


Keywords: fibrosis, inflammation, macrophages, regeneration, necroinflammation, anti-HMGB1, HMGB1 receptors, human scRNA-seq
Abstract
Significance Statement
Cells undergoing necrosis release extracellular high mobility group box (HMGB)-1, which triggers sterile inflammation upon AKI in mice. Neither deletion of HMGB1 from tubular epithelial cells, nor HMGB1 antagonism with small molecules, affects initial ischemic tubular necrosis and immediate GFR loss upon unilateral ischemia/reperfusion injury (IRI). On the contrary, tubular cell-specific HMGB1 deficiency, and even late-onset pharmacological HMGB1 inhibition, increased functional and structural recovery from AKI, indicating that intracellular HMGB1 partially counters the effects of extracellular HMGB1. In vitro studies indicate that intracellular HMGB1 decreases resilience of tubular cells from prolonged ischemic stress, as in unilateral IRI. Intracellular HMGB1 is a potential target to enhance kidney regeneration and to improve long-term prognosis in AKI.
Background
Late diagnosis is a hurdle for treatment of AKI, but targeting AKI-CKD transition may improve outcomes. High mobility group box-1 (HMGB1) is a nuclear regulator of transcription and a driver of necroinflammation in AKI. We hypothesized that HMGB1 would also modulate AKI-CKD transition in other ways.
Methods
We conducted single-cell transcriptome analysis of human and mouse AKI and mouse in vivo and in vitro studies with tubular cell-specific depletion of Hmgb1 and HMGB1 antagonists.
Results
HMGB1 was ubiquitously expressed in kidney cells. Preemptive HMGB1 antagonism with glycyrrhizic acid (Gly) and ethyl pyruvate (EP) did not affect postischemic AKI but attenuated AKI-CKD transition in a model of persistent kidney hypoxia. Consistently, tubular Hmgb1 depletion in Pax8 rtTA, TetO Cre, Hmgb1fl/fl mice did not protect from AKI, but from AKI-CKD transition. In vitro studies confirmed that absence of HMGB1 or HMGB1 inhibition with Gly and EP does not affect ischemic necrosis of growth-arrested differentiated tubular cells but increased the resilience of cycling tubular cells that survived the acute injury to oxidative stress. This effect persisted when neutralizing extracellular HMGB1 with 2G7. Consistently, late-onset HMGB1 blockade with EP started after the peak of ischemic AKI in mice prevented AKI-CKD transition, even when 2G7 blocked extracellular HMGB1.
Conclusion
Treatment of AKI could become feasible when (1) focusing on long-term outcomes of AKI; (2) targeting AKI-CKD transition with drugs initiated after the AKI peak; and (3) targeting with drugs that block HMGB1 in intracellular and extracellular compartments.
Treatment of AKI has remained challenging when focusing on AKI itself because, in most clinical settings, the diagnosis of AKI implies that kidney injury has already occurred and can no longer be significantly modulated. By contrast, focusing on long-term outcomes of AKI in AKI survivors seems more promising but would require drug interventions that are still efficacious when initiated after the peak of AKI. Survivors of an AKI frequently develop CKD because of an irreversible loss of tubular cells in the acute phase and an ongoing loss of tubular cells thereafter.1-4 Adaptation of the remaining tubular cells to an increased metabolic demand involves stress response pathways that promote tubulointerstitial inflammation and fibrosis and ultimately further tubular cell demise, i.e., CKD progression.1 Hypoxia signaling is a well-characterized element of AKI-CKD transition because injury and kidney remodeling induces microvascular dysfunction and rarefaction in the context of an increasing metabolic demand per remaining tubular epithelial cell, as the number of these cells declines.5,6 Indeed, tubulointerstitial hypoxia is a hallmark of AKI-CKD transition.7,8
High mobility group box-1 (HMGB1) is a nucleoprotein interacting with nucleosomes, transcription factors, and histones to organize chromatin structure and to regulate transcription.9 On acetylation, HMGB1 can translocate to the cytosol where it regulates additional cellular processes, e.g., autophagic flux.10 On cell necrosis or the release of neutrophil extracellular traps (NETs), HMGB1 can reach the extracellular space, where it elicits proinflammatory effects as a chemoattractant through CXCR4 or as a damage-associated molecular pattern (DAMP) activating toll-like receptors 2 and 4 and receptor of advanced glycation end products (RAGE).10 Indeed, numerous studies document the role of HMGB1 as a DAMP in models of acute tissue injuries including the kidney.11 For example, neutralizing extracellular HMGB1 with specific antibodies attenuates necroinflammation in ischemic kidneys.12-14 Consistently, HMGB1/TLR4/NF-κB signaling contributes to interstitial fibrosis in more chronic kidney injuries.11
In all this, the relative contribution of these diverse intracellular and extracellular roles of HMGB1 remains elusive and difficult to dissect experimentally. Conventional Hmgb1 KO mice are not viable; hence, only cell type–specific model systems allow for addressing this issue.15 Constitutive hepatocyte-specific deletion of Hmgb1 did not cause a spontaneous liver phenotype but aggravated ischemia-reperfusion injury of the liver in association with an access of mitochondrial dysfunction and increased nuclear instability.16 By contrast, constitutive deletion of Hmgb1 in cardiomyocytes caused small hearts with cardiac dysfunction and consecutive growth retardation.17 The authors attributed this to changes in fatty acid metabolism.
In view of these data, we hypothesized that tubular epithelial cell HMGB1 would contribute in various ways to ischemic kidney injury, e.g., through HMGB1 release as well as through its physiological role as a modulator of intracellular processes. To test this concept, we performed a series of experiments with newly generated mice with tubular epithelial cell–specific deletion of HMGB1 and various antagonists of HMGB1 and performed mechanistic studies with cells from these mice in vitro.
Methods
Animal Studies
Generation of Mice
Pax8 rtTA, TetO Cre, Hmgb1fl/fl (Pax8-Cre; Hmgb1fl/fl) mice in the C57BL/6J background were generated by crossing Pax8 rtTA, TetO Cre mice with Hmgb1fl/fl mice, generated by Tadatsugu Taniguchi, Department of Molecular Immunology, Institute of Industrial Science, University of Tokyo, Japan, and generously provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan.15 We used littermate mice for all experiments. We cohoused male mice, 8–9 weeks of age, in groups of 4–5 in cages under SPF conditions with unlimited access to standard chow (ssniff, Germany) and desalinated water. We sterilized cages, nestlets, food, and water by autoclaving before use.
At age 8 weeks, Pax8-Cre, Hmgb1fl/fl, and Hmgb1fl/fl mice received a 2 mg/ml doxycycline 5% sucrose solution for drinking water to induce Cre expression and recombination of the Hmgb1 flox cassette in Pax8-expressing tubular epithelial cells resulting in an irreversible deletion of HMGB1 exon 2–4 in a cell type–specific manner under steady-state conditions. We changed doxycycline-enriched water every 2 days and for a total duration of 21 consecutive days. We sacrificed the animals at days 7 and 30 after induction or prior of ischemia/reperfusion injury (IRI), as indicated in figure captions. We measured GFR at days 0, 1, 4, 7, 14, 21, and 30 after IRI. We stored one pole of each kidney in RNAlater at −80°C and another pole in 4% paraformaldehyde fixation before embedding the tissue in paraffin. The local government authorities had approved all experimental procedures (AZ: ROB 55.2-2532.Vet_02-20-101).
Drug Interventions
We injected male C57BL/6J mice at age 8–10 weeks (Charles River, Sulzfeld, Germany) every 24 hours intraperitoneal at a dose of 50 mg/kg (glycyrrhizic acid) and 80 mg/kg (ethyl pyruvate) body weight for 14 consecutive days, respectively, starting either 1 hour before surgery (pretreatment) or 72 hours after reperfusion (delayed treatment). Control animals of the same background, age, and sex received 10% DMSO/90% PBS (vehicle) at the same volume and time points. Anti-HMGB1 antibody (2G7) or control IgG was injected intraperitoneal at a dose of 100 µg/mice every second day for 14 days, starting 72 hours after reperfusion (delayed treatment).
Study Design
We based group size calculation for the primary end point on numeric assumptions derived from our previous experience with this animal model.18-20
Block Randomization by Genotype and GFR
We randomized mice according to their baseline excretory kidney function to either treatment or vehicle group so that for each experiment, mean and SD of baseline GFR were identical between the treatment groups in wild-type animals. We excluded mice with temperature readings below or above the target range of 36.5°C–38.5°C prior or during IRI from the study (see below).21 After applying the exclusion criteria, the total number of animals was at least seven wild-type animals in each treatment group, seven Pax8-Cre, Hmgb1fl/fl, and seven Hmgb1fl/fl mice, respectively. We collected data for each treatment group from at least two independent experiments.
