


Keywords: CAKUT, monogenic kidney disease, development, pediatric
Abstract
Significance Statement
About 40 disease genes have been described to date for isolated CAKUT, the most common cause of CKD during childhood. However, mutations in these genes explain only 20% of cases. The authors performed exome sequencing in an international cohort of individuals with CAKUT. They identified genetic variants in ARHGEF6 (a gene on the X chromosome in humans that encodes a guanine nucleotide exchange factor) as a potential novel cause of this disease. Using a multifaceted approach, including cellular and independent animal models, they found evidence that ARHGEF6 variants cause disease, potentially via dysregulation of integrin/parvin/RAC1/CDC42 signaling. These findings further link ARHGEF6 function to integrin/parvin/RAC1/CDC42 signaling, thereby strengthening this pathway's relevance for renal development.
Background
About 40 disease genes have been described to date for isolated CAKUT, the most common cause of childhood CKD. However, these genes account for only 20% of cases. ARHGEF6, a guanine nucleotide exchange factor that is implicated in biologic processes such as cell migration and focal adhesion, acts downstream of integrin-linked kinase (ILK) and parvin proteins. A genetic variant of ILK that causes murine renal agenesis abrogates the interaction of ILK with a murine focal adhesion protein encoded by Parva, leading to CAKUT in mice with this variant.
Methods
To identify novel genes that, when mutated, result in CAKUT, we performed exome sequencing in an international cohort of 1265 families with CAKUT. We also assessed the effects in vitro of wild-type and mutant ARHGEF6 proteins, and the effects of Arhgef6 deficiency in mouse and frog models.
Results
We detected six different hemizygous variants in the gene ARHGEF6 (which is located on the X chromosome in humans) in eight individuals from six families with CAKUT. In kidney cells, overexpression of wild-type ARHGEF6—but not proband-derived mutant ARHGEF6—increased active levels of CDC42/RAC1, induced lamellipodia formation, and stimulated PARVA-dependent cell spreading. ARHGEF6-mutant proteins showed loss of interaction with PARVA. Three-dimensional Madin-Darby canine kidney cell cultures expressing ARHGEF6-mutant proteins exhibited reduced lumen formation and polarity defects. Arhgef6 deficiency in mouse and frog models recapitulated features of human CAKUT.
Conclusions
Deleterious variants in ARHGEF6 may cause dysregulation of integrin-parvin-RAC1/CDC42 signaling, thereby leading to X-linked CAKUT.
Introduction
CAKUT represent one of the most frequent birth defects and is the most common cause of CKD in childhood.1–3 To date, the discovery of approximately 40 monogenic causes of isolated or approximately 200 monogenic causes of syndromic CAKUT has delineated multiple pathways of this disease.1,4 Numerous transcription factors and signaling molecules, including GDNF-RET, Notch, BMP, Hedgehog, or FGF,1,5 but also extracellular matrix (ECM) proteins and their receptors, such as integrins, play an essential role in renal development.1,6,7 Within the kidney, integrins are expressed in a spatially and temporally well-controlled pattern. The loss of specific integrins and ECM components disrupts normal renal development and can lead to CAKUT in humans and mice.7–11 For instance, collecting-duct-specific deletion of Itgb1 (integrin β1) results in severe defects in branching morphogenesis,10 causing a wide range of kidney abnormalities, including bilateral renal agenesis or renal hypoplasia.9 Loss of nephronectin, the functional ligand of integrin α8/β1 in mice12,13 and integrin α8 in mice or humans, leads to renal agenesis.11,14 In addition, a point mutation resulting in an amino acid change in the ATP-binding site of the integrin-linked kinase (ILK) causes murine renal agenesis.8 This variant specifically abrogates the interaction of ILK with α-parvin (encoded by Parva), a focal adhesion protein, leading to CAKUT if mutated in mice.8 On integrin-mediated signaling, the ILK-PARV complex recruits ARHGEF6 (also known as αPIX) to focal adhesion sites where it activates small Rho-like GTPases in HEK293 and CHO-K1 cells.15,16
ARHGEF6 and its paralog ARHGEF7 belong to a family of guanine nucleotide exchange factors (GEFs), which activate small Rho-like GTPases.17 ARHGEF6 is implicated in various biological processes, including cell migration and focal adhesion, and acts downstream of ILK and parvin proteins.16,18 However, its role in renal development remains elusive.
A monogenic causation accounts for approximately 20% of individuals with CAKUT diagnosed before the age of 25 years,4,19 leaving approximately 80% of cases genetically unsolved. To identify novel genes causing CAKUT, if mutated, we performed exome sequencing (ES) in an international cohort of 1265 CAKUT families. We identified six different hemizygous variants in the gene ARHGEF6 in eight individuals from six unrelated families and demonstrate loss of function of those variants in cellular and animal models.
Materials and Methods
Study Approval and Participants
Approval for human individual's research was obtained from the University of Michigan, the Boston Children's Hospital, the Baylor College of Medicine, Clinical Genetics, Erasmus MC, University Medical Center Rotterdam, and Wolfson Medical Center Institutional Review Boards. All participants or their guardians provided written informed consent. After obtaining informed consent, clinical data and blood samples were obtained from individuals with CAKUT on the basis of the standardized questionnaire (http://www.renalgenes.org).
Exome Sequencing and Mutation Calling
Homozygosity mapping, ES, and variant calling were performed as described previously20 (details in the Supplemental Methods section). ES was performed in 1265 individuals with CAKUT from worldwide sources, using Agilent SureSelect human exome capture arrays (Thermo Fisher Scientific) with next-generation sequencing on an Illumina platform. ES at Baylor-Hopkins Center for Mendelian Genomics was performed as published previously.21 Sequence reads were mapped against the human reference genome (NCBI build 37/hg19) and evaluated in Cartagenia or CLC Genomics Workbench (version 6.5.1; CLC bio). Genetic location information is based on the February 2009 Human Genome Browser data, hg19 assembly (http://www.genome.ucsc.edu). Downstream processing of aligned BAM files was done using Picard and samtools,22 and single-nucleotide variant calling was done using GATK5.
Variant calling was performed in line with proposed guidelines,23 and the following criteria were used as previously described2,4: Only rare variants were included, with mean allele frequency <1% in dbSNP147 and with only zero to one homozygotes in the adult genome database gnomAD. Furthermore, variants were nonsynonymous and/or located within splice sites. Subsequently, variant severity was stratified based on protein effects (truncating frameshift or nonsense variants, essential or extended splice-site variants, and missense variants). Splice-site variants were assessed by in silico tools, MaxEnt and NNSPLICE splice-site variant prediction scores,24,25 and conservation across human splice-sites as described previously.26 Missense variants were assessed based on SIFT, MutationTaster, PolyPhen 2.0 prediction, REVEL, and CADD scores,27–31 and evolutionary conservation evaluated based on manually derived multiple sequence alignments. Whenever possible, trio evaluation was performed. Identified variants were confirmed via Sanger sequencing.
Plasmids, siRNAs, Cell Culture, Transfection, and Transduction
Information on plasmids, cell lines, and reagents can be found in the Supplemental Methods section.
Immunoblotting, Immunoprecipitation, and Immunofluorescence Staining
Immunoblotting, immunoprecipitation, and immunofluorescence staining were performed as described previously.20 Summary of antibodies used in this study is provided in the Supplemental Methods section.
Cell Spreading Assay
Coverslips were coated with 10 µg/ml fibronectin (FN; F2006, Sigma) in PBS overnight at 4°C and washed with PBS. Cells were kept under serum-deprived conditions overnight, detached with Trypsin-EDTA (0.25%; Thermo Fisher Scientific), washed with PBS, and seeded on FN-coated coverslips in DMEM/10% FBS. After 4 hours at 37°C, samples were fixed with 4% PFA for 10 minutes at room temperature and, subsequently, permeabilized (PBS/0.1% Triton X100) and treated with blocking solution (1% BSA, 10% donkey serum in PBS), followed by overnight incubation in primary antibody solution and 1 h incubation in secondary antibody solution (sc-53711 AF647, Santa Cruz Biotechnology, Inc.). After washing with PBS, cells on a cover slip were mounted in ProLong Diamond Antifade Mountant with DAPI on microscope slides (P36962, Life Technologies).
G-LISA Small G-Protein Activation Assays
HEK293T cells were transfected with cDNA representing ARHGEF6 wild-type (WT) or proband-derived variants using Lipofectamine 2000. Transfected cells were incubated in DMEM medium with 10% fetal bovine serum for 24 h and then in serum-free medium for 24 hours. RAC1 or CDC42 activity was determined using a G-LISA Activation Assay Biochem Kit (Cytoskeleton, CO) according to the manufacturer's instructions.
3D Madin-Darby Canine Kidney Spheroid Culture and Analysis
For 3D cyst culture, single cells in a final concentration of 2 × 104/ml were resuspended on ice in a collagen I solution containing 2.5 mg/ml bovine skin collagen I (PureCol, Advanced Biomatrix), neutralized with 0.23% NaHCO3 wt/vol, 20 mM HEPES (pH 7.6), and supplemented with growth medium to the required collagen concentration. In total, 150 μl of cells in collagen were plated in an imaging 96-well plate (Greiner Bio One, PS, F-bottom, μClear, Black, CELLSTAR). The collagen was allowed to gel, overlayed with medium, and grown for 12 days.
