

ABSTRACT
Molecular surveillance for malaria has great potential to support national malaria control programs (NMCPs). To bridge the gap between research and implementation, several applications (use cases) have been identified to align research, technology development, and public health efforts. For implementation at NMCPs, there is an urgent need for feasible and cost-effective tools. We designed a new highly multiplexed deep sequencing assay (Pf AmpliSeq), which is compatible with benchtop sequencers, that allows high-accuracy sequencing with higher coverage and lower cost than whole-genome sequencing (WGS), targeting genomic regions of interest. The novelty of the assay is its high number of targets multiplexed into one easy workflow, combining population genetic markers with 13 nearly full-length resistance genes, which is applicable for many different use cases. We provide the first proof of principle for hrp2 and hrp3 deletion detection using amplicon sequencing. Initial sequence data processing can be performed automatically, and subsequent variant analysis requires minimal bioinformatic skills using any tabulated data analysis program. The assay was validated using a retrospective sample collection (n = 254) from the Peruvian Amazon between 2003 and 2018. By combining phenotypic markers and a within-country 28-single-nucleotide-polymorphism (SNP) barcode, we were able to distinguish different lineages with multiple resistance haplotypes (in dhfr, dhps, crt and mdr1) and hrp2 and hrp3 deletions, which have been increasing in recent years. We found no evidence to suggest the emergence of artemisinin (ART) resistance in Peru. These findings indicate a parasite population that is under drug pressure but is susceptible to current antimalarials and demonstrate the added value of a highly multiplexed molecular tool to inform malaria strategies and surveillance systems.
IMPORTANCE While the power of next-generation sequencing technologies to inform and guide malaria control programs has become broadly recognized, the integration of genomic data for operational incorporation into malaria surveillance remains a challenge in most countries where malaria is endemic. The main obstacles include limited infrastructure, limited access to high-throughput sequencing facilities, and the need for local capacity to run an in-country analysis of genomes at a large-enough scale to be informative for surveillance. In addition, there is a lack of standardized laboratory protocols and automated analysis pipelines to generate reproducible and timely results useful for relevant stakeholders. With our standardized laboratory and bioinformatic workflow, malaria genetic surveillance data can be readily generated by surveillance researchers and malaria control programs in countries of endemicity, increasing ownership and ensuring timely results for informed decision- and policy-making.
KEYWORDS: DNA sequencing, Plasmodium falciparum, drug resistance, genetic epidemiology, hrp2 and hrp3 deletions, malaria, surveillance studies
INTRODUCTION
The Global Technical Strategy for Malaria 2016–2030 identified the use of high-quality surveillance data for decision-making as an essential pillar for malaria elimination (1). In view of the current challenges in malaria control (e.g., the spread of drug resistance, changing transmission intensity, risk of malaria importation into malaria-free areas, and rapid diagnostic test [RDT] failures), genetic epidemiology is increasingly being recognized for its potential to inform national malaria control programs (NMCPs). By aligning multiple scientific fields in support of NMCP priorities, “use cases” were specified to create scenarios where genetic surveillance can be informative for decision-making (2, 3) and can be used to develop and implement new technologies.
Molecular markers associated with drug resistance are reliable predictors of treatment responses (2, 4, 5), can provide warning for emerging resistance, guide treatment policies (6), and even replace therapeutic efficacy studies in low-malaria-transmission areas (4, 5, 7, 8). The value of these markers was demonstrated in Southeast Asia, where Plasmodium falciparum resistance to artemisinin (ART) was associated with mutations in the Kelch protein 13 gene (K13) (9–12). Resistance mutations in the same gene recently emerged in Africa (13).
In addition, monitoring population genetic markers can guide control and elimination strategies by identifying drug resistance gene flow, distinguishing imported from autochthonous cases, and estimating the source of reintroduced cases in malaria-free areas (2, 14–16). Parasite genetics can identify underlying patterns of transmission and transmission intensity (17).
The gene products of P. falciparum hrp2 and hrp3 are detected by RDTs, and gene deletions resulting in false-negative RDT results were first detected in Peru 10 years ago (18). Since then, hrp2 and hrp3 deletions have been reported in many areas (19–24), raising concerns about the viability of histidine-rich protein 2 (HRP2)-based RDTs. The WHO recommends assessing the prevalence of hrp2 and hrp3 mutants among malaria patients and changing case management strategies when more than 5% of infections contain deletions (25).
Multiple genetic markers are required to address the above-mentioned challenges and are most efficiently targeted in a single genomic tool (3, 26). Whole-genome sequencing (WGS) can provide the most information content per sample; however, implementing WGS for surveillance is constrained by the limited access to high-throughput sequencers and the lack of technical and bioinformatic skills in countries of endemicity. As a result, sequencing is often performed through genomic consortia, delaying the turnaround time from sample collection to genetic data and limiting its usefulness to NMCPs. In addition, obtaining high-quality WGS data from low-density infections and dried blood spots (DBSs) remains challenging. Consequently, it is rare for genomic surveillance to be used beyond research (27–29).
Targeted next-generation sequencing (NGS) tools are emerging that combine numerous targets of interest into one workflow, being more time- and cost-efficient than WGS or traditional PCR-based approaches with amplicons detected by different techniques (10, 30–33). Laboratory protocols for NGS assays are well standardized, sequencing is feasible on benchtop sequencers, and automated analysis pipelines are available (2, 6), for example, the Malaria Resistance Surveillance (MaRS) protocol (34), which targets multiple drug resistance markers. While microsatellites (MSs) have frequently been used for population genetic surveillance as they are not under evolutionary pressure, these markers are not easily translated to NGS due to the short repetitive nature of the variation. Instead, single-nucleotide polymorphism (SNP) barcodes can be successfully targeted by NGS (35–37) and are informative for connectivity between populations (38, 39) and predictions of infection origins (40, 41). The SpotMalaria platform combining drug resistance markers with a SNP barcode was helpful in tracking the rapid spread of resistant parasites in the Greater Mekong subregion (42).
Currently, there is no multifunctional tool that includes a combination of more than two types of markers (i.e., SNP barcodes and drug resistance, etc.) to serve several use cases. The characterization of hrp2 and hrp3 deletions still relies on PCR assays that classify deletions based on the failure to amplify targets (23). The few existing tools that combine population markers with drug resistance target short regions around validated resistance SNPs, missing the potential to detect novel resistance-associated mutations. Furthermore, many SNP panels were designed from genomes across the world and lack the resolution to study subtle patterns on a smaller geographical scale. Therefore, we designed a targeted NGS assay (Pf AmpliSeq) that combines a specifically designed barcode for the target country, 13 full-length resistance-associated genes, hrp2 and hrp3, and an ama1 microhaplotype region. The assay is applicable to DBSs and adaptable to different settings. The AmpliSeq technology (43) applies multiplex PCR to simultaneously amplify a high number of targets in a rapid procedure and allows overlapping amplicons to cover large genes.
We applied the Pf AmpliSeq assay to Peruvian samples for assay validation. P. falciparum in Peru makes a good case study as malaria elimination is on the regional agenda (44), with countries first targeting P. falciparum elimination due to its lower case load than that of Plasmodium vivax but also in fear of emerging ART resistance. South America has been a hot spot for chloroquine (CQ) and sulfadoxine-pyrimethamine (SP) resistance evolution (45). More recently, K13-mediated ART resistance emerged in Guyana (46, 47), and K13-independent delayed parasite clearance after ART treatment was reported in Suriname (46, 48, 49). P. falciparum elimination is also threatened by the increasing prevalence and spread of hrp2 and hrp3 deletions (21, 23).
Peru reduced its malaria case incidence by ≥40% in 2010 (Fig. 1), and after a resurgence in 2016, cases have been decreasing in the past 5 years after the introduction of focal screening and treatment (50). Past high levels of CQ and SP resistance (51–54) led Peru to become one of the first countries in the Americas to introduce artemisinin combination therapy (ACT) in 1999 (54–56). However, ACT efficacy and drug resistance markers have not been monitored in Peru since 2009. In 1998 to 2001, up to 20% of infections in Peru had an hrp2 deletion, increasing to 53% with hrp2 deletions and 37% with deletions of both genes in 2012 to 2014 (18, 19). In this study, we demonstrate the added value of molecular surveillance in Peru with different use cases using our Pf AmpliSeq assay and explore the changes in the parasite population in the past 2 decades.
FIG 1.

Annual P. falciparum cases in Peru (black line) from 1992 to 2020, with first-line treatment policy changes (data from the Centro Nacional de Epidemiología, Prevención y Control de Enfermedades [2020] Sistema de Atención de Solicitudes de Acceso a la Información Pública vía Internet del Ministerio de Salud [available at http://www.minsa.gob.pe/portada/transparencia/solicitud/]). Artemisinin combination therapies (ACTs) were first introduced into the Amazon region in Peru in 1999 and the North Coast in 2001, contributing to a dramatic decline in the number of cases. The combination of artesunate, mefloquine, and primaquine (AS+MQ+PQ) was the recommended first-line treatment in the Amazon region from the start (51), with continued high efficacy (107), and the combination of AS plus sulfadoxine-pyrimethamine (SP) was used on the North Coast. In 2015, AS+MQ+PQ became the recommended first-line treatment in the whole country (108). Peru reduced the malaria case incidence by ≥40% by 2010 using passive case detection (PCD), diagnosis by light microscopy (LM), and treatment with AS+MQ as the main components of the control program (109). In 2015, with the realization that PCD missed asymptomatic infections (110), focal screening and treatment were introduced, resulting in a further reduction in the malaria incidence (50). CQ+PQ, chloroquine plus primaquine; QN, quinine.
RESULTS
Pf AmpliSeq design and performance.
A panel of 13 P. falciparum drug resistance-associated genes (Table 1) and corresponding variants of interest, i.e., (putative) resistance-associated mutations (see Data Set S1 in the supplemental material), were included in the assay design. The selected genes included validated mutations (4, 7) or variants relevant for Peru, i.e., those previously reported or associated with historically and currently used drugs. Additional targets were the hrp2 and hrp3 genes, the apical membrane antigen 1 gene (ama1), the conserved Plasmodium membrane protein gene (cpmp) (57), and the microsatellites (MSs) poly-alpha, ARAII, TA81, and PfPK2 (58). We designed a barcode of 28 SNPs with in-country resolution in Peru, as explained in detail in the supplemental material. Primers for an AmpliSeq custom panel targeting the desired regions were designed using DesignStudio software by the Illumina concierge team. This design process makes individual amplicon testing (by PCR) before multiplexing unnecessary and resulted in two primer pools ready for library preparation. The final assay included >85% of the desired regions in 233 amplicons (Data Set S1) with various amplicon lengths (55 to 323 bp). Target regions where primer design was not possible within the constraints of the multiplex assay due to AT-rich sequences or high genetic variability were the cpmp region and a small section of the crt gene, including crt variant I218F.
TABLE 1.
Genes of interest for drug resistance molecular surveillance of malaria in Peru
| Gene ID | Chromosome | Gene with resistance-associated SNP(s) | Drug(s) associated with resistance (reference[s])a |
|---|---|---|---|
| PF3D7_1218300 | 12 | ap2-mu | ART (68, 72, 111, 112), QN (111) |
| PF3D7_1251200 | 12 | coronin | DHA (60, 76) |
| PF3D7_0709000 | 7 | crt | CQ (113–117), PPQ (96, 118–121), AMQ (122) |
| mal_mito_3 | Mitochondrial | Cytochrome b | ATQ (123–125) |
| PF3D7_0417200 | 4 | dhfr | PYR (61, 79, 126–129), PG (130) |
| PF3D7_0810800 | 8 | dhps | SULF (61, 79, 126, 127, 129) |
| PF3D7_1362500 | 13 | Exonuclease | PPQ (131) |
| PF3D7_1343700 | 13 | K13 | ART (9, 46) |
| PF3D7_0523000 | 5 | mdr1 | CQ (132, 133); PPQ (66); MQ (65); QN, HF, ART (61, 63, 65, 66, 127, 134); AMQ (66, 122); LF (134, 135) |
| PF3D7_0112200 | 1 | mrp1 | ART, MQ, LF (66, 136) |
| PF3D7_1408000 | 14 | Plasmepsin II | PPQ (64, 131, 137) |
| PF3D7_API04900 | Apicoplast | 23S rRNA | CM (138) |
| PF3D7_0104300 | 1 | ubp-1 | ART (68, 71, 111, 112) |
The list of drugs to which resistance has been observed is nonexhaustive. Validated mutations are listed in the WHO Report on Antimalarial Drug Efficacy, Resistance and Response: 10 Years of Surveillance (2010–2019) (7). AMQ, amodiaquine; ART, artesunate; ATQ, atovaquone; CM, clindamycin; CQ, chloroquine; DHA, dihydroartemisinin; HF, halofantrine; LF, lumefantrine; MQ, mefloquine; PG, proguanil; PPQ, piperaquine; PYR, pyrimethamine; SULF, sulfadoxine; QN, quinine.
We validated the assay with P. falciparum cases (n = 312) (Fig. 2) from multiple previous studies in Peru, laboratory strains (n = 5), and previously genotyped samples (n = 6) as controls. A high number of reads was generated in the assay (mean of 217,037 ± 490,478 paired reads/sample after trimming low-quality reads), with a median of 99.6% (range, 3.9 to 99.9%) of trimmed reads aligning to the P. falciparum 3D7 (Pf3D7) genome. A mean of 1,336 ± 3,627 paired reads (range, 20 to 43,795) were generated per library (including all samples and controls [also negative controls]). To improve the quality of the sequences (percentage of base calls with quality score above 30 [%Q30]), we increased the nucleotide diversity in the run (30% GC content in the libraries) with a 20% spike-in of a PhiX library. However, while the overall %Q30 increased, adding 20% PhiX is not recommended as it did not decrease the proportion of low-quality reads that were excluded (9.7% ± 10.6% of reads trimmed without a spike-in versus 10.3% ± 7.3% with a spike-in), with a 20% reduction of the total read number.
FIG 2.