Ischemia Reperfusion Injury
We induced kidney IRI as described.21 In brief, anesthesia included a mixture of medetomidine, midazolam, and fentanyl to achieve analgesia, amnesia, and hypnosis before surgery. The model comparison study (sham surgery, unilateral IRI, bilateral IRI, uninephrectomy) also involved C57BL/6N mice, with 40 minutes unilateral and 20 minutes bilateral ischemia time, respectively. We subsequently optimized the ischemia times for the respective genetic background and survival of the animals under our specific experimental conditions, namely a better temperature control by prewarming the animals, which allowed stabilizing the body core temperature between 36.5°C and 38.5°C during the procedure as assured by rectal temperature recording for every mouse from after the onset of complete anesthesia until wound closure.21 Unilateral ischemia time in all mice of C57BL/6J background was set to 17 minutes after reperfusion to obtain similar results. After clamp removal and kidney repositioning, wounds were closed using absorbable sutures for peritoneal and cutaneous layer and analgesic treatment with buprenorphine was started 30 minutes before narcosis antagonization. Dosages and treatment regimens of narcosis, antagonization, and analgesia are listed in Supplemental Table 1. For adjustment of fluid losses, we administered 200 µl of saline into the peritoneal cavity. We handled sham-operated mice in the same manner, except for clamping.
Uninephrectomy
Protocols for animal preparation, narcosis, antagonization, and pain management were identical to those of IRI. Instead of clamping, we ligated the renal pedicle using nonabsorbable sutures before resecting the kidney.
Transcutaneous Measurement of GFR
For GFR measurement, we anesthetized mice with isoflurane to attach a miniaturized imager device made of two light-emitting diodes, a photodiode, and a battery (MediBeacon, Mannheim, Germany) using a double-sided adhesive tape onto the shaved animals' neck.22 For the duration of recording (approximately 1.5 hours), each animal remained conscious and mobile in a single cage. Before the intravenous injection of 150 mg/kg FITC-sinistrin, we recorded the skin's background signal for 5 minutes. After removing the imager device, we analyzed the data using MPD Lab software (MediBeacon, Mannheim, Germany). We calculated GFR (µl/min) from the decline of fluorescence intensity over time (i.e., plasma half-life of FITC-sinistrin) using a two-compartment model, the animals' body weight and an empirical conversion factor.22
Secondary End Points
Kidney Weight
At day 30, we determined kidney weight as a marker of kidney atrophy right after sacrifice and decapsulation.
Histology
Ischemic and contralateral kidneys stored in 4% buffered formalin for 18–24 hours were embedded in paraffin, and 2–4 µm sections were prepared for periodic acid–Schiff (PAS), Picro-Sirius Red staining for collagen I and III (Sigma-Aldrich, no. 365548) or immunostaining by deparaffinization (xylene), rehydration (ethanol series), and blockade of endogenous peroxidase (3% H2O2 in methanol). For antigen retrieval, we used either proteinase K or microwave heated unmasking solution at pH6. Sections were blocked with avidin and biotin for 20 minutes each before staining. After the slides were washed in PBS, they were incubated with the primary and secondary antibodies as indicated in Supplemental Table 2.23
Low power field (40×) images were taken for each staining, and the positive signal for Lotus tetragonolobus lectin (LTL), Tamm-Horsfall protein (THP), or nitrotyrosine was measured in pixels and normalized to the whole kidney section using ImageJ 1.53f52.24 Ischemic tubular injury was scored by assessing the percentage of tubules in the outer stripe of outer medulla (OSOM) that displayed cast formation, brush border loss, tubular dilatation, or cell infiltrates in 15 randomly chosen high power fields (100×) and expressed as an injury score 1–10.25 All assessments involved a blinded observer. HMGB1 immunohistochemical staining in Figure 1C was obtained from the Human Protein Atlas (HPA003506, Patient ID: 2165, http://www.proteinatlas.org).
Figure 1.

HMGB1 expression in mouse and human kidneys. (A) UMAP plot (left) or dot plot (right) of published snRNA-seq dataset showing Hmgb1 expression pattern in control mouse kidneys (n=3). The diameter of the dot corresponds to the proportion of cells expressing the indicated gene, and the density of the dot corresponds to average expression relative to all cell types. TEC, tubular epithelial cells; Uro, urothelium; Pod, podocytes; PEC, parietal epithelial cells; EC, endothelial cells; Fib, fibroblasts; Per, pericytes; Mø, macrophages; Tcell; T cells. (B) UMAP plot (left) or violin plot (right) of published snRNA-seq dataset showing HMGB1 expression in healthy human kidneys (n=5). PODO, podocyte; ENDO, endothelial cells; MES, mesangial cells; FIB, fibroblasts; LEUK, leukocytes. (C) Dot plot of published snRNA-seq dataset showing Hmgb1 expression pattern in the proximal tubular cell subtypes along with the time course after bilateral ischemic reperfusion injury (IRI). PT (S1), S1 segment of proximal tubule; PT (S2), S2 segment of proximal tubule; PT (S3), S3 segment of proximal tubule. The diameter of the dot corresponds to the proportion of cells expressing the indicated gene, and the density of the dot corresponds to average expression relative to all proximal tubular cells in the whole dataset.
Analysis of Murine HMGB1 Expression in snRNA-seq Data Set
HMGB1 expression was analyzed with the published snRNA-seq dataset for mouse kidneys with ischemic reperfusion injury (GSE139107)26 or human kidneys (GSE151302)27 on R package Seurat (v4.0.0).28 Proximal tubular cells (S1, S2, and S3) were extracted with subset function and reanalyzed to visualize change in the Hmgb1 expression pattern along with time course for each subtype after ischemic reperfusion injury. The data were visualized with DimPlot, DotPlot, or ViolinPlot functions.
Analysis of HMGB1 Expression in Biopsies and Single Urinary Cells of Patients with AKI
HMGB1 expression was analyzed in 40 urine samples of 32 patients with AKI as defined by Kidney Disease: Improving Global Outcomes (KDIGO) criteria (GSE199321). Sequencing data were analyzed using R and Seurat; for detailed information, see the original publication preprint (Klocke et al. Urinary single-cell sequencing captures intrarenal injury and repair processes in human acute kidney injury, https://doi.org/10.1101/2022.02.15.479234).
Murine HMGB1 Sandwich ELISA
At the indicated time points, plasma was collected in the presence of heparin, centrifuged at 1000g and 4°C for 15 minutes, and stored at −80°C until analysis. Reagents, working standards, and samples were diluted (1:200) and prepared according to the manufacturer's instructions (Mouse HMGB1 ELISA kit, Cusabio, CSB-E08225m, China).
Transmission Electron Microscopy
Samples were fixed in freshly prepared Karnovsky Fixative (Electron Microscopy Sciences; 5% glutaraldehyde and 4% formaldehyde in 0.064 M sodium phosphate buffer according to the manufacturer's protocol, no. 15720) for at least 4 hours. Thereafter, glutaraldehyde was removed, and samples were washed three times with 0.1 M sodium cacodylate buffer, pH 7.4. Postfixation and prestaining were performed for 45–60 minutes with 1% osmium tetroxide (Electron Microscopy Sciences). Samples were washed three times with ddH2O and dehydrated with an ascending ethanol series (15 minutes with 30%, 50%, 70%, and 90%, respectively, and two times 10 minutes with 100%). Subsequently, samples were embedded in Epon (3.61 M glycid ether 100; Serva Electrophoresis), 1.83 M methyl nadic anhydride (Serva Electrophoresis GmbH), 0.92 M dodecenylsuccinic anhydride (Serva Electrophoresis GmbH), and 5.53 mM 2,4,6-tris(dimethylaminomethyl)phenol (Serva Electrophoresis GmbH). 70-nm ultrathin sections were cut at the Reichard-Jung Ultracut E (Darmstadt, Germany) microtome. Ultrathin sections were collected on formvar coated copper grids (Plano, Germany) and automatically stained with UranyLess EM Stain (Electron Microscopy Sciences) and 3% lead citrate (Leica, Wetzlar, Germany) using the contrasting system Leica EM AC20 (Leica, Wetzlar, Germany). Imaging was performed using the JEOL-1200EX II transmission electron microscope (JEOL, Akishima, Tokyo) at 80 kV. Images were taken using a digital camera (KeenViewII; Olympus, Germany) and processed with the iTEM software package (analySIS Five; Olympus, Germany).
In Vitro Studies
Cell Isolation and Culture of Primary Murine Kidney Tubular Cells
Renal tubular cell isolation from mice was performed based on previously described protocols.29,30 In brief, we extracted kidneys from 5- to 6-week-old Pax8-Cre, Hmgb1fl/fl, Hmgb1fl/fl, or C57BL/6J wild-type (Charles River) mice before decapsulation, mashing into small pieces with sterile instruments and digestion in 2 mg/ml collagenase D for 30 min/37°C. The material was pushed through a 70-µm pore diameter preseparation filter and washed and diluted in 10 ml of phosphate buffered saline (PBS). We separated the tubular segments using 31% Percoll density centrifugation at 800 rcf/10 min per 4°C. The pellet of tubular segments formed at the bottom of the tube was collected and washed twice with PBS at 300 rcf/5 min per 4°C. We cultured the renal tubular cells (mTEC) under sterile conditions at 37°C and 5% CO2 (culture medium recipe in Supplemental Table 5). For experiments, the cells were either used right after isolation, where the tubular cells are still organized in segments, or after > 4 days in culture, after they grew out to form a monolayer of cells.
