

Abstract
Natural killer (NK) cells are potent cytotoxic innate lymphocytes that can be used for cancer immunotherapy. Since the balance of signals from activating and inhibitory receptors determines the activity of NK cells, their anti-tumor activity can be potentiated by overexpressing activating receptors or knocking out inhibitory receptors via genome engineering, such as chimeric antigen receptor (CAR) transgenesis and CRISPR-Cas9-mediated gene editing, respectively. Here, we report the development of a one-step strategy for CRISPR-Cas9-mediated gene knockout and CAR transgenesis in NK cells using retroviral particles. We generated NK cells expressing anti-epidermal growth factor receptor (EGFR)-CAR with simultaneous TIGIT gene knockout using single transduction and evaluated the consequence of the genetic modifications in vitro and in vivo. Taken together, our results demonstrate that retroviral particle-mediated engineering provides a strategy readily applicable to simultaneous genetic modifications of NK cells for efficient immunotherapy.
Keywords: natural killer cells, genetic engineering, CRISPR-Cas9, virus-like-particles, retroviral particles, chimeric antigen receptors, TIGIT, EGFR, flow virometry, immunotherapy
Graphical abstract
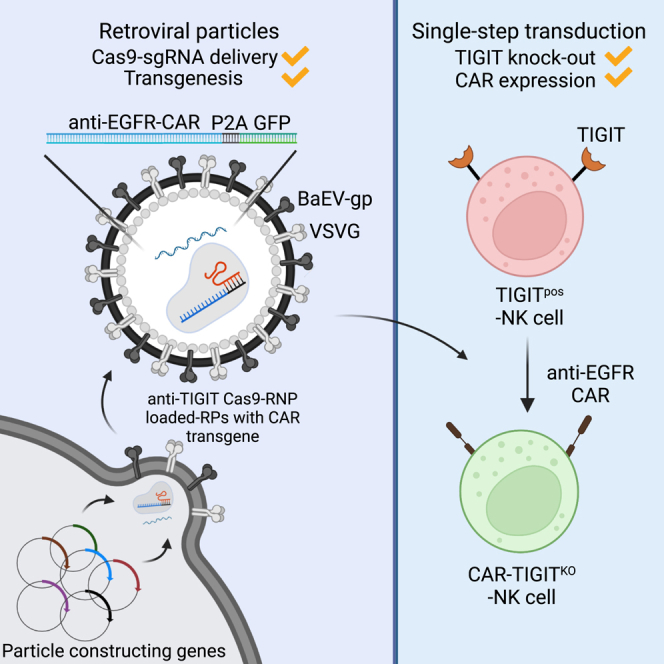
Jo and colleagues found that the CRISPR-Cas9 protein delivery system using retroviral particles is efficient for NK cell gene engineering, allowing simultaneous gene knockout and CAR transgenesis. This technique will provide a strategy readily applicable to potentiate NK cells for efficient immunotherapy.
Introduction
Chimeric antigen receptors (CARs) are hybrid antigen receptors that are composed of an extracellular antigen binding domain and intracellular signaling domains.1,2 Even though CAR-T therapies have shown dramatic efficacy in the treatment of B cell malignancies, strong CAR-T cell responses against tumors can drive the production of an overwhelming amount of cytokines, known as cytokine release syndrome (CRS).2 In addition, the threat of graft-versus-host disease (GvHD) in allogeneic T cell infusions impedes the enthusiasm for an off-the-shelf CAR-T immunotherapy. Natural killer (NK) cells are an ideal cell type for CAR therapy without causing severe GvHD or CRS.3,4,5,6,7 Unlike CAR-T cells, CAR-NK cells retain diverse tumor-specific activating receptors, which decrease the likelihood of tumor cell immunoevasion through downregulation of the CAR target antigen.4,8 Furthermore, NK cells spare healthy cells by using inhibitory receptors that recognize major histocompatibility complex (MHC) class I.8,9 Recent publications have demonstrated that CAR-NK cells could be a highly effective anti-tumor therapy that can be transferred safely in an allogeneic setting,7,10 thereby decreasing cost and increasing the accessibility of CAR therapies. Therefore, these characteristics place CAR-NK as one of the most promising cell immunotherapies for cancer.
Many studies of cancer cells have shown that overexpression of the epidermal growth factor receptor (EGFR) is one of the most common cancer-associated factors, making it one of the most frequently targeted antigens in CAR therapy.11 Activation of EGFR by ligand binding recruits a series of downstream signaling pathways, such as phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and Ras/Raf/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK), responsible for mediating cell proliferation, apoptosis, angiogenesis, and tumorigenesis.12 Specifically, EGFR levels are relatively high on triple-negative breast cancer (TNBC) cells in 45%–70% of patients with poor prognoses.13,14,15,16 Moreover, EGFR mutations rarely occur in TNBC patients, which further promotes the testing of several immunotherapies such as anti-EGFR monoclonal antibodies in TNBC patients.11,15,16 While EGFR is a proven target for immunotherapy, balancing activity and toxicity remains a critical consideration because of the lower levels of EGFR expressions on many healthy cells. Since NK cells can dismiss healthy cells based on the missing self hypothesis,17,18 EGFR-targeted CAR-NK cells may have potential benefits over an EGFR-targeted CAR-T therapy, which has shown mixed results in limited clinical trials thus far.19,20,21
T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif (ITIM) domain (TIGIT) is a receptor of the Ig superfamily that is specifically expressed in immune cells, where it functions as a co-inhibitory receptor.22 TIGIT binds to two ligands, CD155 (PVR) and CD112 (PVRL2), which are expressed on antigen-presenting cells, T cells, and a variety of non-hematopoietic cell types, including tumor cells.23 TIGIT is a checkpoint receptor expressed by both T cells and NK cells and was originally found to induce T cell exhaustion in tumor microenvironments.24,25 Emerging studies have shown that the blockade of TIGIT might be a promising complement to existing immunotherapies.26,27 Recently, it has been shown that the blockade of TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity.28 Therefore, genetically engineering simultaneous overexpression of anti-EGFR-CAR and the knockout of TIGIT can serve as a logical strategy for which CAR-NK cells can be designed to elicit potent anti-tumor activity.
The clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein (Cas9) system has revolutionized biomedical research by providing a powerful genome-editing tool.29,30 Nonetheless, engineering has been hindered in NK cells due to the inefficient delivery of the large-sized CRISPR-Cas9 plasmid.31,32 To overcome this, delivery of Cas9-single guide RNA (sgRNA) complexes by electroporation has been established in NK cells31,32,33; however, electroporation of the Cas9-sgRNA complex requires a convoluted technique. Recently, delivering Cas9-sgRNA complexes using virus-like-particles (VLPs) has shown efficient genetic knockout in CD4+, CD8+, CD34+, and mouse bone marrow cells.34,35 Moreover, the VLP system can induce transient Cas9-mediated recombination and reduce concerning off-target effects from the constitutive expression of the Cas9-sgRNA. Such a gene knockout technology is valuable for removing genes responsible for inhibitory signaling, thereby creating a robust NK cell response in immunotherapy.36 Even though the overexpression of CAR molecules and the knockout of inhibitory receptors seems promising to enhance NK cell recognition and cytotoxicity, recurring genetic engineering in primary NK (pNK) cells is challenging and undesirable due to the limited window for genetic modification of pNK cells. Here, we took advantage of retroviral particles loaded with Cas9-sgRNA complexes to induce efficient anti-EGFR-CAR transgenesis and TIGIT gene knockout simultaneously in NK cells. Retroviral particles efficiently delivered Cas9-sgRNA complexes along with the CAR transgene, proving that this dual engineering technology can be effectively implemented for CAR-NK cell immunotherapy.
