Yaffe, M. B. (2018). Blood clotting and traumatic injury with shock mediates complement‐dependent neutrophil priming for extracellular ROS, ROS‐dependent organ injury and coagulopathy. Clinical and Experimental Immunology, 194(1), 103–117. 10.1111/cei.13166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellander, B. M. , Olafsson, I. H. , Ghatan, P. H. , Bro Skejo, H. P. , Hansson, L. O. , Wanecek, M. , & Svensson, M. A. (2011). Secondary insults following traumatic brain injury enhance complement activation in the human brain and release of the tissue damage marker S100B. Acta Neurochirurgica, 153(1), 90–100. 10.1007/s00701-010-0737-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, P. J. Jr. , McCriskin, B. J. , Sieg, R. N. , Burks, R. , & Schoenfeld, A. J. (2012). Combat wounds in Iraq and Afghanistan from 2005 to 2009. Journal of Trauma and Acute Care Surgery, 73(1), 3–12. 10.1097/TA.0b013e318250bfb4 [DOI] [PubMed] [Google Scholar]
- Bortolotti, P. , Faure, E. , & Kipnis, E. (2018). Inflammasomes in tissue damages and immune disorders after trauma. Frontiers in Immunology, 9, 1900. 10.3389/fimmu.2018.01900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohi, K. , Gruen, R. L. , & Holcomb, J. B. (2019). Why are bleeding trauma patients still dying? Intensive Care Medicine, 45(5), 709–711. 10.1007/s00134-019-05560-x [DOI] [PubMed] [Google Scholar]
- Burk, A. M. , Martin, M. , Flierl, M. A. , Rittirsch, D. , Helm, M. , Lampl, L. , Bruckner, U. , Stahl, G. L. , Blom, A. M. , Perl, M. , Gebhard, F. , & Huber‐Lang, M. (2012). Early complementopathy after multiple injuries in humans. Shock, 37(4), 348–354. 10.1097/SHK.0b013e3182471795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, J. C. , Li, Y. , van Amersfoort, E. , Relan, A. , Dubick, M. , Sheppard, F. , Pusateri, A. , Niemeyer, D. , Tsokos, G. C. , & Dalle Lucca, J. J. (2016). C1 inhibitor limits organ injury and prolongs survival in swine subjected to battlefield simulated injury. Shock, 46(3 Suppl 1), 177–188. 10.1097/SHK.0000000000000677 [DOI] [PubMed] [Google Scholar]
- Cernak, I. (2010). The importance of systemic response in the pathobiology of blast‐induced neurotrauma. Frontiers in Neurology, 1, 151. 10.3389/fneur.2010.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, S. , Karasu, E. , & Huber‐Lang, M. (2018). Complement after trauma: Suturing innate and adaptive immunity. Frontiers in Immunology, 9, 2050. 10.3389/fimmu.2018.02050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion, H. R. , Holcomb, J. B. , Lawnick, M. M. , Kelliher, T. , Spott, M. A. , Galarneau, M. R. , Jenkins, D. H. , West, S. A. , Dye, J. , Wade, C. E. , Eastridge, B. J. , Blackbourne, L. H. , & Shair, E. K. (2010). Improved characterization of combat injury. The Journal of Trauma, 68(5), 1139–1150. 10.1097/TA.0b013e3181d86a0d [DOI] [PubMed] [Google Scholar]
- Chavko, M. , Adeeb, S. , Ahlers, S. T. , & McCarron, R. M. (2009). Attenuation of pulmonary inflammation after exposure to blast overpressure by N‐acetylcysteine amide. Shock, 32(3), 325–331. 10.1097/SHK.0b013e31819c38f1 [DOI] [PubMed] [Google Scholar]
- Ciesla, D. J. , Moore, E. E. , Johnson, J. L. , Burch, J. M. , Cothren, C. C. , & Sauaia, A. (2005). A 12‐year prospective study of postinjury multiple organ failure: Has anything changed? Archives of Surgery, 140(5), 432–438, discussion 438‐440. 10.1001/archsurg.140.5.432 [DOI] [PubMed] [Google Scholar]