Cell Isolation and Culture of Primary Murine Bone Marrow–Derived Macrophages
Pax8-Cre, Hmgb1fl/fl, and Hmgb1fl/fl mice were sacrificed by cervical dislocation, and the whole body was immersed thoroughly with 75% ethanol for 10 minutes before femur and tibia were isolated. The bones were flushed with 75% ethanol and sterile PBS for 1 minute. The residual tissue was removed, and the tibia was separated from the femur by bending slightly at the knee joint. The ends of the bones were removed, and a 25-G needle was used to flush out the bone marrow with sterile DMEM. The flow through was collected and centrifuged at 300 rcf for 5 minutes. Erythrocytes were lysed with 0.155 M NH4Cl for 10 minutes on ice, and the residual cells were washed with PBS and centrifuged for 5 minutes at 300 rcf. The cell pellet was resuspended and passed through a 70-µM cell strainer. The flow through solution was collected and centrifuged at 300 rcf for 5 minutes. The pellet was resuspended in DMEM and seeded in a plate supplemented with 100 ng/ml M-CSF and 10% FCS. Fresh medium was added at days 2 and 5 after isolation, and cells were used for experiments at day 7 after isolation.
RNA Isolation and qPCR.
We extracted total RNA from renal tissue using the pure Link RNA Mini Kit and stored samples in RNAlater according to the manufacturer's instructions. cDNA was synthesized from 2 µg of total RNA by reverse transcription polymerase chain reaction (PCR) using the reagents listed in Supplemental Table 3. The reaction was performed in the presence and absence of reverse transcriptase for each sample to correct for residual genomic DNA contamination during qPCR. After initial denaturation at 65°C for 10 minutes, reverse transcription was performed at 42°C for 90 minutes using a Mastercycler pro (Eppendorf, Germany). Quantitative real-time PCR (qPCR) from cDNA was performed using the reagents listed in Supplemental Table 3 and primers listed in Supplemental Table 4. 18s rRNA was used as a reference transcript for relative quantification; hence, qPCR data for genes of interest were normalized to 18 seconds. Controls consisting of ddH2O were negative for targets and reference genes. Each amplification step included an initiation phase at 95°C, annealing phase at 60°C, and amplification phase at 72°C and was repeated for 40 cycles.31 We designed primers to be cDNA-specific and to target most CCDS-approved transcripts. We considered all samples that did not exceed background fluorescence as undetectable. We analyzed melting curve profiles for every sample to detect unspecific products and primer dimers. In part, we used agarose gels to visualize PCR products.
SDS-PAGE and Western Blot
TECs or bone marrow–derived macrophages (BMDMs) were harvested with trypsin/EDTA and washed with cold PBS. The cell pellet was lysed with the cold RIPA buffer containing protease and phosphatase inhibitors and incubated for 30 minutes at 4°C and 120 rpm horizontal shaking. The suspension was centrifuged at 16,000g and 4°C for 20 minutes, and the supernatant was stored at −80°C until further preparation. The Bradford assay was used to determine the protein concentration of each lysate. At a final concentration of 1.95 mg/ml total protein, the samples were boiled at 95°C for 5 minutes and centrifuged at 16,000g for 1 minute. Equal amounts of protein and appropriate molecular weight markers were loaded into the 12% SDS-PAGE gel pockets, and SDS-PAGE was run 1.5 hours at 100 V. The semidry transfer to PVDF membrane was conducted at 10 V for 50 minutes. The membrane was blocked in 5% nonfatty milk (blocking buffer) for 1 hour at room temperature before incubation with the primary antibody (anti-HMGB1 antibody, no. ab18256, Abcam, 1:200; anti-b-actin antibody, no. 4967, Cell Signaling, 1:500) overnight at 4°C with horizontal agitation. The membrane was washed with TBST three times for 5 minutes and incubated with the secondary antibody (anti-rabbit IgG, HRP-linked antibody, no. 7074, Cell Signaling, 1:5000) diluted in blocking buffer for 1 hour at room temperature. Washing with TBST was repeated, and ECL substrate kit (#ab133406, Abcam) was used to detect the secondary antibody signal. ImageJ software was used to quantify the HMGB1 band thickness and opacity in relation to β-actin. Intracellular immunofluorescence staining for HMGB1. TECs or BMDMs were seeded in 8-well chamber slides and fixed with 4% fresh paraformaldehyde. The slides were incubated with anti-HMGB1 antibody (#ab18256, Abcam) at a 1: 200 dilution in blocking buffer (PBS, 3% BSA, 0.1% saponin) at 4°C overnight. After washing with PBS, the slides were incubated with the secondary Alexa 647-tagged antibody (no. A27040, ThermoFisher Scientific) at a 1:100 dilution in blocking buffer (PBS, 0.1% saponin) at room temperature for 30 min. After washing with PBS, the slides were mounted with DAPI/mounting medium (#VEC-H-1200, Vector laboratories) and covered with coverslips. The slides were kept at 4°C before image acquisition using a Leica DM IL fluorescence microscope (Leica Microsystems, Germany) and a Nikon DS-Qi2 camera (Nikon, Japan).
Treatment with Small Molecules Anti-HMGB1 Antibody
Cells were seeded in 96-well plates and treated with cell death inhibitors Z-VAD (10 µM) and Nec-1f (20 µM) or HMGB1 inhibitors glycyrrhizic acid (12 nM) or ethyl pyruvate (1 mM) and/or 2G7 (20 µg/ml) or combination of 2G7 and HMGB1 inhibitor. After 30 minutes of incubation, hydrogen peroxide (H2O2) at a final concentration of 1 mM was added for 24 hours.
Time-Lapse Imaging
Time-lapse imaging was performed using a 10× objective (Nikon Eclipse Ti2 microscope) and an incubation chamber at 37°C, 5% CO2, and humidity control. Bright field and fluorescence images were obtained using a Nikon DS-Qi2 camera (Nikon, Japan). Cells were imaged every 30 minutes for 24 hours, and cell membrane integrity was assessed using 1-µM Helix NP green stain (#425303, BioLegend). ImageJ was used to quantify cell number and Helix NP fluorescence signal intensity.
Metabolic Activity Assessment and Cytotoxicity
We measured metabolic activity of primary murine cells using the CellTiter 96 Non-Radioactive Cell Proliferation Assay. We incubated 10×103 cells/100 μl with 1 mM H2O2 for 3 or 24 hours in the absence of FCS. After removing the supernatant and washing twice with PBS, we incubated cells for 4 hours with 100 µl medium and 15 µl dye solution. The reaction was stopped thereafter by adding 100 µl Solubilization/Stop Solution, and absorbance of each well was measured at 570 nm using a plate reader. To assess cytotoxicity, supernatants of H2O2-treated cells were assessed for the presence of lactate dehydrogenase (LDH) using the CellTiter 96 Assay (Roche), following the manufacturer's protocol. Tween at 0.1% final concentration was used as a positive control for cell death (100%) and normal culture medium without FBS as a negative control.
Statistical Analysis
We either report data as individual measurements with superimposed boxplots or aggregated as mean±SD. Before every other statistical analysis, we tested for normal distribution (Shapiro–Wilk test), homoscedasticity (Levene test), and outliers (Grubb test). Normally distributed and homoscedastic datasets were tested for statistically significant differences using ANOVA and post hoc Bonferroni correction for multiple comparisons. We corrected heteroscedastic data following the Games-Howell post hoc test. We compared not normally distributed datasets using Kruskal–Wallis and Nemenyi testing. We considered P < 0.05 to indicate statistical significance. P values are indicated as P > 0.05 n.s., P < 0.05 * (or #), P < 0.01 ** (or ##), and P < 0.001 *** (or ###), where hashtags indicate P values comparing a group with the negative control (sham, vehicle, wild-type), and asterisks indicate P values comparing two treatment groups.
Ethics Statement
All experimental procedures were carried out according to the German Animal Care and Ethics legislation and were approved by the local governmental authorities (i.e., the Ethical Committee of the Regierung von Oberbayern, permit no. AZ 55.2-1-54-2532-175-14).
Results
HMGB1 Is Ubiquitously Expressed in the Healthy Kidney and during AKI in Mice and Humans
To define the expression pattern of HMGB1 in healthy kidney, we queried three single-nucleus RNA-sequencing (snRNA-seq) datasets: murine kidneys before and after AKI (GSE139107, Figure 1A), healthy human kidney biopsies (GSE151302, Figure 1B), and human urine–derived cells from critical ill patients with AKI (Supplemental Figure 2). In both mouse (Figure 1A) and human (Figure 1B) healthy kidneys, HMGB1 was broadly expressed throughout tubular epithelial cells, stromal cells, and podocytes, with lower expression in endothelial cells in both human and murine kidneys. Both the mouse and human datasets suggested HMGB1 expression in macrophages as well, with lesser expression in human macrophages, although the human dataset contained fewer macrophages overall.26,27 In mouse IR injury, HMGB1 was downregulated at early time points (4 and 12 hours) but was re-expressed approaching baseline levels by 48 hours after injury (Figure 1C). Single-cell RNA sequencing (scRNAseq) analysis of cells excreted into the urine of critically ill patients with AKI confirmed the ubiquitous expression pattern of HMGB1 (Supplemental Figure 2A, H). HMGB1 receptors (TLR2, TLR4, AGER, CXCR4) were mainly expressed on immune cells, with fewer expressions by TECs (Supplemental Figure 1A–D, Supplemental Figure 2C–F, I–L). Thus, HMGB1 is expressed in multiple cell types of the healthy kidney before and along the phases of AKI.
