

Abstract
Pancreatic cancer remains one of the greatest challenges in oncology for which therapeutic intervention is urgently needed. We previously demonstrated that the intra-tumoral gene transfer of somatostatin receptor 2, to combat tumor aggressiveness, or of deoxycytidine kinase and uridylate monophosphate kinase, to sensitize to gemcitabine chemotherapy, has anti-tumoral potential in experimental models of cancer. Here, we describe the development of the CYL-02 non-viral gene therapy product that comprises a DNA-plasmid encoding for the three aforementioned genes, which expression is targeted to tumor cells, and complexed with polyethyleneimine non-viral vector. We performed pre-clinical toxicology, bio-distribution, and therapeutic activity studies of CYL-02 in two rodent models of pancreatic cancer. We found that CYL-02 is safe, does not increase gemcitabine toxicity, is rapidly cleared from blood following intravenous administration, and sequestered in tumors following intra-tumoral injection. CYL-02 drives the expression of therapeutic genes in cancer cells and strongly sensitizes tumor cells to gemcitabine, both in vitro and in vivo, with significant inhibition of tumor cells dissemination. This study was instrumental for the later use of CYL-02 in patients with advanced pancreatic cancer, demonstrating that rigorous and thorough preclinical investigations are informative for the clinical transfer of gene therapies against this disease.
Keywords: pancreatic cancer, non-viral gene therapy, resistance to treatment, chemotherapy, preclinical regulatory experiments
Graphical abstract
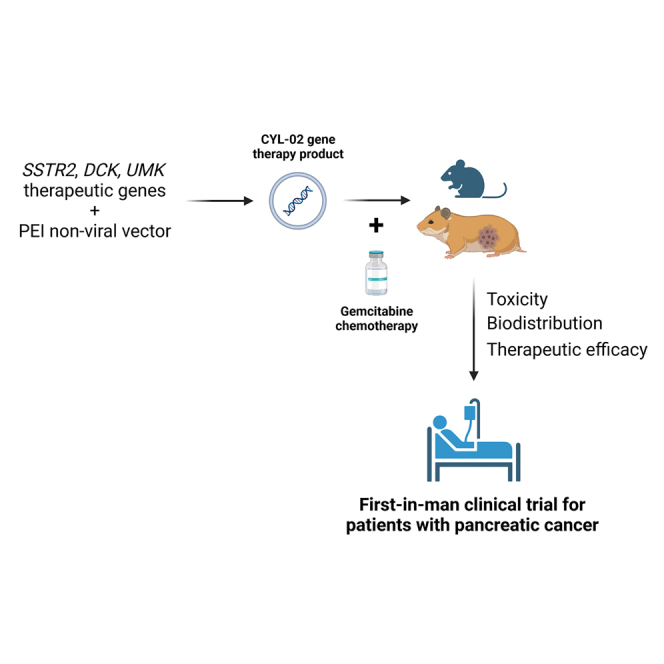
Cordelier et al. describe the preclinical development of a non-viral gene therapy product for patients with pancreatic cancer that encodes genes that inhibit tumor growth and sensitize to chemotherapy, from biodistribution to toxicity and therapeutic efficacy. This work stemmed for first-in-man clinical trials.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) accounts for 5% of cancer deaths worldwide and is projected to become the third leading cause of cancer death by 2025 because of an increasing incidence and stable mortality rate in countries with high human development index.1 Because of the long, silent clinical phase, during tumor development, and the absence of early markers that delay diagnosis, the prognosis of this cancer is very poor. When surgery is not possible, therapeutic options are few and ineffective.2 Patients are treated with Folfirinox or gemcitabine chemotherapy, which ameliorates few clinical parameters including pain intensity, analgesic consumption, and weight loss. Tumor volume is nearly unchanged and survival remains extremely short. Thus, more effective treatments are urgently needed to improve the prognosis of PDAC. However, the moderate activity of standard therapies strongly encourages new translational research programs such as gene therapy to potentiate the anti-tumoral effect of chemotherapy.
In this context, we demonstrated during the last two decades the anti-tumoral potential of the somatostatin receptor subtype 2 (SSTR2) in PDAC experimental models. SSTR2 expression is lost in 95% of PDAC3 and restoring SSTR2 using non-viral gene transfer results in a strong bystander anti-tumoral effect that is antiproliferative, pro-apoptotic, anti-angiogenic, and anti-metastatic.4,5,6,7,8,9 In addition, SSTR2 was found by others to sensitize tumor cells to gemcitabine.10 In parallel, we identified that the expression of deoxycytidine kinase (DCK), which phosphorylates gemcitabine to gemcitabine monophosphate in a rate-limiting step, is often lost in PDAC. Furthermore, DCK loss of expression is associated with acquired resistance to gemcitabine in pancreatic cancer cells, in preclinical models,11 and in patients.12 We demonstrated that using non-viral gene transfer to restore the expression of DCK, but also of uridylate monophosphate kinase (UMK, also named NMPK for nucleoside monophosphate kinase), which generates gemcitabine diphosphate gemcitabine monophosphate, overcomes PDAC-derived cells resistance to gemcitabine.13 Thus, there is a strong rationale to deliver SSTR2, DCK, and UMK coding sequences to sensitize tumors to chemotherapy and to treat PDAC with gemcitabine standard of care. In this work, we describe the preclinical development of the CYL-02 gene therapy product, encoding for SSTR2, DCK, and UMK delivered by a non-viral vector, that was later used in combination with gemcitabine in phase 114 and phase 2 (NCT02806687) clinical trials in patients with PDAC.
Results
Preclinical assessment of the toxicity of the gene therapy product
For this study, we generated plasmid DNA encoding for SSTR2, DCK, and UMK, the expression of which is targeted to tumor cells as described in the materials and methods. Therapeutic DNA is complexed with 22-kDa polycationic polyethyleneimine (in vivo jetPEI) as described before.14 We generated two pre-guanosine monophosphate (GMP) and one GMP batches as part of the pharmaceutical development of the gene therapy product, which we named CYL-01. We first aimed to determine the greatest amount of CYL-01 at which no detectable adverse effects occur in animal models (no observable adverse effect level), starting with 7,500 μg/kg as the toxic dose of PEI in mice.15 Thus, female and male C57Bl/6 mice were injected intravenously (i.v.) with 250 μg/kg, 1,500 μg/kg, and a maximal dose of 7,500 μg/kg the gene therapy product CYL-01. Control mice received CYL-01 excipient (5% glucose) as placebo; this is the resuspension solution of CYL-01, and mice were monitored for 5 weeks (Figure S1A). No mortality, no weight loss, no change in aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase (LDH), or creatinine levels were found in mice receiving an i.v. injection of 250 μg/kg or 1,500 μg/kg of the gene therapy product (Figures S2A–S2I). In contrast, one-half of the mice, two male mice (40%) and three female mice (60%), died within 24 h following i.v. injection of the highest dose of CYL-01. We performed a pathology analysis to better characterize the toxic effect following injection of CYL-01 in mice. Three of 15 (20%), and 4 of 30 (13%) male and female mice show mononuclear cells aggregates in the liver, respectively, regardless of the dose of CYL-01 (Figure 1A). Alveolar atelectasis was identified in the lungs of 11 of 30 (37%) and of 5 of 30 (17%) of male and female mice, respectively, regardless of the dose of CYL-01 (Figure 1B). Last, one mouse that received the highest dose of CYL-01 showed evidence of kidney congestion and small intestine autolysis (Figures 1C and 1D), as a result of agony before mouse killing. Taken together, i.v. injection of CYL-01 is safe in mice with a maximal tolerated dose (MTD) of 1,500 μg/kg.
