

Abstract
Objective
To assess the long‐term safety, tolerability, and efficacy of bimekizumab in active psoriatic arthritis (PsA).
Methods
Adult patients with active PsA who completed the double‐ and dose‐blind periods of the BE ACTIVE randomized controlled trial were eligible to enroll in the open‐label extension (OLE) study at week 48, after which patients received 160 mg of bimekizumab every 4 weeks. Safety and efficacy results are presented through 152 weeks.
Results
At week 152, 161 of 206 patients (78.2%) remained in the study. From weeks 0–152, 184 of 206 patients experienced ≥1 treatment‐emergent adverse event (126.4 per 100 patient‐years). The most frequent events were nasopharyngitis (7.6 per 100 patient‐years), upper respiratory tract infection (6.8 per 100 patient‐years), bronchitis (3.5 per 100 patient‐years), and oral candidiasis (3.5 per 100 patient‐years). Additionally, 47 of 206 patients had mild to moderate localized fungal infections (9.7 per 100 patient‐years), including 24 of 206 patients who had Candida infections (4.6 per 100 patient years) and 19 of 206 patients who had oral candidiasis (3.5 per 100 patient years). Four patients had serious infections (0.7 per 100 patient‐years); there were no reported cases of active tuberculosis, adjudicated major adverse cardiac events, or deaths. Efficacy demonstrated at week 48 was sustained in the OLE study. At week 152, nonresponder imputation analysis showed that 52.9% of patients (69.4% of observed cases) achieved the American College of Rheumatology criteria for 50% improvement, 57.7% (73.8% of observed cases) achieved 100% skin clearance per the Psoriasis Area and Severity Index, and 51.5% (67.5% of observed cases) achieved minimal disease activity. Patients also maintained improvements in pain, physical function, and health‐related quality of life.
Conclusion
The safety profile of bimekizumab was consistent with previous reports, with no new safety signals identified. Sustained joint and efficacy responses were observed over 3 years.
Short abstract
INTRODUCTION
Psoriatic arthritis (PsA) is a chronic, immune‐mediated inflammatory disease with varied musculoskeletal and dermatological manifestations, including joint inflammation, enthesitis, dactylitis, and skin disease (1). Due to the substantial and enduring impact of psoriatic disease on patients (2, 3), it is critical to evaluate the long‐term safety and efficacy of treatments. In recent years, the interleukin (IL)‐17 cytokine superfamily has emerged as a target of new monoclonal antibody therapies for a range of inflammatory diseases. Currently approved therapies for PsA that target IL‐17 cytokines include secukinumab and ixekizumab, both of which inhibit IL‐17A, one of the 6 isoforms of IL‐17 (4). The safety of IL‐17A inhibitor treatment has been reported up to 5 years in patients with PsA (5).
Bimekizumab is a humanized monoclonal IgG1 antibody that selectively inhibits IL‐17F in addition to IL‐17A. Previous studies have shown that the dual neutralization of IL‐17A and IL‐17F results in rapid clinical improvements in patients with PsA (phase IIb) and ankylosing spondylitis (AS) (phase IIb), sustained up to 48 weeks, and plaque psoriasis (phase III), sustained up to 56 weeks (6, 7, 8). Given that PsA is a chronic disease with the potential for lasting complications, including irreversible joint damage, an increased risk of comorbidities, impaired quality of life, and substantial psychosocial burden (9, 10), it is important to establish the long‐term safety and efficacy of treatments in this patient population. While studies have examined the safety and efficacy of IL‐17A inhibitors in PsA patients receiving treatment for up to 5 years (5), the BE ACTIVE trial and its open‐label extension (OLE) study provide the first long‐term safety and efficacy data specific to the mechanism of dual neutralization of IL‐17A and IL‐17F in PsA patients.
In the BE ACTIVE trial, treatment with 160 mg or 320 mg of bimekizumab every 4 weeks for up to 48 weeks was shown to be well tolerated, with demonstrated efficacy in joint and skin outcomes (6). Here, we describe the long‐term safety, tolerability, and efficacy of up to 3 years of treatment with bimekizumab in adult patients with PsA.
PATIENTS AND METHODS
Study design and participants
The BE ACTIVE trial (ClinicalTrials.gov identifier: NCT02969525) was a 48‐week randomized phase IIb dose‐ranging study, double‐blind and placebo‐controlled from the beginning of the study to week 12, then dose‐blind to week 48. It was conducted at 41 sites across 5 countries in Europe and the US (6). Patients who completed 48 weeks of treatment in the double‐ and dose‐blind periods and gave separate informed consent were eligible to enter the OLE study (ClinicalTrials.gov identifier: NCT03347110) for a further 104 weeks of treatment, up to a total of 152 weeks. A safety follow‐up visit took place at week 168, 20 weeks after the last dose of bimekizumab (see Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.42280). For patients who did not complete treatment to week 152, the safety follow‐up visit occurred 20 weeks after their last dose of bimekizumab. Key inclusion and exclusion criteria have been reported previously (6).
Randomization and blinding
At baseline of the double‐blind period, 206 patients were randomized 1:1:1:1:1 to receive subcutaneous bimekizumab at a dose of 16 mg every 4 weeks, 160 mg every 4 weeks with a 320 mg loading dose, 160 mg every 4 weeks, 320 mg every 4 weeks, or placebo. At week 12, patients initially randomized to receive 16 mg of bimekizumab every 4 weeks or placebo were re‐randomized 1:1 to receive bimekizumab at 160 mg or 320 mg every 4 weeks through week 48, while patients initially randomized to receive bimekizumab at 160 mg or 320 mg every 4 weeks continued their dosing to week 48 (6). At week 48, patients completing the dose‐blind period on bimekizumab were eligible to enroll in the OLE study. All patients in the OLE study received 160 mg of bimekizumab every 4 weeks, regardless of prior dosing regimen (Supplementary Figure 1). All study timepoints are hereafter reported relative to baseline (week 0) of the initial randomized controlled study.
Study procedures and outcomes
During the OLE study, safety was assessed at study entry (week 48), then every 4 weeks through week 60, followed by every 12 weeks through week 144, then every 4 weeks through week 152 and subsequently at the safety follow‐up (week 168). Efficacy was assessed at OLE study entry, then every 12 weeks through week 144, and then every 4 weeks through week 152.
The prespecified primary objective of the OLE study was the long‐term safety of bimekizumab treatment over 152 weeks, measured by the incidence of treatment‐emergent adverse events (TEAEs) and serious TEAEs. All TEAEs were classified using the Medical Dictionary for Regulatory Activities (MedDRA) version 19.0. Prespecified safety topics of interest included serious, fungal, and opportunistic infections, including tuberculosis (TB). Prespecified safety topics of interest also included liver enzyme elevation, major adverse cardiac events (MACE), malignancies, inflammatory bowel disease (IBD), neutropenia, hypersensitivity, suicidal ideation and behavior (SIB), and depression. MACE, IBD, and SIB events were adjudicated by independent committees. TEAEs leading to withdrawal from the study were a secondary safety endpoint.
Efficacy endpoints were prespecified via protocol as secondary and included the following: the proportion of patients achieving ≥20%, ≥50%, or ≥70% improvement in disease activity based on the American College of Rheumatology criteria (ACR20, ACR50, ACR70) at week 96 (11); the proportion of patients (among those with ≥3% body surface area [BSA] affected by psoriasis at baseline of the double‐blind period) achieving ≥75% or ≥90% improvement in the Psoriasis Area and Severity Index (PASI75, PASI90) at week 96 (12); and the change from baseline of the double‐blind period in the Leeds Dactylitis Index (LDI, among those with LDI scores >0 at baseline) (13) and the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES, among those with a MASES >0 at baseline) (14) at week 96. Due to a data collection error and lack of patient data for skin outcomes at the week 96 visit, data are not reported at this timepoint for PASI75/PASI90, nor for 100% improvement in the PASI.