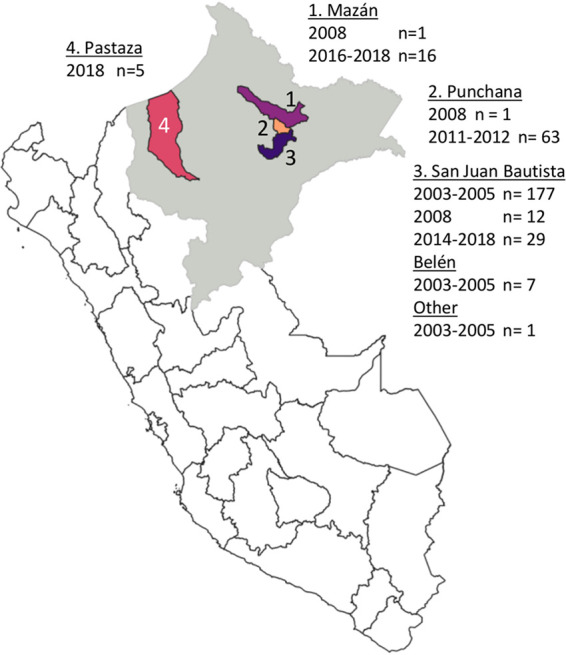
Map of study sites of retrospective sample collection from the Peruvian Amazon region Loreto (gray). The majority of the samples were collected in Maynas Province (areas 1, 2, and 3): samples were collected in or near the San Juan Bautista district (area 3), which covers part of the urban area of Loreto’s capital Iquitos and periurban communities south of Iquitos (n = 226), and the Mazán district (area 1) (n = 17) and the Punchana district (area 2) (n = 64) with remote communities living near forest rivers such as the Amazon River and the Napo River, north of Iquitos. A final collection of samples from 2018 was available from the Northern Amazon region in Datem del Marañón Province, Pastaza district (area 4) (n = 5). P. falciparum-positive samples from previous studies were selected based on geographical representation and parasite density (≥100 p/μL by PCR; geometric mean density of 4,300 ± 3.1 p/μL).
Depth of coverage.
The median amplicon depth of coverage of aligned high-quality reads past the filter (DP) was 82.9 (interquartile range [IQR], 42.9 to 116.1) (Fig. S1). Fifteen (6.4%) amplicons had low DP values (<10) (Table S1), while 6 (2.6%) had high DP values (>150) (Table S2). Among these, 4 hrp2 and hrp3 amplicons had lower DP values in the study samples, but not in the controls, due to the high prevalence of deletions in these genes. The lower DP values for other amplicons are likely due to sequence variations in primer binding sites, resulting in poor amplification and large amounts of missing data for these regions, e.g., A220S in crt. In contrast, 2 hrp2 amplicons had higher depths than the average, possibly due to cross-alignment between hrp2 and hrp3 repetitive regions. For 23/233 amplicons, no variants (i.e., only reference alleles) were detected despite good amplification and sequencing (Table S3), and these amplicons were located in conserved regions of drug resistance genes.
Uninfected controls.
Primer specificity was tested with four uninfected human blood samples (negative controls), and none resulted in genotypes in the assay target region. We did observe background sequences with a very low number of reads corresponding to multiple unspecific hits to Plasmodium and human sequences; all of them were outside the assay target regions and below the filtering threshold.
Eight stored previous library preparations (including control samples and one human negative control) were included to test replicability; however, the human negative control was contaminated with P. falciparum sequences, probably acquired after library preparation and during storage; thus, replicability could not be assessed.
Parasite density limit and selective whole-genome amplification.
The DP decreased while the missingness increased with decreasing parasite density in a dilution of 3D7 (6,000 to 6 parasites/μL [p/μL]) at a DNA concentration mimicking that of a DBS sample. At the lowest density (6 p/μL), the median DP was only 10, and 60% of loci were missing. Selective whole-genome amplification (sWGA) prior to library preparation improved the mean DP and the number of high-quality reads at parasite densities of ≤60 p/μL (Fig. S2), with 10-fold more reads at 60 p/μL and 90-fold more reads at 6 p/μL.
Error rate and reproducibility.
Sequencing accuracy was higher for biallelic SNPs (mean error rate of 0.008% ± 0.004%) than for indels (0.02% ± 0.005%) (Table S4). At parasite densities of >6 p/μL, sWGA is not recommended, as error rates were higher with (0.13% ± 0.06%) than without (0.05% ± 0.01%) sWGA, with little gain in coverage.
The reproducibility of the assay was tested, with a median difference of 4 SNPs (0.6% of SNP loci; 0.007% of 57,445 bp targeted in the assay) between sample and control replicates observed. With indels and multiallelic variants, a median difference of 50 alleles (2.8% of loci; 0.09% of 57,445 bp) was observed.
In comparison to previous results with laboratory strains and control samples, 97.6% of genotypes were accurate (Table S5). For the control samples, 7.9% additional minor alleles in heterozygous genotypes were detected by AmpliSeq, which were likely to be undetected in the SpotMalaria pipeline and WGS data due to lower sequencing depth.
Complexity of infection.
Minority clones were detected by the Pf AmpliSeq assay with up to an 80:20 3D7-Dd2 mixture with 43.8% (28/64) of Dd2 loci detected (Table S6). Below a ratio of 95:5, few Dd2 genotypes were detected.
The complexity of infection (COI) estimates the number of genetically distinct clones in an infection, which can be used as a proxy for transmission intensity (16, 59). We applied different methods to determine the COI from NGS data, with results varying considerably by method (Table S7). Therefore, we determined the most frequent value (mode) of the COI, which showed an increasing proportion of multiple-clone infections in Peru over time (13.8% in 2003 to 2005, 28.8% in 2008 to 2012, and 33.3% in 2014 to 2018 [P = 0.0005 by a χ2 test]) (Fig. S3).
Use case of hrp2 and hrp3 gene deletions.
Compared to previous PCR classifications, all samples (10/10) were correctly classified as “RDT failure” (deletion of both genes) versus “RDT detectable” (presence of one or both genes) using the read depths of hrp2 and hrp3 amplicons. For 6/10 samples, the classification was correct for both genes, but for 4 samples, the hrp2 classification was inconclusive (Table S8) due to discrepancies in classifications between amplicons.
With the Pf AmpliSeq assay, 94.9% (241/254) of the study samples could be classified as RDT failure or RDT detectable. The proportion of individuals with RDT failure increased from 17.7% in 2003 to 2005 to 42.6% in 2008 to 2012 and 73.3% in 2014 to 2018 (P > 0.001 by a χ2 test). While hrp3 presence versus absence could be determined for most samples (96.5%), 26.8% of hrp3-positive (hrp3+) samples were inconclusive for hrp2 (Fig. S9), but since they were not hrp3 deleted, they were classified as RDT detectable.
Barcode performance.
All 28 barcode SNPs were detected by the assay (Table S9), although 1 (Pf3D7_12_1127001) was genotyped as an indel. In the study samples, the population minor allele frequencies (MAFs) of the barcode SNPs ranged between 0.35 and 0.50 at 9 loci and between 0.1 and 0.34 at 12 loci and were <0.1 at 7 loci. We did not observe the minor allele of one of the barcode SNPs (Pf3D7_05_921893) in the study samples, although it was detected in laboratory strains (n = 5) and 5/6 control samples.
Use case of transmission intensity: genetic diversity and differentiation.
The similarity of the samples was explored over space (district) and time (year of collection) using principal-component analysis (PCA), which showed clustering by year rather than by collection site (Fig. S4). Therefore, the samples were divided into three time periods for subsequent analyses: (i) the early time period (2003 to 2005), (ii) the intermediate time period (2008 to 2012), and (iii) the most recent period (2014 to 2018), where little variability was observed.
The genetic diversity (expected heterozygosity [He]) of the parasite population was lower in 2008 to 2012 (P = 0.003) and in 2014 to 2018 (P = 0.00004) than in 2003 to 2005 (Fig. 3A). The total population size was small, as the observed heterozygosity (Hobs) was lower than the He (P < 0.005), indicating that genetic diversity was lower than expected from a population in Hardy-Weinberg equilibrium. The parasites in 2008 to 2012 and 2014 to 2018 showed great genetic differentiation (FST > 0.25) compared to parasites in 2003 to 2005 (Fig. 3B). The parasite population changed considerably since 2008, resulting in a less diverse and smaller population.
FIG 3.

Population genetic statistics. (A) Genetic diversity (expected heterozygosity) for each barcode locus (black dots) grouped by time period. Box plots show a decrease in the median (interquartile range) He after the first time period (2003 to 2005). Since differences in He values might be due to unequal sampling in districts over time, we assessed He within the San Juan Bautista district, which was sampled at all three time periods. This confirmed the decreasing diversity over time, with a pronounced decrease in He in 2014 to 2018 (P > 0.001) (see Fig. S5 and Table S10 in the supplemental material). (B) Genetic differentiation of the three populations measured as the FST (100) using the R package diveRsity. The numbers of individuals for each population were 118 in 2003 to 2005, 65 in 2008 to 2012, and 38 in 2014 to 2018. He values by time and district are plotted in Fig. S5, and the differentiation measures G′ST (101) and Jost’s D (102) are plotted in Fig. S6.
Use case for connectivity of parasite populations: barcode multilocus lineages and population structure.
Using the 28-SNP barcode, we identified 36 multilocus (ML) lineages with distinct barcodes and investigated their dynamics in the study population over time (Fig. 4) and districts (Table 2). The distinct lineages clarify the significant linkage disequilibrium (LD) in the barcode observed for all time periods and districts (Table S11). In 2003 to 2005, we observed 23 lineages, and we observed only 11 in 2008 to 2012, offering an explanation for the decrease in genetic diversity.
FIG 4.

Dynamics of 28-SNP-barcode multilocus lineages. An area plot shows the proportions of each ML lineage detected in the three different time periods in study samples (n = 254) from Peru. ML lineages were identified, clustered using the genetic distance between barcodes with the poppr package in R, and given an identification number. ML lineages differed by multiple SNPs (≥4 SNPs), enabling lineage classification even when barcodes had some missing or heterozygous genotypes. Lineages are characterized by unique barcodes and resistance genotypes and are listed in Table 2.
TABLE 2.
Barcode multilocus lineage characteristicsa
| Barcode multilocus lineage | Sequence | No. of samples |
Both HRP2 and HRP3 deleted (RDT failure) (no. of samples) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2003–2005 |
2008–2012 |
2014–2018 |
Variant(s) of interest for drug resistance (no. of samples) |
||||||||||||||
| Unknown district | Belen | San Juan Bautista | Punchana | San Juan Bautista | Mazan | San Juan Bautista | Pastaza | dhfr | dhps | crt | mdr1 | coronin | K13 | ubp1 category | |||
| 53 | GGTGTCTATCTCATCGCGGGGTTTCATC | 0 | 1 | 19 | 0 | 0 | 0 | 0 | 0 | Single mutant | wt | CVMNT | NDFCDD | V424I | K189T | Q107L and/or K1193T variant | No |
| 142 | GGTGTCTATTTCGGTTAGGAGTTTCATC | 0 | 0 | 19 | 0 | 0 | 0 | 0 | 0 | Single mutant | Single syn mutation at position 540 | CVMNT | NDFCDD | V424I | K189T | R1133S variant | No |
| 148 | AGTGTCCGTCCAATCGCGGGATTTTTTC | 0 | 0 | 16 | 0 | 0 | 0 | 0 | 0 | Double mutant | wt | SVMNT | NGFSDD | wt | wt | R1133S variant | No |
| 141 | GGCGCCCGTCCAATCGCCGGATTACTACC | 0 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | Double mutant | Double mutant | SVMNT | NDFCDY | wt | K189T | R1133S + E1011K variant | No (9)/yes (1) |
| 124 | GGTGCCCGTCCCATCGCCGGATTTTTTC | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | Double mutant | Double mutant/triple mutant | SVMNT | NDFCDY | V424I/wt | wt | Q107L and/or K1193T variant | No |
| 140 | GGTGTCTATCTCATCGCGGGACTTCATC | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | Single mutant | wt | CVMNT | NGFSDD | V424I | K189T | Q107L and/or K1193T variant | No |
| 146 | GGTXXCTATCTCATCGXGGGGCTTCATC | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | Single mutant | wt incomplete | CVMNT | Various | V424I | wt incomplete | Q107L and/or K1193T variant | No |
| 161 | GGTGCCCXTCCCATCGCCGGATTATTACC | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | Double mutant | Double mutant/triple mutant | SVMNT | NDFCDY | V424I/wt | K189T | No | |
| 62 | GGCGCCTATCTCATCGCGGGATTTTACC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Single mutant | wt | CVMNT | NDFCDY | V424I | K189T | No | |
| 63 | GGCGCCTATCTAATXGCCGGATTATNATC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Single mutant | Incomplete mutant | CVMNT | Various | V424I | K189T | Various | |
| 100 | GGXGXCTGXCCXGGXNCXGGATTANTANC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Single mutant | Incomplete mutant | CVMNT | NDFCDD | V424I | K189T | Various | No |
| 104 | GGXGXCTATCTXGGTTXXGAGTTCCACT | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Single mutant | wt | CVMNT | NDFCDD | V62M | K189T | R1133S variant | No |
| 119 | GGXXXCTXTNTXNTNGXXXXGTTXCXTC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Single mutant | NDFCDD | V424I | G449C | Incomplete only wt | No | ||
| 121 | GGXXXCCGTTTXATTGXXGGATTACCACC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Double mutant | wt | SVMNT | NGFSDD | V62M | K189T | R1133S variant | No |
| 131 | GAXAXTTAXCCXGGCTXXGAACTATCATT | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | wt | wt | CVMNT | NDFSDD | V62M | wt incomplete | R1133S + E1011K variant | No |
| 133 | GXXGXCTAXTTXATCGNXGGGTTTCATC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Single mutant | wt | CVMNT | NDFCDD | V424I | K189T | Incomplete only wt | No |
| 136 | GXXXXCTAXTTXGTTXXXGAGXTCCAXC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | wt incomplete | CVMNT | Incomplete | V62M | wt incomplete | Incomplete only wt | Yes | |
| 144 | XGTXCXTXTXXCXTCTXCXGATTAXCXTC | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Incomplete mutant | Double mutant | Incomplete | wt | wt incomplete | No | ||
| 162 | XACXCXTXTXTCXGTTXGXAGCTAXCXTT | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | Single mutant | Single syn mutation at position 540 | NG_SDD | V62M | No | |||
| 163 | XAXXXCNXTTXXNTTTXGXAGTTXTXTC | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | wt | wt incomplete | Various | V62M | wt incomplete | Incomplete only wt | No | |
| 157 | GGCGCCTATCCCATCGCCGGGTTACTACC | 0 | 0 | 29 | 0 | 2 | 0 | 0 | 0 | Single mutant | wt | SVMNT | NDFCDD | V424I | K189T | Q107L and/or K1193T variant | No |
| 46 | GGTGCCTATTTAGTTGAGGAGTTACCACC | 1 | 0 | 22 | 1 | 3 | 0 | 0 | 0 | Single mutant | Single mutant/wt | CVMNT | NGFSDD | V62M | K189T | Q107L and/or K1193T variant | Yes |
| 158 | GGCGXCCGTCCXATCGXCGXATTACTTTC | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | Double mutant/triple mutant | Double mutant | SVMNT | NDFCDY | V424I/wt | K189T | R1133S + E1011K variant | Yes (2)/no (1) |
| 155 | GGCGCCCATCTAATCGCCGGATTACTTTC | 0 | 0 | 0 | 24 | 0 | 11 | 21 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | V424I (and 1 wt) | K189T/wt (1) | R1133S + E1011K variant | Yes (51)/no (4) |
| 29 | NNCGCCCATCTAATCGCCGGATTACTTTC | 0 | 0 | 0 | 11 | 0 | 0 | 0 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | V424I | K189T/wt (1) | R1133S + E1011K variant | No |
| 95 | GGCGCCNATCTAATCGCCGGATTACTTTC | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | V424I | K189T | Yes | |
| 14 | AACGTCCGTCCAATCGCGGGATTACTTCC | 0 | 0 | 0 | 16 | 2 | 0 | 2 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | wt | wt/K189T (2) | R1133S + E1011K variant | No |
| 130 | GACGTCCATCCAATCGCGGGATTACTTTC | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | Triple mutant | Triple mutant | SVMNT | NDFCDY | V424I/wt | K189T/wt | R1133S + E1011K variant | No (2)/yes (1) |
| 7 | AACGTCCATCTAATCGCGGGATTACTTTC | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | V424I | wt/G449C | R1133S + E1011K variant | No |
| 57 | GGNGCCNATTTAATTGCGGAGTTACCACC | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | Single mutant | CVMNT/SVMNT | NGFSDD | V62M | K189T | Various | Yes | |
| 132 | GAXXXCCATCTXATCGCXGGATTACTTTC | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | V424I | K189T | No | |
| 149 | GGCGCCCGTCCAATTGCGGAGTTACCACC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | Triple mutant | wt | SVMNT | NDFCDY | wt | K189T | Q107L and/or K1193T variant | No |
| 109 | GGCGCCCGTCTAATTGAGGGATTACTTTC | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | wt | K189T (1)/wt (1) | R1133S + E1011K variant | No |
| 24 | AXXXXCCNXCCXATCNXXGGATTACTTCC | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | Triple mutant | Incomplete mutant | SVMNT | NDFCDY | V424I | wt incomplete | R1133S + E1011K variant | No |
| 58 | GGCGTCCATTTAATTGCCGGATTACTATC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | Triple mutant | Triple mutant | SVMNT | NDFCDY | wt | K189T | R1133S + E1011K variant | No |
| 59 | GGCGNCCNTCTAATXGCNGGATTACTTNC | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | Triple mutant | Triple mutant | SVMNT | NDFCDY | V424I | K189T | R1133S + E1011K variant | No |
ML lineages were identified, clustered using the genetic distance between barcodes with the poppr package in R, and given a number as an identifier. Heterozygote genotypes in the barcode were included, resulting in the creation of some polyclonal genotypes in barcodes, which included barcodes from other lineages. Lineages 29 and 95 are, in fact, the same as lineage 155, with additional minor clones. wt, wildtype (3D7) genotype; syn, synonymous.
