


Keywords: whole-exome sequencing, reverse phenotyping, genetic renal disease, chronic kidney disease, cost analysis, workflow, cost
Abstract
Significance Statement
To optimize the diagnosis of genetic kidney disorders in a cost-effective manner, we developed a workflow based on referral criteria for in-person evaluation at a tertiary center, whole-exome sequencing, reverse phenotyping, and multidisciplinary board analysis. This workflow reached a diagnostic rate of 67%, with 48% confirming and 19% modifying the suspected clinical diagnosis. We obtained a genetic diagnosis in 64% of children and 70% of adults. A modeled cost analysis demonstrated that early genetic testing saves 20% of costs per patient. Real cost analysis on a representative sample of 66 patients demonstrated an actual cost reduction of 41%. This workflow demonstrates feasibility, performance, and economic effect for the diagnosis of genetic kidney diseases in a real-world setting.
Background
Whole-exome sequencing (WES) increases the diagnostic rate of genetic kidney disorders, but accessibility, interpretation of results, and costs limit use in daily practice.
Methods
Univariable analysis of a historical cohort of 392 patients who underwent WES for kidney diseases showed that resistance to treatments, familial history of kidney disease, extrarenal involvement, congenital abnormalities of the kidney and urinary tract and CKD stage ≥G2, two or more cysts per kidney on ultrasound, persistent hyperechoic kidneys or nephrocalcinosis on ultrasound, and persistent metabolic abnormalities were most predictive for genetic diagnosis. We prospectively applied these criteria to select patients in a network of nephrology centers, followed by centralized genetic diagnosis by WES, reverse phenotyping, and multidisciplinary board discussion.
Results
We applied this multistep workflow to 476 patients with eight clinical categories (podocytopathies, collagenopathies, CKD of unknown origin, tubulopathies, ciliopathies, congenital anomalies of the kidney and urinary tract, syndromic CKD, metabolic kidney disorders), obtaining genetic diagnosis for 319 of 476 patients (67.0%) (95% in 21 patients with disease onset during the fetal period or at birth, 64% in 298 pediatric patients, and 70% in 156 adult patients). The suspected clinical diagnosis was confirmed in 48% of the 476 patients and modified in 19%. A modeled cost analysis showed that application of this workflow saved 20% of costs per patient when performed at the beginning of the diagnostic process. Real cost analysis of 66 patients randomly selected from all categories showed actual cost reduction of 41%.
Conclusions
A diagnostic workflow for genetic kidney diseases that includes WES is cost-saving, especially if implemented early, and is feasible in a real-world setting.
Podcast
This article contains a podcast at https://dts.podtrac.com/redirect.mp3/www.asn-online.org/media/podcast/JASN/2023_04_03_JASN2022060725.mp3
Introduction
Providing an accurate genetic diagnosis is an important component of personalized medicine. A conclusive diagnosis allows defining appropriate diagnostic procedures, helps avoiding unnecessary treatments, improves prognosis prediction, clarifies necessary follow-up intervals, allows counseling of other family members, and defines risk constellations for living-donor kidney transplantation.1-6 However, bottlenecks to broader implementation of genomics in routine clinical practice include factors such as lacking awareness of inherited diseases, accessibility to genetic testing, interpretation quality, and cost concerns.3,6,7
Genetic testing with whole-exome sequencing (WES) revealed that genetic kidney disorders account for a significant proportion of cases with CKD in children and adults.1,2,6–9 Initially, WES was used mostly as an exploratory research tool to investigate pathomechanisms of kidney diseases. The progressive decline in sequencing costs led this technology to subsequently spread also in clinical settings to unravel the etiology of kidney phenotypes, although consensus criteria on which patients should undergo WES have not been reached yet.10-18 Studies on the utility of WES in patients with kidney diseases mostly relate to highly selected cohorts of patients (e.g., steroid-resistant nephrotic syndrome, distal renal tubular acidosis) in single-center or registry-based approaches. Selection criteria were usually stringent and phenotype-centered.15,19-27 However, genetic testing can provide diagnostic certainty also in less well-defined settings such as in advanced CKD where biopsy is usually no longer performed or only reveals nonspecific features of kidney atrophy (CKD of unknown origin [CKDu]).3,28 Recently, testing with WES in a large cross-section cohort of unselected patients with kidney diseases across all age groups showed an overall diagnostic yield of approximately 10%,29 questioning cost-effectiveness for health care systems and hence a broader implementation into daily clinical practice in unselected cohorts of patients. Recent recommendations on genetic testing in kidney diseases define cost-effectiveness studies as a knowledge gap.4
We hypothesized that a much higher diagnostic yield and cost savings could be achieved by implementing a workflow that aims to solve three problems: (1) define selection criteria on which patients should undergo genetic testing, (2) increase the rate and accuracy of genetic testing, and (3) identify those kidney diseases for which early genetic testing is economically advantageous for health care providers based on prespecified clinical criteria for tertiary center referral.
Methods
Assessment of Clinical Criteria for Patient Selection and Genetic Testing Prioritization
To establish and validate clinical criteria for patient selection and genetic testing prioritization, we retrospectively revised the results of genetic testing with WES in all the probands referred to the laboratory of Medical Genetics of Meyer Children' Hospital IRCCS of Florence (Italy) from 2015 (when WES was first adopted as a sequencing strategy) to 2018, with kidney diseases suspected to be of genetic origin (Supplemental Table 1). Patients belonged to all clinical categories of nephropathies. Clinical data were retrospectively collected by medical records and verified by nephrologists with expertise in genetic kidney diseases. We analyzed the rough diagnostic rate in different clinical categories, and we evaluated clinical features enriching the diagnostic yield in each category by univariable logistic regression analysis. We tested clinical variables potentially predicting a positive genetic testing by WES (see Supplemental Methods for details). Features resulting to predict a genetic diagnosis from this analysis were adopted as criteria for prospective patient selection, referral, and prioritization for genetic testing.
Study Population
Starting from 2018, we developed and prospectively applied a workflow for personalized diagnosis of genetic kidney disorders (Figure 1). Patients were referred from nephrology units in the whole Tuscany region to the Rare Genetic Kidney Diseases outpatient service at the tertiary center Meyer Children's Hospital IRCCS of Florence (Italy) or directly accessed the tertiary center. Enrollment stopped on January 2022. Referral was ordered by the treating nephrologist if at least one of the selection criteria established was matched, irrespective of clinical phenotype and kidney biopsy results (Figure 1). At the tertiary center, patients were evaluated by a multidisciplinary team. During pre-WES consultation, all relevant information (demographic, self-reported ancestry, clinical, laboratory, instrumental) were collected to frame clinically suspected diagnoses that were classified according to eight broad categories (Supplemental Figure 1): (1) podocytopathies (defined as in Ref. 30), (2) collagenopathies and other glomerular basement membrane disorders (defined as in Refs. 31,32), (3) CKDu (defined as in Refs. 7,28), (4) tubulopathies (defined as in Ref. 33), (5) ciliopathies (including autosomal dominant polycystic kidney disease and other cystic kidney diseases, as in Refs. 34,35), (6) congenital anomalies of the kidney and urinary tract (CAKUT, defined as in Refs. 36,37), (7) syndromic CKD (defined as the association of kidney involvement with extrarenal manifestations7), and (8) metabolic kidney disorders (including monogenic urinary stone disorders, defined as in Refs. 8,38). Clinical data were collected from direct interview of patients, families, and referring nephrologists and from medical records. Comprehensive phenotypic information and a detailed family history were collected and directly revised by the multidisciplinary team. Exclusion criteria were (1) demonstration of secondary causes of kidney phenotype (e.g., drug exposure, infections, malignancies, other recognized causes), (2) absence of clinical information, and (3) genetic diagnosis already established and confirmed at the tertiary center. Genetic testing with WES was offered to all eligible patients referred who were followed up until June 2022. First-degree relatives (parents and siblings) were either included before the study or asked to participate after the identification of potentially causative variants in the probands. All the patients and their families were counseled regarding the diagnostic algorithm (i.e., indication to genetic testing, basic technical clues, post-sequencing bioinformatic analysis and clinical interpretation, limitations), and all patients or their legal representatives gave written informed consent. Ancestry and ethnic groups were self-reported by the patients and collected to establish different risks of disease. The local Ethics Committee of the Meyer University Hospital of Florence approved the study (Approval number 50/2020). This study was conducted according to the Declaration of Helsinki.