Figure 1.

Toxicology study following a single i.v. injection of CYL-01 in mice
We administered 250 μg/kg, 1,500 μg/kg, and 7,500 μg/kg of CYL-01 i.v. in a total of 45 C57Bl/6 mice from either sex. Five weeks later, mice were killed and analyzed for toxic events associated with gene therapy administration such as (A) mononuclear cells aggregates in the liver of mice receiving 7,500 μg/kg CYL-01, (B) alveolar atelectasis in the lung of mice receiving 250 μg/kg CYL-01, (C) kidney congestion in mice receiving 7,500 μg/kg CYL-01, and (D) small intestine autolysis in mice receiving 7,500 μg/kg of CYL-01.
We then extended the toxicity study to an another animal model following the guidelines of the International Council for Harmonisation (ICH) of technical Requirements for Pharmaceuticals for Human Use (ICH M3; S6 and S9). According to regulatory guidelines, the dose conversion factor for MTD between mouse and hamster is 1.7,16 so that the calculated MTD for hamsters is 900 μg/kg. We generated orthotopic pancreatic tumors in Syrian golden hamsters as described in the materials and methods and Figure S1B. Seven days later, CYL-01 was administrated in tumors (i.t.), as the intended route of administration in humans, with a starting dose of 500 μg/kg pre-GMP grade CYL-01. Control animals were injected with 5% glucose (CYL-01 excipient). Animals received 80 mg/kg gemcitabine by intraperitoneal (i.p.) route on days 2, 4, and 6 following intra-tumoral gene transfer. No animal died during the experiment, with only mild body weight loss in hamsters receiving gemcitabine, or CYL-01 and gemcitabine (−21% ± 9%, p < 0.01) (Figure 2A). We next evaluated the toxicity associated with repeated injections of CYL-01, as this may induce unwanted adverse immune responses in patients. Thus, both male and female C57B/6 mice were injected i.v. with 250 μg/kg CYL-01 on days 0, 15, and 25 (Figure S1A). During this experiment, all animals injected survived and no organ-specific toxicity was detected. We next performed sequential injections in mice of 250 μg/kg CYL-01 i.v., followed by subcutaneous injection of 250 μg/kg of the gene therapy product on days 15, 25, and 40. Here again, animals were examined for systemic and cutaneous toxicities following each injection; no local adverse reactions were recorded.
Figure 2.

Toxicology study following a single intra-tumoral injection of the gene therapy product in orthotopic pancreatic tumors in Syrian golden hamsters
Experimental orthotopic PDAC tumors were induced as described in materials and methods. Eight days later, 500 μg/kg CYL-01 (A) or 900 μg/kg CYL-02 (B) were administered in exponentially growing tumors, when control animals received 5% glucose. Gemcitabine was given at 80 mg/kg i.p. every 2 days for a week. NaCl9°/00 was given i.p. as control. Hamster body weight was monitored from tumor induction, up to 7 days following treatment. n = 5 hamsters were used per group. White blood cells (C) and alkaline phosphatase (ALP) (D) monitoring in hamsters receiving an intra-tumoral injection of CTL-02 combined with gemcitabine treatment. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005.
In CYL-01, the resistance gene for bacterial selection (NEO) is co-expressed with SSTR2 cDNA using an internal ribosomal entry site. To minimize the risk of expressing antibiotic resistance gene in humans, we generated a second version of the gene therapy product, namely, CYL-02, in which the NEO gene expression is driven by a separate bacterial promoter. We next performed bridging toxicology studies to address whether CYL-01 and CYL-02 share the same toxicity profile in hamsters. Thus, experimental tumors were injected i.t. with pre-GMP grade CYL-02 at the theoretical MTD (900 μg/kg). Blood and urine were sampled from three animals from four independent experimental groups 15 min, 30 min, 1 h, 3 h, 6 h, 12 h, 18 h, and 24 h after injection. We did not observe any evidence of acute or systemic toxicities following CYL-02 intra-tumoral injection in experimental tumors in hamsters, and conclude that CYL-02 is safe in hamsters.
As for CYL-01, we next injected GMP-grade CYL-02 in exponentially growing pancreatic tumors in hamsters in combination with gemcitabine to fully capture the toxicity of the cell gene therapy product combined with chemotherapy. CYL-02 was not associated with animal death, with the exception of one hamster that also received gemcitabine (1/5 [20%]). This death was preceded by diarrhea and by the alteration of the general condition resulting in a significant weight loss (−15%) and lethargy. Autopsy revealed significant tumor invasion by regional extension in the spleen, stomach, liver, bowel, and colon. As shown in Figure 2B, animals receiving either CYL-02 or placebo gained weight with a similar trend (+9% ± 2.8% vs +8.9% ± 6.2%, respectively). In contrast, significant weight loss was measured when gemcitabine was administered alone (−4.1% ± 2%; p < 0.05) or in combination with CYL-02 (−3.6% ± 1%; p < 0.05). We identified leukopenia in the gemcitabine group and in combination with CYL-02, as white blood cells count significantly dropped as compared with control (Figure 2C) (−89% ± 2%, p < 0.005 and −94% ± 3%, p < 0.005, respectively). To a lesser extent, we also measured a significant decrease in alkaline phosphatase in hamsters treated by gemcitabine only (Figure 4D) (−56% ± 11%; p < 0.05). No changes in red blood cells or platelets counts, or in the level of AST, ALT, or creatinine were identified between the different groups (Figures S3A–S3D). Pathological examination of the organs during the autopsy did not reveal significant alterations in the brain, lung, heart, bladder, or muscles of the animals between the different study groups. However, we identified peritoneal alterations in all animals, which were probably caused by repeated laparotomies and i.p. injections. In addition, all study groups evidenced testicular inflammation, as a marker of abdominal tumor development. Last, one hamster of the five (20%) receiving gemcitabine alone demonstrated renal parenchyma alteration and central testicular necrosis without bladder involvement.