Other efficacy endpoints included the following: the proportion of patients over time achieving ACR20, ACR50, ACR70, PASI75, PASI90, PASI100, and minimal disease activity (MDA) (15); change from baseline in LDI, MASES, Disease Activity Score in 28 joints using the C‐reactive protein level (DAS28‐CRP) (16), Psoriatic Arthritis Impact of Disease in 9 domains (PsAID‐9) (17) total score, and Short Form 36‐item health survey (SF‐36) physical component summary (PCS) and mental component summary (MCS) scores (18); and the proportion of patients achieving depression and anxiety status “normal” (score < 8) as defined by the Hospital Anxiety and Depression Scale (HADS) anxiety and HADS depression subscales (19). The classification criteria for MDA and very low disease activity (VLDA) have been reported previously (15, 20).
Post hoc analyses included assessment of the proportion of patients achieving a response based on the ACR50 + PASI100 composite outcome, the proportion of patients over time achieving VLDA, and the proportion of patients over time achieving remission as defined by the Disease Activity Index for Psoriatic Arthritis (DAPSA) score (21). While not prespecified endpoints, change from baseline in Health Assessment Questionnaire disability index (HAQ DI) (22) and Patient's Assessment of Arthritis Pain (PtAAP) total scores are also reported to show the effects of treatment on patient health‐related quality of life (HRQoL) and pain.
Statistical analysis
In the OLE study, the safety set consisted of all randomized study participants who received ≥1 dose of bimekizumab in weeks 48–152. The full analysis set consisted of all randomized study participants who received ≥1 dose of bimekizumab in weeks 48–152 and had a valid measurement of ≥1 efficacy variable after OLE study entry. The full analysis and safety sets for the double‐ and dose‐blind periods have been described previously (6). Of the 206 patients included in the safety set, only 204 received bimekizumab due to the withdrawal of 2 patients originally allocated to the placebo group; both patients completed the double‐blind period and discontinued before receiving bimekizumab.
Primary and secondary safety analyses are presented for the safety sets. The summary of TEAEs from weeks 48–152 included all patients who received bimekizumab during the OLE study. Exposure‐adjusted incidence rates (EAIRs) per 100 patient‐years are reported for the BE ACTIVE (6) and OLE safety sets.
Secondary and other efficacy variables were summarized for all subjects in the BE ACTIVE full analysis set. Responder variables were derived relative to baseline of the double‐blind period and were summarized descriptively. Missing binary efficacy variables were imputed in the most conservative manner, using nonresponder imputation (NRI), with patients who did not enter the OLE study considered nonresponders from week 48 onwards. Missing continuous efficacy variables were imputed using multiple imputation based on the assumption that data were missing at random. Nonmonotone missing data were imputed with the Markov chain Monte Carlo (MCMC) method; monotone missing data were imputed with a monotone regression model. All computations and generation of outputs were done in SAS version 9.3 or later.
Ethics approval
The BE ACTIVE trial and its OLE study were conducted in accordance with the Declaration of Helsinki and the International Conference for Harmonisation Guidelines for Good Clinical Practice. Ethical approval was obtained from the relevant institutional review boards at all participating sites. All patients provided written informed consent in accordance with local requirements, with additional written informed consent required for enrollment in the OLE study. All the results presented in this article are in aggregate form, and no personally identifiable information was used for this study.
RESULTS
Patient disposition and baseline characteristics
At the end of the dose‐blind period (week 48), 184 of 206 patients (89.3%) enrolled in the OLE study and 183 received ≥1 dose of bimekizumab (Supplementary Figure 1, http://onlinelibrary.wiley.com/doi/10.1002/art.42280). Patient retention during the OLE study comprised 161 of 184 patients (87.5%) who completed treatment to week 152. Thus, 78.2% of all patients who started at week 0 completed the full 152 weeks of treatment. Kaplan‐Meier curves of patient retention rates are provided in Supplementary Figure 2 (http://onlinelibrary.wiley.com/doi/10.1002/art.42280).
There were 12 discontinuations during the double‐ and dose‐blind periods: 5 due to adverse events, 5 due to withdrawn consent, and 2 due to other reasons. An additional 4 patients discontinued due to adverse events after completing the double‐blind period but prior to re‐randomization in the dose‐blind period. A total of 184 patients entered the OLE study; however, 1 patient discontinued from the study prior to any dosing in the OLE study. There were 22 discontinuations during the OLE study: 9 due to adverse events, 2 due to loss of efficacy, 9 due to withdrawn consent (not due to adverse events), 1 due to loss to follow‐up, and 1 due to other reasons (Supplementary Figure 3, http://onlinelibrary.wiley.com/doi/10.1002/art.42280). Patient demographics and characteristics at baseline of the double‐blind period are reported in Table 1.
Table 1.
Demographics and baseline characteristics of psoriatic arthritis patients*
| Group A | Group B | Total BKZ | |
|---|---|---|---|
| (n = 124)† | (n = 82)‡ | (N = 206) | |
| Age, mean ± SD years | 50.5 ± 12.6 | 47.4 ± 12.0 | 49.3 ± 12.4 |
| Male | 55 (44.4) | 50 (61.0) | 105 (51.0) |
| Weight, mean ± SD kg | 83.8 ± 18.4 | 88.5 ± 18.5 | 85.7 ± 18.5 |
| BMI, mean ± SD kg/m2 | 29.3 ± 5.9 | 30.1 ± 6.2 | 29.6 ± 6.0 |
| Time since first diagnosis of PsA, mean ± SD years | 7.9 ± 9.1 | 6.0 ± 6.3 | 7.1 ± 8.2 |
| TJC, mean ± SD | 20.6 ± 14.3 | 23.3 ± 15.9 | 21.7 ± 15.0 |
| SJC, mean ± SD | 10.9 ± 7.5 | 12.5 ± 9.5 | 11.5 ± 8.4 |
| hsCRP, median (range) mg/liter | 5.7 (0.1–99.9) | 5.9 (0.3–85.2) | 5.9 (0.1–99.9) |
| HAQ DI score, mean ± SD | 1.0 ± 0.6 | 1.0 ± 0.6 | 1.0 ± 0.6 |
| PsAID‐9 total score, mean ± SD | 4.5 ± 1.9 | 4.9 ± 2.0 | 4.6 ± 1.9 |
| PtAAP score, mean ± SD | 50.8 ± 22.7 | 54.3 ± 23.4 | 52.2 ± 23.0 |
| SF‐36 score, mean ± SD | |||
| PCS | 37.0 ± 9.1 | 36.0 ± 9.0 | 36.6 ± 9.1 |
| MCS | 56.0 ± 8.6 | 55.4 ± 9.1 | 55.8 ± 8.7 |
| Psoriasis | |||
| BSA <3% | 45 (36.3) | 22 (26.8) | 67 (32.5) |
| BSA ≥3–<10% | 45 (36.3) | 33 (40.2) | 78 (37.9) |
| BSA ≥10% | 34 (27.4) | 25 (30.5) | 59 (28.6) |
| Dactylitis | 34 (27.4) | 25 (30.5) | 59 (28.6) |
| Enthesitis | 65 (52.4) | 42 (51.2) | 107 (51.9) |
| Prior TNFi therapy | 23 (18.5) | 16 (19.5) | 39 (18.9) |
| Concomitant therapies | |||
| NSAIDs | 81 (65.3) | 52 (63.4) | 133 (64.6) |
| Methotrexate | 75 (60.5) | 56 (68.3) | 131 (63.6) |
| Steroids | 23 (18.5) | 23 (28.0) | 46 (22.3) |
Except where indicated otherwise, values are the number (%) of patients. Patients were randomized to receive bimekizumab (BKZ) or placebo at any dose at the beginning of the double‐blind period (weeks 0–12) of the phase IIb randomized controlled trial BE ACTIVE. In the dose‐blind period, patients initially randomized to receive 16 mg of BKZ or placebo every 4 weeks were rerandomized to receive BKZ at 160 mg or 320 mg every 4 weeks, and patients initially randomized to receive BKZ at 160 mg or 320 mg every 4 weeks continued their dosing (weeks 12–48). BMI = body mass index; PsA = psoriatic arthritis; TJC = tender joint count; SJC = swollen joint count; hsCRP = high sensitivity C‐reactive protein; HAQ DI = Health Assessment Questionnaire Disability Index; PsAID‐9 = Psoriatic Arthritis Impact of Disease in 9 domains; PtAAP = Patient's Assessment of Arthritis Pain; SF‐36 = Short Form 36‐item health survey; PCS = physical component summary; MCS = mental component summary; BSA = body surface area; TNFi = tumor necrosis factor inhibitor; NSAID = nonsteroidal antiinflammatory drug.
