

ABSTRACT
Streptococcus dysgalactiae subsp. equisimilis is a bacterial pathogen that is increasingly recognized as a cause of severe human infections. Much less is known about the genomics and infection pathogenesis of S. dysgalactiae subsp. equisimilis strains compared to the closely related bacterium Streptococcus pyogenes. To address these knowledge deficits, we sequenced to closure the genomes of seven S. dysgalactiae subsp. equisimilis human isolates, including six that were emm type stG62647. Recently, for unknown reasons, strains of this emm type have emerged and caused an increasing number of severe human infections in several countries. The genomes of these seven strains vary between 2.15 and 2.21 Mbp. The core chromosomes of these six S. dysgalactiae subsp. equisimilis stG62647 strains are closely related, differing on average by only 495 single-nucleotide polymorphisms, consistent with a recent descent from a common progenitor. The largest source of genetic diversity among these seven isolates is differences in putative mobile genetic elements, both chromosomal and extrachromosomal. Consistent with the epidemiological observations of increased frequency and severity of infections, both stG62647 strains studied were significantly more virulent than a strain of emm type stC74a in a mouse model of necrotizing myositis, as assessed by bacterial CFU burden, lesion size, and survival curves. Taken together, our genomic and pathogenesis data show the strains of emm type stG62647 we studied are closely genetically related and have enhanced virulence in a mouse model of severe invasive disease. Our findings underscore the need for expanded study of the genomics and molecular pathogenesis of S. dysgalactiae subsp. equisimilis strains causing human infections.
IMPORTANCE Our studies addressed a critical knowledge gap in understanding the genomics and virulence of the bacterial pathogen Streptococcus dysgalactiae subsp. equisimilis. S. dysgalactiae subsp. equisimilis strains are responsible for a recent increase in severe human infections in some countries. We determined that certain S. dysgalactiae subsp. equisimilis strains are genetically descended from a common ancestor and that these strains can cause severe infections in a mouse model of necrotizing myositis. Our findings highlight the need for expanded studies on the genomics and pathogenic mechanisms of this understudied subspecies of the Streptococcus family.
KEYWORDS: Streptococcus dysgalactiae, genomics, pathogenesis, emerging clone
INTRODUCTION
Streptococcus dysgalactiae subsp. equisimilis is a bacterial pathogen that causes diverse infections in humans; in recent years, it has been reported as the cause of increasing numbers of severe invasive infections (1–15). At the species level, S. dysgalactiae subsp. equisimilis is most closely related to Streptococcus pyogenes, a common cause of human infections, such as pharyngitis, skin and soft tissue infections, bacteremia, and severe invasive infections including necrotizing fasciitis, -myositis, and -pneumonia. Although generally perceived as less virulent, S. dysgalactiae subsp. equisimilis causes a spectrum of noninvasive and invasive human infections similar to that of S. pyogenes. S. dysgalactiae subsp. equisimilis is an increasingly recognized cause of serious invasive infections, and some strains have been documented to be resistant to beta-lactam antibiotics (16), a concerning development that may lead to substantial treatment difficulties and public health problems.
Compared to S. pyogenes, relatively few studies of the genomics and molecular pathogenic mechanisms of S. dysgalactiae subsp. equisimilis have been conducted, and thus much remains to be learned. For example, although there are 41,249 publicly available whole-genome sequencing run files for S. pyogenes, for S. dysgalactiae subsp. equisimilis there are only 648 (NCBI Sequence Read Archive as of 8 September 2022). Similarly, there are 2,223 publicly available S. pyogenes whole-genome assemblies with 257 closed, whereas for S. dysgalactiae subsp. equisimilis there are only 67 with 21 closed (NCBI Microbial Genome Database as of 8 September 2022). On average, the genome sequence of S. pyogenes strains is approximately 1.8 Mbp. In contrast, the genomes of S. dysgalactiae subsp. equisimilis strains are larger, averaging approximately 2.1 Mbp (8, 9, 17–24).
Similar to the relative lack of information about S. dysgalactiae subsp. equisimilis genomics, very little is known about the molecular basis of S. dysgalactiae subsp. equisimilis pathogenesis. S. dysgalactiae subsp. equisimilis strains share extensive gene content with S. pyogenes (25), but very few S. dysgalactiae subsp. equisimilis genes have been shown unambiguously to encode virulence factors experimentally proven to participate in pathogenesis based on analysis of isogenic mutant strains (19, 26–31). Also compared to S. pyogenes, only very limited pathogenesis work has been conducted with animal infection models, including necrotizing soft tissue infections (19, 27, 28, 30–32). Thus, in the aggregate, an understanding of S. dysgalactiae subsp. equisimilis pathogenic capacity is severely hampered by basic biology knowledge deficits, including population genomic diversity, gene expression, regulatory networks, virulence factors, and molecular interactions with the host.
The work presented here had two goals. First, we sought to investigate the genomics of emm type stG62647 S. dysgalactiae subsp. equisimilis strains cultured from patients with infections in Brittany, France. In recent years, strains of this emm type have been an increasingly important cause of severe infections in several geographic areas (7, 9, 11, 13, 33–36). The emm type stG62647 strains have been reported to cause an increased number of osteoarticular infections in humans compared to other S. dysgalactiae subsp. equisimilis emm types, although the cause is unknown (33, 37). Second, we sought to evaluate the relative virulence of the emergent stG62647 lineage by comparing two clonally related stG62647 strains with a genetically divergent strain of emm type stC74a using an established mouse model of necrotizing myositis (38–42). Our results provided important new information about the genetic relatedness and enhanced virulence of this emergent clone of S. dysgalactiae subsp. equisimilis and delineated a framework for expanded genomics and molecular pathogenesis analyses of understudied S. dysgalactiae subsp. equisimilis strains.
RESULTS AND DISCUSSION
Overview of genomes.
To establish a genetic foundation for investigating the molecular pathogenic capacity of S. dysgalactiae subsp. equisimilis strains to cause disease, the genomes of seven human infection isolates collected in French Brittany between 2010 and 2018 were determined by a combination of Illumina paired-end short-read and Oxford Nanopore long-read sequencing. The seven isolates included six group C strains of emm type stG62647 (MGCS35823, MGCS35922, MGCS35957, MGCS36044, MGCS36083, and MGCS36089), which were selected because strains with these characteristics have recently emerged in multiple countries as a cause of increased numbers of severe infections (9, 36, 43). One group G stC74a strain (MGGS36055) was selected as a comparator, because strains with these characteristics have been reported to be an abundant cause of human infections in multiple epidemiological studies (7, 11, 15, 34, 44–46). The depth of short-read sequencing coverage averaged 587-fold (range, 204- to 1,539-fold) and long-read coverage averaged 94-fold (range, 21- to 239-fold), for a combined average of 681-fold (range, 337- to 1,560-fold) (see Table S1 in the supplemental material). The assembled and closed genomes had an average size of 2,196,360 bp (range, 2,151,694 to 2,233,554) with an average 2,130 protein-encoding genes (range, 2,075 to 2,209) and an average G+C content of 39.4% (range, 39.3% to 39.6%) (Table 1 and Table S2). Although some closed S. dysgalactiae subsp. equisimilis genomes differed in their overall architecture due to large regions of sequence inversion (for example, NCTC7136 versus ATCC 12394), these seven genomes were all colinear and had the same chromosomal architecture as reference strain ATCC 12394. These genome size and composition characteristics are consistent with those of the 21 publicly available closed S. dysgalactiae subsp. equisimilis genomes (Table S3). Consistent with the close species genetic relatedness, 1,280 of the 2,075 (61.7%) coding DNA sequences (CDSs) for the genome of strain MGCS36044 had a reciprocal Blastp best hit with a CDS of S. pyogenes emm1 strain MGAS2221 genome (GenBank accession number GCA_012572265.1).
TABLE 1.
Strains and closed genome characteristics
| Strain | Yr | Invasive | Group antigen | MLST | emm type | Size (bp) | CDSs |
|---|---|---|---|---|---|---|---|
| MGCS35823 | 2015 | Pos | C | 20 | stG62647 | 2,189,610 | 2,140 |
| MGCS35922a | 2016 | Neg | C | 20 | stG62647 | 2,223,912 | 2,145 |
| MGCS35957 | 2017 | Neg | C | 20 | stG62647 | 2,233,554 | 2,209 |
| MGCS36044 | 2018 | Pos | C | 20 | stG62647 | 2,153,748 | 2,075 |
| MGCS36083 | 2018 | Pos | C | 20 | stG62647 | 2,212,841 | 2,140 |
| MGCS36089 | 2018 | Pos | C | 20 | stG62647 | 2,151,694 | 2,077 |
| MGGS36055 | 2018 | Pos | G | 17 | stC74a | 2,204,518 | 2,126 |
Strain MGCS35922 had additional gene content of 47,051 bp that assembled extrachromosomally.
Regions of difference and putative mobile genetic elements.
