


Keywords: unfolded protein response, polycystic kidney disease, polycystin-1, Ire1α-XBP1 pathway, protein folding, chaperone, genetic renal disease, mutation
Abstract
Significance Statement
XBP1 activation in neonatal and adult doxycycline-inducible murine models of ADPKD due to a hypomorphic polycystin-1 missense mutation orthologous to human PC1R2220W delays cyst formation. Activating XBP1s, a pro-chaperone inducer of the endoplasmic reticulum stress response, can improve steady-state expression, ciliary trafficking, and cleavage of the mutant protein, providing initial in vivo proof of concept that modulating levels of poorly functioning hypomorphic PC1 alleles can slow progression of kidney cyst formation in ADPKD.
Background
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in Pkd1 and Pkd2. They encode the polytopic integral membrane proteins polycystin-1 (PC1) and polycystin-2 (PC2), respectively, which are expressed on primary cilia. Formation of kidney cysts in ADPKD starts when a somatic second hit mechanism inactivates the wild-type Pkd allele. Approximately one quarter of families with ADPDK due to Pkd1 have germline nonsynonymous amino acid substitution (missense) mutations. A subset of these mutations is hypomorphic, retaining some residual PC1 function. Previous studies have shown that the highly conserved Ire1α-XBP1 pathway of the unfolded protein response can modulate levels of functional PC1 in the presence of mutations in genes required for post-translational maturation of integral membrane proteins. We examine how activity of the endoplasmic reticulum chaperone-inducing transcription factor XBP1 affects ADPKD in a murine model with missense Pkd1.
Methods
We engineered a Pkd1 REJ domain missense murine model, Pkd1R2216W, on the basis of the orthologous human hypomorphic allele Pkd1R2220W, and examined the effects of transgenic activation of XBP1 on ADPKD progression.
Results
Expression of active XBP1 in cultured cells bearing PC1R2216W mutations increased levels and ciliary trafficking of PC1R2216W. Mice homozygous for Pkd1R2216W or heterozygous for Pkd1R2216W in trans with a conditional Pkd1fl allele exhibit severe ADPKD following inactivation in neonates or adults. Transgenic expression of spliced XBP1 in tubule segments destined to form cysts reduced cell proliferation and improved Pkd progression, according to structural and functional parameters.
Conclusions
Modulating ER chaperone function through XBP1 activity improved Pkd in a murine model of PC1, suggesting therapeutic targeting of hypomorphic mutations.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary disease that primarily affects the kidney, occurring in 1:500–1:1000 individuals.1 It accounts for >90% of all hereditary cystic kidney diseases and is characterized by bilateral fluid-filled cysts that expand and increase in number over decades. This results in increased total kidney volume and is accompanied by CKD that leads to ESKD in at least 50% of affected individuals by the age of 60.2,3 Mutations in Pkd1 and Pkd2, respectively encoding the proteins polycystin-1 (PC1) and polycystin-2 (PC2), are responsible for the occurrence of most cases of ADPKD associated with risk for ESKD. Nearly 78% of families with ADPKD have defined mutations in Pkd1.4 PC1 has 11 membrane-spanning domains, with a large extracellular portion that self-cleaves at the G protein–coupled receptor proteolytic site (GPS).2 PC2, which bears similarity with the last six transmembrane domains of PC1, is a member of the transient receptor potential channel family, and it interacts with PC1 to form a putative receptor-channel complex5 on the membrane of primary cilia.6
Although inherited as a dominant trait, cyst initiation in ADPKD occurs at least in part by a “two-hit” mechanism following somatic mutations affecting the normal allele that results in homozygous inactivation of either Pkd1 or Pkd2 in kidney tubule cells.7–11 Other mechanisms have been described that impact the progression of polycystic kidney disease, including noncell autonomous effects on cells still expressing polycystins,12 the developmental timing of Pkd1 inactivation,13 the effects of the inflammatory response,14,15 and the milder effects of PC1 hypomorphic missense mutations compared with complete loss of function.16–19 The latter studies have highlighted the potential importance of protein folding as a means of enhancing the function of PC1 hypomorphs and alleviating polycystic disease progression. In support of this idea, a genetically distinct but phenotypically related disorder (isolated polycystic liver disease [PCLD]) is caused by mutations in genes encoding proteins expressed in the endoplasmic reticulum (ER) that are active in post-translational protein processing of integral membrane and secreted proteins.20–22 Loss of function in these PCLD genes results in polycystic kidney and liver phenotypes specifically due to impaired functionally effective maturation and dosage of PC1.20–22
Our previous studies showed that increasing the housekeeping chaperone repertoire by upregulation of active, spliced X-box binding protein-1 (XBP1s), a major pro-chaperone transcriptional effector of the Ire1α-dependent ER stress response, exerts a beneficial effect on the levels of PC1 and severity of polycystic kidney disease in a PCLD model of defective N-glycosylation.23 We now sought to investigate whether enhanced chaperone expression via XBP1s can also modulate the levels and polycystic disease severity in orthologous models of an incompletely penetrant hypomorphic human PC1 variant, p.R2220W (Arg2220Trp).24 The steady-state expression levels, GPS cleavage, and cilia trafficking of murine ortholog PC1R2216W is impaired, and mice homozygous for this mutant have early-onset postnatal severe polycystic kidney disease. Expression of XBP1s increased steady-state expression levels and cilia location of PC1R2220W protein in vitro and significantly reduced the severity of polycystic kidney disease in both perinatal and adult onset ADPKD murine models based on this hypomorphic variant. These findings show the potential beneficial role of enhanced chaperone function via XBP1s activation in modulating polycystic kidney disease progression due to at least some hypomorphic missense mutations in PC1.
Methods
Murine Lines
All experiments were conducted in accordance with Yale University Institutional Animal Care and Use Committee guidelines and procedures. The mice described in this study are on a C57BL6/129 mixed background. Mice of both sexes were used in this study. The genetic models Pkd1fl,25 Xbp1fl,26 Pkhd1-cre,27 ROSA26-XBP1s,23 Pax8rtTA,27 and tet-OCre27 have been described previously. Mice were euthanized, and tissues were processed for histology, immunocytochemistry, and immunoblotting. Blood was collected for BUN measurements, which were performed by the George M. O’Brien Kidney Center at Yale University.