- Cole, E. , Gillespie, S. , Vulliamy, P. , Brohi, K. , Akkad, H. , Apostolidou, K. , Ardley, R. , Aylwin, C. , Bassford, C. , Bonner, S. , Brooks, A. , Cairns, T. , Cecconi, M. , Clark, F. , Dempsey, G. , Denison Davies, E. , Docking, R. , Eddlestone, J. , Ellis, D. , … Welters, I. (2020). Multiple organ dysfunction after trauma. The British Journal of Surgery, 107(4), 402–412. 10.1002/bjs.11361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRASH‐2 trial collaborators , Shakur, H. , Roberts, I. , Bautista, R. , Caballero, J. , Coats, T. , Dewan, Y. , el‐Sayed, H. , Gogichaishvili, T. , Gupta, S. , Herrera, J. , Hunt, B. , Iribhogbe, P. , Izurieta, M. , Khamis, H. , Komolafe, E. , Marrero, M. A. , Mejía‐Mantilla, J. , Miranda, J. , … Yutthakasemsunt, S. (2010). Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH‐2): A randomised, placebo‐controlled trial. Lancet, 376(9734), 23–32. 10.1016/S0140-6736(10)60835-5 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle Lucca, J. J. , Chavko, M. , Dubick, M. A. , Adeeb, S. , Falabella, M. J. , Slack, J. L. , McCarron, R. , & Li, Y. (2012). Blast‐induced moderate neurotrauma (BINT) elicits early complement activation and tumor necrosis factor alpha (TNFalpha) release in a rat brain. Journal of the Neurological Sciences, 318(1–2), 146–154. 10.1016/j.jns.2012.02.002 [DOI] [PubMed] [Google Scholar]
- Dalle Lucca, J. J. , Li, Y. , Simovic, M. , Pusateri, A. E. , Falabella, M. , Dubick, M. A. , & Tsokos, G. C. (2012). Effects of C1 inhibitor on tissue damage in a porcine model of controlled hemorrhage. Shock, 38(1), 82–91. 10.1097/SHK.0b013e31825a3522 [DOI] [PubMed] [Google Scholar]
- Dalle Lucca, J. J. , Li, Y. , Simovic, M. O. , Slack, J. L. , Cap, A. , Falabella, M. J. , Dubick, M. , Lebeda, F. , & Tsokos, G. C. (2013). Decay‐accelerating factor limits hemorrhage‐instigated tissue injury and improves resuscitation clinical parameters. The Journal of Surgical Research, 179(1), 153–167. 10.1016/j.jss.2012.10.017 [DOI] [PubMed] [Google Scholar]
- Dalle Lucca, J. J. , Simovic, M. , Li, Y. , Moratz, C. , Falabella, M. , & Tsokos, G. C. (2011). Decay‐accelerating factor mitigates controlled hemorrhage‐instigated intestinal and lung tissue damage and hyperkalemia in swine. The Journal of Trauma, 71(1 Suppl), S151–S160. 10.1097/TA.0b013e318221aa4c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danobeitia, J. S. , Ziemelis, M. , Ma, X. , Zitur, L. J. , Zens, T. , Chlebeck, P. J. , Van Amersfoort, E. S. , & Fernandez, L. A. (2017). Complement inhibition attenuates acute kidney injury after ischemia‐reperfusion and limits progression to renal fibrosis in mice. PLoS ONE, 12(8), e0183701. 10.1371/journal.pone.0183701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Blasio, D. , Fumagalli, S. , Orsini, F. , Neglia, L. , Perego, C. , Ortolano, F. , Zanier, E. R. , Picetti, E. , Locatelli, M. , Stocchetti, N. , Longhi, L. , Garred, P. , & De Simoni, M. G. (2019). Human brain trauma severity is associated with lectin complement pathway activation. Journal of Cerebral Blood Flow and Metabolism, 39(5), 794–807. 10.1177/0271678X18758881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk, S. , Neher, M. D. , Messerer, D. A. C. , Wiegner, R. , Nilsson, B. , Rittirsch, D. , Nilsson‐Ekdahl, K. , Weckbach, S. , Ignatius, A. , Kalbitz, M. , Gebhard, F. , Weiss, M. E. , Vogt, J. , Radermacher, P. , Kohl, J. , Lambris, J. D. , & Huber‐Lang, M. S. (2017). Complement C5a functions as a master switch for the pH balance in neutrophils exerting fundamental Immunometabolic effects. Journal of Immunology, 198(12), 4846–4854. 10.4049/jimmunol.1700393 [DOI] [PubMed] [Google Scholar]
- Duehrkop, C. , & Rieben, R. (2014). Ischemia/reperfusion injury: Effect of simultaneous inhibition of plasma cascade systems versus specific complement inhibition. Biochemical Pharmacology, 88(1), 12–22. 10.1016/j.bcp.2013.12.013 [DOI] [PubMed] [Google Scholar]