One lineage (155) became predominant after 2008 in coexistence with other (related) lineages within district and period (Table 2 and Fig. S7). This explains the increasing number of barcode alleles that became fixed (2 alleles in 2003 to 2005, 8 in 2008 to 2012, and 14 in 2014 to 2018) (Table S12). The ML lineages in later periods (2008 to 2018) are distinct from those observed in 2003 to 2005 (Fig. S7), paralleling the observed FST.
With discriminant analysis of principal components (DAPC), which is similar to PCA but maximizes the differences between predefined populations, we increased the resolution to all biallelic variants (nloci = 772), and we were able to determine the underlying causes of the observed differentiation (Fig. 5). The highest-contributing alleles along the first axis (2003 to 2005 versus 2008 to 2018) were the drug resistance mutations dhfr C50R and N51I, mdr1 D1246Y, and dhps K540E (Table S13). Contributors to the differentiation along the second axis (2014 to 2018 and Pastaza) were seven barcode SNPs and dhfr, mdr1, coronin, and ubp1 variants.
FIG 5.
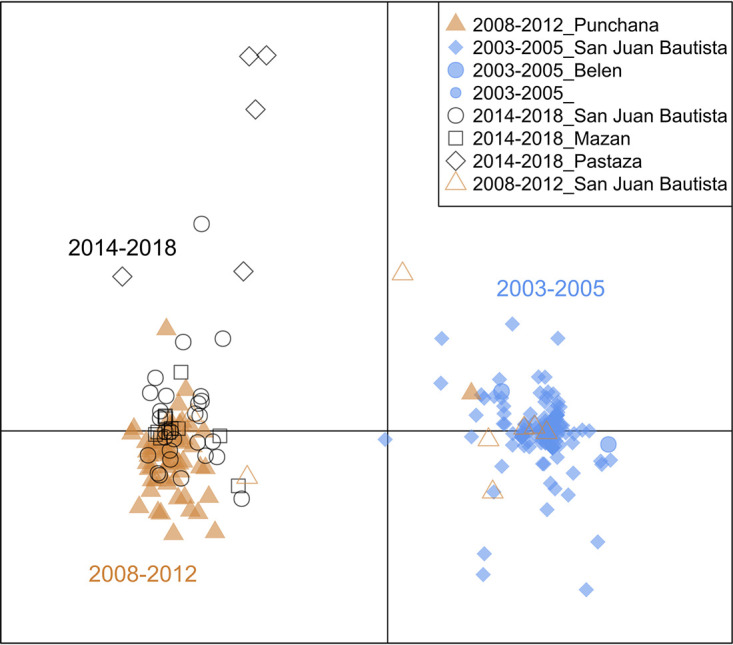
Discriminant analysis of principal components (DAPC). A scatterplot of discriminant analysis eigenvalues 1 and 2 using all biallelic SNPs in the core targeted region (excluding amplicons targeting repetitive regions and nonnuclear targets) shows the differentiation between Peruvian P. falciparum samples collected before 2008 and those collected afterward (x axis) (PC1) and a subsequent separation along the y axis (PC2) from 2014 to more recent years and along geographic distance. DAPC calculates the discriminant components using predefined populations in such a way that samples from the same population will be grouped, while it simultaneously maximizes the distance with samples from other populations. SNPs contributing most to the DAPC are listed in Table S13 in the supplemental material. DAPC was performed with 40 principal components and 7 discriminants as determined by cross-validation. The increased resolution of the DAPC allowed the detection of greater genetic variation in samples from 2014 to 2018 (black shapes) than with the lineage analysis.
Use case for drug resistance: artemisinin resistance-associated genes.
The full-length K13 gene was amplified with 12 overlapping amplicons. Although ART resistance-validated SNPs were detected in control samples, we did not observe these SNPs (F446I, N458Y, M476I, Y493H, R539T, I543T, P553L, R561H, P574L, and C580Y) in the study samples. We observed several low-frequency mutations in the K13 propellor region (V445A, I448K, G449C, V581I, and a premature stop codon at position 613) (Table 3), always as minor alleles in polyclonal infections. Outside the propeller region, the K189T mutation was observed at a high frequency (63% to 84%).
TABLE 3.
Haplotype frequencies in ART and partner drug resistance-associated genes observed in at least 1 sampleb
| Drug(s) and gene haplotype(s) | 2003–2005 |
2008–2012 |
2014–2018 |
χ2 P value | |||
|---|---|---|---|---|---|---|---|
| No. of samples with haplotype/total no. of samples | % of samples with haplotype (95% CI) | No. of samples with haplotype/total no. of samples | % of samples with haplotype (95% CI) | No. of samples with haplotype/total no. of samples | % of samples with haplotype (95% CI) | ||
| ART | |||||||
| K13 | |||||||
| G449Ca | 1/141 | 0.7 (0.1–4.9) | 0/68 | 0 | 1/45 | 2.2 (0.3–73.4) | 0.03621 |
| K189T | 94/141 | 66.7 (58.4–74.0) | 42/68 | 61.8 (49.6–73.6) | 25/45 | 55.6 (40.7–73.4) | |
| wt | 15/141 | 10.6 (6.5–17.0) | 16/68 | 23.5 (14.9–73.6) | 3/45 | 6.7 (2.1–73.4) | |
| wt incomplete | 29/141 | 20.6 (14.6–28.1) | 10/68 | 14.7 (8.0–73.6) | 14/45 | 31.1 (19.2–73.4) | |
| Missing | 2/141 | 1.4 (0.4–5.6) | 0/68 | 0 | 2/45 | 4.4 (1.1–73.4) | |
| ubp1 | |||||||
| Q107L and/or K1193T type | 60/141 | 42.6 (34.6–50.9) | 6/68 | 8.8 (4.0–18.4) | 3/45 | 6.7 (2.1–19.0) | <2.2e−16 |
| R1133S type | 21/141 | 14.9 (9.9–21.8) | 7/68 | 10.3 (4.9–20.2) | 1/45 | 2.2 (0.3–14.6) | |
| R1133S + E1011K type | 33/141 | 23.4 (17.1–31.2) | 53/68 | 77.9 (66.4–86.3) | 41/45 | 91.1 (78.3–96.7) | |
| Polyclonal Q107L/K1193T+ R1133S | 3/141 | 2.1 (0.7–6.4) | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | |
| wt or wt incomplete | 24/141 | 17.0 (11.6–24.2) | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | |
| coronin | |||||||
| V424I | 78/141 | 55.3 (47.0–63.4) | 41/68 | 60.3 (48.2–71.3) | 37/45 | 82.2 (68.0–91.0) | 0.002743 |
| V62M | 29/141 | 20.6 (14.6–28.1) | 5/68 | 7.4 (3.1–16.6) | 0/45 | 0 | |
| wt | 30/141 | 21.3 (15.3–28.9) | 20/68 | 29.4 (19.7–41.4) | 8/45 | 17.8 (9.0–32.0) | |
| Polyclonal | 0/141 | 0 | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | |
| Incomplete | 4/141 | 2.8 (1.1–7.4) | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | |
| SP | |||||||
| dhfr | |||||||
| Single mutant (S108N) | 98/141 | 69.5 (61.4–76.6) | 6/68 | 8.8 (4.0–18.4) | 0/45 | 0 | <2.2e−16 |
| Double mutant + BR + I164L | 32/141 | 22.7 (16.5–30.4) | 0/68 | 0 | 0/45 | 0 | |
| Triple mutant | 0/141 | 0 | 61/68 | 89.7 (79.8–95.1) | 44/45 | 97.8 (85.4–99.7) | |
| Incomplete | 3/141 | 2.1 (0.7–6.4) | 0/68 | 0 | 1/45 | 2.2 (0.3–14.6) | |
| Missing | 6/141 | 4.3 (1.9–9.2) | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | |
| dhps | |||||||
| Single mutant K540K | 16/141 | 11.3 (7.0–17.8) | 0/68 | 0 | 0/45 | 0 | <2.2e−16 |
| Single mutant A437G | 3/141 | 2.1 (0.7–6.4) | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | |
| Double mutant | 14/141 | 9.9 (5.9–16.1) | 0/68 | 0 | 0/45 | 0 | |
| Triple mutant | 2/141 | 1.4 (0.4–5.6) | 48/68 | 70.6 (58.6–80.3) | 30/45 | 66.7 (51.6–79.0) | |
| Incomplete mutant | 7/141 | 5.0 (2.4–10.1) | 13/68 | 19.1 (11.4–30.4) | 12/45 | 26.7 (15.7–41.6) | |
| wt | 80/141 | 56.7 (48.4–64.7) | 4/68 | 5.9 (2.2–14.8) | 2/45 | 4.4 (1.1–16.4) | |
| wt incomplete | 19/141 | 13.5 (8.7–20.2) | 2/68 | 2.9 (0.7–11.2) | 1/45 | 2.2 (0.3–14.6) | |
| CQ and MQ | |||||||
| crt (positions 72–76) | |||||||
| CVMNT | 65/141 | 46.1 (38.0–54.4) | 2/68 | 2.9 (0.7–11.2) | 0/45 | 0 | 8.37e−16 |
| SVMNT | 49/141 | 34.8 (27.3–43.0) | 60/68 | 88.2 (78.0–94.1) | 32/45 | 71.1 (56.1–82.6) | |
| CVMNT/SVMNT polyclonal | 0/141 | 0 | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | |
| Incomplete | 27/141 | 19.1 (13.4–26.6) | 5/68 | 7.4 (3.1–16.6) | 13/45 | 28.9 (17.4 to 43.9) | |
| mdr1 (positions 86, 144, 184, 1034, 1042, 1246) | |||||||
| NDFCDD | 61/141 | 43.3 (35.3–51.6) | 1/68 | 1.5 (0.2–9.9) | 0/45 | 0 | <2.2e−16 |
| ND_CDD | 2/141 | 1.4 (0.4–5.6) | 0/68 | 0 | 0/45 | 0 | |
| NDFCDY | 17/141 | 12.1 (7.6–18.6) | 60/68 | 88.2 (78.0–94.1) | 42/45 | 93.3 (81.0–97.9) | |
| NGFSDD | 43/141 | 30.5 (23.4–38.6) | 5/68 | 7.4 (3.1–16.6) | 0/45 | 0 | |
| NG_SDD | 4/141 | 2.8 (1.1–7.4) | 0/68 | 0 | 0/45 | 0 | |
| NDFSDD | 1/141 | 0.7 (0.1–4.9) | 0/68 | 0 | 0/45 | 0 | |
| Polyclonal | 5/141 | 3.5 (1.5–8.3) | 0/68 | 0 | 0/45 | 0 | |
| Incomplete | 8/141 | 5.7 (2.8–11.0) | 2/68 | 2.9 (0.7–11.2) | 3/45 | 6.7 (2.1–19.0) | |
One out of 2 samples with G449C was also accompanied by K189T; both cases were polyclonal infections.