Figure 4.

Biodistribution study following intra-tumoral injection of CYL-02 in orthotopic pancreatic tumors in Syrian golden hamsters
Experimental orthotopic PDAC tumors were induced as described in materials and methods. (A) We administered 900 μg/kg CYL-02 in exponentially growing tumors, and urine and blood were sampled at the indicated time after gene transfer. As control, tumors were sampled and analyzed 24 h following intra-tumoral gene transfer of CYL-02. CYL-02 DNA was detected by qPCR for neomycin gene. Data are expressed as Ct of 3 biological replicates. (B) We administered 900 μg/kg CYL-02 at days 0 and 7 in exponentially growing tumors, when control animals received 5% glucose. Gemcitabine was given at 80 mg/kg i.p. every 2 days for 1 week following each injection. NaCl9°/00 was given i.p. as control. CYL-02 DNA was detected in the indicated organs and in tumors by qPCR for neomycin gene. Data are expressed as Ct ± SD for neomycin gene of five biological replicates per group with three experimental replicates. ∗∗∗p < 0.005.
We then questioned the safety of repeated intra-tumoral injection of CYL-02 combined with treatment with gemcitabine. Thus, pre-GMP grade CYL-02 was administered into tumors on days 0 and 7, and gemcitabine was given i.p. on days 2, 4, and 6 days (first cycle) and 9, 11, and 13 days (second cycle). While CYL-02 injection alone was safe, we found that animal survival was significantly shortened when receiving gemcitabine (p < 0.01) or CYL-02 + gemcitabine (p < 0.01) as compared to with controls (Figure 3A). Gemcitabine toxicity also translated into significant body weight loss when administered alone (−17% ± 10%; p < 0.05) or in combination with CYL-02 (−20% ± 4%; p < 0.01), as compared with controls (Figure 3B). Here again, CYL-02 is safe; it did not alter animal body weight (Figure 3B). Gemcitabine and CYL-02 combined with gemcitabine treatment resulted in leukopenia (Figure 3C), and we also identified low platelet count in hamsters receiving the chemotherapy only (Figure 3D). Red blood cell counts and the levels of AST, ALT, LDH, and creatinine remained unchanged between the different groups (Figures S4A–S4E). Thus, these data demonstrate the safety of the CYL-02 gene therapy product in hamster following two intra-tumoral injections. When combined with gemcitabine, CYL-02 did not aggravate chemotherapy toxicity. Collectively, toxicity studies reveal that the MTD for CYL-02 in mice is 1500 μg/Kg corresponding and 900μg/kg in Syrian golden hamsters. In both animal models, CYL-02 is safe, even following repeated administration, and does not aggravate gemcitabine toxicity.
Figure 3.

Toxicology study following two intra-tumoral injection of CYL-02 in orthotopic pancreatic tumors in Syrian golden hamsters
Experimental orthotopic PDAC tumors were induced as described in the materials and methods. We administered 500 μg/kg CYL-02 at days 0 and 7 in exponentially growing tumors, when control animals received 5% glucose. Gemcitabine was given at 80 mg/kg i.p. every 2 days for a week following each injection. NaCl9°/00 was given i.p. as control. Hamster body weight was monitored up to 14 days following the first intra-tumoral injection of the gene therapy product. n = 5 hamsters were used per group. (A) Probability of survival between the different experimental groups. (B) Hamster body weight, 15 days following the first intra-tumoral gene transfer. White blood cells (C) and platelets (D) monitoring in hamsters receiving two cycles of intra-tumoral injection of CYL-02 combined with gemcitabine treatment. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.001.
Preclinical study of the biodistribution of the CYL-02 gene therapy product
We performed a first set of studies to address the biodistribution of CYL-01 in female and male mice receiving 250, 1,500, or 7,500 μg/kg of the gene therapy product i.v. Eleven organs were analyzed by quantitative PCR (qPCR) for CYL-01 DNA detection, up to 6 weeks following injection. Table 1 shows that CYL-01 DNA is readily detectable in all organs tested, 6-h post-injection of female mice. Twenty-four hours later, we sampled blood and urine from animals receiving 250 μg/kg of the gene therapy product and found that only one-fourth of the urine samples tested were positive for CYL-01, when all blood samples were negative (data not shown). After 7 days, CYL-01 was detected inconsistently in the lung, liver, kidney, and the heart of animals that received 250 and 1,500 μg/kg of the gene therapy product. In addition, female mice receiving the highest dose of CYL-01 (7,500 μg/kg) showed a positive signal in muscles and brain. By 2 weeks, most male and female mice were free of CYL-01 (Table 1 and 2). Collectively, these data show that CYL-01 is only transiently detectable in lung, heart, liver, and pancreas when injected i.v.
Table 1.
Analysis of the biodistribution of CYL-01 in female mice using qPCR following i.v. injection at the indicated dose
| Lung | Kidney | Bowel | Heart | Muscle | Brain | Peritoneum | Pancreas | Spleen | Liver | Bladder | Gonads | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 250 μg/kg | ||||||||||||
| D7 | – | – | – | – | – | – | – | – | – | – | – | |
| D14 | – | – | – | – | – | – | – | – | – | – | – | – |
| D21 | – | – | – | – | – | – | – | – | – | – | – | + |
| D28 | – | – | – | – | – | – | – | + | – | + | – | – |
| D35 | – | – | – | + | – | + | – | – | – | – | – | – |
| 1,500 μg/kg | ||||||||||||
| D7 | ++ | ++ | – | ++ | – | – | – | – | + | – | + | |
| D14 | + | – | – | – | – | – | – | – | – | + | – | + |
| D21 | – | – | – | + | – | – | – | – | – | + | – | + |
| D28 | + | – | – | + | – | – | – | – | – | – | – | – |
| D35 | – | – | – | – | – | – | – | – | – | – | – | – |
| 7,500 μg/gg | ||||||||||||
| H6 | ++++ | +++ | ++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | + | +++ |
| D7 | ++ | – | – | + | + | ++ | – | – | – | – | – | – |
| D14 | – | – | – | – | – | – | – | – | – | – | – | – |
–, negative; +, 2.5 < ddCt<5; ++, 5 < ddCt<10; +++, 10 < ddCt<20; ++++, ddCt>20. ddCt = [(Ctneo-CtGAPDH) in organ from the injected group] – [(Ctneo-CtGAPDH) in organ from the non-injected group).
