

ABSTRACT
The increasing integration of genomics into routine clinical diagnostics requires reliable computational tools to identify determinants of antimicrobial resistance (AMR) from whole-genome sequencing data. Here, we developed PorinPredict, a bioinformatic tool that predicts defects of the Pseudomonas aeruginosa outer membrane porin OprD, which are strongly associated with reduced carbapenem susceptibility. PorinPredict relies on a database of intact OprD variants and reports inactivating mutations in the coding or promoter region. PorinPredict was validated against 987 carbapenemase-negative P. aeruginosa genomes, of which OprD loss was predicted for 454 out of 522 (87.0%) meropenem-nonsusceptible and 46 out of 465 (9.9%) meropenem-susceptible isolates. OprD loss was also found to be common among carbapenemase-producing isolates, resulting in even further increased MICs. Chromosomal mutations in quinolone resistance-determining regions and OprD loss commonly co-occurred, likely reflecting the restricted use of carbapenems for multidrug-resistant infections as recommended in antimicrobial stewardship programs. In combination with available AMR gene detection tools, PorinPredict provides a robust and standardized approach to link P. aeruginosa phenotypes to genotypes.
IMPORTANCE Pseudomonas aeruginosa is a major cause of multidrug-resistant nosocomial infections. The emergence and spread of clones exhibiting resistance to carbapenems, a class of critical last-line antibiotics, is therefore closely monitored. Carbapenem resistance is frequently mediated by chromosomal mutations that lead to a defective outer membrane porin OprD. Here, we determined the genetic diversity of OprD variants across the P. aeruginosa population and developed PorinPredict, a bioinformatic tool that enables the prediction of OprD loss from whole-genome sequencing data. We show a high correlation between predicted OprD loss and meropenem nonsusceptibility irrespective of the presence of carbapenemases, which are a second widespread determinant of carbapenem resistance. Isolates with resistance determinants to other antibiotics were disproportionally affected by OprD loss, possibly due to an increased exposure to carbapenems. Integration of PorinPredict into genomic surveillance platforms will facilitate a better understanding of the clinical impact of OprD modifications and transmission dynamics of resistant clones.
KEYWORDS: Pseudomonas aeruginosa, meropenem resistance, outer membrane porin, inactivating mutations, antimicrobial resistance, whole-genome sequencing, surveillance, carbapenem resistance, genotype-phenotype predictions, outer membrane porin OprD
INTRODUCTION
Carbapenems are last-line antibiotics reserved for the treatment of severe bacterial infections. The emergence of carbapenem-resistant (CR) pathogens has thus created a major challenge for public health. Of particular concern is the spread of carbapenem-resistant Acinetobacter baumannii (CRAB), Enterobacteriaceae (CRE; including Klebsiella pneumoniae, Escherichia coli, and Enterobacter spp.), and Pseudomonas aeruginosa (CRPsA), which are major causes of nosocomial and multidrug-resistant infections and associated with high mortality rates (1).
In clinical CRE and CRAB isolates, carbapenem resistance is typically conferred by plasmid-acquired carbapenem-hydrolyzing enzymes (carbapenemases) (2, 3). In contrast, among clinical CRPsA isolates, carbapenemase positivity rates vary considerably between survey sites: rates are below 5% in most European countries and in North America, while rates of 30% to 70% have been reported from studies in Asia (4–11). Among these carbapenemases, VIM and IMP show the highest prevalence; others such as FIM, GES, GIM, KPC, OXA-48, and SPM are often associated with specific geographic regions or high-risk clones, i.e., clones associated with multidrug resistance and global spread (12).
In carbapenemase-negative CRPsA, carbapenem resistance is primarily associated with the inactivation of the OprD outer membrane porin (11, 13). OprD, a monomeric 18-stranded β-barrel, is responsible for the passive uptake of basic amino acids and small peptides and serves as a diffusion channel for carbapenems such as meropenem and imipenem, which structurally resemble dipeptides (14, 15). OprD defects can be mediated by indels, recombination events such as the insertion of insertion sequence (IS) elements, nonsense mutations, start- or stop-loss mutations, promoter disruptions, or amino acid substitutions affecting the porin’s pore width, charge, or structural integrity (7, 11, 16). Other genetic determinants contributing to decreased carbapenem susceptibility include the overexpression of multidrug efflux pumps and the overexpression of the intrinsic beta-lactamase AmpC (17, 18).
Pathogen surveillance has greatly benefited from the recent integration of genomics: the timely and accurate identification of pandemic clones and transmission events is critical to guide public health responses (19). Whole-genome sequencing has also become an integral part of antimicrobial resistance (AMR) surveillance, complementing phenotypic antimicrobial resistance testing by providing valuable information on resistance mechanisms and evolutionary events that led to their acquisition (20). Whereas reliable databases and computational tools are available for the detection of carbapenemase genes from whole-genome sequencing data (summarized by Hendriksen et al. [21]), there is a lack of robust and standardized approaches for the detection of porin loss-mediated carbapenem resistance. The identification of the various possible mutations leading to OprD deficiency currently relies on technical expertise and laborious curation and is thus often omitted from routine genotype-phenotype analyses. To address this, we investigated the genetic diversity of OprD in 2,088 P. aeruginosa genomes and compiled a database of intact variants. This was integrated into PorinPredict, a tool that predicts OprD porin loss from whole-genome sequencing data. PorinPredict was validated using 1,124 additional genomes (including 987 carbapenemase-negative genomes) and facilitated considerably improved genotype-phenotype predictions for P. aeruginosa meropenem resistance.
RESULTS AND DISCUSSION
Diversity of OprD in P. aeruginosa.
A total of 2,088 high-quality genome assemblies of P. aeruginosa isolates (NCBI data set) covering the phylogenetic clades A (n = 1,500), B (n = 541), C1 (n = 25), and C2 (n = 14) and a C1-C2 intermediate clade (n = 8) (Fig. 1) were screened for oprD. The oprD gene or fragments thereof were detected in 2,087 (99.95%) assemblies. Despite a phylogenetically diverse data set, few OprD protein variants were identified: of 1,483 isolates with a presumptively intact oprD open reading frame, 1432 (96.6%) encoded 1 of 15 distinct OprD protein variants (exact match), and 51 (3.4%) encoded one of these 15 dominant variants with 1 to 4 variable amino acid substitutions (no length variation). Eight variants were found in isolates belonging to clade A (OprD_1, OprD_2, OprD_3, OprD_4, OprD_5, OprD_6, OprD_7, and OprD_9) and five variants in clade B (OprD_1, OprD_2, OprD_3, OprD_5, and OprD_8). Six (OprD_10 to OprD_15) of the 15 intact variants were only detected in clade C (Fig. 1; see Table S1 in the supplemental material).
FIG 1.