Note that the ubp1 haplotype with R1133S and E1011K was frequently observed with additional SNP mutations (K764N, K774N, and D777G) and a large insert (EQKY) between amino acid positions 2826 and 2827. dhps double mutant indicates the A437G A581G mutant; dhps triple mutant indicates the A437G A581G K540E mutant. wt, wild type; CI, confidence interval.
Forty-four amplicons covered the ubp1 gene (91.8% of the gene), including all variants of interest. Previously reported variants (including R3138H) were not detected, but we observed a change in the predominant ubp1 haplotype. In 2003 to 2005, a haplotype of Q107L, commonly with K1193T, was most frequently observed (42.6%) (Table 3). In later years, R1133S haplotypes (often linked with E1011K) replaced the Q107L haplotypes.
Eleven amplicons amplified the full-length coronin gene. We identified two novel mutations, V62M and V424I, and none of the in vitro-associated variants (60). V62M was seen at 20.6% in 2003 to 2005 but decreased over time (Table 3). V424I increased from 55.4% in 2003 to 2005 to 80.9% in 2014 to 2018 (Table 3). While V62M is located in the WD-40 beta-propeller domain (containing the resistance-associated mutations G50E, R100K, and E107V), V424I is located just outside this region and is not expected to contribute to ACT resistance.
Use case for drug resistance: CQ and ACT partner drug resistance-associated genes.
The SP resistance-associated genes were each targeted with 9 amplicons, covering 100% of the dhfr gene and 87% of dhps, including all variants of interest. For 2003 to 2005, we observed predominantly single (69.5%) and double (22.7%) dhfr mutants and wild-type (wt) dhps (≥56.7%) (Table 3). Although SP use was discontinued in 1999, after 2008, SP resistance increased, with a high frequency of triple mutants in dhfr and dhps. Consistent with previous reports (45, 61), we did not detect dhfr C59R in Peru. Therefore, the “sextuple” mutant here is different from the African sextuple mutant, with C59R instead of C50R, but with a similar superresistance phenotype (62).
Sixteen amplicons covered the crt gene (91.1%), missing one variant of interest (I218F) associated with piperaquine (PPQ) resistance. The CQ resistance (CQR) crt haplotypes CVMNT (CQR) and SVMNT (highly CQR) increased over time (Table 3), with 100% SVMNT in 2014 to 2018. Mutation I356V, whose contribution to CQR is unclear, was frequent (>93%) in all time periods.
Eighteen amplicons covered the mdr1 gene (87%), targeting all variants of interest associated with CQ and mefloquine (MQ) resistance (7). While the mdr1 haplotypes NDFCDD and NGFSDD were predominant in 2003 to 2005, in later years, mainly the NDFCDY haplotype was found (Table 3). There are conflicting results on the association of NDFCDY with susceptibility to MQ, artesunate (AS), and CQ (61, 63). The mdr1 D144G SNP has so far been reported only in Peru (annotated as D142G previously [61]) and has been decreasing in recent years.
Copy number variations (CNVs) in the mdr1 and plasmepsin II genes associated with PPQ (64) and MQ (65) resistance, respectively, were not previously observed in Peru (66). We confirmed single copies of both genes by quantitative PCR (qPCR) in a subset of 78 samples (Fig. S8). While both genes were sequenced in our assay, the lack of increased copy numbers resulted in insufficient data to validate CNV detection by Pf AmpliSeq.
We detected 10.6% S160N mutants in 2003 to 2005 in the ap2-mu gene (9 amplicons covering 99.1% of the gene), which decreased in later periods (1.5% and 2.2%). No variants of interest were observed in the pfmrp1 (95.1% covered), P. falciparum exonuclease (98.6% covered), cytochrome b (100% covered), and 23S rRNA (100% covered) genes.
Use case for drug resistance gene flow: drug resistance and parasite lineage evolution.
Between 2003 and 2005, we observed lineages with predominantly single and double dhfr mutants, wt dhps, CVMNT and SVMNT crt haplotypes, multiple mdr1 haplotypes, and the presence of hrp2 and hrp3 deletions (Table 2). From 2008, the majority of parasite lineages harbored sextuple dhfr/dhps mutants, crt SVMNT, mdr1 NDFCDY, and hrp2 and/or hrp3 deletions. Parasites with this dhps/dhfr/crt/mdr1 resistance haplotype and both hrp2 and hrp3 deleted were first reported in a few cases in Peru in 2006 (61) and were called the BV1 lineage. Here, we observed many parasites with the same “BV1-type” resistance haplotype although not always with hrp2 and hrp3 deletions. With the higher resolution of the Pf AmpliSeq assay, we observed many related lineages with this resistance haplotype that differed in their barcode genotypes, the presence of hrp2 and hrp3 deletions, and coronin SNPs (Table 2). This indicates gene exchange between lineages, in contrast to the idea of isolated clonal lineages in Peru.
The BV1-type lineages (lineage 155 and others) seen after 2008 are most closely related to earlier lineages 158, 141, and 63 in our study samples (Fig. S7). However, the triple dhfr mutants in the BV1 lineage are not accompanied by secondary mutations (I164L and the “Bolivian repeat” [BR], a silent 5-amino-acid insertion before codon 30), indicating that they did not emerge from the dhfr double mutant lineages circulating in earlier years. In conclusion, the combination of different markers in the Pf AmpliSeq assay allowed the identification of multiple recombining P. falciparum lineages in Peru that share increasing SP and CQ resistance, as well as RDT failure, in more recent years.
DISCUSSION
We designed a new highly multiplexed sequencing assay (Pf AmpliSeq) targeting 13 antimalarial resistance genes, combined with a country-specific 28-SNP barcode for population genetic analysis in Peru and hrp2 and hrp3 genes to detect deletions that cause false-negative RDT results. The novelty and strength of this assay are in the many different types of markers multiplexed in one easy workflow, making it suitable for many surveillance use cases. Compared to other amplicon sequencing assays (30, 34, 67) and genetic surveillance platforms (42), the Pf AmpliSeq assay includes the highest number of drug resistance-associated genes combined with an in-country SNP barcode as well as diagnostic resistance markers.
In many remote areas, sample collection for malaria molecular surveillance is possible with DBSs only, which are easily transported at room temperature to a central laboratory for further processing and ideally stored at −20°C to maintain DNA integrity. This assay performs well on DBS samples with parasite densities of ≥60 p/μL and allows high-accuracy sequencing at a higher depth of coverage (100- to 1,000-fold versus 50- to 100-fold) and a lower cost (see Table S14 in the supplemental material) than WGS. At densities of <60 p/μL, sWGA prior to the Pf AmpliSeq assay increases the number of reads but also increases the error rate and cost. In addition, the Pf AmpliSeq assay is less sensitive to DNA degradation than methods targeting long amplicons.
Initial raw data processing can be performed automatically with the software of the sequencer using the manifest file available at bio-protocol. However, here, we used a Linux-based pipeline that gave us more flexibility and insight during the validation process. Subsequent variant analysis can be performed with minimal bioinformatic skills using any software that can analyze tabulated data. An automated analysis environment would be of great benefit for the rapid output of genetic reports and easy interpretation, increasing the actionable use of data to enable programmatic decision-making.
Only nonvalidated K13 SNPs were detected at low frequencies in the Peruvian samples, suggesting that ART resistance has not reached Peru, at least in recent years (2003 to 2018). Mutations in other genes in the endocytosis pathway have been associated with decreased sensitivity to ART in in vitro studies, and changes in ubp1 haplotypes paralleled the emergence of K13 markers in Thailand (60, 68–76). However, the observed ubp1 and coronin mutations here are uncharacterized. Genetic variants in KIC7, Eps15, and Formin2 associated with in vitro ART resistance (70–72, 74, 77) were not yet reported when the assay design was being completed. These genes will be included in future versions of the assay.
One main advantage of the Pf AmpliSeq assay is that the panel of targeted amplicons can be easily adapted either to update the panel with newly identified drug resistance-associated genes or other makers of interest or to adapt the assay to a different country or region by exchanging the SNP barcode for in-country resolution. The assay in its current form is limited to South America.
This is the first proof of principle of an NGS amplicon assay targeting hrp2 and hrp3 deletions. From the analyzed study samples, 56.3% had both hrp2 and hrp3 deleted, but this might be an underestimation since infections containing polyclonal strains with and without deletions will be classified as RDT detectable. For hrp2, 22% of samples could not be classified due to conflicting results between hrp2 amplicons. The difference in read depths among hrp2 amplicons suggests that only part of the gene is deleted, with part of the first exon still being present. As was recently done in Ethiopia (78), a better characterization of the genomic structure of hrp2 deletions in Peru will improve amplicon design. In addition, copy number calling tools could be explored for more systematic classifications of hrp2 and hrp3 deletions.
With the combination of phenotypic markers and the 28-SNP barcode in the Pf AmpliSeq assay, we were able to detect changing allele frequencies in resistance-associated genes, but we were also able to identify temporal genetic differentiation in the parasite population, alongside a decrease in genetic diversity. The barcode served to distinguish different lineages in multiple resistance haplotypes (in dhfr, dhps, crt and mdr1) circulating in recent years and enabled the investigation of drug resistance evolution.
SP and CQ resistance in South America was reportedly spread from a single origin in the lower Amazon (45), with five distinct clonal lineages circulating in Peru before 2006 (19, 61, 79), followed by frequent outcrossing and lineage evolution (79). With the Pf AmpliSeq assay, we observe the same SP/CQ-resistant lineages and their evolution over time, with increased resolution and additional variants detected in other genes.
After 2008, the parasite population in Peru was increasingly dominated by BV1-type lineages, which are highly SP/CQ resistant and can frequently escape HRP2-based RDTs (HRP2-based RDTs are not commonly used in Peru). The BV1-type lineages seem to be sensitive to ART-MQ (absence of K13 variants and the mdr1 CNV), while the high level of CQR (in crt and mdr1) could be the result of CQ treatment of coendemic P. vivax cases. Coinfections with very low P. falciparum densities (80) are frequently misdiagnosed and treated as P. vivax monoinfection (79). When the BV1-type lineages first appeared (2011), we observed strong population differentiation and the loss of secondary dhfr mutations, characteristic of Peruvian and Bolivian SP-resistant parasites (45), which supports suggestions from others that this lineage was introduced from other parts of the Amazon (81, 82), possibly Colombia (20, 21) or Suriname (49). Analysis of samples from neighboring countries could shed light on the evolution and spread of resistance and hrp2 and hrp3 deletions in the region; however, genomes from South America are heavily underrepresented in publicly available genomic databases.
The COI results varied depending on the analysis method used, and it was difficult to determine which method approached the true COI. The variant calling tool used here assumes a diploid genotype with balanced within-sample allele frequencies, which is frequently not the case in malaria complex infections, and this can complicate COI analyses. Alternative haplotype-based analysis approaches (e.g., SeekDeep [83], HaplotypR [57], and DADA2 [84]) might have a greater ability to unravel complex infections. These tools use a different error-handling approach and thereby can resolve more fine-scale variation and are more suitable for high-resolution relatedness analysis (in combination with identity by descent [IBD] [85]) but are less suitable for confidently calling drug-resistant variants. However, applying these methods to the Pf AmpliSeq data is complicated by the overlapping amplicons in drug resistance genes and requires further validation. While it is possible to perform IBD analysis with the AmpliSeq data, in this study with sampling by convenience, it was deemed less appropriate.
Half of the SNPs in the barcode became fixed in the most recent years, reflecting a true decrease in diversity (as the sample size was sufficient) with closely related clonal lineages. Although our study sample size is comparable to those of previous reports from Peru (e.g., 220 samples in 1999 [79], 104 samples from 1999, and 62 samples from 2006 to 2007 [61]), it represents a limited proportion of the Peruvian P. falciparum population. In the future, a more systematic sampling approach, ideally performed at regular times and covering all regions where malaria is endemic, would increase genetic surveillance intelligence to inform malaria control and elimination strategies in Peru.
Concluding remarks.
We have demonstrated the validity of the Pf AmpliSeq assay for several use cases in malaria molecular surveillance in Peru. With this tool, we have identified P. falciparum lineages with an increasing accumulation of drug and diagnostic resistance-associated mutations that can become a serious threat to P. falciparum elimination in the Amazon region. The Pf AmpliSeq assay also has the potential to perform well to characterize malaria outbreaks in Peru (81, 86) and predict the origin of imported infections (40, 41, 87).
In Peru, we have established a molecular surveillance network (GENMAL) with the aims of sharing tools, samples, and data; coordinating malaria research efforts; and stimulating dialogue among malaria stakeholders in the region. The network promotes the implementation and use of NGS tools to inform decision-making for malaria elimination and is a platform to support the efficient translation of results into policy and practice. The Pf AmpliSeq assay can be adapted to serve malaria molecular surveillance in the Amazon region and South America, filling the void of genetic data from this region.
MATERIALS AND METHODS
Study sites, samples, and controls.
P. falciparum qPCR-positive samples (n = 312) from previous studies were selected based on geographical representation (Fig. 2) and parasite density (≥100 p/μL by qPCR). Samples from 2003 to 2017 (n = 269) were from published (18, 88) and ongoing studies led by the Universidad Peruana Cayetano Heredia (UPCH) and U.S. Naval Medical Research Unit 6 (NAMRU-6) (n = 63). Samples from 2018 (n = 5) were P. falciparum-positive samples collected in Loreto in a collaboration between the Zero Malaria Programme and UPCH.
DNA from all samples was newly extracted from DBSs using the E.Z.N.A. blood DNA minikit (Omega Bio-Tek, GA, USA) according to the manufacturer’s instructions. Per sample, two pieces of ~0.5 cm2 were used, and DNA was eluted in a final volume of 100 μL. P. falciparum parasitemia was quantified by qPCR (89). The median parasite density was lower in 2014 to 2018 than in earlier periods (geometric mean density of 2,953 [adjusted P value {Padj} of 0.0087 by a pairwise t test with Benjamini-Hochberg corrections for multiple testing {90}]).