Generation of the PC1R2216W Murine Allele
The PC1R2216W allele is orthologous to the human PC1R2220W mutation. Two single guide RNAs (sgRNAs; CCCACAGGTAGTGCCAGCCG for PC1R2216W and CCTAGGGCCTAAGTGCTGCT for V5 tag insertion) were designed with CRISPR Design (http://crispr.mit.edu/). For precise genome editing, two single-stranded oligodeoxynucleotide donor (ssODN) templates, one with the point mutation (PC1R2216W), the other with V5 tag in-frame at the C terminus immediately before the termination codon, each flanked with two respective homology arms, were made by Integrated DNA Technologies (Coralville, IA). The plasmids containing the sgRNA were linearized, and in vitro transcription of sgRNAs was done using a MEGAshortscript Kit (Thermo Fisher Scientific, Waltham, MA). The in vitro transcribed sgRNAs, Cas9 protein (New England Biolabs, Ipswich, MA), and the two ssODN donor templates were microinjected into fertilized zygotes, and blastocysts were transferred into the uterus of pseudopregnant ICR females by the Yale Genome Editing Center. Mice were genotyped for the presence of the V5 epitope tag sequence and the PC1R2216W/R2216W mutation by PCR and Sanger sequencing, and two founders were identified. In addition, one founder (Pkd1V5) containing the V5 tag on a wild-type Pkd1 allele was also obtained.
Cell Lines
Pkd1V5/+, Pkd1R2216W/flox, and Pkd1R2216W/flox;ROSA26-XBP1s parental kidney cell lines were isolated by tubule microdissection23 and immortalized via lentiviral transduction of SV40 large T antigen (Addgene #58993). The Pkd1flox alleles were converted to their null counterparts while also turning on XBP1 expression in the lines containing the ROSA26-floxSTOPflox-XBP1s transgene by infection with Adeno-Cre-recombinase (Vector Biolabs, Malvern, PA). Clonal cell lines (two independent lines) were produced by limiting dilution matched with isogenic parental controls.
Generation of Pkd1-KO IMCD3 Cells via Clustered Regularly Interspaced Short Palindromic Repeats/Cas9
To maximize the effect of gene disruption, gRNA sequences near the exon 2 and exon 3 of mouse Pkd1 genomic DNA were used (i.e., sgRNAs GATAGTCAGGAGACGAGCTC and CTACTTCAGACGCTGGACAT for Pkd1 exon 2 editing; sgRNAs CCCTGCAAGAAAGAAAGATG and AGGATTTCTACATTAGAAGA for Pkd1 exon 3 editing). The sgRNAs were cloned into pGL3-U6-sgRNA-PGK-hygromycin plasmid, which was obtained from the modified pGL3-U6-sgRNA-PGK-puromycin plasmid (Addgene #51133). Cas9-D10A plasmid (CMV-hspCas9[D10A]-T2A-Puro; SBI CASLV100PA-1), pMDLg/pRRE, pRSV-Rev, and pMD2.G were transfected into HEK293T cell lines with lipofectamine 2000 (#11668019; Invitrogen, Waltham, MA) to generate Cas9-D10A lentivirus. Infected IMCD3 cells were selected with puromycin to obtain stable Cas9-D10A lines. pGL3-U6-sgRNA-PGK-hygromycin with Pkd1-specific sgRNAs were transfected into Cas9-D10A IMCD3 stable cells followed by selection, cloning, and expansion. Final Pkd1 knockout IMCD3 cell lines carrying frameshift insertions and deletions in exon 2 and exon 3 were determined by PCR and sequencing. Knockout cell lines were further confirmed via Western blot using PC1 antisera (7E12; Santa Cruz Biotechnology, Dallas, TX; sc-130554).
Immunohistochemistry and Immunofluorescent Staining
Mice were anesthetized by an intraperitoneal injection of ketamine/xylazine and fixed in situ by perfusion through the heart with 4% paraformaldehyde in 1× PBS for 3 minutes. Sections (5–7 μm) were used for immunohistochemical studies according to standard procedures.27 Immunofluorescence staining and the quantification of cystic indices were carried out using a Nikon TE2000U microscope and MetaMorph software (Molecular Devices, San Jose, CA). Primary antibodies and lectins used were fluorescein-labeled Lotus tetragonolobus lectin (Vector Laboratories, Burlingame, CA; FL-1321; 1:500), fluorescein-labeled Dolichos biflorus lectin (DBA; Vector Laboratories; FL-1031; 1:100), rabbit anti-Arl13b (Proteintech, Chicago, IL; #17711–1; 1:100), mouse anti-Arl13b (Proteintech; #66739–1; 1:100), rabbit anti-XBP1s (Novus Biologicals, Littleton, CO; NB100–78403; 1:100), mouse anti-V5 (Invitrogen; mA5–15253; 1:200), and Alexa Fluor-conjugated acetylated tubulin (Santa Cruz Biotechnology; sc-23950; 1:500).
Proliferation analysis was performed by immunohistochemistry using a rabbit anti-Ki67 (Sigma–Aldrich, St. Louis, MO; Ab-254; 1:200) monoclonal antibody. Apoptosis analysis was carried out by terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) staining according to the manufacturer’s instructions (Roche, Basel, Switzerland; #11684795910). Sections were also stained with DAPI and DBA, and the number of Ki67- or TUNEL-positive nuclei in at least 1000 DBA-positive nuclei per kidney were counted to determine the rates for proliferation and apoptosis, respectively. Sodium 4-phenylbutyrate (4-PBA) was purchased from Sigma-Aldrich (CAS 1716-12-7).
Protein Preparation and Immunoblot Analysis
Tissues were extracted and homogenized with a motor driven Teflon pestle homogenizer in ice-cold buffer (250 mM sucrose, 1 mM EGTA, 25 mM Tris, pH 7.4 containing protease inhibitors) as previously described.23 The homogenates were centrifuged twice at 500 g. The resulting supernatant was analyzed as total lysate. Cells were harvested and lysed in RIPA buffer containing complete protease inhibitor cocktails (Roche). Immunoblotting was performed using rabbit anti-Hsp90 (Santa Cruz Biotechnology; sc-7947; 1:3000), mouse anti-V5 (Invitrogen; mA5–15253; 1:1000), rabbit anti-XBP1s (Novus Biologicals; NB100–78403; 1:1000), mouse anti-Lamin A/C (Cell Signaling Technology, Danvers, MA; #2032; 1:1000). Adeno-XBP1s (mouse) was a kind gift from Dr. Ann-Hwee Lee (Weill Cornell/Regeneron). Secondary antibodies included anti-mouse/rabbit/rat HRP-conjugated antibodies (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA) were incubated with the membrane for 1 hour at room temperature. Western Lightning Plus-ECL (PerkinElmer, Waltham, MA; #34579) or SuperSignal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific; #34094) was used for chemiluminescence detection. The volume of individual immunoblot bands, in pixels, was determined by optical densitometry using ImageJ.