Results
Efficient Cas9-sgRNA delivery to NK cells through BaEV-TR and VSV-G envelope glycoprotein-pseudotyped retroviral particles
We adopted the retroviral particle (RP) system, called Nanoblades, from an original report.35 Production of RPs requires plasmids coding for murine leukemia virus (MLV) structural proteins (Gag-Pol), viral envelope glycoprotein (gp), gag-conjugated Cas9 (Gag-Cas9), and sgRNA. NK cells were found refractory to transduction with classic vesicular stomatitis virus glycoprotein (VSV-G, herein referred to as V)-pseudotyped lentiviral vectors (LVs) because they lack the low-density lipoprotein (LDL) receptor.37 Recently, the Baboon retroviral envelope glycoprotein (BaEV)-pseudotyped lentiviral vectors greatly enhanced the transduction of NK cells.37,38 Two types of synthetic BaEVs, non-syncytia-forming BaEV-TR (T) and syncytia-forming BaEVRless (R), were previously reported to improve lentiviral vector production and lentiviral transduction in NK cells.37,38,39 Interestingly, a combination of V and R enhanced the packaging of RPs compared with individual V- or R-pseudotyped RPs.34,35 Thus, we reasoned that efficient delivery of RPs to NK cells might also be achieved by optimizing the viral envelopes.
To test this, Lenti-X 293T cells were transfected with plasmids encoding Gag-Pol, Gag-Cas9, sgRNA, and various envelope glycoproteins (V, R, and T envelopes alone, along with their combinations). RPs were harvested 48 h post transfection and used for transducing NK cells. As proof of concept, we decided to use an immortalized human NK cell line (NK92) expressing enhanced green fluorescent protein (EGFP) (EGFP-NK92 cells) and perform sgRNA-targeted knockout of EGFP using RPs pseudotyped with V, R, T, V + R, V + T, and R + T envelopes. We used a plasmid coding for anti-EGFP sgRNA, which was previously validated for effectively knocking out EGFP in EGFP+ mouse bone marrow cells.35 EGFP-NK92 cells were transduced with various preparations of RPs, and the efficiency of EGFP knockout was evaluated by flow cytometry 5 days post transduction. Consistent with the previous paper,34,35 we observed that the efficiency of EGFP knockout was higher in V + R-pseudotyped RPs (up to 45% EGFP-negative populations) compared with V alone (up to 8%) and R alone (up to 15%). Interestingly, T alone showed similar EGFP knockout efficiency (up to 49%) to the V + R RPs. Furthermore, the efficiency was greatly improved with the pseudotyped combination of VSV-G and non-syncytia-forming BaEV (V + T), resulting in up to 78% EGFP loss by flow cytometry (Figures 1A and 1B). Therefore, this result suggests that RP-mediated delivery of the Cas9-sgRNA ribonucleoprotein (RNP) is efficient in NK92 cells.
Figure 1.

VSV-G- and BaEV-TR-envelope-pseudotyped RPs efficiently targeted the EGFP gene in EGFP-NK92 cells
(A) Representative flow cytometry of EGFP expression in EGFP-NK92 cells transduced with RPs pseudotyped with various envelope glycoproteins. (B) The EGFP knockout efficiency in EGFP-NK92 cells transduced by RPs pseudotyped with various envelope glycoproteins (n = 3). (C) Flow virometry analysis of variously pseudotyped RPs. The numbers indicate the total number of viral particles in the gated area. NC, negative control; V, VSV-G; T, BaEV-TR; R, BaEVRless; VSSC, violet side scatter.
To investigate the differences in the knockout efficiency among RPs pseudotyped with distinct envelope glycoproteins, we performed flow virometry to characterize and quantitate RPs using a violet side scatter.40 To discern background signals, we included phosphate-buffered saline (PBS) and supernatant from cells received transfection reagents without plasmids (mock transfection). Ten microliters of 200-fold-diluted supernatant was acquired by flow virometry (Figure 1C). We identified particle populations, distinguishable from background noise, and measured the total number of particles in a gated area. Interestingly, although EGFP was slightly reduced in EGFP-NK92 cells that were transduced with V-pseudotyped RPs, we found the most particles from V-pseudotyped RPs (≈121,000 RPs/gate). This inconsistency may be due to the lack of LDL-R expressions on NK cells, as previously described.37 Although R- and V + R-pseudotyped RPs were found in a similar amount (≈52,000 RPs/gate), pseudotyping with the combination of V and R enhanced the formation of a distinct population in the dot plot. Notably, T-pseudotyped RPs showed clear separation from background signals observed in PBS and mock controls. Moreover, the number of V + T-pseudotyped RPs (≈115,000 RPs/gate) was increased from that of T-pseudotyped RPs (≈85,000 RPs/gate). The number of T + R-pseudotyped RPs was found to be the lowest (≈51,000 RPs/gate). Overall, the results from flow virometry were consistent with the knockout efficiencies in EGFP-NK92 cells by RPs.
Cas9-sgRNA delivery to pNK cells using RPs efficiently abrogates TIGIT expression
To test whether RP-mediated delivery of the Cas9-sgRNA RNP is also effective in human pNK cells, we decided to knock out the TIGIT inhibitory receptor using a previously validated sgRNA targeting the first exon of the TIGIT gene (GenBank: NM_173799) in ex vivo expanded human pNK cells.32 pNK cells were enriched from human peripheral blood mononuclear cells (PBMCs), and further expanded by ex vivo co-culture with irradiated K562 feeder cells expressing membrane-bound IL-21 and 4-1BBL.41,42 pNK cells were transduced with anti-TIGIT V + T-pseudotyped RPs, and TIGIT expression was evaluated by flow cytometry. Anti-EGFP RPs were used as a negative control. NK cells transduced with anti-TIGIT RPs showed reduced TIGIT expression almost to the basal levels based on TIGIT mean fluorescence intensity (MFIs) by flow cytometry (Figure 2A). No abnormal expression of other NK cell surface receptors was observed, supporting that RPs specifically targeted genes guided by the Cas9-loaded sgRNAs (Figure 2B). To further determine the number of RPs required for pNK cell modification, we performed volume-based transduction to correlate the number of RPs for each volume and the knockout efficiency. We reasoned that 2.7 × 108 RPs are required to modify ≈89.02% of the pNK population (Figures S1A and S1B). Additionally, we calculated the size of anti-TIGIT RPs by flow virometry. We normalized the data based on MFIs of violet side scatter signals from different-sized polystyrene beads and the previously validated MLV refractive index (1.455) using FCMPASS software.43 The average size of the RPs was 120.7 ± 9.8 nm (Figure S2).
Figure 2.

Anti-TIGIT RPs abrogated TIGIT expression in primary human NK cells
(A) Representative mean fluorescence intensity (MFI) of TIGIT surface expression in human pNK cells transduced with RPs targeting the TIGIT gene (n = 3). Data represent mean ± SD. (B) Representative histograms depicting the expression of various surface receptors on pNK cells transduced by RPs. (C) Sequences of the whole TIGIT PCR fragments from pNK cells treated with RPs. The arrow indicates a mutation starting site analyzed by Sanger sequencing. (D) ICE analysis of sequenced TIGIT PCR fragments from three donors that received CRISPR-Cas9 and sgRNA-loaded RPs. The upper chromatograms show sequencing results (Cas9-RNP-RP samples, top; non-treated controls, bottom). The sequence signal plots show the discordant sequences between control (orange) and RP-received samples (green). The right bar graphs indicate frequencies of indel mutations. Eff, editing efficiency; ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Cas9-sgRNA usually induces targeted DNA double-strand breaks (DSBs), leading to error-prone non-homologous end-joining (NHEJ) DNA repair and indel mutations at the target site.29 To confirm gene editing caused by the Cas9-sgRNA RNP complex at the genomic level, we PCR amplified the targeted genomic region from a pool of NK cells treated with RPs containing either EGFP sgRNA or TIGIT sgRNA and analyzed the sequence of amplicons. As a non-treated control, amplicons from wild-type NK cells were used. From the sequencing results, we confirmed gene mutations in the sgRNA-targeted TIGIT locus (Figure 2C). Additionally, we analyzed the sequenced TIGIT PCR fragments using Inference of CRISPR Edits (ICE; Synthego) to computationally validate editing efficiencies.44 As expected, we found a series of indel mutations and aberrant sequences from all TIGITKO-pNK samples (Figure 2D). On average, 89% of pNK cells were modified. Thus, these results indicate that Cas9-loaded RPs can be used for efficient gene knockout in pNK cells.