The seven genomes differed in size primarily due to discrete regions of difference (RODs) in gene content attributable to integration of putative mobile genetic elements (MGEs), with gene content characteristic of integrative conjugative elements (ICEs) (47) and phages (48) (Fig. 1). These RODs are the major source of genetic diversity and gene content difference among the seven genomes, constituting on average 267.4 kbp or 12.2% of each genome sequence. In comparing gene content among the seven genomes using ST20/stG62647 strain MGCS36044 as the reference, there were nine RODs (comprising 215.4 kbp) that had gene content that was largely present among all six of the ST20/stG62647 genomes but was largely absent from the ST17/stC74a strain MGGS36055 genome (Fig. 1A and Table S4). Conversely, using ST17/stC74a strain MGGS36055 as the reference, there were 11 RODs (comprising 270.2 kbp) that had gene content that was present in the stC74a strain MGGS36055 genome but was largely absent from the six stG62647 genomes (Fig. 1B and Table S5). Cumulatively, these 20 RODs constituted 485.6 kbp of genetic content differing between strains MGCS36044 and MGGS36055. These RODs ranged in size from 5.2 to 70.7 kbp, and each encoded 4 to 70 inferred genes. Nearly all of these RODs encoded a site-specific integrase or recombinase gene, most of which were terminally located. Moreover, the G+C content of most of these RODs was on average 3% to 7% lower than the overall average of the genome. These characteristics—variable presence among the strains, differing from the core genome in nucleotide composition, and encoding an integrase gene—were all consistent with these RODs being integrative mobile genetic elements that had been acquired exogenously. Although not systematically evaluated, much of the gene content of the RODs shared sequence similarity with MGEs (phages and ICEs) of other streptococci, in particular S. pyogenes and S. dysgalactiae subsp. dysgalactiae. The larger (≥30 kbp) RODs had mobilization and structural gene content characteristic of ICEs and phages, whereas many of the smaller RODs encoded an integrase- or recombinase-like gene but lacked other gene content commonly found in ICEs (e.g., genes encoding conjugal transfer proteins) and phages (e.g., genes encoding coat and tail structural proteins), making the nature of their putative mobilization cryptic. These smaller RODs may represent integrative and mobilizable elements (IMEs) (49) or satellite prophages (48). Some of the smaller RODs, which lacked an integrase gene, were flanked terminally by insertion sequences which were prevalent in the S. dysgalactiae subsp. equisimilis genomes (average, 95) and may have represented composite transposons.
FIG 1.
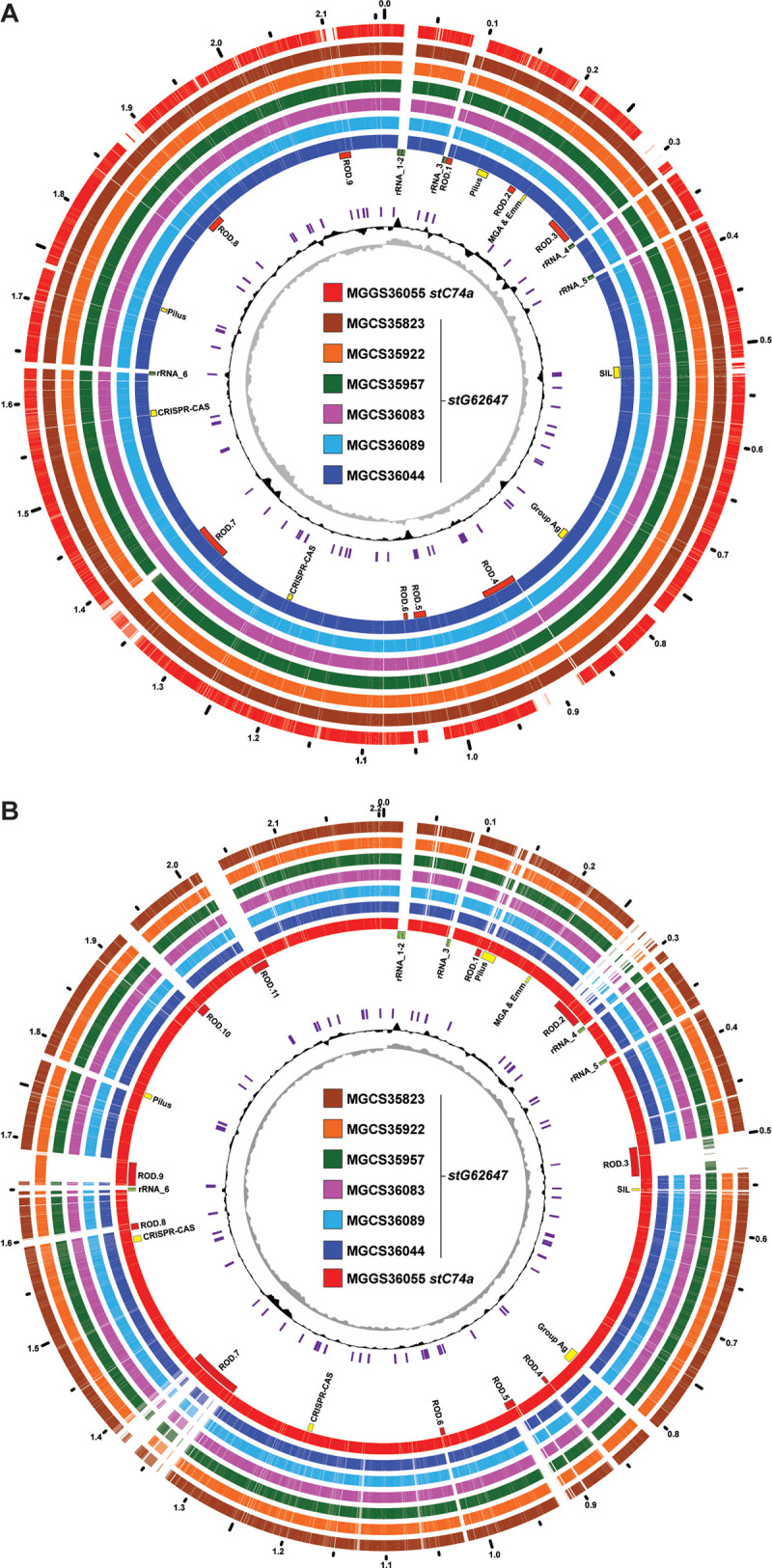
Comparative genome atlases. (A) Illustrated is the 2,158,391-bp genome of the GCS, ST20, stG62647 strain MGCS36044 and the genetic content it shares with the six other strains of the cohort, as determined by Blastn comparison of CDSs (at 80% identity) relative to the MGCS36044 genome. The common CDS content of the seven strains is shown in the outermost rings, colored as indicated in the central index. The GC skew, indicating the forward and reverse replicores, is plotted in the innermost ring in gray. The G+C percent above and below the genome average (in a sliding window of 10,000 bp) is plotted in the adjacent ring in black. Insertion sequences are shown in purple. Genomic landmarks are indicated as labeled in the ring adjacent to the strain MGCS36044 genome. Of note are nine regions of difference constituting 215.4 kbp, which contain gene content that is largely present in the six stG62647 genomes but is largely absent from the stC74a comparator strain MGGS36055 genome (see Table S3 in the supplemental material). (B) Illustrated is the 2,204,518-bp genome of GGS, ST17, stC74a strain MGGS36055 and the genetic content it shares with the six other strains of the cohort. Of note, there are 11 regions of difference constituting 270.7 kbp which contain gene content present in the stC74a MGGS36055 genome but largely absent in the six stG62647 genomes (Table S4). For both strain MGCS36044 and strain MGGS36055, many of the RODs encode an integrase and have a G+C content that is 3% to 7% lower than that of the genome average (approximately 39.4%), consistent with these regions potentially being integrative mobile genetic elements of exogenous origin.
An extrachromosomal ICE in stG62647 strain MGCS35922.
In addition to the 20 RODs that largely differed in all six stG62647 strains from stC74a strain MGGS36055 and vice versa, there were several additional RODs with gene content largely present in only one stG62647 strain of the cohort (Table S6). Among these strain-specific RODs, we identified a 47-kb circular element that Unicycler assembled in its entirety extrachromosomally from the stG62647 strain MGGS35922 closed genome. Because of the concern that this might represent an assembly error, the strain MGGS35922 extrachromosomal (35922ec) circularized element was checked for consistency with the short- and long-read sequencing data by read mapping. No locus was found where there was discontinuity in either the paired mapping of short reads or disrupted mapping of long reads to indicate that the element was inappropriately assembled as an extrachromosomal closed circularized sequence. Moreover, 35922ec had gene content encoding conjugal transfer and mobilization proteins and a serine-type site-specific integrase or recombinase flanked by an attachment site sequence homologous to (i.e., targeting integration to) the chromosomally encoded 23S RNA methyltransferase gene rlmD, all characteristics consistent with it being an ICE (50) (Fig. 2). Additionally, 35922ec encoded a RepA-like replication protein and a chromosome segregation ATPase protein, findings consistent with the capacity for episomal replication (47). Compared to the seven closed genomes, the 35922ec ICE had sequence similarity with elements integrated at rmlD and corresponding to ROD.7, as defined relative to the strain MGCS36044 genome (Fig. 1A) and strain MGGS36055 genomes (Fig. 1B). 35922ec had partial gene content that was present in the genomes of all seven strains of the cohort, including the MGGS35922 genome (Fig. 2). As an additional test of the genome assembly validity, PCR was used to check the structure at the putative extrachromosomal element 35922ec putative attachment site and chromosomal attachment and integration sites flanking 35922_ROD.7 (Fig. S1). All PCR amplicons were consistent with the genome architecture produced by the Unicycler hybrid sequencing assembly. 35922ec and 35922_ROD.7, although having some ICE-like gene content in common, differed in sequence and overall length by 13 kbp. We hypothesized that the 35922ec ICE did not integrate into the strain MGGS35922 chromosome because the rlmD integration site was already occupied by 35922_ROD.7 ICE (Fig. 2). Experimental studies are required to test this hypothesis.
