


Keywords: focal segmental glomerulosclerosis, genetic kidney disease, genetics and development, glomerular disease, glomerulonephritis, glomerulopathy
Abstract
Significance Statement
Pathogenic structural genetic variants, also known as genomic disorders, have been associated with pediatric CKD. This study extends those results across the lifespan, with genomic disorders enriched in both pediatric and adult patients compared with controls. In the Chronic Renal Insufficiency Cohort study, genomic disorders were also associated with lower serum Mg, lower educational performance, and a higher risk of death. A phenome-wide association study confirmed the link between kidney disease and genomic disorders in an unbiased way. Systematic detection of genomic disorders can provide a molecular diagnosis and refine prediction of risk and prognosis.
Background
Genomic disorders (GDs) are associated with many comorbid outcomes, including CKD. Identification of GDs has diagnostic utility.
Methods
We examined the prevalence of GDs among participants in the Chronic Kidney Disease in Children (CKiD) cohort II (n=248), Chronic Renal Insufficiency Cohort (CRIC) study (n=3375), Columbia University CKD Biobank (CU-CKD; n=1986), and the Family Investigation of Nephropathy and Diabetes (FIND; n=1318) compared with 30,746 controls. We also performed a phenome-wide association analysis (PheWAS) of GDs in the electronic MEdical Records and GEnomics (eMERGE; n=11,146) cohort.
Results
We found nine out of 248 (3.6%) CKiD II participants carried a GD, replicating prior findings in pediatric CKD. We also identified GDs in 72 out of 6679 (1.1%) adult patients with CKD in the CRIC, CU-CKD, and FIND cohorts, compared with 199 out of 30,746 (0.65%) GDs in controls (OR, 1.7; 95% CI, 1.3 to 2.2). Among adults with CKD, we found recurrent GDs at the 1q21.1, 16p11.2, 17q12, and 22q11.2 loci. The 17q12 GD (diagnostic of renal cyst and diabetes syndrome) was most frequent, present in 1:252 patients with CKD and diabetes. In the PheWAS, dialysis and neuropsychiatric phenotypes were the top associations with GDs. In CRIC participants, GDs were associated with lower serum magnesium, lower educational achievement, and higher mortality risk.
Conclusion
Undiagnosed GDs are detected both in children and adults with CKD. Identification of GDs in these patients can enable a precise genetic diagnosis, inform prognosis, and help stratify risk in clinical studies. GDs could also provide a molecular explanation for nephropathy and comorbidities, such as poorer neurocognition for a subset of patients.
Podcast
This article contains a podcast at https://dts.podtrac.com/redirect.mp3/www.asn-online.org/media/podcast/JASN/2023_04_03_JASN2022060725.mp3
Introduction
CKD afflicts 15% of adults in the United States, where dialysis accounts for close to 1% of the total federal spending. Genetic testing-based precise diagnosis and intervention can help prevent CKD progression to ESKD. Heritability of quantitative measures of kidney function, such as eGFR, has been estimated to be approximately 30%–60%,1,2 and 10%–29% of adult patients with ESKD report a positive family history.3,4 Sequencing studies have recently demonstrated that Mendelian forms of CKD constitute 10% of all adult patients with ESKD,5–7 and >70% of pediatric CKD.8,9 These data indicate that genetic factors play an important role in CKD, but a significant proportion of the genetic risk of CKD remains unexplained.
Copy number variants (CNVs) are a type of structural genetic variant defined as loss (deletions) or gain (duplications or higher) of genomic DNA ranging from kilobase to megabase in size, which contribute significantly to human genetic variation.10–12 In contrast with the more general term genetic disorders, genomic disorders (GDs) have been defined as diseases resulting from specific rare, recurrent, pathogenic CNVs,13 such as the archetypal duplication in chromosome 17p causing Charcot-Marie-tooth disease type 1A.14 GD can be used to refer both to the disease or syndrome itself and the underlying pathogenic CNV. GDs affect multiple systems and display neurologic, cardiac, and renal manifestations, among others.10,15–20 We previously demonstrated that large pathogenic CNVs are detected in 5%–10% of pediatric patients, with congenital defects of the kidney and urinary tract, but also other forms of CKD, such as nephronophthisis or cystinosis17,21–24; the detection of GDs can provide a unifying explanation for the association of kidney disease and associated comorbidities such as neurocognitive impairment25; and CNV analysis can help identify novel causal gene-disease associations.16,23,26 Recent studies in the UK Biobank, an adult cohort, have shown that rare pathogenic CNVs associate with multiple comorbidities and increase the risk of mortality.27 Here, we extend our pediatric CKD study, investigate the frequency of GDs in adults with CKD, and examine the spectrum of phenotypic associations.