Ischemic Necroinflammation Followed by Persistent Hypoxia Leads to Kidney Atrophy
Kidney injury is usually bilateral, but in contrast to humans where kidney replacement therapy can assure survival, severe bilateral AKI/AKD is lethal in mice.32 Nonlethal ischemic AKI in mice induced by transient bilateral kidney pedicle clamping (20 minutes ischemia time) resulted only in moderate loss of renal epithelium that largely recovers without measurable kidney atrophy and only slightly reduced GFR 30 days after reperfusion (Supplemental Figure 3A–D). By contrast, unilateral ischemia-reperfusion injury, even at longer ischemia times (40 min), was nonlethal and reliably produces AKI-CKD transition with profound atrophy of the ischemic kidney at the same readout time points (Supplemental Figure 3A–D). Unilateral kidney pedicle clamping was associated with persistent kidney hypoxia (Supplemental Figure 3E) due to persistent vasoconstriction and a reduction in renal blood flow.33 Thus, unilateral kidney pedicle clamping is a reproducible model of persistent kidney hypoxia as a driver of AKI-CKD transition and kidney atrophy.
HMGB1 Blockers Glycyrrhizic Acid and Ethyl Pyruvate Attenuate AKI-CKD Transition
Previous studies using HMGB1-neutralizing antibodies or HMGB1 inhibitors have demonstrated a proinflammatory role of extracellular HMGB1 during the acute and subacute phase after ischemic AKI.13,14,34-37 Whereas neutralizing antibodies target exclusively extracellular HMGB1, small molecule antagonists such as glycyrrhizic acid (Gly) and ethyl pyruvate (EP) also target intracellular functions of HMGB1. Gly induces conformational changes interfering with HMGB1 DNA-binding ability in the nucleus, HMGB1 phosphorylation in the cytosol, and HMGB1 receptor-binding ability in the extracellular space.38 EP selectively inhibits translocation of HMGB1 from the nucleus, which suppresses its functions in the cytosol and active HMGB1 secretion on cell activation, albeit not its passive release on cell necrosis.39
In our model of postischemic AKI-CKD transition, plasma HMGB1 levels peaked early and declined thereafter, although plasma levels remained elevated up to day 30, consistent with ongoing (kidney) cell death (Figure 2B). Preemptive Gly treatment (Figure 2A) recovered kidney function on day 4, although GFR declined again (Figure 2, C and D) without significantly reducing the circulating levels of HMGB1 (Figure 2E). The kidney injury score (Figure 2F) of Gly-treated mice at day 30 after reperfusion was significantly lower due to a better recovery of the proximal tubule (Figure 2, G and H). Preemptive EP treatment recovered normal GFR within 4 days, which persisted up to day 30 (Figure 2C) together with a significant reduction in plasma HMGB1 levels (Figure 2E), probably because EP significantly reduced the kidney injury score with better recovery of proximal and distal tubules (Figure 2, F–H). Notably, both treatments had no effect on the initial GFR decline (Figure 2C). Still, both Gly and EP protected mice from AKI-CKD transition as seen by significantly reduced interstitial matrix deposits at day 30 post-IRI (Figure 2I). Taken together, Gly and EP have renoprotective effects beyond the early injury phase.
Figure 2.
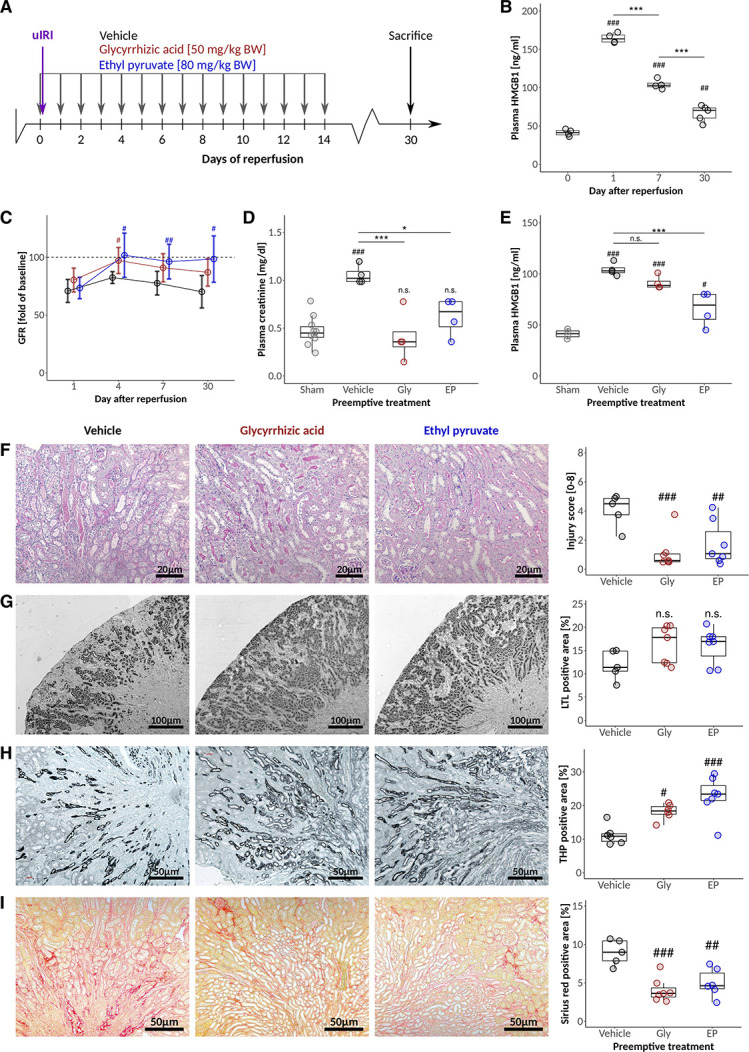
HMGB1 blockers glycyrrhizic acid and ethyl pyruvate attenuate AKI-CKD transition. (A) C57BL6/J mice were challenged with 17 minutes unilateral IRI and treated with Gly and EP at the indicated time points. (B) Time course of plasma HMGB1 levels in untreated animals at the indicated times after reperfusion. (C) Changes in GFR (presurgery values for each mouse defined as 100%) after IRI in response to vehicle, Gly, and EP treatment. (D) Plasma creatinine and (E) plasma HMGB1 levels at day 7 after IRI. Quantification and representative images for (F) semiquantitative injury scoring on the basis of PAS stainings, (G) proximal and (H) distal tubular mass assessment, and (I) fibrosis 30 days after IRI. */#P<0.05, **/##P<0.01, ***/###P<0.001. Data presented in this figure are derived from two independent experiments.
Genetic Depletion of Hmgb1 from Tubules of Adult Mice Shows No Spontaneous Phenotype
In light of the above findings and with tubular cells being both the primary target of postischemic injury and putatively major source of extracellular HMGB1, we aimed to address the putative role of tubular cell HMGB1. We generated conditional knockout mice under the control of the Pax8 promoter that is expressed by all cells of the kidney tubules (Pax8-Cre, Hmgb1fl/fl, Figure 3A).40 By introducing the rTta and TetOCre element, the Cre recombinase-mediated Hmgb1 exon 2–4 exposure to doxycycline can induce Hmgb1 deficiency after completion of kidney development and kidney growth in adult mice. Indeed, primary tubular epithelial cells isolated from induced mice showed reduced Hmgb1 mRNA expression and lost nuclear and Western blot positivity for HMGB1, documenting tubular cell-specific deletion of HMGB1 at high efficacy (Figure 3, B–D). By contrast, we did not detect either of the above in bone marrow–derived macrophages (Figure 3, B–D). Pax8-Cre, Hmgb1fl/fl mice bred normally at Mendelian distribution, matching size, body weight, and weight gain similar to their WT littermates (not shown). GFR and histology were identical and unremarkable in doxycycline-induced Pax8-Cre, Hmgb1fl/fl mice and Hmgb1fl/fl controls at age 20 weeks (Supplemental Figure 4A–D). Ultrastructural analysis of kidney tubules with transmission electron microscopy (TEM) did not reveal obvious abnormalities in the subcellular structures of proximal and distal tubules between both groups (Supplemental Figure 4C). Together, mice that specifically lack HMGB1 in the epithelial cells of kidney tubules do not show a spontaneous phenotype even at the ultrastructural level.
Figure 3.

Characterization of renal Pax8-positive tubular cell Hmgb1-deficient mice. (A) The transactivator rtTA lies under the control of the Pax8 promoter, while the TetOCre recombinase responds to rtTA only in the presence of doxycycline. Fourteen consecutive days of doxycycline treatment using drinking water will result in the excision of exons 2–4 of the Hmbg1 flox allele. (B) Ex vivo qPCR, (C) Western blot, and (D) immunofluorescence staining for cell nuclei (DAPI) and HMGB1 of primary TECs and BMDMs, respectively, from doxycycline-induced animals. ***/###P<0.001.