Patients received 160 mg of BKZ every 4 weeks continuously from week 12 to week 152 (includes those originally assigned to BKZ 160 mg every 4 weeks with a loading dose).
Patients were dose‐reduced from 320 mg of BKZ every 4 weeks to 160 mg every 4 weeks at OLE study entry (week 48).
Among all patients randomized at week 0 (N = 206), the mean ± SD time since first diagnosis of PsA was 7.1 ± 8.2 years. The mean ± SD tender joint count (TJC) and swollen joint count (SJC) were 21.7 ± 15.0 and 11.5 ± 8.3, respectively, and the median serum high‐sensitivity C‐reactive protein level was 5.9 mg/liter (range 0.1–99.9 mg/liter). At the double‐blind period baseline, patients had a mean SF‐36 PCS score of 36.6 (compromised physical function, increased bodily pain, and decreased general health) and a mean SF‐36 MCS score of 55.8 (unimpaired mental function) (18). At the end of the dose‐blind period, the mean TJC and mean SJC among the randomized patients had decreased to 5.3 and 1.9, respectively, and the mean SF‐36 PCS score had increased to 45.7. At baseline, approximately two‐thirds of patients (66.5%) had ≥3% BSA with psoriasis involvement, approximately one‐quarter (28.6%) had dactylitis, and slightly more than one‐half (51.9%) had enthesitis. Resolution of dactylitis and enthesitis at the end of the dose‐blind period are reported as part of the efficacy results (see below).
Safety
Exposure to bimekizumab over 152 weeks among all patients randomized at baseline was 570.1 patient‐years, including 392.7 patient‐years of exposure during the OLE study. From the double‐blind period baseline through 152 weeks of therapy, 184 of 206 patients (89.3%) experienced ≥1 TEAE (EAIR 126.4 per 100 patient‐years), and 22 of 206 patients (10.7%) experienced ≥1 serious TEAE (EAIR 4.1 per 100 patient‐years) (Table 2). Seventeen patients (8.3%) permanently discontinued bimekizumab due to adverse events during the double‐ or dose‐blind periods or during the OLE study. Cellulitis and oral fungal infection resulted in discontinuation of 2 patients each, and all other TEAEs leading to discontinuation were reported for a single patient. There were no deaths reported throughout the double‐ and dose‐blind periods or throughout the OLE study (weeks 0–152).
Table 2.
Safety of bimekizumab treatment in psoriatic arthritis patients across the BE ACTIVE randomized controlled trial and the OLE study (safety set)*
| Weeks 0–48† | Weeks 48–152 | Weeks 0–152‡ | ||
|---|---|---|---|---|
| 160 mg BKZ every 4 weeks | 320 mg BKZ every 4 weeks | Total BKZ | Total BKZ | |
| (n = 126; 113.2 patient‐years) | (n = 80; 72.9 patient‐years) | (N = 183; 392.7 patient‐years) | (N = 206; 570.1 patient‐years) | |
| Any TEAE | 94 (74.6) (177.6) | 57 (71.3) (165.9) | 148 (80.9) (94.3) | 184 (89.3) (126.4) |
| Serious TEAEs | 8 (6.3) (7.9) | 0 | 14 (7.7) (3.8) | 22 (10.7) (4.1) |
| Severe TEAEs | 5 (4.0) (4.6) | 2 (2.5) (2.9) | 8 (4.4) (2.1) | 14 (6.8) (2.5) |
| Withdrawal due to TEAEs | 6 (4.8) (5.9) | 2 (2.5) (3.1) | 9 (4.9) (2.3) | 17 (8.3) (3.0) |
| Drug‐related TEAEs | 43 (34.1) (52.7) | 29 (36.3) (57.0) | 60 (32.8) (20.0) | 97 (47.1) (26.4) |
| Deaths | 0 | 0 | 0 | 0 |
| Most frequently reported TEAEs (≥5%) by MedDRA preferred term | ||||
| Nasopharyngitis | 12 (9.5) (12.0) | 11 (13.8) (18.4) | 19 (10.4) (5.2) | 37 (18.0) (7.6) |
| Upper respiratory tract infection | 12 (9.5) (12.0) | 8 (10.0) (13.2) | 20 (10.9) (5.5) | 34 (16.5) (6.8) |
| Bronchitis | 7 (5.6) (6.9) | 3 (3.8) (4.8) | 11 (6.0) (2.9) | 19 (9.2) (3.5) |
| Oral candidiasis | 6 (4.8) (6.0) | 4 (5.0) (6.4) | 13 (7.1) (3.5) | 19 (9.2) (3.5) |
| Pharyngitis | 4 (3.2) (3.9) | 7 (8.8) (11.6) | 10 (5.5) (2.7) | 17 (8.3) (3.2) |
| Sinusitis | 6 (4.8) (5.9) | 4 (5.0) (6.5) | 10 (5.5) (2.6) | 17 (8.3) (3.2) |
| Psoriasis | 2 (1.6) (1.9) | 2 (2.5) (3.1) | 14 (7.7) (3.7) | 16 (7.8) (2.9) |
| Psoriatic arthropathy | 2 (1.6) (1.9) | 1 (1.3) (1.6) | 12 (6.6) (3.1) | 16 (7.8) (2.9) |
| Respiratory tract infection | 8 (6.3) (8.0) | 2 (2.5) (3.2) | 4 (2.2) (1.0) | 15 (7.3) (2.8) |
| Oral fungal infection | 3 (2.4) (2.9) | 3 (3.8) (4.7) | 9 (4.9) (2.4) | 14 (6.8) (2.6) |
| Tonsillitis | 6 (4.8) (5.9) | 2 (2.5) (3.2) | 6 (3.3) (1.6) | 14 (6.8) (2.6) |
| ALT increased | 6 (4.8) (6.0) | 3 (3.8) (4.7) | 6 (3.3) (1.6) | 13 (6.3) (2.4) |
| Safety topics of interest | ||||
| Serious infections | 3 (2.4) (2.9) | 0 | 1 (0.5) (0.3) | 4 (1.9) (0.7) |
| Fungal infections | 17 (13.5) (17.8) | 10 (12.5) (16.7) | 32 (17.5) (9.2) | 47 (22.8) (9.7) |
| Candida infections | 9 (7.1) (9.1) | 5 (6.3) (8.1) | 16 (8.7) (4.3) | 24 (11.7) (4.6) |
| Oral candidiasis | 6 (4.8) (6.0) | 4 (5.0) (6.4) | 13 (7.1) (3.5) | 19 (9.2) (3.5) |
| Skin candidiasis | 1 (0.8) (1.0) | 0 | 1 (0.5) (0.3) | 2 (1.0) (0.4) |
| Vulvovaginal candidiasis | 0 | 0 | 1 (0.5) (0.3) | 1 (0.5) (0.2) |
| Genital candidiasis | 1 (0.8) (1.0) | 0 | 1 (0.5) (0.3) | 1 (0.5) (0.2) |