Methods
Cohorts, Genotyping, and Sequencing
All aspects of this study involving human research participants adhered to the principles of the Declaration of Helsinki and the protocols were approved by the Institutional Review Boards of Columbia University Medical Center, and each participating recruitment site from the different studies described below. Except for the CKD-affected participants from the Columbia University Biobank, all other data were obtained for this study in anonymized, deidentified form.
The Chronic Kidney Disease in Children Study (CKiD) is a longitudinal observational study of children with all-cause CKD (eGFR between 30 and 90 ml/min per 1.73 m2).28 CKiD cohort II comprises children with mildly impaired kidney function (eGFR between 45 and 90 ml/min per 1.73 m2).29 We generated high-quality CNV calls for 248 CKiD II participants that were genotyped on Illumina Omni2.5 arrays.
The longitudinal Chronic Renal Insufficiency Cohort (CRIC) study recruited patients with nephropathy and with or without diabetes, with age-related entry criteria for eGFR. CRIC participants in the All Health Conditions consent group were genotyped on the Illumina Omni1 array.30 We generated high-quality CNV calls for 3375 participants. CRIC patients with CKD were compared with a large multiethnic dataset from a total of 24,765 convenience controls that we previously described.23,24 Briefly, of those 24,765 samples, 518 were genotyped on Affymetrix SNP6.0 arrays from the control set of an Irish study on vesicoureteral reflux31; 1890 on Illumina Hap550, from primary care and well-child clinic practices within the Children's Hospital of Philadelphia Health Care Network32; 4345 on Illumina 610/660w, from studies on Parkinson’s disease,33 hypertension34 and IgA nephropathy35 (controls only); 2371 on Illumina 1M, from a study on addiction36; 5384 on Illumina 1M-Duo, from studies on visceral adiposity and hypertension37; 6990 on Illumina Omni1, from studies on Parkinson’s disease,38 melanoma,39 and blood clotting40; and 3267 on Illumina MEGA1.0/1.1, which include unaffected controls from our biobank and participants from the Population Architecture using Genomics and Epidemiology Consortium.41 All Illumina arrays have a significant number of overlapping probes (>300,000 shared probes between microarray platforms in this study) and we performed all data processing in a standardized fashion, making all variants calls ab initio in the same way for both patients and controls.
Patients in CKiD were compared with the subset of 1890 pediatric controls from Children's Hospital of Philadelphia listed above.
The Family Investigation of Nephropathy and Diabetes (FIND) is a multiethnic, multicenter study designed to identify genetic determinants of diabetic nephropathy.42 Patients were selected if they met study criteria for diabetic nephropathy, or met inclusion criteria on the basis of elevated serum creatinine levels and abnormal urine protein excretion. Controls were long-term patients with diabetes with otherwise normal kidney function. CNV calls were generated simultaneously for 1318 patients in FIND and 1013 FIND controls genotyped on Affymetrix SNP6.0 arrays.42
The Columbia Chronic Kidney Disease cohort recruited patients with a CKD diagnosis on the basis of clinical parameters, and, whenever available, results of renal imaging studies or biopsies previously performed for clinical indications, following the Kidney Disease: Improving Global Outcomes guidelines. CNV calls for 1986 participants were on the basis of whole-exome sequencing (WES) data, obtained as described by Groopman et al.43 The WES control dataset consisted of 4968 unrelated individuals without kidney disease, sequenced at the Institute for Genomic Medicine, Columbia University.44
The electronic MEdical Records and GEnomics Network Phase I (eMERGE I) linked DNA specimens with electronic medical records from participants across five US centers, with the purpose of performing GWAS for common traits and developing phenotyping algorithms.45 We performed CNV analysis on 11,146 eMERGE I participants genotyped on Illumina 660W arrays, for whom both high-quality microarray intensity level data and International Classification of Diseases (ICD) 9 and 10 codes records were available.46,47 These eMERGE I participants were not selected on the basis of a CKD phenotype, but comprised a wide range of phenotypes, including CKD.