FIG 2.
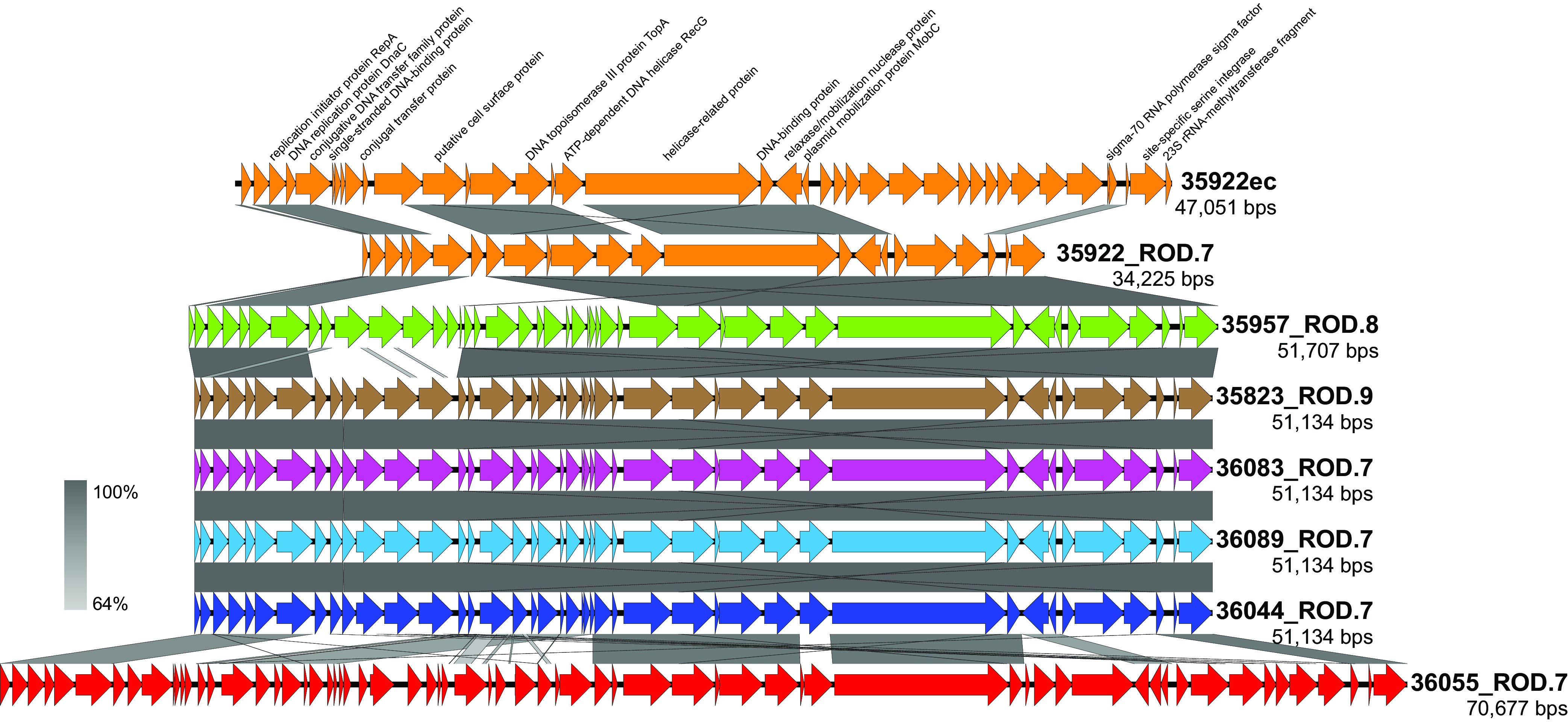
Comparison of the ROD.7 and 35922ec ICEs. Illustrated is an alignment of the 47-kb 35922ec extrachromosomal element in the strain MGCS35922 genome with the ROD.7 equivalent elements of all seven isolates of the cohort. Shown in a gradient of gray, indicating percent sequence identity, are strain-to-strain aligned regions. These elements have conjugal transfer gene content indicating that they are ICEs. All have a PinE superfamily site-specific serine integrase at their 3′ end which directs integration into the 23S rRNA methyltransferase gene rmlD (aka rumA). The 5′ end of rmlD is part of the 35922ec ICE. These ICEs are most closely related among the six stG62647 strains, being 100% identical in sequence among strains MGCS35823, MGCS36044, MGCS36083, and MGCS36089 throughout their 51.1-kbp lengths, indicating a very recent common ancestor. The ICE in stC74a strain MGGS36055 has 52.6% of its sequence in common with that of strain MGCS36044 ROD.7. Most distant is the 35922ec ICE, which has 45.9% of its sequence in common with that of strain MGCS36044 ROD.7.
Additional sources of genetic content diversity.
In addition to the RODs with characteristics of MGEs, there were several additional regions of lower variation in genetic content that differed between the six stG62647 strains and stC74a strain MGGS36055. These regions included gene content encoding pilus, M protein, carbohydrate group antigen, and the extended streptococcal invasion locus (SIL) (each indicated as a landmark in Fig. 1). These were not regions of complete gene content difference. That is, they did not simply differ by strain-to-strain gene presence or absence, but rather had partial gene content that was conserved and partial content that varied. The regions had in common that each encoded one or more inferred secreted or surface-attached molecules. In principle, these proteins and sugar polymers interact with the host immune system, and genetic variation at these loci may reflect diversifying evolutionary selection for polymorphisms that contribute to evasion of the host immune response. Oppegaard et al. (9) reported that among the 18 ST20/stG62647 isolates collected in western Norway that they studied, the SIL quorum-sensing regulon (51–53) was disrupted by insertion of IS1548 into the silB gene encoding the two-component system sensor histidine kinase SilB. The investigators postulated that disruption of the silB gene contributed to ST20/stG62647 isolates causing severe infections. All six of the stG62647 isolates collected in French Brittany that we studied also had the same transposon insertion (IS1548) disrupting silB, whereas stC74a strain MGGS36055 lacked all six SIL genes (silA, -B, -C, -CR, -D, and -E) (Fig. 3). Among the 21 closed S. dysgalactiae subsp. equisimilis genomes in GenBank, the SIL genes are absent in 5 but present in 16 (Table S3). However, in 5 of the 16 genomes with the SIL genes present, one or more of the SIL genes were disrupted, including silB in the strain TPCH-A19. Clearly the SIL region, not to mention the additional bacteriocin-like gene content flanking it and constituting the extended SIL, was fairly variable among S. dysgalactiae subsp. equisimilis genomes. The function of SIL has not been experimentally characterized in S. dysgalactiae subsp. equisimilis, and thus the genes it regulates and its role in S. dysgalactiae subsp. equisimilis pathogenesis are currently unknown.
FIG 3.