| Oropharyngeal candidiasis | 1 (0.8) (1.0) | 0 | 0 | 1 (0.5) (0.2) |
| Fungal infections NEC | 9 (7.1) (9.0) | 4 (5.0) (6.3) | 17 (9.3) (4.6) | 25 (12.1) (4.7) |
| Oral fungal infection | 3 (2.4) (2.9) | 3 (3.8) (4.7) | 9 (4.9) (2.4) | 14 (6.8) (2.6) |
| Tongue fungal infection | 3 (2.4) (2.9) | 0 | 4 (2.2) (1.0) | 5 (2.4) (0.9) |
| Fungal skin infection | 0 | 1 (1.3) (1.6) | 3 (1.6) (0.8) | 4 (1.9) (0.7) |
| Fungal esophagitis | 1 (0.8) (1.0) | 1 (1.3) (1.6) | 1 (0.5) (0.3) | 3 (1.5) (0.5) |
| Vulvovaginal mycotic infection | 2 (1.6) (1.9) | 0 | 0 | 2 (1.0) (0.4) |
| Onochomycosis | 0 | 0 | 2 (1.1) (0.5) | 2 (1.0) (0.4) |
| Fungal pharyngitis | 0 | 0 | 1 (0.5) (0.3) | 1 (0.5) (0.2) |
| Tinea infections | 0 | 1 (1.3) (1.6) | 1 (0.5) (0.3) | 2 (1.0) (0.4) |
| Tinea pedis | 0 | 1 (1.3) (1.6) | 0 | 1 (0.5) (0.2) |
| Tineas cruris | 0 | 0 | 1 (0.5) (0.3) | 1 (0.5) (0.2) |
| Serious hypersensitivity reactions | 0 | 0 | 0 | 0 |
| Opportunistic infections | 1 (0.8) (1.0) | 1 (1.3) (1.6) | 1 (0.5) (0.3) | 3 (1.5) (0.5) |
| Active tuberculosis | 0 | 0 | 0 | 0 |
| Liver enzyme elevation | ||||
| ALT increased | 6 (4.8) (6.0) | 3 (3.8) (4.7) | 6 (3.3) (1.6) | 13 (6.3) (2.4) |
| AST increased | 4 (3.2) (4.0) | 2 (2.5) (3.1) | 6 (3.3) (1.6) | 10 (4.9) (1.8) |
| Hepatic enzymes increased | 2 (1.6) (1.9) | 1 (1.3) (1.6) | 1 (0.5) (0.3) | 4 (1.9) (0.7) |
| MACE | 0 | 0 | 0 | 0 |
| Malignancies | 1 (0.8) (1.0) | 0 | 0 | 1 (0.5) (0.2) |
| IBD | 0 | 0 | 1 (0.5) (0.3) | 1 (0.5) (0.2) |
| Microscopic colitis | 0 | 0 | 1 (0.5) (0.3) | 1 (0.5) (0.2) |
| Anterior uveitis | 0 | 0 | 0 | 0 |
| Neutropenia | 0 | 1 (1.3) (1.6) | 5 (2.7) (1.3) | 6 (2.9) (1.1) |
| Drug hypersensitivity | 2 (1.6) (1.9) | 0 | 1 (0.5) (0.3) | 3 (1.5) (0.5) |
| Injection site reactions | 0 | 3 (3.8) (4.9) | 0 | 3 (1.5) (0.5) |
| SIB | 1 (0.8) (1.0) | 0 | 0 | 1 (0.5) (0.2) |
| Depression | 1 (0.8) (1.0) | 1 (1.3) (1.6) | 2 (1.1) (0.5) | 4 (1.9) (0.7) |
Values are the number (%) of patients (exposure‐adjusted incidence rate per 100 patient‐years). The safety set consisted of all randomized study participants who received ≥1 dose of BKZ in weeks 48–152 of the OLE study. After week 48, all patients received 160 mg of BKZ every 4 weeks, regardless of prior dosing regimen. All oral candidiasis treatment‐emergent adverse events (TEAEs) were mild to moderate (no serious cases), and all fungal infections were mild to moderate and localized, not systemic. Two patients reported 3 opportunistic events (2 fungal esophagitis, 1 oropharyngeal candidiasis) in weeks 0–48, 1 patient reported 2 events (fungal pharyngitis, fungal esophagitis) in weeks 48–152. No Hy's law cases reported. Suicidal ideation and behavior (SIB) events were adjudicated by an independent committee. One malignant melanoma in situ case was reported. No drug hypersensitivity reactions were anaphylactic. OLE = open‐label extension; MedDRA = Medical Dictionary for Regulatory Activities; ALT = alanine aminotransferase; AST = aspartate aminotransferase; MACE = major adverse cardiac events; IBD = inflammatory bowel disease. See Table 1 for other definitions.
Patients re‐randomized 1:1 at week 12 from placebo or 16 mg of BKZ every 4 weeks to 160 mg BKZ every 4 weeks or 320 mg BKZ every 4 weeks; 2 patients completing the double‐blind period on placebo were re‐randomized but did not receive BKZ. Two patients were included in both groups due to a dosing error, allocation done per actual treatment.
Includes safety follow‐up to possible 168 weeks total for some patients.
The most frequently reported TEAEs (incidence ≥5%) by MedDRA preferred term in the double‐ and dose‐blind periods and the OLE study included nasopharyngitis (18.0%), upper respiratory tract infection (16.5%), bronchitis (9.2%), oral candidiasis (9.2%), pharyngitis (8.3%), sinusitis (8.3%), psoriasis (7.8%), psoriatic arthropathy (7.8%), respiratory tract infection (7.3%), oral fungal infection (6.8%), tonsillitis (6.8%), and alanine aminotransferase (ALT) increased (6.3%) (Table 2).
Safety topics of interest
Four patients (1.9%) experienced TEAEs of serious infections (1 case each of cellulitis, chronic otitis media, hepatitis E, and chronic sinusitis) during the double‐ and dose‐blind periods and the OLE study. There was 1 case of microscopic colitis reported during the OLE study in a patient without prior history of IBD, which was adjudicated as IBD by an independent committee; the event was classified as moderate in intensity and was unrelated to treatment with bimekizumab. Throughout the double‐ and dose‐blind periods and the OLE study, no reported events were adjudicated as MACE, and there were no reported cases of anterior uveitis. No further cases of malignancies or injection site reactions were reported during the OLE study; 1 case of malignant melanoma in situ and 3 cases of injection site reactions had been reported previously (6).