Cas9-RNP-loaded RPs can induce TIGIT gene knockout and anti-EGFR-CAR transgenesis simultaneously
Since RPs efficiently deliver Cas9-sgRNA RNP complexes into NK cells, we reasoned that combining Cas9-RNP RPs with a transgene could be used to simultaneously induce transgenesis and genetic knockout in NK cells. We used a CAR structure containing an anti-EGFR nanobody, a hinge, a CD28 transmembrane and intracellular domain, and a CD3ζ intracellular domain (Figure 3A). This camelid nanobody-based anti-EGFR-CAR has previously been shown to have strong activity in CAR-T cells but had not yet been tested in NK cells prior to this report.45 Surface expression of the EGFR-CAR was confirmed using a broadly cross-reactive anti-nanobody to stain NK cells.46 To test the anti-tumor functionality of the anti-EGFR-CAR in NK cells, we generated NK92 cells expressing the CAR using BaEV-TR pseudotyped LVs, resulting in CAR expression in ≈38% of NK92 cells (CAR-NK92 cells) (Figure 3B). CAR-NK92 cells were co-cultured with either the TNBC cell line, MDA-MB-231 (EGFRhigh), or an estrogen receptor-positive (ER+) breast cancer cell line, MCF7 (EGFRlow)47 (Figure S3A), for 4 h and analyzed for expression of the CD107a degranulation marker by flow cytometry (Figure 3A). CAR-negative (CARneg) and CAR-positive (CARpos) NK92 cells showed <6% CD107a expression when co-incubated with EGFRlow MCF7 cells, while robust CD107a expression (≈67.6%) was observed only in CARpos NK92 cells when co-incubated with EGFRhigh MDA-MB-231 cells (Figure 3B). These results indicate that the CAR structure is functional in NK cells, eliciting a robust anti-tumor response.
Figure 3.

Simultaneous TIGIT gene knockout with CAR integration into an NK cell genome by Cas9-RNP-loaded RPs
(A) Graphical summary depicting anti-EGFR-CAR-induced tumor killing in NK cells. (B) Representative dot plots and bar graphs of the enhanced anti-tumor activity of CAR-NK92 cells against the EGFRhigh TNBC cell line, MDA-MB-231. (C) Graphical summary depicting the strategy by which RPs engineer NK cells with anti-EGFR-CAR transgenesis and TIGIT gene knockout. (D) TIGIT, CAR, and GFP expression on pNK cells from three different donors that were transduced by RPs. ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
To enhance the anti-tumor activity of the CAR, we decided to simultaneously knock out TIGIT, an inhibitory receptor shown to suppress the anti-tumor activity of NK cells in a mouse model.23 Since the Cas9-RNP RPs were established in the retroviral system, we cloned anti-EGFR-CAR into the pMIG retroviral backbone. Lenti-X 293T cells were transfected with plasmids encoding Gag-Pol, Gag-Cas9, anti-TIGIT sgRNA, VSV-G, BaEV-TR, and anti-EGFR-CAR to prepare V + T-pseudotyped RPs encoding CAR and anti-TIGIT Cas9-RNPs (Figure 3C). As controls, RPs targeting only TIGIT or CAR were also prepared. The RPs were used to transduce expanded pNK cells from three different donors. Transduced pNK cells were analyzed for the efficacy of TIGIT knockout and CAR expression by flow cytometry. pNK cells transduced with RPs targeting only TIGIT showed a great reduction of TIGIT expression. Notably, pNK cells transduced with RPs simultaneously targeting TIGIT and delivering a CAR gene showed significantly reduced TIGIT and induced CAR expression post transduction (Figure 3D).
TIGIT knockout does not improve human NK cell function in vitro and in vivo
We next investigated whether the TIGIT knockout in pNK cells enhances NK cell degranulation. The inhibitory receptor TIGIT expressed on NK cells recognizes the CD112 and CD155 proteins, suggesting the functional inhibition in NK cells through ITIM and immunoglobulin tail tyrosine (ITT)/ITT-like domains.23,48 We first assessed the expression of TIGIT ligands, CD112 and CD155, on EGFRhigh TNBC cells (MDA-MB-231 and SUM149PT), EGFRlow ER+ breast cancer cells (MCF7), and EGFRneg B cell lymphoma cells (Raji). All three breast cancer cell lines showed surface expression of CD112 and CD155 receptors (Figure S3B). Neither CD112 nor CD155 was observed on Raji EGFRneg cells. To investigate the effect of TIGIT knockout on anti-tumor activity, CAR-pNK and CAR-TIGITKO-pNK cells were prepared from three different donors and co-cultured with various target cells. After 4 h of co-incubation, we measured CD107a and interferon gamma (IFN-γ) expression in GFP-positive (CARpos) and GFP-negative (CARneg) cells (Figure 4A). As expected, CARpos cells showed higher CD107a and IFN-γ expression than CARneg cells when stimulated with EGFRhigh target cells, MDA-MB-231 and SUM149PT, in all three donors. CAR expression did not enhance NK cell effector function against EGFRlow and EGFRneg target cells, MCF-7, and Raji cells. Unexpectedly, we did not observe enhanced NK cell function in TIGITKO CARpos cells relative to CARpos cells with normal TIGIT expression (Figure 4B).
Figure 4.

TIGIT knockout failed to enhance the anti-tumor activity of human NK cell function in vitro and in vivo
(A) Representative histograms show the gating strategy to measure CD107a and IFN-γ expression on NK cells in regards to CAR expression. (B) The proportions of CD107a+ and IFN-γ+ cells among the total NK cell population upon stimulation with various target cells (n = 3). Expanded pNK cells from three donors were analyzed. Data represent mean ± SD. (C) NK cell phenotypes used in the first dose on the day of injection. TIGIT, CAR, and GFP expression on CD3−CD56+ cells were analyzed. (D) Schematic of experimental procedures for the evaluation of the anti-tumor activity of genetically modified pNK cells in vivo using an MDA-MB-231 intraperitoneal xenograft mouse model (n = 5). (E) Tumor burden of each group was measured using total bioluminescence values from control pNK, CAR-pNK, or CAR-TIGITKO-pNK cells. (F) Bioluminescence images were acquired on days 2, 9, 16, 23, and 30 by IVIS. ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. I.P., intraperitoneal injection.
To further investigate whether TIGIT knockout might enhance the anti-tumor activity of CAR-NK cells for an extended time that may not be obvious in our short-term in vitro assessments, we next proceeded to an in vivo model of human breast cancer. We first injected 1 × 105 EGFRhigh MDA-MB-231 cells expressing firefly luciferase (MDA-MB-231-Fluc) intraperitoneally (i.p.) into immunocompromised NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. After 3 days post tumor engraftment, the mice received two doses of 1 × 107 engineered pNK cells separated by a 2-day interval (Figures 4C and 4D). Luciferase signals were measured every week by in vivo imaging system (IVIS) to assess tumor growth in vivo (Figure 4D). Consistent with our in vitro data, CAR expression on pNK cells greatly enhanced tumor control in both groups that received either CAR or CAR-TIGITKO-pNK cells. However, TIGIT knockout did not further enhance the tumor control in vivo (Figures 4E and 4F). These results indicated that TIGIT knockout in CAR-NK cells did not enhance anti-tumor activity against TNBC cells.