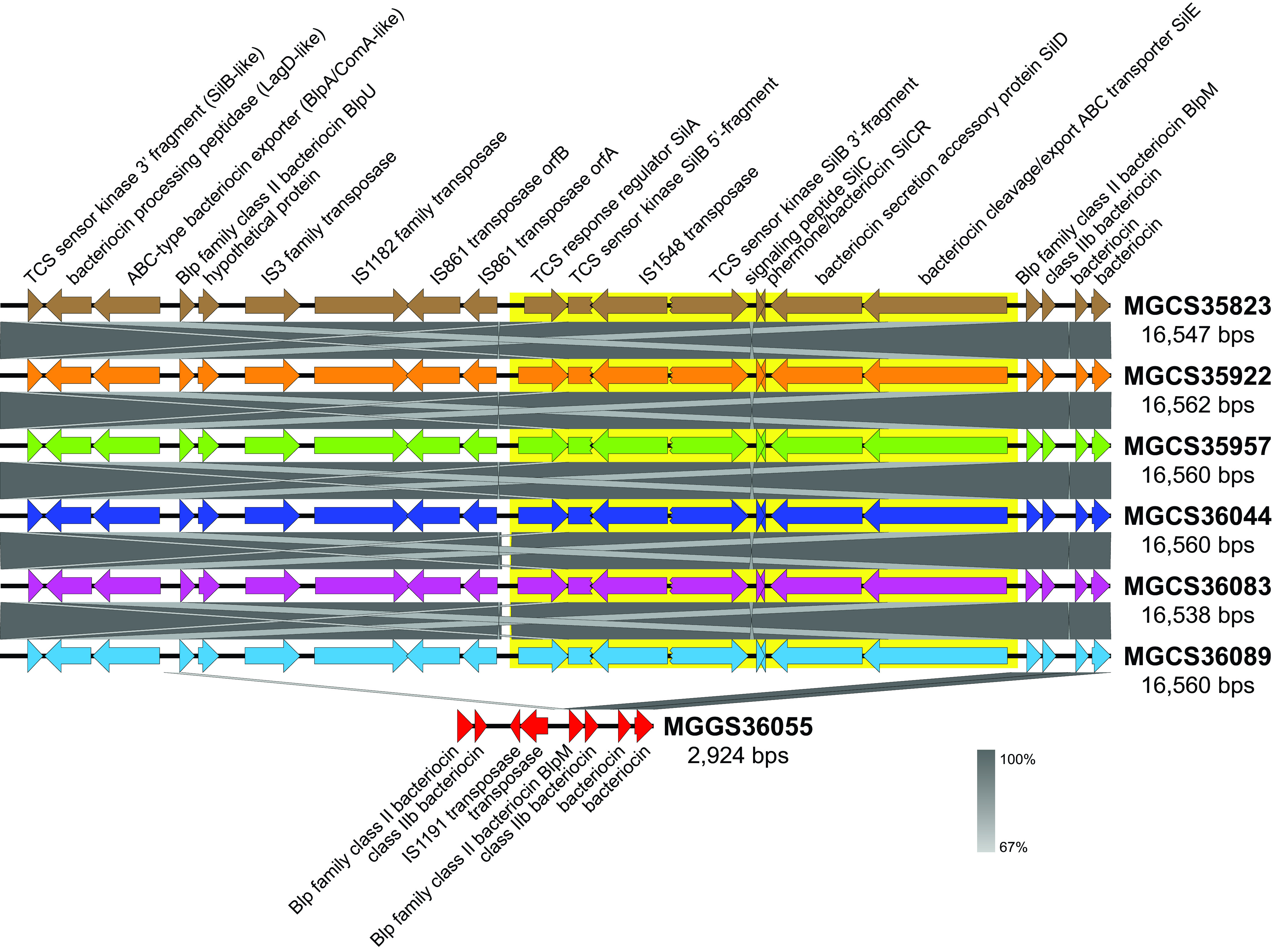
Variation in the streptococcal invasion locus. Illustrated is an alignment of the streptococcal invasion locus between all seven isolates of the cohort. Shown in a gradient of gray, indicating percent sequence identity, are strain-to-strain aligned regions. The SIL is composed of six genes: silA, -B, -C, -CR, -D, and -E (highlighted in yellow), which are variably present among S. dysgalactiae subsp. equisimilis genomes. In multiple streptococcal species, the SIL genes are flanked by additional variably present genes with similarity to genes involved in competence quorum sensing and bacteriocin production. Together, the SIL and flanking competence/bacteriocin-like genes constitute the extended SIL. The extended SIL among the six ST20/stG62647 genomes is 16.5 kb, encodes 21 genes which, strain to strain, share greater than 99.4% nucleotide identity. SilB is disrupted by insertion of IS1548 in all six stG62647 genomes. In contrast, the corresponding locus of the ST17/stC74a strain MGGS36055 genome is only 2.9 kbp and lacks all six SIL genes. Sequence similarity between stC674a strain MGGS36055 and the six stG62647 strains is limited primarily to the four bacteriocin genes at the 3′ end of the extended SIL.
Phylogenetic analysis of the seven genomes.
Phylogenetic analysis of the seven genomes based on core-genome single-nucleotide polymorphisms (SNPs) found that the six stG64647 strains we studied were closely genetically related, differing from each other on average by only 495 core chromosomal SNPs (Fig. 4). These six isolates (collected in Brittany, France between 2015 and 2018) were of the same multilocus sequence type clonal complex, CC20 (CC20 is composed of ST20 and its single-locus variants), as were 18 of the 19 stG62647 strains recently reported by Oppegaard et al. (9) as an emergent cause of severe infections in western Norway. Phylogenetically, these two sets of isolates clustered together (Fig. 4), consistent with them being clonally derived from an evolutionarily recent common progenitor. In contrast, the stC74a comparator strain MGGS36055 along with reference strain RE378 and Norway isolate T666 were multilocus sequence type CC17 and differed on average from the CC20 isolates by 12,050 core SNPs (Fig. 4).
FIG 4.
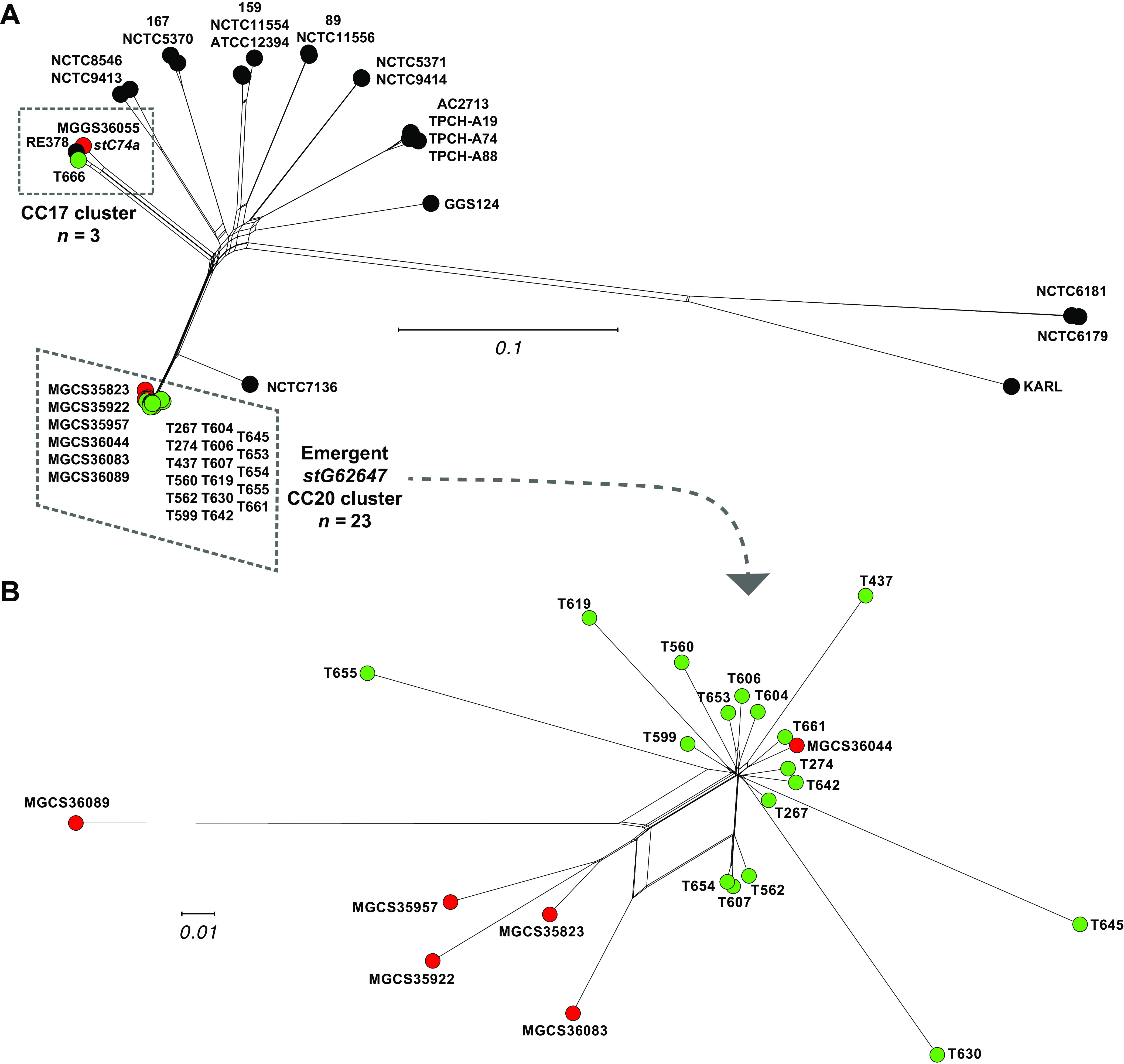
(A) Genetic relationships among 46 S. dysgalactiae subsp. equisimilis isolates inferred by neighbor-network based on 84,926 core chromosomal SNPs. Illustrated are three strain sets: 21 closed reference genomes from the National Center for Biotechnology Information (NCBI) Microbial Genome Database (MGDB) are in black, 18 stG62647 isolates collected in Norway are in green (whole genome sequenced by Oppegaard et al.), and seven isolates (six stG62647 and one stC74a) collected in French Brittany are in red. (B) Genetic relationships among 23 (17 Norway and 6 French Brittany) clustered and closely related S. dysgalactiae subsp. equisimilis stG62647 isolates of human infections inferred by neighbor-network based on 3,369 core chromosomal SNPs. The strain-to-strain mean genetic distance (MGD, i.e., core SNP difference) between the 21 closed reference genomes is 20,428 (range,137 to 42,147), the MGD between the 17 Norway emergent stG62647 cluster isolates is 291 (range, 42 to 776), the MGD between the 6 French Brittany stG62647 closed genomes is 495 (range, 237 to 847), the MGD between all 23 isolates of the emergent stG62647 genetic cluster is 379 (range, 42 to 1,080), and the MGD between the 6 French Brittany stG62647/ST20 isolates and stC74a/ST17 strain MGGS36055 is 11,022 (range, 10,976 to 11,097).