Figure 1.
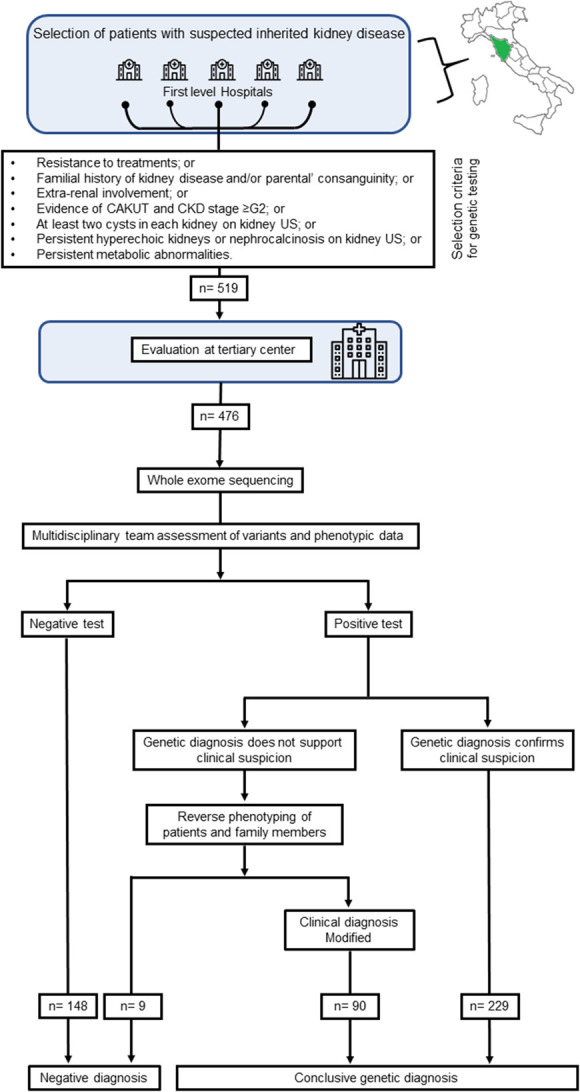
Diagnostic workflow for the diagnosis of kidney diseases. CAKUT, congenital anomalies of the kidney and urinary tract; US, ultrasound scanning.
Whole-Exome Sequencing and Assessment of Genetic Diagnosis
Genomic DNA was extracted from peripheral blood. The clinically accredited laboratory of Meyer University Hospital (Florence) performed WES and bioinformatic variant prioritization for genes reported in the Online Mendelian Inheritance in Man and/or Human Gene Mutation Database or in scientific literature in association with kidney diseases or phenotype, as previously described26 and detailed in Supplemental Methods. The results of genetic testing were evaluated by a multidisciplinary team for assessment of genetic diagnosis (Supplemental Figure 2). If the results of genetic testing were consistent with the clinical diagnosis, the latter was considered confirmed. If the results were not in line with suspected diagnosis, we used the label term “modified” (see Supplemental Methods for details).
Modeled Cost Analysis
We conducted a modeled cost analysis considering the use of WES in different phases of the diagnostic trajectory on the study population. We modeled two possible diagnostic trajectories: (1) late WES model, considering a complete diagnostic pathway and late use of WES, and (2) early WES model, considering an early use of WES. In both models, genomic and nongenomic investigations were divided into three tiers (tier 1, 2, 3), based on increasingly complex and/or expensive diagnostic examinations, as detailed in Supplemental Methods. The list of examinations included in the three tiers was drafted according to guidelines, available literature, current routine clinical practice, and recommended models of care for each clinical category. In the late WES model, only patients who remained undiagnosed after a complete diagnostic workup including even genomic investigations (i.e., targeted gene sequencing, panel sequencing) underwent WES. Results are expressed as cost per diagnosis according to the late WES and early WES models for the whole study population.
One-Way Sensitivity Analysis
To test the robustness of the results, we performed a deterministic sensitivity analysis, by using a distribution of values instead of fixed estimates for each key parameter of the simulation (see Supplemental Methods).
Real-Life Cost Analysis
To test the results of the modeled analysis, we performed a real descriptive cost analysis on a subgroup of patients. These patients were randomly selected among those with available information concerning the entire diagnostic pathway before genetic testing with WES from all the clinical categories. Two reviewers blinded for the clinical diagnosis, for the genetic diagnosis obtained by WES, and for the results of the other reviewer randomly collected retrospective clinical data of ten patients from each clinical category, plus seven for CAKUT and nine for metabolic kidney disorders. All the categories were represented in the analysis. Actual costs of the diagnostic pathway were compared with those that would have resulted from applying the early WES model in each of these patients. For each patient, we retrospectively evaluated the total expenses for the entire diagnostic pathway before undergoing WES. We included costs for laboratory and radiologic examinations, procedures (including kidney biopsy), consultancies, and other genomic investigations excluding WES (i.e., Sanger sequencing, gene panels).
Definition of Costs
In all the cost analyses performed, we considered only direct medical costs (i.e., laboratory and radiologic tests, procedures, outpatient consultations, and potential previous genetic tests). According to the local policy and reimbursement system, each cost is comprehensive of supplies, staff, results reporting, and overheads. Costs were calculated based on the regional health reimbursement system. Prices are expressed in Euros (2022), and the time horizon is 1 year. The analysis was conducted from a regional health care system perspective.
Statistical Analysis
Statistical analysis was performed using SPSS software v27.0 (SPSS, Inc., Evanston, IL). Quantitative data are shown as median and interquartile range (IQR). Categorical variables are expressed as numbers or percentages for each item. The chi square test was used for categorical variables. Univariable logistic regression was used to identify selection criteria for WES (see Supplemental Methods). The level of statistical significance was set at a value of P<0.05.
Results
Assessment of Clinical Criteria for Patient Selection and Genetic Testing Prioritization
To establish clinical criteria for patient selection, we retrospectively analyzed the results of genetic testing performed since 2015 (when WES was adopted as a diagnostic sequencing strategy) to 2018 in all the 392 probands referred to the laboratory of Medical Genetics of our hospital with a clinical suspect of genetic kidney diseases (Supplemental Table 1). Overall, WES permitted a diagnostic yield of 35.7% (140/392).
We then evaluated the ability of different clinical features to predict a genetic diagnosis by WES using the possible predictors listed in Supplemental Table 2. Univariable analysis showed that, among those, only the following could predict a genetic diagnosis in our cohort: (1) resistance to treatments, (2) familial history of kidney disease and/or parental consanguinity, (3) extrarenal involvement, (4) evidence of CAKUT and CKD stage ≥G2,39 (5) at least two cysts in each kidney on kidney ultrasound scanning, (6) persistent hyperechoic kidneys or nephrocalcinosis on kidney ultrasound scanning, and (7) persistent metabolic abnormalities. Conversely, age (as a continuous variable), nephrotic syndrome, hypertension, CAKUT without kidney function impairment, and isolated complement abnormalities did not predict a genetic diagnosis (Supplemental Table 2). Of note, the application of at least one among these clinical criteria significantly increased the diagnostic yield in comparison with the unselected population (35.1% versus 47.2%, P=0.003). Patients without any clinical criterion had a diagnostic rate of 2.5%. Then, we shared these criteria for referral with all the centers adhering to our regional network to set up a standardized diagnostic workflow for the genetic diagnosis of kidney diseases (Figure 1).