Table 2.
Analysis of the biodistribution of CYL-01 in male mice using qPCR following i.v. injection at the indicated dose
| Lung | Kidney | Bowel | Heart | Muscle | Brain | Peritoneum | Pancreas | Spleen | Liver | Bladder | Gonads | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 250 μg/kg | ||||||||||||
| D7 | + | – | – | – | – | + | – | – | – | – | – | – |
| D14 | – | – | – | – | – | – | – | – | – | – | – | – |
| D21 | – | – | – | – | – | – | – | – | – | – | – | – |
| D28 | – | – | – | – | – | – | – | – | – | – | – | – |
| D35 | – | – | – | – | – | – | – | – | – | – | – | – |
| 1,500 μg/kg | ||||||||||||
| D7 | – | – | – | – | – | – | – | – | – | – | – | |
| D14 | – | – | – | – | – | – | – | – | – | – | – | – |
| D21 | – | – | – | + | – | – | – | – | – | – | – | – |
| D28 | – | – | – | – | – | – | – | – | – | – | – | – |
| D35 | – | – | – | – | – | – | – | – | – | – | – | – |
| 7,500 μg/kg | ||||||||||||
| D7 | ++ | + | – | – | – | – | + | – | + | + | + | + |
| D14 | – | – | – | – | – | – | – | – | – | – | – | – |
–, negative; +, 2.5 < ddCt<5; ++, 5 < ddCt<10; +++, 10 < ddCt<20; ++++, ddCt>20. ddCt = [(Ctneo-CtGAPDH) in organ from the injected group] – [(Ctneo-CtGAPDH) in organ from the non-injected group).
We further analyzed the biodistribution of CYL-01 in hamsters following intra-tumoral injection of 500 μg/kg of the gene therapy product (day 0), followed by treatment with gemcitabine that was given i.p. on days 2, 4, and 6. Organs (spleen, liver, lung, kidney, and bowel) and tumors were sampled 7 days following gene transfer. Table 3 shows that the level of CYL-01 is high in tumors, and in the liver and, to a lesser extent, the spleen of animals receiving CYL-01 and gemcitabine. We then moved to CYL-02 as mentioned before. Hamster pancreatic tumors were injected with 900 μg/kg pre-GMP CYL-02, and urine and blood were sampled 15 min, 30 min, 1 h, 3 h, 6 h, 12 h, 18 h, and 24 h later. Tumors were also collected 24 h following the intra-tumoral injection of the gene therapy product. Figure 4A shows that CYL-02 is not detected in urine (0/24), and occasionally in the blood 6 h (1/3 hamsters, corresponding with 0.1% of the dose injected) or 24 h (1/3 hamsters, corresponding with 2.3% of the dose injected) following intra-tumoral injection. However, experimental tumors showed high levels of the gene therapy product. We conclude that CYL-02 is largely sequestered in the tumor with an inconstant and delayed presence of a small quantity of the gene therapy product in the vascular compartment. We then performed repeated intra-tumoral injection of pre-GMP CYL-02 in pancreatic tumors in hamsters followed by i.p. injection of gemcitabine. Fourteen days later, the hamsters were killed and CYL-02 was detected by qPCR in the tumor, pancreas, lung, liver, bowel, gonad, kidney, bladder, spleen, heart, striated muscle, brain, and peritoneum. As shown in Figure 4B, a significant increase in CYL-02 was identified in tumors from animal receiving the gene therapy product up to 2 weeks following intra-tumoral injection (+8.75 ± 1.3-fold increase; p < 0.001). Thus, we demonstrate that CYL-02 is cleared from organs 1 month following i.v. injection and is sequestered in tumors up to 7 days following intra-tumoral injection with minimal, early, exposure in the blood.
Table 3.
Analysis of the biodistribution of CYL-01 in hamsters using qPCR, 7 days following intra-tumoral injection of the GTP
| Lung | Kidney | Bowel | Spleen | Liver | Tumor | |
|---|---|---|---|---|---|---|
| CYL-01 | – | – | – | + | ++ | ++ |
| CYL-01 + gemcitabine | – | + | – | ++ | +++ | +++ |
–, negative; +, 2.5 < ddCt<5; ++, 5 < ddCt<10; +++, 10 < ddCt<20; ++++, ddCt>20. ddCt = [(Ctneo-CtGAPDH) in organ from the injected group] – [(Ctneo-CtGAPDH) in organ from the non-injected group).
Characterization of CYL-02 activity in preclinical models of PDAC
We next analyzed SSTR2, DCK, and UMK expression following the in vitro transfection of hamster pancreatic cancer cells. Thus, PC.1-0 cells were transfected with 1 μg CYL-02. Control cells received 5% glucose. Forty-height hours later, therapeutic gene expression was quantified by reverse transcription followed by qPCR (RT-qPCR), as described before.14 Figure 5A demonstrates that CYL-02 strongly increases SSTR2 (844-fold ± 307; p < 0.01), DCK (249-fold ± 123; p < 0.01), and UMK expression (516-fold ± 406; p < 0.05) in transfected cells as compared with control cells.
Figure 5.

Pre-clinical characterization of CYL-02 activity and anti-tumoral efficacy in orthotopic pancreatic tumors in Syrian golden hamsters
(A) PC.1-0 hamster pancreatic cancer cells were transfected with CYL-02 as described in materials and methods, and SSTR2, DCK, and UMK gene expression analysis was performed as described elsewhere.14 Results are mean of n = 11 independent experiments performed in duplicate and expressed as mean fold change ± SD between control and transfected cells, using 18S as an internal control. (B) PC.1-0 hamster pancreatic cancer cells were transfected with CYL-02 and treated by gemcitabine as described in the materials and methods. Three days later, cells were counted. Results are mean ± SD of 9 (control, gemcitabine) or 36 (CYL-02, CYL-02 + gemcitabine) independent experiments. (C) Experimental orthotopic PDAC tumors were induced as described in the materials and methods. Eight days later, 900 μg/kg CYL-02 were administered in exponentially growing tumors, when control animals received 5% glucose. Gemcitabine was given at 80 mg/kg i.p. every 2 days for a week. NaCl9°/00 was given i.p. as control. At the end of the experiments, mice were killed and DCK::UMK and SSTR2 gene expression was measured in control tumors and CYL-02-treated tumors, 8 days following gene therapy. Data are means ± SD of five biological replicates per group with three experimental replicates and expressed as arbitrary units (2−ΔCt with ΔCt = CT(DCK::UMK or SSTR2) – CT(18S)). D. SSTR2 protein was detected by immunochemistry in control and CYL-02-treated experimental tumors 8 days following gene therapy. Sixteen fields were analyzed per condition. Data are representative of four biological replicates per group with three experimental replicates. All images: original magnification, ×20. Scale bar, 30 μm (valid for all images). (E) Mean ± SD of the % of SSTR2-positive cells per field. Tumor progression (F) and dissemination (G) at endpoint between the different groups. Results are mean fold ± SD of tumor progression in n = 23–24 animals per group, sum of 5 independent experiments, and of tumor dissemination (n = 20 for each condition, sum of 4 independent experiments). The dashed line indicates no progression (fold = 1). Following autopsy, tumors were sampled and analyzed for PCNA expression (H) and terminal uridine nick-end labeling (TUNEL) assay (J). Fifteen fields were analyzed per condition. Data are representative of six biological replicates from two experimental replicates. All images: original magnification, ×40. Scale bar, 60 μm (valid for all images). Mean ± SD of the % of PCNA-(I) or TUNEL (K)-positive cells per fields. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.001. ####p < 0.001 gemcitabine vs CYL-02 + gemcitabine.