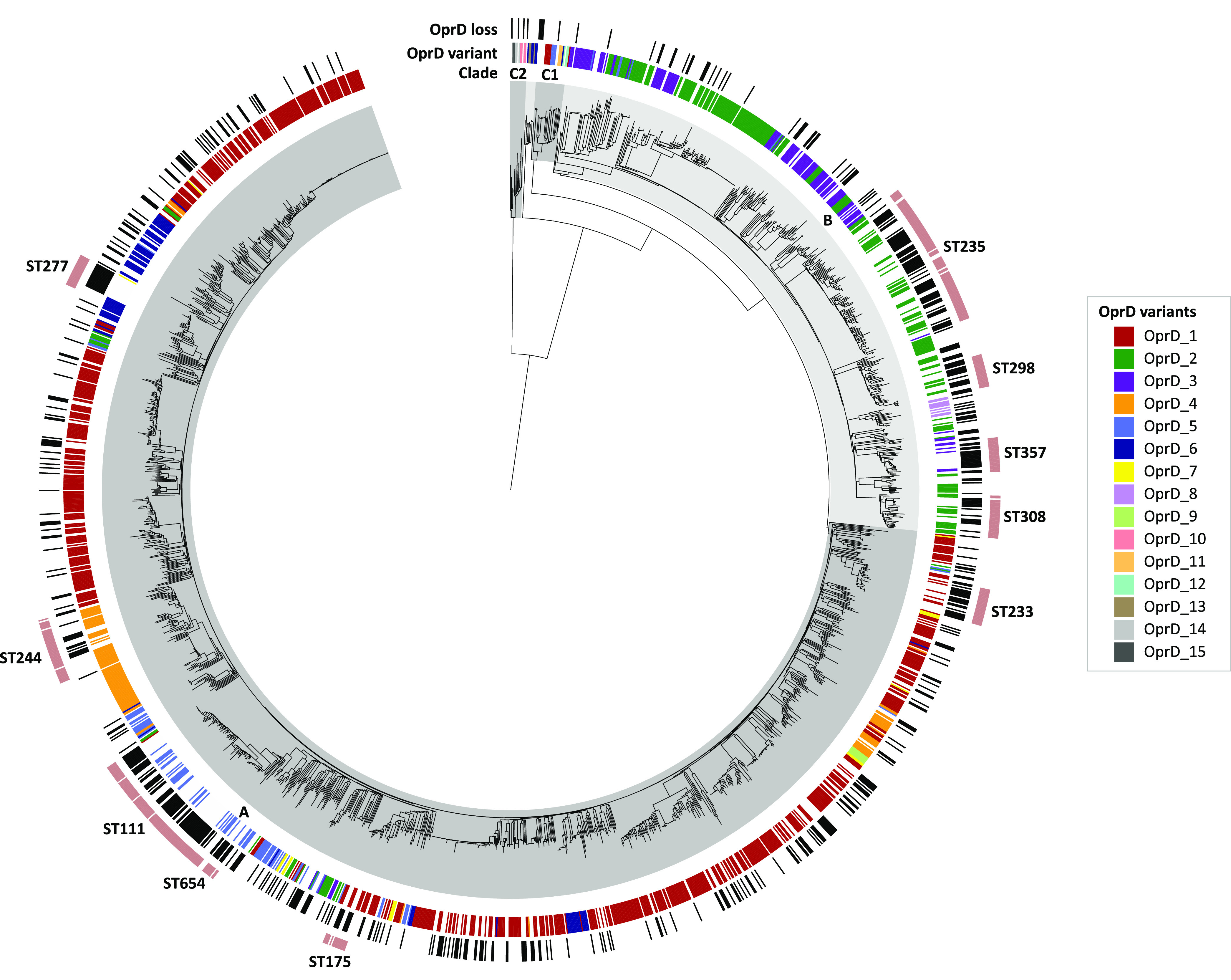
Distribution of OprD variants and predicted OprD loss across the P. aeruginosa phylogeny. Assemblies of 2,088 isolates (NCBI data set) were included in the analysis. Presence of OprD variants (exact amino acid sequence match) is labeled according to the legend. Putative OprD loss is shown in black bars. Intact nonexact matches (n = 51) and putative OprD losses due to promoter disruptions (n = 12) are not indicated. Isolate affiliation to clades (shaded in gray) and high-risk clones (outermost ring) is annotated. Phylogenetic distances were estimated using MASH and visualized in iTOL (46).
The 15 OprD variants consisted of 441, 443, or 446 amino acids and contained one of four C-terminal loop L8 (according to references 15 and 22; formerly L7) sequences, which determined the variants’ overall length, including the previously described L810 (formerly L710; 372-VDSSSSYAGL-381) and L812 (formerly L712; 372-MSDNNVGYKNYG-383) (23), as well as two novel L8 regions detected in clade C2 isolates, L815 (372-VSANSAYVKEDGSPL-386) and L810b (372-VDSSSAYAGL-381) (Fig. 2). The latter differed from L810 by only one amino acid substitution (S377A). Isolates with the L810 type OprD were previously associated with hypersusceptibility to meropenem, but not to imipenem (23, 24). OprD variants of the same L8 group were polyphyletically distributed across the P. aeruginosa population: both L810- and L812-containing variants were detected in isolates of clades A, B, C1, and the C1-C2 intermediate (Fig. 1).
FIG 2.
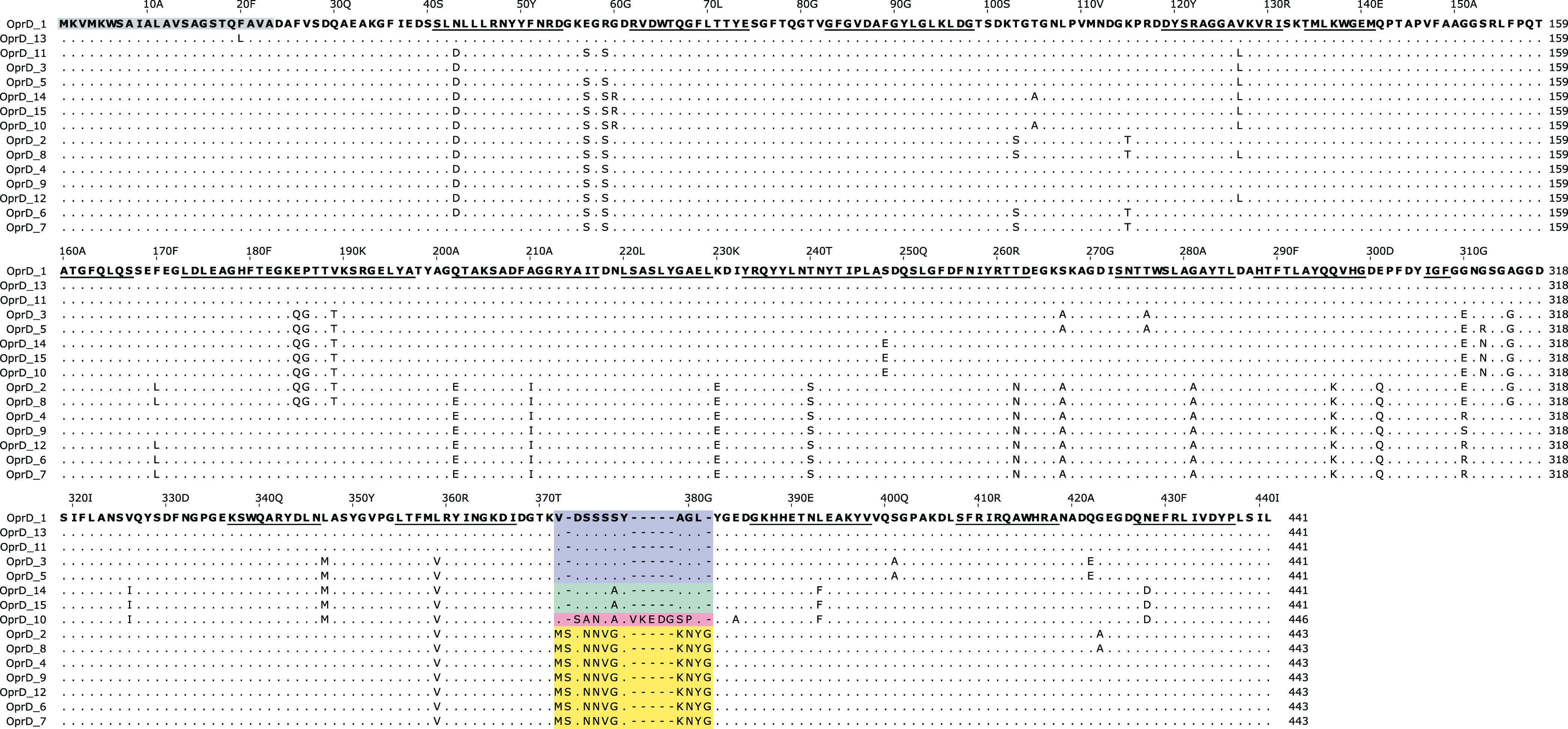
Alignment of P. aeruginosa OprD variants. The variable region within loop 8 is colored (L810, blue; L810b, green; L812, yellow; L815, red). Scale numbers above the alignment refer to amino acid positions in variant OprD_1 and include the signal peptide (23 amino acids, gray box). β-Strands (according to PDB structure 3SY7) are underlined.