The Diagnostic Workflow Increases the Rate of Genetic Diagnosis in Kidney Diseases
Subsequently, a total of 519 consecutive patients matching the prespecified clinical criteria were referred to, or directly selected by, the tertiary center. After the application of exclusion criteria, 476 probands from 430 families were selected for genetic testing (Figure 1; Supplemental Table 3). Two hundred ninteen patients were female (46.0%), and 257 were male (54.0%). One hundred fifty-six patients (32.8%) were 18 years or older. Most patients self-reported European ancestry (94.1%), and consanguinity was declared in 3.9% of patients (Table 1). Based on their clinical phenotype, patients could be grouped into the following eight disease categories: (1) podocytopathies (n=133), (2) collagenopathies and other glomerular basement membrane disorders (n=48), (3) CKDu (n=34), (4) tubulopathies (n=109), (5) ciliopathies (n=69), (6) CAKUT (n=15), (7) syndromic CKD (n=32), and (8) metabolic kidney disorders (n=36). The list of clinical diagnoses included in each category is detailed in Supplemental Table 3. On WES, reverse phenotyping, and multidisciplinary workup, we arrived at a definite molecular diagnosis in 319 of 476 patients (67.0%) (Figure 2A; Supplemental Tables 4-11; Supplemental Figure 3). These results suggest that preselecting patients based on specific clinical criteria substantially increases the rate of genetic diagnosis using WES.
Table 1.
Baseline characteristics of patients included in the study
| Parameter | Podocytopathies (n=133) | Collagenopathies (n=48) | CKDu (n=34) | Tubulopathies (n=109) | Ciliopathies (n=69) | CAKUT (n=15) | Syndromic CKD (n=32) | Metabolic Kidney Disorders (n=36) | Study Population (n=476) |
|---|---|---|---|---|---|---|---|---|---|
| Diagnostic categories | |||||||||
| Congenital, n (%) | 0 | 0 | 0 | 12 (11.0) | 5 (7.2) | 2 (13.3) | 1 (3.1) | 1 (2.8) | 21 (4.4) |
| Age <18 yr, n (%) | 82 (61.7) | 33 (68.8) | 16 (47.1) | 63 (57.8) | 42 (60.9) | 11 (73.3) | 23 (71.9) | 29 (80.6) | 299 (62.8) |
| Age ≥18 yr, n (%) | 51 (38.3) | 15 (31.3) | 18 (52.9) | 34 (31.2) | 22 (31.9) | 2 (13.3) | 8 (25.0) | 6 (16.7) | 156 (32.8) |
| Age at referral, (yr), median (IQR) | 15 (6–23) | 13 (8–34) | 21 (11–40) | 8 (2–22) | 9 (4–24) | 1 (0–5) | 8 (1–18) | 7 (2–15) | 11 (4,0–23,0) |
| Female, n (%) | 62 (44.6) | 27 (56.3) | 11 (32.4) | 56 (51.4) | 26 (37.7) | 5 (33.3) | 18 (56.3) | 14 (38.9) | 219 (46,0) |
| Europeans (self-reported), n (%) | 119 (89.5) | 43 (89.6) | 28 (82.4) | 98 (88.3) | 67 (97.1) | 13 (86.7) | 26 (81.3) | 31 (86.1) | 425 (89.2) |
| Selection criteria | |||||||||
| Resistance to treatments, n (%) | 128 | 3 | 0 | 0 | 0 | 0 | 3 | 0 | 134 (28.2) |
| Family history of kidney disease and/or parental’ consanguinity, n (%) | 32 | 42 | 28 | 28 | 37 | 8 | 15 | 22 | 212 (44.5) |
| Extrarenal involvement, n (%) | 26 | 11 | 6 | 28 | 13 | 4 | 32 | 5 | 125 (26.3) |
| CAKUT and CKD stage G2 or higher, n (%) | 2 | 0 | 0 | 1 | 2 | 15 | 12 | 4 | 36 (7.6) |
| At least two cysts in each kidney on kidney US, n (%) | 0 | 0 | 1 | 1 | 60 | 0 | 3 | 8 | 73 (15.3) |
| Persistent hyperechoic kidneys or nephrocalcinosis on kidney US, n (%) | 1 | 0 | 0 | 32 | 4 | 1 | 5 | 27 | 70 (14.7) |
| Persistent metabolic abnormalities, n (%) | 0 | 0 | 0 | 103 | 0 | 0 | 2 | 7 | 112 (23.5) |
| Diagnostic yield, % (n) | 54.1 (72/133) | 69.2 (29/48) | 17.6 (6/34) | 86.2 (94/109) | 87 (60/69) | 46.7 (7/15) | 78.1 (25/32) | 72.2 (26/36) | 67 (319/476) |
CKDu, CKD of unknown origin; IQR, interquartile range; CAKUT, congenital anomalies of the kidney and urinary tract; US, ultrasound scanning.
Figure 2.

Diagnostic rate and disease reclassification. (A) Percentage of diagnosis confirmed (gray), negative (white), and modified (reticulated) by WES and reverse phenotyping. (B) Inner pie chart: distribution of patients according to the eight clinical categories based on pre-WES clinical evaluation (podocytopathies, collagenopathies, CKDu, tubulopathies, ciliopathies, CAKUT, syndromic CKD, metabolic kidney disorders). Outer pie chart: percentage of diagnosis confirmed (solid), modified (reticulated), and negative (white) in patients belonging to the eight clinical categories. (C) Percentage of diagnosis obtained with WES alone (light green), WES coupled with reverse phenotyping in the patient (striped), and WES coupled with reverse phenotyping in the patient and in the family (double striped) of patients belonging to the eight clinical categories. (D) On the left side, the suspected and on the right side, the genetic diagnosis in those patients that underwent disease reclassification after WES. CAKUT, congenital anomalies of the kidney and urinary tract; CKDu, CKD of unknown origin; RPF, reverse phenotyping in the family; RPP, reverse phenotyping in the patient; WES, whole-exome sequencing.
The Diagnostic Workflow Increases the Accuracy of Genetic Diagnosis of Kidney Diseases
Among the 319 patients with positive genetic testing, we confirmed the suspected clinical diagnosis in 229 (48.0%) (Figure 2A). By contrast, we revised the clinical diagnosis in 90 patients (19.0%), with disease reclassification occurring in 68 patients (Figure 2, A and B; Supplemental Table 12). In detail, category-specific diagnostic rates were as follows: podocytopathies 54.1% (72/133), collagenopathies 60.4% (29/48), CKDu 17.6% (6/34), tubulopathies 86.2% (94/109), ciliopathies 86.9% (60/69), CAKUT 46.7% (7/15), syndromic CKD 78.1% (25/32), and metabolic kidney disorders 72.2% (26/36) (Figure 2B). Reverse phenotyping in the probands permitted diagnosis in 57 patients, particularly those with podocytopathies and CAKUT (Figure 2C). On the other hand, reverse phenotyping in the family, including clinical reassessment and segregation analysis, provided evidence for a genetic diagnosis in 35 patients of the entire cohort. This extended workup especially supported a definite molecular diagnosis of ciliopathies and metabolic kidney disorders (Figure 2C). Interestingly, disease reclassification encompassed all clinical categories, and the final diagnosis ranged over all genetic groups (Figure 2D). These results suggest that the proposed diagnostic workflow increases not only the overall diagnostic rate but also the accuracy of genetic diagnosis of kidney diseases.