Nuclear Preparation
To prepare nuclear lysates, cells were trypsinized and pelleted. Cell pellets were resuspended in lysis buffer (100 mM Tris, pH 7.4, 20 mM NaCl, 10 mM EDTA, 0.1% TX100) and incubated on ice for 5 minutes. Cells were then vortexed and centrifuged at 21,000 g for 5 minutes. The supernatant obtained was collected and considered as the cytosolic fraction. The remaining pellet (nuclear) was resuspended in 1× red loading buffer (Cell Signaling Technology) and sonicated at power 6 three times using a probe sonicator (Misonix, Farmingdale, NY; XL-2000). All the lysates were quantified using a BCA kit (Thermo Fisher Scientific). Immunoblotting was performed using rabbit anti-Lamin B (Cell Signaling Technology; #12586; 1:1000).
Statistical Analyses
Comparisons of three or more groups were performed using one-way ANOVA followed by Tukey’s multiple group comparison post test. Comparison of two groups was performed using the two-tailed t test. A P value of <0.05 was considered the threshold for statistical significance. Data are presented as the mean±SEM.
Results
Our previous studies showed that increased XBP1s expression can modulate expression of PC1 in glycosylation-defective backgrounds.23 We wanted to expand these studies and investigate whether enhanced chaperone expression via increased expression of active XBP1s can also modulate the levels and cystic disease severity caused by a PC1 missense mutant. To answer this question, we investigated a human ADPKD substitution mutation, PC1R2220W (Arg2220Trp), an incompletely penetrant hypomorphic variant that occurred in trans with another missense mutation (PC1R3277C) in an early-onset ADPKD family.24 The PC1R2220W receptor for egg jelly (REJ) domain mutant is a hypomorphic missense variant that leads to a partial defect in PC1 cleavage.28 HEK293 cells transiently expressing either epitope tagged PC1-V5 or PC1R2220W-V528 were transduced with XBP1s adenovirus (mouse adeno-XBP1s). In the absence of overexpressed XBP1s, cells expressing the PC1R2220W mutant showed reduced levels of the intramembranous PC1 C-terminal cleavage fragment (PC1-CTF) compared with wild type (Supplemental Figure 1, A and B). In the presence of XBP1s, an increase in levels of PC1R2220W-V5-CTF was detected by anti-V5 immunoblotting, indicating that XBP1s expression can increase the steady-state expression of PC1R2220W in vitro (Supplemental Figure 1, A and B).
We next tested the effect of XBP1s on the functioning of PC1R2220W in vivo. We introduced the hypomorphic PC1R2216W mutation, the orthologous mutation to human PC1R2220W, into the murine germline using clustered regularly interspaced short palindromic repeats/Cas9 gene editing. We simultaneously introduced a V5 epitope tag at the C terminus of the endogenous PC1 protein in both wild-type and PC1R2216W alleles. Pkd1R2216/R2216W homozygous mice displayed a significant cystic phenotype at postnatal day 11 (P11) and were severely affected at P16 (Figure 1, A–C), with significantly increased kidney-to-body-weight (KW/BW) ratio, cystic index, and BUN levels (Figure 1, D–H). Pkd1R2216/R2216W mice did not typically survive past P20. Kidney cysts in this germline mutant model arose from both proximal and collecting duct segments (Figure 1I). At P16, the livers of Pkd1R2216/R2216W mice displayed early bile duct cystic dilations and portal inflammation (Figure 1J). Finally, by taking advantage of the knock-in V5 tag, expression of PC1 could be detected in the lung, kidney, and heart (Figure 2K, left panel) as had previously been shown in a HA epitope knock-in model.29 The PC1R2216W mutation resulted in a relative reduction in GPS cleavage in vivo. Kidney lysates from PC1R2216W/R2216W mice showed a shift in the densitometric ratio of PC1-CTF to PC1 full-length (PC1-FL) from around 2.5 in wild-type PC1V5 kidneys to a reduced ratio of around 1 in homozygous PC1R2216W kidneys (Figure 1K, right panel). This in vivo finding is consistent with a relative reduction in the GPS cleavage efficiency that had been reported in vitro.28
Figure 1.

Pkd1R2216W/R2216W mice display early severe cystic disease. (A and B) Representative images of kidney sections from mice with the indicated genotypes at (A) P11 and (B) P16. Pkd1R2216W/R2216W display significant cystic disease at P11 compared with heterozygous Pkd1R2216W/+ controls. Disease progresses rapidly whereby by P16 cysts populate the entire kidney parenchyma. (C) Higher power hematoxylin and eosin–stained images corresponding to the genotypes in (A) and (B). Scale bar, 500 μm. (D and E) Quantification of KW/BW ratio and cystic index at P11; symbol colors correspond to genotypes in (A). *P=0.02; ***P<0.001. (F–H) Quantification of KW/BW ratio, cystic index, and BUN at P16; symbol colors correspond to genotypes in (A); ***P<0.001; **P=0.007. Comparisons were performed using the two-tailed t test. A P value of <0.05 was considered significant. (I) Cysts in Pkd1R2216W/R2216W kidneys at P16 arise from both proximal tubules (Lotus tetragonolobus agglutinin, LTA, green) and collecting ducts (Dolichos biflorus agglutinin, DBA, red). Scale bar, 20 μm. (J) Hematoxylin and eosin histology of liver tissues from Pkd1R2216W/R2216W mice reveals large portal tracts with cystic dilation of bile ducts (arrow heads) and abundant periportal inflammation that are absent in Pkd1R2216W/+ controls. Scale bar, 100 μm. (K) Left panel: anti-V5 Western blotting marking the endogenous wild-type PC1 protein expression in the lung (L), kidney (K), and heart (H). Right panel: the homozygous Pkd1R2216W/R2216W mice display an altered GPS cleavage pattern seen by anti-V5 whole lysate immunoblotting and anti–V5-IP followed by immunoblotting. PC1-CTF-to-PC1-FL densitometric ratios displayed below each lane show decreased PC1-CTF-to-PC1-FL ratios in mutant PC1R2216W mice compared with PC1 (PC1V5/V5) mice.
Figure 2.