Lancefield carbohydrate group identification.
S. dysgalactiae subsp. equisimilis strains can be of streptococcal carbohydrate group antigen types G, C, or A. Bioinformatic analysis of the region of the genome encoding the Lancefield carbohydrate group antigen synthesis genes of the six clonally related stG62647 strains indicated that these organisms are group C and that the stC74a strain MGGS36055 is group G. This bioinformatic determination was confirmed experimentally with a commonly used immunologic assay, as described in Materials and Methods (Table 1).
MICs for beta-lactam antibiotics.
Decreased susceptibility of beta-hemolytic streptococci to beta-lactam antibiotics has been reported with increased frequency in recent years (3, 16, 39, 54–58). Concerningly, Fuursted et al. (16) described four clonally related S. dysgalactiae subsp. equisimilis strains resistant to penicillin in vitro and reported that these strains had multiple amino acid changes in PBP2X, the primary target of penicillin in streptococci. Thus, we evaluated MICs in our seven S. dysgalactiae subsp. equisimilis strains for five commonly used beta-lactam antibiotics, including amoxicillin, cefotaxime, cefoxitin, meropenem, and penicillin G. None of the seven S. dysgalactiae subsp. equisimilis strains had MICs deviating from the susceptible range for the five beta-lactam antibiotics tested (Table 2). Consistent with our in vitro susceptibility data, the PBP2X gene in the seven isolates studied did not have amino acid changes expected to result in substantial increases in MICs to these antibiotics. That is, there were no amino acid changes in the PBP2X transpeptidase domain and catalytic motifs that have been associated with reduced beta-lactam susceptibility in streptococci (data not shown).
TABLE 2.
Beta-lactam antibiotic MICs for the seven S. dysgalactiae subsp. equisimilis strains studied
| Strain | MIC (mg/liter) |
||||
|---|---|---|---|---|---|
| Amoxicillin | Cefotaxime | Cefoxitin | Meropenem | Penicillin G | |
| MGCS35823 | 0.032 | 0.023 | 0.75 | 0.012 | 0.012 |
| MGCS35922 | 0.032 | 0.023 | 1.00 | 0.008 | 0.012 |
| MGCS35957 | 0.032 | 0.023 | 0.50 | 0.008 | 0.012 |
| MGCS36044 | 0.032 | 0.023 | 1.00 | 0.012 | 0.016 |
| MGCS36083 | 0.032 | 0.032 | 1.00 | 0.012 | 0.016 |
| MGCS36089 | 0.016 | 0.023 | 1.00 | 0.012 | 0.012 |
| MGGS36055 | 0.016 | 0.023 | 0.75 | 0.006 | 0.012 |
stG62647 strains are significantly more virulent than a genetically divergent strain of emm type stC74a.
Epidemiological surveillance reports and outbreak investigations from diverse locations suggest that emm type stG62647 strains may cause more virulent human infections than S. dysgalactiae subsp. equisimilis strains of other emm types, but this has not been experimentally shown. To test the hypothesis that emm type stG62647 strains of the ST20 genetic lineage are more virulent than other S. dysgalactiae subsp. equisimilis genetic lineages that cause abundant human infections, we compared the virulence of ST20/stG62647 strains MGCS36044 and MGCS36089 relative to that of ST17/stC74a strain MGGS36055 using a well-established mouse model of necrotizing myositis (38–41, 59). These three selected strains were of genetic lineages that cause abundant infections and were also temporally, geographically, and disease manifestation matched. The strains were recovered between March and June 2018 from cases of invasive osteitis infection that occurred in Brittany, France. Of note, although stG62647 strains MGCS36044 and MGCS36089 were virtually identical in gene content, they differed by 755 core chromosomal SNPs, which was greater than the average of 379 SNPs that separated the 23 strains of the emergent stG62647 cluster (Fig. 4), consistent with MGCS36044 and MGCS36089 having an evolutionarily less recent common ancestor. Compared to stC74a strain MGGS36055, the two stG62647 strains, MGCS36044 and MGCS36089, led to significantly higher bacterial burdens in infected limbs and caused significantly more mortality (Fig. 5A to C). The stG62647 strains also caused significantly larger lesions with more tissue destruction (Fig. 2D). Together, these data demonstrated that the two stG62647 strains studied were significantly more virulent than the comparator stC74a strain studied and supported the contention (9) that strains of the emergent ST20/stG62647 clonal lineage cause infections with a more aggressive clinical course.
FIG 5.

Virulence assessment of emm type stG62647 strains MGCS36044 and MGCS36089 and stC74a strain MGGS36055 in a mouse model of necrotizing myositis. (A) Kaplan-Meier survival curves are shown. Survival was monitored for 7 days (log-rank test, n = 20 mice per strain). (B and C) CFU recovery from infected limbs was performed on day 1 and day 2 after infection. Replicate data are expressed as means ± SEM (Kruskal-Wallis test, n = 20 mice per strain). (D) Gross and microscopic examination of infected limbs on day 2 postinfection. Lesion size and necrotic tissue are highlighted (black boxes).
The data presented here show that the six Brittany, France strains of type ST20/stG62647 we studied were clonally derived. These are the only isolates of ST20/stG62647 for which genomes have been sequenced to closure. Analogous to important findings presented by Oppegaard et al. (9) for strains from western Norway, members of this clone also have IS1548 inserted in the silB open reading frame. We also demonstrated that members of this clone have enhanced virulence in a commonly used mouse model of serious invasive disease, thereby adding new information about this emergent clone. However, our study was not designed to address the important topics of the molecular genetic events underlying emergence of the ST20/st62647 clone or the molecular mechanisms bearing on enhanced virulence of members of this clone. Our findings underscore the need for substantially expanded genomics and molecular pathogenesis investigations of S. dysgalactiae subsp. equisimilis strains causing human infections.
MATERIALS AND METHODS
Strains.
The seven strains analyzed were part of a comprehensive prospective study of S. dysgalactiae subsp. equisimilis human infections conducted from 2010 through 2018 in Brittany, France (33, 60). The study was conducted similarly to an investigation of S. pyogenes infections in Brittany, France (60) and will be described in detail elsewhere. Briefly, S. dysgalactiae subsp. equisimilis isolates were identified by standard diagnostic procedures used in the clinical microbiology laboratory, University Hospital of Rennes, France. Culture diagnosis was also conducted at the University Hospital, Rennes, France and analyzed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (Bruker Daltonics GmbH, Germany). Isolates were subcultured at 37°C with 5% CO2 on Columbia blood agar plates containing 5% sheep blood (Bio-Rad, France) and stored at −80°C.
Whole-genome sequencing.
The genomes of the S. dysgalactiae subsp. equisimilis strains were sequenced with methods described previously for S. pyogenes (17, 38, 54, 61, 62). Briefly, strains were grown at 37°C with 5% CO2 on tryptic soy agar with 5% sheep blood (Becton, Dickinson, Franklin Lakes, NJ) or in Todd-Hewitt broth with 2% (wt/vol) yeast extract (THY; Difco Laboratories, Franklin Lakes, NJ). Chromosomal DNA for Illumina short-read sequencing was isolated with the RNAdvance viral kit (Beckman Coulter, Brea, CA) and a BioMek i7 instrument (Beckman Coulter). Libraries were prepared with the NexteraXT kit (Illumina, San Diego, CA) and sequenced with a NovaSeq instrument (Illumina) using a 2 × 250-bp protocol. Chromosomal DNA for long-read sequencing was isolated with a DNeasy blood and tissue kit (Qiagen, Germantown, MD). Libraries were prepared with either a native barcoding kit or rapid barcoding kit (Oxford Nanopore Technologies, United Kingdom) and sequenced with a GridION instrument using version R10.4 or R9.4.1 flow cells (Oxford Nanopore Technologies), respectively.
Genome assembly, closure, and annotation.
Illumina paired-end short reads were base call error corrected with Karect (63), and Oxford Nanopore long reads were corrected with the FM index long read corrector (64). Hybrid assemblies of the preprocessed short- and long-read sequencing were generated with Unicycler (65). Unitigs from genomic assemblies that did not close were ordered against a closely related reference genome (or closed assembly) with progressiveMauve (66). Gaps between the ordered unitigs (mostly rRNA operons) were manually spanned or closed by splicing in corresponding sequences from one or more closely related closed genomes with Sequencher (Gene Codes, Ann Arbor, MI). These closed draft genome assemblies were checked for consistency with the Illumina PE short-read sequencing data by read mapping with SMALT (67) coupled with polymorphism detection with FreeBayes (68) and for structural inconsistencies with NucBreak (69). Consistency between the closed draft assemblies and the Oxford Nanopore long-read sequencing data was checked by read mapping with MiniMap2 (70). Correspondence of the mapped short and long reads with the draft closed genomes was visually inspected with Tablet (71). This reference-assisted and -directed genome closure process was iterated until discrepancies between the sequence data and the draft genomes were resolved. The resultant finished genomes were annotated de novo with RAST (72) and via transfer using PROKKA with S. dysgalactiae subsp. equisimilis strain ATCC 12394 as the reference (73). Annotations were merged and manually curated with Artemis (74). Multilocus sequence types of the isolates were determined by BLAST relative to the PubMLST database (https://pubmlst.org/), emm types were determined relative to the CDC emm type database (https://www2.cdc.gov/vaccines/biotech/strepblast.asp), and insertion sequence types were determined relative to the ISfinder database (https://isfinder.biotoul.fr/). Comparative genome atlases were generated with GView (75), and sequence alignment comparisons were generated with EasyFig (76).