Tubular HMGB1 Deficiency Does Not Attenuate Ischemic AKI but AKI/CKD Transition
To study the effect of Hmgb1 deficiency in kidney tubules during the early necroinflammation phase of ischemic AKI/AKD, we analyzed mice at different days after kidney pedicle clamping (Figure 4A). The initial drop of GFR was comparable between mice with or without tubular HMGB1 (Figure 4B). Seven days after reperfusion, kidney injury score, proximal, and distal tubule staining indicated comparable levels of early injury (Supplemental Figure 5A–C, E), which was consistent with the results of the aforementioned antagonist experiments. To study the role of tubular cell HMGB1 during AKI-CKD transition and kidney atrophy associated with persistent hypoxia, we analyzed later time points, too. While control mice displayed a persistent loss of GFR compared with baseline, the GFR of mice with tubular Hmgb1 deficiency started to recover after day 7, reaching baseline values at days 21 and 30 (Figure 4B). This was associated with an indistinguishable kidney weight between contralateral (CO) and postischemic (IRI) kidney at 30 days after AKI (Figure 4C), implying that lack of tubular HMGB1 prevented ischemic kidney atrophy and compensatory hypertrophy of the contralateral kidney. Plasma HMGB1 levels were identical at day 30 after reperfusion independent of the genotype (Supplemental Figure 4B). The kidney injury score (Figure 4E) of HMGB1-deficient mice at day 30 after reperfusion was significantly lower due to a better recovery of the proximal tubule (Figure 4, F and G), with significantly reduced interstitial matrix deposits (Figure 4H) and less expression of the Ngal (Figure 4D) at day 30 post-IRI. These results confirm that tubular cell HMGB1 contributes to AKI/CKD transition in the context of persistent kidney hypoxia.
Figure 4.
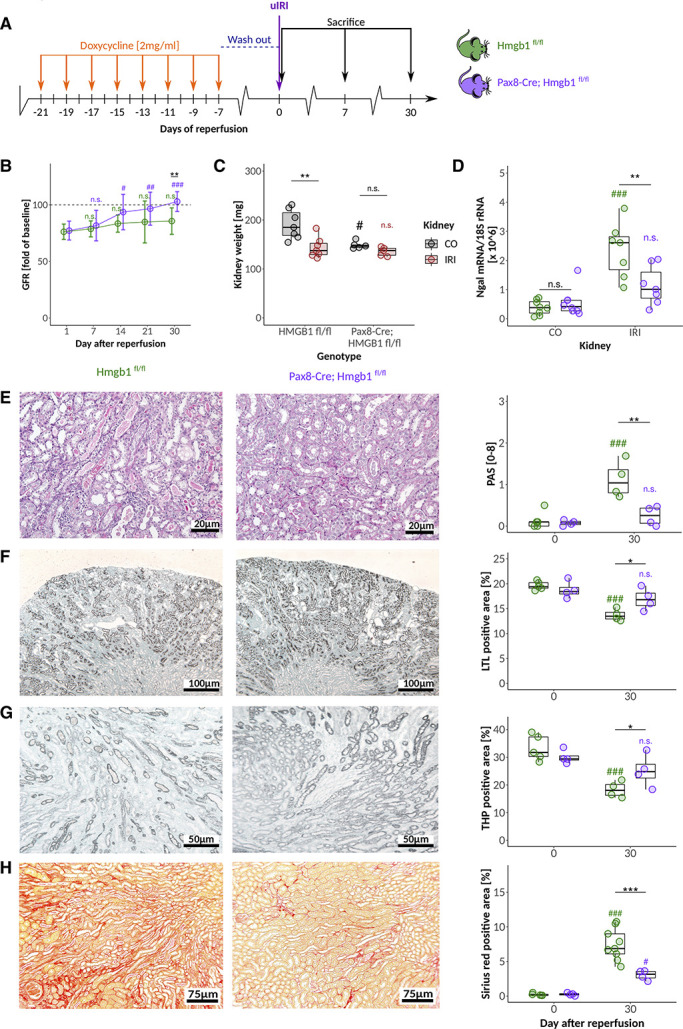
Tubular HMGB1 deficiency does not attenuate ischemic AKI but AKI/CKD transition. (A) After doxycycline induction Hmgb1fl/fl and Pax8-Cre, Hmgb1fl/fl mice were challenged with 17 minutes of unilateral IRI. (B) Changes in GFR (presurgery values for each mouse defined as 100%) after IRI in response to the genotype. (C) Delta kidney weight (mg) as an indicator of renal atrophy and compensatory hypertrophy of the contralateral kidney in response to genotype. (D) mRNA expression level of Ngal in contralateral (CO) and postischemic (IRI, day 30) whole kidney tissue. Quantification and representative images at day 30 after reperfusion for (E) semiquantitative injury scoring on the basis of PAS stainings, (F) proximal and (G) distal tubular mass assessment, and (H) fibrosis 30 days after IRI. */#P<0.05, **/##P<0.01, ***/###P<0.001. Data presented in this figure are derived from at least two independent experiments.
HMGB1 Affects the Susceptibility of Tubular Cells to Prolonged Oxidative Stress
Acute ischemia-related kidney necroinflammation involves necrosis of tubular epithelial cells, a major determinant of AKI severity. To understand the direct contribution of intracellular HMGB1 to the survival of primary tubular cells exposed to ischemic stress, we investigated the survival rate of freshly isolated tubular segments (Supplemental Figure 6A, Supplemental Video 1), which by itself mimics features of acute injury to terminally differentiated tubular cells. As expected, the isolation itself led to the death of a significant number of cells within tubular segments. Notably, the segments of differentiated tubular cells isolated from Pax8-Cre, Hmgb1fl/fl, and Hmgb1fl/fl mice with the absence or presence of HMGB1, respectively, displayed no difference in resilience to the stress at 4 hours. Next, we compared primary tubular cell outgrowth from both genotypes left to proliferate in culture for more than 4 days, thereby positively selecting for cells entering the cell cycle and completing mitosis. In this study, we exposed the cells to 1mM H2O2 for 24 hours to mimic persistent oxidative stress during the postinjury phase (Figure 5, A–C, Supplemental Video 2). Initially, cells from both genotypes reacted similarly to this oxidative stress with subsequent release of LDH (Figure 5C) and loss of metabolic activity (Supplemental Figure 6B). Live cell imaging using Helix NP green as a marker of cell death revealed that within 10 hours of incubation, Hmgb1-deficient cells showed a greater resilience to prolonged oxidative stress (Figure 5, A and B, Supplemental Video).
Figure 5.

HMGB1 affects the susceptibility of tubular cells to prolonged oxidative stress. (A and B) Helix NP green cell death assay from time lapse images of primary murine TECs (after 4 days in culture) and representative images at 24 hours after 1 mM H2O2 stimulation in response to genotype. (C) Cytotoxicity as per LDH measurement of primary TECs of both genotypes in dependence of time after treatment with 1 mM H2O2. (D) Cytotoxicity as per LDH measurement of primary murine TECs at 24 hours after 1 mM H2O2 stimulation with or without 2G7 antibody in response to genotype. (E) Cytotoxicity as per LDH measurement of primary murine wild-type TECs at 24 hours after 1 mM H2O2 stimulation and treatment with 2G7 or IgG in combination with or without Gly or EP. Cytotoxicity of 1 mM H2O2 treatment in response to (F) EP and (G) Gly pretreatment at given concentrations using primary wild-type TECs. (H) Comparison of the protective effect of different pretreatments after 1 mM H2O2 stimulation of primary TECs using the Helix NP green cell death assay. */#P<0.05, **/##P<0.01, ***/###P<0.001.
To test for HMGB1 functions beyond the known extracellular role of HMGB1 as a DAMP, we treated H2O2-stimulated cells from both genotypes with either control IgG or the HMGB1-neutralizing antibody 2G7 at a dose documented to induce maximal inhibition of HMGB1 in vitro.41-44 Although neutralization of extracellular HMGB1 was beneficial, knockdown of HMGB1 provided additional protection (Figure 5D). These findings were confirmed by cotreatment experiments with IgG/2G7 and Gly/EP using primary wild-type TECs (Figure 5E). Both Gly and EP offered additional protection beyond neutralization of extracellular HMGB1 with 2G7.
Taken together, these results are in line with our in vivo findings and strengthen the notion that HMGB1 contributes to the vulnerability of cycling tubular epithelial cells under conditions of oxidative stress through a mechanism beyond the DAMP function of extracellular HMGB1 during the early necroinflammation phase of AKI. This additional contribution of HMGB1 to AKI-CKD transition operates rather in the late/subacute phase when a subset of tubular epithelial cells proliferates and regenerates injured tubules.45
Gly and EP reduced ROS-mediated cytotoxicity in wild-type tubular epithelial cells in a dose-dependent manner (Figure 5, F and G). It remains speculative how HMGB1 inhibition rescues the cells in response to ROS exposure. The direct comparison of HMGB1 inhibitors with potent cell death inhibitors Nec-1f (inhibitor of necroptosis and ferroptosis) and Z-VAD (pan-caspase inhibitor) alone or in combination revealed that Gly and EP are more potent than either of these drugs in blocking cell stress/cell death in response to prolonged ROS exposure (Figure 5H). Hence, we conclude that the absence or inhibition of HMGB1 increases the resilience of tubular cells to prolonged oxidative stress.