CNV Calling and Annotation
Microarray-based CNV calling was done using PennCNV (Illumina arrays), and Affymetrix Power Tools and PennCNV-Affy software (Affymetrix arrays). Only data samples that passed stringent quality control filters applied with Plink48 software and PennCNV49 were included in the CNV analysis, as previously described.23 WES-based CNV calling was performed using XHMM software.50 CNVs with PennCNV confidence scores <30 were excluded from the analysis.17 To annotate rare variants, we have developed a bioinformatics tool,51 which compares patient and control CNVs, precisely defines the degree of overlap, calculates their frequencies, and annotates them with RefSeq, OMIM, and manually curated sets of genes and loci. In particular, we surveyed CNV calls for GDs, defined as CNVs that had at least 70% overlap of their span with a set of known pathogenic CNVs’ coordinates obtained from the Database of Genomic Variation and Phenotype in Humans using Ensembl Resources52 and the literature,15,17,53,54 in agreement with American College of Medical Genetics guidelines.55,56
Statistical Analyses
Association of GDs with CKD
GD enrichment in patients with CKD was compared with controls for all cohorts combined and for each separate cohort, using Cochran-Mantel-Haenszel χ2 and Fisher’s exact tests, respectively.
Tests for Associations of GD Carrier Status with Baseline Phenotypic Variables in the CRIC Cohort
Categorical variables were tested using Fisher’s exact test. Continuous variables were tested with the nonparametric Wilcoxon test; Cohen’s d was also calculated to provide an effect size estimate as a standardized difference of phenotypic variables’ means between carriers and noncarriers. Tests with P<0.05, uncorrected for multiple testing, were considered evidence of “nominal” association.
Outcomes in the survival analyses in the CRIC cohort were defined as previously described by Yang et al.57 The survival and survminer R packages were used to generate Kaplan–Maier curves, perform multivariable Cox proportional hazards regression analyses, and test for independence between scaled Schoenfeld residuals and time.
The phenome-wide association study in the eMERGE cohort was performed using the PheWAS R package,58 testing for associations between GD carrier status and each of 1487 phenotype codes (phecodes), for which there were at least 20 “patients.” For a phecode to be assigned to a subject, at least two International Classification of Diseases codes59 had to support it. The phenotypic control groups for each phenotype are predefined for each phecode, in the package. Sex-specific phenotypes were restricted by sex. All phecodes were tested with logistic regression with each phecode patient-control status as an outcome and GD carrier status, adjusted for sex, and three principal components of ancestry as a predictor. For an association to be considered statistically significant, the total number of phecodes tested was considered (P<0.05/1487=3.36×10−5). All other statistical analyses were performed with R 3.6.3.
Results
High Burden of GDs among Children with CKD
We previously detected GDs in 19 out of 419 (4.5%) cohort I participants in the CKiD study.22 To follow up on these findings, we examined the prevalence of GD in 248 children from CKiD cohort II28,29 (ranging from 1 to 17 years of age; mean 11 years; Figure 1 and Supplemental Figure 1). We detected GD in nine out of 248 children from cohort II (3.6%) compared with 22 out of 1890 (1.2%) pediatric controls22,32 (odds ratio [OR], 3.19; 95% confidence interval [95% CI], 1.28 to 7.32, P=6.61×10−3). There were no significant differences in GD prevalence between CKID cohorts I and II. Combining cohorts I and II, 28 out of 667 (4.2%) CKiD participants carried a GD (OR, 3.72; 95% CI, 2.03 to 6.87, P=1.01×10−5, in comparison with pediatric controls). Recurrent GDs observed in both CKiD cohorts included deletions in chr17q12 (diagnostic of renal cyst and diabetes syndrome, RCAD) and homozygous deletions in chr2q13 (NPHP1) and chr17p13.13 (CTNS). In four of nine GD carriers in cohort II (Supplemental Table 1), the GD finding confirmed the clinical diagnosis (one patient with branchio-oto-renal syndrome, one with juvenile nephronophthisis, and two patients with cystinosis). In other patients, the GD reclassified the disease (NPHP1 deletion in a child diagnosed with reflux nephropathy) or provided a precise molecular explanation for the clinical diagnosis (RCAD syndrome in patients with dysplastic or hypoplastic kidneys), whereas in the remaining two patients, the GD, although not incompatible with the patients’ diagnoses, would have warranted further clinical assessment.
Figure 1.