The Diagnostic Workflow Improves the Rate and Accuracy of Genetic Diagnosis in All Classes of Age
We separately evaluated the performance of our diagnostic workflow in congenital (disease onset during fetal period or at birth), pediatric (<18 years: IQR, 3–12 years), and adult (≥18 years: IQR, 23–48 years) patients (Figure 3A). Overall, the rate of genetic diagnosis was as high as 95% in 21 patients with congenital disease, 64% in 299 pediatric patients, and 70% in 156 adult patients (Figure 3B), without a significant difference between patients younger than and older than 18 years (chi square=0.856, P=0.355). Notably, age confirmed not to predict a genetic diagnosis (logistic regression, P=0.22, odds ratio, 1.01; 95% confidence interval, 0.99 to 1.02). Reverse phenotyping increased the diagnostic yield, irrespective of the group of age analyzed (Figure 3B). Tubulopathies represented the most frequent genetic diagnosis in patients with congenital disease, followed by ciliopathies, CAKUT, and syndromic CKD (Figure 3C). Genetic diagnosis of CAKUT was more relevant in the congenital and pediatric groups, likely related to selection of only patients with CKD stage ≥G2, who show more severe phenotypes, leading to early diagnosis (Figure 3C). Not only was the overall rate of genetic diagnosis similar in pediatric and adult patients but also the prevalence of different genetic diagnoses was almost uniformly distributed (Figure 3, A and C). Taken together, these results suggest that application of the proposed diagnostic workflow yields a high likelihood of a genetic diagnosis across all ages.
Figure 3.

Diagnostic rate and genetic findings in different age groups. (A) Distribution of genetic diagnosis according to age in the eight clinical categories (podocytopathies, collagenopathies, CKDu, tubulopathies, ciliopathies, CAKUT, syndromic CKD, metabolic kidney disorders). Each dot represents a patient. Dots are colored according to the genetic diagnosis. Age is reported in years. (B) Diagnostic rate according to age in the study population (476 patients). For each age group (congenital, pediatric, adults), we show confirmed (gray), modified (reticulated), and negative (white) results. Age is reported in years. (C) Genetic findings according to age in 319 patients with a genetic diagnosis. Age is reported in years. CAKUT, congenital anomalies of the kidney and urinary tract; CKDu, CKD of unknown origin.
The Diagnostic Workflow Improves Clinical Management
We next evaluated the overall clinical effect of obtaining a genetic diagnosis at a single-patient level (Table 2). In detail, genetic diagnosis and disease reclassification informed (1) prognosis (i.e., risk of progression to kidney failure); (2) cascade family member screening, additional workup, and surveillance; (3) treatment (including withdrawal of unnecessary therapies or switch to appropriate ones); (4) approach to kidney transplantation (donor selection, inscription to waiting list); (5) eligibility for clinical trials; (6) access to disease-specific registries; and (7) familial planning and reproductive counseling. Our strategy enabled us to reclassify 90 patients, with implications on the accuracy of prognosis prediction. Genetic testing was offered as cascade screening to 91 relatives from 74 families, providing a genetic diagnosis in 79 family members with a previously unsuspected or unspecified kidney disorder. The clinical workup changed and was redirected on average in 52.0% of patients. In 11.9% of patients, the results of genetic testing helped in guiding kidney transplant decisions. In addition, the genetic diagnosis informed treatment in 29.2% of patients. A further ten patients in the study received an additional molecular diagnosis (Supplemental Table 13). Moreover, in three patients, we identified the biallelic polymorphic G1/G2 risk alleles in APOL1 and in one patient, the CFHR1-CFHR3 homozygous deletion. These further diagnoses affected patient management but were not included in the total diagnostic yield reported in the study. These results suggest that the application of our diagnostic workflow affected patient management by changing workup, personalizing therapy, and orienting clinical counseling.
Table 2.
Clinical utility of genetic diagnosis in 319 patients
| Genetic Diagnosis | Informing Prognosis, n (%) | Cascade Family Members Screening, n (%) | Additional Workup/Changed Surveillance, n (%) | Tailoring Treatments, n (%) | Kidney Transplant Implications, n (%) | Enrollment in Clinical Trials, n (%) | Reproductive Counseling, n (%) | Additional Genetic Diagnosis, n (%) | Eligibility for Disease-Specific Registries, n (%) |
|---|---|---|---|---|---|---|---|---|---|
| Podocytopathies (n=40) | 3 (7.5) | 6 (15.0) | 6 (15.0) | 15 (37.5) | 16 (40.0) | 0 | 7 (17.5) | 1 (2.5) | 34 (100) |
| Collagenopathies (n=42) | 16 (38.1) | 25 (59.5) | 10 (29.4) | 21 (50.0) | 7 (16.7) | 1 (2.8) | 11 (27.0) | 0 | 34 (100) |
| Tubulopathies (n=96) | 23 (24.0) | 8 (8.3) | 38 (39.6) | 11 (11.5) | 1 (1.0) | 0 | 22 (22.9) | 3 (3.1) | 87 (100) |
| Ciliopathies (n=67) | 16 (23.8) | 16 (23.8) | 33 (49.3) | 6 (8.9) | 8 (11.9) | 1 (1.5) | 24 (29.8) | 3 (4.5) | 60 (100) |
| CAKUT (n=8) | 5 (62.5) | 4 (50.0) | 7 (87.5) | 2 (25.0) | 1 (12.5) | 0 | 1 (12.5) | 0 | 7 (100) |
| Syndromic CKD (n=37) | 15 (40.5) | 8 (21.6) | 30 (81.1) | 18 (48.6) | 5 (13.5) | 0 | 5 (13.5) | 2 (5.4) | 13 (100) |
| Metabolic kidney disorders (n=26) | 9 (34.6) | 5 (19.2) | 9 (34.6) | 15 (69.2) | 0 | 1 (3.8) | 6 (23.1) | 1 (3.8) | 23 (100) |
| Other nephropathies (n=3) | 3 (100) | 2 (66.7) | 2 (66.7) | 2 (66.7) | 0 | 0 | 1 (33.3) | 0 | 34 (100) |
| Total (n=319) | 90 (28.2) | 74 (23.2) | 167 (52.3) | 93 (29.2) | 38 (11.9) | 3 (0.1) | 77 (24.1) | 10 (3.1) | 319 (100) |
For each genetic group of diagnosis (podocytopathies, collagenopathies, tubulopathies, ciliopathies, CAKUT, syndromic CKD, metabolic kidney disorders and other nephropathies), we evaluated the effect of the genetic results (genetic diagnosis) in the clinical management.
Informing prognosis refers to the number (and percentage) of patients who received clarification about prognosis (progression toward kidney failure). Cascade family members screening is the number (and percentage) of families who were offered genetic testing after the results in the patient. Additional workup/changed surveillance is the number (and percentage) of patients who have been referred to other consultants for extrarenal workup or presymptomatic monitoring. Tailoring treatments is the number (and percentage) of patients who changed treatments (including withdrawal of unnecessary therapies or switching to appropriate ones). Kidney transplant implications is the number (and percentage) of patients who received information about donor selection or inscription to the waiting. Enrollment in clinical trials is the number (and percentage) of patients who were enrolled in clinical trials. Reproductive counseling is the number (and percentage) of patients who received dedicated information about the risk of recurrence in the progeny. Additional diagnosis is the number (and percentage) of patients who received additional genetic diagnosis. Eligibility for disease-specific registries is the number (and percentage) of patients whose information can be added in registries recording data about specific diseases. CAKUT, congenital anomalies of the kidney and urinary tract.