During the OLE study, 12 patients (6.6%) had reports of elevated liver enzymes, including increases in ALT, aspartate aminotransferase (AST), gamma glutamyltransferase (GGT), and blood bilirubin. Four patients (2.2%) reported elevations of ALT or AST >3 times the upper limit of normal during the OLE study. None of these patients had a history of fatty liver or other hepatic dysfunction, and 2 of the 4 patients had concomitant methotrexate use. Three patients had confirmed normalization of laboratory values by the study conclusion. One patient did not complete final study assessments due to fear of COVID‐19 infection, and therefore, no laboratory values were available for this patient. No patients discontinued bimekizumab due to elevated liver enzymes in the OLE study; 2 discontinuations occurred during weeks 0–48. No Hy's Law cases were reported.
A total of 47 of 206 patients (EAIR 9.7 per 100 patient‐years) had fungal infections over 152 weeks (Table 2). All fungal infections were assessed as mild to moderate in intensity by the study investigator and none were serious. Of these, 24 patients with ≥1 infection had Candida infections (EAIR 4.6 per 100 patient‐years), with the majority of these (19 of 24; EAIR 3.5 per 100 patient‐years) being oral candidiasis. Twenty‐five patients had fungal infections not elsewhere classified (EAIR 4.7 per 100 patient‐years), and the majority (14 of 25; EAIR 2.6 per 100 patient‐years) were oral infections. Infections at other sites were low in frequency, including vulvovaginal infections (1 Candida infection [0.5%] and 2 fungal infections [1.0%]) and skin infections (2 Candida infections [1.0%] and 4 fungal infections [1.9%]) (Table 2). Two patients discontinued due to oral fungal infections. Baseline steroid use, sex, and presence of diabetes mellitus were not clear risk factors for susceptibility to Candida infections. All fungal infections, including Candida infections, were localized, not systemic, and the majority resolved and were easily treatable with systemic or topical antifungal treatments such as fluconazole, nystatin, itraconazole, and miconazole (Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.42280). A total of 23 of 206 patients (11.2%) had >1 episode of a fungal infection over 152 weeks, of which 12 of 206 (5.8%) had >1 episode of a Candida infection. All Candida infections resolved without sequelae. No opportunistic infections were reported, except localized fungal events consisting of one case each of fungal pharyngitis and fungal esophagitis. There were no reported cases of active TB.
No cases of SIB were reported during the OLE study; 1 case was previously reported during the dose‐blind period (6). Among patients in the OLE study with available data (N = 181), 92.3% had HADS anxiety and HADS depression scores of <8 (“normal”) at baseline of the double‐blind period, which was sustained through week 48 to 92.1% at week 152. Prespecified safety topics of interest and other events across weeks 0–152 by randomized dose at baseline are provided in Supplementary Table 1 (http://onlinelibrary.wiley.com/doi/10.1002/art.42280).
Efficacy
Efficacy at weeks 12 and 48 of the BE ACTIVE trial has been reported previously (6). There was no worsening of disease in patients whose dose of bimekizumab was decreased from 320 mg to 160 mg in the OLE.
NRI analysis of joint efficacy outcomes showed that ACR20, ACR50, and ACR70 response rates were sustained, with 64.1%, 52.9%, and 39.3% of patients, respectively, meeting criteria at week 152, compared to 72.3%, 57.3%, and 39.8% of patients, respectively, meeting criteria at week 48 (Figures 1A–C). For skin efficacy outcomes, NRI analysis showed that PASI75, PASI90, and PASI100 responder rates were sustained, with 69.3%, 64.2%, and 57.7% of patients, respectively, meeting criteria at week 152, compared to 81.0%, 73.7%, and 64.2% of patients meeting criteria at week 48 (Figure 1D).
Figure 1.
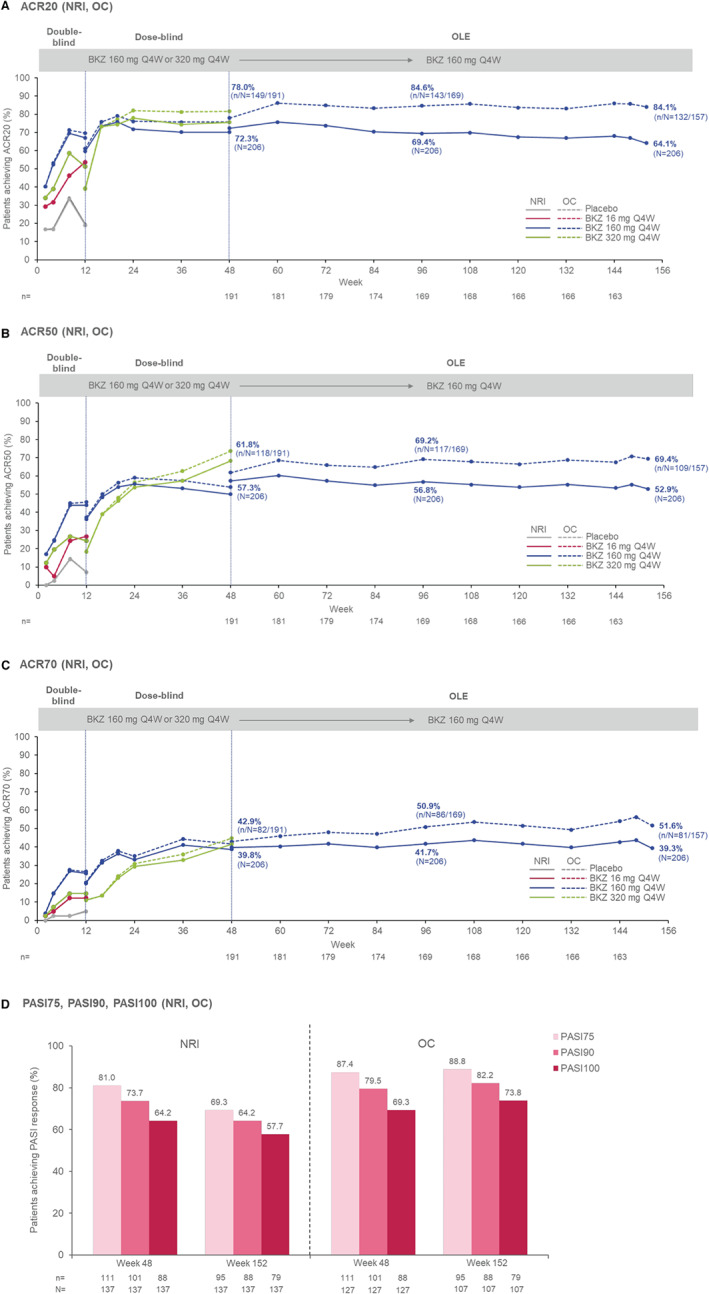
Key efficacy outcomes in psoriatic arthritis patients from baseline (week 0) of the double‐blind period of the BE ACTIVE randomized controlled trial to week 152 (full analysis set). Nonresponder imputation (NRI; solid lines) and observed case (OC; dashed lines) data are shown for the percentages of patients achieving ≥20%, ≥50%, and ≥70% improvement in disease activity based on the American College of Rheumatology criteria for all timepoints from weeks 0–152 (A–C), as well as for the percentages of patients achieving 75%, 90%, and 100% improvement in disease activity based on the Psoriasis Area and Severity Index (PASI) (D). Patients randomized to receive placebo (gray lines), 16 mg of bimekizumab (BKZ; red lines) every 4 weeks, or 320 mg of BKZ (green lines) every 4 weeks through the double‐blind period are shown, and patients assigned to 160 mg of BKZ every 4 weeks with a 320 mg loading dose or 160 mg of BKZ every 4 weeks at double‐blind period entry are combined for weeks 0–12 (blue lines). Percentages in the dose‐blind period include those assigned to 160 mg of BKZ every 4 weeks, 160 mg of BKZ every 4 weeks with a 320 mg loading dose, or 320 mg of BKZ every 4 weeks at double‐blind period entry, as well as those assigned to placebo or 16 mg of BKZ every 4 weeks who were re‐randomized to 160 mg or 320 mg of BKZ every 4 weeks. All open‐label extension (OLE) study patients received 160 mg of BKZ every 4 weeks regardless of prior dosing regimen; 157 patients had an efficacy assessment at week 152. At baseline of the double‐blind period, 137 patients had ≥3% body surface area affected by psoriasis; due to a data collection error and lack of data from the study visit, week 96 data are not reported for PASI. Circles represent timepoints at which patients were assessed.