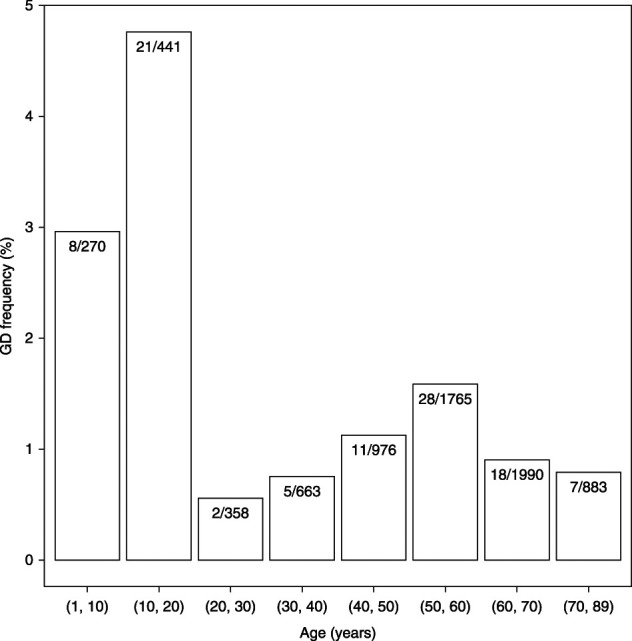
GD in CKD across the lifespan. GD frequency (n GD carriers/n subjects×100) distribution by age (years) of enrollment among patients with CKD in the combined CKiD I and II, FIND, CRIC, and CU-CKD cohorts (n=7346). The youngest and oldest GD carriers were 2 and 78 years old, respectively.
GDs in Adult CKD
We next analyzed CNVs in 6679 adult CKD patients, ranging from 18 to 89 years of age (mean age=55 years; Figure 1); from three multiethnic US studies, the CRIC cohort60,61 (n=3375; mean age=58 years), the FIND42 study cohort (n=1318; mean age=61 years), and the Columbia University Chronic Kidney Disease Biobank cohort43 (CU-CKD; n=1986; mean age=46 years). We compared these adults with CKD with a total of 30,746 controls (Supplemental Figure 1). In all, 72 (1.1%) out of 6679 patients with CKD carried a known GD compared with 199 out of 30,746 (0.7%) of controls (Cochran–Mantel–Haenszel χ2 test stratified by cohort: OR, 1.66; 95% CI, 1.25 to 2.20, P=3.40×10−4; Table 1). Recurrent GD comprised pathogenic CNV in the 1q21.1, 16p11.2, 17q12, and 22q11.2 loci. Among individual CNV, the 17q12 deletion/duplication (RCAD) was the most frequent, with nine deletions and seven duplications (0.2% of patients) compared with seven duplications in controls (0.02%) (OR, 10.57; 95% CI, 4.11 to 30.40, P=6.92×10−8). Notably, the 17q12 deletion/duplication was detected in 1:252 patients with CKD and diabetes. Although there were no statistically significant differences in GD burden between CKD cohorts (P>0.05), subcohort analyses demonstrated that the GD signal was mostly driven from the larger CRIC cohort (Supplemental Note, Supplemental Figure 1). Finally, in the CU-CKD cohort, which had exome sequence data available,43 GDs were detected only in patients without a previously known monogenic kidney disorder, indicating that CNV detection increased the diagnostic yield of exome sequencing.
Table 1.