In the 605 genomes of the NCBI data set with a presumptively inactivated OprD coding sequence, the open reading frame contained premature stop codons (n = 150), frameshift mutations (n = 297), other indels or truncations (n = 155), start-loss mutations (n = 2), or was not detected (n = 1). Disrupted promoter regions (but intact OprD coding sequences) were identified in nine assemblies, which all contained contig breaks within 200 bp upstream of oprD. In all nine cases, sequences matching with various IS elements (IS1394, ISPa82, ISPpu1, ISPpu21, ISPsme1, ISPsp2, ISPst7, or IS5 family) were identified between the contig break and oprD (Table S3). IS elements typically occur in multiple copies in a genome, preventing a resolved assembly of their surrounding region from short-read sequencing data.
The high proportion (28.9%) of genomes with an inactivated OprD coding sequence in this data set likely reflects an overrepresentation of sequenced clinical isolates which had been exposed to antibiotics. Of the isolates, 391 (18.7%) belonged to the high-risk clones sequence type 111 (ST111), ST175, ST233, ST235, ST244, ST277, ST298, ST308, ST357, or ST654, which are known for their increased virulence and multidrug resistance, often associated with various carbapenemases and extended-spectrum beta-lactamases (12). Overall, 239 (61.1%) of all high-risk clone isolates were predicted to be OprD deficient, compared to 375 of 1,697 (22.1%) isolates not belonging to high-risk clones (Fig. 1).
Evaluation of PorinPredict: concordance of predicted OprD loss and carbapenem resistance.
PorinPredict detects putative OprD defects by querying genomes against a database consisting of the 15 identified intact OprD variants and against an oprD promoter database. Truncated or absent oprD, indels, premature stop codons, stop-loss and start-loss mutations, and disrupted promoter sequences are reported as putative OprD porin loss. Missense mutations are reported but not per se classified as porin loss.
To evaluate PorinPredict regarding genotype-phenotype associations, 1,124 genomes of P. aeruginosa isolates with known phenotypic meropenem susceptibility (validation data set) were screened for a putative OprD loss and the presence of carbapenemase-encoding genes. OprD loss was common and strongly associated with meropenem resistance: OprD loss was predicted for 556 of 653 (85.1%) nonsusceptible isolates (including 484 of 551 [87.8%] resistant and 72 of 102 [70.5%] intermediately resistant isolates), as well as 47 of 471 (10.0%) susceptible isolates (Fig. 3; Table S2). Carbapenemase-encoding genes were identified in 132 (20.2%) of the nonsusceptible isolates, often (n = 102, 83.1%) in combination with an inactivated OprD. Midlevel resistance conferred by carbapenemases may predispose for OprD loss, synergistically decreasing carbapenem susceptibility: isolates with concomitant OprD loss and carbapenemase presence showed a median meropenem MIC of >32 mg/L (interquartile range [IQR], >8 to 128) compared to 12 mg/L (IQR, 4 to >32) among isolates with a carbapenemase alone (Fig. 3b). A similar synergy of carbapenemases and outer membrane porin modifications has been described in carbapenem-resistant K. pneumoniae (25).
FIG 3.

Correlation between predicted OprD loss, presence of carbapenemases, and meropenem resistance phenotype. (a) Intersection of P. aeruginosa isolates with (black circles) or without (white circles) carbapenemase and/or a disrupted OprD. Assemblies of 1,124 isolates (validation data set) were included in the analysis. (b) Approximations of meropenem MICs among a subset of 1,078 isolates with reported values. Due to differing test ranges, MIC values reported as 512, 128, 64, >32, ≥32, 24, or >16 mg/L are here summarized as >16 mg/L; >8 or ≥16 mg/L are shown as 16 mg/L, and ≤0.25 mg/L and <0.25 mg/L are shown as 0.25 mg/L and 0.1 and 0.02 mg/L, respectively, thus representing an approximation.
In the subset of carbapenemase-negative isolates, OprD loss was predicted for 454 of 522 (87.0%) nonsusceptible and 46 of 465 (9.9%) susceptible isolates (Table 1). Among the 79 isolates obtained at the University Hospital Basel, only 1 (1.3%) isolate contained a carbapenemase; in the remaining (carbapenemase-negative) isolates, OprD loss was predicted for 22 of 23 (95.7%) meropenem-nonsusceptible and 2 of 53 (3.8%) susceptible (MIC = 0.75 mg/L each) isolates.
TABLE 1.
Genotype-phenotype associations for meropenem resistance and causes of putative OprD loss among 987 carbapenemase-negative P. aeruginosa isolates
| Genotype-phenotype association | No. of susceptible isolates (n = 465) | No. of intermediately resistant isolates (n = 97) | No. of resistant isolates (n = 425) |
|---|---|---|---|
| Intact OprD (exact match)a | 408 | 22 | 28 |
| Intact OprD (nonexact match)a | 11 | 3 | 15 |
| SNAP2 prediction: effect | 0 | 3 | 12 |
| SNAP2 prediction: neutral | 11 | 0 | 3 |
| OprD loss | 46 | 72 | 382 |
| Frameshift mutation | 13 | 33 | 177 |
| Premature stop | 17 | 22 | 111 |
| Gene truncation | 14 | 11 | 87 |
| Other indels | 0 | 2 | 3 |
| Incomplete promoterb | 2 | 3 | 2 |
| Start-loss | 0 | 0 | 2 |
| Stop-loss | 0 | 1 | 0 |
Intact variants with disrupted promoter regions are counted as OprD loss.
Counted only if oprD coding region was intact.