- Eastridge, B. J. , Mabry, R. L. , Seguin, P. , Cantrell, J. , Tops, T. , Uribe, P. , Mallett, O. , Zubko, T. , Oetjen‐Gerdes, L. , Rasmussen, T. E. , Butler, F. K. , Kotwal, R. S. , Holcomb, J. B. , Wade, C. , Champion, H. , Lawnick, M. , Moores, L. , & Blackbourne, L. H. (2012). Death on the battlefield (2001‐2011): Implications for the future of combat casualty care. Journal of Trauma and Acute Care Surgery, 73(6 Suppl 5), S431–S437. 10.1097/TA.0b013e3182755dcc [DOI] [PubMed] [Google Scholar]
- Fluiter, K. , Opperhuizen, A. L. , Morgan, B. P. , Baas, F. , & Ramaglia, V. (2014). Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. Journal of Immunology, 192(5), 2339–2348. 10.4049/jimmunol.1302793 [DOI] [PubMed] [Google Scholar]
- Ganter, M. T. , Brohi, K. , Cohen, M. J. , Shaffer, L. A. , Walsh, M. C. , Stahl, G. L. , & Pittet, J. F. (2007). Role of the alternative pathway in the early complement activation following major trauma. Shock, 28(1), 29–34. 10.1097/shk.0b013e3180342439 [DOI] [PubMed] [Google Scholar]
- Gentile, L. F. , Cuenca, A. G. , Efron, P. A. , Ang, D. , Bihorac, A. , McKinley, B. A. , Moldawer, L. L. , & Moore, F. A. (2012). Persistent inflammation and immunosuppression: A common syndrome and new horizon for surgical intensive care. Journal of Trauma and Acute Care Surgery, 72(6), 1491–1501. 10.1097/TA.0b013e318256e000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, J. , Motamedi, V. , Osier, N. , Dell, K. , Arcurio, L. , Carr, W. , Walker, P. , Ahlers, S. , Lopresti, M. , & Yarnell, A. (2017). Moderate blast exposure results in increased IL‐6 and TNFalpha in peripheral blood. Brain, Behavior, and Immunity, 65, 90–94. 10.1016/j.bbi.2017.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruys, E. , Toussaint, M. J. , Niewold, T. A. , & Koopmans, S. J. (2005). Acute phase reaction and acute phase proteins. Journal of Zhejiang University. Science. B, 6(11), 1045–1056. 10.1631/jzus.2005.B1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullstrand, B. , Martensson, U. , Sturfelt, G. , Bengtsson, A. A. , & Truedsson, L. (2009). Complement classical pathway components are all important in clearance of apoptotic and secondary necrotic cells. Clinical and Experimental Immunology, 156(2), 303–311. 10.1111/j.1365-2249.2009.03896.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, R. F. , & Ward, P. A. (2005). Role of C5a in inflammatory responses. Annual Review of Immunology, 23, 821–852. 10.1146/annurev.immunol.23.021704.115835 [DOI] [PubMed] [Google Scholar]
- Held, K. , Thiel, S. , Loos, M. , & Petry, F. (2008). Increased susceptibility of complement factor B/C2 double knockout mice and mannan‐binding lectin knockout mice to systemic infection with Candida albicans. Molecular Immunology, 45(15), 3934–3941. 10.1016/j.molimm.2008.06.021 [DOI] [PubMed] [Google Scholar]
- Hellerud, B. C. , Orrem, H. L. , Dybwik, K. , Pischke, S. E. , Baratt‐Due, A. , Castellheim, A. , Fure, H. , Bergseth, G. , Christiansen, D. , Nunn, M. A. , Espevik, T. , Lau, C. , Brandtzaeg, P. , Nielsen, E. W. , & Mollnes, T. E. (2017). Combined inhibition of C5 and CD14 efficiently attenuated the inflammatory response in a porcine model of meningococcal sepsis. Journal of Intensive Care, 5, 21. 10.1186/s40560-017-0217-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepburn, N. J. , Williams, A. S. , Nunn, M. A. , Chamberlain‐Banoub, J. C. , Hamer, J. , Morgan, B. P. , & Harris, C. L. (2007). In vivo characterization and therapeutic efficacy of a C5‐specific inhibitor from the soft tick Ornithodoros moubata. The Journal of Biological Chemistry, 282(11), 8292–8299. 10.1074/jbc.M609858200 [DOI] [PubMed] [Google Scholar]