Quantification of lumen phenotypes was done by phase contrast microscopy (50–150 cysts/condition) by manual counting. Prior analysis by confocal microscopy showed that over 99% of cysts were GFP-positive.
For tubulogenesis, 9-day-old cysts were stimulated with 5× diluted conditioned medium containing hepatocyte growth factor (HGF).32 Assessment of cysts is further described in the Supplemental Methods section.
In Situ Hybridization Analysis and RNAScope
In situ hybridization (ISH) on 10-µm paraffin sections was performed with a digoxigenin-labeled antisense riboprobe of ARHGEF6 mRNA following a standard procedure.33 RNAScope ISH was performed on 1.5-µm FFPE sections using the RNAScope multiplex fluorescent V2 assay (Advanced Cell Diagnostics, Inc.) according to the manufacturer's instructions. The following probes were used: Arhgef6 (Cat.: 574371), Wt1 (Cat.: 432711-C2), and Parva (319811-C3).
Mouse Breeding, Maintenance, and CAKUT Phenotype Evaluation
Animal care and experimental protocols were approved by the Institutional Animal Care and Use Committee at the Boston Children's Hospital (no. 18-12-3826R, no.00001522). Additional information on the mice, breeding, or genotyping is found in the Supplemental Methods section.
Generation of Xenopus Models
All experiments with Xenopus embryos (CRISPR/cas9 and morpholino) were conducted in accordance with local laws and institutional regulations and approved by the regulatory authorities (Kantonales Veterinäramt Zürich). Guide RNAs were designed, generated, and delivered as previously described.34 (Oligos and detailed information can be found in the supplemental information.)
Statistics
Graphpad Prism 9.2.0 software was used to perform statistical testing. For comparison of groups of n>2, one-way ANOVA or nonparametric Kruskal–Wallis test was used. For statistical testing between two experimental groups, nonparametric Mann–Whitney U test was used. P values are annotated as *P<0.05, **P<0.01, and ***P<0.001.
Results
Hemizygous Variants in ARHGEF6 in Six Unrelated Families with CAKUT
We performed ES in a cohort of 1265 families with CAKUT. Sequencing data were first interrogated for variants in the >200 known isolated or syndromic CAKUT genes and then evaluated for potential novel disease genes.
In family A5124, we detected a hemizygous missense variant (c.1331T>A, p.Ile444Asn) in ARHGEF6 that was shared by all affected individuals (A5123-11/-32/-33) but absent or heterozygous in unaffected family members (Figure 1A, Supplemental Figure 1, Table 1). All three affected individuals presented with unilateral renal hypoplasia (Table 1), proband A5123-32 additionally with bladder diverticula. The identified variant was not present in homozygous state and was very rare in hemizygous state in the control database gnomAD and yielded strong in silico deleteriousness prediction scores (Table 1). Mutated isoleucine residue was evolutionarily conserved to Xenopus tropicalis (Figure 1B).
Figure 1.
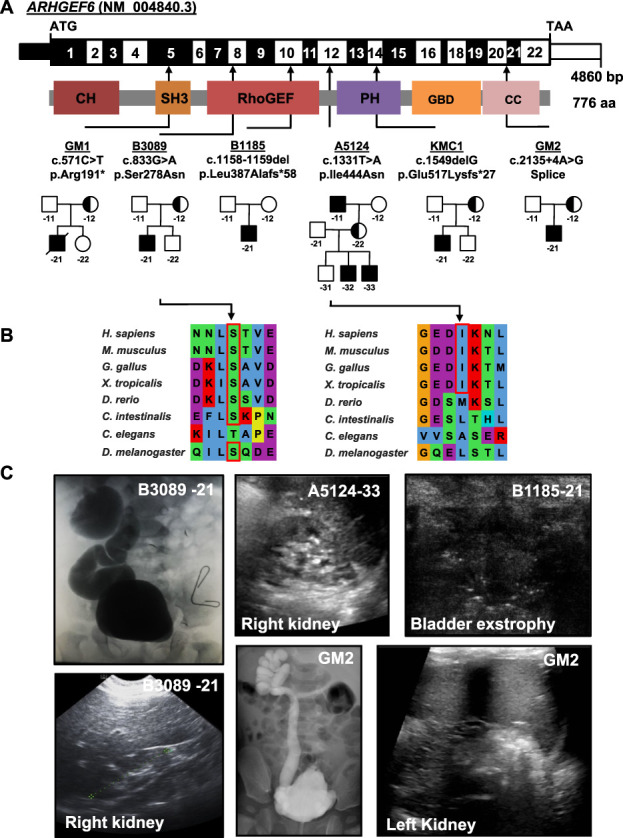
Exome sequencing identifies X-linked recessive variants in the gene ARHGEF6 in six families with CAKUT. (A) Exon structure (upper bar) and protein domains (lower bar) of human ARHGEF6. Positions of start codon (ATG) and stop codon (TAA) are indicated. Exon numbers are marked on an alternating black or white background. Protein domain lengths are shown by the colored boxes in respect to encoding exons. Positions of variants are indicated by black arrows in relation to the exon and the protein domain (see also Table 1). Pedigrees of families with genetic variants in ARHGEF6 are depicted below each family number. (B) CLUSTAL-generated amino acid sequence alignments of ARHGEF6 orthologous proteins are shown for the regions surrounding sites of missense variants. (C) Representative clinical images of individuals with ARHGEF6 variants. Upper row (from left to right): Voiding cysto-urethrogram showing vesico-uretral reflux grade IV or V on the right (B3089-21), renal ultrasound image of proband A5124-33 showing renal hypoplasia, and bladder ultrasound images of B1185-21 indicating bladder exstrophy. Lower row (from left to right): Renal ultrasound of right kidney (B3089-21) showing renal hypoplasia on the right. Voiding cysto-urethrogram showing vesico-uretral reflux grade V on the right (GM2) and renal ultrasound of left kidney of GM2 indicating hypoplastic kidney.
Table 1.
Identification of six different X-linked recessive variants in the gene ARHGEF6 (NM_004840.3) in eight male individuals with CAKUT
| Family Individual | Self-Reported Ethnicity, Race, or Origin | Con-san-guinity | Nucleotide Change AA Change | hg19 Position | Exon (Segregation) Zygostiy | PP2/SIFT/MT | CADD/REVEL | AA Conservation | gnomAD Allele Frequencies (hom/hemi/het/Total Count) | Renal Phenotype | Extrarenal Features |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GM1 | White | No | c.571C>T p.Arg191* | chrX: 135825834 | 5 (m), hemi | NA | NA | NA | NP | Bilateral renal agenesis | CM, termination of pregnancy at 21 wk |
| B3089-21 | Mace donia | No | c.833G>A p.Ser278Asn | chrX: 135790924 | 8 (m), hemi | 0.36/Del/Dis | 20.3/0.092 | D.m. | 0/7/19/204,748 | Hypoplastic kidney with VUR IV-V ri, VUR II le | — |
| B1185-21 | White | No | c.1158_ 1159del p.Leu387 Alafs*58 |
chrX: 135772799_135772800 | 10, hemi | NA | NA | NA | NP | Bladder exstrophy with VUR I le, II-III ri, medullary nephrocalcinosis, hypospadias | Kyphosis, extra vertebrate, attention deficit disorder |
| A5124 -11 -32 -33 |
Ashkenazi | No | c.1331T>A p.Ile444Asn | chrX: 135767897 | 12 (m), hemi | 0.94/Del/Dis | 23.1/0.44 | X.t. | 0/4/10/183,437 | 11: renal hypoplasia le 32: renal hypoplasia le, bladder diverticula 33: renal hypoplasia ri |
-11: Deafness |
| KMC1 | Ashkenazi/Russian nonjewish | No | c.1549delG p.Glu517 Lysfs*27 |
chrX: 135764058 | 14, hemi | NA | NA | NA | NP | Unilateral renal agenesis ri | Autism, sleeping disturbances recurrent vomiting |
| GM2 | African American | No | c.2135+4 A>G p.? |
chrX: 135754175 | i20 (m), hemi | NA | NA | NA | 0/0/4/205,321 | Bilateral hydrouretero-nephrosis, VUR grade V le, hypodysplastic le kidney, PUV (ablated), trabeculated bladder. | — |
AA, amino acid change; PP2, Polyphen Prediction score Humvar; SIFT Sorting Intolerant From Tolerant; MT, mutationtaster; CADD, Combined Annotation Dependent Depletion; REVEL, Rare Exome Variant Ensemble Learner; hom, homozygous; hemi, hemizygous; het, heterozygous; NA, not applicable; NP, not present; CM, cardiomyopathy; Del, deleterious; Dis, disease causing; D.m., Drosophila melanogaster; VUR, vesicoureteral reflux; ri, right; le, left; X.t., Xenopus tropicalis; m, mother; M, male; PUV, posterior urethral valves.