For a total of 43 (7.8%) meropenem-resistant and 25 (24.5%)-intermediately resistant isolates, the phenotype could not be explained by carbapenemase-encoding genes or OprD loss. However, OprD variants with missense mutations were significantly more common among these isolates (15/43, 34.9% and 3/25, 12.0%) than among meropenem-susceptible isolates with intact OprD (11/419, 2.6%, P < 0.001, respectively), possibly affecting the structural or functional integrity of OprD (Table 1). Missense mutations in nonsusceptible isolates affected residue R154 (corresponding to R131 when excluding the 23-amino-acid signal sequence), a critical site known to determine pore width (15); H288 and S403, which are both conserved across different members of the OprD family (15); multiple sites in β-strands (S278, Q340, L359) replaced by proline, which disrupts secondary structures; and G316, which forms a hydrogen bond to R391 (R393 in L812), part of the characteristic basic ladder in the OprD pore constriction (15) (Table 2). The R154, G316, S278P, and S403P substitutions were previously associated with carbapenem resistance (16). Using the neural network-based classifier SNAP2, most (15/18) missense mutations in nonsusceptible isolates were predicted to have functional effects (Table 2). Missense mutations in susceptible isolates were typically located in the N-terminal region, more conservative, and predicted to be neutral. The position of the amino acid substitutions in the OprD protein structure is shown in Fig. S1.
TABLE 2.
Missense mutations in OprD in 29 isolates of the validation data set and associated meropenem susceptibility
| Isolate | Meropenem susceptibility | OprD reference variant | Missense mutation(s) | Predicted functional effect (SNAP2) | Expected prediction accuracy (SNAP2 [%]) |
|---|---|---|---|---|---|
| 287.12499_PA | Intermediate | OprD_3 | R154H | Effect | 95 |
| 287.12621_PA | Intermediate | OprD_1 | L359P | Effect | 75 |
| 287.1101_PA | Intermediate | OprD_2 | L434R (in L812) | Effect | 80 |
| 287.5955_PA | Resistant | OprD_11 | L127V | Neutral | 93 |
| 287.12587_PA | Resistant | OprD_4 | R154C | Effect | 95 |
| 287.8031_PA | Resistant | OprD_5 | S278P | Effect | 91 |
| 287.7799_PA | Resistant | OprD_1 | S278P | Effect | 85 |
| 287.1095_PA | Resistant | OprD_5 | S278P | Effect | 91 |
| 287.12616_PA | Resistant | OprD_1 | H288R | Effect | 85 |
| 287.12490_PA | Resistant | OprD_1 | L292Q | Effect | 80 |
| 287.5956_PA | Resistant | OprD_1 | G307D | Neutral | 57 |
| 287.8490_PA | Resistant | OprD_6 | G316D | Neutral | 61 |
| 287.12754_PA | Resistant | OprD_1 | G316D | Effect | 71 |
| 287.12755_PA | Resistant | OprD_1 | G316D | Effect | 71 |
| 287.12669_PA | Resistant | OprD_1 | Q340P | Effect | 80 |
| 287.5971_PA | Resistant | OprD_1 | G383D (in L810) | Effect | 80 |
| 287.12734_PA | Resistant | OprD_2 | S403P (in L812) | Effect | 63 |
| 287.12639_PA | Resistant | OprD_6 | L409P (in L812) | Effect | 76 |
| 287.860_PA | Susceptible | OprD_2 | A8S | Neutral | 97 |
| 502940-8-20 | Susceptible | OprD_1 | K34N | Neutral | 82 |
| 502940-9-20 | Susceptible | OprD_1 | K34N | Neutral | 82 |
| 502940-3-20 | Susceptible | OprD_1 | K34N, A247S | Neutral (each) | 82, 97 |
| 287.7801_PA | Susceptible | OprD_4 | D43N | Neutral | 95 |
| 501539-19 | Susceptible | OprD_5 | V63A | Neutral | 93 |
| 500658-19 | Susceptible | OprD_6 | T66S | Neutral | 97 |
| 721748-19 | Susceptible | OprD_6 | T66S | Neutral | 97 |
| 287.1066_PA | Susceptible | OprD_1 | V86I | Neutral | 93 |
| 287.1043_PA | Susceptible | OprD_2 | G98A, V129I | Neutral (each) | 82, 97 |
| 287.1190_PA | Susceptible | OprD_2 | V129I | Neutral | 97 |
OprD loss and carbapenemase-encoding genes were identified in 47 (10.0%) and 5 (1.1%) out of 470 meropenem-susceptible isolates, respectively, and resulted in increased MIC values (median, 2 mg/L) compared to those isolates without any detected carbapenem resistance determinants (median, 0.5 mg/L) (Fig. 3b).
Possible reasons for inconsistent genotype-phenotype associations include mixed bacterial cultures, fragmented or absent (carbapenemase) gene assemblies from short-read data, alternative carbapenem-resistance mechanisms such as mutations affecting the functionality and overexpression of intrinsic beta-lactamases or efflux-pumps, technical errors during the susceptibility testing or sequencing, interlaboratory differences in MIC readings, or transcription errors.
Overall, PorinPredict improved the accuracy of genotype-phenotype predictions for meropenem nonsusceptibility from 53.1% (when predicted based on the presence of carbapenemase-encoding genes alone; sensitivity and specificity of 20.2% and 98.9%, respectively) to 89.6% (when predicted based on presence of carbapenemase-encoding genes and/or porin loss; sensitivity and specificity of 89.1% and 89.4%, respectively). Among the 79 University Hospital Basel (USB) isolates, for which MIC measurements were performed as part of this study, the accuracy was 96.2% (sensitivity and specificity of 95.8% and 96.4%, respectively). Very major discrepancies (resistant isolates predicted to be susceptible) were found for 1 of 21 (4.7%) and 42 of 430 (9.8%) isolates of the USB and PATRIC data sets, respectively. Considering amino acid substitutions at critical sites as inactivating mutations would further improve accuracies and discrepancy rates for genotype-phenotype predictions.
Role of regulators affecting MexAB and AmpC expression.
Carbapenem susceptibility can be affected by additional genetic factors, including the expression levels of the intrinsic genes blaPDC (AmpC beta-lactamase) and mexA and mexB (MexAB multidrug efflux pump) (17, 18). Here, loss (truncations, premature stop codons, frameshifts, or stop loss) of the MexAB repressors MexR, NalC, and NalD was predominantly found among isolates that simultaneously harbored primary carbapenem resistance determinants (OprD loss, OprD missense mutations, or presence of carbapenemase genes) (Table 3). MexR-, NalC-, and NalD-deficient isolates exhibited 1.5- to 4-fold-increased meropenem MICs compared to those with intact repressor genes. In the absence of carbapenemase genes or OprD loss, these isolates were predominantly classified as susceptible. MexR loss was, however, detected in 11 out of the 43 isolates with very major genotype-phenotype discrepancies, sometimes (n = 4) coinciding with OprD missense mutations (Table S2). Loss of the AmpC repressor AmpR (in combination with an intact blaPDC) was detected in 20 isolates, which often (n = 11; 55.0%) coharbored primary carbapenem resistance determinants. The nine isolates with AmpR loss alone showed no increased MICs (median, 0.5 mg/L; IQR, 0.5 to 1 mg/L). Missense mutations in repressor genes or changes in their binding sites may also affect expression levels but were not further investigated here. Overall, our data suggest that the derepression of efflux pumps and intrinsic beta-lactamase genes plays a minor role in meropenem resistance of clinical P. aeruginosa isolates. Similar results were recently reported for other carbapenems (11).