The Diagnostic Workflow Is Cost-Saving
To assess the economic effect of our workflow, we conducted a modeled cost analysis. We chose a modeled analysis to bypass the heterogeneity of clinical data of such a large cohort. Indeed, most of the patients arrived at our center after a “diagnostic odyssey” across different nephrology units even from different countries, hampering our ability to retrieve for all of them the diagnostic trajectories they went through. We analyzed 442 patients of the study population (patients with CKDu were not included because of the lack of guidelines allowing us to model the diagnostic trajectory for this clinical condition).
We compared two different modeled approaches (Figure 4A): (1) early use of WES and reverse phenotyping in the diagnostic workup (early WES model) and (2) the standard clinical practice that saves WES only for otherwise undiagnosed patients (late WES model). In both models, diagnostic investigations were grouped into three tiers (Supplemental Table 14), followed by WES and reverse phenotyping at different timings (early versus late WES; see Supplemental Methods for details). Tiers included increasingly complex and/or expensive diagnostic examinations (Supplemental Table 14). The cost analysis showed that the early WES model led to a cost reduction of 20% per diagnosis in comparison with the late WES model (Figure 4B). Overall, the mean cost per diagnosis in the late WES model was estimated at 7530€ versus 6033€ in the early WES model, leading to a cost saving of 1497€ per patient tested (Figure 4B). One-way sensitivity analysis confirmed the results of the base case analysis (Table 3), with the early WES model maintaining cost reduction in comparison with the late WES model, except for changes in costs of genetic investigations (i.e., increase in costs of WES or decrease in costs of other genetic tests).
Figure 4.

Cost analysis. (A) Modeled diagnostic trajectories for patients with suspected genetic kidney diseases based on clinical categories. In the late WES model, the patient undergoes investigations included in tiers 1, 2, and 3 until reaching a final diagnosis. The diagnostic workup is based on current guidelines, available literature, and local clinical practice for each clinical category. Tier 1 includes baseline investigations that allow clinicians to suspect inherited nephropathies as belonging to broad clinical categories (podocytopathies, collagenopathies, tubulopathies, ciliopathies, syndromic CKD, metabolic kidney disorders). Tier 2 and 3 include increasingly complex and/or expensive investigations. Tier 3 includes first-choice genetic testing for each clinical category, excluding WES. Genomic investigations were modeled according to literature, current guidelines, and the genetic architecture of each clinical category of kidney diseases. In the late WES model, if the diagnostic workup (including genomic and nongenomic investigations of tiers 2 and 3) turns out negative, the patient undergoes WES. In the early WES model, the patient goes through only tier 1 and then directly to WES. In both models, patients are selected with the clinical criteria adopted in this study. (B) Comparison between mean costs per diagnosis of the late WES model versus the early WES model in the study population. We excluded patients belonging to the CKDu category because of the lack of guidelines allowing us to model the diagnostic trajectory for this clinical condition. (C) Strategy applied for real-life costs analysis and comparison with the early WES model. Real-life diagnostic pathway included all the real costs retrieved from the clinical history of a subgroup of patients (n=66) with all available clinical information and recorded data in the administrative database. The clinical categories (podocytopathies, collagenopathies, tubulopathies, ciliopathies, syndromic CKD, metabolic kidney disorders) were equally represented. Costs of the real-life diagnostic pathway were compared with those derived from the hypothetical application of the early WES model to the same patients. (D) Comparison between mean costs per diagnosis of the real-life diagnostic pathway versus the early WES model in 66 patients. Icons included in Panels A and C are from the website Noun Project (thenounproject.com) and the credits go to their creators. CKDu, CKD of unknown origin; CAKUT, congenital anomalies of the kidney and urinary tract; WES, whole-exome sequencing.
Table 3.
One-way sensitivity analysis
| Base Case Analysis: Using Early WES versus Late WES Model Saves 1497 € per Diagnosis | |||||
|---|---|---|---|---|---|
| Sensitivity Analysis | First Alternative Assumption | Second Alternative Assumption | |||
| Parameter | Base Case Estimate | Lower Value | Cost per Diagnosis Variation | Upper Value | Cost per Diagnosis Variation |
| WES cost | 3670€ | 466€ | −3859€ | 6670€ | +586€ |
| Gene panel cost | 1646€ | 350€ | +281€ | 5060€ | −6109€ |
| Biopsy cost | 2198€ | 1480€ | −1139€ | 4366€ | −2583€ |
| Reverse phenotyping mean cost per patient | 115€ | 90€ | −1525€ | 230€ | −1341€ |
| Proportion of patients needing biopsy | 1 (for required cases) | 0.5 | −76€ | 0.7 | −177€ |
| Proportion of patients needing reverse phenotyping after WES | 0.2 | 0.1 | −1544€ | 0.4 | −1423€ |
| Proportion of patients with podocytopathies | 0.3 | 0.2 | −1032€ | 0.4 | −1980€ |
One-way sensitivity analysis shows how the results of the modeled analysis change using a range of values of each uncertain model parameter while holding other parameters fixed to their base case scenario. Base case estimate represents costs used in the modeled analysis. Lower and upper values represent the variations in costs of the base case parameter.
WES, whole-exome sequencing.
To test the strength of our model, we compared the results obtained with the early WES model with those obtained retrieving real costs of the diagnostic pathway in a representative subgroup of 66 patients randomly selected among those with all available clinical information and recorded data in administrative databases (Figure 4C). The cost analysis showed that actual costs in these patients were higher than those inferred from the early WES model (6805€ versus4009 €; Figure 4D), leading to a cost reduction of 41% and validating our modeled analysis on the whole cohort. Taken together, these results suggest that WES is cost-saving for the diagnosis of kidney diseases when implemented in a diagnostic workflow guided by selection criteria.
Discussion
In this study, we established a diagnostic workflow applicable in daily clinical practice intended to facilitate a cost-saving and high-rate diagnosis of genetic kidney diseases. We showed that (1) our workflow increases the diagnostic rate and accuracy of genetic diagnosis of kidney diseases; (2) when this workflow is applied, the diagnostic success in children and adults is similar; (3) the application of our diagnostic workflow can affect clinical management; and (4) our approach has the potential to reduce health care spending in patients with kidney diseases.
Setting up service delivery models that empower timely and cost-efficient access to genetic testing and counseling is important but challenging. Several studies described such service delivery models in clinics for genetic kidney diseases,10-12,15 reporting diagnostic yields of 10%–40% and benefits at the single-patient level, but low cost-effectiveness.10,29 In this study, we report on a workflow that led to a diagnostic rate as high as 67.0% in patients of all ages affected by different types of kidney diseases and was ultimately cost-saving. To achieve cost-effectiveness, it is mandatory to include not only obvious but also less likely genetic cases, to avoid missing too many genetic diagnoses undiscovered. To this aim, we optimized the standard diagnostic workflow at three different levels: (1) Nephrologist level: identifying criteria for referral of patients that avoided subjectivity and could increase the probability of selecting patients with genetic nephropathies; (2) Diagnostic center level: referring the patients to a single diagnostic center where WES equipment and multiple specialist expertise for reverse phenotyping are available, optimizing a procedure for patient re-evaluation and WES interpretation; and (3) Health care provider level: minimizing costs with an organized procedure that centralized WES analysis in a single center to increase the diagnostic rate, a requirement for reaching cost-effectiveness and allocative efficiency. This multiple-step workflow distinguishes our procedure from those previously reported and likely explains the substantially higher rate of genetic diagnosis reported in our study.