Within our cohort, we then identified two additional families (B1185, B3089) with affected individuals carrying hemizygous variants in ARHGEF6. In B1185-21, we identified a variant in exon 10 (c.1158-1159del, p.Leu387Alafs*58, Figure 1A, Supplemental Figures 1–2, Table 1). The patient presented with bladder exstrophy and hypospadias, which was subjected to complete repair as a neonate. Later, residual symptoms of mild bladder dysfunction with incomplete voiding were described. The variant was predicted to lead to truncation of the protein and was not reported in gnomAD.
Proband B3089-21 carried a hemizygous missense variant (c.833G>A, p.Ser278Asn, Figure 1A, Supplemental Figures 1–2, Table 1) affecting the RhoGEF domain of the protein. This variant yielded strong in silico prediction scores, and the mutated residue was evolutionarily conserved to invertebrate C. intestinalis (Table 1, Figure 1B). The proband presented with bilateral vesicoureteral reflux (VUR) accompanied by a right hypoplastic kidney.
Through collaboration with the Baylor College of Medicine, we identified family GM2 with a hemizygous splice site variant in ARHGEF6 (c.2135+4A>G, Figure 1, Supplemental Figure 1, Table 1). Proband GM2 was born with posterior urethral valves and developed a trabeculated bladder wall, bilateral hydroureteronephrosis with VUR, and a hypodysplastic left kidney. The variant was predicted to disrupt the SRp40 binding site, potentially leading to skipping of exon 20 or (partial) retention of intron 20 (MaxEnt −52.5%, NNSPLICE −60.3%).
Using the online genetic matching tool GeneMatcher,35 we identified two additional families (GM1, KMC1) carrying ARHGEF6 variants (Figure 1, Supplemental Figure 1, Table 1), both not reported in gnomAD. In family GM1, pregnancy was terminated at 21 2/7 weeks due to bilateral renal agenesis and severe cardiomyopathy of the male fetus (GM1-21). ES revealed a nonsense variant in ARHGEF6 (c.571C>T, p.Arg191*; Figure 1, Supplemental Figure 1, Table 1). In family KMC1, the proband KMC1-21 carried a variant in ARHGEF6, predicted to cause a frameshift and premature stop (c.1549delG, p.Glu517Lysfs*27, Figure 1, Supplemental Figure 1, Table 1). The proband was listed for intellectual disability on GeneMatcher,35 but, interestingly, reverse phenotyping revealed right-sided renal agenesis.
Truncating ARHGEF6 Variants Lead to Reduced Active Levels of RAC1 and CDC42
Small GTPases, such as RAC1, CDC42, and RHOA, play pivotal roles in kidney development.36,37 As ARHGEF6 encodes a GEF for RAC1 and CDC42, we hypothesized that proband-derived mutants affect active GTPase levels. To examine this potential pathogenic mechanism, G-LISA assays were performed using HEK293T cells transfected with ARHGEF6 cDNA representing either WT or proband-derived variants (Figure 2A, Supplemental Figures 3–4). Compared with empty vector (MOCK), WT significantly increased active levels of RAC1 and CDC42, while protein-truncating mutants did not. However, we did not observe differences between WT and missense variants (Figure 2A).
Figure 2.

ARHGEF6 increases levels of active CDC42 and RAC1, lamellipodia formation, and FN-induced cell spreading in renal cells. (A) HEK293T cells were transfected with ARHGEF6 WT cDNA and cDNA representing proband variants. After 24 hours, transfection active levels of CDC42 and RAC1, respectively, were measured by G-LISA assay and normalized to MOCK. Each color represents an individual experiment. p.Lys712* indicates splice variant of family GM2. **P<0.01; ***P<0.001; ns, not significant, ordinary one-way ANOVA. (B) Human podocytes were transfected with N-MYC ARHGEF6 WT and mutant cDNA representing proband variants. After 24 hours, cells were fixed and stained with anti-MYC (DaM488) and phalloidin (594) representing F-actin. Lamellipodia/membrane ruffles were counted in a blinded way in three independent experiments (approximately 20 cells/group/experiment counted; p.Glu517Lysfs*27 and p.Pro278Ser only two experiments). Lamellipodia/membrane ruffle was defined as convex cell protrusion. P<0.001 ***, n.s., not significant, Kruskal–Wallis test. (C) Representative images for lamellipodia/membrane ruffle formation, quantified in (B). White arrowheads indicate lamellipodia/membrane ruffles. Scale bar 10 µm. (D) Human podocytes were cotransfected with GFP-tagged WT ARHGEF6 cDNA and c-MYC-tagged ILK cDNA (upper panel), c-MYC-tagged PARVA cDNA (middle panel), or c-MYC-tagged PARVB cDNA (lower panel). After 24 hours, cells were fixed and stained with anti-MYC (secondary antibody DaM594). Note that ARHGEF6 colocalizes with ILK, PARVA, and PARVB, especially in the cell periphery (white arrow). Scale bars 25 µm. (E) IMCD3 cells were cotransfected with GFP-tagged ARHGEF6 (WT/proband-derived variants) and c-MYC-tagged PARVA cDNA. Twenty-four hours after transfection, cells were trypsinized, counted, and plated on FN-coated glass slides. Cells were allowed to attach for 5 hours, washed, and then fixed, followed by antibody staining with anti-MYC (secondary antibody DaM594). Cell morphology was assessed in double-positive cells only and in at least 40 cells per condition. Cell spreading was quantified in a dichotomous way (cells spread or not spread). Scale bars 25 µm. (F) Graph shows quantification of (E) as fractions of spread-out cells per conditions. Different colors represent four independent experiments. (G) HEK293T cells were transfected with GFP-tagged ARHGEF6 (WT versus proband mutants) and c-MYC-tagged PARVA cDNA. Note that GFP ARHGEF6 WT coimmunoprecipitates with MYC-tagged PARVA in HEK293T cells, but not with GFP control vector (MOCK), with early truncating variants (p.Arg191* and p.Le387Alafs*58) and only weakly with late truncating variants (p.Glu517Lys*27 and p.Lys712*). Left panel shows input and right panel immunoprecipitate (IP) immunoblots.
Truncating ARHGEF6 Variants Impair Formation of Lamellipodia/Membrane Ruffles
It was demonstrated that expressing ARHGEF6 in CHO-K1 cells leads to cellular protrusions consistent with lamellipodia/membrane ruffles.38,39 Overexpressing ARHGEF6 mutants lacking GEF activity abrogated these morphological changes.38 In addition, ARHGEF6 was shown to stimulate PAK proteins, which act upstream of RAC1 and can induce lamellipodia on binding ARHGEF proteins.40–43 Because we demonstrated that ARHGEF6 WT, but not truncating mutants, increases active levels of RAC1 and CDC42 (Figure 2A), we hypothesized that WT, but not mutants, induces formation of cellular protrusions. Cellular morphology was assessed in human podocytes and IMCD3 cells 24 h after transfection (WT or proband-derived variants) via blinded evaluation (Figure 2, B and C, Supplemental Figure 5). We observed that lamellipodia/membrane ruffles, here defined as convex cellular protrusions (Figure 2C, arrowhead), were more frequent on overexpression of WT ARHGEF6 compared to MOCK conditions. Overexpression of truncating proband variants led to a reduced number of cellular protrusions per cell (Figure 2, B and C, Supplemental Figure 5), indicating loss of function.
ARHGEF6 Variants Lead to Decreased FN-Induced Cell Spreading
Arhgef6 was shown to act downstream of Ilk, encoded by a murine CAKUT gene, and to interact with proteins associated with focal adhesion sites, such as actin-binding parvin proteins.8,16 First, we demonstrated that ARHGEF6 colocalizes, especially in the cellular periphery, with ILK, PARVA, and PARVB on respective coexpression (Figure 2D, Supplemental Figure 6). FN-induced cell spreading is promoted in an integrin/ILK/ARHGEF6-dependent pathway.15 We thus hypothesized that WT, but not proband-derived mutants, promotes FN-induced cell spreading in IMCD3 cells. On coexpression with PARVA, we detected a significant increase in spread-out cells for WT ARHGEF6 compared to MOCK (PARVA only; Figure 2, E and F). By contrast, all cDNAs representing CAKUT proband variants failed to cause a comparable increase in spread-out cells, suggesting loss of function of variants (Figure 2, E and F).
Truncated ARHGEF6 Variants Do Not Interact with PARVA
After demonstrating colocalization of ARHGEF6 with ILK, PARVB, and PARVA, we tested protein-protein interaction. As the interaction of ARHGEF6 with PARVB had recently been shown, we tested the interaction with either PARVA or ILK, and both proteins encoded by murine CAKUT genes.8 Using coimmunoprecipitation, we detected an interaction of GFP-tagged WT ARHGEF6 with myc-tagged PARVA. This interaction was abrogated for early truncating variants (GM1/B1185) and reduced for late truncating variants (KMC1/GM2), but not impaired by missense variants (Figure 2G, Supplemental Figure 7). In addition, we found an interaction of ARHGEF6 with ILK, which was not altered by CAKUT-associated variants (Supplemental Figure 7).