Cas9-RNP containing RPs allow site-specific CAR integration into the NK cell genome
CRISPR-Cas9 genetic modification is mediated by a cellular DNA repair mechanism caused by DSBs induced by Cas9-sgRNA, which has been shown to insert a lentiviral genome at the DSB site when combined with Cas9 and transgene-loaded lentiviral particles (LPs).49 Thus, we assessed whether site-specific CAR integration into a genome also occurred in our CAR-TIGITKO NK cells (Figure 5A). To investigate DSB-mediated genome insertion in NK cells, we extracted genomic DNA from pNK cells transduced with RPs containing the CAR transgene and the TIGIT-targeting Cas9-sgRNA, and amplified a vicinity of the sgRNA binding site that binds to the TIGIT gene using gene-specific primers (set 1, TIGIT-For and TIGIT-Rev), as shown in Figure 5A. All DNA extracted from parental and transduced pNK cells showed the presence of a TIGIT amplicon in three different donors. To assess integrated viral genomes, we amplified a portion of pMIG and pMIG-EGFR-CAR from transduced NK cells using two pMIG targeting primers (set 2, pMIG-F and pMIG-R). This part of the transgene was found in all NK cells receiving pMIG (GFP control), pMIG-EGFR-CAR, and pMIG-EGFR-CAR with anti-TIGIT RPs. To determine whether anti-EGFR-CAR specifically integrated into the TIGIT sgRNA targeting site, we amplified gene fragments using a TIGIT-specific primer and 5′ long terminal repeat (LTR)-specific primer (set 3, TIGIT-For and 5′ LTR-Rev) encompassing the retroviral 5′ LTR. Notably, amplicons from PCR with TIGIT-For and 5′LTR-Rev primers were observed only in the genomic DNA from the CAR-TIGITKO-pNK cells, suggesting targeted CAR transgene insertion into the TIGIT locus (Figure 5B). The amplicons by primer set 2 were more abundant than the amplicons by primer set 3, indicating that random CAR integrations predominantly occurred. To validate the site-specific CAR integration, we cloned the PCR amplicons and Sanger-sequenced seven individual PCR clones. We confirmed the presence of site-specific transgene insertions from all the clones. Interestingly, due to the NHEJ and indel mutation, various genomic modifications such as nucleotide deletion and insertion were observed (Figures 5C and 5D). Taken together, our RPs utilizing Cas9-RNP against the TIGIT locus could allow simultaneous site-specific anti-EGFR-CAR integration in primary human NK cells.
Figure 5.

Site-specific CAR integration into an NK cell genome by Cas9-sgRNA RPs
(A) Illustrative concept of a site-specific CAR integration into a genome by Cas9-sgRNAs. The bottom DNA indicates the genomic modification after the engineering using Cas9-RNP-loaded RPs. Arrows indicate the locations and directions of primers for PCR. (B) PCR amplicons from PCR using primer set 1, set 2, and set 3. Note that the amplicons from PCR with primer set 3 indicate possible site-specific CAR integration into the TIGIT locus. (C) Representative Sanger sequencing results of three independent amplicons from PCR with primer set 3. (D) Summary of various genomic events in seven independent amplicons from PCR with primer set 3.
Discussion
This work aimed to test whether delivery of Cas9-sgRNA RNP complexes via RPs is effective in knockout genes in NK cells. This technology has been successful in engineering mouse bone marrow cells and human T cells, B cells, etc.,35,49 but has not yet been reported for NK cells, which are known to be more refractory to genetic modification than other immune cells.37 By adopting the retroviral particle system, called Nanoblade, in the original report,35 we clearly demonstrated that the RP technology is applicable to NK cell engineering. We established a protocol by which not only Cas9-sgRNA RNP complexes but also CAR transgene could be concurrently delivered to pNK cells by RPs without a convoluted virus precipitation process. The engineered NK cells clearly showed knockout of a target gene and CAR expression, and were further used for investigating the outcome of the genetic modification in the anti-tumor response ex vivo and in vivo.
Baboon envelope retroviral glycoprotein (BaEV) allowed genomic levels of successful modifications in NK cells.34,39 BaEV binds amino acid transporters, ASCT1 and ASCT2, which are highly expressed on activated NK cells, facilitating BaEV-pseudotyped vectors to enter NK cells.37,38 In an earlier report, a baboon envelope protein lacking the fusion-inhibiting R domain (BaEVRless) was used to pseudotype retroviral and lentiviral vectors. Rless viral envelopes can cause conformational changes and may enhance fusion pores for viral entry.50,51 Therefore, BaEVRless expression benefited the production of BaEV-pseudotyped viral vectors compared with wild-type BaEV.39 BaEVRless could also increase the titer of viral proteins in combination with VSV-G pseudotyping.34,35 Despite these reported advantages of the synthetic BaEVRless, we observed a highly cytotoxic effect from syncytia formation among RP-producing Lenti-X 293T cells post transfection. Fusion-mediated producer cell death led us to test non-syncytia-forming BaEV. Previously, the RD114 retroviral envelope was modified to contain a cytoplasmic domain and R peptide from MLV, which was shown to enhance viral vector production.52 The same strategy was applied to BaEV (BaEV-TR) and was shown to increase the scale of viral vector production without causing fusion in viral vector producer cells.39 Here, we tested whether the BaEV-TR is beneficial in RP production. In addition, we applied a novel flow virometry technique to characterize and quantify RP particles. Since violet lasers are much stronger than blue lasers, violet laser-based side scatter (SSC) calibration was used to measure small retroviral particles.53,54 Our EGFP knockout data reproduced the results previously reported using VSV-G and BaEVRless-pseudotyped Cas9-loaded RPs.34,35 Interestingly, we found that VSV-G and BaEV-TR-pseudotyped RPs were abundantly produced compared with those RPs with VSV-G and BaEVRless pseudotyping and showed the greatest knockdown efficiency. RPs produced with BaEV-TR alone could also form a clear retroviral particle population and induced EGFP knockout at high efficiencies. Taken together, our data suggested that the non-syncytia-forming BaEV-TR is beneficial for RP production.
Although pNK cells expanded from PBMCs can be an excellent source of NK cells that can be modified by CAR transduction, performing multiple rounds of genetic manipulation may not be plausible, given the limitations of NK cell expansion. Therefore, a novel strategy, as we have developed here, to simultaneously modify pNK cells with CAR overexpression and with the knockout of a suppressive regulatory factor would be desirable. We combined the RP delivery system and successfully achieved anti-EGFR-CAR overexpression and TIGIT knockout in pNK cells using single transduction, leading to an innovative strategy to generate a large number of gene-edited CAR-NK cells. A recent report showed a similar approach in which T cell receptor knockout and CAR overexpression was achieved in T cells using lentiviral particles.49 Our report is the first report demonstrating the double delivery of a CAR construct and Cas9-sgRNA RNP complexes to pNK cells. In addition to the robust delivery of Cas9-sgRNA complexes and a CAR construct into NK cells, we also showed evidence of site-specific CAR transgene integration into the site targeted by the Cas9-sgRNA RPs. Since the concern of random genomic integration of the CAR transgene is serious in CAR-NK cell-mediated immunotherapy due to accidental insertional oncogenesis, the site-specific integration by RPs would greatly address this safety concern if used with an integrase-deficient viral vector. It should also be possible to leverage endogenous genetic regulatory machinery to drive gene expression of CARs or other transgenes using this approach, similar to what has been done with targeted CAR-knockin at the T cell receptor alpha constant (TRAC) locus of human T cells.55
In earlier studies, titration of Cas9-loaded RPs was performed by conventional Western blot and enzyme-linked immunosorbent assay (ELISA).35,49 Moreover, obtaining the functionality of gene-targeting Cas9 protein requires a sgRNA incorporation.56 Therefore, confirming the Cas9 proteins from the RPs does not ensure the quantity of sgRNA-loaded Cas9 complexes, which are the functional RNP structures. Due to the concern of empty Cas9 proteins, the functional RPs in this study were confirmed by checking for reduced TIGIT expression of post-transduced NK cells by flow cytometry in combination with the number of particles determined by flow virometry. For transgene and knockout combinational modifications, CAR expression was a primary consideration in NK cells to target EGFR-positive cells. Therefore, the titer was mainly estimated based on the expression of GFP and CAR in NK cells. Even though our method could assess the total number of RPs, advanced methods are required to quantitate the number of empty RPs.