Also Delayed-Onset Treatment with HMGB1 Inhibitors Ameliorates AKI-CKD Transition
The above findings suggest that HMGB1 could be a potential target for delayed-onset therapeutic interventions in AKI to improve outcomes of AKI survivors. However, both the early-onset Gly and EP treatment as well as the HMGB1 knockout studies could not dissect the role of HMGB1 during the early and late phases of AKI. To clarify this point, we conducted further IRI experiments in wild-type animals and started Gly and EP treatment only after the peak of AKI and the acute necroinflammation phase (i.e., 72 hours after reperfusion; Supplemental Figure 7A). Both treatments significantly improved GFR to baseline levels at 30 days compared with the persistently reduced GFR in vehicle-treated mice (Supplemental Figure 7B). In addition, 30-day kidney histology scores improved significantly with Gly and EP treatment (Supplemental Figure 7C–F).
Similar to the in vitro studies presented above, it remains elusive whether tubular cell–derived HMGB1 aggravates the postischemic phenotype primarily as an extracellular DAMP or through additional biological effects. Therefore, we conducted delayed-onset cotreatment experiments in postischemic mice using both HMGB1-neutralizing antibody 2G7 and Gly and EP, respectively (Figure 6A). While 2G7 alone did not improve AKI recovery, adding EP improved GFR recovery significantly supporting the concept that HMGB1 promotes AKI-CKD transition beyond its extracellular effects (Figure 6B). For Gly+2G7, the effect did not reach statistical significance compared with 2G7 alone (Figure 6B). Ngal mRNA expression (Figure 6C) as well as histological assessment of injury (Figure 6D), tubular recovery (Figure 6, E and F), and interstitial fibrosis (Figure 6G) gave similar results.
Figure 6.

Delayed-onset treatment with HMGB1 inhibitors ameliorates postischemic phenotypes. (A) C57BL6/J mice were challenged with 17 minutes of unilateral IRI and treated with 2G7, 2G7+Gly, and 2G7+EP at the indicated time points, with the onset of treatment 72 hours after reperfusion. (B) Changes in GFR (presurgery values for each mouse defined as 100%) after IRI in response to vehicle+IgG, 2G7, 2G7+Gly, and 2G7+EP treatment. (C) mRNA expression level of Ngal in contralateral (CO) and postischemic (IRI, day 30) whole kidney tissue. Quantification and representative images for (D) semiquantitative injury scoring on the basis of PAS stainings, (E) proximal tubular, (F) distal tubular mass assessment, and (G) fibrosis 30 days after IRI. */#P<0.05. Data presented in this figure are derived from two independent experiments. Cells undergoing necrosis release extracellular high mobility group box-1 (HMGB1), which triggers sterile inflammation on acute kidney injury (AKI) in mice. Neither deletion of HMGB1 from tubular epithelial cells nor HMGB1 antagonism with small molecules affects initial ischemic tubular necrosis and immediate GFR loss on unilateral IRI. On the contrary, tubular cell–specific HMGB1 deficiency, and even late-onset pharmacological HMGB1 inhibition, increased functional and structural recovery from AKI, indicating that intracellular HMGB1 partially counters the effects of extracellular HMGB1. In vitro studies indicate that intracellular HMGB1 decreases the resilience of tubular cells from prolonged ischemic stress, as in unilateral IRI. Intracellular HMGB1 is a potential target to enhance kidney regeneration and to improve long-term prognosis in AKI.
Together, HMGB1 sensitizes tubular epithelial cells to hypoxia-induced cell death in the AKI-CKD transition phase of unilateral IRI, a model of persistent kidney hypoxia as it occurs in numerous clinical settings also in humans. Neutralizing HMGB1 functions with suitable antagonists that target HMGB1 beyond its DAMP effects could be a novel way to improve long-term kidney outcomes in those who survived an episode of severe AKI.
Discussion
We hypothesized that tubular epithelial cell HMGB1 would contribute in different ways to ischemic kidney atrophy. Our in vivo studies show that neither conditional deletion of HMGB1 from tubular epithelial cells nor HMGB1 antagonism with Gly and EP affects the initial ischemia-induced acute tubular necrosis and the immediate GFR loss on unilateral kidney pedicle clamping. By contrast, lack of tubular HMGB1 and even late-onset HMGB1 inhibition prevent the kidney atrophy after ischemic AKI followed by persistent tissue hypoxia, which offers the perspective of AKI treatment after the peak of AKI already passed. This treatment effect persisted in the presence of an inhibitor of extracellular HMGB1, suggesting that the mechanism of action in this phase of the disease relates to DAMP-unrelated effects of HMGB1. Our aligned in vitro studies consistently demonstrate that lack of HMGB1 does not affect ischemia-induced necrosis of growth-arrested differentiated tubular epithelial cells but attenuates that of proliferating tubular epithelial cells. These data suggest that HMGB1 limits the intrinsic regenerative capacity of injured tubules by increasing the vulnerability of cycling tubular cells within the recovering tubule to persistent oxidative stress. These findings imply an opportunity for a therapeutic HMGB1 blockade even once AKI has already fully established to improve the long-term kidney outcome of AKI survivors.
HMGB1 depletion from tubular epithelial cells in mice did not induce a spontaneous kidney phenotype in tubular ultrastructure, injury, or inflammation. We cannot exclude minor functional alterations on the clearance of certain solutes or the gene expression patterns. However, from what we observed, HMGB1 serves mainly as a modulator of stress responses, rather than physiological conditions. Our findings are in line with selective Hmgb1 depletion from hepatocytes,16,46 hematopoietic cells,15,46 and keratinocytes47 in healthy mice. However, mice with Hmgb1-deficient cardiomyocytes developed small hearts and spontaneous heart failure.17
The early phase of necroinflammation during ischemic AKI is characterized by ischemic cell necrosis of growth-arrested and differentiated tubular epithelial cells.45,48 Previously, it was believed that ischemic cell necrosis would be a passive process, but it has become evident that tubular cell necrosis during ischemic AKI is a highly regulated process involving numerous signaling pathways ultimately leading to plasma membrane rupture.49-51 Nevertheless, these processes neither involve chromatin remodeling inside the nucleus nor transcription; hence, it may not come as a great surprise that the lack of nuclear HMGB1 does neither affect acute ischemic necrosis of differentiated and growth-arrested tubular epithelial cells in vitro nor in vivo. Of note, studies with hepatocyte-specific deletion of HMGB1 during ischemic liver injury arrived at different results, but portal and arterial perfusion of the liver are not necessarily comparable with the kidney that entirely depends from arterial blood flow.16,46 In addition, the genetic and antagonist experiments gave consistent results in the early phase of postischemic AKI, which validates our conclusion. Interestingly, studies using neutralizing HMGB1 antibodies, which selectively block extracellular HMGB1, were protective in the early phase of AKI,12-14 indicating that the different HMGB1 functions partially neutralize each other in an acute loss of GFR and tubular cell necrosis and the net effect is negligible.
Recovery from AKI involves diffuse cell cycle activation in tubular epithelial cells, inside and outside the injured area as indicated by widespread positivity of tubular cells for cell cycle activation markers. Our model did not allow us to dissect between endocycle-driven hypertrophy of uninjured differentiated tubular cells and clonal proliferation of immature progenitor cells scattered along the nephron.45 Persistent hypoxia and inflammatory signaling suppress adequate kidney regeneration and therefore contribute to AKI-CKD transition.52,53 Our in vitro data suggest that HMGB1 impairs the resilience of tubular epithelial cells to persistent hypoxia in the phase of regenerative outgrowth on ischemic tubular cell necrosis. Hmgb1-deficient tubular cells exposed to ROS during the proliferative phase of the cell cycle were rescued by Nec-1f, a blocker of cell death using necroptosis and ferroptosis.51 Similarly, caspase inhibition blocked cell death using apoptosis and pyroptosis.54 All these pathways were entirely blocked by Gly and EP, suggesting that HMGB1 impairs an upstream resilience of tubular cells to these cell death pathways. Of note, also growth-arrested tubular epithelial cells die using these routes in the early injury phase,49,50 while lack of HMGB1 did not interfere with that. Therefore, HMGB1 specifically promotes the death of cells in cycling tubular cells during the recovery phase of AKI. In addition, by preventing HMGB1 secretion from tubular cells, EP also reduces the levels of extracellular HMGB1 and the related necroinflammation.37,55,56
These findings offer a unique opportunity to enhance kidney regeneration and to improve the long-term kidney prognosis in AKI survivors by blocking HMGB1 with suitable small molecule antagonists that also interfere with “physiological” and DAMP functions of HMGB1 in different compartments. Such a strategy would address the unsolved problem that patients with AKI are usually identified only once kidney injury has already occurred and that therapeutic interventions aiming to attenuate AKI itself have largely failed.1 Our approach would rather focus on the long-term consequences of AKI (i.e., AKI-CKD transition and its potential consequences on post-AKI cardiovascular health and possibly even kidney cancer).57 In this context, drug interventions that would help to attenuate AKI-CKD transition although being initiated only once AKI had occurred could be a possible solution.