The studies were approved by local ethical review boards. Protocols were registered in the Decentralized System of Information and Follow-Up to Research (SIDISI) (numbers 52707, 61703, 101645, 66235, and 102725), and one clinical trial was registered at ClinicalTrials.gov (identifier NCT00373607). The NAMRU-6 study was approved by its Institutional Review Board in compliance with all applicable federal regulations governing the protection of human subjects (protocol NMRCD.2007.0004). Individuals were included in this study only if signed informed consent included a future-use clause, and secondary use was approved by the Institutional Review Board of the Institute of Tropical Medicine Antwerp (reference number 1417/20).
P. falciparum laboratory isolates from cultures (91) and uninfected human blood spotted onto DBS cards were included as controls (details are listed in Table S15 in the supplemental material). Control samples (n = 6) with known drug resistance-associated variants from a previous study in Vietnam (92) and genotyped using the SpotMalaria pipeline and WGS were also included. DNA from these controls (100 μL or 3 DBS punches) was extracted using a QIAamp DNA minikit (Qiagen) according to the manufacturer’s instructions. sWGA was performed as previously described (93). Genotypes of the laboratory strains were reported previously (66, 94–96).
SNP barcode design.
A barcode of 28 biallelic SNPs was designed as described in detail in the supplemental material. Briefly, MalariaGEN Plasmodium falciparum Community Project data (97) were used to select SNPs (0.35 ≤ MAF ≤ 0.5) contributing to between-country differentiation using DAPC (98). Two SNPs per chromosome were selected with priority for synonymous SNPs with low pairwise LD.
Pf AmpliSeq workflow and sequencing.
Library preparation was performed using the AmpliSeq library plus for Illumina kit (Illumina), AmpliSeq custom panels (Data Set S2), and AmpliSeq CD Indexes (Illumina) according to the manufacturer’s instructions. The DNA concentrations for the controls and 25 samples were determined using the Qubit v3 high-sensitivity DNA kit (Invitrogen). Controls were diluted to 1 ng input DNA to mimic DBS samples. Study samples (mean DNA concentration of 6.1 ± 0.3 ng/μL) were not diluted prior to library preparation. For each sample, a single library was prepared, except for a subset of samples to determine reproducibility. Target regions were amplified from 7 μL DNA (1 to 150 ng) with adjusted cycling conditions (1 cycle of 99°C for 2 min, 21 cycles of 99°C for 15 s, and 1 cycle of 60°C for 8 min) in two reactions and subsequently combined for final library preparation according to the manufacturer’s guidelines. Sample libraries were quantified using the Kapa library quantification kit for Illumina platforms (Kapa Biosystems), diluted to 2 nM with low-Tris-EDTA buffer, and pooled to generate a final pool containing 2 nM each library. The denatured library pool (diluted to 18 pM) was loaded onto a MiSeq system (Illumina) for 2 × 300-bp paired-end sequencing (MiSeq reagent kit v3; Illumina). A detailed protocol is available at https://doi.org/10.21769/BioProtoc.4621. A 20% PhiX spike-in (Illumina) was used in one sequencing run to determine the effect on the sequencing quality and the trade-off in coverage. The generated demultiplexed FASTQ files were processed with an in-house analysis pipeline on a Unix operating system computer as described in more detail in the supplemental material.
For the final analysis, we selected 254/312 (81%) samples with good-quality data (<50% missing genotype calls and mean coverage of >15) and retained only one library of replicates (with the lowest missingness). A MAF of 0.01 with a precision of 0.02 (2%) can be detected with sample size of 96 samples, while a MAF of 0.01 with 1% precision can be detected (as determined with https://epitools.ausvet.com.au/oneproportion).
Detection of mdr1 and pm2 copy number variations by PCR.
CNVs in pm2 and mdr1 were determined for a subset of samples (n = 78) selected randomly from all time periods and districts using qPCR (92). Samples and controls were tested in triplicate, with 20% of the samples being retested for reproducibility and 100% of the results being in agreement.
Analysis of the complexity of infection.
The COI was estimated using Real McCOIL categorical and proportional methods (99) in R using the following subsets of variants: (i) all biallelic SNPs, (ii) 25 barcode SNPs, and (iii) “core variants,” biallelic variants excluding repetitive regions (i.e., hrp2, hrp3, and MSs) and mitochondrial and apicoplast regions. Default settings were used for a fixed error; for the categorical method, an upper bound of 10 and an initial COI of 5 were used, and for the proportional method, an upper bound of 15 and an initial COI of 7 were used. We allowed 50% missingness for biallelic SNPs, 20% for the barcode, and 40% for core variants.
We also estimated the proportions of single- and multiple-clone infections based on the number of heterozygous genotypes in (i) the 28-SNP barcode, (ii) biallelic SNPs in ama1, and (iii) core variants. Samples with ≥1 heterozygous genotype were considered to contain multiple clones (COI ≥ 2). Finally, we estimated the COI from the MS-amplified regions. As the different methods generated divergent results, the mode (most frequent value for single versus multiple clones) was determined as the final measure for each sample.
Validation of detection of hrp2 and hrp3 deletions.
The performance of Pf AmpliSeq for hrp2 and hrp3 deletion detection was assessed using previous PCR-based hrp2 and hrp3 genotypes of 10 included samples (31). The presence or absence of both genes was determined using the mean read depth of hrp2 and hrp3 amplicons, as explained in more detail in the supplemental material. Due to the repetitive nature and homologies between the hrp2 and hrp3 genes, we used a conservative cutoff value, resulting in a “gray zone” where deletion/presence was left inconclusive when the majority of amplicons were not in accordance (Fig. S9). A final variable for RDT failure (absence of both hrp2 and hrp3) versus RDT detectable (presence of hrp2 and/or hrp3) was created, allowing the classification of samples that were inconclusive for one of the two genes if the other gene was present.
Population genetic analysis.
Population genetic analyses were performed using the 28-SNP barcode, excluding samples missing >7 SNPs (25%), unless specified otherwise. Samples with heterozygous genotypes were included, justified by the little genetic differentiation (FST = 0.05) between samples with single-clone barcodes and samples with heterozygous barcode genotypes. Genetic differentiation was measured as FST (100), G′ST (101), and Jost’s D (102) using 1,000 bootstraps with the R package diveRsity (103). PCA was performed on the genotype matrix of within-sample allele frequencies using the prcomp function (R stats package v4.0.5) using core variants. Prior to PCA, missing genotypes were replaced by the mean allelic frequency at a locus in all samples. The expected heterozygosity (He) was calculated (adegenet package [104, 105] in R) using diploid barcode genotypes. A Wilcoxon signed-rank test with continuity correction was used to compare the mean observed heterozygosity (Hobs) to the He. LD was measured as the standardized index of association () in a multilocus analysis using 999 resamplings (method, permutation of alleles), using the poppr package v2.8.6. (106) in R. ML lineages were defined by grouping isolates with similar barcodes using the Hamming distance and clustered based on the maximum distance (farthest neighbor) using poppr. DAPC (98) was performed with cross-validation, and the associated allele loadings for the first four components were determined using the adegenet package in R.
Analysis of genetic variants.
Barcode allele frequencies were calculated from allele depths to reflect population allele frequencies in complex infections, as described in the supplemental material. Haplotypes were created by combining genotypes of variants of interest (listed in Data Set S1). Frequency tables with confidence intervals were created with the freqtables R package.
Data availability.
Sample metadata, drug resistance and hrp2 and hrp3 haplotypes, barcodes, lineages, locations, and dates, etc., are accessible at https://microreact.org/project/aV7RHNCBmG3sJ2rwzchE4k-peru-2003-2018-molecular-surveillance-with-p-falciparum-ampliseq-assay. Raw data (FASTQ files) are available at the SRA under BioProject accession number PRJNA855317, and individual library accession numbers are listed in the supplemental metadata file at Microreact (see the URL mentioned above). A detailed protocol of the library preparation procedures has been deposited at https://doi.org/10.21769/BioProtoc.4621. It is anticipated that this link will be active by 5 March 2023; until that time, all the data will be available from the corresponding author upon request. Variant files (vcf) and scripts are available upon request. All other data are included in the manuscript and supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank all clinical, microscopy, and field staff who supported the sample collections and all participants for making their material available for malaria studies. We acknowledge the support of the Dirección Regional de Salud de Loreto for the study authorization and sample collection activities in Loreto. The following reagents were obtained through BEI Resources, NIAID, NIH: P. falciparum strain Dd2 (catalogue number MRA-150), contributed by David Walliker; P. falciparum strain Dd2_R539T (catalogue number MRA-1255) and P. falciparum strain CamWT_C580Y (catalogue number MRA-1251), contributed by David A. Fidock; and P. falciparum strain IPC 4912 (catalogue number MRA-1241), contributed by Didier Ménard.
This work was funded by the Belgium Development Cooperation (DGD) under the Framework Agreement Program between the DGD and ITM (FA4 Peru, 2017–2021), and the sample collections in 2018 were supported by VLIR-UOS (project PE2018TEA470A102; University of Antwerp). Funding for the sample collections led by U.S. Naval Medical Research Unit 6 (NAMRU-6) in 2011 and 2012 was provided by the Armed Forces Health Surveillance Division (AFHSD) and its Global Emerging Infections Surveillance and Response (GEIS) section (P0144_20_N6_01, 2020–2021). The funding agencies had no role in the design of the study.
We declare that we have no competing interests.
Some authors of the manuscript are military service members and employees of the U.S. Government. This work was prepared as part of their official duties. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the U.S. Government.
Conception of ideas for the study, A.R.-U., D.G., and J.H.K.; selection of targets and design of the assay, E.R.-V., J.H.K., and N.J.V.D.; sample collections and coordination of field work, C.D.-R., C.F.-M., D.G., H.O.V., and J.-P.V.G.; laboratory experiments, C.F.-M., J.H.K., N.J.V.D., and P.G.; bioinformatics and data analysis, J.H.K., L.L.A., N.J.V.D., and P.M.; writing the first draft of the manuscript, A.R.-U., J.H.K., C.F.-M., and N.J.V.D. All authors reviewed and contributed to the final version of the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Johanna Helena Kattenberg, Email: ekattenberg@itg.be.
Anna Rosanas-Urgell, Email: arosanas@itg.be.
Gemma Moncunill, Barcelona Centre for International Health Research (CRESIB, Hospital Clínic-Universitat de Barcelona).