Mouse model of necrotizing myositis and CFU determination.
The mouse model of necrotizing myositis used in these experiments has been previously described and used extensively (38–41, 59). Animals were infected intramuscularly in the right lower hindlimb with 1 × 108 CFU of the indicated strain and monitored daily for near mortality using standard criteria (77). Survival was graphed as a Kaplan-Meier curve, and statistical differences were determined with a log-rank test (GraphPad Prism V9.4.1; San Diego, CA). CFU from infected muscle were determined by culturing limb homogenates as described elsewhere (38–41, 59). Briefly, each infected limb was amputated, weighed, homogenized (Omni International, Kennesaw, GA) in 1 mL sterile phosphate-buffered saline, serially diluted, plated, and cultured overnight. CFU data were graphed as means ± standard errors of the means (SEM), and statistical differences were determined with a Kruskal-Wallis test. For histopathology examination, amputated limbs were photographed with a Solo8 Hovercam (Pathway Innovations, Las Vegas, NV) and processed with standard pathology laboratory methods.
(i) Ethics statement. Mouse experiments were approved by the Institutional Animal Care and Use Committee of Houston Methodist Research Institute (protocol IS00006169).
Identification of Lancefield group carbohydrate.
The Lancefield carbohydrate group antigen of the seven strains studied was determined with a latex agglutination method (BBL Streptocard enzyme latex test; Becton, Dickinson, Franklin Lakes, NJ) on overnight growth harvested from blood agar plates.
Determination of MICs for antimicrobial agents.
MICs for beta-lactam antibiotics, including amoxicillin, cefotaxime, cefoxitin, meropenem, and penicillin G, were determined by the MIC test strip method (Liofilchem, Waltham, MA) according to the manufacturer’s instructions. Strains were grown overnight on tryptic soy agar with 5% sheep blood (Becton, Dickinson), and fresh colonies were collected with the BBL prompt inoculation system (Becton, Dickinson) and plated onto Mueller-Hinton agar (Remel Microbiology Products, Lenexa, KS) with the antibiotic test strips. MICs (in milligrams per liter) were read after 24 h of incubation independently by two technologists.
Data availability.
The annotated closed genome assemblies and short- and long-read sequencing data for the seven S. dysgalactiae subsp. equisimilis strains studied have been submitted to the National Center for Biotechnology Information (NCBI) under Bioproject accession number PRJNA925803.
ACKNOWLEDGMENTS
We declare no conflict of interest.
These studies were funded in part by the Fondren Foundation. We thank Alma Amaya, Akanksha Batajoo, Jessica Cambric, Ryan Gadd, Nicole Kanellopoulos, Shelby Kvinta, Regan Mangham, Eleanor Nichols, Jordan Pachuca, Sindy Pena, Kristina Reppond, Matthew Ojeda Saavedra, Madison Shyer, and Rashi Thakur for expert technical assistance, and Sasha Pejerrey and Heather McConnell for editorial assistance.
Footnotes
Supplemental material is available online only.
Contributor Information
James M. Musser, Email: jmmusser@houstonmethodist.org.
Shannon D. Manning, Michigan State University
REFERENCES
- 1.Ahmad Y, Gertz RE, Jr, Li Z, Sakota V, Broyles LN, Van Beneden C, Facklam R, Shewmaker PL, Reingold A, Farley MM, Beall BW. 2009. Genetic relationships deduced from emm and multilocus sequence typing of invasive Streptococcus dysgalactiae subsp. equisimilis and S. canis recovered from isolates collected in the United States. J Clin Microbiol 47:2046–2054. doi: 10.1128/JCM.00246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandt CM, Spellerberg B. 2009. Human infections due to Streptococcus dysgalactiae subspecies equisimilis. Clin Infect Dis 49:766–772. doi: 10.1086/605085. [DOI] [PubMed] [Google Scholar]
- 3.Chochua S, Rivers J, Mathis S, Li Z, Velusamy S, McGee L, Van Beneden C, Li Y, Metcalf BJ, Beall B. 2019. Emergent invasive group A Streptococcus dysgalactiae subsp. equisimilis, United States, 2015–2018. Emerg Infect Dis 25:1543–1547. doi: 10.3201/eid2508.181758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashikawa S, Iinuma Y, Furushita M, Ohkura T, Nada T, Torii K, Hasegawa T, Ohta M. 2004. Characterization of group C and G streptococcal strains that cause streptococcal toxic shock syndrome. J Clin Microbiol 42:186–192. doi: 10.1128/JCM.42.1.186-192.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ikebe T, Murayama S, Saitoh K, Yamai S, Suzuki R, Isobe J, Tanaka D, Katsukawa C, Tamaru A, Katayama A, Fujinaga Y, Hoashi K, Watanabe H, Working Group for Streptococci in Japan . 2004. Surveillance of severe invasive group-G streptococcal infections and molecular typing of the isolates in Japan. Epidemiol Infect 132:145–149. doi: 10.1017/s0950268803001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambertsen LM, Ingels H, Schonheyder HC, Hoffmann S, Danish Streptococcal Surveillance Collaboration Group . 2014. Nationwide laboratory-based surveillance of invasive beta-haemolytic streptococci in Denmark from 2005 to 2011. Clin Microbiol Infect 20:O216–O223. doi: 10.1111/1469-0691.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leitner E, Zollner-Schwetz I, Zarfel G, Masoud-Landgraf L, Gehrer M, Wagner-Eibel U, Grisold AJ, Feierl G. 2015. Prevalence of emm types and antimicrobial susceptibility of Streptococcus dysgalactiae subsp. equisimilis in Austria. Int J Med Microbiol 305:918–924. doi: 10.1016/j.ijmm.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Oppegaard O, Mylvaganam H, Kittang BR. 2015. Beta-haemolytic group A, C and G streptococcal infections in western Norway: a 15-year retrospective survey. Clin Microbiol Infect 21:171–178. doi: 10.1016/j.cmi.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 9.Oppegaard O, Mylvaganam H, Skrede S, Lindemann PC, Kittang BR. 2017. Emergence of a Streptococcus dysgalactiae subspecies equisimilis stG62647-lineage associated with severe clinical manifestations. Sci Rep 7:7589. doi: 10.1038/s41598-017-08162-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinho MD, Melo-Cristino J, Ramirez M. 2006. Clonal relationships between invasive and noninvasive Lancefield group C and G streptococci and emm-specific differences in invasiveness. J Clin Microbiol 44:841–846. doi: 10.1128/JCM.44.3.841-846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rantala S. 2014. Streptococcus dysgalactiae subsp. equisimilis bacteremia: an emerging infection. Eur J Clin Microbiol Infect Dis 33:1303–1310. doi: 10.1007/s10096-014-2092-0. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi T, Ubukata K, Watanabe H. 2011. Invasive infection caused by Streptococcus dysgalactiae subsp. equisimilis: characteristics of strains and clinical features. J Infect Chemother 17:1–10. doi: 10.1007/s10156-010-0084-2. [DOI] [PubMed] [Google Scholar]
- 13.Trell K, Nilson B, Rasmussen M. 2016. Species and emm-type distribution of group C and G streptococci from different sites of isolation. Diagn Microbiol Infect Dis 86:467–469. doi: 10.1016/j.diagmicrobio.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Vahakuopus S, Vuento R, Siljander T, Syrjanen J, Vuopio J. 2012. Distribution of emm types in invasive and non-invasive group A and G streptococci. Eur J Clin Microbiol Infect Dis 31:1251–1256. doi: 10.1007/s10096-011-1436-2. [DOI] [PubMed] [Google Scholar]
- 15.Wajima T, Morozumi M, Hanada S, Sunaoshi K, Chiba N, Iwata S, Ubukata K. 2016. Molecular characterization of invasive Streptococcus dysgalactiae subsp. equisimilis, Japan. Emerg Infect Dis 22:247–254. doi: 10.3201/eid2202.141732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuursted K, Stegger M, Hoffmann S, Lambertsen L, Andersen PS, Deleuran M, Thomsen MK. 2016. Description and characterization of a penicillin-resistant Streptococcus dysgalactiae subsp. equisimilis clone isolated from blood in three epidemiologically linked patients. J Antimicrob Chemother 71:3376–3380. doi: 10.1093/jac/dkw320. [DOI] [PubMed] [Google Scholar]
- 17.Beres SB, Sesso R, Pinto SW, Hoe NP, Porcella SF, Deleo FR, Musser JM. 2008. Genome sequence of a Lancefield group C Streptococcus zooepidemicus strain causing epidemic nephritis: new information about an old disease. PLoS One 3:e3026. doi: 10.1371/journal.pone.0003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishihara H, Ogura K, Miyoshi-Akiyama T, Nakamura M, Kaya H, Okamoto S. 2020. Prevalence and genomic characterization of group A Streptococcus dysgalactiae subsp. equisimilis isolated from patients with invasive infections in Toyama Prefecture, Japan. Microbiol Immunol 64:113–122. doi: 10.1111/1348-0421.12760. [DOI] [PubMed] [Google Scholar]