Limitations of this study include that the precise mechanisms by which HMGB1 promotes the demise of cycling tubular epithelial cells beyond its extracellular effects remain unclear. In addition, our in vitro assay probably included more tubular cell necrosis than what occurs in vivo during AKI-CKD transition because extracellular HMGB1 blockade with 2G7 significantly reduced tubular cell death in vitro, but 2G7 monotherapy had no effect on AKI-CKD transition in vivo.
In summary, HMGB1 has a role in AKI beyond danger signaling. Tubular HMGB1 impairs the intrinsic regenerative capacity of the kidney by increasing the susceptibility of cycling tubular cells to persistent oxidative stress. We therefore conclude that treatment of AKI could become feasible when (1) focusing on long-term outcomes of AKI, (2) targeting AKI-CKD transition with drugs initiated after the AKI peak, and (3) with drugs that block HMGB1 in intracellular and extracellular compartments.
Disclosures
J.A. Marschner reports Speakers Bureau: MediBeacon Inc. A. Linkermann reports Consultancy: Alexion, Genentech, and HBM; Research Funding: Apogenix, Else Kröner-Fresenius Foundation, Fresenius, Novartis, Pfizer, and Wilhelm Sander Foundation; Honoraria: Alexion, Genentech, and Novartis; and Patents or Royalties: patent for Nec-1f, a combined inhibitor of necroptosis and ferroptosis (no. 20160943.5). P. Enghard reports Consultancy: Advisory Board Glaxo Smith Klein GSK; Ownership Interest: Gilead Stocks; Honoraria: Akademie der Nieren, AstraZeneca, BDI, GlaxoSmithKline, and NAWBerlin; and Patents or Royalties: submitted several patents on urinary cells as biomarkers and of a solution for conserving urinary cells for subsequent analysis using flow cytometry. B.D. Humphreys reports Consultancy: Chinook Therapeutics, Janssen, and Pfizer; Ownership Interest: Chinook Therapeutics; Research Funding: Chinook Therapeutics, Janssen, and Pfizer; Honoraria: 10X Genomics; Patents or Royalties: Evotec, AG; and Advisory or Leadership Role: Seminars in Nephrology Editorial Board, Kidney International Editorial Board, JCI Insight Editorial Board, American Journal of Physiology Renal Physiology Editorial Board, RegMed XB, Regenerative Medicine Crossing Borders SAB, ASCI President-elect, Chinook Therapeutics SAB, and Board of Scientific Advisors NIDDK. P. Romagnani reports Research Funding: AstraZeneca. H. Anders reports Consultancy: AstraZeneca, Bayer, GSK, Janssen, Kezar, Novartis, Previpharma, Sanofi, and Vifor; Research Funding: Boehringer-Ingelheim; Honoraria: Lilly, and Otsuka; and Advisory or Leadership Role: JASN, and NDT. Because H.-J. Anders is an associate editor of the Journal of the American Society of Nephrology, he was not involved in the peer review process for this manuscript. A guest editor oversaw the peer review and decision-making process for this manuscript.
Funding
This work was funded by the Chinese Scholarship Council (award 201706170071) to Z.B. Zhao, Deutsche Forschungsgemeinschaft (grants AN372/14-4, AN372/30-1, CRC TRR332, project A7 to H.-J. Anders to H.J.A., INST 86/1851-1 FUGG), and the National Institute of Diabetes and Digestive and Kidney Diseases (grant UC2DK126024) to B.D. Humphreys.
Supplementary Material
ACKNOWLEDGMENTS
The expert technical support of Yvonne Minor, Jana Mandelbaum, and Anna Anfimiadou is gratefully acknowledged. Prof.MarthaMerrow and Dr. Francesca Sartor of the LMUgenerously provided access to and expertise regarding theNikon EclipseTi2 microscope system (funded by DFG GZ: INST 86/1851-1 FUGG).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Author Contributions
H.-J. Anders, A. Linkermann, J.A. Marschner, P. Romagnani, and Z.B. Zhao conceptualized the study; J.A. Marschner was responsible for data curation; H.-J. Anders, P. Enghard, B.D. Humphreys, J. Klocke, J.A. Marschner, Y. Muto, and Z.B. Zhao were responsible for formal analysis; P. Enghard, J. Klocke, J.A. Marschner, and Z.B. Zhao were responsible for visualization; J.A. Marschner and Z.B. Zhao were responsible for project administration; H.-J. Anders, J.A. Marschner, P. Romagnani, and Z.B. Zhao wrote the original draft; H.-J. Anders was responsible for funding acquisition; H. Erlandsson-Harris and A. Linkermann were responsible for resources; H.-J. Anders provided supervision; P. Enghard, H. Erlandsson-Harris, B.D. Humphreys, J. Klocke, A. Linkermann, J.A. Marschner, Y. Muto, B. Popper, and Z.B. Zhao were responsible for methodology; and B.D. Humphreys, T. Iwakura, M. Kuang, C. Li, J. Marschner, M. Motrapu, Y. Muto, and Z.B. Zhao were responsible for investigation.
Data Sharing Statement
All data used in this study are available in this article.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/A653.
Supplemental Table 1. Narcosis, antagonization, and analgesia.
Supplemental Table 2. Antibodies and reagents for immunohistochemical and immunofluorescence staining.
Supplemental Table 3. PCR reaction mixtures.
Supplemental Table 4. Primer pairs used for qPCR and genotyping.
Supplemental Table 5. Primary murine tubular cell culture medium composition.
Supplemental Table 6. Reagents, antibodies, and kits.
Supplemental Figure 1. Expression of HMGB1 receptors in murine kidneys after AKI.
Supplemental Figure 2. Expression of HMGB1 and its receptors in urine derived human cells.
Supplemental Figure 3. Ischemic necroinflammation followed by persistent hypoxia leads to kidney atrophy.
Supplemental Figure 4. Genetic depletion of Hmgb1 from tubules of adult mice shows no spontaneous phenotype.
Supplemental Figure 5. Tubular HMGB1 deficiency does not attenuate ischemic AKI but AKI/CKD transition.
Supplemental Figure 6. HMGB1 affects the susceptibility of tubular cells to prolonged oxidative stress.
Supplemental Figure 7. Delayed onset treatment with HMGB1 inhibitors ameliorates postischemic phenotypes.
Supplemental Video 1. Time-lapse images of the experiments conducted and presented in Supplemental Figure 6A.
Supplemental Video 2. Time-lapse images of the experiments conducted and presented in Figure 5A.