Genomic disorders in adult patients with CKD and controls
| Chromosome | Start (Mb, hg19) | End (Mb, g19) | Genomic Disorder | Total Number of Carriers | |
|---|---|---|---|---|---|
| Patients with CKD (n=6679) | Controls (n=30,746) | ||||
| All GD (%) | 72 (1.08) | 199 (0.65) | |||
| 1 | 0.01 | 12.84 | 1p36 duplication | 1 (0.015) | 1 (0.0033) |
| 1 | 146.53 | 147.88 | 1q21.1 recurrent microduplication | 2 (0.030) | 12 (0.039) |
| 1 | 146.53 | 147.88 | 1q21.1 recurrent microdeletion | 0 (0.00) | 5 (0.016) |
| 1 | 145.39 | 145.75 | 1q21.1 TAR Syndrome deletion | 3 (0.045) | 10 (0.033) |
| 1 | 145.39 | 145.75 | 1q21.1 TAR Syndrome region duplication | 4 (0.060) | 12 (0.039) |
| 2 | 96.73 | 97.68 | 2q11.2 deletion | 1 (0.015) | 1 (0.0033) |
| 2 | 96.73 | 97.68 | 2q11.2 duplication | 1 (0.015) | 1 (0.0033) |
| 2 | 100.69 | 108.44 | 2q11.2-q13 duplication | 1 (0.015) | 0 (0.00) |
| 5 | 0.06 | 10.91 | 5p distal duplication | 1 (0.015) | 0 (0.00) |
| 5 | 112.04 | 112.18 | 5q21.3-q22.3 deletion | 1 (0.015) | 1 (0.0033) |
| 7 | 6.86 | 7.30 | 7p22.1 interstitial duplication | 1 (0.015) | 2 (0.0065) |
| 9 | 14.82 | 14.99 | 9p22.3 deletion | 1 (0.015) | 1 (0.0033) |
| 10 | 81.90 | 88.80 | 10q23 duplication | 1 (0.015) | 0 (0.00) |
| 15 | 31.13 | 32.48 | 15q13.3 duplication | 1 (0.015) | 12 (0.039) |
| 15 | 30.91 | 32.45 | 15q13.3 microdeletion syndrome | 2 (0.030) | 3 (0.0098) |
| 16 | 15.48 | 16.30 | 16p13.11 recurrent microdeletion | 2 (0.030) | 4 (0.013) |
| 16 | 15.48 | 16.30 | 16p13.11 recurrent microduplication | 11 (0.16) | 43 (0.14) |
| 16 | 29.65 | 30.20 | 16p11.2 deletion | 3 (0.045) | 6 (0.020) |
| 16 | 29.65 | 30.20 | 16p11.2 duplication | 0 (0.00) | 1 (0.0033) |
| 17 | 14.10 | 15.47 | CMT1A duplication | 3 (0.045) | 10 (0.033) |
| 17 | 14.10 | 15.47 | HNPP deletion | 3 (0.045) | 16 (0.052) |
| 17 | 34.82 | 36.21 | 17q12 RCAD syndrome deletion | 9 (0.13) | 0 (0.00) |
| 17 | 34.82 | 36.21 | 17q12 RCAD syndrome region duplication | 7 (0.10) | 7 (0.023) |
| 17 | 57.66 | 58.08 | 17q23 deletion | 1 (0.015) | 0 (0.00) |
| 21 | 10.70 | 48.12 | Down syndrome | 1 (0.015) | 0 (0.00) |
| 22 | 18.89 | 23.65 | 22q11.21 deletion | 2 (0.030) | 4 (0.013) |
| 22 | 18.89 | 21.47 | 22q11.21 duplication | 3 (0.045) | 7 (0.023) |
| X | 107.68 | 107.94 | COL4A5 hemizygous deletion | 1 (0.015) | 0 (0.00) |
| X | 153.29 | 153.36 | Xq28 heterozygous deletion (Rett syndrome) | 1 (0.015) | 1 (0.0033) |
| X | 6.46 | 8.13 | Xp22.31 hemizygous deletion (STS) | 1 (0.015) | 3 (0.0098) |
| X | 0.00 | 155.27 | triple X | 5 (0.075) | 2 (0.0065) |
| other GD | 0 (0.00) | 35 (0.11) | |||
Genomic coordinates on the basis of UCSC genome build hg19 of all known GD found in patients with CKD are listed, together with GD carriers counts (and %) for patients with CKD and controls. Counts of GD found in controls only (other GD) are also shown. Counts of all GD carriers are smaller than the sum of individual GD carriers counts because two patients and one control carried two GDs each. GD, genomic disorder; TAR, thrombocytopenia-absent radius syndrome; COL4A5, collagen type IV alpha 5 chain gene; CMT1A, Charcot-Marie-tooth disease type 1A; HNPP, hereditary neuropathy with pressure palsies; STS, steroid sulfatase deficiency.