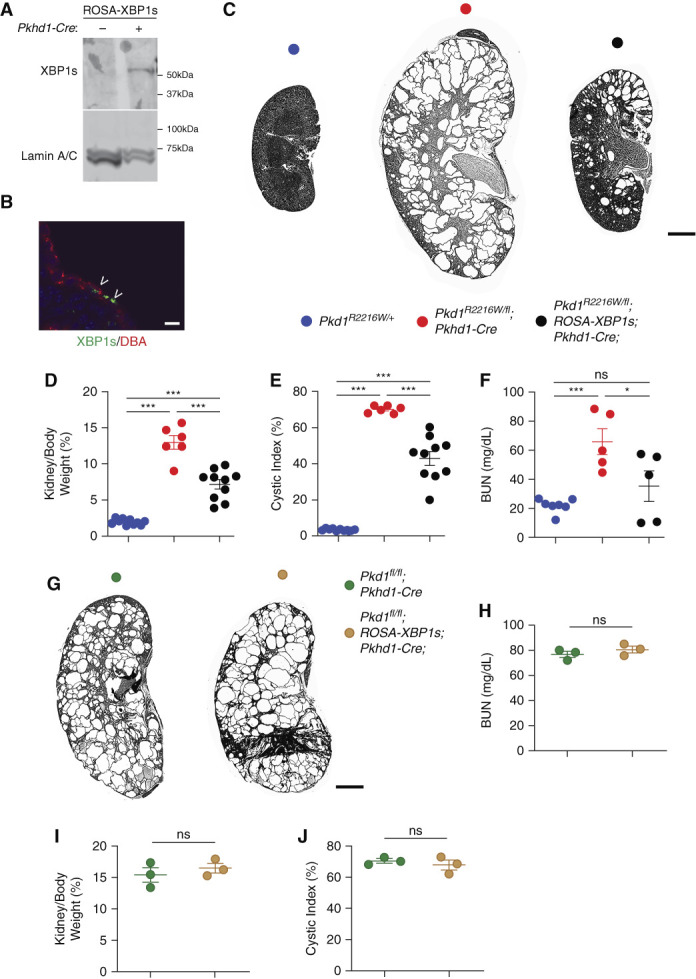
XBP1s expression slows down disease progression in Pkd1R2216W/flox;Pkhd1Cre mice. (A and B) Nuclear XBP1s expression in the kidneys of Pkd1R2216W/flox in the absence or presence of Pkhd1-Cre at P16 observed by immunoblotting of nuclear fraction from (A) kidney tissue lysates or (B) immunofluorescence (green, indicated by arrowheads) in DBA-positive (red) collecting duct segments. Scale bar, 10 μm. (C) Representative images of kidneys with the genotypes indicated by the color key at P16. Scale bar, 2 mm. (D–F) Aggregate data from mice with the genotypes indicated by the color codes from (C) at P16 showing significant reversal in kidney weight/body weight ratio (KW/BW), cystic index, and BUN levels in the presence XBP1s expression; ***P<0.001; **P<0.01; *P<0.05. Comparisons among groups were performed using one-way ANOVA followed by Tukey’s multiple group comparison post test. (G) Representative images of kidneys with the genotypes indicated by the color key at P24. Size bar, 2 mm. (H–J) Aggregate data from mice with the genotypes indicated by colors in (G).
To achieve expression of XBP1s in vivo, we made use of a murine line in which the active spliced form of human XBP1 (i.e., XBP1s) is knocked into the ubiquitous ROSA26 locus but is silenced by a floxed stop cassette (ROSA-XBP1s).23 In the presence of Pkhd1-Cre, nuclear XBP1s expression is turned on in collecting ducts as evidenced by immunoblotting of the nuclear fraction from whole kidney lysates and immunostaining of kidney sections (Figure 2, A and B). To examine the effect of XBP1s in the presence of the PC1R2216W mutation, we generated Pkd1R2216W/flox;Pkhd1-Cre mice with or without the ROSA-XBP1s. At P16, Pkd1R2216W/flox;Pkhd1-Cre mice displayed severe cyst formation with increased in KW/BW ratio, cystic index, and BUN compared with noncystic Pkd1R2216W/+ control mice (Figure 2, C–F). Expression of XBP1s in the same segments where PC1 was inactivated in Pkd1R2216W/flox;ROSA-XBP1s;Pkhd1-Cre mice led to a significant decrease in the cystic burden compared with the mutant animals not expressing XBP1s (Figure 2, D–F; all kidney images and sex of each are shown in Supplemental Figure 2).
To establish whether the effect of XBP1s is dependent on the presence of the PC1R2220W allele, we generated Pkd1flox/flox;ROSA-XBP1s;Pkhd1-Cre mice in which no PC1 protein is present in segments expressing Cre recombinase. We did not observe any difference in phenotype severity between Pkd1fl/fl;Pkhd1-Cre mice with or without XBP1s (Figure 2, G–J). We next examined the contributions of apoptosis and proliferation to the observed beneficial effect of XBP1s by performing TUNEL and Ki67 staining in DBA-positive segments where Cre is active. Although induction of XBP1s in the collecting duct did not impact apoptosis (Figure 3, A and B), it led to a significant reduction in proliferation (Figure 3, C and D), suggesting that the reduced severity of the cystic phenotype in the Pkd1R2216W/flox;ROSA-XBP1s;Pkhd1-Cre animals at P16 is due at least in part to a slowdown of cyst growth.
Figure 3.
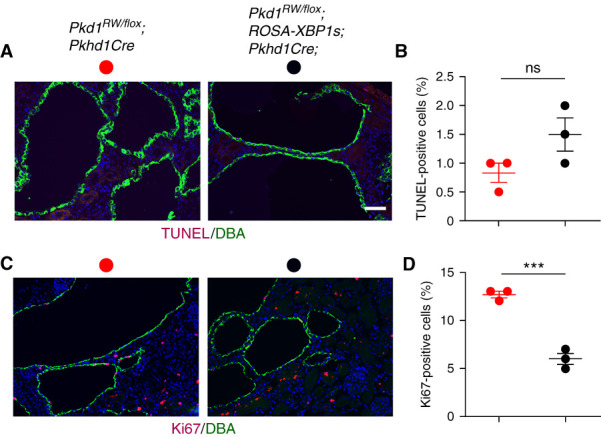
The in vivo effect of XBP1s occurs via a decrease in cell proliferation. Expression of XBP1s in the cystic epithelia of Pkd1R2216W/flox;Pkhd1Cre mice at P16 leads to a significant reduction of proliferation with no impact on apoptosis. (A) Representative images of TUNEL staining (apoptosis; red) in DBA-positive collecting duct segments (green) of Pkd1R2216W/flox;Pkhd1Cre and Pkd1R2216W/flox;ROSA-XBP1s;Pkhd1Cre mice. Sections were counterstained with DAPI (blue). Scale bar, 20 μm. (B) Quantitation of apoptosis for the genotypes described in (A). (C) Representative images of Ki67 staining (proliferation; red) in DBA-positive collecting duct segments (green) of Pkd1R2216W/flox;Pkhd1Cre and Pkd1R2216W/flox;ROSA-XBP1s;Pkhd1Cre mice. Sections were counterstained with DAPI (blue). Scale bar, 20 μm. (D) Quantification of proliferation indexes for genotypes described in (C). Apoptosis and proliferation rates were determined by counting >1000 DBA-positive collecting duct cells per kidney from three kidneys per each genotype for both TUNEL and Ki67. Results are shown as the mean±SEM (t test); n.s. P=0.09; ***P<0.001.