Standardizing selection criteria for genetic testing of patients with nephropathies is essential to optimize financial and personnel resources when applying WES in clinical practice. Defined clinical criteria can simplify patient referral from nephrologists of peripheral centers and ensure unrestricted access to the latest technology of genetic testing in a tertiary center. From a clinical perspective, this can allow clinicians to significantly reduce the risk of missing patients who would benefit from genetic testing. The criteria for patient selection were tested for the ability to increase the likelihood of genetic diagnosis. Although they included well-established ones,7,26,30,39-50 in this study, we applied all these criteria at the same time to all the patients, avoiding clinician's subjectivity. Currently, age is among the most widely used criterion for ordering genetic testing in clinical practice based on the concept that pediatric patients are more frequently affected by genetic kidney diseases than adults do.51 However, in this study, the success rate of genetic diagnosis was similar across age suggesting that, when the right clinical selection criteria are applied, age loses relevance in predicting the diagnosis in patients selected by clinical criteria.
Another step of prioritization before genetic testing was the thorough clinical (re-)evaluation of the patient by nephrologists specialized in genetic kidney disorders. Indeed, the constantly increasing number of genes identified as possible causes of kidney disorders, most very rare, together with the rapidly evolving knowledge in the field, requires personnel trained in genetics of rare kidney diseases. Moreover, we integrated genetic and clinical data in multidisciplinary board discussions, including nephrologists with genetic expertise, pathologists, and nephrology-trained geneticists, which broadens the spectrum of expertise inputs for each patient. Finally, we re-evaluated patients based on genetic results and multidisciplinary board indication, that is, by reverse phenotyping. Reverse phenotyping is currently used in clinical practice in different fields of medicine,52 and we have established it successfully in steroid-resistant nephrotic syndrome.26 Very recently, a collaborative work pointed out the role of reverse phenotyping also in CAKUT.53 Obviously, our workflow requires availability of staff with multiple expertise and WES technology, implying higher costs compared with genetic testing on shipped DNA samples. However, a centralized straightforward clinical workup and WES analysis avoided the costs for multiple referrals and consultations. In addition, it further standardized the access of patients to an evaluation for genetic kidney diseases, avoiding missing diagnosis. Finally, cascade diagnosis involved 79 family members who did not know to be affected, emphasizing that diagnosis of genetic nephropathies is a dynamic and iterative process. This should be explained to patients and families by clinicians during all counseling sessions.54
The high costs of WES are considered a major hurdle for a broader implementation of genetic testing in clinical practice.28,51,55,56 We showed that diagnostic strategies using WES could save health care spendings when guided by this diagnostic workflow. This is true even when considering early introduction of genomic investigations in the diagnostic pathway (early WES model), in line with previous results on glomerular diseases.57 The potential monetary benefits are mainly due to obtaining diagnosis in previously unsolved cases and disease reclassification that avoids unnecessary diagnostic investigations and therapeutic interventions.58 Of note, our analysis focused on a limited period and used a very conservative approach, likely underestimating cost savings. Indeed, we did not consider patients undergoing genomic and nongenomic investigations repeatedly in cases in which a precise diagnosis could not be established. Moreover, we did not consider potential sources of economic benefits either because of indirect medical costs savings or to cascade diagnosis and surveillance. The real cost analysis performed in a subset of patients who had all the clinical information available suggests that this could indeed be the case and fully validates our approach. Indeed, the cost reduction was even higher in the 66 patients where the real costs were evaluated and directly compared with those that would have come from the early WES model. Of note, this study was conducted from the perspective of a fully public health care system, but the results can be relevant also in private or semiprivate settings.
This study has some limitations. First, our study population is restricted geographically (89% of patients were Europeans, mostly coming from Italy), although comparable with those reported in previous studies.10,12-15 Second, we could not perform either cost-effectiveness analysis or cost-utility analysis modeling quality-adjusted life-years that are used in economic settings as an outcome measure of health benefits.58 Consequently, future studies are needed to address these issues. Third, excluding patients not satisfying our selection criteria from access to genetic testing could represent a source of health care inequity. However, our workflow does not aim at excluding patients but rather to prioritize those with a stringent indication as for other diagnostic investigation (e.g., kidney biopsy). Finally, our assumptions do not exclude that disease reclassification may be also successfully performed when our clinical workflow is applied with extended genetic panels including genes that can give heterogeneous clinical phenotypes multiple times across different panels, rather than using WES.
In conclusion, in this study, we provide evidence of the performance and feasibility of a service delivery model based on a network of nephrology units applying defined clinical preselection criteria and working side by side with a tertiary referral center for patient selection, diagnosis, and management of rare kidney diseases. Altogether, this study demonstrates that personalized nephrology is feasible and affordable in real-world clinical settings and that such an approach could finally lead to apply genomic medicine in routine care of patients with kidney diseases.
Supplementary Material
Acknowledgments
The patient association “Associazione Malattie Renali Toscana (A.Ma.R.T.I)” supported this project. P. Romagnani, F. Becherucci, S.Landini, V. Palazzo, L.Cirillo, and A. Vaglio are members of the European Reference Network for Rare Kidney Diseases (ERKNet).
Footnotes
S.L., V.P., and L.C. contributed equally to this work.
See related editorial, “Kidney Genetics: Continuing Discoveries and a Roadmap to the Clinic,” on pages 519–520.
Disclosures
S. Landini reports Other Interests or Relationships: member of the ERKNet Pediatric Unit, Institution Florence Meyer Children's Hospital. M. Allinovi reports Advisory or Leadership Role: Editorial Board member of the following journals: British Medical Journal Case Reports, BMC Nephrology, and Frontiers in Pharmacology. D. Giannese reports Advisory or Leadership Role: GSK. L. Cirami reports Consultancy: Alexion-AstraZeneca, AMICUS, Biotec, Sanofi-Genzyme, and Takeda-Shire; Research Funding: Alexion, Amicus, Bayer, Hospira, Modify, Morphosys, Otsuka, Protalix, Pfizer, Retrophin, Sandoz, Sangamo, and Sanofi; Advisory or Leadership Role: AstraZeneca-Alexion, Biotec, and Sanofi-Genzyme; and Other Interests or Relationships: Member of the Italian Society of Nephrology, Member of the American Society of Nephrology, and Member of Amnesty International. H.-J. Anders reports Consultancy: AstraZeneca, Bayer, GSK, Janssen, Kezar, Novartis, Previpharma, Sanofi, and Vifor; Research Funding: Boehringer-Ingelheim; Honoraria: Lilly and Otsuka; and Advisory or Leadership Role: JASN and Nephrology Dialysis Transplantation. P. Romagnani reports Research Funding: AstraZeneca. Because H.-J. Anders is an Associate Editor of the JASN, he was not involved in the peer-review process for this manuscript. A guest editor oversaw the peer review and decision-making process for this manuscript.
Funding
This research project was funded by the Tuscany region government to PR at Meyer University Hospital with the grant “Bando Ricerca Salute 2018.“ Project title: Setup of a platform for personalized diagnosis of rare kidney disease. Project acronym: NIKE. Italian Society of Nephrology to F. Becherucci at Meyer University Hospital with the grant “Adotta un progetto di ricerca 2018.” Project title: Whole-exome sequencing for personalized diagnosis of idiopathic nephrotic syndrome. Project acronym: WESsn.