Effects of ARHGEF6 Variants on Lumen Clearance and Polarity in 3D Madin-Darby Canine Kidney Cell Cultures
Branching morphogenesis is an essential step during nephron formation,44,45 and disruption can lead to numerous kidney diseases, including CAKUT.46 Madin-Darby canine kidney (MDCK) cells are successfully used to study ureteric branching morphogenesis.47 Branching requires coordinated ECM/integrin adhesion signaling and can be induced by mesenchymal growth factors, such as HGF.48 In the absence of these factors, expression of a kinase-dead PAK1 mutant induces spontaneous tubulogenesis in an ARHGEF-dependent way.47,49 We therefore evaluated branching morphogenesis in MDCK cysts stably expressing either WT or three representative proband variants (p.Arg191*/p.Leu387Alafs*58/p.Ile444Asn) with and without HGF-conditioned media (HGF-CM). Under stable expression of WT alone or after adding HGF-CM, we observed no increase in branching compared to controls. However, we found that cells expressing proband-derived variants, compared to WT, exhibited an altered lumen clearance pattern that was shifted toward intermediate (50%–90% clearance) or disorganized (<50% clearance) phenotypes under native conditions (Figure 3, A–C). This finding suggests a role of ARHGEF6 during the initial steps of tubulogenesis. Lumen clearance of fluid-filled MDCK cysts depends on correct apical-basal polarization, which is regulated by Rac-dependent deposition of laminin in the basement membrane, and downstream of β1-integrin.50 We therefore analyzed localization of laminin in MDCK cysts expressing either WT or proband-derived ARHGEF6 variants (Figure 3, B and D). To test if a laminin phenotype was independent of lumen clearance efficiency, we carefully selected cysts of all categories (normal, intermediate, and disorganized) for each genotype (Supplemental Figure 8). In WT-ARHGEF6-expressing cultures, laminin staining appeared as a continuous thin layer at the periphery of cysts, indicative of normal basement membrane assembly at the basal surface. Interestingly, laminin localization was abnormal in the vast majority of cultures expressing proband-derived variants, independent of lumen clearance (Figure 3, B and D, Supplemental Figure 8).
Figure 3.

Consequences of ARHGEF6 variants on lumen clearance and cell polarity in MDCK 3D cell cultures. (A) Branching morphogenesis was studied in 3D cultures from MDCK cells stably expressing either ARHGEF6 WT or one CAKUT proband variant (GM1, c.571C>T, p.Arg191*; B1185, c.1158_1159del, p.Leu387Alafs*58; A5124, c.1331T>A, p.Ile444Asn). Cysts were then evaluated and classified as normal (>90% cleared), intermediate (50–90% cleared), or abnormal/disorganized (<50% cleared). Clearance and distribution of ARHGEF6 protein are further depicted by immunofluorescence staining using podocalyxin (PODXL) as an apical marker. Scale bars 10 µm. (B) Lumen clearance of fluid-filled MDCK cysts depends on correct apical-basal polarization. To assess polarization and correct basement membrane assembly, cysts were stained for F-actin and laminin. The panel shows representative images for WT ARHGEF6 and each of the following proband-derived variants: GM1, c.571C>T, p.Arg191*; B1185, c.1158_1159del, p.Leu387Alafs*58; A5124, c.1331T>A, p.Ile444Asn. Scale bars 20 µm. (C) Lumen clearance was assessed as described in (A) and then quantified for GFP control, WT ARHGEF6, and the following proband-derived variants: GM1, c.571C>T, p.Arg191*; B1185, c.1158_1159del, p.Leu387Alafs*58; A5124, c.1331T>A, p.Ile444Asn. All cysts from six independent experiments were evaluated in a blinded manner (approximately 500–700 per condition). The majority of cysts expressing WT ARHGEF6 showed predominantly normal (>90% clearance) or intermediate phenotypes (50%–90% clearance) while cysts expressing any of the tested proband-derived ARHGEF6 variants showed a distribution that was shifted toward intermediate or abnormal/disorganized phenotypes (<50% clearance). (D) Laminin distribution in cysts shown in (B) was evaluated as either normal or abnormal for WT ARHGEF6 and the following proband-derived variants: GM1, c.571C>T, p.Arg191*; B1185, c.1158_1159del, p.Leu387Alafs*58; A5124, c.1331T>A, p.Ile444Asn. Abnormal laminin distribution included absent staining, high luminal staining, irregular or patchy staining, or a combination of these patterns. For each ARHGEF6 variant, cysts of all clearance categories (normal, intermediate, and disorganized) were analyzed. Laminin distribution for cysts expressing WT ARHGEF6 was almost completely normal, whereas cysts expressing any of the tested proband ARHGEF6 variants showed largely abnormal laminin patterns (also see Supplemental Figure 8).
Arhgef6 Deficiency Leads to CAKUT in Mice
Arhgef6 has been previously shown to be expressed in mouse kidneys51 and in cap mesenchyme of fetal human kidneys.52 When interrogating single-cell RNA sequencing databases52–54 of adult mouse or fetal human kidneys, we could only find low overall expression (Supplemental Figure 9). To further evaluate Arhgef6 expression during embryogenesis, we therefore performed ISH studies of the urogenital system of E11.5/E12.5/E14.5/E18.5 mice, representing early and midstages of murine kidney development (Supplemental Figure 10). Because of inconclusive results, we next used highly sensitive and specific RNAScope ISH. This analysis revealed specific signals of the tested Arhgef6 probe in WT kidneys during embryogenesis (E11-16) that were not detected in Arghef6-deficient mice (Supplemental Figure 11). At Theiler stage 20 (TS20), the punctuate Arhgef6 signals were present throughout the kidney in both metanephric mesenchyme and ureteric tree regions. At TS22-TS24, Arhgef6 signals were still detected throughout the developing kidney with enrichments within later-stage nephrons (immature glomerular structures; red arrowheads) over earlier-stage nephrons (cap mesenchyme or renal vesicles; white arrows) and Wt1-negative regions (Supplemental Figure 11). Genes, important for renal development, are expressed in a spatially and temporally tightly controlled manner. Although the RNAscope results do not allow us to decern the cell type of expression, the absence of signal in Arghef6-deficient mice and the detected regions of Arghef6 signal enrichment prompted us to next test if the detected Arghef6 signal conveys biological relevance for kidney development in vivo. We therefore next studied hemizygous Arhgef6 knockout mice as a model for the all-male individuals with CAKUT in which we had initially detected ARGHEF6 variants (Supplemental Figure 12). Maintenance breedings showed no overt lethality and normal sex and Mendelian ratios. Assessment of CAKUT phenotypes of mice was performed in a blinded two-step fashion (Figure 1, Supplemental Figure 12). Litters were dissected at days 3–4 of life (P3-4) and macroscopically assessed according to the following criteria: suspected duplex kidney (Figure 4A), numeric abnormality, hydronephrosis or hydroureter, hypoplasia (Figure 4B), and suspected dysplasia (side difference in color/opacity; Figure 4, C and D). In total, 43 of 423 specimens fulfilled those criteria and were processed for Masson's trichrome staining. Microscopic evaluation was then performed according to the following criteria: duplex kidney, hydronephrosis/hydroureter, and hypodysplasia (size difference >10%, cystic structures, very heterogeneous parenchyma, loss of zonal compartmentalization; Figure 4D). In total, 27 of 43 specimens were classified as pathologic and subsequently genotyped, and 11.5% of Arhgef6−/Y mice and 3.6% of Arhgef6+/Y mice showed any form of CAKUT, respectively (Figure 4E). Female mice (heterozygous and homozygous) did not show a markedly increased frequency of CAKUT (5%) compared with male WT mice (Supplemental Figure 13A).
Figure 4.

Increased rate of duplex kidneys and renal hypodysplasia in Arhgef6-deficient mice. Heterozygous Arhgef6+/− were mated with hemizygous Arhgef6−/Y and litters sacrificed at day 4 postnatally. Kidneys and urinary tract have been evaluated macroscopically and dissected on suspected CAKUT phenotype. Specimens have then been processed for Masson's trichrome (MT) staining and subsequent blinded evaluation of multiple sections of each specimen. (A) Two exemplary kidney pairs from Arhgef6−/− and Arhgef6−/Y mice, respectively, are shown demonstrating duplex kidneys. Red arrowhead points to fissure: whole organ overview (upper) and MT-stained section (lower). (B) One exemplary kidney pair of an Arhgef6−/Y mouse is shown demonstrating a left hypoplastic kidney—whole organ overview. (C) One exemplary kidney of an Arhgef6−/Y mouse is shown demonstrating a dysplastic kidney—MT-stained section. (D) Graph shows relative frequency of three different observed CAKUT phenotypes (duplex kidney, hypodysplasia, and hydronephrosis) for wild-type Arhgef6+/Y and hemizygous Arhgef6−/Y animals from macroscopic evaluation. Unpaired t test; *P<0.05. (E) Graph shows relative frequency of total observed CAKUT for wild-type Arhgef6+/Y and hemizygous Arhgef6−/Y animals from macroscopic evaluation. Unpaired t test; *, P<0.05. (F) In a second evaluation, whole litters from Arhgef6+/− × Arhgef6−/Y matings were evaluated at 4 weeks of age independent of macroscopic CAKUT. Kidneys were processed for histology, and parenchyma was scored for dysplastic features (cystic structures, very heterogeneous parenchyma, loss of zonal compartmentalization). Bar graph shows relative frequency of dysplasia in wild-type Arhgef6+/Y and hemizygous Arhgef6−/Y animals. Unpaired t test; *P<0.05. Nephron quantity was estimated by counting the number of glomeruli in longitudinal midline sections of kidneys. Scatter dot plot shows numbers of glomeruli for wild-type Arhgef6+/Y and hemizygous Arhgef6−/Y animals. Each animal is represented by two data points, one for each kidney. Mann–Whitney test; ***P<0.001.