We then performed cell proliferation studies to document the activity of CYL-02. Thus, PC.1-0 cells were transfected with 1 μg CYL-02 and treated or not by gemcitabine 48 h later. Control cells received either 5% glucose and/or NaCl 90/00 as controls. Forty-eight hours later, cells were counted. Results shown in Figure 5B demonstrate that gemcitabine significantly inhibits the proliferation of PC.1-0 cells (−71% ± 12%; p < 0.0001). Remarkably, CYL-02 transfection strongly inhibits hamster PDAC cells proliferation (−94% ± 5%; p < 0.0001), and combination of CYL-02 and gemcitabine treatment led to nearly complete eradication of the tumor cells population (−97% ± 6%; p < 0.0001).
We next generated PDAC experimental orthotopic tumors following engraftment of PC1.-0 cells in the pancreas of immune competent Syrian golden hamsters. Seven days later, tumors received an intra-tumoral injection of 900 μg/kg GMP CYL-02. Animals were treated on days 9, 11, and 13 following engraftment with 80 mg/kg gemcitabine, when control animals received NaCl 90/00. Hamsters were killed 15 days following tumor engraftment. SSTR2, DCK, and UMK expression were monitored by qPCR in tumors as previously described.14 Results shown in Figure 5C demonstrate that CYL-02 transfection strongly increases the intra-tumoral expression of the therapeutic genes (p < 0.005). We next performed immunohistochemistry for SSTR2 expression (Figure 5D), and found that 16.25% ± 7.4% of tumor cells expressed the therapeutic transgene (Figure 5E). Last, we questioned the anti-tumoral activity of CYL-02 intra-tumoral injection, in the presence or not of gemcitabine. Thus, experimentally growing tumors were injected with GMP CYL-02, then treated with gemcitabine as described before. Hamsters were killed and tumors were measured and sampled 8 days following gene transfer. We found that gemcitabine resulted in the significant inhibition of the tumor growth (−74% ± 12%; p < 0.001). Four of 24 animals (16%) showed tumor regression. CYL-02 significantly inhibited tumor growth (−66% ± 12%; p < 0.005), with 1 of 23 animals (4%) showing tumor regression. Remarkably, combining CYL-02 and gemcitabine showed stronger inhibition of tumor growth as compared with controls (−89% ± 12%; p < 0.001) or chemotherapy alone (p < 0.001). Combination therapy resulted in tumor regression in 15 of 24 animals (63%). We obtained similar results using pre-GMP CYL-02 (Figure S5A). We next analyzed the metastatic propensity of tumors from the different groups. We found that 86.5% ± 3% of animals from the control group presented with metastasis, mainly in the liver and in the peritoneum (Figure 5F). Gemcitabine or CYL-02 administration significantly reduced tumor dissemination (−41% ± 11% and −49% ± 27%, respectively; p < 0.05). Here again, the combination of CYL-02 and gemcitabine demonstrate stronger inhibition of tumor dissemination (−81.5% ± 8%; p < 0.005) and antimetastatic activity when compared with chemotherapy alone (p < 0.005). Collectively, these results demonstrate that CYL-02 significantly sensitizes tumor cells to chemotherapy, with more frequent tumor regression and less metastatic dissemination as compared with gemcitabine or CYL-02 administration alone. Last, we investigated the molecular mechanisms involved in the anti-tumoral activity of CYL-02 gene therapy when combined to gemcitabine chemotherapy. We found that tumor cell proliferation was significantly inhibited (proliferating cell nuclear antigen [PCNA] labeling; −59 ± 5%; p < 0.05) (Figure 5G), when cancer cell death by apoptosis was significantly increased (terminal uridine nick-end labeling assay; 11.25 ± 0.16-fold increase; p < 0.001) (Figure 5H). Collectively, we demonstrate herein that CYL-02 that encodes for SSTR2, DCK, and UMK cDNA complexed with PEI non-viral vector is safe and shows promising anti-tumoral and antimetastatic potential for the non-viral gene therapy of patients with PDAC.
Discussion
PDAC is characterized by a unique ability to withstand therapeutic aggression, and current treatments, mainly chemotherapies, are ineffective; they increase survival times of patients only by weeks to months.2 In previous studies, we demonstrated that SSTR2, DCK, and UMK genes have strong potential to limit cancer cell proliferation, dissemination, and sensitize tumors to gemcitabine chemotherapy, respectively. This was achieved following in vivo intra-tumoral gene transfer using PEI non-viral vector in very aggressive PDAC experimental models.4,5,13 These findings advocated for clinical exploration, but required proper development and characterization of the candidate gene therapy product.
Meanwhile, we deliberately excluded viral vectors that may improve therapeutic gene transduction and expression, but that come with limitations, such as cloning capacity for adeno-associated virus, or even threats, illustrated by the risk of insertional mutagenesis caused by host genome integration by lentiviral vectors, or supra-physiological inflammation and risk of pancreatitis due to adenoviral transduction. As long-term gene expression is not a priority in cancer gene therapy, and considering the unrivalled safety of non-viral vectors, we confirmed PEI as the gene delivery vehicle to be used for the production of the gene therapy product. Another technical innovation to improve safety is the use of promoters to limit transgene expression to target tumor cells. Glucose-regulated protein 78 (GRP78) and 94 (GRP94) are chaperone proteins that are induced at the transcriptional level following endoplasmic reticulum stress to regulate unfolded protein response and apoptosis.17 Both proteins were shown to be involved in PDAC early stages or late dissemination.18 Thus, we selected GRP78 and GRP94 promoters to drive DCK::UMK and SSTR2 expression, respectively. These promoters were also chosen as they share common regulatory factors and are coordinately regulated into cells,19 so as to avoid transcriptional competition.