REFERENCES
- 1.World Health Organization. 2019. World malaria report 2019. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Malaria Policy Advisory Committee. 2019. Technical consultation on the role of parasite and anopheline genetics in malaria surveillance. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Dalmat R, Naughton B, Kwan-Gett TS, Slyker J, Stuckey EM. 2019. Use cases for genetic epidemiology in malaria elimination. Malar J 18:163. doi: 10.1186/s12936-019-2784-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. 2018. Malaria surveillance, monitoring & evaluation: a reference manual. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 5.World Health Organization. 2009. Methods for surveillance of antimalarial drug efficacy. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 6.World Health Organization. 2016. Minutes of the Evidence Review Group meeting on the emergence and spread of multidrug-resistant Plasmodium falciparum lineages in the Greater Mekong subregion. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 7.World Health Organization. 2020. Report on antimalarial drug efficacy, resistance and response: 10 years of surveillance (2010-2019). World Health Organization, Geneva, Switzerland. [Google Scholar]
- 8.Wangdi K, Pasaribu AP, Clements ACA. 2021. Addressing hard-to-reach populations for achieving malaria elimination in the Asia Pacific Malaria Elimination Network countries. Asia Pac Policy Stud 8:176–188. doi: 10.1002/app5.315. [DOI] [Google Scholar]
- 9.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Menard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Menard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, Rahim-Awab G, Barnadas C, Berry A, Boum Y, Bustos MD, Cao J, Chen J-H, Collet L, Cui L, Thakur G-D, Dieye A, Djalle D, Dorkenoo MA, Eboumbou-Moukoko CE, Espino F-E-CJ, Fandeur T, Ferreira-da-Cruz M-F, Fola AA, Fuehrer H-P, Hassan AM, Herrera S, Hongvanthong B, Houze S, Ibrahim ML, Jahirul-Karim M, Jiang L, Kano S, Ali-Khan W, Khanthavong M, Kremsner PG, Lacerda M, Leang R, Leelawong M, Li M, Lin K, Mazarati J-B, Menard S, Morlais I, Muhindo-Mavoko H, Musset L, Na-Bangchang K, Nambozi M, Niare K, Noedl H, et al. 2016. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med 374:2453–2464. doi: 10.1056/NEJMoa1513137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium . 2008. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med 359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 12.Woodrow CJ, White NJ. 2017. The clinical impact of artemisinin resistance in Southeast Asia and the potential for future spread. FEMS Microbiol Rev 41:34–48. doi: 10.1093/femsre/fuw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balikagala B, Fukuda N, Ikeda M, Katuro OT, Tachibana S-I, Yamauchi M, Opio W, Emoto S, Anywar DA, Kimura E, Palacpac NMQ, Odongo-Aginya EI, Ogwang M, Horii T, Mita T. 2021. Evidence of artemisinin-resistant malaria in Africa. N Engl J Med 385:1163–1171. doi: 10.1056/NEJMoa2101746. [DOI] [PubMed] [Google Scholar]
- 14.WHO Global Malaria Programme. 2019. WHO Malaria Policy Advisory Committee (MPAC) meeting—meeting report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 15.Mita T, Tanabe K, Kita K. 2009. Spread and evolution of Plasmodium falciparum drug resistance. Parasitol Int 58:201–209. doi: 10.1016/j.parint.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Auburn S, Barry AE. 2017. Dissecting malaria biology and epidemiology using population genetics and genomics. Int J Parasitol 47:77–85. doi: 10.1016/j.ijpara.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 17.Escalante AA, Ferreira MU, Vinetz JM, Volkman SK, Cui L, Gamboa D, Krogstad DJ, Barry AE, Carlton JM, van Eijk AM, Pradhan K, Mueller I, Greenhouse B, Pacheco MA, Vallejo AF, Herrera S, Felger I. 2015. Malaria molecular epidemiology: lessons from the International Centers of Excellence for Malaria Research Network. Am J Trop Med Hyg 93:79–86. doi: 10.4269/ajtmh.15-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gamboa D, Ho M-F, Bendezu J, Torres K, Chiodini PL, Barnwell JW, Incardona S, Perkins M, Bell D, McCarthy J, Cheng Q. 2010. A large proportion of P. falciparum isolates in the Amazon region of Peru lack pfhrp2 and pfhrp3: implications for malaria rapid diagnostic tests. PLoS One 5:e8091. doi: 10.1371/journal.pone.0008091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akinyi S, Hayden T, Gamboa D, Torres K, Bendezu J, Abdallah JF, Griffing SM, Quezada WM, Arrospide N, De Oliveira AM, Lucas C, Magill AJ, Bacon DJ, Barnwell JW, Udhayakumar V. 2013. Multiple genetic origins of histidine-rich protein 2 gene deletion in Plasmodium falciparum parasites from Peru. Sci Rep 3:2797. doi: 10.1038/srep02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murillo Solano C, Akinyi Okoth S, Abdallah JF, Pava Z, Dorado E, Incardona S, Huber CS, Macedo de Oliveira A, Bell D, Udhayakumar V, Barnwell JW. 2015. Deletion of Plasmodium falciparum histidine-rich protein 2 (pfhrp2) and histidine-rich protein 3 (pfhrp3) genes in Colombian parasites. PLoS One 10:e0131576. doi: 10.1371/journal.pone.0131576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorado EJ, Okoth SA, Montenegro LM, Diaz G, Barnwell JW, Udhayakumar V, Murillo Solano C. 2016. Genetic characterisation of Plasmodium falciparum isolates with deletion of the pfhrp2 and/or pfhrp3 genes in Colombia: the Amazon region, a challenge for malaria diagnosis and control. PLoS One 11:e0163137. doi: 10.1371/journal.pone.0163137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rachid Viana GM, Akinyi Okoth S, Silva-Flannery L, Lima Barbosa DR, Macedo de Oliveira A, Goldman IF, Morton LC, Huber C, Anez A, Dantas Machado RL, Aranha Camargo LM, Costa Negreiros do Valle S, Marins Póvoa M, Udhayakumar V, Barnwell JW. 2017. Histidine-rich protein 2 (pfhrp2) and pfhrp3 gene deletions in Plasmodium falciparum isolates from select sites in Brazil and Bolivia. PLoS One 12:e0171150. doi: 10.1371/journal.pone.0171150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agaba BB, Yeka A, Nsobya S, Arinaitwe E, Nankabirwa J, Opigo J, Mbaka P, Lim CS, Kalyango JN, Karamagi C, Kamya MR. 2019. Systematic review of the status of pfhrp2 and pfhrp3 gene deletion, approaches and methods used for its estimation and reporting in Plasmodium falciparum populations in Africa: review of published studies 2010-2019. Malar J 18:355. doi: 10.1186/s12936-019-2987-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson R, Parr JB, Cheng Q, Chenet S, Perkins M, Cunningham J. 2020. Prevalence of Plasmodium falciparum lacking histidine-rich proteins 2 and 3: a systematic review. Bull World Health Organ 98:558–568F. doi: 10.2471/BLT.20.250621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.World Health Organization. 2021. Statement by the Malaria Policy Advisory Group on the urgent need to address the high prevalence of pfhrp2/3 gene deletions in the Horn of Africa and beyond. World Health Organization, Geneva, Switzerland. https://www.who.int/news/item/28-05-2021-statement-by-the-malaria-policy-advisory-group-on-the-urgent-need-to-address-the-high-prevalence-of-pfhrp2-3-gene-deletions-in-the-horn-of-africa-and-beyond. [Google Scholar]
- 26.Noviyanti R, Miotto O, Barry A, Marfurt J, Siegel S, Thuy-Nhien N, Quang HH, Anggraeni ND, Laihad F, Liu Y, Sumiwi ME, Trimarsanto H, Coutrier F, Fadila N, Ghanchi N, Johora FT, Puspitasari AM, Tavul L, Trianty L, Utami RAS, Wang D, Wangchuck K, Price RN, Auburn S. 2020. Implementing parasite genotyping into national surveillance frameworks: feedback from control programmes and researchers in the Asia-Pacific region. Malar J 19:271. doi: 10.1186/s12936-020-03330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fola AA, Harrison GLA, Hazairin MH, Barnadas C, Hetzel MW, Iga J, Siba PM, Mueller I, Barry AE. 2017. Higher complexity of infection and genetic diversity of Plasmodium vivax than Plasmodium falciparum across all malaria transmission zones of Papua New Guinea. Am J Trop Med Hyg 96:630–641. doi: 10.4269/ajtmh.16-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fola AA, Nate E, Abby Harrison GL, Barnadas C, Hetzel MW, Iga J, Siba P, Mueller I, Barry AE. 2018. Nationwide genetic surveillance of Plasmodium vivax in Papua New Guinea reveals heterogeneous transmission dynamics and routes of migration amongst subdivided populations. Infect Genet Evol 58:83–95. doi: 10.1016/j.meegid.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 29.Waltmann A, Koepfli C, Tessier N, Karl S, Fola A, Darcy AW, Wini L, Harrison GLA, Barnadas C, Jennison C, Karunajeewa H, Boyd S, Whittaker M, Kazura J, Bahlo M, Mueller I, Barry AE. 2018. Increasingly inbred and fragmented populations of Plasmodium vivax associated with the eastward decline in malaria transmission across the Southwest Pacific. PLoS Negl Trop Dis 12:e0006146. doi: 10.1371/journal.pntd.0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor SM, Parobek CM, Aragam N, Ngasala BE, Martensson A, Meshnick SR, Juliano JJ. 2013. Pooled deep sequencing of Plasmodium falciparum isolates: an efficient and scalable tool to quantify prevailing malaria drug-resistance genotypes. J Infect Dis 208:1998–2006. doi: 10.1093/infdis/jit392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nsanzabana C, Djalle D, Guérin PJ, Ménard D, González IJ. 2018. Tools for surveillance of anti-malarial drug resistance: an assessment of the current landscape. Malar J 17:75. doi: 10.1186/s12936-018-2185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duraisingh MT, Curtis J, Warhurst DC. 1998. Plasmodium falciparum: detection of polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes by PCR and restriction digestion. Exp Parasitol 89:1–8. doi: 10.1006/expr.1998.4274. [DOI] [PubMed] [Google Scholar]
- 33.Eldin de Pecoulas P, Basco LK, Abdallah B, Dje MK, Le Bras J, Mazabraud A. 1995. Plasmodium falciparum: detection of antifolate resistance by mutation-specific restriction enzyme digestion. Exp Parasitol 80:483–487. doi: 10.1006/expr.1995.1060. [DOI] [PubMed] [Google Scholar]
- 34.Talundzic E, Ravishankar S, Kelley J, Patel D, Plucinski M, Schmedes S, Ljolje D, Clemons B, Madison-Antenucci S, Arguin PM, Lucchi NW, Vannberg F, Udhayakumar V. 2018. Next-generation sequencing and bioinformatics protocol for malaria drug resistance marker surveillance. Antimicrob Agents Chemother 62:e02474-17. doi: 10.1128/AAC.02474-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baniecki ML, Faust AL, Schaffner SF, Park DJ, Galinsky K, Daniels RF, Hamilton E, Ferreira MU, Karunaweera ND, Serre D, Zimmerman PA, Sa JM, Wellems TE, Musset L, Legrand E, Melnikov A, Neafsey DE, Volkman SK, Wirth DF, Sabeti PC. 2015. Development of a single nucleotide polymorphism barcode to genotype Plasmodium vivax infections. PLoS Negl Trop Dis 9:e0003539. doi: 10.1371/journal.pntd.0003539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniels R, Volkman SK, Milner DA, Mahesh N, Neafsey DE, Park DJ, Rosen D, Angelino E, Sabeti PC, Wirth DF, Wiegand RC. 2008. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J 7:223. doi: 10.1186/1475-2875-7-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fola AA, Kattenberg E, Razook Z, Lautu-Gumal D, Lee S, Mehra S, Bahlo M, Kazura J, Robinson LJ, Laman M, Mueller I, Barry AE. 2020. SNP barcodes provide higher resolution than microsatellite markers to measure Plasmodium vivax population genetics. Malar J 19:375. doi: 10.1186/s12936-020-03440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daniels RF, Schaffner SF, Wenger EA, Proctor JL, Chang H-H, Wong W, Baro N, Ndiaye D, Fall FB, Ndiop M, Ba M, Milner DA, Taylor TE, Neafsey DE, Volkman SK, Eckhoff PA, Hartl DL, Wirth DF. 2015. Modeling malaria genomics reveals transmission decline and rebound in Senegal. Proc Natl Acad Sci USA 112:7067–7072. doi: 10.1073/pnas.1505691112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor AR, Schaffner SF, Cerqueira GC, Nkhoma SC, Anderson TJC, Sriprawat K, Pyae Phyo A, Nosten F, Neafsey DE, Buckee CO. 2017. Quantifying connectivity between local Plasmodium falciparum malaria parasite populations using identity by descent. PLoS Genet 13:e1007065. doi: 10.1371/journal.pgen.1007065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preston MD, Campino S, Assefa SA, Echeverry DF, Ocholla H, Amambua-Ngwa A, Stewart LB, Conway DJ, Borrmann S, Michon P, Zongo I, Ouédraogo J-B, Djimde AA, Doumbo OK, Nosten F, Pain A, Bousema T, Drakeley CJ, Fairhurst RM, Sutherland CJ, Roper C, Clark TG. 2014. A barcode of organellar genome polymorphisms identifies the geographic origin of Plasmodium falciparum strains. Nat Commun 5:4052. doi: 10.1038/ncomms5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trimarsanto H, Amato R, Pearson RD, Sutanto E, Noviyanti R, Trianty L, Marfurt J, Pava Z, Echeverry DF, Lopera-Mesa TM, Montenegro LM, Tobón-Castaño A, Grigg MJ, Barber B, William T, Anstey NM, Getachew S, Petros B, Aseffa A, Assefa A, Rahim AG, Chau NH, Hien TT, Alam MS, Khan WA, Ley B, Thriemer K, Wangchuck S, Hamedi Y, Adam I, Liu Y, Gao Q, Sriprawat K, Ferreira MU, Barry A, Mueller I, Drury E, Goncalves S, Simpson V, Miotto O, Miles A, White NJ, Nosten F, Kwiatkowski DP, Price RN, Auburn S. 2019. A molecular barcode and online tool to identify and map imported infection with Plasmodium vivax. bioRxiv. doi: 10.1101/776781. [DOI]
- 42.Jacob CG, Thuy-Nhien N, Mayxay M, Maude RJ, Quang HH, Hongvanthong B, Vanisaveth V, Ngo Duc T, Rekol H, van der Pluijm RW, von Seidlein L, Fairhurst RM, Nosten FH, Hossain MA, Park N, Goodwin S, Ringwald P, Chindavongsa K, Newton PN, Ashley EA, Phalivong S, Maude RR, Leang R, Huch C, Dong LT, Nguyen K-T, Nhat TM, Hien TT, Nguyen H, Zdrojewski N, Canavati SE, Sayeed AA, Uddin D, Buckee C, Fanello CI, Onyamboko M, Peto T, Tripura R, Amaratunga C, Myint Thu A, Delmas G, Landier J, Parker DM, Chau NH, Lek D, Suon S, Callery JJ, Jittamala P, Hanboonkunupakarn B, Pukrittayakamee S, et al. 2021. Genetic surveillance in the Greater Mekong subregion and South Asia to support malaria control and elimination. Elife 10:e62997. doi: 10.7554/eLife.62997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Illumina. 2018. AmpliSeq for Illumina custom and community panels: reference guide. Illumina, San Diego, CA. [Google Scholar]