Phenotypic Associations in the CRIC and eMERGE studies
We first tested for univariate associations of GDs with clinical variables in the CRIC cohort. GD carrier status among CRIC participants was nominally associated with lower serum magnesium (Mg; Cohen’s d = −0.62; 95% CI, −0.92 to −0.33; Wilcoxon test P=2.22×10−3; Figure 2A), with 47% of GD carriers in the first quartile of serum Mg concentration (≤1.8 mg/dl). In agreement with our previous findings in pediatric CKD,25 GDs were also associated with lower performance in the Modified Mini Mental State Exam (d=−0.58, 95% CI, −0.88 to −0.29; P=1.42×10−2; Figure 2B) and low educational achievement (OR, 2.88; 95% CI, 1.48 to 5.88, Fisher’s exact test P=7.50×10−4; Figure 2C). Among diabetic participants, GD were also associated with lower Mg (d=−1.11; 95% CI, −1.53 to −0.68; P=8.52×10−5). Of note, the chr17q12 deletion/duplication was detected in nine CRIC participants and was associated with diabetes (eight out of nine carriers, OR, 8.45; 95% CI, 1.13 to 374.73, P=1.85×10−2) and insulin therapy (seven out of nine carriers, OR, 10.49; 95% CI, 1.99 to 103.78, P=1.37×10−3), and six out of nine of carriers were in the first quartile of serum Mg concentration. After excluding the nine chr17q12 CNV carriers, GDs were still nominally associated with lower serum Mg only among CRIC participants with diabetes (d=−0.78; 95% CI, −0.25 to −1.30; P=1.13×10−2). Moreover, Mg level was also nominally lower in 22q11.2 CNV carriers among Black CRIC participants (d=−1.11, 95% CI, −2.09 to −0.13; P=1.16×10−2). All GD nominal associations found in CRIC are shown in Supplemental Table 2. Next, we performed survival analyses in CRIC to test associations with all-cause mortality and kidney failure outcomes. Multivariable-adjusted Cox proportional hazards analysis showed GD carrier status was associated with a higher risk of death (GD carrier status hazard ratio, 1.70; 95% CI, 1.03 to 2.75, P=3.11×10−2; Figure 2D; Supplemental Table 3), but not of ESKD or halving of eGFR.
Figure 2.

GD phenotypic associations in the CRIC cohort. Lower serum Mg++ concentrations (A) (P=2.22×10−3), lower Modified Mini Mental State Exam scores. (B) (P=1.42×10−2), a smaller proportion of higher education level. (C) (P=7.50×10−4), and a higher rate of death. (D) (P=3.11×10−2) in GD carriers compared with noncarriers in the CRIC cohort (n=3375).
Finally, we searched for phenotypic associations for GD carrier status in an independent cohort. We conducted a phenome-wide association (PheWAS) study in 11,146 participants of the eMERGE Network46,47 (Figure 3), for which microarray intensity level data were available. These eMERGE participants comprise patients with a wide range of phenotypes, including CKD. The eMERGE cohort was not part of our patient-control CKD analysis, and we did not select eMERGE participants on the basis of a CKD phenotype. Altogether, we identified GDs in 89 (0.8%) of eMERGE cohort participants, with recurrent CNVs detected at the 1q21.1, 16p13.11, 22q11.21, 16p11.2, and 17p12 loci (Supplemental Table 4). Among 1487 phenotypes surveyed, we found that dialysis was the top phenome-wide significant phenotypic association with GD carrier status (OR, 6.90; 95% CI, 2.80 to 14.66, P=3.43×10−6), followed by borderline personality disorders (OR, 12.06; 95% CI, 3.43 to 32.82, P=9.28×10−6) and congenital anomalies of skin (OR, 9.02; 95% CI, 3.06 to 21.39, P=6.07×10−6; Supplemental Table 5). The associations with dialysis and personality disorders were supported by other nominal kidney and neuropsychiatric related associations, such as renal osteodystrophy (OR, 6.16; 95% CI, 1.83 to 15.63, P=6.53×10−4), type 2 diabetes with renal manifestations (OR, 2.92; 95% CI, 1.49 to 5.37, P=9.45×10−4), ESKD (OR, 3.60; 95% CI, 1.23 to 8.39, P=7.52×10−3), schizophrenia (OR, 9.78; 95% CI, 2.28 to 28.85, P=2.63×10−4), suicidal ideation (OR, 8.02; 95% CI, 1.27 to 28.03, P=5.48×10−3), and epilepsy (OR, 2.60; 95% CI, 1.24 to 4.91, P=5.75×10−3).
Figure 3.

GDs pheWAS in eMERGE. Manhattan plot of association tests between GD carrier status and 1487 phenotypes in 11,146 eMERGE participants. The −log(P value) as a function of phenotypic code is shown. Only top signals are annotated.