Nearly half of the observed CAKUT phenotypes in Arhgef6−/Y mice presented as hypodysplasia, which could easily be missed during initial macroscopic evaluation. We therefore performed a second round of breedings for unbiased histological analysis of all kidneys, focusing on qualitative and quantitative assessment of the parenchyma. In this study, we found a total of 10% (2/19) of Arhgef6+/+ mice exerting features of renal dysplasia while Arhgef6−/Y mice scored positive in 37.5% (6/16) of cases (Figure 4F). Similarly, female Arhgef6+/− and Arhgef6−/− mice also showed an increased percentage of dysplastic kidneys compared with WT male mice (Supplemental Figure 13B). In the second step, we quantified the nephron number by counting glomeruli in longitudinal sections, revealing a significantly lower number in Arhgef6−/Y compared with Arhgef6+/Y mice (Figure 4F).
In summary, male mice, deficient for Arhgef6, recapitulated human phenotypes by showing different features of mild CAKUT.
Arhgef6 Deficiency Leads to Renal Hypoplasia in Frogs
We next investigated a second in vivo model with Arhgef6 deficiency. Xenopus is an effective model to study kidney development as embryos develop externally, are easy to manipulate, and have functional kidneys after approximately 2 days. In addition, Xenopus expresses orthologs of approximately 80% of human CAKUT genes.55 We first tested the efficiency of CRISPR/Cas9 editing after bilateral injection of two-cell embryos and demonstrated in vivo arhgef6 gene editing at high efficiency (86%±10%), yielding high knockout scores (78%±10%; Figure 5A). Next, we evaluated the consequences of global arhgef6 knockout by bilateral injection in the two-cell stage. Global knockout led to a dramatic reduction of survival of embryos compared to noninjected or slc45a2 crispants, which serve as an additional control56 leading to albinism in the frog (Figure 5B). The majority of arhgef6 crispants failed to develop beyond the gastrula and neurula stages. For all surviving embryos, kidney size was determined at late stage 39. Interestingly, arhgef6 crispants showed significantly reduced kidney sizes compared with both noninjected and slc45a2 crispants (Figure 5, C and D). To perform side comparisons and bypass the observed early lethality, we performed unilateral injections in the ventral blastomere of four-cell embryos. Here, arhgef6 crispants, concordantly, showed reduced kidney size on the injected side, when compared to either the noninjected side or injected side of tyrosinase control crispants, another albinism target57 (Figure 5, C and E, F, Supplemental Video 1). Of note, besides kidney hyperplasia, no gross morphologic differences were observed of arhgef6 crispants (Figure 5C, Supplemental Video 1). As an alternative strategy, we performed morpholino-mediated knockdown of arhgef6 using unilateral ventral injections (four-cell state). In direct phenocopy of the arhgef6 crispants, a significantly reduced kidney size on the injected side was observed, compared with either noninjected or control morpholino animals (Figure 5, C and G). In summary, both arhgef6 knockout and knockdown during early embryogenesis affect proper renal development in Xenopus, leading to significantly reduced kidney sizes.
Figure 5.

Knockdown or knockout of arhgef6 impairs renal development in Xenopus larvae. (A) Three distinct gRNAs targeting arhgef6 were separately and bilaterally injected in both blastomeres of two-cell X. tropicalis embryos. Embryos were then lysed one day postfertilization, and CRISPR/Cas9 editing efficiencies were determined using Sanger sequencing and ICE deconvolution analysis. This demonstrated in vivo arhgef6 gene editing at high efficiency (86%±10%), enriched toward frameshift variants, yielding high knockout score (78%±10%) for all three gRNAs. (B) Bar graph shows relative frequency of effect on survival from global knockout of arhgef6 compared to noninjected embryos or embryos injected with slc45a2 gRNA, leading to albinism in the frog. (C) Fluorophore-conjugated lectin (LEL) was then used to stain proximal nephron parts to qualitatively compare kidney development in Xenopus embryos from different injections. Exemplary images are shown for arhgef6 gRNA, arghef6 morpholino, and control morpholino injections. LEL, Lycopersicon esculentum Lectin. White scale bar, 100 µm; black scale bar, 20 µm. (D) Scatter plot shows quantification of kidney size from two-cell stage bilateral injections, comparing noninjected, arghef6 gRNA and control slc45a2 gRNA-injected animals. Kruskal–Wallis test; NI, noninjected animal; *P<0.05; ***P<0.001. (E) Side comparison of kidney size from arghef6 gRNA unilateral four-cell stage ventral injection is shown. NI, noninjected side of same animal; Mann–Whitney test; ***P<0.001. (F) Side comparison of kidney size from arghef6 gRNA and tyrosinase gRNA in unilateral four-cell stage ventral injection is shown. Mann–Whitney test; ***P<0.001. (G) Scatter plot shows comparison of kidney size from morpholino-mediated knockdown of arghef6 using unilateral four-cell stage ventral injections using the following conditions: noninjected, arghef6 morpholino, control morpholino. Kruskal–Wallis test; NI, noninjected side; MO, morpholino; ***P<0.001.
Discussion
In this study, we describe the discovery and functional characterization of hemizygous variants in ARHGEF6 detected in six families with CAKUT (Figure 6). Consistent with its known function as a GEF, we show that WT ARHGEF6, but not truncated mutants, increases active levels of small GTPases, the formation of cellular protrusions, or FN-induced cell-spreading. We show that ARHGEF6 WT interacts with ILK and PARVA, both proteins encoded by genes, known to cause murine CAKUT if mutated. The interaction with PARVA was abrogated for truncating variants. When testing lumen clearance of MDCK cysts, proband-derived variants displayed an impaired clearance and basement membrane assembly compared with ARHGEF6 WT. Finally, mouse and Xenopus Arhgef6 knockout models replicated a CAKUT phenotype, supporting the role of ARHGEF6 during renal development (Figure 6).
Figure 6.
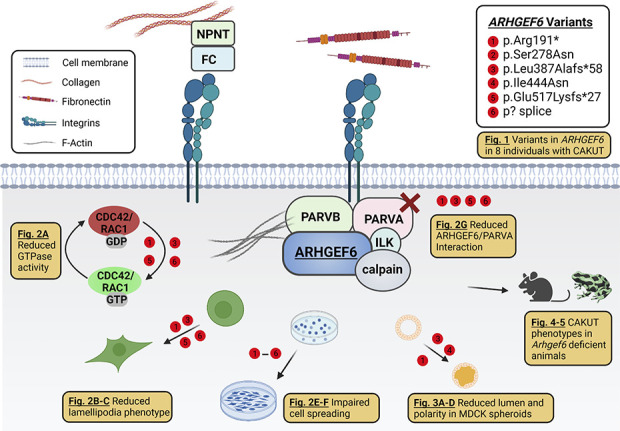
Variants in ARHGEF6 lead to CAKUT in humans, mice, and frogs. This graphical summary depicts (1) basic aspects of the ARHGEF6/integrin/ILK/parvin/CDC42/RAC1-dependent pathway and (2) different facets of ARHGEF6 loss of function studied in this work. First, we identified six different X-linked recessive variants in ARHGEF6 as a novel cause of monogenic CAKUT in humans (Fig. 1). On the basis of our findings, ARHGEF6 variants reduce active levels of the small GTPases CDC42 and RAC1 (Fig. 2A). Mutants lead to defective lamellipodia/membrane ruffle formation (Fig. 2, B and C) and decreased FN-induced cell spreading (Fig. 2, E and F). Furthermore, mutants show a reduced binding to PARVA (Fig. 2G) and impaired lumen clearance and cell polarity in MDCK cell 3D spheroids (Fig. 3, A–D). Finally, we replicate human CAKUT phenotypes in Arhgef6 deficient mice (Fig. 4) and frogs (Fig. 5). NPNT, nephronectin; FC, Fraser complex (consisting of FRAS1, FREM2, and FREM1). Created with biorender.com.