Safety has long been a primary concern in gene therapy research, particularly after the death of a gene therapy trial participant.20 During this work, we identified that the MTD of the gene therapy product in mice was of 1,500 μg/kg of body weight. Pathological analysis further revealed mononuclear aggregates in the liver, which are generally found in mice and should not be attributed to the injection of a toxic substance, kidney congestion, and bowel autolysis, due to dying mice, and alveolar atelectasis, which usually indicates a defect in tissue fixation. We confirmed the lack of toxicity and the excellent safety profile of CYL-02 in Syrian golden hamsters; no animal death was recorded and control and CYL-02 injected animals grew similarly. However, significant body weight loss, leukopenia, and animal death were recorded when gemcitabine was combined or not with the gene therapy. However, we found that CYL-02 administration did not aggravate chemotherapy toxicity. We then explored whether the gene therapy product may induce unwanted immune responses that could hamper translation to the clinic. Repeated injection of CYL-01 or CYL-02 did not result in acute or systemic reaction following, regardless of the route of administration. Collectively, we demonstrate here that the gene therapy product is safe, with an MTD of 1,500 μg/kg in mice corresponding with 900 μg/kg in hamsters when associated with chemotherapy, respectively. To calculate the human equivalent dose (HED), the MTD in mice was divided by 12.3 according to regulatory guidelines16 to give 122 μg/kg. Dividing HED values by a safety or uncertainty factor is the common procedure to calculate a maximum recommended starting dose (MRSD) and decrease the risk of possible adverse effects in humans.16,21 We used an uncertainty factor of 30 as the gene therapy product classifies as a nanoparticle, so that the MRSD for CYL-02 in humans is 4 μg/kg. As one hamster receiving 900 μg/kg CYL-02 showed frailty, we added an additional safety factor of 2, to define 2 μg/kg as the starting dose of CYL-02 in humans. Extrapolating by body weight, the minimum injected DNA dose to be tested in the first injection in humans was 125 μg for a 60-kg male or female.14
We next addressed the biodistribution of CYL-02 in mice and in preclinical models of PDAC. Importantly, PEI the amplification of the plasmid DNA by PCR (data not shown). This indicates that positive signal by PCR is tangible proof of the delivery and of the release of the therapeutic DNA into cells. We demonstrate that CYL-02 is transiently detected in organs following i.v. injection and is sequestered in the tumor following intra-tumoral injection with low diffusion in the vascular compartment (<2.5% of the initial dose). Importantly, CYL-02 was not detected in urine from hamsters following i.v. injection. This suggests that CYL-02 is not free, probably intra-globular, because it is not eliminated in the urine. This was later confirmed in humans receiving the gene therapy product.14 Interestingly, when used alone, CYL-02 is not detected in the lung, kidney, or bowel. In association with gemcitabine, kidney and bowel show low levels of the gene therapy product, in sharp contrast with high levels of CYL-02 in spleen. We speculate that this may reflect the microsomal release of the gene therapy product caused by tumor cell lysis in the presence of gemcitabine, which is in line with the expected pharmacological action of the combination. Although toxicity studies showed CYL-02 to be safe, and although tissue biodistribution studies and respiratory system monitoring did not show pulmonary toxicity, we nevertheless recommended pulmonary function monitoring in follow-up clinical studies, because the complex injected is considered as a nano-vector, so that it can eventually forms micro-aggregates. Last, we recommended the monitoring of cardiac, renal, and hepatic function; we found traces of the gene therapy product in these organs when injected at the highest dose.
In this work, we addressed for the first time the anti-tumoral potential of combining SSTR2, DCK, and UMK gene transfer all together with chemotherapy treatment. We found that CYL-02 delivery resulted in comparable levels of expression of the three therapeutic genes, in cell lines and tumors, even if we identified a non-significant trend for lower expression of DCK and UMK as compared with SSTR2. This validates the choice of the two promoters that drive therapeutic gene expression. We found that almost 20% of cancer cells were transfected in vivo using CYL-02, which is in line with our previous experience with commercial PEI.4 This validates that CYL-02 processing does not alter therapeutic gene delivery or expression into cancer cells. Next, we demonstrated that CYL-02 treatment strongly sensitizes PDAC cells to chemotherapy, both in vitro and in vivo, with increased anti-tumoral efficacy and more frequent tumor regression as compared with gemcitabine alone. Although comparisons are difficult to make, this surpasses the anti-tumoral effect following SSTR2 gene transfer alone,4 or combination of DCK::UMK gene transfer with gemcitabine treatment.13 In addition, CYL-02 therapy decreased the capacity of tumor cells to disseminate, as previously found with SSTR2 monotherapy.22 The latter strongly suggests that the anti-tumoral properties of SSTR2 and DCK and UMK are complementary to inhibit the growth of very aggressive PDAC tumors and are preserved in the CYL-02 gene therapy product. However, proper evaluation of possible synergism of the four active agents would have required matrix designs23 and/or synergy scoring models,24 as the results may have further influenced the dosing regimen for the clinical trial. Last, we found that CYL-02 combined with gemcitabine inhibits tumor cell proliferation and induces cell death by apoptosis. Still, one of the major limitation of this work is that we did not use during this study transgenic animal models of PDAC, such as KPC mice,25,26 as they were not available at the time the experiments were performed. These mice would also be instrumental to question the potential of CYL-02 and gemcitabine to induce an immune response against tumors.
Collectively, we provide during this work evidence that the CYL-02 gene therapy product generated under pre-GMP and GMP conditions for a pharmaceutical development is safe in pre-clinical models and strongly inhibits PDAC experimental growth when combined with gemcitabine. In addition, we determined the MTD that was later used to define the starting dose of CYL-02 that was administered in humans.14 In addition, this work generated analytical procedures to explore the biodistribution of the gene therapy product in patients and also recommendation for organs monitoring during and after intra-tumoral gene delivery. Based on these findings, 57 patients with PDAC were treated by CYL-02 gene therapy combined with gemcitabine and no adverse events directly related to the gene therapy drug were recorded. In some patients, CYL-02 and chemotherapy treatment resulted in tumor control, which advocated for a phase II study that was closed this year. Taken together, we demonstrate herein that rigorous and thorough preclinical investigations are essential for the clinical transfer of gene therapy against PDAC.
Materials and methods
Animal studies
Experimental procedures performed on mice and hamsters were approved by the ethical committee of INSERM CREFRE US006 animal facility and authorized by the French Ministry of Research: APAFIS#3600-2015121608386111v3. C57Bl/6 mice were obtained from Charles River and Syrian Golden hamsters from Harlan.