- 44.World Health Organization. 2020. World malaria report 2020. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 45.Cortese JF, Caraballo A, Contreras CE, Plowe CV. 2002. Origin and dissemination of Plasmodium falciparum drug-resistance mutations in South America. J Infect Dis 186:999–1006. doi: 10.1086/342946. [DOI] [PubMed] [Google Scholar]
- 46.Chenet SM, Akinyi Okoth S, Huber CS, Chandrabose J, Lucchi NW, Talundzic E, Krishnalall K, Ceron N, Musset L, Macedo de Oliveira A, Venkatesan M, Rahman R, Barnwell JW, Udhayakumar V. 2016. Independent emergence of the Plasmodium falciparum Kelch propeller domain mutant allele C580Y in Guyana. J Infect Dis 213:1472–1475. doi: 10.1093/infdis/jiv752. [DOI] [PubMed] [Google Scholar]
- 47.Mathieu LC, Cox H, Early AM, Mok S, Lazrek Y, Paquet JC, Ade MP, Lucchi NW, Grant Q, Udhayakumar V, Alexandre JS, Demar M, Ringwald P, Neafsey DE, Fidock DA, Musset L. 2020. Local emergence in Amazonia of Plasmodium falciparum k13 C580Y mutants associated with in vitro artemisinin resistance. Elife 9:e51015. doi: 10.7554/eLife.51015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vreden SG, Jitan JK, Bansie RD, Adhin MR. 2013. Evidence of an increased incidence of day 3 parasitaemia in Suriname: an indicator of the emerging resistance of Plasmodium falciparum to artemether. Mem Inst Oswaldo Cruz 108:968–973. doi: 10.1590/0074-0276130167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chenet SM, Okoth SA, Kelley J, Lucchi N, Huber CS, Vreden S, Macedo de Oliveira A, Barnwell JW, Udhayakumar V, Adhin MR. 2017. Molecular profile of malaria drug resistance markers of Plasmodium falciparum in Suriname. Antimicrob Agents Chemother 61:e02655-16. doi: 10.1128/AAC.02655-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosas-Aguirre A, Erhart A, Llanos-Cuentas A, Branch O, Berkvens D, Abatih E, Lambert P, Frasso G, Rodriguez H, Gamboa D, Sihuincha M, Rosanas-Urgell A, D’Alessandro U, Speybroeck N. 2015. Modelling the potential of focal screening and treatment as elimination strategy for Plasmodium falciparum malaria in the Peruvian Amazon region. Parasit Vectors 8:261. doi: 10.1186/s13071-015-0868-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anonymous. 1999. Política nacional de medicamentos para el control de la malaria en el Perú. Ministerio de Salud del Perú, Lima, Perú. [Google Scholar]
- 52.Marquino W, MacArthur JR, Barat LM, Oblitas FE, Arrunategui M, Garavito G, Chafloque ML, Pardave B, Gutierrez S, Arrospide N, Carrillo C, Cabezas C, Ruebush TK, II.. 2003. Efficacy of chloroquine, sulfadoxine-pyrimethamine, and mefloquine for the treatment of uncomplicated Plasmodium falciparum malaria on the North Coast of Peru. Am J Trop Med Hyg 68:120–123. doi: 10.4269/ajtmh.2003.68.120. [DOI] [PubMed] [Google Scholar]
- 53.Aramburú Guarda J, Ramal Asayag C, Witzig R. 1999. Malaria reemergence in the Peruvian Amazon region. Emerg Infect Dis 5:209–215. doi: 10.3201/eid0502.990204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruebush TK, II, Neyra D, Cabezas C. 2004. Modifying national malaria treatment policies in Peru. J Public Health Policy 25:328–345. doi: 10.1057/palgrave.jphp.3190032. [DOI] [PubMed] [Google Scholar]
- 55.Williams HA, Vincent-Mark A, Herrera Y, Chang OJ. 2009. A retrospective analysis of the change in anti-malarial treatment policy: Peru. Malar J 8:85. doi: 10.1186/1475-2875-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marquino W, Huilca M, Calampa C, Falconi E, Cabezas C, Naupay R, Ruebush TK, II.. 2003. Efficacy of mefloquine and a mefloquine-artesunate combination therapy for the treatment of uncomplicated Plasmodium falciparum malaria in the Amazon Basin of Peru. Am J Trop Med Hyg 68:608–612. doi: 10.4269/ajtmh.2003.68.608. [DOI] [PubMed] [Google Scholar]
- 57.Lerch A, Koepfli C, Hofmann NE, Messerli C, Wilcox S, Kattenberg JH, Betuela I, O’Connor L, Mueller I, Felger I. 2017. Development of amplicon deep sequencing markers and data analysis pipeline for genotyping multi-clonal malaria infections. BMC Genomics 18:864. doi: 10.1186/s12864-017-4260-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, Bockarie M, Mokili J, Mharakurwa S, French N, Whitworth J, Velez ID, Brockman AH, Nosten F, Ferreira MU, Day KP. 2000. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol 17:1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- 59.Koepfli C, Mueller I. 2017. Malaria epidemiology at the clone level. Trends Parasitol 33:974–985. doi: 10.1016/j.pt.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Demas AR, Sharma AI, Wong W, Early AM, Redmond S, Bopp S, Neafsey DE, Volkman SK, Hartl DL, Wirth DF. 2018. Mutations in Plasmodium falciparum actin-binding protein coronin confer reduced artemisinin susceptibility. Proc Natl Acad Sci USA 115:12799–12804. doi: 10.1073/pnas.1812317115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bacon DJ, McCollum AM, Griffing SM, Salas C, Soberon V, Santolalla M, Haley R, Tsukayama P, Lucas C, Escalante AA, Udhayakumar V. 2009. Dynamics of malaria drug resistance patterns in the Amazon Basin region following changes in Peruvian national treatment policy for uncomplicated malaria. Antimicrob Agents Chemother 53:2042–2051. doi: 10.1128/AAC.01677-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sridaran S, McClintock SK, Syphard LM, Herman KM, Barnwell JW, Udhayakumar V. 2010. Anti-folate drug resistance in Africa: meta-analysis of reported dihydrofolate reductase (dhfr) and dihydropteroate synthase (dhps) mutant genotype frequencies in African Plasmodium falciparum parasite populations. Malar J 9:247. doi: 10.1186/1475-2875-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sidhu AB, Valderramos SG, Fidock DA. 2005. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol 57:913–926. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 64.Witkowski B, Duru V, Khim N, Ross LS, Saintpierre B, Beghain J, Chy S, Kim S, Ke S, Kloeung N, Eam R, Khean C, Ken M, Loch K, Bouillon A, Domergue A, Ma L, Bouchier C, Leang R, Huy R, Nuel G, Barale JC, Legrand E, Ringwald P, Fidock DA, Mercereau-Puijalon O, Ariey F, Menard D. 2017. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype-genotype association study. Lancet Infect Dis 17:174–183. doi: 10.1016/S1473-3099(16)30415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, Nosten F, Krishna S. 2004. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364:438–447. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Veiga MI, Dhingra SK, Henrich PP, Straimer J, Gnadig N, Uhlemann AC, Martin RE, Lehane AM, Fidock DA. 2016. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun 7:11553. doi: 10.1038/ncomms11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tessema SK, Hathaway NJ, Teyssier NB, Murphy M, Chen A, Aydemir O, Duarte EM, Simone W, Colborn J, Saute F, Crawford E, Aide P, Bailey JA, Greenhouse B. 2022. Sensitive, highly multiplexed sequencing of microhaplotypes from the Plasmodium falciparum heterozygome. J Infect Dis 225:1227–1237. doi: 10.1093/infdis/jiaa527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henriques G, Hallett RL, Beshir KB, Gadalla NB, Johnson RE, Burrow R, van Schalkwyk DA, Sawa P, Omar SA, Clark TG, Bousema T, Sutherland CJ. 2014. Directional selection at the pfmdr1, pfcrt, pfubp1, and pfap2mu loci of Plasmodium falciparum in Kenyan children treated with ACT. J Infect Dis 210:2001–2008. doi: 10.1093/infdis/jiu358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cerqueira GC, Cheeseman IH, Schaffner SF, Nair S, McDew-White M, Phyo AP, Ashley EA, Melnikov A, Rogov P, Birren BW, Nosten F, Anderson TJC, Neafsey DE. 2017. Longitudinal genomic surveillance of Plasmodium falciparum malaria parasites reveals complex genomic architecture of emerging artemisinin resistance. Genome Biol 18:78. doi: 10.1186/s13059-017-1204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie SC, Ralph SA, Tilley L. 2020. K13, the cytostome, and artemisinin resistance. Trends Parasitol 36:533–544. doi: 10.1016/j.pt.2020.03.006. [DOI] [PubMed] [Google Scholar]
- 71.Birnbaum J, Scharf S, Schmidt S, Jonscher E, Hoeijmakers WAM, Flemming S, Toenhake CG, Schmitt M, Sabitzki R, Bergmann B, Frohlke U, Mesen-Ramirez P, Blancke Soares A, Herrmann H, Bartfai R, Spielmann T. 2020. A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 367:51–59. doi: 10.1126/science.aax4735. [DOI] [PubMed] [Google Scholar]
- 72.Henrici RC, Edwards RL, Zoltner M, van Schalkwyk DA, Hart MN, Mohring F, Moon RW, Nofal SD, Patel A, Flueck C, Baker DA, John ARO, Field MC, Sutherland CJ. 2020. The Plasmodium falciparum artemisinin susceptibility-associated AP-2 adaptin mu subunit is clathrin independent and essential for schizont maturation. mBio 11:e02918-19. doi: 10.1128/mBio.02918-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Behrens HM, Schmidt S, Spielmann T. 2021. The newly discovered role of endocytosis in artemisinin resistance. Med Res Rev 41:2998–3022. doi: 10.1002/med.21848. [DOI] [PubMed] [Google Scholar]
- 74.Sutherland CJ, Henrici RC, Artavanis-Tsakonas K. 2021. Artemisinin susceptibility in the malaria parasite Plasmodium falciparum: propellers, adaptor proteins and the need for cellular healing. FEMS Microbiol Rev 45:fuaa056. doi: 10.1093/femsre/fuaa056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Velavan TP, Nderu D, Agbenyega T, Ntoumi F, Kremsner PG. 2019. An alternative dogma on reduced artemisinin susceptibility: a new shadow from east to west. Proc Natl Acad Sci USA 116:12611–12612. doi: 10.1073/pnas.1907142116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sharma AI, Shin SH, Bopp S, Volkman SK, Hartl DL, Wirth DF. 2020. Genetic background and PfKelch13 affect artemisinin susceptibility of PfCoronin mutants in Plasmodium falciparum. PLoS Genet 16:e1009266. doi: 10.1371/journal.pgen.1009266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oberstaller J, Zoungrana L, Bannerman CD, Jahangiri S, Dwivedi A, Silva JC, Adams JH, Takala-Harrison S. 2021. Integration of population and functional genomics to understand mechanisms of artemisinin resistance in Plasmodium falciparum. Int J Parasitol Drugs Drug Resist 16:119–128. doi: 10.1016/j.ijpddr.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feleke SM, Reichert EN, Mohammed H, Brhane BG, Mekete K, Mamo H, Petros B, Solomon H, Abate E, Hennelly C, Denton M, Keeler C, Hathaway NJ, Juliano JJ, Bailey JA, Rogier E, Cunningham J, Aydemir O, Parr JB. 2021. Plasmodium falciparum is evolving to escape malaria rapid diagnostic tests in Ethiopia. Nat Microbiol 6:1289–1299. doi: 10.1038/s41564-021-00962-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Griffing SM, Mixson-Hayden T, Sridaran S, Alam MT, McCollum AM, Cabezas C, Marquino Quezada W, Barnwell JW, De Oliveira AM, Lucas C, Arrospide N, Escalante AA, Bacon DJ, Udhayakumar V. 2011. South American Plasmodium falciparum after the malaria eradication era: clonal population expansion and survival of the fittest hybrids. PLoS One 6:e23486. doi: 10.1371/journal.pone.0023486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Recht J, Siqueira AM, Monteiro WM, Herrera SM, Herrera S, Lacerda MVG. 2017. Malaria in Brazil, Colombia, Peru and Venezuela: current challenges in malaria control and elimination. Malar J 16:273. doi: 10.1186/s12936-017-1925-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baldeviano GC, Okoth SA, Arrospide N, Gonzalez RV, Sanchez JF, Macedo S, Conde S, Tapia LL, Salas C, Gamboa D, Herrera Y, Edgel KA, Udhayakumar V, Lescano AG. 2015. Molecular epidemiology of Plasmodium falciparum malaria outbreak, Tumbes, Peru, 2010-2012. Emerg Infect Dis 21:797–803. doi: 10.3201/eid2105.141427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Villena FE, Lizewski SE, Joya CA, Valdivia HO. 2021. Population genomics and evidence of clonal replacement of Plasmodium falciparum in the Peruvian Amazon. Sci Rep 11:21212. doi: 10.1038/s41598-021-00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hathaway NJ, Parobek CM, Juliano JJ, Bailey JA. 2018. SeekDeep: single-base resolution de novo clustering for amplicon deep sequencing. Nucleic Acids Res 46:e21. doi: 10.1093/nar/gkx1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Henden L, Lee S, Mueller I, Barry A, Bahlo M. 2018. Identity-by-descent analyses for measuring population dynamics and selection in recombining pathogens. PLoS Genet 14:e1007279. doi: 10.1371/journal.pgen.1007279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okoth SA, Chenet SM, Arrospide N, Gutierrez S, Cabezas C, Matta JA, Udhayakumar V. 2016. Molecular investigation into a malaria outbreak in Cusco, Peru: Plasmodium falciparum BV1 lineage is linked to a second outbreak in recent times. Am J Trop Med Hyg 94:128–131. doi: 10.4269/ajtmh.15-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schmedes SE, Patel D, Kelley J, Udhayakumar V, Talundzic E. 2019. Using the Plasmodium mitochondrial genome for classifying mixed-species infections and inferring the geographical origin of P. falciparum parasites imported to the U.S. PLoS One 14:e0215754. doi: 10.1371/journal.pone.0215754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grande T, Bernasconi A, Erhart A, Gamboa D, Casapia M, Delgado C, Torres K, Fanello C, Llanos-Cuentas A, D’Alessandro U. 2007. A randomised controlled trial to assess the efficacy of dihydroartemisinin-piperaquine for the treatment of uncomplicated falciparum malaria in Peru. PLoS One 2:e1101. doi: 10.1371/journal.pone.0001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mangold KA, Manson RU, Koay ES, Stephens L, Regner M, Thomson RB, Jr, Peterson LR, Kaul KL. 2005. Real-time PCR for detection and identification of Plasmodium spp. J Clin Microbiol 43:2435–2440. doi: 10.1128/JCM.43.5.2435-2440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289–300. [Google Scholar]