In the NRI analysis, 51.0% and 26.2% of bimekizumab‐treated patients reached MDA and VLDA responses, respectively, at week 48, with the proportions increasing to 51.5% and 30.1% at week 152 (Figure 2A). Among patients with dactylitis at baseline (LDI score > 0), 45 of 59 patients (76.3%) had resolution at week 48 in the NRI analysis, and 42 of 59 patients (71.2%) were dactylitis‐free at week 152 (Figure 2B). Due to lack of convergence in the analytical model, only observed case data were generated for change from baseline in LDI over 152 weeks. The analysis showed that the mean ± SD change from baseline (SD) in LDI score following treatment with bimekizumab was –8.0 ± 69.5 at week 48, with even greater improvement in LDI score at week 152 (mean ± SD –16.2 ± 45.5) (Supplementary Table 3). Among patients with enthesitis at baseline (MASES >0), 61 of 107 patients (57.0%) had achieved resolution at week 48 in the NRI analysis, and 67 of 107 patients (62.6%) were enthesitis‐free at week 152 (Figure 2C). Multiple imputation analysis showed that the mean ± SEM change from baseline in MASES following treatment with bimekizumab was –2.6 ± 0.3 at week 48, with even greater improvement in MASES at week 152 (mean ± SEM –3.3 ± 0.3) (Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.42280).
Figure 2.
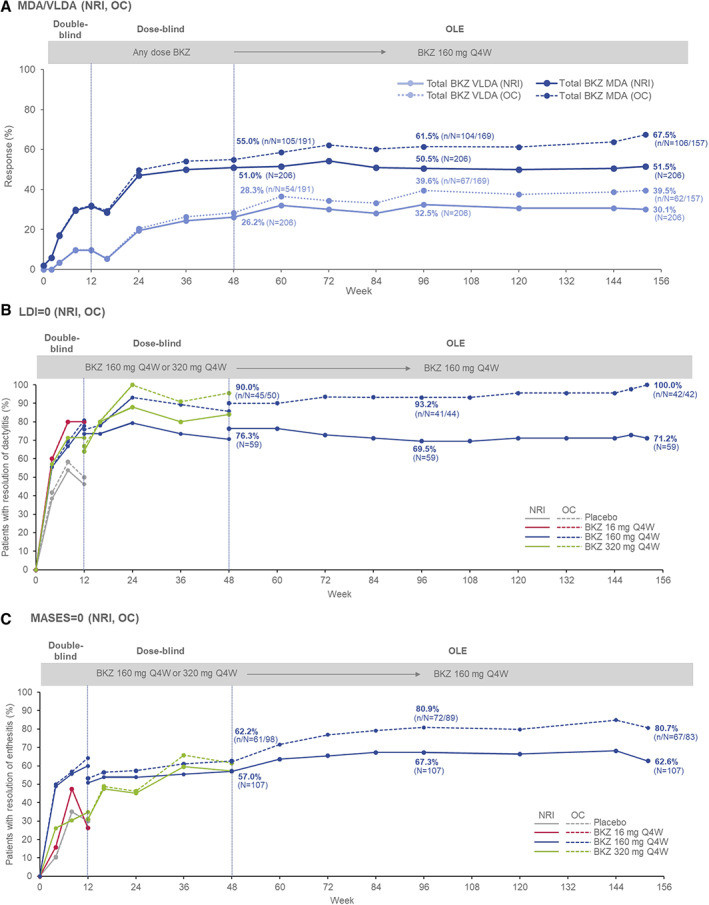
Additional efficacy outcomes in psoriatic arthritis patients from baseline (week 0) of the double‐blind period of the BE ACTIVE randomized controlled trial to week 152 (full analysis set). NRI and OC data are shown for the percentages of patients achieving minimal disease activity (MDA) or very low disease activity (VLDA) (A), percentages of patients achieving resolution of dactylitis based on Leeds Dactylitis Index (LDI) score (includes patients with LDI score >0 at baseline [n = 59]) (B), and percentages of patients achieving resolution of enthesitis based on the Maastricht Ankylosing Spondylitis Enthesitis Index (MASES) (includes patients with MASES score >0 at baseline [n = 107]) (C). Patients were classified as having MDA or VLDA when they met 5 of 7 or 7 of 7, respectively, of the following criteria: tender joint count score ≤1, swollen joint count score ≤1, PASI ≤1 or ≤3% body surface area affected by psoriasis, visual analog scale (VAS) score ≤15 for pain, VAS score ≤20 for patient global activity, Health Assessment Questionnaire disability index score ≤0.5, and tender entheseal points score ≤1. See Figure 1 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42280/abstract.
In addition, 35.0% of patients achieved remission based on the DAPSA score at week 48, increasing to 38.8% at week 152. In NRI analysis, 46.0% of bimekizumab‐treated patients who had ≥3% BSA with psoriasis involvement at baseline of the double‐blind period achieved a response based on the ACR50 + PASI100 composite outcome at week 48 and week 152 (Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.42280). Patients also maintained improvements in pain, physical function, and HRQoL from week 48 to week 152, as measured by PtAAP, SF‐36 PCS, PsAID‐9, and HAQ DI scores (Supplementary Table 3).
DISCUSSION
Patients with PsA require effective long‐term treatment of both joint and skin symptoms. The proportion of patients with PsA who achieve an ACR50 response during treatment with currently licensed IL‐17A inhibitors is ~30–50% at 16–24 weeks, while the proportion achieving PASI90 with IL‐17A inhibitors is ~30–70% at 16–24 weeks, demonstrating the need for more effective treatments (23, 24). Furthermore, sustaining this efficacy over the long‐term is important, as patients may lose clinical benefits of treatment over time (2). In patients with PsA, 3‐year extension data from studies of IL‐17A inhibitors showed similar or slightly lower ACR50 and PASI90 response rates at 156 weeks compared to ACR50 and PASI90 response rates at 24 weeks (25, 26). Similar trends have been observed out to 5 years with IL‐17A inhibitor therapy (27).
In the phase IIb BE ACTIVE trial and its OLE study, bimekizumab was found to be well tolerated in PsA patients up to 3 years of treatment, with no new safety signals identified compared to the first 48 weeks of treatment (6). Findings from the BE ACTIVE trial showed that both joint and skin outcomes improved from baseline of the double‐blind period through week 12 to week 24, after which responses were sustained through the dose‐blind period to week 48. Here, we show that efficacy outcomes were sustained at 3 years.
Patient retention supported long‐term tolerability of bimekizumab, with treatment completed up to week 48 by 91.7% of patients, and up to week 152 by 78.2% of patients (87.5% of those who enrolled in the OLE study). This finding supports the long‐term tolerability and efficacy of treatment with bimekizumab in adult patients with PsA.