For the method development work performed here, we decided to knock out the TIGIT gene in NK cells due to the robust tumor control previously observed in a TIGITKO mouse model.28 TIGIT controls NK cell function by competing with CD226 and CD96. Although CD226 and TIGIT share their ligands, CD112 and CD155, CD96 and TIGIT bind to CD155.23 A previous study investigating this TIGIT blockade showed increased NK cell degranulation and IFN-γ production in targeting ovarian cancer cells.57 Our results suggest that breast cancer cells do not enhance TIGITKO NK cell function in vitro. Furthermore, we tested the anti-tumor activity of TIGITKO NK cells in combination with CAR overexpression against EGFR-positive breast cancer cells. CAR expression significantly increased NK cell degranulation and cytokine production against EGFR-positive cells. In contrast to our expectations and previous literature, we found that TIGITKO was not beneficial to further enhance the function of EGFR-targeting CAR-NK cells against both the triple-negative and the ER-positive breast cancer cells in vitro and in vivo. While these results should not be too broadly interpreted, it appears that, in the context of CAR-NK cells, TIGIT may not have a strong inhibitory effect on NK cell function. Given the efficiency of our approach, it would be interesting to combine multiple sgRNAs targeting NK-inhibitory receptors (including TIGIT) to assess their combinatorial effect in the future.
In conclusion, CRISPR-Cas9 is a powerful tool to study the genomic to the proteomic level of NK cell biology. Here, we demonstrated that the Cas9-sgRNA complex-loaded RPs can greatly enhance Cas9 delivery in NK cells in a cost-effective and reproducible manner. Furthermore, Cas9-sgRNA complex RPs loaded with transgenic cargo will allow precise genome editing in combination with transgenic integration at a targeted site, giving significant therapeutic potential.
Materials and methods
Culture of human cell lines
TNBC cell lines, MDA-MB-231 and SUM149PT cells, and ER+ breast cancer cell line, MCF7 cells, were obtained from ATCC and cultured in DMEM/F12 media (319-075-CL; Wisent) containing 10% heat-inactivated fetal bovine serum (HI-FBS) (12484028; Gibco) and 100U/mL penicillin and 100 μg/mL streptomycin (Pen/Strep) (SV30010; HyClone). Raji cells were obtained from ATCC and cultured with RPMI-1640 media (350-000-CL; Wisent) containing 10% HI-FBS, 100 μg/mL Pen/Strep, 55 μM b-mercaptoethanol (21985023; Gibco), and 20 mM HEPES (CA12001-708; VWR) (RP10 medium). Lenti-X 293T cells were purchased from Takara (632180) and cultured with 10% HI-FBS and 100 μg/mL Pen/Strep supplemented high-glucose DMEM medium (319-005-CL; Wisent). NK92 cells were cultured with RP10 medium supplemented with 200 U/mL human recombinant interleukin (IL)-2 (NCI Preclinical Repository, USA).
Isolation and culture of human pNK cells
Healthy adult whole-blood collection was approved by the Ottawa Health Science Network Research Ethics Board (# 20200527-01H) and the University of Ottawa (# H-01-21-6568). PBMCs were isolated by Ficoll (45001750; Cytiva) gradient centrifugation. Briefly, approximately 35 mL of whole blood was layered gently over 15 mL of Ficoll and centrifuged at 2,500 rpm, acceleration 1, brake 0 at room temperature, followed by pipette separation of the PBMC layer. CD56-positive pNK cells were isolated by negative magnetic selection using a MojoSort human NK cell isolation kit (480054; BioLegend). Isolated pNK cells were immediately cultured 1:2 with irradiated membrane-bound IL-21 and 4-1BBL expressing K562 feeder cells (gifted from CYTOSEN) and 100 U/mL IL-2 based on the previously reported protocol for 5 days,42 and the partially expanded pNK cells were frozen at −80°C with freezing medium containing 90% FBS and 10% DMSO (BP231-100; Fisher BioReagents).
Plasmid construction
BaEV-TR and BaEVRless sequences were previously published.39 Codon-optimized EcoRI-flanking BaEV-TR and BaEVRless were purchased from Thermo Fisher Scientific (GeneArt) and digested with EcoRI (FD0274; Thermo Fisher Scientific). The fragment was cloned into an EcoRI-digested pMD2.G plasmid (gifted from Didier Trono; 12259; Addgene). To generate a retroviral pMIG-anti-EGFR-CAR-mNeonGreen plasmid, EcoRI-anti-EGFR-CAR-mNeonGreen-PacI fragments were amplified from a pSLQC5-anti-EGFR-CAR-mNeonGreen plasmid and digested with EcoRI and PacI (FD2204; Thermo Fisher Scientific). The fragment was then cloned into an EcoRI and PacI-treated pMIG plasmid (gifted from William Hahn; 9044; Addgene). The anti-TIGIT sgRNA (TCCTCCTGATCTGGGCCCAG) containing plasmid was generated using an Esp3I (FD0454; Thermo Fisher Scientific) digested Superblade5 plasmid35 (gifted from Philippe Mangeot, Théophile Ohlmann & Emiliano Ricci; 134913; Addgene), based on the Zhang lab Golden gate cloning protocol.58
BaEV pseudotyped lentiviral vector production
To produce LVs, 1.5 × 106 Lenti-X 293T cells were plated per well on a six-well plate in 2 mL of Opti-MEM medium (31985070; Gibco) containing 5% HI-FBS and 100 μg/mL Pen/Strep. The next day, the Lenti-X 293T cells were transfected using Lipofectamine 3000 (L3000015; Invitrogen). Briefly, 1,200 ng of transfer plasmids, pLenti-CMV-MCS-GFP-SV-puro59 (for EGFP-NK92) (gifted from Paul Odgren; 73582; Addgene), or pSLQC5-anti-EGFR-CAR-mNeonGreen (for CAR-NK92 cells) (gifted from National Research Council of Canada), 1,200 ng of pSPAX2 (gifted from Didier Trono; 12260; Addgene), and 200 ng of BaEV-TR were combined with Lipofectamine 3000 (6 μL of P3000 and 7 μL of Lipofectamine 3000) in 500μL of serum-free Opti-MEM. Then 1 mL of medium was removed from each well and replaced with the Lipofectamine/plasmid mix. After a 4-h incubation, all medium was removed from each well and replaced with fresh, complete Opti-MEM medium. Without concentrating the LVs, viral supernatant was collected at 48 and 96 h and filtered using a low-protein-binding polyethersulfone (PES) filter (83.1826; Sarstedt) and kept at −80°C.
Cas9-RNP and transgene-loaded RP production
To produce Cas9-RNP RPs, 1.2 × 106 Lenti-X 293T cells were plated per well on a six-well plate in 2 mL of serum-containing Opti-MEM medium. The next day, the cells received 600 ng of BIC-Gag-Cas935 (gifted from Philippe Mangeot, Théophile Ohlmann & Emiliano Ricci; 119942; Addgene), 1,200 ng of MLV-gag-pol (gifted from Marceline Côté, University of Ottawa), 1,200 ng of anti-EGFP sgRNA plasmid (BLADE-182)35 (gifted from Philippe Mangeot, Théophile Ohlmann & Emiliano Ricci; 134914; Addgene) or anti-TIGIT sgRNA plasmid, 100 ng of pMD2.G, and 100 ng of BaEV-TR using Lipofectamine 3000 (6 μL of P3000 and 7 μL of Lipofectamine 3000). To optimize the envelope combinations, a total of 200 ng of envelope plasmids were used (200 ng for a single envelope or 100 + 100 ng for two envelopes). For simultaneous CAR transgenesis and genetic TIGIT knockout, 1,200 ng of pMIG-anti-EGFR-CAR-mNeouGreen was also added with additional Lipofectamine 3000 (7.7 μL of P3000 and 9 μL of Lipofectamine 3000). After a 4-h incubation, all medium was removed from each well and replaced with fresh, complete Opti-MEM media. Without concentrating the RPs, supernatant was collected at 48 h, filtered using a low-protein-binding PES filter, and kept at −80°C.