- 19.Matsue M, Ogura K, Sugiyama H, Miyoshi-Akiyama T, Takemori-Sakai Y, Iwata Y, Wada T, Okamoto S. 2020. Pathogenicity characterization of prevalent-type Streptococcus dysgalactiae subsp. equisimilis strains. Front Microbiol 11:97. doi: 10.3389/fmicb.2020.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porcellato D, Smistad M, Skeie SB, Jorgensen HJ, Austbo L, Oppegaard O. 2021. Whole genome sequencing reveals possible host species adaptation of Streptococcus dysgalactiae. Sci Rep 11:17350. doi: 10.1038/s41598-021-96710-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimomura Y, Okumura K, Murayama SY, Yagi J, Ubukata K, Kirikae T, Miyoshi-Akiyama T. 2011. Complete genome sequencing and analysis of a Lancefield group G Streptococcus dysgalactiae subsp. equisimilis strain causing streptococcal toxic shock syndrome (STSS). BMC Genomics 12:17. doi: 10.1186/1471-2164-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki H, Lefebure T, Hubisz MJ, Pavinski Bitar P, Lang P, Siepel A, Stanhope MJ. 2011. Comparative genomic analysis of the Streptococcus dysgalactiae species group: gene content, molecular adaptation, and promoter evolution. Genome Biol Evol 3:168–185. doi: 10.1093/gbe/evr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe S, Kirikae T, Miyoshi-Akiyama T. 2013. Complete genome sequence of Streptococcus dysgalactiae subsp. equisimilis 167 carrying Lancefield group C antigen and comparative genomics of S. dysgalactiae subsp. equisimilis strains. Genome Biol Evol 5:1644–1651. doi: 10.1093/gbe/evt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishihara H, Ogura K, Nguyen VA, Miyoshi-Akiyama T, Okamoto S, Takemoto N. 2021. Comparative genome analysis of three group A Streptococcus dysgalactiae subsp. equisimilis strains isolated in Japan. J Med Microbiol 70. doi: 10.1099/jmm.0.001322. [DOI] [PubMed] [Google Scholar]
- 25.Lo HH, Cheng WS. 2015. Distribution of virulence factors and association with emm polymorphism or isolation site among beta-hemolytic group G Streptococcus dysgalactiae subspecies equisimilis. APMIS 123:45–52. doi: 10.1111/apm.12305. [DOI] [PubMed] [Google Scholar]
- 26.Humar D, Datta V, Bast DJ, Beall B, De Azavedo JC, Nizet V. 2002. Streptolysin S and necrotising infections produced by group G Streptococcus. Lancet 359:124–129. doi: 10.1016/S0140-6736(02)07371-3. [DOI] [PubMed] [Google Scholar]
- 27.Ikebe T, Otsuka H, Chiba K, Kazawa Y, Yamaguchi T, Okuno R, Date Y, Sasaki M, Isobe J, Ohnishi M, Akeda Y. 2022. Natural mutation in the regulatory gene (srrG) influences virulence-associated genes and enhances invasiveness in Streptococcus dysgalactiae subsp. equisimilis strains isolated from cases of streptococcal toxic shock syndrome. EBioMedicine 81:104133. doi: 10.1016/j.ebiom.2022.104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogura K, Okumura K, Shimizu Y, Kirikae T, Miyoshi-Akiyama T. 2018. Pathogenicity induced by invasive infection of Streptococcus dysgalactiae subsp. equisimilis in a mouse model of diabetes. Front Microbiol 9:2128. doi: 10.3389/fmicb.2018.02128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steiner K, Malke H. 2002. Dual control of streptokinase and streptolysin S production by the covRS and fasCAX two-component regulators in Streptococcus dysgalactiae subsp. equisimilis. Infect Immun 70:3627–3636. doi: 10.1128/IAI.70.7.3627-3636.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe S, Shimomura Y, Ubukata K, Kirikae T, Miyoshi-Akiyama T. 2013. Concomitant regulation of host tissue-destroying virulence factors and carbohydrate metabolism during invasive diseases induced by group g streptococci. J Infect Dis 208:1482–1493. doi: 10.1093/infdis/jit353. [DOI] [PubMed] [Google Scholar]
- 31.Yoshida H, Takahashi T, Nakamura M, Overby A, Takahashi T, Ubukata K, Matsui H. 2016. A highly susceptible CD46 transgenic mouse model of subcutaneous infection with Streptococcus dysgalactiae subspecies equisimilis. J Infect Chemother 22:229–234. doi: 10.1016/j.jiac.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Bisno AL, Gaviria JM. 1997. Murine model of recurrent group G streptococcal cellulitis: no evidence of protective immunity. Infect Immun 65:4926–4930. doi: 10.1128/iai.65.12.4926-4930.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boukthir S, Moullec S, Vincent P, Faili A, Kayal S. 2019. Osteoarticular infections caused by Streptococcus dysgalactiae subsp. equisimilis and Streptococcus pyogenes: a five year prospective study in French Brittany. ASM Microbe 2019, ASM Press, Washington, DC. [Google Scholar]
- 34.Rojo-Bezares B, Toca L, Azcona-Gutierrez JM, Ortega-Unanue N, Toledano P, Saenz Y. 2021. Streptococcus dysgalactiae subsp. equisimilis from invasive and non-invasive infections in Spain: combining epidemiology, molecular characterization, and genetic diversity. Eur J Clin Microbiol Infect Dis 40:1013–1021. doi: 10.1007/s10096-020-04119-9. [DOI] [PubMed] [Google Scholar]
- 35.Ruppen C, Rasmussen M, Casanova C, Sendi P. 2017. A 10-year observational study of Streptococcus dysgalactiae bacteraemia in adults: frequent occurrence among female intravenous drug users. Swiss Med Wkly 147:w14469. doi: 10.4414/smw.2017.14469. [DOI] [PubMed] [Google Scholar]
- 36.Traverso F, Blanco A, Villalon P, Beratz N, Saez NJ, Lopardo H, Related B, National Collaborative Group for the Study of Streptococci and Related Bacteria . 2016. Molecular characterization of invasive Streptococcus dysgalactiae subsp. equisimilis. Multicenter study: Argentina 2011–2012. Rev Argent Microbiol 48:279–289. doi: 10.1016/j.ram.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 37.Oppegaard O, Skrede S, Mylvaganam H, Kittang BR. 2016. Temporal trends of beta-haemolytic streptococcal osteoarticular infections in western Norway. BMC Infect Dis 16:535. doi: 10.1186/s12879-016-1874-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kachroo P, Eraso JM, Beres SB, Olsen RJ, Zhu L, Nasser W, Bernard PE, Cantu CC, Saavedra MO, Arredondo MJ, Strope B, Do H, Kumaraswami M, Vuopio J, Grondahl-Yli-Hannuksela K, Kristinsson KG, Gottfredsson M, Pesonen M, Pensar J, Davenport ER, Clark AG, Corander J, Caugant DA, Gaini S, Magnussen MD, Kubiak SL, Nguyen HAT, Long SW, Porter AR, DeLeo FR, Musser JM. 2019. Integrated analysis of population genomics, transcriptomics and virulence provides novel insights into Streptococcus pyogenes pathogenesis. Nat Genet 51:548–559. doi: 10.1038/s41588-018-0343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olsen RJ, Zhu L, Mangham RE, Faili A, Kayal S, Beres SB, Musser JM. 2022. A chimeric penicillin binding protein 2X significantly decreases in vitro beta-lactam susceptibility and increases in vivo fitness of Streptococcus pyogenes. Am J Pathol 192:1397–1406. doi: 10.1016/j.ajpath.2022.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu L, Olsen RJ, Beres SB, Eraso JM, Saavedra MO, Kubiak SL, Cantu CC, Jenkins L, Charbonneau ARL, Waller AS, Musser JM. 2019. Gene fitness landscape of group A Streptococcus during necrotizing myositis. J Clin Invest 129:887–901. doi: 10.1172/JCI124994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu L, Olsen RJ, Lee JD, Porter AR, DeLeo FR, Musser JM. 2017. Contribution of secreted NADase and streptolysin O to the pathogenesis of epidemic serotype M1 Streptococcus pyogenes infections. Am J Pathol 187:605–613. doi: 10.1016/j.ajpath.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu L, Olsen RJ, Nasser W, de la Riva Morales I, Musser JM. 2015. Trading capsule for increased cytotoxin production: contribution to virulence of a newly emerged clade of emm89 Streptococcus pyogenes. mBio 6:e01378-15. doi: 10.1128/mBio.01378-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oppegaard O, Mylvaganam H, Skrede S, Kittang BR. 2018. Exploring the arthritogenicity of Streptococcus dysgalactiae subspecies equisimilis. BMC Microbiol 18:17. doi: 10.1186/s12866-018-1160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bruun T, Kittang BR, de Hoog BJ, Aardal S, Flaatten HK, Langeland N, Mylvaganam H, Vindenes HA, Skrede S. 2013. Necrotizing soft tissue infections caused by Streptococcus pyogenes and Streptococcus dysgalactiae subsp. equisimilis of groups C and G in western Norway. Clin Microbiol Infect 19:E545–E550. doi: 10.1111/1469-0691.12276. [DOI] [PubMed] [Google Scholar]