Experimental tumor induction in Syrian golden hamsters
Hamster PDAC-derived PC-1.0 cells are grown in RPMI medium supplemented with 10% fetal calf serum, L-glutamine, antibiotics, antimycotics (Life Technologies), and Plasmocin (InvivoGen) in a humidified incubator at 37°C in 5% CO2. Six-week-old male Syrian golden hamsters were anesthetized by intraperitoneal injection of pentobarbital (80 mg/kg) diluted in NaCl 90/00, supplemented with oral anesthesia using oxygen/isoflurane (2.5 mixture), and PC-1.0 cells were implanted in the tail of pancreas as previously described.27
Gene therapy product
CYL-02 is a complex of plasmid DNA and linear polymers of polyethyleneimine (in vivo-jetPEI 22-kDa from Polyplus), prepared in 5% w/v glucose with a PEI nitrogen to DNA phosphate ratio of 8–10. The plasmid within the gene therapy product encodes for DCK:UMK cDNAs (separated by the self-cleaving FMDV 2A peptide), the expression of which is driven by the glucose-regulated protein 78 (GRP78) promoter, and the human SSTR2 cDNA, the expression of which is driven by the glucose-regulated protein 94 (GRP94) promoter. Briefly, GRP78 and GRP94 are chaperone proteins that are induced following endoplasmic reticulum stress to regulate unfolded protein response and apoptosis,17 that were shown to be involved in PDAC early stages or late dissemination.18 These promoters share common regulatory factors and are coordinately regulated into cells,19 so to avoid transcriptional competition. The prokaryotic promoter-driven neomycin gene is used for bacterial selection and biodistribution and pharmacokinetic studies. The gene therapy product is assembled and lyophilized by InvivoGen following good medical product guidelines. Lyophilized CYL-02 was reconstituted 10 min before use by adding 2.5 mL sterile water for the injectable preparation. Therapeutic DNA and RNA were detected as described elsewhere.14
In vitro analysis of CYL-02 activity and therapeutic efficacy
We incubated 5 × 10e5 PC.1-0 cells with 1 μg CYL-02 and plated in 100-mm dishes. Three days later, the culture medium was removed, cells were washed using PBS, and cellular RNA was extracted using the RNAeasy kit from Qiagen. SSTR2, DCK, and UMK expression was quantified by RT-qPCR as described before.14 For proliferation studies, 1 × 10e5 PC.1-0 cells were plated in 6-well plates in the presence of 1 μg CYL-02. Two days later, cells were treated with 20 nM gemcitabine and cells were counted 3 days later using the Z1 Coulter (Beckman).
In vivo delivery of CYL-02 gene therapy product
Following laparotomy, tumors were measured using a caliper and randomized with mean = 100 mm3 in size as described before.27 CYL-02 was administrated in exponentially growing orthotopic tumors 8 days following tumor induction as previously described.27 Control animals received 5% glucose. Gemcitabine (80 mg/kg) was injected i.p. every 2 days for 1 week, 48 h after CYL-02 intra-tumoral injection. Control animals received NaCl 90/00 i.p. At 8 days after CYL-02 injection, animals were killed, primary tumors were measured using a caliper, and tumor volume was calculated as described before.27 Lung, liver, and peritoneal macro-metastases were counted.
Experimental tumor analysis
PC.1-0 tumors were harvested and fixed in formalin. Four-micrometer-thick sections were prepared from paraffin-embedded sections and rehydrated. DNA fragmentation was performed using In situ Apoptosis Detection Kit according to the manufacturer’s instructions (Takara Bio Inc.). For immunostaining, sections were incubated for 10 min in Protein Block, Serum-free reagent following antigen retrieval to reduce background staining (DakoCytomation). Slides were next incubated overnight at 4°C with anti-PCNA (DakoCytomation, clone PC10, ref M0879, dilution: 1:100), or SSTR2 antibodies (AbCAM clone [UMB1] ref ab134152, dilution: 1:100) diluted in Antibody diluent (DakoCytomation). Slides were washed and incubated in 3% H2O2 for 30 min at room temperature for endogenous peroxidase inhibition, quickly rinsed in distilled water, washed twice in PBS, and incubated for 30 min at room temperature with Envision+ system-HRP (DakoCytomation). After one wash in distilled water, slides were incubated in AEC+ reagent and counterstained with Mayer’s hematoxylin. Immunostaining was recorded with an optical microscope and quantified using a VisioLab2000 image analyzer (Biocom). For each sample, 15 fields were analyzed.
Statistical analysis
Unpaired Student’s t test or Wilcoxon Mann-Whitney tests were used to determine the statistical significance of differences between two groups using GraphPad Prism 9 software with the default settings. Methods of statistical analysis are indicated in the figure captions. Values are presented as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.0001. Error bars are SEM unless otherwise stated. The experiments were performed with a sample size ngreater than or equal to three replicates. When monitoring tumor growth, the investigators were blinded to the group allocation but were aware of group allocation when assessing the outcome. No data were excluded from the analyses.
Data availability statement
Most of the data are included in the manuscript. Additional data supporting the findings of this study are available from the corresponding author upon request.
Acknowledgments
In loving memory of Prof. Gérard Tiraby (Invivogen), who was instrumental to this project. The authors thank Annie Souque for technical counseling and Severine Joubert for supervision counseling. Financial support provided by Région Midi-Pyrénées APRTCN 2006 N° 0401401 and APRTCN 2011 N° 12050667, ANR-RIB 07, Inserm Cossec, and INVIVOGEN.
Author contributions
Study concept and design: L.B., P.C., F.G., and H.B.T.; experimental procedures: O.B., H.L., N.H., F.V., and G.C.; data collection: L.B., F.G., P.C., O.B., and H.L.; data analysis: L.B., F.G., P.C., O.B., and H.B.T.; supervision: L.B., P.C., F.G., M.T., and S.J.; data interpretation: L.B., F.G., P.C., and H.B.T.; manuscript writing: P.C.; manuscript revision: F.G., L.B., and P.C.
Declaration of interests
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.03.005.
Contributor Information
Fabian Gross, Email: gross.f@chu-toulouse.fr.