Cell spreading is a complex and dynamic process that depends on a variety of pathways including the integrin/ILK/parvins/ARHGEF6-dependent activation of small GTPases.15,58 Here, we demonstrate reduced levels of active RAC1/CDC42 for proband-derived truncating but not missense variants, potentially causative for the reduced cellular protrusion formation and cell spreading phenotype. It has been suggested that ARHGEF6 has a dual function for cell spreading.38 Impaired cell spreading in cells expressing proband variants might thus only be partially explained by deficient GTPase activity. Cell spreading can also be induced by a GEF-independent association through interaction with calpain-4 via an unknown signaling cascade.16 As both missense variants are located within ARHGEF6's predicted binding site to Calpain-4, a potential loss of interaction could explain the here-observed phenotype.38 However, future studies are warranted to test this hypothesis. Integrins and their ligands, such as nephronectin, ILK, and PARVA, are expressed in metanephric mesenchyme and/or ureteric bud at early developmental stages in mice and are thus important for cross talk during early kidney morphogenesis.6–8,11,12 Loss of function of these proteins can lead to different renal anomalies indicating an essential role of this pathway for the CAKUT development.6–8,11,12 FN, another integrin ligand, is detected in glomeruli and interstitium of developing kidneys.59 As mice lacking FN die in utero and no kidney-specific deletion has been performed to date, its role for renal development remains unclear. However, as FN is important for branching morphogenesis during salivary and mammary gland development, its role during kidney formation might be comparable.60 As ARHGEF6 acts downstream of FN/integrins, ILK, and parvins,15,16 it is conceivable that ARHGEF6 also plays a role in kidney development. We confirm that ARHGEF6 colocalizes with ILK and parvins at the cellular periphery in renal cell lines. ISH and RNAScope experiments of Arhgef6 in mouse embryonic tissue at different developmental time points, however, remained inconclusive. Thus, further experiments are warranted to determine the expression pattern of Arhgef6 during embryonic development. We further tested if ARHGEF6 also regulates integrin trafficking but did not detect significant differences in cell surface or internalized β1 integrin between WT and proband-derived variants (Supplemental Figure 14). ARHGEF6 directly binds to PARVB,39 and by Co-IP, we additionally demonstrate interaction of ARHGEF6 with PARVA, encoded by a murine CAKUT gene and a paralog of PARVB. This interaction was abrogated by proband-derived truncating variants including the variant present in GM2. The site of interaction of ARHGEF6 with PARVB was mapped to the CH domain.39 It was demonstrated that deletion of the coiled-coil domain of ARHGEF6, important for homodimerization, similarly reduced binding to PARVB.39 This suggests that both dimerization and presence of an intact CH domain are required for proper interaction of ARHGEF6 with parvins, potentially explaining the here-observed phenotype.
Cell polarization is necessary for the physiologic function of tubular organs, including transport across luminal epithelia or barrier function. However, correct cell polarity is also indispensable for tubulogenesis because the development of a basement membrane is a pre-requisite for apical-basal polarization and lumen formation, preceding branching morphogenesis.61 Defects of lumen clearance can thereby be caused by loss of cell polarity. MDCK cysts are a well-established 3D model used to study renal tubulogenesis. In this study, we demonstrate reduced lumen clearance efficiency for three representative ARHGEF6 mutants compared to WT and control in MDCK cysts, accompanied by a dysregulated laminin assembly. Thus, ARHGEF6 might affect lumen clearance, the initial step in tubulogenesis through cell-matrix-dependent signaling, potentially via a dysregulated ECM adhesion/integrin pathway.
We further show that loss of Arhgef6 in mice and Xenopus partially recapitulates the human CAKUT phenotype. A comparatively low percentage of CAKUT during initial evaluation of Arhgef6-deficient mice might be explained through variable expressivity and incomplete penetrance, which makes interpretation of CAKUT genetics challenging in general. Furthermore, the background frequency of congenital abnormalities in inbred mouse strains, which is described to be up to 10%,62 additionally complicates the evaluation. We initially prescreened mice for macroscopic phenotypes. Thus, milder forms of CAKUT (e.g., dysplasia) might have been missed. The second evaluation of complete litters at 4 weeks of age indicates a higher percentage of mild CAKUT (37.5%), dysplasia specifically, in hemizygous and to a lesser extent in homozygous mice, but also in heterozygous females. To identify the exact developmental stages, Arghef6 is involved in, generation of conditional knockout mice using Cre reporter lines, for selective deletion of Arhgef6 (Gdnf, Six2, or Tcf21) could be more conclusive. We observed reduced numbers of nephrons in all allele-carrying mice compared with WT males. Similarly, as for the dysplasia phenotype, we did not see differences between heterozygous and homozygous females. However, because nephron count is described to be higher in females than in males, especially on C57BL/6 background,63–65 additional breedings yielding WT females are necessary to confirm this observation. Considering the observed duplex phenotype and reduced nephron number in Arhgef6-deficient mice, it is conceivable that Arhgef6 plays a role in the initial generation of progenitor cells or branching morphogenesis. However, reduced nephron number, especially in light of the enriched Arhgef6 signal (RNAscope; Supplemental Figure 11) within immature glomerular structures, could also be explained by Arhgef6 function during glomerulus maturation. Future studies should more specifically assess Arhgef6 expression at additional developmental time points and in coexpression with other markers (e.g., Odd1, Six2, Wnt9b, Wnt4, Jag1, Fgf8). Because X-inactivation happens before most of the nephrogenic development occurs and branching is initiated within small groups of cells of the ureteric bud, cellular mosaicism resulting from X-inactivation might affect nephrogenesis.66,67 Further studies are warranted to identify the initial number of progenitor cells and pattern of mosaicism within the kidney to understand effects of inherited X-chromosomal variants. In Xenopus, arhgef6 resides on an autosome and gene inactivation, via CRISPR/Cas9 or morpholinos technology, allows direct reduction of Arhgef6 levels during early embryonic development independent of sex. Our Xenopus manipulations culminate in hypoplasia of pronephros, suggesting a role of Arhgef6 during early kidney development.
As a biologically relevant X-chromosomal gene, ARHGEF6 is significantly constraint against loss of function (LOF) variants. The ARHGEF6 gene is expected to contain 35 LOF variants, but only one is observed in the population database gnomAD, yielding an o/e score of 0.03 (CI, 0.01 to 0.14). This suggests that loss of ARHGEF6 strongly interferes with either survivability or reproductive fitness. As missense variants in ARHGEF6 are not constraint in general (o/e score of 0.77; CI, 0.69 to 0.87), missense variants might be tolerated across the whole gene/protein. However, this does not exclude that certain amino acid positions are constraint against variation or that particular amino acid changes can be deleterious. Regarding our conducted cell culture experiments and the low constraint against missense variants, the potential pathogenicity of the missense variants and their consequence for the individuals carrying those variants remains uncertain and still has to be critically discussed. An additional challenge in vetting the relevance of the detected variants is inherent to the study of potential novel rare disease genes as the very low numbers of families do not allow testing for statistical significance. Thus, the identification of additional CAKUT families especially with ARHGEF6 missense variants and future studies are needed to delineate the role of ARHGEF6 during renal morphogenesis and disease development.
In summary, our data suggest that deleterious ARHGEF6 variants cause dysregulation of integrin-parvin-RAC1/CDC42 signaling. Moreover, deficiency of Arhgef6 leads to abnormal renal development in mice and Xenopus. Therefore, we conclude that variants in ARHGEF6 are a novel cause of monogenic CAKUT.
Supplementary Material
Acknowledgments
We are grateful to the families and study individuals for their contribution. F. Hildebrandt is the William E. Harmon Professor of Pediatrics. Microscopy was partially performed at the Research Technology Center Microscopy at the Radboud University Medical Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank the National Institutes of Health (5R01DK076683-13 [F. Hildebrandt], F32 DK122766 [A.C. Onuchic-Whitford], 5K12HD052896-13 and 1K08DK125768-01A1 [A.J. Majmundar]), the Deutsche Forschungsgemeinschaft (403877094 [V. Klämbt], 404527522 [F. Buerger], 442070894 [S. Seltzsam], 394046635 [T. Sieckmann, K.M. Kirschner]), the Else-Kröner Fresenius Stiftung (Else-Kröner Memorial Grant [V. Klämbt]), the American Society of Nephrology (Carl W. Gottschalk Research Scholar Grant [F. Buerger], Norman Siegel Research Scholar Grant [A.J. Majmundar]), and the Manton Center for Orphan Disease Research (A.J. Majmundar). We also are thankful to the Wilhelm Sander Stiftung (2018.015.1 [T. Sieckmann]), the BIH Charité Clinician Scientist Program by the Charité – Universitätsmedizin Berlin, and the Berlin Institute of Health at Charité (V. Klämbt), the Bundesministerium für Bildung und Forschung (BMBF, GeNeRARe, TP6, funding code 01GM1902E [T. Naert, G. Rosenberger]), the American College of Medical Genetics and Genomics Foundation (ACMG/Takeda Next-Generation Biochemical Genetics Award [C.-H.W. Wu]) and the Swiss National Science Foundation (NCCR Kidney.CH, Project grant 310030_189102/1 [S.S. Lienkamp]). We are grateful to the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement (891127 [T. Naert]), the ISF (Clinical Scientist grant 2773/19 [A. Vivante]), the National Human Genome Research Institute (UM1HG006504 Yale Center for Mendelian Genomics), and also the GSP Coordinating Center (U24 HG008956) that contributed to cross-program scientific initiatives and provided logistical and general study coordination. We also thank the National Heart, Lung, and Blood Institute/National Eye Institute.
Footnotes
V.K. and F.B. contributed equally to this work.
Disclosures
F. Hildebrandt is a cofounder and S.A.B. member of Goldfinch Biopharma Inc. F. Hildebrandt also reports Research Funding: NIH; Honoraria: Sanofi; Patents or Royalties: NPHP1; and Advisory or Leadership Role: Goldfinch-Bio. K.M. Kirschner reports Ownership Interest: Bio-Techne. E. Banne reports Consultancy: Pronto Diagnostics, Israel; and Research Funding: Rhythm Pharmaceuticals. R.P. Lifton reports Advisory or Leadership Role: Roche Board of Directors, and Genentech Board of Directors. A.J. Majmundar reports Consultancy: Judo Bio, Inc. C.-H.W. Wu reports Advisory or Leadership Role: GENE (journal), and The Association of Chinese Geneticists in America (ACGA). All remaining authors have nothing to disclose.