- Hofer, J. , Forster, F. , Isenman, D. E. , Wahrmann, M. , Leitner, J. , Holzl, M. A. , Kovarik, J. J. , Stockinger, H. , Bohmig, G. A. , Steinberger, P. , & Zlabinger, G. J. (2016). Ig‐like transcript 4 as a cellular receptor for soluble complement fragment C4d. The FASEB Journal, 30(4), 1492–1503. 10.1096/fj.15-275594 [DOI] [PubMed] [Google Scholar]
- Hoth, J. J. , Wells, J. D. , Jones, S. E. , Yoza, B. K. , & McCall, C. E. (2014). Complement mediates a primed inflammatory response after traumatic lung injury. Journal of Trauma and Acute Care Surgery, 76(3), 601–608, discussion 608‐609. 10.1097/TA.0000000000000129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, W. Y. , Kemp, T. J. , Pfeiffer, R. M. , Pinto, L. A. , Hildesheim, A. , & Purdue, M. P. (2017). Impact of freeze‐thaw cycles on circulating inflammation marker measurements. Cytokine, 95, 113–117. 10.1016/j.cyto.2017.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber‐Lang, M. , Barratt‐Due, A. , Pischke, S. E. , Sandanger, O. , Nilsson, P. H. , Nunn, M. A. , Denk, S. , Gaus, W. , Espevik, T. , & Mollnes, T. E. (2014). Double blockade of CD14 and complement C5 abolishes the cytokine storm and improves morbidity and survival in polymicrobial sepsis in mice. Journal of Immunology, 192(11), 5324–5331. 10.4049/jimmunol.1400341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber‐Lang, M. , Lambris, J. D. , & Ward, P. A. (2018). Innate immune responses to trauma. Nature Immunology, 19(4), 327–341. 10.1038/s41590-018-0064-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber‐Lang, M. , Sarma, J. V. , Zetoune, F. S. , Rittirsch, D. , Neff, T. A. , McGuire, S. R. , Lambris, J. D. , Warner, R. L. , Flierl, M. A. , Hoesel, L. M. , Gebhard, F. , Younger, J. G. , Drouin, S. M. , Wetsel, R. A. , & Ward, P. A. (2006). Generation of C5a in the absence of C3: A new complement activation pathway. Nature Medicine, 12(6), 682–687. 10.1038/nm1419 [DOI] [PubMed] [Google Scholar]
- Huber‐Lang, M. S. , Ignatius, A. , Kohl, J. , Mannes, M. , & Braun, C. K. (2020). Complement in trauma‐traumatised complement? British Journal of Pharmacology, 178, 2863–2879. 10.1111/bph.15245 [DOI] [PubMed] [Google Scholar]
- Jaffer, U. , Wade, R. G. , & Gourlay, T. (2010). Cytokines in the systemic inflammatory response syndrome: A review. HSR Proceedings in Intensive Care and Cardiovascular Anesthesia, 2(3), 161–175. https://www.ncbi.nlm.nih.gov/pubmed/23441054 [PMC free article] [PubMed] [Google Scholar]
- Karasu, E. , Nilsson, B. , Kohl, J. , Lambris, J. D. , & Huber‐Lang, M. (2019). Targeting complement pathways in Polytrauma‐ and sepsis‐induced multiple‐organ dysfunction. Frontiers in Immunology, 10, 543. 10.3389/fimmu.2019.00543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenawy, H. I. , Boral, I. , & Bevington, A. (2015). Complement‐coagulation cross‐talk: A potential mediator of the physiological activation of complement by low pH. Frontiers in Immunology, 6, 215. 10.3389/fimmu.2015.00215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshari, R. S. , Silasi, R. , Popescu, N. I. , Patel, M. M. , Chaaban, H. , Lupu, C. , Coggeshall, K. M. , Mollnes, T. E. , DeMarco, S. J. , & Lupu, F. (2017). Inhibition of complement C5 protects against organ failure and reduces mortality in a baboon model of Escherichia coli sepsis. Proceedings of the National Academy of Sciences of the United States of America, 114(31), E6390–E6399. 10.1073/pnas.1706818114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinhase, I. , Holers, V. M. , Thurman, J. M. , Harhausen, D. , Schmidt, O. I. , Pietzcker, M. , Taha, M. E. , Rittirsch, D. , Huber‐Lang, M. , Smith, W. R. , Ward, P. A. , & Stahel, P. F. (2006). Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neuroscience, 7, 55. 10.1186/1471-2202-7-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Chavko, M. , Slack, J. L. , Liu, B. , McCarron, R. M. , Ross, J. D. , & Dalle Lucca, J. J. (2013). Protective effects of decay‐accelerating factor on blast‐induced neurotrauma in rats. Acta Neuropathologica Communications, 1, 52. 10.1186/2051-5960-1-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Yang, Z. , Chavko, M. , Liu, B. , Aderemi, O. A. , Simovic, M. O. , Dubick, M. A. , & Cancio, L. C. (2018). Complement inhibition ameliorates blast‐induced acute lung injury in rats: Potential role of complement in intracellular HMGB1‐mediated inflammation. PLoS ONE, 13(8), e0202594. 10.1371/journal.pone.0202594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Yang, Z. , Liu, B. , Valdez, C. , Chavko, M. , & Cancio, L. C. (2019). Low‐level primary blast induces Neuroinflammation and neurodegeneration in rats. Military Medicine, 184(Suppl 1), 265–272. 10.1093/milmed/usy330 [DOI] [PubMed] [Google Scholar]
- Li, Y. , Zhao, Q. , Liu, B. , Dixon, A. , Cancio, L. , Dubick, M. , & Dalle Lucca, J. (2019). Early complementopathy predicts the outcomes of patients with trauma. Trauma Surgery & Acute Care Open, 4(1), e000217. 10.1136/tsaco-2018-000217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. , Cirino, G. , Docherty, J. R. , George, C. H. , Insel, P. A. , Izzo, A. A. , Ji, Y. , Panettieri, R. A. , Sobey, C. G. , Stefanska, B. , Stephens, G. , Teixeira, M. , & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord, J. M. , Midwinter, M. J. , Chen, Y. F. , Belli, A. , Brohi, K. , Kovacs, E. J. , Koenderman, L. , Kubes, P. , & Lilford, R. J. (2014). The systemic immune response to trauma: An overview of pathophysiology and treatment. Lancet, 384(9952), 1455–1465. 10.1016/S0140-6736(14)60687-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, X. , Li, Y. , Simovic, M. O. , Peckham, R. , Wang, Y. , Tsokos, G. C. , & Dalle Lucca, J. J. (2011). Decay‐accelerating factor attenuates C‐reactive protein‐potentiated tissue injury after mesenteric ischemia/reperfusion. The Journal of Surgical Research, 167(2), e103–e115. 10.1016/j.jss.2009.10.021 [DOI] [PubMed] [Google Scholar]
- Lundberg, C. , Marceau, F. , & Hugli, T. E. (1987). C5a‐induced hemodynamic and hematologic changes in the rabbit. Role of cyclooxygenase products and polymorphonuclear leukocytes. The American Journal of Pathology, 128(3), 471–483. https://www.ncbi.nlm.nih.gov/pubmed/3115110 [PMC free article] [PubMed] [Google Scholar]
- Lusczek, E. R. , Muratore, S. L. , Dubick, M. A. , & Beilman, G. J. (2017). Assessment of key plasma metabolites in combat casualties. Journal of Trauma and Acute Care Surgery, 82(2), 309–316. 10.1097/TA.0000000000001277 [DOI] [PubMed] [Google Scholar]
- Mannucci, P. M. , & Levi, M. (2007). Prevention and treatment of major blood loss. The New England Journal of Medicine, 356(22), 2301–2311. 10.1056/NEJMra067742 [DOI] [PubMed] [Google Scholar]
- Muroya, T. , Kannan, L. , Ghiran, I. C. , Shevkoplyas, S. S. , Paz, Z. , Tsokos, M. , Dalle Lucca, J. J. , Shapiro, N. I. , & Tsokos, G. C. (2014). C4d deposits on the surface of RBCs in trauma patients and interferes with their function. Critical Care Medicine, 42(5), e364–e372. 10.1097/CCM.0000000000000231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson, B. , Ekdahl, K. N. , Mollnes, T. E. , & Lambris, J. D. (2007). The role of complement in biomaterial‐induced inflammation. Molecular Immunology, 44(1–3), 82–94. 10.1016/j.molimm.2006.06.020 [DOI] [PubMed] [Google Scholar]