Flow virometry
For the mock control, 1.2 × 106 Lenti-X 293T cells were plated and the Cas9-RNP RP production procedure without plasmids was followed. Produced RP supernatants and mock control (50 μL) were fixed with 50 μL of 2% paraformaldehyde (PFA) for 25 min at room temperature. The fixed particles (30 μL) were diluted with 1× PBS (20012050; Thermo Fisher Scientific) to obtain 5 × 10−3 dilution. A total of 10 μL of each sample was acquired using the Cytoflex (Beckman) with a violet SSC (VSSC) laser at the Flow Virometry Core, University of Ottawa, and analyzed using Kaluza 2.1 Analysis software (Beckman Coulter). For addressing viral particle sizes, analyzed data were further normalized using the FCMPASS v4.1.6 software (National Cancer Institute) to create a new axis (nanometers).43 The MFI (VSSC) of various sizes of polystyrene beads (Thermo Fisher Scientific) and previously validated refractive index of MLV, 1.455, were applied to convert VSSC signal from RPs to a new axis (nanometers).
NK92 cell transduction
For the functional titer calculation, 5 × 104 NK92 cells were incubated on a 96-well U-bottom plate with serial diluted RP supernatants, 4 μg/mL polybrene (TR-1003-G; MilliporeSigma), and 200 U/mL IL-2. The cells were centrifuged for 30 min at 1,500 × g, 32°C followed by a 3-day incubation at 37°C and 5% CO2. After 3 days of incubation, transgene expression or protein reduction was acquired using the Attune NxT flow cytometer (Thermo Fisher Scientific). Functional titer was calculated by using an equation from Addgene (https://www.addgene.org/protocols/fluorescence-titering-assay/).
For NK92 transduction, NK92 cells were plated on a Retronectin (20 μg/mL, T100B, Takara) coated 96-well flat-bottom plate with viral supernatant, 4 μg/mL polybrene, and 200 U/mL IL-2. The cells were spun down for 30 min at 1,500 × g at 32°C. The cells were incubated overnight at 37°C and 5% CO2. The next day, the viral supernatant was removed and the cells were resuspended in RP10 medium containing 200 U/mL IL-2. Post transduction, GFP expression was observed using the Attune NxT flow cytometer (Thermo Fisher Scientific) and ZOE fluorescent cell imager (Bio-Rad).
To obtain single-copy integrated EGFP-NK92 cells for the EGFP knockout study, less than 10% NK92 cells were transduced and the EGFP+ cells were sorted by the SH800 sorter (SONY) at the Flow core, University of Ottawa.
Human primary NK cell transduction
Five-day expanded and previously frozen pNK cells were thawed and rested overnight in RP10 medium containing 100 U/mL IL-2. The next day, the pNK cells were transduced in a Retronectin coated 48-well flat-bottom plate with RP supernatant (MOI 1 for 40% or MOI 2 for 60% transduction efficiencies), and 100 U/mL IL-2. The cells were spun down at 1200 × g for 90 min at 32°C and cultured overnight at 37°C and 5% CO2. The next day, the cells were stimulated with irradiated feeder cells in 1:5 ratio and 100 U/mL IL-2. The pNK cells were further expanded for nine more days by replacing culture medium every 2 days without an additional feeder cell stimulation. Expanded NK cells were used for phenotyping, functional assays, genomic analysis, and in vivo studies.
Flow cytometry and antibodies
The following monoclonal antibodies were used: anti-TIGIT (PerCP-710; 46-9500-42), anti-CTLA4 (FITC; 11-1529-42), anti-NKG2D (PerCP-710; 46-5878-42), anti-CD112 (PE; 12-1128-42), anti-CD155 (FITC; 11-1550-41), and Live/Dead Fixable Aqua Dead Cell Stain (L34966) from Invitrogen; anti-CD56 (BV421; 318328), anti-NKG2A (APC; 375108), anti-KIR3DL1 (BV421; 312713), anti-CD107a (PE; 328608), and anti-EGFR (PE; 352904) from BioLegend; and anti-CD56 (BV786; 564058), anti-IL18Rα (PE; 564675), anti-TIM3 (BV421; 565562), and anti-CD107a (APC; 560664) from BD Biosciences. An anti-sdCAR antibody was generated at the National Research Council.46 For surface antigen staining, cells were washed once with 150 μL of staining buffer (SB) (PBS containing 2% HI-FBS). The cells were then resuspended in 50 μL of SB and incubated with fluorophore-conjugated antibodies for 25 min at 4°C. After the incubation, cells were washed once with 150 μL of SB and fixed with 100 μL of 2% PFA for 10 min at 4°C. The PFA was washed using 200 μL of SB. The cells were resuspended in 200 μL of SB and data were acquired using the Attune NxT flow cytometer (Thermo Fisher Scientific) and analyzed using Kaluza 2.1 Analysis software (Beckman Coulter). Intracellular staining of IFN-γ was carried out using an anti-IFN-γ antibody (BV785; 502542; BioLegend) and a BD Cytofix/Cytoperm kit (554714). Briefly, cells were fixed with 100 μL of BD Cytofix/Cytoperm buffer after the surface staining procedure. The cells were washed once with 200 μL of Wash Buffer (WB) (554714) and resuspended in 50 μL of WB containing IFN-γ antibody. Intracellular staining was performed for 25 min at 4°C. The cells were then washed once with 200 μL of SB. For the data acquisition, the cells were resuspended in 200 μL of SB.
NK cell functional assay
For CD107a and IFN-γ assay, 3 × 104 target cells were plated on a 96-well U-bottom plate and rested overnight at 37°C and 5% CO2. The next day, expanded 3 × 104 pNK cells were added to the target cell-containing wells with an anti-CD107a antibody (1 μL) and 1× brefeldin A (00-4506-51; Invitrogen). After 4 h of co-incubation, the cells were washed with SB and stained with an anti-CD56 antibody and a live/dead fixable Aqua dye. Intracellular IFN-γ proteins were stained according to the manufacturer’s protocol with an anti-IFN-γ antibody as previously described.
Genomic analysis
Genomic DNA (gDNA) was isolated from expanded pNK cells using a Monarch Genomic DNA Purification Kit (T3010S) from New England Biolab (NEB). The TIGIT fragment was amplified using a forward TIGIT primer (TIGIT-For) (TCTTGTGGCTCACCCTATGC) and reverse TIGIT primer (TIGIT-Rev) (AAGCTGGAGCAGGAATGAGC) from the prepared gDNAs and analyzed by sequencing. The sequencing files from TIGIT fragments were further analyzed by ICE (Synthego) by comparing non-RP treated controls with Cas9-RNP-RP-received cells.44 For integration analysis, a portion of pMIG fragment was amplified using a pMIG forward primer (pMIG-For) (TGACGAGTTCTGAACACCCG) and a pMIG reverse primer (pMIG-Rev) (CAGTCAGACAGAGACAACAC). For targeted integration analysis, TIGIT-5′ LTR was amplified using the forward TIGIT primer and reverse 5′ LTR primer (5′LTR-Rev) (CAGCAAGAGGCTTTATTGGGAA). A bacterial library was generated by putting the amplicons into a plasmid using a PCR cloning kit (E1202S; NEB). All PCR was performed by using a Q5 high-fidelity polymerase (NEB) and primers were annealed at 67°C for 20 s. PCR amplicons were analyzed by Sanger sequencing. All sequencing data were obtained from the Ottawa Hospital Research Institute (OHRI) DNA sequencing facility. Snapgene (Dotmatics) was used to check the sequencing results.
In vivo tumor control
NSG mice were used for the injection of TNBC cancer xenografts and NK cell treatment studies. A breeding pair of NSG mice was purchased from Jackson Laboratories and the colony was maintained in the specific-pathogen-free animal facility at the University of Ottawa in agreement with guidelines and regulations of the Canadian Council on Animal Care. NSG female mice (8–14 weeks old) received 1 × 105 firefly luciferase-expressing MDA-MB-231 cells intraperitoneally and the mice were randomized to each group (n = 5/group). On days 3 and 5 post target cell injection, mice received 1 × 107 GFP-pNK, CAR-pNK, or CAR-TIGITKO-pNK cells with 2,000 U of IL-2 intraperitoneally. Luciferase signal was measured once a week using an animal imaging system, IVIS (PerkinElmer), and the data were analyzed using Aura software 3.2 (Spectral Instruments Imaging). Mouse weight was measured once per week. Mice were sacrificed based on the luciferase signal, movement ability, reactivity, and weight loss. All procedures were approved by and conducted in accordance with the animal guidelines of the University of Ottawa.