Funding
This work was supported by the National Institutes of Health (grant 5R01DK088767-09), the Swiss National Science Foundation (grant 310030_189102/1), the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement (grant 891127), ISF—Clinical Scientist grant 2773/19, the Deutsche Forschungsgemeinschaft (grants 403877094, 404527522 and 442070894), the Else-Kröner Fresenius Stiftung (2020_EKMS.13 Else-Kröner Memorial Grant), and the Bundesministerium für Bildung und Forschung (grant 01GM1902E).
Author Contributions
F. Buerger, F. Hildebrandt, and V. Klämbt conceptualized the study; E. Banne, M.R. Bekheirnia, F. Buerger, D.M. Connaughton, R. Dai, M. Joosten, K.M. Kirschner, V. Klämbt, E. Lai, S.S. Lienkamp, A.J. Majmundar, N. Mann, T. Naert, T. Nauth, A.C. Onuchic-Whitford, K. Richter, G. Rosenberger, L. Schierbaum, S. Schneider, S. Seltzsam, O. Shlomovitz, S. Shril, T. Sieckmann, A. Vivante, C. Wang, A.-C. Weiss, C.-H. Wu, and M.P. Zegers were responsible for data curation; F. Buerger, A. Kispert, and M.P. Zegers were responsible for visualization; V. Klämbt, S. Shril, S.S. Lienkamp, and M.P. Zegers were responsible for formal analysis; K.-D. Fischer, F. Hildebrandt, S.S. Lienkamp, G. Rosenberger, and M.P. Zegers were responsible for funding acquisition; F. Hildebrandt and S.S. Lienkamp were responsible for investigation; F. Buerger was responsible for methodology; F. Buerger, F. Hildebrandt, and V. Klämbt were responsible for project administration; K.-D. Fischer, F. Hildebrandt, R.P. Lifton, and S. Mane were responsible for resources; R.P. Lifton, S. Mane, S. Shril, and A. Vivante were responsible for software; K.-D. Fischer, F. Hildebrandt, A. Kispert, S.S. Lienkamp, S. Mane, and G. Rosenberger provided supervision; F. Buerger and V. Klämbt wrote the original draft; and K.-D. Fischer, F. Hildebrandt, A. Kispert, S.S. Lienkamp, T. Naert, G. Rosenberger, and M.P. Zegers reviewed and edited the manuscript.
Data Sharing Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Materials are available upon request to the corresponding author and may be subject to an MTA. The genetics datasets supporting this study have not been deposited in a public repository due to restriction by patient consent but are available from the corresponding author on request.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/A651 and http://links.lww.com/JSN/D635.
Supplemental Figure 1. Sequencing traces and family pedigrees of individuals with ARHGEF6 variants.
Supplemental Figure 2. Additional clinical images of individuals with ARHGEF6 variants.
Supplemental Figure 3. ARHGEF6 cDNA clone and antibody validation.
Supplemental Figure 4. ARHGEF6 antibody and clone testing on immunofluorescence.
Supplemental Figure 5. ARHGEF6 WT overexpression induces lamellipodia formation in IMCD3 cells.
Supplemental Figure 6. Antibody and clone testing on immunofluorescence.
Supplemental Figure 7. ARHGEF6 interacts with ILK and PARVA.
Supplemental Figure 8. Laminin deposition in basement membrane of MDCK cysts is affected in cells expressing proband-derived variants of ARHGEF6.
Supplemental Figure 9. Expression of Arhgef6 in mouse urogenital development on the basis of single-cell RNA sequencing databases.
Supplemental Figure 10. Expression of Arhgef6 in mouse urogenital development.
Supplemental Figure 11. RNAScope-based expression of Arhgef6 in mouse urogenital development.
Supplemental Figure 12. Generation and workflow of Arhgef6-deficient mouse model.
Supplemental Figure 13. CAKUT phenotype in Arhgef6 mice.
Supplemental Figure 14. Consequences of ARHGEF6 variants on β1 integrin internalization in COS7 cells.
References
- 1.van der Ven AT, Vivante A, Hildebrandt F. Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29(1):36-50. doi: 10.1681/ASN.2017050561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol. 2016;12(3):133-146. doi: 10.1038/nrneph.2015.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol. 2014;29(4):695-704. doi: 10.1007/s00467-013-2684-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Ven AT, Connaughton DM, Ityel H, et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29(9):2348-2361. doi: 10.1681/ASN.2017121265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies J. Intracellular and extracellular regulation of ureteric bud morphogenesis. J Anat. 2001;198(3):257-264. doi: 10.1046/j.1469-7580.2000.19830257.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kreidberg JA, Symons JM. Integrins in kidney development, function, and disease. Am J Physiol Renal Physiol. 2000;279(2):F233-F242. doi: 10.1152/ajprenal.2000.279.2.F233 [DOI] [PubMed] [Google Scholar]
- 7.Mathew S, Chen X, Pozzi A, Zent R. Integrins in renal development. Pediatr Nephrol. 2012;27(6):891-900. doi: 10.1007/s00467-011-1890-1 [DOI] [PubMed] [Google Scholar]
- 8.Lange A, Wickstrom SA, Jakobson M, Zent R, Sainio K, Fassler R. Integrin-linked kinase is an adaptor with essential functions during mouse development. Nature. 2009;461(7266):1002-1006. doi: 10.1038/nature08468 [DOI] [PubMed] [Google Scholar]
- 9.Wu W, Kitamura S, Truong DM, et al. β1-Integrin is required for kidney collecting duct morphogenesis and maintenance of renal function. Am J Physiol Renal Physiol. 2009;297(1):F210-F217. doi: 10.1152/ajprenal.90260.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Mernaugh G, Yang DH, et al. β1 integrin is necessary for ureteric bud branching morphogenesis and maintenance of collecting duct structural integrity. Development. 2009;136(19):3357-3366. doi: 10.1242/dev.036269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller U, Wang D, Denda S, Meneses JJ, Pedersen RA, Reichardt LF. Integrin α8β1 is critically important for epithelial–mesenchymal interactions during kidney morphogenesis. Cell. 1997;88(5):603-613. doi: 10.1016/s0092-8674(00)81903-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brandenberger R, Schmidt A, Linton J, et al. Identification and characterization of a novel extracellular matrix protein nephronectin that is associated with integrin α8β1 in the embryonic kidney. J Cell Biol. 2001;154(2):447-458. doi: 10.1083/jcb.200103069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linton JM, Martin GR, Reichardt LF. The ECM protein nephronectin promotes kidney development via integrinα8β1-mediated stimulation of Gdnf expression. Development. 2007;134(13):2501-2509. doi: 10.1242/dev.005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Humbert C, Silbermann F, Morar B, et al. Integrin alpha 8 recessive mutations are responsible for bilateral renal agenesis in humans. Am J Hum Genet. 2014;94(2):288-294. doi: 10.1016/j.ajhg.2013.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Filipenko NR, Attwell S, Roskelley C, Dedhar S. Integrin-linked kinase activity regulates Rac- and Cdc42-mediated actin cytoskeleton reorganization via alpha-PIX. Oncogene. 2005;24(38):5837-5849. doi: 10.1038/sj.onc.1208737 [DOI] [PubMed] [Google Scholar]
- 16.Rosenberger G, Kutsche K. αPIX and βPIX and their role in focal adhesion formation. Eur J Cell Biol. 2006;85(3-4):265-274. doi: 10.1016/j.ejcb.2005.10.007 [DOI] [PubMed] [Google Scholar]
- 17.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16(13):1587-1609. doi: 10.1101/gad.1003302 [DOI] [PubMed] [Google Scholar]
- 18.Zhou W, Li X, Premont RT. Expanding functions of GIT Arf GTPase-activating proteins, PIX Rho guanine nucleotide exchange factors and GIT-PIX complexes. J Cell Sci. 2016;129(10):1963-1974. doi: 10.1242/jcs.179465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber S, Moriniere V, Knuppel T, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17(10):2864-2870. doi: 10.1681/ASN.2006030277 [DOI] [PubMed] [Google Scholar]
- 20.Braun DA, Sadowski CE, Kohl S, et al. Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet. 2016;48(4):457-465. doi: 10.1038/ng.3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bekheirnia MR, Bekheirnia N, Bainbridge MN, et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet Med. 2017;19(4):412-420. doi: 10.1038/gim.2016.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078-2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469-476. doi: 10.1038/nature13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311-323. doi: 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- 25.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2-3):377-394. doi: 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- 26.Sibley CR, Blazquez L, Ule J. Lessons from non-canonical splicing. Nat Rev Genet. 2016;17(7):407-421. doi: 10.1038/nrg.2016.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877-885. doi: 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310-315. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248-249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361-362. doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 31.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(W1):W452-W457. doi: 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu W, O'Brien LE, Wang F, Bourne H, Mostov KE, Zegers MM. Hepatocyte growth factor switches orientation of polarity and mode of movement during morphogenesis of multicellular epithelial structures. Mol Biol Cell. 2003;14(2):748-763. doi: 10.1091/mbc.e02-06-0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moorman AFM, Houweling AC, de Boer PAJ, Christoffels VM. Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J Histochem Cytochem. 2001;49:1-8. doi: 10.1177/002215540104900101 [DOI] [PubMed] [Google Scholar]