The frequencies of TEAEs and serious TEAEs were not increased with further exposure to bimekizumab and were aligned with prior reporting for the double‐ and dose‐blind periods from this phase IIb study in PsA patients (6). The 3 most frequently reported TEAEs over 3 years by MedDRA preferred term (nasopharyngitis, upper respiratory tract infection, and bronchitis) were consistent with previous 48‐week data from the double‐ and dose‐blind periods, as well as a study of bimekizumab in AS patients (6, 7).
Given the role of the IL‐23/IL‐17 pathway in mucocutaneous protection against a variety of pathogens, in particular extracellular fungal infections, the inhibition of IL‐17A was predicted to increase susceptibility to oral candidiasis (28, 29, 30). In addition, due to the involvement of both IL‐17A and IL‐17F in CD4+ T helper 17 (Th17) cell–mediated mucocutaneous immunity (31), dual inhibition of these cytokines is theoretically expected to further increase the risk of fungal infection compared with inhibition of IL‐17A alone. In the OLE study, EAIRs of Candida infection remained stable compared with the first 48 weeks of treatment. No clear risk factors for Candida infection were identified in the BE ACTIVE trial or its OLE study; however, further analyses in phase III studies of bimekizumab in PsA with larger patient populations are needed to confirm whether any potential risk factors for Candida infection can be distinguished. Just over one‐tenth of patients with fungal infections, including Candida infections, reported recurrence of infection at any time during the double‐ and dose‐blind periods or the OLE study, whereas among patients who reported Candida infections, one‐half experienced a recurrence of infection. However, the majority of Candida cases over the course of the study were easily managed and resolved with topical or systemic antifungal treatment.
There were no MACE or cases of SIB during the OLE study. There was 1 moderate case of microscopic colitis adjudicated as IBD in a patient without prior history of IBD; this event was unrelated to treatment with bimekizumab. No cases of uveitis, additional injection site reactions, or nonfungal opportunistic infections were reported in the OLE study. However, results from phase III studies, including larger patient groups, are needed to further establish the long‐term safety profile of bimekizumab in PsA patients.
The improvements in key joint (articular) and skin efficacy outcomes observed in patients receiving bimekizumab over 48 weeks were generally maintained through 3 years of treatment. MDA has been previously identified as a desirable target in treat‐to‐target strategies for PsA, defining a satisfactory state of disease activity that encompasses all aspects of the disease (15). At week 152, more than one‐half of patients achieved MDA, including with NRI. Furthermore, over one‐half of patients achieved ACR50 or PASI100 for those with ≥3% BSA with psoriasis involvement at the double‐blind period baseline responses individually, and the proportion achieving both ACR50 and PASI100 responses remained stable at just under one‐half of bimekizumab‐treated patients between week 48 and week 152.
Notwithstanding the improvements seen in clinically assessed PsA disease outcomes, patients receiving bimekizumab also reported improvements in selected patient‐reported outcome measures that were maintained over 3 years of treatment, including reduced pain, increased physical function, lower disease impact, and improved quality of life.
The 3‐year duration of this phase IIb study provides the most comprehensive evidence to date of the long‐term safety and efficacy of bimekizumab in patients with PsA. A limitation of the present analysis relates to the lack of an active comparator, or placebo comparator beyond week 12. After week 12, all patients received bimekizumab at 160 mg or 320 mg every 4 weeks. Efficacy was similar at week 48 among patients receiving bimekizumab at 160 mg or 320 mg, and no appreciable decrease in efficacy was observed in patients who reduced their dose from 320 mg to 160 mg at the start of the OLE study. Additionally, as a phase IIb study, the limited sample size of ~200 patients restricts the interpretation of the results. There was also no evaluation of structural inhibition in the present study. Results from phase III studies in larger patient populations are therefore awaited to further investigate the safety, tolerability, and efficacy of bimekizumab in PsA patients during long‐term treatment, including structural inhibition. There are currently 2 phase III studies underway, the BE OPTIMAL trial (ClinicalTrials.gov identifier: NCT03895203) and the BE COMPLETE trial (ClinicalTrials.gov identifier: NCT03896581), as well as their combined OLE study, BE VITAL (ClinicalTrials.gov identifier: NCT04009499), after which more robust conclusions will likely be possible.
In conclusion, the safety of bimekizumab in patients with PsA over 3 years of treatment was consistent with the previous 48‐week results, as well as other recently published studies of IL‐17 inhibitors in PsA patients. High thresholds of disease control were achieved within the first year of treatment and sustained through 3 years. Despite the limited sample size, the present study supports the further development of bimekizumab to address an unmet need for improved and sustained efficacy on skin and joint disease in patients with PsA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Coates had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Coates, McInnes, Merola, Warren, Kavanaugh, Gottlieb, Assudani, Bajracharya, Coarse, Ink, Ritchlin.
Acquisition of data
Coarse, Ink.
Analysis and interpretation of data
Coates, McInnes, Merola, Warren, Kavanaugh, Gottlieb, Gossec, Assudani, Bajracharya, Coarse, Ink, Ritchlin.
ROLE OF THE STUDY SPONSOR
UCB Pharma facilitated the study design, funded writing assistance for the manuscript, and reviewed and approved the manuscript prior to submission. The authors independently collected the data, interpreted the results, and had the final decision to submit the manuscript for publication. Publication of this article was contingent upon approval by UCB Pharma.
Supporting information
Disclosure Form
Supplementary Table 1 Safety across BE ACTIVE and the OLE by randomized dose at baseline (safety set)
Supplementary Table 2. Further information on Candida infections in BE ACTIVE and the OLE (safety set)
Supplementary Table 3. Efficacy and patient‐reported outcomes to Week 152 (full analysis set)
Supplementary Figure 1 BE ACTIVE and OLE study design
Supplementary Figure 2 Patient retention in BE ACTIVE and the OLE (full analysis set)
Supplementary Figure 3 Patient disposition in BE ACTIVE and the OLE
Supplemental Video S1
ACKNOWLEDGMENTS
The authors thank the patients and the investigators and their teams who took part in this study. The authors also acknowledge Heather Edens, PhD (UCB Pharma, Smyrna, GA) for publication coordination and Aaron Keeling, BA (Costello Medical, Boston, MA) and Hannah Brechka, PhD (Costello Medical, Cambridge, UK), for medical writing and editorial assistance based on the authors’ input and direction. Support for third‐party writing assistance for this article, provided by Aaron Keeling, BA, and Hannah Brechka, PhD, was funded by UCB Pharma in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
A video abstract of this article can be found at: https://players.brightcove.net/3806881048001/default_default/index.html?videoId=6309214290112
Clinicaltrials.gov identifiers: NCT02969525 and NCT03347110.
The views expressed herein are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Supported by UCB Pharma. Dr. Coates' work was supported by an NIHR Clinician Scientist award and the NIHR Oxford Biomedical Research Centre. Dr. Warren is supported by the NIHR Manchester Biomedical Research Centre.