References
- 1.Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders H-J. Acute kidney injury. Nat Rev Dis Primers. 2021;7(1):52. doi: 10.1038/s41572-021-00284-z [DOI] [PubMed] [Google Scholar]
- 2.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371(1):58-66. doi: 10.1056/nejmra1214243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. 2015;26(8):1765-1776. doi: 10.1681/ASN.2015010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81(5):442-448. doi: 10.1038/ki.2011.379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka S, Tanaka T, Nangaku M. Hypoxia as a key player in the AKI-to-CKD transition. Am J Physiol Renal Physiol. 2014;307(11):F1187-F1195. doi: 10.1152/ajprenal.00425.2014 [DOI] [PubMed] [Google Scholar]
- 6.Bábíčková J, Klinkhammer BM, Buhl EM, et al. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017;91(1):70-85. doi: 10.1016/j.kint.2016.07.038 [DOI] [PubMed] [Google Scholar]
- 7.Ullah MM, Basile DP. Role of renal hypoxia in the progression from acute kidney injury to chronic kidney disease. Semin Nephrol. 2019;39(6):567-580. doi: 10.1016/j.semnephrol.2019.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mimura I, Nangaku M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol. 2010;6(11):667-678. doi: 10.1038/nrneph.2010.124 [DOI] [PubMed] [Google Scholar]
- 9.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799(1-2):101-113. doi: 10.1016/j.bbagrm.2009.09.008 [DOI] [PubMed] [Google Scholar]
- 10.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29(1):139-162. doi: 10.1146/annurev-immunol-030409-101323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Z, Hu Z, Zeng R, Yao Y. HMGB1 in kidney diseases. Life Sci. 2020;259:118203. doi: 10.1016/j.lfs.2020.118203 [DOI] [PubMed] [Google Scholar]
- 12.Miura K, Sahara H, Sekijima M, et al. Protective effect of neutralization of the extracellular high-mobility group box 1 on renal ischemia-reperfusion injury in miniature swine. Transplantation. 2014;98(9):937-943. doi: 10.1097/tp.0000000000000358 [DOI] [PubMed] [Google Scholar]
- 13.Li J, Gong Q, Zhong S, et al. Neutralization of the extracellular HMGB1 released by ischaemic damaged renal cells protects against renal ischaemia-reperfusion injury. Nephrol Dial Transplant. 2011;26(2):469-478. doi: 10.1093/ndt/gfq466 [DOI] [PubMed] [Google Scholar]
- 14.Wu H, Ma J, Wang P, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010;21(11):1878-1890. doi: 10.1681/ASN.2009101048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yanai H, Matsuda A, An J, et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci USA. 2013;110(51):20699-20704. doi: 10.1073/pnas.1320808110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H, Nace GW, McDonald K-A, et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology. 2014;59(5):1984-1997. doi: 10.1002/hep.26976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu P, Liu M, Zhang B, et al. Cardiomyocyte-restricted high-mobility group box 1 (HMGB1) deletion leads to small heart and glycolipid metabolic disorder through GR/PGC-1α signalling. Cell Death Discov. 2020;6:106. doi: 10.1038/s41420-020-00340-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulkarni OP, Hartter I, Mulay SR, et al. Toll-like receptor 4-induced IL-22 accelerates kidney regeneration. J Am Soc Nephrol. 2014;25(5):978-989. doi: 10.1681/ASN.2013050528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakazawa D, Kumar SV, Marschner J, et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol. 2017;28(6):1753-1768. doi: 10.1681/ASN.2016080925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salei N, Rambichler S, Salvermoser J, et al. The kidney contains ontogenetically distinct dendritic cell and macrophage subtypes throughout development that differ in their inflammatory properties. J Am Soc Nephrol. 2020;31(2):257-278. doi: 10.1681/ASN.2019040419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marschner JA, Schäfer H, Holderied A, Anders H-J. Optimizing mouse surgery with online rectal temperature monitoring and preoperative heat supply. Effects on post-ischemic acute kidney injury. PLoS One. 2016;11(2):e0149489. doi: 10.1371/journal.pone.0149489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreiber A, Shulhevich Y, Geraci S, et al. Transcutaneous measurement of renal function in conscious mice. Am J Physiology Renal Physiol. 2012;303(5):F783-F788. doi: 10.1152/ajprenal.00279.2012 [DOI] [PubMed] [Google Scholar]
- 23.Lech M, Avila-Ferrufino A, Allam R, et al. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J Immunol. 2009;183(6):4109-4118. doi: 10.4049/jimmunol.0900118 [DOI] [PubMed] [Google Scholar]
- 24.Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671-675. doi: 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulay SR, Thomasova D, Ryu M, Anders H-J. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int. 2012;81(12):1199-1211. doi: 10.1038/ki.2011.482 [DOI] [PubMed] [Google Scholar]
- 26.Kirita Y, Wu H, Uchimura K, Wilson PC, Humphreys BD. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci USA. 2020;117(27):15874-15883. doi: 10.1073/pnas.2005477117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muto Y, Wilson PC, Ledru N, et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat Commun. 2021;12(1):21902021. doi: 10.1038/s41467-021-22368-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573-3587.e29. doi: 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terryn S, Jouret F, Vandenabeele F, et al. A primary culture of mouse proximal tubular cells, established on collagen-coated membranes. Am J Physiol Renal Physiol. 2007;293(2):F476-F485. doi: 10.1152/ajprenal.00363.2006 [DOI] [PubMed] [Google Scholar]
- 30.Hagemann JH, Thomasova D, Mulay SR, Anders H-J. Nrf2 signalling promotes ex vivo tubular epithelial cell survival and regeneration via murine double minute (MDM)-2. Nephrol Dial Transplant. 2013;28(8):2028-2037. doi: 10.1093/ndt/gft037 [DOI] [PubMed] [Google Scholar]
- 31.Lech M, Anders H-J. Expression profiling by real-time quantitative polymerase chain reaction (RT-qPCR). Methods Mol Biol. 2014;1169:133-142. doi: 10.1007/978-1-4939-0882-0_13 [DOI] [PubMed] [Google Scholar]
- 32.Fu Y, Tang C, Cai J, Chen G, Zhang D, Dong Z. Rodent models of AKI-CKD transition. Am J Physiology Renal Physiol. 2018;315(4):F1098–F1106. doi: 10.1152/ajprenal.00199.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn WF. Enhanced recovery from postischemic acute renal failure. Micropuncture studies in the rat. Circ Res. 1980;46(3):440-448. doi: 10.1161/01.res.46.3.440 [DOI] [PubMed] [Google Scholar]
- 34.Lau A, Wang S, Liu W, Haig A, Zhang Z-X, Jevnikar AM. Glycyrrhizic acid ameliorates HMGB1-mediated cell death and inflammation after renal ischemia reperfusion injury. Am J Nephrol. 2014;40(1):84-95. doi: 10.1159/000364908 [DOI] [PubMed] [Google Scholar]
- 35.Chung KY, Park JJ, Kim YS. The role of high-mobility group box-1 in renal ischemia and reperfusion injury and the effect of ethyl pyruvate. Transplant Proc. 2008;40(7):2136-2138. doi: 10.1016/j.transproceed.2008.06.040 [DOI] [PubMed] [Google Scholar]
- 36.Rabadi MM, Ghaly T, Goligorksy MS, Ratliff BB. HMGB1 in renal ischemic injury. Am J Physiol Renal Physiol. 2012;303(6):F873-F885. doi: 10.1152/ajprenal.00092.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo MS, Kim HJ, Kim H, Park SW. Ethyl pyruvate directly attenuates active secretion of HMGB1 in proximal tubular cells via induction of heme oxygenase-1. J Clin Med. 2019;8(5):629. doi: 10.3390/jcm8050629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakamoto R, Okano M, Takena H, Ohtsuki K. Inhibitory effect of glycyrrhizin on the phosphorylation and DNA-binding abilities of high mobility group proteins 1 and 2 in vitro. Biol Pharm Bull. 2001;24(8):906-911. doi: 10.1248/bpb.24.906 [DOI] [PubMed] [Google Scholar]
- 39.Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881-892. doi: 10.1083/jcb.200911078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bouchard M, Souabni A, Busslinger M. Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis. 2004;38(3):105-109. doi: 10.1002/gene.20008 [DOI] [PubMed] [Google Scholar]
- 41.Schierbeck H, Lundbäck P, Palmblad K, et al. Monoclonal anti-HMGB1 (high mobility group box chromosomal protein1) antibody protection in two experimental arthritis models. Mol Med. 2011;17(9-10):1039-1044. doi: 10.2119/molmed.2010.00264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lundbäck P, Lea JD, Sowinska A, et al. A novel high mobility group box 1 neutralizing chimeric antibody attenuates drug-induced liver injury and postinjury inflammation in mice. Hepatology. 2016;64(5):1699-1710. doi: 10.1002/hep.28736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang J, Xia Y, Wasserloos K, et al. Cyclic stretch induced IL-33 production through HMGB1/TLR-4 signaling pathway in murine respiratory epithelial cells. PLoS One. 2017;12(9):e0184770. doi: 10.1371/journal.pone.0184770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JD, Liu N, Levin SC, et al. Therapeutic blockade of HMGB1 reduces early motor deficits, but not survival in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2019;16(1):45. doi: 10.1186/s12974-019-1435-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazzeri E, Angelotti ML, Peired A, et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat Commun. 2018;9(1):1344. doi: 10.1038/s41467-018-03753-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huebener P, Pradere J-P, Hernandez C, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125(2):539-550. doi: 10.1172/jci76887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Zhou H, Zheng H, et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation. Autophagy. 2021;17(2):529-552. doi: 10.1080/15548627.2020.1725381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulay SR, Linkermann A, Anders H-J. Necroinflammation in kidney disease. J Am Soc Nephrol. 2016;27(1):27-39. doi: 10.1681/ASN.2015040405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25(12):2689-2701. doi: 10.1681/ASN.2014030262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belavgeni A, Meyer C, Stumpf J, Hugo C, Linkermann A. Ferroptosis and necroptosis in the kidney. Cell Chem Biol. 2020;27(4):448-462. doi: 10.1016/j.chembiol.2020.03.016 [DOI] [PubMed] [Google Scholar]
- 51.Tonnus W, Meyer C, Steinebach C, et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun. 2021;12(1):4402. doi: 10.1038/s41467-021-24712-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders HJ. Immune system modulation of kidney regeneration—mechanisms and implications. Nat Rev Nephrol. 2014;10(6):347-358. doi: 10.1038/nrneph.2014.68 [DOI] [PubMed] [Google Scholar]
- 53.Lech M, Gröbmayr R, Ryu M, et al. Macrophage phenotype controls long-term AKI outcomes—kidney regeneration versus atrophy. J Am Soc Nephrol. 2014;25(2):292-304. doi: 10.1681/ASN.2013020152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Man SM, Kanneganti T-D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16(1):7-21. doi: 10.1038/nri.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin J-H, Lee H-K, Lee H-B, Jin Y, Lee J-K. Ethyl pyruvate inhibits HMGB1 phosphorylation and secretion in activated microglia and in the postischemic brain. Neurosci Lett. 2014;558:159-163. doi: 10.1016/j.neulet.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 56.Davé SH, Tilstra JS, Matsuoka K, et al. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J Leukoc Biol. 2009;86(3):633-643. doi: 10.1189/jlb.1008662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peired AJ, Lazzeri E, Guzzi F, Anders H-J, Romagnani P. From kidney injury to kidney cancer. Kidney Int. 2021;100(1):55-66. doi: 10.1016/j.kint.2021.03.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study are available in this article.