Author Contributions
F. Becherucci, L. Cirillo, B. Mazzinghi, and P. Romagnani conceptualized the study; M. Allinovi, H.-J. Anders, R. Artuso, F. Becherucci, S. Bellelli, G. Campolo, L. Cirami, L. Cirillo, P.C. Dattolo, E. Dirupo, D. Giannese, S. Landini, J. Lomi, G. Lugli, T. Mazzierli, B. Mazzinghi, V. Palazzo, V. Raglianti, F. Ravaglia, P. Romagnani, A. Rosati, G. Sansavini, C. Somma, G. Spatoliatore, L. Tiberi, A. Vaglio, and D. Vergani were responsible for data curation; F. Becherucci, L. Cirillo, G. Lugli, B. Mazzinghi, and P. Romagnani were responsible for formal analysis; F. Becherucci, L. Cirillo, S. Landini, G. Lugli, B. Mazzinghi, V. Raglianti,V. Palazzo, and P. Romagnani were responsible for investigation; F. Becherucci,S. Bellelli, L. Cirillo, S. Landini, G. Lugli, B. Mazzinghi, V. Palazzo, V. Raglianti, and L. Tiberi were responsible for methodology; H.-J. Anders, F. Becherucci, S. Bellelli, B. Mazzinghi, and P. Romagnani provided supervision; H.-J. Anders, F. Becherucci, L. Cirillo, S. Landini, B. Mazzinghi, V. Palazzo, and P. Romagnani were responsible for validation; F. Becherucci, E. Lazzeri, L. Papi, and P. Romagnani were responsible for resources; F. Becherucci, E. Lazzeri, L. Papi, and P. Romagnani were responsible for funding acquisition; E. Lazzeri and P. Romagnani were responsible for project administration; B. Mazzinghi was responsible for visualization; F. Becherucci, and P. Romagnani wrote the original draft; M. Allinovi, H.-J. Anders, F. Becherucci, S. Bellelli, G. Campolo, L. Cirami, P.C. Dattolo, L. De Chiara, E. Lazzeri, J. Lomi, T. Mazzierli, L. Papi, F. Ravaglia, P. Romagnani, A. Rosati, G. Sansavini, C. Somma, G. Spatoliatore, and A. Vaglio reviewed and edited the manuscript.
Data Sharing Statement
All data used in this study are available in this article.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/D721.
Supplemental Table 1. Clinical diagnosis in the historical cohort.
Supplemental Table 2. Univariable logistic regression analysis.
Supplemental Table 3. Clinical diagnosis of patients included in the study.
Supplemental Table 4. Genetic findings in patients with Podocytopathies.
Supplemental Table 5. Genetic findings in patients with Collagenopathies.
Supplemental Table 6. Genetic findings in patients with CKDu.
Supplemental Table 7. Genetic findings in patients with Tubulopathies.
Supplemental Table 8. Genetic findings in patients with Ciliopathies.
Supplemental Table 9. Genetic findings in patients with CAKUT.
Supplemental Table 10. Genetic findings in patients with Syndromic CKD.
Supplemental Table 11. Genetic findings in patients with Metabolic Kidney Disorders.
Supplemental Table 12. Patients with modified clinical diagnosis.
Supplemental Table 13. Additional genetic diagnosis.
Supplemental Table 14. List of the investigations required for the diagnosis of kidney diseases.
Supplemental Figure 1. Selection criteria and clinical categories.
Supplemental Figure 2. Workflow for genetic testing results interpretation.
Supplemental Figure 3. Common genetic findings.
References
- 1.Connaughton DM, Hildebrandt F. Personalized medicine in chronic kidney disease by detection of monogenic mutations. Nephrol Dial Transplant. 2020;35(3):390-397. doi: 10.1093/ndt/gfz028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groopman EE, Rasouly HM, Gharavi AG. Genomic medicine for kidney disease. Nat Rev Nephrol. 2018;14(2):83-104. doi: 10.1038/nrneph.2017.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stokman MF, Renkema KY, Giles RH, Schaefer F, Knoers NVAM, van Eerde AM. The expanding phenotypic spectra of kidney diseases: insights from genetic studies. Nat Rev Nephrol. 2016;12(8):472-483. doi: 10.1038/nrneph.2016.87 [DOI] [PubMed] [Google Scholar]
- 4.Knoers N Antignac C Bergmann C, et al. Genetic testing in the diagnosis of chronic kidney disease: recommendations for clinical practice. Nephrol Dial Transplant. 2022;37(2):239-254. doi: 10.1093/ndt/gfab218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murray SL Dorman A Benson KA, et al. Utility of genomic testing after renal biopsy. Am J Nephrol. 2020;51(1):43-53. doi: 10.1159/000504869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torra R, Furlano M, Ortiz A, Ars E. Genetic kidney diseases as an underrecognized cause of chronic kidney disease: the key role of international registry reports. Clin Kidney J. 2021;14(8):1879-1885. doi: 10.1093/ckj/sfab056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cocchi E, Nestor JG, Gharavi AG. Clinical genetic screening in adult patients with kidney disease. Clin J Am Soc Nephrol. 2020;15(10):1497-1510. doi: 10.2215/CJN.15141219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol. 2016;12(3):133-146. doi: 10.1038/nrneph.2015.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connaughton DM Kennedy C Shril S, et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019;95(4):914-928. doi: 10.1016/j.kint.2018.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayasinghe K Stark Z Kerr PG, et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet Med. 2021;23(1):183-191. doi: 10.1038/s41436-020-00963-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto E Vairo F Prochnow C Kemppainen JL, et al. Genomics integration into nephrology practice. Kidney Med. 2021;3(5):785-798. doi: 10.1016/j.xkme.2021.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas CP Freese ME Ounda A, et al. Initial experience from a renal genetics clinic demonstrates a distinct role in patient management. Genet Med. 2020;22(6):1025-1035. doi: 10.1038/s41436-020-0772-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alkanderi S, Yates LM, Johnson SA, Sayer JA. Lessons learned from a multidisciplinary renal genetics clinic. QJM. 2017;110(7):453-457. doi: 10.1093/qjmed/hcx030 [DOI] [PubMed] [Google Scholar]
- 14.Chen J Lin F Zhai Y, et al. Diagnostic and clinical utility of genetic testing in children with kidney failure. Pediatr Nephrol. 2021;36(11):3653-3662. doi: 10.1007/s00467-021-05141-5 [DOI] [PubMed] [Google Scholar]
- 15.Mann N Braun DA Amann K, et al. Whole-exome sequencing enables a precision medicine approach for kidney transplant recipients. J Am Soc Nephrol. 2012019;30(2):201-215. doi: 10.1681/ASN.2018060575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domingo-Gallego A Pybus M Bullich G, et al. Clinical utility of genetic testing in early-onset kidney disease: seven genes are the main players. Nephrol Dial Transplant. 2022;37(4):687-696. doi: 10.1093/ndt/gfab019 [DOI] [PubMed] [Google Scholar]
- 17.de Haan A, Eijgelsheim M, Vogt L, Knoers NVAM, de Borst MH. Diagnostic yield of next-generation sequencing in patients with chronic kidney disease of unknown etiology. Front Genet. 2019;10:1264. doi: 10.3389/fgene.2019.01264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schrezenmeier E Kremerskothen E Halleck F, et al. The underestimated burden of monogenic kidney disease in adults waitlisted for kidney transplantation. Genet Med. 2021;23(7):1219-1224. doi: 10.1038/s41436-021-01127-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trautmann A, Lipska-Ziętkiewicz BS, Schaefer F. Exploring the clinical and genetic spectrum of steroid resistant nephrotic syndrome: the PodoNet registry. Front Pediatr. 2018;6:200. doi: 10.3389/fped.2018.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warejko JK Tan W Daga A, et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2018;13(1):53-62. doi: 10.2215/CJN.04120417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bierzynska A McCarthy HJ Soderquest K, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91(4):937-947. doi: 10.1016/j.kint.2016.10.013 [DOI] [PubMed] [Google Scholar]