Discussion
GDs are associated with multiple developmental disorders such as neuropsychiatric disease, congenital heart disease, and congenital defects of the kidney and urinary tract, but often remain undiagnosed.15–19,21,24,25 We examined the prevalence of GDs among children and adults with CKD and found significant enrichment across both pediatric and adult age groups. The youngest and oldest GD carriers in this study were 2 and 78 years of age, respectively, indicating that GDs can be encountered across the lifespan. In agreement with prior reports,17,21–23 GDs were more prevalent in children with CKD, detected in 4% of patients. The frequency of GDs in adults was lower (Figure 1), likely reflecting the health burden conferred by GDs, which leads to attrition among adults. We could speculate that the lower numbers of adult carriers in research studies is attributable to higher mortality in childhood, and/or reduced enrollment bias in adulthood, due to increased risk of neuropsychiatric disease. Consistent with this hypothesis, we found that GDs were associated with higher mortality in the CRIC study, even after adjusting for demographic and other clinical risk factors, replicating recent data from the UK Biobank.27 We also replicated prior associations with impaired cognition, which can reduce the ability to participate in clinical studies. Because GDs are associated with multiple comorbidities,15–19,24,25,62–66 their identification can provide an alternative explanation for outcomes that are often causally attributed to CKD, such as metabolic defects or neurocognitive performance.25 Systematic detection of GDs can thus provide a precise genetic diagnosis in both adults and children, enable risk stratification, and inform associations with multiple comorbidities.
Although we have made every effort to harmonize different datasets, pooling together data from different genotyping and sequencing platforms in an approach that we and others have used successfully15,23,67; this is, nevertheless, a limitation of our study. Another limitation is that to establish causality for specific GD whose link to CKD has not been previously proven would require much larger datasets, and confirmation with animal models and functional studies.
Recent studies have shown that massive parallel sequencing modalities such as exome sequencing provide a 10%–20% diagnostic yield in patients with CKD.43,68,69 Our data suggest that GDs can explain approximately 4% of pediatric and approximately 1% of adult patients. Thus, as demonstrated in the analysis of the Columbia cohort, a search for pathogenic CNVs may improve the diagnostic yield of genetic testing in patients where no pathogenic SNVs have been detected by massive parallel sequencing, and there is still a suspicion of genetic disease. Because large CNVs are usually associated with kidney and urologic malformations and extrarenal features,16,23–25,63,70 a search for GDs should be specially considered in patients with syndromic presentations. We note that deletions and duplications at the 17q12 locus, which have been associated with RCAD,63,71 were the most frequently identified GDs, detected in 1:252 of adults with CKD and diabetes. As expected, these patients also had a high prevalence of hypomagnesemia. These data suggest that nephrologists should become aware of the common clinical manifestations of RCAD and have a high index of suspicion among patients with diabetes with CKD, particularly in the presence of hypomagnesemia or other extrarenal features.
We used multiple approaches, including patient-control association studies and PheWAS to confirm phenotypic associations of GDs with CKD and other phenotypes. In particular, analysis of the CRIC and eMERGE cohorts confirmed prior reported association of GDs with neuropsychiatric disease.25,64–66,72 Patients with CKD are at higher risk of impaired neurocognition and depression, often postulated to be due to the metabolic derangements, physical decline, or medication side effects.73–77 Here we show that in a subset of patients with CKD, the neuropsychiatric symptoms may be due to an underlying genetic syndrome that can also explain CKD. In addition, although some of the GDs may not be directly causal of CKD, they still explain some of the comorbidities, such as neuropathy for carriers of CMT1A duplications or HNPP deletions. These data further demonstrate the utility of genetic testing for clarifying the causal link between CKD and prevalent comorbidities.
Although GDs have been associated with many medical conditions, they also have variable penetrance and are detected in 0.2%–0.5% of control populations.78,79 Because GDs have overall low population frequencies, association studies of GDs are affected by cohort size, recruitment criteria (such as diabetic status), and detection method. The clinical manifestations in adulthood may also be quite variable or even subclinical. For example, screening for CNVs in Estonian and Icelandic populations has shown evidence of mild, but statistically significant, impaired neurocognition in GD carriers.78,79 However, a recent analysis of the UK Biobank has shown a broad spectrum of clinical associations with GD, including CKD and mortality.10,27 The large sample size enabled analysis of individual CNVs with various phenotype, enabling detection of the association of 17q12 and 16p11.2 CNVs with renal failure. Although we clearly observed a statistically significant enrichment of 17q12 CNVs in adults with CKD, we did not detect any of the other individual GD associations reported in the UK Biobank study, likely due to comparatively reduced power among our adult populations. These data emphasize the need for a large sample size to obtain a precise estimate of GD prevalence among patients with CKD and better define the association of individual GDs with CKD and their comorbidities.