We next examined the biochemical impact of XBP1s on the expression and subcellular localization of the PC1R2216W mutant. Because in vivo biochemical investigations of PC1 are not very reliable, we performed our analysis in kidney cells generated by microdissection of kidney tubules from Pkd1R2216W/flox or Pkd1R2216W/flox;ROSA-XBP1s mice. We immortalized the cells with SV40 large T antigen and transduced them with Cre to generate Pkd1R2216W/– or Pkd1R2216W/–;XBP1s cell lines. Immunoblotting of nuclear fractions from cell lysates and immunocytochemistry with anti-XBP1s confirmed nuclear expression of the active XPB1s in Pkd1R2216W/–;XBP1s cells (Figure 4, A and B). We also confirmed XBP1s activation by upregulation of its transcription and that of its canonical target chaperone Erdj4 in the Pkd1R2216W/–;XBP1s cells (Figure 4, C and D). We compared wild-type V5-epitope tagged PC1V5 from microdissected tubules of mice heterozygous for the V5 wild-type allele (Pkd1V5/+) with PC1R2216W from Pkd1R2216W/– mutant cells without or with XBP1s. In the absence of XBP1s, PC1R2216W displayed a relative defect in GPS cleavage compared with the PC1V5 control (Figure 4E). PC1R2216W showed a marked decrease in expression of PC1-CTF and a mild increase in PC1-FL compared with the nonmutant PC1V5, as reported previously.28 Increased degradation of the mutant protein may also contribute to the decrease in steady-state levels of PC1-CTF in the Pkd1R2216W/– cells because proteasome inhibition with MG132 results in increased mutant PC1-CTF steady-state levels (Supplemental Figure 3). The presence of activated XBP1s in the Pkd1R2216W/–;XBP1s cells results in an increase steady state in both PC1-CTF and PC1-FL compared with the Pkd1R2216W/– cells alone (Figure 4, E and F). This was associated with a small but significant increase in the PC1-CTF-to-PC1-FL ratio in the presence of XBP1s (Figure 4F). Taken together, the data indicate that XBP1s, presumably through its target chaperone gene products, increases steady-state levels of mutant PC1R2216W and also improves the GPS cleavage process that is thought to occur co-translationally as the PC1 protein folds in the ER.
Figure 4.

Expression of XBP1s improves expression and trafficking of PC1R2216W. (A and B) Immortalized tubule kidney cells isolated from Pkd1R2216W/flox or Pkd1R2216W/flox;ROSA-XBP1s mice were converted to their null counterparts via Cre infection. Nuclear expression of XBP1s is present in the Pkd1R2216W/–;XBP1s versus Pkd1R2216W/– cells as seen via (A) anti-XBP1s Western blotting and (B) immunofluorescence (co-localization of XBP1s and DAPI indicated by arrowheads); XBP1s (+) control represents Pkd1R2216W/– cells treated with the canonical ER stress inducer, tunicamycin; size bar, 10 μm. (C and D) Expression of (C) XBP1s and (D) its canonical downstream target Erdj4 message is increased in the Pkd1R2216W/–;XBP1s versus Pkd1R2216W/– cells as seen via quantitative PCR; **P=0.005 (XBP1s); **P=0.002 (Erdj4). (E) When compared with wild-type PC1 (Pkd1V5/+), the PC1R2216W/– mutant displays a pronounced cleavage defect, with a decrease in PC1-CTF and a concomitant increase in PC1-FL as seen by anti-V5 immunoblotting. When XBP1s is turned on, an increase in both the PC1-CTF and PC1-FL species is observed. (F) Quantification of the results in (E); the band intensity of PC1-CTF and PC1-FL was normalized by the Hsp90 loading control. The amount of PC1-CTF and PC1-FL is significantly increased in the PC1R2216W/– cells expressing XBP1s (***P<0.001). Furthermore, the PC1-CTF/FL ratio shows a small increase in the Pkd1R2216W/–;XBP1s versus Pkd1R2216W/– cells (P=0.02). (G) The PC1V5/+, Pkd1R2216W/–, and Pkd1R2216W/–;XBP1s cells were ciliated and anti-V5 staining was used to assess PC1 expression in cilia (using acetylated tubulin, Ac-Tub as the ciliary marker). Although PC1V5/+ cells display robust ciliary V5 staining (arrowheads), the PC1R2216W/– mutant exhibits very low ciliary V5 immunoreactivity; expression of XBP1s leads to an increased mutant PC1 expression in cilia (arrowheads); size bar, 10 μm. (H) Quantification of the IF experiment described in (G); percent positive V5 in cilia is shown for PC1V5/+ (84 V5 positive out of 107 cilia quantified), PC1R2216W/– (15/150), and PC1R2216W/– with XBP1s (73/141).
Next, we investigated the effect of XBP1s on the ciliary expression of PC1R2216W. In the absence of XBP1s in Pkd1R2216W/– cells, 10% of cilia had anti-V5 reactive protein compared with around 80% of cilia in cells expressing wild-type PC1-V5 (Figure 4, G and H). This finding was also recapitulated in IMCD3 Pkd1-KO cells transfected with either PC1-V5 or PC1R2220W-V5 (Supplemental Figure 4, A and B). When XBP1s expression was turned on, an anti–V5-immunoreactive ciliary signal was detected in around 50% of Pkd1R2216W/− cells (Figure 4, G and H). These data suggest that expression of XBP1s and its downstream chaperone targets (e.g., Erdj4) is associated with increased expression of PC1R2216W in cilia. To further support a potential role of enhanced ER protein folding in ameliorating the cilia expression defect of the Pkd1R2216W/− cells, we used the chemical chaperone 4-phenyl butyrate (4-PBA) that promotes ER release and trafficking of mutant forms of other integral membrane proteins, e.g., CFTR30. Treatment of Pkd1R2216W/− cells with 4-PBA led to an increase in the ciliary expression of the mutant species compared with untreated cells (Supplemental Figure 5). The combined effect of XBP1s in increasing the steady state and improving GP S cleavage and cilia expression of mutant P C1 suggests that its global chaperone-related function may benefit polycystic kidney disease progression due to PC1R2216W.