Statistic analysis and graph generation
The mean values were tested using two-way ANOVA by comparing cell means regardless of rows and columns. For two-sample comparisons, the mean values were tested by t test using GraphPad Prism 9 (Dotmatics) (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Data represent mean ± SD. All graphs were generated and analyzed using GraphPad Prism 9.
Acknowledgments
We thank CYTOSEN for kindly providing the K562 feeder cells expressing 4-1BBL (CD137L) and membrane-bound IL-21 (mIL-21). The rhIL-2 cytokine was obtained from NCI Preclinical Repository. We thank Vera Tang from the University of Ottawa flow cytometry core for the help with the flow virometry. We appreciate Nusrah Rajabalee and Jim Sun for their insight into the CRISPR-Cas9 technique. Last, we acknowledge the StemCore Laboratories Genomics Core Facility (OHRI) and Preclinical Imaging Core (PCIC) (RRID: SCR_012601 and SCR_021832). Illustrations were created with Biorender.com. This work was funded by the National Research Council Canada Disruptive Technology Solutions for Cell and Gene Therapy Challenge Program (CGT-501-2) and the Canadian Institutes of Health Research (PJT-178197).
Author contributions
D.-H.J., S.K., O.S., S.M., and S.-H.L. contributed to designing and performing the experiments and analyzing the data. D.-H.J., S.K., and S.-H.L. wrote the manuscript. D.-H.J., S.K., L.W., J.C., S.M., and S.-H.L. contributed to the discussion. S.-H.L. obtained funding.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.03.006.
Supplemental information
Data availability
Data are available upon request.
References
- 1.Rafiq S., Hackett C.S., Brentjens R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roselli E., Frieling J.S., Thorner K., Ramello M.C., Lynch C.C., Abate-Daga D. CAR-T engineering: optimizing signal transduction and effector mechanisms. BioDrugs. 2019;33:647–659. doi: 10.1007/s40259-019-00384-z. [DOI] [PubMed] [Google Scholar]
- 3.Fehniger T.A., Cooper M.A. Harnessing NK cell memory for cancer immunotherapy. Trends Immunol. 2016;37:877–888. doi: 10.1016/j.it.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guillerey C., Huntington N.D., Smyth M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016;17:1025–1036. doi: 10.1038/ni.3518. [DOI] [PubMed] [Google Scholar]
- 5.Liu E., Tong Y., Dotti G., Shaim H., Savoldo B., Mukherjee M., Orange J., Wan X., Lu X., Reynolds A., et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimasaki N., Jain A., Campana D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020;19:200–218. doi: 10.1038/s41573-019-0052-1. [DOI] [PubMed] [Google Scholar]
- 7.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee S.H., Miyagi T., Biron C.A. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol. 2007;28:252–259. doi: 10.1016/j.it.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. Functions of natural killer cells. Nat. Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 10.Xiao L., Cen D., Gan H., Sun Y., Huang N., Xiong H., Jin Q., Su L., Liu X., Wang K., et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol. Ther. 2019;27:1114–1125. doi: 10.1016/j.ymthe.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaft N. The landscape of CAR-T cell clinical trials against solid tumors-A comprehensive overview. Cancers. 2020;12:2567. doi: 10.3390/cancers12092567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakajima H., Ishikawa Y., Furuya M., Sano T., Ohno Y., Horiguchi J., Oyama T. Protein expression, gene amplification, and mutational analysis of EGFR in triple-negative breast cancer. Breast Cancer. 2014;21:66–74. doi: 10.1007/s12282-012-0354-1. [DOI] [PubMed] [Google Scholar]
- 13.Hoadley K.A., Weigman V.J., Fan C., Sawyer L.R., He X., Troester M.A., Sartor C.I., Rieger-House T., Bernard P.S., Carey L.A., Perou C.M. EGFR associated expression profiles vary with breast tumor subtype. BMC Genom. 2007;8:258. doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masuda H., Zhang D., Bartholomeusz C., Doihara H., Hortobagyi G.N., Ueno N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012;136:331–345. doi: 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Secq V., Villeret J., Fina F., Carmassi M., Carcopino X., Garcia S., Metellus I., Boubli L., Iovanna J., Charpin C. Triple negative breast carcinoma EGFR amplification is not associated with EGFR, Kras or ALK mutations. Br. J. Cancer. 2014;110:1045–1052. doi: 10.1038/bjc.2013.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tilch E., Seidens T., Cocciardi S., Reid L.E., Byrne D., Simpson P.T., Vargas A.C., Cummings M.C., Fox S.B., Lakhani S.R., Chenevix Trench G. Mutations in EGFR, BRAF and RAS are rare in triple-negative and basal-like breast cancers from Caucasian women. Breast Cancer Res. Treat. 2014;143:385–392. doi: 10.1007/s10549-013-2798-1. [DOI] [PubMed] [Google Scholar]
- 17.Ljunggren H.G., Kärre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol. Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 18.Portillo A.L., Hogg R., Poznanski S.M., Rojas E.A., Cashell N.J., Hammill J.A., Chew M.V., Shenouda M.M., Ritchie T.M., Cao Q.T., et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience. 2021;24:102619. doi: 10.1016/j.isci.2021.102619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei J., Liu Y., Wang C., Zhang Y., Tong C., Dai G., Wang W., Rasko J.E.J., Melenhorst J.J., Qian W., et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct. Target. Ther. 2020;5:134. doi: 10.1038/s41392-020-00256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanber K., Savani B., Jain T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: the diametric opposition of T cells. Br. J. Haematol. 2021;195:660–668. doi: 10.1111/bjh.17544. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y., Zhang Z., Ding Y., Fang Y., Wang P., Chu W., Jin Z., Yang X., Wang J., Lou J., Qian Q. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021;147:3725–3734. doi: 10.1007/s00432-021-03613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson A.C., Joller N., Kuchroo V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chauvin J.M., Zarour H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer. 2020;8:e000957. doi: 10.1136/jitc-2020-000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X., Harden K., Gonzalez L.C., Francesco M., Chiang E., Irving B., Tom I., Ivelja S., Refino C.J., Clark H., et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 25.Okwor C.I.A., Oh J.S., Crawley A.M., Cooper C.L., Lee S.H. Expression of inhibitory receptors on T and NK cells defines immunological phenotypes of HCV patients with advanced liver fibrosis. iScience. 2020;23:101513. doi: 10.1016/j.isci.2020.101513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnston R.J., Comps-Agrar L., Hackney J., Yu X., Huseni M., Yang Y., Park S., Javinal V., Chiu H., Irving B., et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26:923–937. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Chauvin J.M., Pagliano O., Fourcade J., Sun Z., Wang H., Sander C., Kirkwood J.M., Chen T.h.T., Maurer M., Korman A.J., Zarour H.M. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Invest. 2015;125:2046–2058. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q., Bi J., Zheng X., Chen Y., Wang H., Wu W., Wang Z., Wu Q., Peng H., Wei H., et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018;19:723–732. doi: 10.1038/s41590-018-0132-0. [DOI] [PubMed] [Google Scholar]