TABLE 3.
Effect of defective MexAB repressors on meropenem susceptibility among 1,055 isolates with presumptively intact MexA and MexB
| Genotype | Predicted OprD loss, OprD missense mutations, or carbapenemase gene presence | Median MIC (IQRa [mg/L]) | No. (%) of nonsusceptible isolates |
|---|---|---|---|
| MexR, NalC, and NalD intact (n = 878) | Yes (n = 501) | 8 (8–16) | 461 (92.0) |
| No (n = 377) | 0.5 (0.25–1) | 38 (10.1) | |
| MexR loss (n = 65) | Yes (n = 47) | 16 (8–32) | 40 (85.1) |
| No (n = 18) | 2 (1–8) | 7 (38.9) | |
| NalC loss (n = 38) | Yes (n = 24) | 12 (8–28) | 22 (91.2) |
| No (n = 14) | 1 (0.25–2.5) | 3 (21.4) | |
| NalD loss (n = 78) | Yes (n = 64) | 16 (8–32) | 62 (96.7) |
| No (n = 14) | 2 (1.75–2) | 2 (14.3) |
IQR, interquartile range.
Convergent OprD-inactivating mutations and spread of oprD-deficient clones.
We observed identical OprD-inactivating mutations in distantly related isolates, suggesting independent acquisitions at vulnerable sites. In the NCBI data set, these included a premature stop at position W277* identified in different OprD variants (OprD_1, OprD_2, OprD_8, and OprD_15) of 28 isolates belonging to 21 distinct (and 3 unknown) sequence types (Table S1). Likewise, the frameshift mutation c.1199_1200insC (in L810 variants [p.Q400fs]) or the corresponding c.1205_1206insC (in L812 variants [p.Q402fs]) was found in multiple OprD variants (OprD_1, OprD_2, OprD_4, and OprD_8) of 38 isolates belonging to at least 22 distinct (and 2 unknown) sequence types.
Identical disruptive mutations were occasionally identified among closely related isolates, pointing to the spread of OprD-deficient clones. A phylogenetic cluster of 18 isolates within ST277 carried the frameshift mutation c.380_381delTG (p.E127fs) within OprD_6 (Table S1). These isolates were obtained between 1997 and 2019 at various geographic locations in Brazil (n = 16) and the United States (n = 2), corresponding with reports of a Brazilian endemic ST277 clone carrying this stable OprD-inactivating mutation (26). A subclone of the high-risk clone ST175 carrying a characteristic premature stop codon (Q142*) in OprD_1 and detected in Spanish and French hospitals over at least a decade (27, 28) was represented in the validation data set (17 isolates) (Table S2).
Compensatory mutations and coacquisition of AMR determinants.
The presence of relatively conserved OprD throughout the P. aeruginosa population suggests an important metabolic role of this porin, with OprD loss possibly leading to fitness defects. To investigate potential compensatory mutations acquired by OprD-deficient isolates, we performed a kmer-based genome-wide association study (using DBGWAS) for a subset of 700 isolates with (n = 206) versus without (n = 494) predicted porin loss (NCBI data set; Table S1). While mutations in oprD itself were not captured by DBGWAS, three genetic determinants reached genome-wide significance (q [Benjamini-Hochberg adjusted P] < 0.05) for associations with OprD-loss, (i) the chromosomal mutation gyrA T83I (qmin = 6.6E-18) and (ii) parC S87L (qmin = 1.6E-7) (both contributing to ciprofloxacin resistance), and (iii) a component (qmin = 1.4E-5) representing intI1, sul1 (sulfonamide resistance), qacEΔ1 (antiseptic resistance), and an acetyltransferase-encoding gene, which are typically colocated on a class I integron (Table S4). Rather than representing compensatory mutations or gene acquisitions, these results likely reflect an accumulation of antimicrobial resistance mechanisms during patient treatment due to the use of carbapenems as a last-resort antibiotic for multidrug-resistant clones, as commonly recommended in antibiotic stewardship programs. In the entire NCBI data set of 2,088 isolates, 65.3% of the OprD-deficient isolates (n = 614) carried a mutation linked to quinolone resistance (gyrA T83I, gyrA D87G, gyrA D87H, gyrA D87N, parC S87L, or parE A473V) compared to 18.3% (P < 0.001; odds ratio [OR], 8.4; 95% confidence interval [CI, 6.8 to 10.4]) among the isolates with an intact OprD (n = 1,474). The sul1 gene was found in 47.7% of the OprD-deficient isolates, compared to 10.3% (P < 0.001; OR, 7.9; 95% [CI, 6.3 to 10.0]) of the isolates with intact OprD.
Conclusion.
In conclusion, we developed, evaluated, and validated PorinPredict, a much-needed tool that predicts OprD-inactivating mutations in P. aeruginosa. Among carbapenemase-negative isolates, predicted OprD loss was overall highly consistent with phenotypic meropenem nonsusceptibility. Independent prospective validation is, however, necessary to provide evidence suitable for in vitro diagnostic regulations (IVDR). Although relatively rare, the effects of OprD missense mutations often remain difficult to interpret. The increasing availability of whole-genome sequencing and associated susceptibility data will enable an extension of the database covering additional variants. Given the increased application of whole-genome sequencing in clinical laboratories, PorinPredict will be valuable for detecting and tracking resistance within patients and communities.
MATERIALS AND METHODS
OprD database generation.
For the generation of a database of intact OprD sequences, 2,186 randomly selected P. aeruginosa genome assemblies were retrieved from the NCBI Reference Sequence Database (see “NCBI data set” in Table S1 in the supplemental material), corresponding to approximately 30% of the available RefSeq assemblies (searched in April 2021) and including both clinical and environmental isolates. Species affiliation was confirmed using ribosomal multilocus sequence typing (rMLST) (version 2022-06-23) (29) and sequence types assigned using mlst 2.19.0 (https://github.com/tseemann/mlst). Assembly quality was assessed using QUAST 5.0.2 (30) and CheckM 1.1.3 (31). After removal of low-quality assemblies (N50 < 15 kb, assembly length < 5.9 Mb, CheckM completeness < 97%, CheckM contamination < 3%), 2,088 assemblies were available to investigate the diversity of OprD. OprD amino acid sequences were extracted using DIAMOND 2.0.7 (32) (reference, GenPept accession no. AKQ14499, BLASTx mode, 70% sequence identity and coverage) and bedtools 2.30.0 (33), and variants were deduplicated using CD-HIT 4.8.1 (34). Deduplicated variants (n = 364) with inactivating mutations (n = 309; C- or N-terminal deletions; frameshift, nonsense, start-loss, or stop-loss mutations) and rare variants (n = 40; identified in <5 isolates of the dominant clades A and B or in <2 isolates of clade C) were excluded. Protein alignments of the remaining 15 intact variants were generated using MAFFT version 7 (35) and visualized using Jalview version 2.11.2.0 (36).
Development of PorinPredict.