Finally, given the role of PC2 as a critical modulator of PC1 stability,16 we intercrossed a three-copy PC2-BAC22 transgenic mouse with Pkd1R2216W/flox;Pkhd1-Cre mice to determine whether increased PC2 levels would lead to improved polycystic kidney disease severity in vivo. The presence of the Pkd2-BAC transgene in Pkd1R2216W/flox;Pkhd1-Cre;Pkd2-BAC mice resulted in significantly improved cystic disease as indicated by decreased KW/BW ratio and BUN (Supplemental Figure 6).
Although the in vivo experiments provided evidence for a disease-modifying role of XBP1s, they were done in a neonatal ADPKD model, which does not recapitulate the slow progressive nature of ADPKD. We made use of a previously described doxycycline-inducible kidney tubule selective digenic Cre/lox system27 to generate Pkd1R2216W/fl;Pax8rtTA;Tet-OCre and Pkd1R2216W/fl;Pax8rtTA;Tet-OCre;ROSA-XBP1s mice. Mice were induced with oral doxycycline between P28 and P42 and examined at 18 weeks of age. The Pkd1R2216W/fl;Pax8rtTA;Tet-OCre mice displayed significant cyst burden at 18 weeks compared with the Pkd1R2216W/+ controls (Figure 5, A–D). The presence of XBP1s led to an improvement in KW/BW ratio, cystic index, and BUN (Figure 5, B–D), which extended the in vivo findings from the neonatal to the adult model of ADPKD. The complete kidney panel along with the sex of each mouse used in the analysis is shown in Supplemental Figure 7. No sex difference trend in the KW/BW ratio was observed in the Pkd1R2216W/fl;Pax8rtTA;Tet-OCre mice (Supplemental Figure 8A). Finally, because by chance all the Pkd1R2216W/fl;Pax8rtTA;Tet-OCre;ROSA-XBP1s mice used in the analysis were female, we compared the KW/BW ratio between the two cystic groups with and without XBP1s using only female mice (Supplemental Figure 8B) and found a statistically significant difference similar to that seen for the comparison with both sexes (Figure 5B). In aggregate, our data support the hypothesis that global chaperone therapies that can increase steady-state expression levels of pathogenic missense PC1 mutants may be able to slow the progression of ADPKD.
Figure 5.
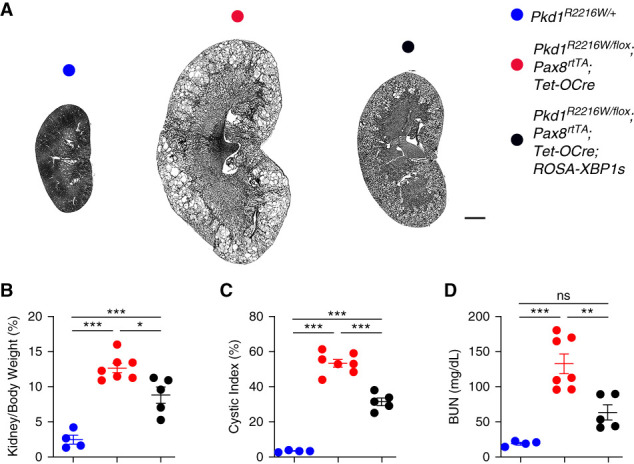
XBP1s slows down disease progression in adult Pkd1R2216W/flox mice. Expression of XBP1s significantly improves cystic disease in an adult inducible model of ADPKD. (A) Representative images of kidney sections from mice with the indicated genotypes at 18 weeks of age. Mice were given doxycycline water between 4 and 6 weeks of age to inactivate Pkd1 and turn on XBP1s and were examined 12 weeks later. Scale bar, 2 mm. (B) Aggregate data from mice with the indicated genotypes for KW/BW ratio, cystic index, and BUN levels; results are shown as the mean±SEM (ANOVA); ***P<0.001; **P<0.01; *P<0.05.
Discussion
This study describes the in vivo effect of XBP1 activation on the progression of polycystic kidney disease due to a pathogenic missense mutation in PC1, that is, mouse PC1R2216W orthologous to human PC1R2220W. By using both biochemical and genetic models, we found that XBP1s, one of the main pro-chaperone inducers of the ER stress response, can improve the steady-state levels, GPS cleavage, and ciliary trafficking of the PC1R2220W mutant. This effect leads to a significant delay in cyst formation in both a rapid progressing neonatal model and an adult doxycycline-inducible model. This supports the notion that the effect of XBP1s is independent of the developmental stage at which it is turned on. Importantly, the impact of XBP1s is entirely contingent on the presence of the Pkd1R2216W mutation because cyst formation is not impacted on a complete PC1 loss-of-function background. Our data suggest that XBP1s is modulating the overall steady-state levels of the hypomorphic PC1R2220W mutant in addition to a small effect on GPS cleavage; the increased dosage combined with a partial correction in the GPS cleavage of mutant PC1 is likely leading to a higher functional output in the primary cilium, which leads to an improvement in the cystic phenotype. Our studies provide initial proof of concept that enhancing steady-state levels and GPS cleavage of poorly functioning hypomorphic PC1 alleles can improve the progression of kidney cyst formation in ADPKD. Further work will be necessary to elucidate the precise impact of XBP1s on the biogenesis and trafficking of the PC1-CTF and/or PC1-NTF fragments.
A previous knock-in murine model of a clinically proposed incompletely penetrant Pkd1 allele, p.R3277C (RC), has been described.28 At the molecular level, the authors found a slight impact on PC1 cleavage and defective folding and trafficking; in vivo, the phenotype of the Pkd1RC/RC mice was very mild, with a small increase in the KW/BW ratio at 3 months and no impact on BUN until 9 months. When Pkd1RC/null mice were generated, they showed enlarged kidneys by P1, with very severe disease by P18. From a severity standpoint, the Pkd1RC/null mice are similar to the homozygous Pkd1R2216W/R2216W phenotype in both morphologic and functional parameters. Given the more severe impact of the R2216W mutation on PC1 processing compared with R3277C, half dosage of the latter is needed to elicit a phenotype similar to two alleles for the former. This may pose an advantage for utilizing the Pkd1R2216W (“RW”) allele in trans with a flox allele in adult inducible conditional models to study various genetic and therapeutic interventions. Interestingly, the PC1R2216W mutant mice do not exhibit an overt sex dimorphism observed with complete loss-of-function Pkd1 alleles.