- Ort, M. , Dingemanse, J. , van den Anker, J. , & Kaufmann, P. (2020). Treatment of rare inflammatory kidney diseases: Drugs targeting the terminal complement pathway. Frontiers in Immunology, 11, 599417. 10.3389/fimmu.2020.599417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckham, R. M. , Handrigan, M. T. , Bentley, T. B. , Falabella, M. J. , Chrovian, A. D. , Stahl, G. L. , & Tsokos, G. C. (2007). C5‐blocking antibody reduces fluid requirements and improves responsiveness to fluid infusion in hemorrhagic shock managed with hypotensive resuscitation. Journal of Applied Physiology (Bethesda, MD: 1985), 102(2), 673–680. 10.1152/japplphysiol.00917.2006, 10.1152/japplphysiol.00917.2006 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , Browne, W. J. , Clark, A. , Cuthill, I. C. , Dirnagl, U. , Emerson, M. , Garner, P. , Holgate, S. T. , Howells, D. W. , Karp, N. A. , Lazic, S. E. , Lidster, K. , MacCallum, C. J. , Macleod, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. British Journal of Pharmacology, 177(16), 3617–3624. 10.1111/bph.15193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak, M. Z. , Pedziwiatr, D. , Cymer, M. , Kucia, M. , Kucharska‐Mazur, J. , & Samochowiec, J. (2018). Sterile inflammation of brain, due to activation of innate immunity, as a culprit in psychiatric disorders. Frontiers in Psychiatry, 9, 60. 10.3389/fpsyt.2018.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin, D. , & Lambris, J. D. (2007). Complement‐targeted therapeutics. Nature Biotechnology, 25(11), 1265–1275. 10.1038/nbt1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritenour, A. E. , Blackbourne, L. H. , Kelly, J. F. , McLaughlin, D. F. , Pearse, L. A. , Holcomb, J. B. , & Wade, C. E. (2010). Incidence of primary blast injury in US military overseas contingency operations: A retrospective study. Annals of Surgery, 251(6), 1140–1144. 10.1097/SLA.0b013e3181e01270 [DOI] [PubMed] [Google Scholar]
- Rittirsch, D. , Redl, H. , & Huber‐Lang, M. (2012). Role of complement in multiorgan failure. Clinical & Developmental Immunology, 2012, 962927. 10.1155/2012/962927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyam, A. , Andreo, K. , Lapchak, P. H. , Dalle Lucca, J. J. , Davis, R. B. , Tsokos, M. G. , Shapiro, N. I. , & Tsokos, G. C. (2020). Complement deposition on the surface of RBC after trauma serves a biomarker of moderate trauma severity: A prospective study. Shock, 53(1), 16–23. 10.1097/SHK.0000000000001348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillesen, M. , Rasmussen, L. S. , Jin, G. , Jepsen, C. H. , Imam, A. , Hwabejire, J. O. , Halaweish, I. , DeMoya, M. , Velmahos, G. , Johansson, P. I. , & Alam, H. B. (2014). Assessment of coagulopathy, endothelial injury, and inflammation after traumatic brain injury and hemorrhage in a porcine model. Journal of Trauma and Acute Care Surgery, 76(1), 12–19. discussion 19‐20. 10.1097/TA.0b013e3182aaa675 [DOI] [PubMed] [Google Scholar]
- Skjeflo, E. W. , Sagatun, C. , Dybwik, K. , Aam, S. , Urving, S. H. , Nunn, M. A. , Fure, H. , Lau, C. , Brekke, O. L. , Huber‐Lang, M. , Espevik, T. , Barratt‐Due, A. , Nielsen, E. W. , & Mollnes, T. E. (2015). Combined inhibition of complement and CD14 improved outcome in porcine polymicrobial sepsis. Critical Care, 19, 415. 10.1186/s13054-015-1129-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, K. V. , Deng, M. , & Ting, J. P. (2019). The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nature Reviews. Immunology, 19(8), 477–489. 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou, M. , Hughes, T. R. , Morgan, B. P. , & Triantafilou, K. (2016). Complementing the inflammasome. Immunology, 147(2), 152–164. 10.1111/imm.12556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valparaiso, A. P. , Vicente, D. A. , Bograd, B. A. , Elster, E. A. , & Davis, T. A. (2015). Modeling acute traumatic injury. The Journal of Surgical Research, 194(1), 220–232. 10.1016/j.jss.2014.10.025 [DOI] [PubMed] [Google Scholar]