PorinPredict was designed to detect mutations leading to a putative OprD loss or OprD amino acid substitutions from preassembled genomes. PorinPredict uses OprD nucleotide and protein sequence databases in combination with BLASTn 2.5.0+ (37) and DIAMOND 2.0.7 (32), respectively, to assess OprD integrity. BLASTn 2.5.0+ (37) is used with a 95% sequence identity threshold and the options “-max_target_seqs 10 -evalue 1E-20 -culling_limit 1.” The BLASTn hit with the lowest E value is evaluated. DIAMOND is run in BLASTx mode with sequence identity and coverage thresholds of 95% and 60%, respectively, and the options “--query-gencode 11 -b 6 --max-target-seqs 1 --sensitive --masking 0 -c1.” Amino acid substitutions are identified using the R package Biostrings (v3.14) (38). PorinPredict additionally assesses the integrity of the oprD promoter region by screening input assemblies against a database consisting of oprD promoter regions from five isolates representing the distinct P. aeruginosa clades (A, B, C1, C2, and C1-2 intermediate; defined according to Ozer et al. [39]). These sequences consist of the 200-bp region upstream of oprD comprising the ArgR binding site, −35 and −10 region, and Shine-Dalgarno sequence (40) and 10 bp of the N-terminal oprD sequence. For the promoter screening, BLASTn 2.5.0+ (37) is used as described above. BLASTn and DIAMOND results are filtered and summarized in an interpretable output table. PorinPredict reports (i) exact matches (same length, no amino acid substitutions) to intact OprD variants in the database; (ii) nonexact matches, i.e., intact same-length variants with amino acid substitutions (missense mutations); (iii) mutations leading to a presumptive OprD loss, including premature stop codons (nonsense mutations), truncations, frameshift mutations and other indels, loss of stop codons (stop-loss), loss of start codons (start-loss), and absence of oprD in the assembly (no hit); and (iv) putatively disrupted oprD promoter regions (BLASTn matches with <98% coverage).
Evaluation of PorinPredict.
PorinPredict 1.0.0 was evaluated using a second, independent data set consisting of 1,124 P. aeruginosa genome assemblies that had associated meropenem susceptibility data available and passed the above-defined CheckM and QUAST quality criteria. This collection (“validation data set” in Table S2) included 79 assemblies from isolates of the University Hospital Basel (USB) sequenced as part of this study and 1,045 assemblies accessed from PATRIC (41). For short-read sequencing of in-house isolates, genomic DNA was extracted using the DNeasy blood and tissue kit (Qiagen) and sequenced on the Illumina NextSeq 500 or Illumina MiSeq platforms. Draft genomes were assembled using Unicycler version 0.3.0b (42) after read trimming with Trimmomatic version 0.38 (43).
Meropenem susceptibly of the USB isolates was determined using the microdilution assay Vitek 2 (n = 64; bioMérieux) or Etest (n = 15; Apteq) (Table S2). Meropenem susceptibility values of the PATRIC data originated from various studies and were determined based on MICs (n = 1,001) or disk diffusion (inhibitory zone diameter [IZD]; n = 44). When available, MICs and IZDs were interpreted according to CLSI guideline M100 (susceptible, ≤2 mg/L or ≥19 mm; intermediate, 4 mg/L or 16 to 18 mm; resistant, ≥8 mg/L or ≤15 mm) (44). For 29 PATRIC assemblies, only phenotypic classifications (interpreted according to CLSI criteria) but no MIC/IZD values were available (Table S2).
Genomic analyses.
Phylogenetic distances were estimated using Mashtree 1.2.0 (45) in accurate mode (--mindepth 0), and clades (A, B, C1, C2) were assigned based on phylogenetic clustering with previously typed (39) genomes. Phylogenetic trees were visualized with iTOL version 6 (46). Antimicrobial resistance genes and mutations in quinolone resistance-determining regions were detected using AMRfinder 3.10.24 with default parameters (47). Beta-lactamases were classified as carbapenemases based on antibiotic subclass associations listed in the AMRfinder database. Genome-wide association studies for isolates with versus without predicted OprD loss were performed using DBGWAS version 0.5.4 (options, –maf 0.05 -q100) (48). Due to high computational requirements, the analysis was performed on a subset of 700 randomly selected assemblies of the NCBI data set irrespective of their phylogenetic distribution or susceptibility (Table S4). For selected assemblies, the genetic context of oprD was manually investigated in CLC Main Workbench 22.0.1. Insertion sequence (IS) elements were identified using ISFinder (49). Assemblies containing the oprD frameshift mutation c.1199_1200insC were identified using BLASTn 2.10.1+ (37) in short-sequence mode with a 38-bp partial oprD sequence of NCBI accession nos. USI81034 (intact) and CP050331 (frameshifted) as references. Functional effects of OprD amino acid substitutions were predicted using SNAP2 (50). Assemblies were queried for MexR (references, GenPept accession nos. NP_249115 and WP_003153084), MexA (reference, GenPept accession no. NP_249116), MexB (reference, GenPept accession no. NP_249117), AmpR (references, GenPept accession nos. NP_252798, WP_024915537, and WP_058145575), NalC (references, GenPept accession nos. NP_252410 and WP_033984356), and NalD (reference, GenPept accession no. NP_252264) using DIAMOND 2.0.7 (32) in BLASTx mode (minimum sequence coverage and identity, 50% and 90%, respectively). Assemblies with no hit, incomplete hits (<98% alignment coverage on the amino acid level), or hits with internal stop codons were considered defective for the respective protein. Plots were generated in R 4.0.3 using the ComplexUpset 1.3.3 package (https://zenodo.org/record/5762625#.YrWOVXZByUk).
Statistical tests.
Frequency counts were compared using a two-tailed Fisher’s exact test. P values of <0.05 were considered to reflect statistical significance.
Data and software availability.
Sequencing data generated as part of this study are available under BioProject accession no. PRJEB54973. Accession numbers of included assemblies are listed in Table S1 (NCBI data set) and Table S2 (validation data set) in the supplemental material. PorinPredict is available under the terms of the GNU General Public License 3.0 at https://github.com/MBiggel/PorinPredict/.
ACKNOWLEDGMENTS
We thank Daniel Gander, Christine Kiessling, Magdalena Schneider, Elisabeth Schultheiss, Clarisse Straub, and Rosa-Maria Vesco (University Hospital Basel) for excellent technical assistance with sequencing. We thank Alfredo Mari for extensive helpful discussions on the implementation of PorinPredict.
Footnotes
Supplemental material is available online only.
Contributor Information
Michael Biggel, Email: michael.biggel@uzh.ch.
Helena M. B. Seth-Smith, Email: hsethsmith@imm.uzh.ch.
Ana Paula D'Alincourt Carvalho-Assef, Instituto Oswaldo Cruz.