XBP1s is a bZIP transcription factor that binds to the CRE-like promoter element to activate a group of UPR target genes such as BiP, Erdj4, Sec61p, and Herp.31–33 The genes upregulated by XBP1s are part of the ER folding machinery and the ER-associated degradation pathway, among others, with its overall effect thought to result in an adaptive survival advantage under conditions of ER stress. We have previously shown that on a Prkcsh-deficient background, where the glycosylation and folding of PC1 is impacted in the absence of a direct mutation in Pkd1,23 XBP1s can promote PC1 processing partly via ERdj4; the effect of ERdj4 in this case was far weaker than that elicited by XBP1s itself, suggesting that the impact from XBP1s results from the cooperative action of several its transcriptional targets, of which ERdj4 is one. It would be important to investigate whether Erdj4 and other related XBP1s-dependent transcriptional effectors are modulating the enhanced steady state and GPS cleavage effect we see in the Pkd1R2216W cells with or without XBP1s. Pinpointing those targets would lead to a better understanding of the folding requirements for achieving enhanced PC1 function under hypomorphic missense mutant backgrounds and potentially inform future chaperone-based therapies. Along these lines, approaches to increase protein homeostasis and trafficking using chemical chaperones such as 4-PBA or TUDCA34 may show utility for a particular set of PC1 missense mutants. Finally, the previously described effect of PC2 on the steady-state levels of PC1-CTF16 suggests that modulation of PC2 levels may prove beneficial in the context of select PC1 mutants. Our initial data in an early, fast progression model suggests a potential beneficial role of enhanced PC2 in the context of the PC1R2216W mutation. The effect of enhanced PC2 levels on the severity of cystic disease due to a hypomorphic Pkd1 mutation has also been recently shown in the context of the Pkd1RC allele.35
The Ire1α-XBP1 pathway itself may represent a target on backgrounds of hypomorphic PC1 activity via specific agonists that activate its endoribonuclease function.36,37 Although Ire1α signaling has been shown to promote protection against different types of pathologic insults associated with multiple diseases,36 in the setting of its chronic activation, damaging effects such as increased apoptosis, inflammation, and oncogenic signaling have been observed, potentially limiting the therapeutic applications of XBP1s activating molecules. Missense mutations in Pkd1 occur throughout the gene, and it is hard to predict which ones will benefit the most from an enhanced protein folding approach. Nevertheless, specific in vitro testing of missense variants in “hot spot” regions of the protein (e.g., the REJ/GAIN domain, accounting for 9% of all missense mutations, including R2220W) via PC1 biogenesis and functional assays (expression, cilia trafficking, cilia calcium read-outs, etc) may provide initial insight into the amenability of a particular molecular lesion to a chaperone-type therapy. It is important to keep in mind that a PC1 modulator may not be efficacious in the case of missense mutations that disrupt its interaction with other proteins as part of a functional complex (e.g., PC1/PC2) or for instances where too little PC1 output is present via either defective GPS cleavage and/or impaired ciliary trafficking.
Overall, these studies provide evidence for a beneficial role of XBP1s on the steady-state levels, GP S cleavage,trafficking, and polycystic kidney disease progression due to a PC1 human pathogenic missense mutation. Futurestudies on the effect of increased folding on other polycystin 1/2 pathogenic missense mutations may form the basis of personalized therapies targeted for select classes of hypomorphic mutants.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lonnette Diggs in the George M. O’Brien Kidney Center at Yale (P30 DK079310) for BUN measurements. We are grateful to Peter Harris (Mayo Clinic) for the PC1-WT-V5 and PC1-R2220W-V5 constructs. We thank Dhanpat Jain for assessment of the liver phenotype in the Pkd1R2216W/R2216W animals. We are grateful for the generous and steadfast support from the Amy P. Goldman Foundation. M. Krappitz is a participant in the BIH Charité Clinician Scientist Program funded by the Charité—Universitätsmedizin Berlin and the Berlin Institute of Health at Charité (BIH).
Footnotes
M.K. and R.B. contributed equally to this work.
Published online ahead of print. Publication date available at www.jasn.org.
Disclosures
Y. Cai reports royalties from Santa Cruz (through Albert Einstein College of Medicine) and being a member of the editorial board (unpaid) of the Open Access Journal of Nephrology and Urology, Open Access Journal of Internal Medicine, and World Journal of Nephrology. S. Fedeles reports being an employee of Critical Path Institute, with an appointment as an adjunct faculty at Yale University; honoraria from Vertex Pharmaceuticals; and patents from Yale University. A.R. Gallagher reports ownership interest in Goldfinch Bio and other interests or relationships with Pkd Foundation (volunteer). M. Krappitz reports consultancy for Maze Therapeutics and honoraria from Maze Therapeutics. S. Somlo reports consultancy for BridgeBio Pharma, Goldfinch Bio, and Maze Therapeutics; ownership interest in Goldfinch Bio; and patents or royalties from Yale University. All remaining authors have nothing to disclose.
Funding
This work was supported by National Institutes of Health/National Institute of Diabetes and Digestive Kidney Diseases grants R01 DK54053 and DK100592 to S. Somlo and PKD Foundation grant 202g16a to S. Fedeles.
Author Contributions
R. Bhardwaj, Y. Cai, A. R. Gallagher, M. Krappitz, T.A. Nguyen, C. Pioppini, D. Ruemmele, T. Staudner, P. Westergerling, D.E. Yilmaz, T.A. Hollmann, K. Dong, and S. Fedeles were responsible for the investigation; M. Krappitz, S. Somlo, and S. Fedeles reviewed and edited the manuscript; M. Krappitz, R. Bhardwaj, S. Somlo, and S. Fedeles were responsible for the methodology; S. Fedeles was responsible for the conceptualization and project administration and wrote the original draft of the manuscript; S. Somlo and S. Fedeles were responsible for funding acquisition; and S. Somlo and S. Fedeles were responsible for supervision.
Data Sharing Statement
All data used in this study are available in this article.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/D626.