- 91.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 92.Rovira-Vallbona E, Van Hong N, Kattenberg JH, Huan RM, Hien NTT, Ngoc NTH, Guetens P, Hieu NL, Mai TT, Duong NTT, Duong TT, Phuc BQ, Xa NX, Erhart A, Rosanas-Urgell A. 2020. Efficacy of dihydroartemisinin/piperaquine and artesunate monotherapy for the treatment of uncomplicated Plasmodium falciparum malaria in central Vietnam. J Antimicrob Chemother 75:2272–2281. doi: 10.1093/jac/dkaa172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oyola SO, Ariani CV, Hamilton WL, Kekre M, Amenga-Etego LN, Ghansah A, Rutledge GG, Redmond S, Manske M, Jyothi D, Jacob CG, Otto TD, Rockett K, Newbold CI, Berriman M, Kwiatkowski DP. 2016. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J 15:597. doi: 10.1186/s12936-016-1641-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Straimer J, Gnädig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Ménard D, Fidock DA. 2015. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miotto O, Sekihara M, Tachibana S-I, Yamauchi M, Pearson RD, Amato R, Gonçalves S, Mehra S, Noviyanti R, Marfurt J, Auburn S, Price RN, Mueller I, Ikeda M, Mori T, Hirai M, Tavul L, Hetzel MW, Laman M, Barry AE, Ringwald P, Ohashi J, Hombhanje F, Kwiatkowski DP, Mita T. 2020. Emergence of artemisinin-resistant Plasmodium falciparum with kelch13 C580Y mutations on the island of New Guinea. PLoS Pathog 16:e1009133. doi: 10.1371/journal.ppat.1009133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ross LS, Dhingra SK, Mok S, Yeo T, Wicht KJ, Kumpornsin K, Takala-Harrison S, Witkowski B, Fairhurst RM, Ariey F, Menard D, Fidock DA. 2018. Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat Commun 9:3314. doi: 10.1038/s41467-018-05652-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.MalariaGEN. 2015. MalariaGEN Plasmodium falciparum community project. MRC Centre for Genomics and Global Health, University of Oxford, Oxford, United Kingdom. https://www.malariagen.net/apps/pf/4.0/. [Google Scholar]
- 98.Jombart T, Devillard S, Balloux F. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chang H-H, Worby CJ, Yeka A, Nankabirwa J, Kamya MR, Staedke SG, Dorsey G, Murphy M, Neafsey DE, Jeffreys AE, Hubbart C, Rockett KA, Amato R, Kwiatkowski DP, Buckee CO, Greenhouse B. 2017. THE REAL McCOIL: a method for the concurrent estimation of the complexity of infection and SNP allele frequency for malaria parasites. PLoS Comput Biol 13:e1005348. doi: 10.1371/journal.pcbi.1005348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 101.Hedrick PW. 2005. A standardized genetic differentiation measure. Evolution 59:1633–1638. [PubMed] [Google Scholar]
- 102.Jost L. 2008. G(ST) and its relatives do not measure differentiation. Mol Ecol 17:4015–4026. doi: 10.1111/j.1365-294x.2008.03887.x. [DOI] [PubMed] [Google Scholar]
- 103.Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA. 2013. diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. doi: 10.1111/2041-210X.12067. [DOI] [Google Scholar]
- 104.Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 105.Jombart T, Ahmed I. 2011. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kamvar ZN, Tabima JF, Grunwald NJ. 2014. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. doi: 10.7717/peerj.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.de Oliveira AM, Chavez J, de Leon GP, Durand S, Arrospide N, Roberts J, Cabezas C, Marquino W. 2011. Efficacy and effectiveness of mefloquine and artesunate combination therapy for uncomplicated Plasmodium falciparum malaria in the Peruvian Amazon. Am J Trop Med Hyg 85:573–578. doi: 10.4269/ajtmh.2011.11-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Anonymous. 2015. Norma téchnica de salud para la atencion de la malaria y malaria grave en el Peru. Ministerio de Salud Perú, Direccion Regional de Salud Loreto, Iquitos, Peru. [Google Scholar]
- 109.Rosas-Aguirre A, Speybroeck N, Llanos-Cuentas A, Rosanas-Urgell A, Carrasco-Escobar G, Rodriguez H, Gamboa D, Contreras-Mancilla J, Alava F, Soares IS, Remarque E, D’Alessandro U, Erhart A. 2015. Hotspots of malaria transmission in the Peruvian Amazon: rapid assessment through a parasitological and serological survey. PLoS One 10:e0137458. doi: 10.1371/journal.pone.0137458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Branch O, Casapia WM, Gamboa DV, Hernandez JN, Alava FF, Roncal N, Alvarez E, Perez EJ, Gotuzzo E. 2005. Clustered local transmission and asymptomatic Plasmodium falciparum and Plasmodium vivax malaria infections in a recently emerged, hypoendemic Peruvian Amazon community. Malar J 4:27. doi: 10.1186/1475-2875-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Henriques G, van Schalkwyk DA, Burrow R, Warhurst DC, Thompson E, Baker DA, Fidock DA, Hallett R, Flueck C, Sutherland CJ. 2015. The mu subunit of Plasmodium falciparum clathrin-associated adaptor protein 2 modulates in vitro parasite response to artemisinin and quinine. Antimicrob Agents Chemother 59:2540–2547. doi: 10.1128/AAC.04067-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Adams T, Ennuson NAA, Quashie NB, Futagbi G, Matrevi S, Hagan OCK, Abuaku B, Koram KA, Duah NO. 2018. Prevalence of Plasmodium falciparum delayed clearance associated polymorphisms in adaptor protein complex 2 mu subunit (pfap2mu) and ubiquitin specific protease 1 (pfubp1) genes in Ghanaian isolates. Parasit Vectors 11:175. doi: 10.1186/s13071-018-2762-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martin RE, Marchetti RV, Cowan AI, Howitt SM, Broer S, Kirk K. 2009. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science 325:1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- 114.Sidhu AB, Verdier-Pinard D, Fidock DA. 2002. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE. 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nagesha HS, Casey GJ, Rieckmann KH, Fryauff DJ, Laksana BS, Reeder JC, Maguire JD, Baird JK. 2003. New haplotypes of the Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene among chloroquine-resistant parasite isolates. Am J Trop Med Hyg 68:398–402. doi: 10.4269/ajtmh.2003.68.398. [DOI] [PubMed] [Google Scholar]
- 117.Roepe PD. 2009. Molecular and physiologic basis of quinoline drug resistance in Plasmodium falciparum malaria. Future Microbiol 4:441–455. doi: 10.2217/fmb.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dhingra SK, Redhi D, Combrinck JM, Yeo T, Okombo J, Henrich PP, Cowell AN, Gupta P, Stegman ML, Hoke JM, Cooper RA, Winzeler E, Mok S, Egan TJ, Fidock DA. 2017. A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. mBio 8:e00303-17. doi: 10.1128/mBio.00303-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hamilton WL, Amato R, van der Pluijm RW, Jacob CG, Quang HH, Thuy-Nhien NT, Hien TT, Hongvanthong B, Chindavongsa K, Mayxay M, Huy R, Leang R, Huch C, Dysoley L, Amaratunga C, Suon S, Fairhurst RM, Tripura R, Peto TJ, Sovann Y, Jittamala P, Hanboonkunupakarn B, Pukrittayakamee S, Chau NH, Imwong M, Dhorda M, Vongpromek R, Chan XHS, Maude RJ, Pearson RD, Nguyen T, Rockett K, Drury E, Gonçalves S, White NJ, Day NP, Kwiatkowski DP, Dondorp AM, Miotto O. 2019. Evolution and expansion of multidrug-resistant malaria in Southeast Asia: a genomic epidemiology study. Lancet Infect Dis 19:943–951. doi: 10.1016/S1473-3099(19)30392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pelleau S, Moss EL, Dhingra SK, Volney B, Casteras J, Gabryszewski SJ, Volkman SK, Wirth DF, Legrand E, Fidock DA, Neafsey DE, Musset L. 2015. Adaptive evolution of malaria parasites in French Guiana: reversal of chloroquine resistance by acquisition of a mutation in pfcrt. Proc Natl Acad Sci USA 112:11672–11677. doi: 10.1073/pnas.1507142112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van der Pluijm RW, Imwong M, Chau NH, Hoa NT, Thuy-Nhien NT, Thanh NV, Jittamala P, Hanboonkunupakarn B, Chutasmit K, Saelow C, Runjarern R, Kaewmok W, Tripura R, Peto TJ, Yok S, Suon S, Sreng S, Mao S, Oun S, Yen S, Amaratunga C, Lek D, Huy R, Dhorda M, Chotivanich K, Ashley EA, Mukaka M, Waithira N, Cheah PY, Maude RJ, Amato R, Pearson RD, Gonçalves S, Jacob CG, Hamilton WL, Fairhurst RM, Tarning J, Winterberg M, Kwiatkowski DP, Pukrittayakamee S, Hien TT, Day NP, Miotto O, White NJ, Dondorp AM. 2019. Determinants of dihydroartemisinin-piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect Dis 19:952–961. doi: 10.1016/S1473-3099(19)30391-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Folarin OA, Bustamante C, Gbotosho GO, Sowunmi A, Zalis MG, Oduola AM, Happi CT. 2011. In vitro amodiaquine resistance and its association with mutations in pfcrt and pfmdr1 genes of Plasmodium falciparum isolates from Nigeria. Acta Trop 120:224–230. doi: 10.1016/j.actatropica.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother 44:2100–2108. doi: 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Akhoon BA, Singh KP, Varshney M, Gupta SK, Shukla Y, Gupta SK. 2014. Understanding the mechanism of atovaquone drug resistance in Plasmodium falciparum cytochrome b mutation Y268S using computational methods. PLoS One 9:e110041. doi: 10.1371/journal.pone.0110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Peters JM, Chen N, Gatton M, Korsinczky M, Fowler EV, Manzetti S, Saul A, Cheng Q. 2002. Mutations in cytochrome b resulting in atovaquone resistance are associated with loss of fitness in Plasmodium falciparum. Antimicrob Agents Chemother 46:2435–2441. doi: 10.1128/AAC.46.8.2435-2441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Griffing SM, Viana GM, Mixson-Hayden T, Sridaran S, Alam MT, de Oliveira AM, Barnwell JW, Escalante AA, Povoa MM, Udhayakumar V. 2013. Historical shifts in Brazilian P. falciparum population structure and drug resistance alleles. PLoS One 8:e58984. doi: 10.1371/journal.pone.0058984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Costa GL, Amaral LC, Fontes CJF, Carvalho LH, de Brito CFA, de Sousa TN. 2017. Assessment of copy number variation in genes related to drug resistance in Plasmodium vivax and Plasmodium falciparum isolates from the Brazilian Amazon and a systematic review of the literature. Malar J 16:152. doi: 10.1186/s12936-017-1806-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Basco LK, Ringwald P. 2000. Molecular epidemiology of malaria in Yaounde, Cameroon. VI. Sequence variations in the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene and in vitro resistance to pyrimethamine and cycloguanil. Am J Trop Med Hyg 62:271–276. doi: 10.4269/ajtmh.2000.62.271. [DOI] [PubMed] [Google Scholar]
- 129.Hyde JE. 2007. Drug-resistant malaria—an insight. FEBS J 274:4688–4698. doi: 10.1111/j.1742-4658.2007.05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Staines HM, Burrow R, Teo BH-Y, Chis Ster I, Kremsner PG, Krishna S. 2018. Clinical implications of Plasmodium resistance to atovaquone/proguanil: a systematic review and meta-analysis. J Antimicrob Chemother 73:581–595. doi: 10.1093/jac/dkx431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Amato R, Lim P, Miotto O, Amaratunga C, Dek D, Pearson RD, Almagro-Garcia J, Neal AT, Sreng S, Suon S, Drury E, Jyothi D, Stalker J, Kwiatkowski DP, Fairhurst RM. 2017. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect Dis 17:164–173. doi: 10.1016/S1473-3099(16)30409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. 2000. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 403:906–909. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- 133.Pickard AL, Wongsrichanalai C, Purfield A, Kamwendo D, Emery K, Zalewski C, Kawamoto F, Miller RS, Meshnick SR. 2003. Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob Agents Chemother 47:2418–2423. doi: 10.1128/AAC.47.8.2418-2423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mbaye A, Dieye B, Ndiaye YD, Bei AK, Muna A, Deme AB, Yade MS, Diongue K, Gaye A, Ndiaye IM, Ndiaye T, Sy M, Diallo MA, Badiane AS, Ndiaye M, Seck MC, Sy N, Koita O, Krogstad DJ, Nwakanma D, Ndiaye D. 2016. Selection of N86F184D1246 haplotype of Pfmrd1 gene by artemether-lumefantrine drug pressure on Plasmodium falciparum populations in Senegal. Malar J 15:433. doi: 10.1186/s12936-016-1490-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, Price RN, Martensson A, Rosenthal PJ, Dorsey G, Sutherland CJ, Guerin P, Davis TME, Menard D, Adam I, Ademowo G, Arze C, Baliraine FN, Berens-Riha N, Bjorkman A, Borrmann S, Checchi F, Desai M, Dhorda M, Djimde AA, El-Sayed BB, Eshetu T, Eyase F, Falade C, Faucher JF, Froberg G, Grivoyannis A, Hamour S, Houze S, Johnson J, Kamugisha E, Kariuki S, Kiechel J-R, Kironde F, Kofoed P-E, LeBras J, Malmberg M, Mwai L, Ngasala B, Nosten F, Nsobya SL, Nzila A, Oguike M, Otienoburu SD, Ogutu B, et al. 2014. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg 91:833–843. doi: 10.4269/ajtmh.14-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dahlstrom S, Ferreira PE, Veiga MI, Sedighi N, Wiklund L, Martensson A, Farnert A, Sisowath C, Osorio L, Darban H, Andersson B, Kaneko A, Conseil G, Bjorkman A, Gil JP. 2009. Plasmodium falciparum multidrug resistance protein 1 and artemisinin-based combination therapy in Africa. J Infect Dis 200:1456–1464. doi: 10.1086/606009. [DOI] [PubMed] [Google Scholar]
- 137.Bopp S, Magistrado P, Wong W, Schaffner SF, Mukherjee A, Lim P, Dhorda M, Amaratunga C, Woodrow CJ, Ashley EA, White NJ, Dondorp AM, Fairhurst RM, Ariey F, Menard D, Wirth DF, Volkman SK. 2018. Plasmepsin II-III copy number accounts for bimodal piperaquine resistance among Cambodian Plasmodium falciparum. Nat Commun 9:1769. doi: 10.1038/s41467-018-04104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dharia NV, Plouffe D, Bopp SE, Gonzalez-Paez GE, Lucas C, Salas C, Soberon V, Bursulaya B, Kochel TJ, Bacon DJ, Winzeler EA. 2010. Genome scanning of Amazonian Plasmodium falciparum shows subtelomeric instability and clindamycin-resistant parasites. Genome Res 20:1534–1544. doi: 10.1101/gr.105163.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Set S1. Download spectrum.00960-22-s0001.xlsx, XLSX file, 0.3 MB (291.1KB, xlsx)
Supplemental material. Download spectrum.00960-22-s0002.pdf, PDF file, 0.8 MB (774.4KB, pdf)
Data Availability Statement
Sample metadata, drug resistance and hrp2 and hrp3 haplotypes, barcodes, lineages, locations, and dates, etc., are accessible at https://microreact.org/project/aV7RHNCBmG3sJ2rwzchE4k-peru-2003-2018-molecular-surveillance-with-p-falciparum-ampliseq-assay. Raw data (FASTQ files) are available at the SRA under BioProject accession number PRJNA855317, and individual library accession numbers are listed in the supplemental metadata file at Microreact (see the URL mentioned above). A detailed protocol of the library preparation procedures has been deposited at https://doi.org/10.21769/BioProtoc.4621. It is anticipated that this link will be active by 5 March 2023; until that time, all the data will be available from the corresponding author upon request. Variant files (vcf) and scripts are available upon request. All other data are included in the manuscript and supplemental material.