- Wagner‐Golbs, A. , Neuber, S. , Kamlage, B. , Christiansen, N. , Bethan, B. , Rennefahrt, U. , Schatz, P. , & Lind, L. (2019). Effects of long‐term storage at −80 degrees C on the human plasma metabolome. Metabolites, 9(5), 99. 10.3390/metabo9050099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. M. , & Chen, J. (2016). Damage of vascular endothelial barrier induced by explosive blast and its clinical significance. Chinese Journal of Traumatology, 19(3), 125–128. 10.1016/j.cjtee.2016.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, P. A. , Fattahi, F. , & Bosmann, M. (2016). New insights into molecular mechanisms of immune complex‐induced injury in lung. Frontiers in Immunology, 7, 86. 10.3389/fimmu.2016.00086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Wise, L. , & Fukuchi, K. I. (2020). TLR4 cross‐talk with NLRP3 Inflammasome and complement signaling pathways in Alzheimer's disease. Frontiers in Immunology, 11, 724. 10.3389/fimmu.2020.00724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Aderemi, O. A. , Zhao, Q. , Edsall, P. R. , Simovic, M. O. , Lund, B. J. , Espinoza, M. D. , Woodson, A. M. , Li, Y. , & Cancio, L. C. (2019). Early complement and fibrinolytic activation in a rat model of blast‐induced multi‐organ damage. Military Medicine, 184(Suppl 1), 282–290. 10.1093/milmed/usy412 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Correlations of complement and inflammatory mediators/cytokines/chemokines. Abbreviations: FGF basic, basic fibroblast growth factor; G‐CSF, granulocyte‐colony stimulating factor; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; MCP‐1, monocyte chemoattractant protein‐1; MPO, myeloperoxidase; MIP, macrophage inflammatory protein; PDGF‐bb, platelet‐derived growth factor‐BB; RANTES, regulated on activation, normal T cell expressed and secreted; VEGF, vascular endothelial growth factor. n/a, not applicable. The correlation analyses were performed by Spearman's rank correlation. A significant correlation (p < .05) is indicated by boldface type.
Table S2. Correlations between complement/inflammatory cytokines/chemokines and clinical outcomes. Abbreviations: MPO, myeloperoxidase; FGF basic, basic fibroblast growth factor; G‐CSF, granulocyte‐colony stimulating factor; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor. MCP‐1, monocyte chemoattractant protein‐1 (also known as CCL2); MIP, macrophage inflammatory protein (also known as CCL3); PDGF‐bb, platelet‐derived growth factor‐BB; RANTES, regulated on activation, normal T cell expressed and Secreted (also known as CCL5); VEGF, vascular endothelial growth factor. n/a, not applicable. The correlation analyses were performed by Spearman's rank correlation. A significant correlation (p < .05) is indicated by boldface type.
Table S3. Blast wave parameters from pilot and main (treatment) studies. Legend: B + H group = blast + haemorrhage; NOM group = nomacopan i.v. + blast + haemorrhage + nomacopan s.c.; P0 (peak pressure) in kPa (the kilopascal, a unit of pressure); t + [the positive‐pressure phase duration in milliseconds (ms)]; I [impulse (kPa‐ms)].
Table S4. Blood chemistry changes in control and nomacopan treatment (NOM_15′) groups. Legend: Data are expressed as mean ±SD; statistical analyses were performed by Mann–Whitney U test; * = p < .05 vs. the vehicle (saline); # = p < .05 vs. baseline (0 hours).