- 45.Davies MR, McMillan DJ, Beiko RG, Barroso V, Geffers R, Sriprakash KS, Chhatwal GS. 2007. Virulence profiling of Streptococcus dysgalactiae subspecies equisimilis isolated from infected humans reveals 2 distinct genetic lineages that do not segregate with their phenotypes or propensity to cause diseases. Clin Infect Dis 44:1442–1454. doi: 10.1086/516780. [DOI] [PubMed] [Google Scholar]
- 46.McDonald M, Towers RJ, Andrews RM, Carapetis JR, Currie BJ. 2007. Epidemiology of Streptococcus dysgalactiae subsp. equisimilis in tropical communities, northern Australia. Emerg Infect Dis 13:1694–1700. doi: 10.3201/eid1311.061258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ambroset C, Coluzzi C, Guedon G, Devignes MD, Loux V, Lacroix T, Payot S, Leblond-Bourget N. 2015. New insights into the classification and integration specificity of Streptococcus integrative conjugative elements through extensive genome exploration. Front Microbiol 6:1483. doi: 10.3389/fmicb.2015.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rezaei Javan R, Ramos-Sevillano E, Akter A, Brown J, Brueggemann AB. 2019. Prophages and satellite prophages are widespread in Streptococcus and may play a role in pneumococcal pathogenesis. Nat Commun 10:4852. doi: 10.1038/s41467-019-12825-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coluzzi C, Guedon G, Devignes MD, Ambroset C, Loux V, Lacroix T, Payot S, Leblond-Bourget N. 2017. A glimpse into the world of integrative and mobilizable elements in streptococci reveals an unexpected diversity and novel families of mobilization proteins. Front Microbiol 8:443. doi: 10.3389/fmicb.2017.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson CM, Grossman AD. 2015. Integrative and conjugative elements (ICEs): what they do and how they work. Annu Rev Genet 49:577–601. doi: 10.1146/annurev-genet-112414-055018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eran Y, Getter Y, Baruch M, Belotserkovsky I, Padalon G, Mishalian I, Podbielski A, Kreikemeyer B, Hanski E. 2007. Transcriptional regulation of the sil locus by the SilCR signalling peptide and its implications on group A streptococcus virulence. Mol Microbiol 63:1209–1222. doi: 10.1111/j.1365-2958.2007.05581.x. [DOI] [PubMed] [Google Scholar]
- 52.Hidalgo-Grass C, Dan-Goor M, Maly A, Eran Y, Kwinn LA, Nizet V, Ravins M, Jaffe J, Peyser A, Moses AE, Hanski E. 2004. Effect of a bacterial pheromone peptide on host chemokine degradation in group A streptococcal necrotising soft-tissue infections. Lancet 363:696–703. doi: 10.1016/S0140-6736(04)15643-2. [DOI] [PubMed] [Google Scholar]
- 53.Hidalgo-Grass C, Ravins M, Dan-Goor M, Jaffe J, Moses AE, Hanski E. 2002. A locus of group A Streptococcus involved in invasive disease and DNA transfer. Mol Microbiol 46:87–99. doi: 10.1046/j.1365-2958.2002.03127.x. [DOI] [PubMed] [Google Scholar]
- 54.Beres SB, Zhu L, Pruitt L, Olsen RJ, Faili A, Kayal S, Musser JM. 2022. Integrative reverse genetic analysis identifies polymorphisms contributing to decreased antimicrobial agent susceptibility in Streptococcus pyogenes. mBio 13. doi: 10.1128/mbio.03618-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Musser JM, Beres SB, Zhu L, Olsen RJ, Vuopio J, Hyyrylainen HL, Grondahl-Yli-Hannuksela K, Kristinsson KG, Darenberg J, Henriques-Normark B, Hoffmann S, Caugant DA, Smith AJ, Lindsay DSJ, Boragine DM, Palzkill T. 2020. Reduced in vitro susceptibility of Streptococcus pyogenes to beta-lactam antibiotics associated with mutations in the pbp2x gene is geographically widespread. J Clin Microbiol 58. doi: 10.1128/JCM.01993-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olsen RJ, Zhu L, Musser JM. 2020. A single amino acid replacement in penicillin-binding protein 2X in Streptococcus pyogenes significantly increases fitness on subtherapeutic benzylpenicillin treatment in a mouse model of necrotizing myositis. Am J Pathol 190:1625–1631. doi: 10.1016/j.ajpath.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Southon SB, Beres SB, Kachroo P, Saavedra MO, Erlendsdottir H, Haraldsson G, Yerramilli P, Pruitt L, Zhu L, Musser JM, Kristinsson KG. 2020. Population genomic molecular epidemiological study of macrolide-resistant Streptococcus pyogenes in Iceland, 1995 to 2016: identification of a large clonal population with a pbp2x mutation conferring reduced in vitro beta-lactam susceptibility. J Clin Microbiol 58. doi: 10.1128/JCM.00638-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vannice KS, Ricaldi J, Nanduri S, Fang FC, Lynch JB, Bryson-Cahn C, Wright T, Duchin J, Kay M, Chochua S, Van Beneden CA, Beall B. 2020. Streptococcus pyogenes pbp2x mutation confers reduced susceptibility to beta-lactam antibiotics. Clin Infect Dis 71:201–204. doi: 10.1093/cid/ciz1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu L, Olsen RJ, Nasser W, Beres SB, Vuopio J, Kristinsson KG, Gottfredsson M, Porter AR, DeLeo FR, Musser JM. 2015. A molecular trigger for intercontinental epidemics of group A Streptococcus. J Clin Invest 125:3545–3559. doi: 10.1172/JCI82478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boukthir S, Moullec S, Cariou ME, Meygret A, Morcet J, Faili A, Kayal S. 2020. A prospective survey of Streptococcus pyogenes infections in French Brittany from 2009 to 2017: comprehensive dynamic of new emergent emm genotypes. PLoS One 15:e0244063. doi: 10.1371/journal.pone.0244063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beres SB, Kachroo P, Nasser W, Olsen RJ, Zhu L, Flores AR, de la Riva I, Paez-Mayorga J, Jimenez FE, Cantu C, Vuopio J, Jalava J, Kristinsson KG, Gottfredsson M, Corander J, Fittipaldi N, Di Luca MC, Petrelli D, Vitali LA, Raiford A, Jenkins L, Musser JM. 2016. Transcriptome remodeling contributes to epidemic disease caused by the human pathogen Streptococcus pyogenes. mBio 7. doi: 10.1128/mBio.00403-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Kristinsson KG, Gottfredsson M, Vuopio J, Raisanen K, Caugant DA, Steinbakk M, Low DE, McGeer A, Darenberg J, Henriques-Normark B, Van Beneden CA, Hoffmann S, Musser JM. 2014. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci USA 111:E1768–E1776. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allam A, Kalnis P, Solovyev V. 2015. Karect: accurate correction of substitution, insertion and deletion errors for next-generation sequencing data. Bioinformatics 31:3421–3428. doi: 10.1093/bioinformatics/btv415. [DOI] [PubMed] [Google Scholar]
- 64.Wang JR, Holt J, McMillan L, Jones CD. 2018. FMLRC: hybrid long read error correction using an FM-index. BMC Bioinformatics 19:50. doi: 10.1186/s12859-018-2051-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ponstingl H, Zemin N. 2010. SMALT: a new read mapper for DNA sequencing reads. Intelligent Systems for Molecular Biology 2010, International Society for Computational Biology, San Diego, CA. [Google Scholar]
- 68.Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. arXiv 1207.3907 [q-bio.GN]. https://arXiv.1207.3907.
- 69.Khelik K, Sandve GK, Nederbragt AJ, Rognes T. 2020. NucBreak: location of structural errors in a genome assembly by using paired-end Illumina reads. BMC Bioinformatics 21:66. doi: 10.1186/s12859-020-3414-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. 2010. Tablet–next-generation sequence assembly visualization. Bioinformatics 26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 74.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petkau A, Stuart-Edwards M, Stothard P, Van Domselaar G. 2010. Interactive microbial genome visualization with GView. Bioinformatics 26:3125–3126. doi: 10.1093/bioinformatics/btq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Download spectrum.04550-22-s0001.pdf, PDF file, 0.1 MB (153.7KB, pdf)
Tables S1 to S7. Download spectrum.04550-22-s0002.xlsx, XLSX file, 0.03 MB (27.8KB, xlsx)
Data Availability Statement
The annotated closed genome assemblies and short- and long-read sequencing data for the seven S. dysgalactiae subsp. equisimilis strains studied have been submitted to the National Center for Biotechnology Information (NCBI) under Bioproject accession number PRJNA925803.