REFERENCES
- 1.Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, Ouellette M, Outterson K, Patel J, Cavaleri M, Cox EM, Houchens CR, Grayson ML, Hansen P, Singh N, Theuretzbacher U, Magrini N, AboDerin AO, Al-Abri SS, Awang Jalil N, Benzonana N, Bhattacharya S, Brink AJ, Burkert FR, Cars O, Cornaglia G, Dyar OJ, Friedrich AW, Gales AC, Gandra S, Giske CG, Goff DA, Goossens H, Gottlieb T, Guzman Blanco M, Hryniewicz W, Kattula D, Jinks T, Kanj SS, Kerr L, Kieny M-P, Kim YS, Kozlov RS, Labarca J, Laxminarayan R, Leder K, et al. 2018. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18:318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- 2.David S, Reuter S, Harris SR, Glasner C, Feltwell T, Argimon S, Abudahab K, Goater R, Giani T, Errico G, Aspbury M, Sjunnebo S, Feil EJ, Rossolini GM, Aanensen DM, Grundmann H. 2019. Epidemic of carbapenem-resistant Klebsiella pneumoniae in Europe is driven by nosocomial spread. Nat Microbiol 4:1919–1929. doi: 10.1038/s41564-019-0492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poirel L, Nordmann P. 2006. Carbapenem resistance in Acinetobacter baumannii: mechanisms and epidemiology. Clin Microbiol Infect 12:826–836. doi: 10.1111/j.1469-0691.2006.01456.x. [DOI] [PubMed] [Google Scholar]
- 4.Zhu Y, Chen J, Shen H, Chen Z, Yang Q, Zhu J, Li X, Yang Q, Zhao F, Ji J, Cai H, Li Y, Zhang L, Leptihn S, Hua X, Yu Y. 2021. Emergence of ceftazidime- and avibactam-resistant Klebsiella pneumoniae carbapenemase-producing Pseudomonas aeruginosa in China. mSystems 6:e00787-21. doi: 10.1128/mSystems.00787-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pragasam AK, Veeraraghavan B, Anandan S, Narasiman V, Sistla S, Kapil A, Mathur P, Ray P, Wattal C, Bhattacharya S, Deotale V, Subramani K, Peter JV, Hariharan TD, Ramya I, Iniyan S, Walia K, Ohri VC. 2018. Dominance of international high-risk clones in carbapenemase-producing Pseudomonas aeruginosa: multicentric molecular epidemiology report from India. Indian J Med Microbiol 36:344–351. doi: 10.4103/ijmm.IJMM_18_294. [DOI] [PubMed] [Google Scholar]
- 6.Bocharova Y, Savinova T, Lazareva A, Polikarpova S, Gordinskaya N, Mayanskiy N, Chebotar I. 2020. Genotypes, carbapenemase carriage, integron diversity and oprD alterations among carbapenem-resistant Pseudomonas aeruginosa from Russia. Int J Antimicrob Agents 55:105899. doi: 10.1016/j.ijantimicag.2020.105899. [DOI] [PubMed] [Google Scholar]
- 7.Teo JQ-M, Tang CY, Lim JC, Lee SJ-Y, Tan SH, Koh T-H, Sim JH-C, Tan T-T, Kwa AL-H, Ong RT-H. 2021. Genomic characterization of carbapenem-non-susceptible Pseudomonas aeruginosa in Singapore. Emerg Microbes Infect 10:1706–1716. doi: 10.1080/22221751.2021.1968318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walters MS, Grass JE, Bulens SN, Hancock EB, Phipps EC, Muleta D, Mounsey J, Kainer MA, Concannon C, Dumyati G, Bower C, Jacob J, Cassidy PM, Beldavs Z, Culbreath K, Phillips WE, Hardy DJ, Vargas RL, Oethinger M, Ansari U, Stanton R, Albrecht V, Halpin AL, Karlsson M, Rasheed JK, Kallen A. 2019. Carbapenem-resistant Pseudomonas aeruginosa at US emerging infections program sites, 2015. Emerg Infect Dis 25:1281–1288. doi: 10.3201/eid2507.181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCracken MG, Adam HJ, Blondeau JM, Walkty AJ, Karlowsky JA, Hoban DJ, Zhanel GG, Mulvey MR, Zhanel GG, Hoban DJ, Adam HJ, Baxter MR, Nichol KA, Lagacé-Wiens PRS, Walkty A, Karlowsky JA, Blondeau J, Slinger R, Davidson R, Zhanel G, Hoban D, Delport J, Ellis C, Laverdière M, Loo V, Poutanen S, Fuller J, Roscoe D, Desjardins M, Matukas L, Goyette M, Lee C, Carignan A, Bergevin M, Pelletier R, Canadian Antimicrobial Resistance Alliance (CARA) and CANWARD . 2019. Characterization of carbapenem-resistant and XDR Pseudomonas aeruginosa in Canada: results of the CANWARD 2007–16 study. J Antimicrob Chemother 74:iv32–iv38. doi: 10.1093/jac/dkz285. [DOI] [PubMed] [Google Scholar]
- 10.Kazmierczak KM, de Jonge BLM, Stone GG, Sahm DF. 2020. Longitudinal analysis of ESBL and carbapenemase carriage among Enterobacterales and Pseudomonas aeruginosa isolates collected in Europe as part of the International Network for Optimal Resistance Monitoring (INFORM) Global Surveillance Programme, 2013–17. J Antimicrob Chemother 75:1165–1173. doi: 10.1093/jac/dkz571. [DOI] [PubMed] [Google Scholar]
- 11.Diorio-Toth L, Irum S, Potter RF, Wallace MA, Arslan M, Munir T, Andleeb S, Burnham C-AD, Dantas G. 2022. Genomic surveillance of clinical Pseudomonas aeruginosa isolates reveals an additive effect of carbapenemase production on carbapenem resistance. Microbiol Spectr 10:e00766-22. doi: 10.1128/spectrum.00766-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.del Barrio-Tofiño E, López-Causapé C, Oliver A. 2020. Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int J Antimicrob Agents 56:106196. doi: 10.1016/j.ijantimicag.2020.106196. [DOI] [PubMed] [Google Scholar]
- 13.Stanton RA, Campbell D, McAllister GA, Breaker E, Adamczyk M, Daniels JB, Lutgring JD, Karlsson M, Schutz K, Jacob JT, Wilson LE, Vaeth E, Li L, Lynfield R, Snippes Vagnone PM, Phipps EC, Hancock EB, Dumyati G, Tsay R, Cassidy PM, Mounsey J, Grass JE, Bulens SN, Walters MS, Halpin AL. 2022. Whole-genome sequencing reveals diversity of carbapenem-resistant Pseudomonas aeruginosa collected through CDC’s emerging infections program, United States, 2016–2018. Antimicrob Agents Chemother 66:e00496-22. doi: 10.1128/aac.00496-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hancock REW, Brinkman FSL. 2002. Function of Pseudomonas porins in uptake and efflux. Annu Rev Microbiol 56:17–38. doi: 10.1146/annurev.micro.56.012302.160310. [DOI] [PubMed] [Google Scholar]
- 15.Biswas S, Mohammad MM, Patel DR, Movileanu L, van den Berg B. 2007. Structural insight into OprD substrate specificity. Nat Struct Mol Biol 14:1108–1109. doi: 10.1038/nsmb1304. [DOI] [PubMed] [Google Scholar]
- 16.Richardot C, Plésiat P, Fournier D, Monlezun L, Broutin I, Llanes C. 2015. Carbapenem resistance in cystic fibrosis strains of Pseudomonas aeruginosa as a result of amino acid substitutions in porin OprD. Int J Antimicrob Agents 45:529–532. doi: 10.1016/j.ijantimicag.2014.12.029. [DOI] [PubMed] [Google Scholar]
- 17.Pan Y-P, Xu Y-H, Wang Z-X, Fang Y-P, Shen J-L. 2016. Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch Microbiol 198:565–571. doi: 10.1007/s00203-016-1215-7. [DOI] [PubMed] [Google Scholar]