Supplemental Figure 1. HEK cells were transfected with PC1-V5 or PC1-R2220W-V5 and transduced with XBP1s adenovirus 2 days after transfection.
Supplemental Figure 2. Whole kidney panel showing all the sections used in the comparison among the Pkd1R2216W/+, Pkd1R2216W/flox;Pkhd1Cre, and Pkd1R2216W/flox;ROSA-XBP1s;Pkhd1Cre groups. f, female; m, male. Size bar, 2 mm.
Supplemental Figure 3. Proteasomal inhibition via MG132 treatment leads to an increase in the PC1-CTF levels in PC1R2216W-V5 cells compared with the untreated cells.
Supplemental Figure 4. IMCD3 Pkd1−/− cells were transiently transfected with PC1-V5 or PC1R2220W-V5 mutant.
Supplemental Figure 5. 4-PBA enhances trafficking of the PC1R2216W mutant.
Supplemental Figure 6. Pkd2-BAC leads to a partial rescue of the cystic phenotype on the Pkd1R2216W/flox;Pkhd1Cre background.
Supplemental Figure 7. Whole kidney panel showing all the kidneys used in the comparison among the Pkd1R2216W/+, Pkd1R2216W/flox;Pax8rtTA;tet-OCre, and Pkd1R2216W/flox Pax8rtTA;tet-OCre;ROSA-XBP1s groups.
Supplemental Figure 8. Gender comparison of KW/BW ratios among Pkd1R2216W/flox;Pax8rtTA;tet-OCre mice.
References
- 1.LaRiviere WB, Irazabal MV, Torres VE: Novel therapeutic approaches to autosomal dominant polycystic kidney disease. Transl Res 165: 488–498, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim DY, Park JH: Genetic mechanisms of ADPKD. Adv Exp Med Biol 933: 13–22, 2016 [DOI] [PubMed] [Google Scholar]
- 3.Rossetti S, Hopp K, Sikkink RA, Sundsbak JL, Lee YK, Kubly V, et al. : Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol 23: 915–933, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. : Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 24: 1006–1013, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su Q, Hu F, Ge X, Lei J, Yu S, Wang T, et al. : Structure of the human PKD1-PKD2 complex. Science 361: eaat9819, 2018 [DOI] [PubMed] [Google Scholar]
- 6.Ta CM, Vien TN, Ng LCT, DeCaen PG: Structure and function of polycystin channels in primary cilia. Cell Signal 72: 109626, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian F, Watnick TJ, Onuchic LF, Germino GG: The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 87: 979–987, 1996 [DOI] [PubMed] [Google Scholar]
- 8.Wu G, D’Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, et al. : Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93: 177–188, 1998 [DOI] [PubMed] [Google Scholar]
- 9.Torra R: Autosomal dominant polycystic kidney disease, type 2 (PKD2 disease). Adv Nephrol Necker Hosp 29: 277–287, 1999 [PubMed] [Google Scholar]
- 10.Brasier JL, Henske EP: Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J Clin Invest 99: 194–199, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pei Y, Watnick T, He N, Wang K, Liang Y, Parfrey P, et al. : Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol 10: 1524–1529, 1999 [DOI] [PubMed] [Google Scholar]
- 12.Nishio S, Hatano M, Nagata M, Horie S, Koike T, Tokuhisa T, et al. : Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J Clin Invest 115: 910–918, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG: A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13: 1490–1495, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, et al. : Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 22: 1809–1814, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swenson-Fields KI, Vivian CJ, Salah SM, Peda JD, Davis BM, van Rooijen N, et al. : Macrophages promote polycystic kidney disease progression. Kidney Int 83: 855–864, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai Y, Fedeles SV, Dong K, Anyatonwu G, Onoe T, Mitobe M, et al. : Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest 124: 5129–5144, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJ: Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet 16: 3188–3196, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Jiang ST, Chiou YY, Wang E, Lin HK, Lin YT, Chi YC, et al. : Defining a link with autosomal-dominant polycystic kidney disease in mice with congenitally low expression of Pkd1. Am J Pathol 168: 205–220, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, et al. : Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int 75: 848–855, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, et al. : Genkyst Study Group; HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease: Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 102: 832–844, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, et al. : Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest 127: 3558, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fedeles SV, Tian X, Gallagher AR, Mitobe M, Nishio S, Lee SH, et al. : A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet 43: 639–647, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fedeles SV, So JS, Shrikhande A, Lee SH, Gallagher AR, Barkauskas CE, et al. : Sec63 and Xbp1 regulate IRE1α activity and polycystic disease severity. J Clin Invest 125: 1955–1967, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, et al. : Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol 21: 1097–1102, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, et al. : Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 17: 1505–1516, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee AH, Scapa EF, Cohen DE, Glimcher LH: Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320: 1492–1496, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S: Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45: 1004–1012, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, et al. : Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 122: 4257–4273, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wodarczyk C, Rowe I, Chiaravalli M, Pema M, Qian F, Boletta A: A novel mouse model reveals that polycystin-1 deficiency in ependyma and choroid plexus results in dysfunctional cilia and hydrocephalus. PLoS One 4: e7137, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Woerd WL, Wichers CG, Vestergaard AL, Andersen JP, Paulusma CC, Houwen RH, et al. : Rescue of defective ATP8B1 trafficking by CFTR correctors as a therapeutic strategy for familial intrahepatic cholestasis. J Hepatol 64: 1339–1347, 2016 [DOI] [PubMed] [Google Scholar]
- 31.Clauss IM, Chu M, Zhao JL, Glimcher LH: The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res 24: 1855–1864, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee AH, Iwakoshi NN, Glimcher LH: XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23: 7448–7459, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malhotra JD, Kaufman RJ: The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18: 716–731, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khalaf K, Tornese P, Cocco A, Albanese A: Tauroursodeoxycholic acid: A potential therapeutic tool in neurodegenerative diseases. Transl Neurodegener 11: 33, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakhia R, Ramalingam H, Chang CM, Cobo-Stark P, Biggers L, Flaten A, et al. : PKD1 and PKD2 mRNA cis-inhibition drives polycystic kidney disease progression. Nat Commun 13: 4765, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grandjean JMD, Madhavan A, Cech L, Seguinot BO, Paxman RJ, Smith E, et al. : Pharmacologic IRE1/XBP1s activation confers targeted ER proteostasis reprogramming. Nat Chem Biol 16: 1052–1061, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu J, He GT, Zhang WJ, Xu J, Huang QB: IRE1α signaling pathways involved in mammalian cell fate determination. Cell Physiol Biochem 38: 847–858, 2016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study are available in this article.