- 29.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savić N., Schwank G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl. Res. 2016;168:15–21. doi: 10.1016/j.trsl.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Huang R.S., Lai M.C., Shih H.A., Lin S. A robust platform for expansion and genome editing of primary human natural killer cells. J. Exp. Med. 2021;218:e20201529. doi: 10.1084/jem.20201529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang R.S., Shih H.A., Lai M.C., Chang Y.J., Lin S. Enhanced NK-92 cytotoxicity by CRISPR genome engineering using Cas9 ribonucleoproteins. Front. Immunol. 2020;11:1008. doi: 10.3389/fimmu.2020.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daher M., Basar R., Gokdemir E., Baran N., Uprety N., Nunez Cortes A.K., Mendt M., Kerbauy L.N., Banerjee P.P., Shanley M., et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137:624–636. doi: 10.1182/blood.2020007748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutierrez-Guerrero A., Abrey Recalde M.J., Mangeot P.E., Costa C., Bernadin O., Périan S., Fusil F., Froment G., Martinez-Turtos A., Krug A., et al. Baboon envelope pseudotyped "Nanoblades" carrying Cas9/gRNA complexes allow efficient genome editing in human T, B, and CD34(+) cells and knock-in of AAV6-encoded donor DNA in CD34(+) cells. Front. Genome Ed. 2021;3:604371. doi: 10.3389/fgeed.2021.604371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mangeot P.E., Risson V., Fusil F., Marnef A., Laurent E., Blin J., Mournetas V., Massouridès E., Sohier T.J.M., Corbin A., et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019;10:45. doi: 10.1038/s41467-018-07845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bexte T., Alzubi J., Reindl L.M., Wendel P., Schubert R., Salzmann-Manrique E., von Metzler I., Cathomen T., Ullrich E. CRISPR-Cas9 based gene editing of the immune checkpoint NKG2A enhances NK cell mediated cytotoxicity against multiple myeloma. Oncoimmunology. 2022;11:2081415. doi: 10.1080/2162402X.2022.2081415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bari R., Granzin M., Tsang K.S., Roy A., Krueger W., Orentas R., Schneider D., Pfeifer R., Moeker N., Verhoeyen E., et al. A distinct subset of highly proliferative and lentiviral vector (LV)-Transducible NK cells define a readily engineered subset for adoptive cellular therapy. Front. Immunol. 2019;10:2001. doi: 10.3389/fimmu.2019.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colamartino A.B.L., Lemieux W., Bifsha P., Nicoletti S., Chakravarti N., Sanz J., Roméro H., Selleri S., Béland K., Guiot M., et al. Efficient and robust NK-cell transduction with Baboon envelope pseudotyped lentivector. Front. Immunol. 2019;10:2873. doi: 10.3389/fimmu.2019.02873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Girard-Gagnepain A., Amirache F., Costa C., Lévy C., Frecha C., Fusil F., Nègre D., Lavillette D., Cosset F.L., Verhoeyen E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood. 2014;124:1221–1231. doi: 10.1182/blood-2014-02-558163. [DOI] [PubMed] [Google Scholar]
- 40.Brittain G.C., 4th, Chen Y.Q., Martinez E., Tang V.A., Renner T.M., Langlois M.A., Gulnik S. A novel semiconductor-based flow cytometer with enhanced light-scatter sensitivity for the analysis of biological nanoparticles. Sci. Rep. 2019;9:16039. doi: 10.1038/s41598-019-52366-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denman C.J., Senyukov V.V., Somanchi S.S., Phatarpekar P.V., Kopp L.M., Johnson J.L., Singh H., Hurton L., Maiti S.N., Huls M.H., et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One. 2012;7:e30264. doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Somanchi S.S., Senyukov V.V., Denman C.J., Lee D.A. Expansion, purification, and functional assessment of human peripheral blood NK cells. J. Vis. Exp. 2011 doi: 10.3791/2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welsh J.A., Jones J.C. Small particle fluorescence and light scatter calibration using FCM(PASS) software. Curr. Protoc. Cytom. 2020;94:e79. doi: 10.1002/cpcy.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conant D., Hsiau T., Rossi N., Oki J., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., et al. Inference of CRISPR Edits from sanger trace data. CRISPR J. 2022;5:123–130. doi: 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
- 45.McComb S., Nguyen T., Shepherd A., Henry K.A., Bloemberg D., Marcil A., Maclean S., Gilbert R., Gadoury C., Pon R. Antigenic sensitivity of membrane-proximal targeting chimeric antigen receptors can be fine-tuned through hinge truncation. bioRxiv. 2022 doi: 10.1101/2020.10.30.360925. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McComb S., Nguyen T., Shepherd A., Henry K.A., Bloemberg D., Marcil A., Maclean S., Zafer A., Gilbert R., Gadoury C., et al. Programmable attenuation of antigenic sensitivity for a nanobody-based EGFR chimeric antigen receptor through hinge domain truncation. Front. Immunol. 2022;13:864868. doi: 10.3389/fimmu.2022.864868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao T., Li W., Wang X., Xu H., Yang J., Wu Q., Huang Y., Geradts J., Jiang P., Fei T., et al. Estrogen-regulated feedback loop limits the efficacy of estrogen receptor-targeted breast cancer therapy. Proc. Natl. Acad. Sci. USA. 2018;115:7869–7878. doi: 10.1073/pnas.1722617115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu S., Zhang H., Li M., Hu D., Li C., Ge B., Jin B., Fan Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013;20:456–464. doi: 10.1038/cdd.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamilton J.R., Tsuchida C.A., Nguyen D.N., Shy B.R., McGarrigle E.R., Sandoval Espinoza C.R., Carr D., Blaeschke F., Marson A., Doudna J.A. Targeted delivery of CRISPR-Cas9 and transgenes enables complex immune cell engineering. Cell Rep. 2021;35:109207. doi: 10.1016/j.celrep.2021.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kubo Y., Izumida M., Togawa K., Zhang F., Hayashi H. Cytoplasmic R-peptide of murine leukemia virus envelope protein negatively regulates its interaction with the cell surface receptor. Virology. 2019;532:82–87. doi: 10.1016/j.virol.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Melikyan G.B., Markosyan R.M., Brener S.A., Rozenberg Y., Cohen F.S. Role of the cytoplasmic tail of ecotropic moloney murine leukemia virus Env protein in fusion pore formation. J. Virol. 2000;74:447–455. doi: 10.1128/jvi.74.1.447-455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandrin V., Boson B., Salmon P., Gay W., Nègre D., Le Grand R., Trono D., Cosset F.L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. 2002;100:823–832. doi: 10.1182/blood-2001-11-0042. [DOI] [PubMed] [Google Scholar]
- 53.McVey M.J., Spring C.M., Kuebler W.M. Improved resolution in extracellular vesicle populations using 405 instead of 488 nm side scatter. J. Extracell. Vesicles. 2018;7:1454776. doi: 10.1080/20013078.2018.1454776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gardiner C., Shaw M., Hole P., Smith J., Tannetta D., Redman C.W., Sargent I.L. Measurement of refractive index by nanoparticle tracking analysis reveals heterogeneity in extracellular vesicles. J. Extracell. Vesicles. 2014;3:25361. doi: 10.3402/jev.v3.25361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eyquem J., Mansilla-Soto J., Giavridis T., van der Stegen S.J.C., Hamieh M., Cunanan K.M., Odak A., Gönen M., Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cofsky J.C., Soczek K.M., Knott G.J., Nogales E., Doudna J.A. CRISPR-Cas9 bends and twists DNA to read its sequence. Nat. Struct. Mol. Biol. 2022;29:395–402. doi: 10.1038/s41594-022-00756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maas R.J., Hoogstad-van Evert J.S., Van der Meer J.M., Mekers V., Rezaeifard S., Korman A.J., de Jonge P.K., Cany J., Woestenenk R., Schaap N.P., et al. TIGIT blockade enhances functionality of peritoneal NK cells with altered expression of DNAM-1/TIGIT/CD96 checkpoint molecules in ovarian cancer. Oncoimmunology. 2020;9:1843247. doi: 10.1080/2162402X.2020.1843247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H., et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Witwicka H., Hwang S.Y., Reyes-Gutierrez P., Jia H., Odgren P.E., Donahue L.R., Birnbaum M.J., Odgren P.R. Studies of OC-STAMP in osteoclast fusion: a new knockout mouse model, rescue of cell fusion, and transmembrane topology. PLoS One. 2015;10:e0128275. doi: 10.1371/journal.pone.0128275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request.