- 18.Quale J, Bratu S, Gupta J, Landman D. 2006. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother 50:1633–1641. doi: 10.1128/AAC.50.5.1633-1641.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardy JL, Loman NJ. 2018. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 19:9–20. doi: 10.1038/nrg.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.World Health Organization. 2020. Whole-genome sequencing for surveillance of antimicrobial resistance: Global Antimicrobial Resistance and Use Surveillance System (GLASS). World Health Organization, Geneva, Switzerland. [Google Scholar]
- 21.Hendriksen RS, Bortolaia V, Tate H, Tyson GH, Aarestrup FM, McDermott PF. 2019. Using genomics to track global antimicrobial resistance. Front Public Heal 7:242. doi: 10.3389/fpubh.2019.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eren E, Vijayaraghavan J, Liu J, Cheneke BR, Touw DS, Lepore BW, Indic M, Movileanu L, van den Berg B. 2012. Substrate specificity within a family of outer membrane carboxylate channels. PLoS Biol 10:e1001242. doi: 10.1371/journal.pbio.1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epp SF, Köhler T, Plésiat P, Michéa-Hamzehpour M, Frey J, Pechère JC. 2001. C-Terminal region of Pseudomonas aeruginosa outer membrane porin OprD modulates susceptibility to meropenem. Antimicrob Agents Chemother 45:1780–1787. doi: 10.1128/AAC.45.6.1780-1787.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanbongi Y, Shimizu A, Suzuki T, Nagaso H, Ida T, Maebashi K, Gotoh N. 2009. Classification of OprD sequence and correlation with antimicrobial activity of carbapenem agents in Pseudomonas aeruginosa clinical isolates collected in Japan. Microbiol Immunol 53:361–367. doi: 10.1111/j.1348-0421.2009.00137.x. [DOI] [PubMed] [Google Scholar]
- 25.David S, Wong JLC, Sanchez-Garrido J, Kwong H-S, Low WW, Morecchiato F, Giani T, Rossolini GM, Brett SJ, Clements A, Beis K, Aanensen DM, Frankel G. 2022. Widespread emergence of OmpK36 loop 3 insertions among multidrug-resistant clones of Klebsiella pneumoniae. PLoS Pathog 18:e1010334. doi: 10.1371/journal.ppat.1010334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nascimento APB, Ortiz MF, Martins WMBS, Morais GL, Fehlberg LCC, Almeida LGP, Ciapina LP, Gales AC, Vasconcelos ATR. 2016. Intraclonal genome stability of the metallo-β-lactamase SPM-1-producing Pseudomonas aeruginosa ST277, an endemic clone disseminated in Brazilian hospitals. Front Microbiol 7:1946. doi: 10.3389/fmicb.2016.01946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabot G, López-Causapé C, Ocampo-Sosa AA, Sommer LM, Domínguez MÁ, Zamorano L, Juan C, Tubau F, Rodríguez C, Moyà B, Peña C, Martínez-Martínez L, Plesiat P, Oliver A. 2016. Deciphering the resistome of the widespread Pseudomonas aeruginosa sequence type 175 international high-risk clone through whole-genome sequencing. Antimicrob Agents Chemother 60:7415–7423. doi: 10.1128/AAC.01720-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.del Barrio-Tofiño E, Zamorano L, Cortes-Lara S, López-Causapé C, Sánchez-Diener I, Cabot G, Bou G, Martínez-Martínez L, Oliver A, Galán F, Gracia I, Rodríguez MA, Martín L, Sánchez JM, Viñuela L, García MV, Lepe JA, Aznar J, López-Hernández I, Seral C, Javier Castillo-García F, López-Calleja AI, Aspiroz C, de la Iglesia P, Ramón S, Riera E, Cruz Pérez M, Gallegos C, Calvo J, Dolores Quesada M, Marco F, Hoyos Y, Pablo Horcajada J, Larrosa N, González JJ, Tubau F, Capilla S, Pérez-Moreno MO, Centelles MJ, Padilla E, Rivera A, Mirelis B, Elisa Rodríguez-Tarazona R, Arenal-Andrés N, del Pilar Ortega M, Megías G, García I, Colmenarejo C, González JC, et al. 2019. Spanish nationwide survey on Pseudomonas aeruginosa antimicrobial resistance mechanisms and epidemiology. J Antimicrob Chemother 74:1825–1835. doi: 10.1093/jac/dkz147. [DOI] [PubMed] [Google Scholar]
- 29.Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, Wimalarathna H, Harrison OB, Sheppard SK, Cody AJ, Maiden MCJ. 2012. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology (Reading) 158:1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchfink B, Reuter K, Drost H-G. 2021. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18:366–368. doi: 10.1038/s41592-021-01101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 35.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pagès H, Aboyoun P, Gentleman R, DebRoy S. 2022. Biostrings: efficient manipulation of biological strings. R package version 2.64.0. https://rdrr.io/bioc/Biostrings/.
- 39.Ozer EA, Nnah E, Didelot X, Whitaker RJ, Hauser AR. 2019. The population structure of Pseudomonas aeruginosa is characterized by genetic isolation of exoU+ and exoS+ lineages. Genome Biol Evol 11:1780–1796. doi: 10.1093/gbe/evz119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochs MM, Lu C-D, Hancock REW, Abdelal AT. 1999. Amino acid-mediated induction of the basic amino acid-specific outer membrane porin OprD from Pseudomonas aeruginosa. J Bacteriol 181:5426–5432. doi: 10.1128/JB.181.17.5426-5432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, Chlenski P, Conrad N, Dickerman A, Dietrich EM, Gabbard JL, Gerdes S, Guard A, Kenyon RW, Machi D, Mao C, Murphy-Olson D, Nguyen M, Nordberg EK, Olsen GJ, Olson RD, Overbeek JC, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomas C, VanOeffelen M, Vonstein V, Warren AS, Xia F, Xie D, Yoo H, Stevens R. 2019. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res 48:D606–D612. doi: 10.1093/nar/gkz943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clinical and Laboratory Standards Institute. 2020. Performance standards for antimicrobial susceptibility testing, 30th ed. CLSI supplement M100. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 45.Katz L, Griswold T, Morrison S, Caravas J, Zhang S, Bakker H, Deng X, Carleton H. 2019. Mashtree: a rapid comparison of whole genome sequence files. J Open Source Softw 4. doi: 10.21105/joss.01762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Letunic I, Bork P. 2019. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, Haft DH, Hoffmann M, Pettengill JB, Prasad AB, Tillman GE, Tyson GH, Klimke W. 2021. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci Rep 11:12728. doi: 10.1038/s41598-021-91456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaillard M, Lima L, Tournoud M, Mahé P, van Belkum A, Lacroix V, Jacob L. 2018. A fast and agnostic method for bacterial genome-wide association studies: bridging the gap between k-mers and genetic events. PLoS Genet 14:e1007758. doi: 10.1371/journal.pgen.1007758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hecht M, Bromberg Y, Rost B. 2015. Better prediction of functional effects for sequence variants. BMC Genomics 16:S1. doi: 10.1186/1471-2164-16-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.03588-22-s0001.xlsx, XLSX file, 0.5 MB (486.2KB, xlsx)
Supplemental material. Download spectrum.03588-22-s0002.pdf, PDF file, 0.3 MB (270.9KB, pdf)