- 34.Naert T, Tulkens D, Edwards NA, et al. Maximizing CRISPR/Cas9 phenotype penetrance applying predictive modeling of editing outcomes in Xenopus and zebrafish embryos. Sci Rep. 2020;10(1):14662. doi: 10.1038/s41598-020-71412-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928-930. doi: 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elias BC, Das A, Parekh DV, et al. Cdc42 regulates epithelial cell polarity and cytoskeletal function during kidney tubule development. J Cell Sci. 2015;128(23):4293-4305. doi: 10.1242/jcs.164509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang DY, Kohl S, Fan X, et al. Mutations of the SLIT2-ROBO2 pathway genes SLIT2 and SRGAP1 confer risk for congenital anomalies of the kidney and urinary tract. Hum Genet. 2015;134(8):905-916. doi: 10.1007/s00439-015-1570-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberger G, Gal A, Kutsche K. αPIX associates with calpain 4, the small subunit of calpain, and has a dual role in integrin-mediated cell spreading. J Biol Chem. 2005;280(8):6879-6889. doi: 10.1074/jbc.M412119200 [DOI] [PubMed] [Google Scholar]
- 39.Rosenberger G, Jantke I, Gal A, Kutsche K. Interaction of alphaPIX (ARHGEF6) with beta-parvin (PARVB) suggests an involvement of alphaPIX in integrin-mediated signaling. Hum Mol Genet. 2003;12(2):155-167. doi: 10.1093/hmg/ddg019 [DOI] [PubMed] [Google Scholar]
- 40.Bagrodia S, Bailey D, Lenard Z, et al. A tyrosine-phosphorylated protein that binds to an important regulatory region on the cool family of p21-activated kinase-binding proteins. J Biol Chem. 1999;274(32):22393-22400. doi: 10.1074/jbc.274.32.22393 [DOI] [PubMed] [Google Scholar]
- 41.Daniels RH, Zenke FT, Bokoch GM. αPix stimulates p21-activated kinase activity through exchange factor-dependent and -independent mechanisms. J Biol Chem. 1999;274(10):6047-6050. doi: 10.1074/jbc.274.10.6047 [DOI] [PubMed] [Google Scholar]
- 42.Feng Q, Albeck JG, Cerione RA, Yang W. Regulation of the Cool/Pix proteins: key binding partners of the Cdc42/Rac targets, the p21-activated kinases. J Biol Chem. 2002;277(7):5644-5650. doi: 10.1074/jbc.M107704200 [DOI] [PubMed] [Google Scholar]
- 43.Li Z, Hannigan M, Mo Z, et al. Directional sensing requires Gβγ-mediated PAK1 and PIXα-dependent activation of Cdc42. Cell. 2003;114(2):215-227. doi: 10.1016/s0092-8674(03)00559-2 [DOI] [PubMed] [Google Scholar]
- 44.Michos O. Kidney development: from ureteric bud formation to branching morphogenesis. Curr Opin Genet Develop. 2009;19(5):484-490. doi: 10.1016/j.gde.2009.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pohl M, Stuart RO, Sakurai H, Nigam SK. Branching morphogenesis during kidney development. Annu Rev Physiol. 2000;62(1):595-620. doi: 10.1146/annurev.physiol.62.1.595 [DOI] [PubMed] [Google Scholar]
- 46.Shah MM, Sampogna RV, Sakurai H, Bush KT, Nigam SK. Branching morphogenesis and kidney disease. Development. 2004;131(7):1449-1462. doi: 10.1242/dev.01089 [DOI] [PubMed] [Google Scholar]
- 47.Marrs JA. Branching morphogenesis: Rac signaling “PIX” tubulogenesis. Focus on “Pak1 regulates branching morphogenesis in 3D MDCK cell culture by a PIX and β1-integrin-dependent mechanism.” Am J Physiol Cell Physiol. 2010;299(1):C7–C10. doi: 10.1152/ajpcell.00145.2010 [DOI] [PubMed] [Google Scholar]
- 48.Zegers MM. 3D in vitro cell culture models of tube formation. Semin Cell Develop Biol. 2014. 10;31:132-140. doi: 10.1016/j.semcdb.2014.02.016 [DOI] [PubMed] [Google Scholar]
- 49.Hunter MP, Zegers MM. Pak1 regulates branching morphogenesis in 3D MDCK cell culture by a PIX and β1-integrin-dependent mechanism. Am J Physiol Cell Physiol. 2010;299(1):C21-C32. doi: 10.1152/ajpcell.00543.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu W, Datta A, Leroy P, et al. β1-Integrin orients epithelial polarity via Rac1 and laminin. Mol Biol Cell. 2005;16(2):433-445. doi: 10.1091/mbc.e04-05-0435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kutsche K, Gal A. The mouse Arhgef6 gene: cDNA sequence, expression analysis, and chromosome assignment. Cytogenet Genome Res. 2001;95(3-4):196-201. doi: 10.1159/000059346 [DOI] [PubMed] [Google Scholar]
- 52.Wang P, Chen Y, Yong J, et al. Dissecting the global dynamic molecular profiles of human fetal kidney development by single-cell RNA sequencing. Cell Rep. 2018;24(13):3554-3567.e3. doi: 10.1016/j.celrep.2018.08.056 [DOI] [PubMed] [Google Scholar]
- 53.Park J, Shrestha R, Qiu C, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018;360(6390):758-763. doi: 10.1126/science.aar2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Menon R, Otto EA, Kokoruda A, et al. Single-cell analysis of progenitor cell dynamics and lineage specification in the human fetal kidney. Development. 2018;145(16):dev164038. doi: 10.1242/dev.164038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blackburn ATM, Miller RK. Modeling congenital kidney diseases in Xenopus laevis. Dis Model Mech. 2019;12(4):dmm038604. doi: 10.1242/dmm.038604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeLay BD, Corkins ME, Hanania HL, et al. Tissue-specific gene inactivation in Xenopus laevis: knockout of lhx1 in the kidney with CRISPR/Cas9. Genetics. 2018;208(2):673-686. doi: 10.1534/genetics.117.300468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blitz IL, Biesinger J, Xie X, Cho KW. Biallelic genome modification in F(0) Xenopus tropicalis embryos using the CRISPR/Cas system. Genesis. 2013;51(12):827-834. doi: 10.1002/dvg.22719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGrath JL. Cell spreading: the power to simplify. Curr Biol. 2007;17(10):R357-R358. doi: 10.1016/j.cub.2007.03.057 [DOI] [PubMed] [Google Scholar]
- 59.Kanwar YS, Wada J, Lin S, et al. Update of extracellular matrix, its receptors, and cell adhesion molecules in mammalian nephrogenesis. Am J Physiol Renal Physiol. 2004;286(2):F202-F215. doi: 10.1152/ajprenal.00157.2003 [DOI] [PubMed] [Google Scholar]
- 60.Sakai T, Larsen M, Yamada KM. Fibronectin requirement in branching morphogenesis. Nature. 2003;423(6942):876-881. doi: 10.1038/nature01712 [DOI] [PubMed] [Google Scholar]
- 61.Hogan BLM, Kolodziej PA. Organogenesis: molecular mechanisms of tubulogenesis. Nat Rev Genet. 2002;3(7):513-523. doi: 10.1038/nrg840 [DOI] [PubMed] [Google Scholar]
- 62.Kalter H. Sporadic congenital malformations of newborn inbred mice. Teratology. 1968;1(2):193-199. doi: 10.1002/tera.1420010208 [DOI] [PubMed] [Google Scholar]
- 63.Messow C, Gartner K, Hackbarth H, Kangaloo M, Lunebrink L. Sex differences in kidney morphology and glomerular filtration rate in mice. Contrib Nephrol. 1980;19:51-55 [PubMed] [Google Scholar]
- 64.Murawski IJ, Maina RW, Gupta IR. The relationship between nephron number, kidney size and body weight in two inbred mouse strains. Organogenesis. 2010;6(3):189-194. doi: 10.4161/org.6.3.12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murawski IJ, Maina RW, Malo D, et al. The C3H/HeJ inbred mouse is a model of vesico-ureteric reflux with a susceptibility locus on chromosome 12. Kidney Int. 2010;78(3):269-278. doi: 10.1038/ki.2010.110 [DOI] [PubMed] [Google Scholar]
- 66.Migeon BR. X inactivation, female mosaicism, and sex differences in renal diseases. J Am Soc Nephrol. 2008;19(11):2052-2059. doi: 10.1681/ASN.2008020198 [DOI] [PubMed] [Google Scholar]
- 67.Costantini F. Renal branching morphogenesis: concepts, questions, and recent advances. Differentiation. 2006;74(7):402-421. doi: 10.1111/j.1432-0436.2006.00106.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Materials are available upon request to the corresponding author and may be subject to an MTA. The genetics datasets supporting this study have not been deposited in a public repository due to restriction by patient consent but are available from the corresponding author on request.