Pierre Cordelier, Email: pierre.cordelier@inserm.fr.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Neoptolemos J.P., Kleeff J., Michl P., Costello E., Greenhalf W., Palmer D.H. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018;15:333–348. doi: 10.1038/s41575-018-0005-x. [DOI] [PubMed] [Google Scholar]
- 3.Buscail L., Saint-Laurent N., Chastre E., Vaillant J.C., Gespach C., Capella G., Kalthoff H., Lluis F., Vaysse N., Susini C. Loss of sst2 somatostatin receptor gene expression in human pancreatic and colorectal cancer. Cancer Res. 1996;56:1823–1827. [PubMed] [Google Scholar]
- 4.Carrere N., Vernejoul F., Souque A., Asnacios A., Vaysse N., Pradayrol L., Susini C., Buscail L., Cordelier P. Characterization of the bystander effect of somatostatin receptor sst2 after in vivo gene transfer into human pancreatic cancer cells. Hum. Gene Ther. 2005;16:1175–1193. doi: 10.1089/hum.2005.16.1175. [DOI] [PubMed] [Google Scholar]
- 5.Vernejoul F., Faure P., Benali N., Calise D., Tiraby G., Pradayrol L., Susini C., Buscail L. Antitumor effect of in vivo somatostatin receptor subtype 2 gene transfer in primary and metastatic pancreatic cancer models. Cancer Res. 2002;62:6124–6131. [PubMed] [Google Scholar]
- 6.Rochaix P., Delesque N., Estève J.P., Saint-Laurent N., Voight J.J., Vaysse N., Susini C., Buscail L. Gene therapy for pancreatic carcinoma: local and distant antitumor effects after somatostatin receptor sst2 gene transfer. Hum. Gene Ther. 1999;10:995–1008. doi: 10.1089/10430349950018391. [DOI] [PubMed] [Google Scholar]
- 7.Cordelier P., Bienvenu C., Lulka H., Marrache F., Bouisson M., Openheim A., Strayer D.S., Vaysse N., Pradayrol L., Buscail L. Replication-deficient rSV40 mediate pancreatic gene transfer and long-term inhibition of tumor growth. Cancer Gene Ther. 2007;14:19–29. doi: 10.1038/sj.cgt.7700987. [DOI] [PubMed] [Google Scholar]
- 8.Delesque N., Buscail L., Estève J.P., Saint-Laurent N., Müller C., Weckbecker G., Bruns C., Vaysse N., Susini C. sst2 somatostatin receptor expression reverses tumorigenicity of human pancreatic cancer cells. Cancer Res. 1997;57:956–962. [PubMed] [Google Scholar]
- 9.Guillermet J., Saint-Laurent N., Rochaix P., Cuvillier O., Levade T., Schally A.V., Pradayrol L., Buscail L., Susini C., Bousquet C. Somatostatin receptor subtype 2 sensitizes human pancreatic cancer cells to death ligand-induced apoptosis. Proc. Natl. Acad. Sci. USA. 2003;100:155–160. doi: 10.1073/pnas.0136771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sui C., Ma Q., Nan K., Xiao J., Suo A., Sha H., Zhao L. hSSTR2 expression and octreotide treatment reverses multidrug resistance of BxPC-3 human pancreatic cancer cells. Oncol. Rep. 2009;22:1391–1396. doi: 10.3892/or_00000579. [DOI] [PubMed] [Google Scholar]
- 11.Ohhashi S., Ohuchida K., Mizumoto K., Fujita H., Egami T., Yu J., Toma H., Sadatomi S., Nagai E., Tanaka M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008;28:2205–2212. [PubMed] [Google Scholar]
- 12.Maréchal R., Bachet J.-B., Mackey J.R., Dalban C., Demetter P., Graham K., Couvelard A., Svrcek M., Bardier-Dupas A., Hammel P., et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology. 2012;143:664–674.e6. doi: 10.1053/j.gastro.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Vernejoul F., Ghénassia L., Souque A., Lulka H., Drocourt D., Cordelier P., Pradayrol L., Pyronnet S., Buscail L., Tiraby G. Gene therapy based on gemcitabine chemosensitization suppresses pancreatic tumor growth. Mol. Ther. 2006;14:758–767. doi: 10.1016/j.ymthe.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Buscail L., Bournet B., Vernejoul F., Cambois G., Lulka H., Hanoun N., Dufresne M., Meulle A., Vignolle-Vidoni A., Ligat L., et al. First-in-man phase 1 clinical trial of gene therapy for advanced pancreatic cancer: safety, biodistribution, and preliminary clinical findings. Mol. Ther. 2015;23:779–789. doi: 10.1038/mt.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chollet P., Favrot M.C., Hurbin A., Coll J.-L. Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. J. Gene Med. 2002;4:84–91. doi: 10.1002/jgm.237. [DOI] [PubMed] [Google Scholar]
- 16.Nair A.B., Jacob S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016;7:27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee A.S. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen J., Rangel D.F., Ha D., Lee A.S. New role of endoplasmic reticulum chaperones in regulating metaplasia during tumorigenesis. Mol. Cell. Oncol. 2017;4:e1345350. doi: 10.1080/23723556.2017.1345350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang S.C., Erwin A.E., Lee A.S. Glucose-regulated protein (GRP94 and GRP78) genes share common regulatory domains and are coordinately regulated by common trans-acting factors. Mol. Cell Biol. 1989;9:2153–2162. doi: 10.1128/mcb.9.5.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson J.M. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol. Genet. Metabol. 2009;96:151–157. doi: 10.1016/j.ymgme.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Guidance for Industry on Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; Availability. Federal Register; 2005. https://www.federalregister.gov/documents/2005/07/22/05-14456/guidance-for-industry-on-estimating-the-maximum-safe-starting-dose-in-initial-clinical-trials-for [Google Scholar]
- 22.Qin C., Yang G., Yang J., Ren B., Wang H., Chen G., Zhao F., You L., Wang W., Zhao Y. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol. Cancer. 2020;19:50. doi: 10.1186/s12943-020-01169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mäkelä P., Zhang S.M., Rudd S.G. Drug synergy scoring using minimal dose response matrices. BMC Res. Notes. 2021;14:27. doi: 10.1186/s13104-021-05445-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malyutina A., Majumder M.M., Wang W., Pessia A., Heckman C.A., Tang J. Drug combination sensitivity scoring facilitates the discovery of synergistic and efficacious drug combinations in cancer. PLoS Comput. Biol. 2019;15:e1006752. doi: 10.1371/journal.pcbi.1006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 26.Hingorani S.R., Petricoin E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A., et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 27.Benali N., Cordelier P., Calise D., Pages P., Rochaix P., Nagy A., Esteve J.P., Pour P.M., Schally A.V., Vaysse N., et al. Inhibition of growth and metastatic progression of pancreatic carcinoma in hamster after somatostatin receptor subtype 2 (sst2) gene expression and administration of cytotoxic somatostatin analog AN-238. Proc. Natl. Acad. Sci. USA. 2000;97:9180–9185. doi: 10.1073/pnas.130196697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Most of the data are included in the manuscript. Additional data supporting the findings of this study are available from the corresponding author upon request.