- 22.Palazzo V Provenzano A Becherucci F, et al. The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int. 2017;91(5):1243-1255. doi: 10.1016/j.kint.2016.12.017 [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Garcia SC Emma F Walsh SB, et al. Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant. 2019;34(6):981-991. doi: 10.1093/ndt/gfy409 [DOI] [PubMed] [Google Scholar]
- 24.Palazzo V Raglianti V Landini S, et al. Clinical and genetic characterization of patients with bartter and gitelman syndrome. Int J Mol Sci. 2022;23(10):5641. doi: 10.3390/ijms23105641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snoek R van Jaarsveld RH Nguyen TQ, et al. Genetics-first approach improves diagnostics of ESKD patients <50 years old. Nephrol Dial Transplant. 2022;37(2):349-357. doi: 10.1093/ndt/gfaa363 [DOI] [PubMed] [Google Scholar]
- 26.Landini S Mazzinghi B Becherucci F, et al. Reverse phenotyping after whole-exome sequencing in steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2020;15(1):89-100. doi: 10.2215/CJN.06060519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riedhammer KM Braunisch MC Günthner R, et al. Exome sequencing and identification of phenocopies in patients with clinically presumed hereditary nephropathies. Am J Kidney Dis. 2020;76(4):460-470. doi: 10.1053/j.ajkd.2019.12.008 [DOI] [PubMed] [Google Scholar]
- 28.Hays T, Groopman EE, Gharavi AG. Genetic testing for kidney disease of unknown etiology. Kidney Int. 2020;98(3):590-600. doi: 10.1016/j.kint.2020.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Groopman EE Marasa M Cameron-Christie S, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2019;380(2):142-151. doi: 10.1056/nejmoa1806891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kopp JB Anders H-J Susztak K, et al. Podocytopathies. Nat Rev Dis Primers. 2020;6(1):68. doi: 10.1038/s41572-020-0196-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan C, Rheault MN. Genetic basis of type IV collagen disorders of the kidney. Clin J Am Soc Nephrol. 2021;16(7):1101-1109. doi: 10.2215/CJN.19171220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naylor RW, Morais MRPT, Lennon R. Complexities of the glomerular basement membrane. Nat Rev Nephrol. 2021;17(2):112-127. doi: 10.2215/CJN.13921217 [DOI] [PubMed] [Google Scholar]
- 33.Downie ML, Lopez Garcia SC, Kleta R, Bockenhauer D. Inherited tubulopathies of the kidney: insights from genetics. Clin J Am Soc Nephrol. 2021;16(4):620-630. doi: 10.2215/CJN.14481119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533-547. doi: 10.1038/nrm.2017.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364(16):1533-1543. doi: 10.1056/nejmra1010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murugapoopathy V, Gupta IR. A primer on congenital anomalies of the kidneys and urinary tracts (CAKUT). Clin J Am Soc Nephrol. 2020;15(5):723-731. doi: 10.2215/CJN.12581019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol. 2015;11(12):720-731. doi: 10.1038/nrneph.2015.140 [DOI] [PubMed] [Google Scholar]
- 38.Hildebrandt F. Genetic kidney diseases. Lancet. 2010;375(9722):1287-1295. doi: 10.1016/s0140-6736(10)60236-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stevens PE, Levin A, Bilous RW; Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;31:1-150. [Google Scholar]
- 40.Trautmann A Saleem MA Vivarelli M, et al. IPNA clinical practice recommendations for the diagnosis and management of children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2020;35(8):1529-1561. doi: 10.1007/s00467-020-04519-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pei Y Obaji J Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-212. doi: 10.1681/ASN.2008050507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pei Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2006;1(5):1108-1114. doi: 10.2215/CJN.02190606 [DOI] [PubMed] [Google Scholar]
- 43.Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4(1):50. doi: 10.1038/s41572-018-0047-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khan SR Pearle MS Robertson WG, et al. Kidney stones. Nat Rev Dis Primers. 2016;2(1):16008. doi: 10.1038/nrdp.2016.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trepiccione F Walsh SB Ariceta G, et al. Distal renal tubular acidosis: ERKNet/ESPN clinical practice points. Nephrol Dial Transplant. 2021;36(9):1585-1596. doi: 10.1093/ndt/gfab171 [DOI] [PubMed] [Google Scholar]
- 46.Devuyst O, Thakker RV. Dent’s disease. Orphanet J Rare Dis. 2010;5(1):28. doi: 10.1186/1750-1172-5-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konrad M Nijenhuis T Ariceta G, et al. Diagnosis and management of Bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int. 2021;99(2):324-335. doi: 10.1016/j.kint.2020.10.035 [DOI] [PubMed] [Google Scholar]
- 48.Blanchard A Bockenhauer D Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int. 2017;91(1):24-33. doi: 10.1016/j.kint.2016.09.046 [DOI] [PubMed] [Google Scholar]
- 49.Savige J Ariani F Mari F, et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol. 2019;34(7):1175-1189. doi: 10.1007/s00467-018-3985-4 [DOI] [PubMed] [Google Scholar]
- 50.Türk C Petřík A Sarica K, et al. EAU guidelines on diagnosis and conservative management of urolithiasis. Eur Urol. 2016;69(3):468-474. doi: 10.1016/j.eururo.2015.07.040 [DOI] [PubMed] [Google Scholar]
- 51.Pollak MR, Friedman DJ. The genetic architecture of kidney disease. Clin J Am Soc Nephrol. 2020;15(2):268-275. doi: 10.2215/CJN.09340819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becherucci F, Landini S, Cirillo L, Mazzinghi B, Romagnani P. Look alike, sound alike: phenocopies in steroid-resistant nephrotic syndrome. Int J Environ Res Public Health. 2020;17(22):8363. doi: 10.3390/ijerph17228363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seltzsam S Wang C Zheng B, et al. Reverse phenotyping facilitates disease allele calling in exome sequencing of patients with CAKUT. Genet Med. 2022;24(2):307-318. doi: 10.1016/j.gim.2021.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Goede C Yue WW Yan G, et al. Role of reverse phenotyping in interpretation of next generation sequencing data and a review of INPP5E related disorders. Eur J Paediatric Neurol. 2016;20(2):286-295. doi: 10.1016/j.ejpn.2015.11.012 [DOI] [PubMed] [Google Scholar]
- 55.Bélisle-Pipon J-C, Vayena E, Green RC, Cohen IG. Genetic testing, insurance discrimination and medical research: what the United States can learn from peer countries. Nat Med. 2019;25(8):1198-1204. doi: 10.1038/s41591-019-0534-z [DOI] [PubMed] [Google Scholar]
- 56.Manolio TA Abramowicz M Al-Mulla F, et al. Global implementation of genomic medicine: we are not alone. Sci Transl Med. 2015;7(290):290ps13. doi: 10.1126/scitranslmed.aab0194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jayasinghe K Wu Y Stark Z, et al. Cost-effectiveness of targeted exome analysis as a diagnostic test in glomerular diseases. Kidney Int Rep. 2021;6(11):2850-2861. doi: 10.1016/j.ekir.2021.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Payne K, Gavan SP, Wright SJ, Thompson AJ. Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat Rev Genet. 2018;19(4):235-246. doi: 10.1038/nrg.2017.108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study are available in this article.