Data may be requested by qualified researchers 6 months after product approval in the US and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymized IPD and redacted study documents which may include raw datasets, analysis‐ready datasets, study protocol, blank case report form, annotated case report form, statistical analysis plan, dataset specifications, and clinical study report. Prior to use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password protected portal.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42280&file=art42280‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Coates LC, Helliwell PS. Psoriatic arthritis: state of the art review. Clin Med (Lond) 2017;17:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boehncke WH, Menter A. Burden of disease: psoriasis and psoriatic arthritis. Am J Clin Dermatol 2013;14:377–88. [DOI] [PubMed] [Google Scholar]
- 3. Gudu T, Gossec L. Quality of life in psoriatic arthritis [review]. Expert Rev Clin Immunol 2018;14:405–17. [DOI] [PubMed] [Google Scholar]
- 4. Kurschus FC, Moos S. IL‐17 for therapy. J Dermatol Sci 2017;87:221–7. [DOI] [PubMed] [Google Scholar]
- 5. Deodhar A, Mease PJ, McInnes IB, Baraliakos X, Reich K, Blauvelt A, et al. Long‐term safety of secukinumab in patients with moderate‐to‐severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: integrated pooled clinical trial and post‐marketing surveillance data. Arthritis Res Ther 2019;21:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ritchlin CT, Kavanaugh A, Merola JF, Schett G, Scher JU, Warren RB, et al. Bimekizumab in patients with active psoriatic arthritis: results from a 48‐week, randomised, double‐blind, placebo‐controlled, dose‐ranging phase 2b trial. Lancet 2020;395:427–40. [DOI] [PubMed] [Google Scholar]
- 7. Van der Heijde D, Gensler LS, Deodhar A, Baraliakos X, Poddubnyy D, Kivitz A, et al. Dual neutralisation of interleukin‐17A and interleukin‐17F with bimekizumab in patients with active ankylosing spondylitis: results from a 48‐week phase IIb, randomised, double‐blind, placebo‐controlled, dose‐ranging study. Ann Rheum Dis 2020;79:595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gordon KB, Foley P, Krueger JG, Pinter A, Reich K, Vender R, et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): a multicentre, double‐blind, placebo‐controlled, randomised withdrawal phase 3 trial. Lancet 2021;397:475–86. [DOI] [PubMed] [Google Scholar]
- 9. McArdle A, Pennington S, FitzGerald O. Clinical features of psoriatic arthritis: a comprehensive review of unmet clinical needs [review]. Clin Rev Allergy Immunol 2018;55:271–94. [DOI] [PubMed] [Google Scholar]
- 10. Visalli E, Crispino N, Foti R. Multidisciplinary management of psoriatic arthritis: the benefits of a comprehensive approach. Adv Ther 2019;36:806–16. [DOI] [PubMed] [Google Scholar]
- 11. Mease PJ, Antoni CE, Gladman DD, et al. Psoriatic arthritis assessment tools in clinical trials. Ann Rheum Dis 2005;64 (Suppl 2):ii49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fredriksson T, Pettersson U. Severe psoriasis—oral therapy with a new retinoid. Dermatologica 1978;157:238–44. [DOI] [PubMed] [Google Scholar]
- 13. Helliwell PS, FitzGerald O, Fransen J, et al. Development of an assessment tool for dactylitis in patients with psoriatic arthritis. J Rheumatol 2005; 32:1745–50. [PubMed] [Google Scholar]
- 14. Heuft‐Dorenbosch L, Spoorenberg A, van Tubergen A, et al. Assessment of enthesitis in ankylosing spondylitis. Ann Rheum Dis 2003; 62:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coates LC, Fransen J, Helliwell PS. Defining minimal disease activity in psoriatic arthritis: a proposed objective target for treatment. Ann Rheum Dis 2010;69:48–53. [DOI] [PubMed] [Google Scholar]
- 16. Baker JF, Conaghan PG, Smolen JS, et al. Development and validation of modified disease activity scores in rheumatoid arthritis: superior correlation with magnetic resonance imaging–detected synovitis and radiographic progression. Arthritis Rheumatol 2014;66:794–802. [DOI] [PubMed] [Google Scholar]
- 17. Gossec L, de Wit M, Kiltz U, et al. A patient‐derived and patient‐reported outcome measure for assessing psoriatic arthritis: elaboration and preliminary validation of the Psoriatic Arthritis Impact of Disease (PsAID) questionnaire, a 13‐country EULAR initiative. Ann Rheum Dis 2014;73:1012–9. [DOI] [PubMed] [Google Scholar]
- 18. Ware JE Jr, Snow KK, Kosinski M, et al. SF‐36 health survey: manual and interpretation guide. Boston: The Health Institute, New England Medical Center; 1993. [Google Scholar]
- 19. Zigmond AS, Snaith RP. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand 1983;67:361–70. [DOI] [PubMed] [Google Scholar]
- 20. Lubrano E, Perrotta FM, Scriffignano S, Coates LC, Helliwell P. Sustained very low disease activity and remission in psoriatic arthritis patients. Rheumatol Ther 2019;6:521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schoels M, Aletaha D, Funovits J, et al. Application of the DAREA/DAPSA score for assessment of disease activity in psoriatic arthritis. Ann Rheum Dis 2010;69:1441–7. [DOI] [PubMed] [Google Scholar]
- 22. Fries JF, Spitz P, Kraines RG, et al. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. [DOI] [PubMed] [Google Scholar]
- 23. Mease P, van der Heijde D, Landewe R, Mpofu S, Rahman P, Tahir H, et al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double‐blind, phase III FUTURE 5 study. Ann Rheum Dis 2018;77:890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin‐17A specific monoclonal antibody, for the treatment of biologic‐naive patients with active psoriatic arthritis: results from the 24‐week randomised, double‐blind, placebo‐controlled and active (adalimumab)‐controlled period of the phase III trial SPIRIT‐P1. Ann Rheum Dis 2017;76:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chandran V, van der Heijde D, Fleischmann RM, Lespessailles E, Helliwell PS, Kameda H, et al. Ixekizumab treatment of biologic‐naive patients with active psoriatic arthritis: 3‐year results from a phase III clinical trial (SPIRIT‐P1). Rheumatology (Oxford) 2020;59:2774–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mease PJ, Kavanaugh A, Reimold A, Tahir H, Rech J, Hall S, et al. Secukinumab in the treatment of psoriatic arthritis: efficacy and safety results through 3 years from the year 1 extension of the randomised phase III FUTURE 1 trial. RMD Open 2018;4:e000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reich K, Warren RB, Coates LC, Di Comite G. Long‐term efficacy and safety of secukinumab in the treatment of the multiple manifestations of psoriatic disease. J Eur Acad Dermatol Venereol 2020;34:1161–73. [DOI] [PubMed] [Google Scholar]
- 28. Conti HR, Gaffen SL. IL‐17‐mediated immunity to the opportunistic fungal pathogen candida albicans. J Immunol 2015;195:780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blauvelt A, Lebwohl MG, Bissonnette R. IL‐23/IL‐17A dysfunction phenotypes inform possible clinical effects from anti‐IL‐17A therapies. J Invest Dermatol 2015;135:1946–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin‐17A and ‐17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009;30:108–19. [DOI] [PubMed] [Google Scholar]
- 31. Mengesha BG, Conti HR. The role of IL‐17 in protection against mucosal candida infections. J Fungi (Basel) 2017;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Supplementary Table 1 Safety across BE ACTIVE and the OLE by randomized dose at baseline (safety set)
Supplementary Table 2. Further information on Candida infections in BE ACTIVE and the OLE (safety set)
Supplementary Table 3. Efficacy and patient‐reported outcomes to Week 152 (full analysis set)
Supplementary Figure 1 BE ACTIVE and OLE study design
Supplementary Figure 2 Patient retention in BE ACTIVE and the OLE (full analysis set)
Supplementary Figure 3 Patient disposition in BE ACTIVE and the OLE
Supplemental Video S1