Figure S1. Systemic activation of the complement pathways in battlefield trauma. A‐F, plasma levels of complement factors (C3a, C5a, sC5b‐9, Bb, and C4d) were measured by ELISA in healthy donors and trauma patients at admission to the hospital (0 h), and at 8 and 24 hours after admission. A, plasma levels of C3a for all admitted patients; B‐F, plasma levels of complement components in patients with two major mechanisms of injury ‐ blast or gunshot wounds. The data are expressed as nanograms (C3a, C5a, C5b‐9) or microgram (Bb, C4d) per milligram of total plasma proteins and presented as mean ± SEM, * = p < .05, ** = p < .01, *** = p < .001 vs. Healthy.
Figure S2. Systemic inflammatory response to trauma. Inflammatory factors and cytokines were measured by ELISA and by Bio‐Plex Kits, respectively. Pro‐inflammatory factors/cytokines (A‐H), anti‐inflammatory cytokines (I‐L), and regulatory cytokines (M‐R) from healthy donors (n = 10) and trauma patients on admission (n = 54), and at 8 (n = 23) and 24 hours (n = 9) after admission were presented. The data are expressed as nanogram per milligram plasma protein and presented as mean ± SEM, * = p < .05, ** = p < .01, *** = p < .001 vs. Healthy. Pro‐inflammatory versus anti‐inflammatory response (S). The ratio of systemic inflammatory factors (TNF‐α, IFN‐γ, IL‐1β, IL‐6, IL‐8, MCP‐1, MPO, and GM‐CSF) to IL‐10 on admission is given. C, Healthy controls (n = 10); P, trauma patients (n = 54). * = p < .05 vs. respective control (Unpaired t‐ test with Welch's correction).
Figure S3. Preventive effects of nomacopan treatment on the acidosis, hemodynamics, tissue damage, and survival after blast injury and haemorrhage in rats (Pilot study). (A) Experimental design. Anaesthetised male rats were subjected to a moderate blast overpressure (BOP = 111.65 ± 2 kPa, t+ = 3.16 ± 0.03 ms, impulse = 143 ± 2.26 kPa‐ms) and a controlled haemorrhage (40% blood volume). After 30 min of shock, animals were resuscitated with Plasma‐Lyte A (2 × shed blood volume). Animals were randomized to three study arms: nomacopan (15 mg/kg, n = 3), B + H (saline, n = 6) and Sham (no injury, n = 4). The first dose of nomacopan (7.5 mg/kg, i.v.) and a repeated dose of nomacopan (7.5 mg/kg, s.c.) were given immediately before blast injury and at 11 hours after blast injury, respectively. Blood pressure was monitored and recorded with the BIOPAC MP160 Data Acquisition and Analysis Systems via the carotid arterial catheter. Blood and tissue samples were collected for blood complement/chemistry analysis and histopathological evaluation, respectively. (B‐D) Bar graphs showing serum CH50, and plasma concentrations of C1q and C3, respectively. The data were presented as mean ± SEM, * = p < .05, ** = p < .01, *** = p < .001 vs. Sham, † = p < .05, †† = p < .01 vs. baseline (0 hour; by Mann–Whitney U test). (E‐G) Bar graphs displaying the effect of nomacopan on CH50, BE, and MAP, respectively. The data were presented as mean ± SEM. (H) Effect of nomacopan treatment on survival. (I) Representative H & E images show the effect of nomacopan on histological changes of the organs. Labels: B + H = blast overpressure (BOP) + haemorrhage; NOM = nomacopan i.v. + BOP + haemorrhage + nomacopan s.c.; MAP = mean arterial pressure; BE = base excess/base deficit.
Figure S4. Complement hemolytic activity and nomacopan effect on survival dependent on its administration time. Experimental groups: All animals were subjected to blast and haemorrhage, and treated with vehicle (saline, B + H group) or nomacopan; B + H = blast + haemorrhage; NOM = blast + nomacopan i.v. + haemorrhage + nomacopan s.c. in resuscitation phase; NOM‐Late = nomacopan i.v., with the first dose was infused at the end of hemorrhagic shock, immediately before fluid resuscitation + nopmacopan s.c. with the second dose was given in the resuscitation phase; i.v. = intravenous; s.c. = subcutaneous. A, the CH50 test data throughout the observation period; B, the survival distribution for three experimental groups was compared using the log‐rank Mantel‐Cox test; * = p < .05. NOM, nomacopan.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.