Supplementary Material
ACKNOWLEDGMENTS
We thank all participants and investigators of the CKiD, CRIC, FIND, eMERGE, and CU-CKD studies. This manuscript does not necessarily reflect the opinions or views of the CKiD study, the NIDDK Central Repository, or the NIDDK. Computing resources were provided, in part, by the Wake Forest School of Medicine Center for Public Health Genomics. FIND funders had no role in study design, data collection and analysis, decision to publish, or preparation of this manuscript. Dr. Robert P. Igo Jr. passed away in July 2020, before the submission of this paper. He was responsible for raw data processing of Affymetrix SNP6.0 genotyping for the FIND study, worked with Dr. Miguel Verbitsky and Dr. Sudha K. Iyengar in CNV calling in this cohort, and provided invaluable guidance in the analysis of this dataset.
Footnotes
Deceased
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Kidney Genetics: Continuing Discoveries and a Roadmap to the Clinic,” on pages 519–520.
Disclosures
A.G. Gharavi reports having consultancy agreements with Actio Biosciences, AstraZeneca Center for Genomics Research, Goldfinch Bio, Natera, Novartis, and Travere; reports having an ownership interest in Actio Biosciences; reports receiving research funding from Natera and the Renal Research Institute; reports receiving honoraria from Alnylam and Sanofi; and reports having an advisory or leadership role with the editorial boards of JASN and the Journal of Nephrology. A. Parsa reports having an advisory or leadership role with the National Institutes of Health (NIH). B. Levy reports having consultancy agreements with Igenomix, Myome, Myriad, and Natera; reports having an ownership interest in Natera; and reports having an advisory or leadership role with American College of Medical Genetics Foundation, College of American Pathologists Cytogenetics committee, Cancer Genomics Consortium, International Society for Prenatal Diagnosis, and Wiley Prenatal Diagnosis. B.A. Warady reports having consultancy agreements with Amgen, Bayer, Lightline Medical Reata, Relypsa, and UpToDate; reports receiving research funding from Baxter Healthcare; reports receiving honoraria from Amgen, Bayer, Reata, Relypsa, and UpToDate; and reports having an advisory or leadership role with Midwest Transplant Network Governing Board, National Kidney Foundation, North American Pediatric Renal Trials and Collaborative Studies, and NTDS Board of Directors. C.S. Wong reports having an advisory or leadership role with the Steering Committee for National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)–funded CKiD study, participated in the past year on the ad hoc advisory committee to the National Kidney Foundation, board of the New Mexico Pediatric Society in Albuquerque, NM; and reports having other interests or relationships with a contract with NephCure International as part of the CurGN study (participating site). D.R. Crosslin reports having consultancy agreements with UnitedHealth Group; reports having an ownership interest in SoundBiology LLC; reports receiving research funding from UnitedHealth Group; and reports having an advisory or leadership role with the UnitedHealth Group. A.Khan received funding from NIH National Center for Advancing Translational Sciences grant UL1TR001873 and NIH NIDDK grant K25DK128563. E.E. Kenny reports having consultancy agreements with and Forsite Labs Inc. and Galatea Inc.; reports having an ownership interest in Galatea Inc, and Forsite Labs Inc.; reports receiving honoraria from 23&Me, Illumina Inc., and Regeneron Pharmaceuticals; reports having an advisory or leadership role with the editorial boards of American Journal of Human Genetics, Cell Genomics, Frontiers in Ecology and Evolution, and G3; reports being on the Advisory Boards of the American Society of Human Genetics (un-paid), Forsite Labs Inc. (paid), and Galatea Inc., Chair of Science Foundation Ireland Center for Research Training in Genomics Data Science Board (un-paid); and reports receiving speakers bureau from 23&Me, Illumina Inc, and Regeneron, Inc. H.I. Feldman reports having consultancy agreements with Kyowa Hakko Kirin and the National Kidney Foundation; reports receiving honoraria from InMed Inc.; reports having an advisory or leadership role as the American Journal of Kidney Disease Editor in Chief, Member of Advisory Board of the National Kidney Foundation, and Steering Committee Chair of the CRIC Study; and reports receiving speakers bureau from InMed, Inc. K. Kiryluk reports having consultancy agreements with Calvariate, and HiBio; and reports receiving research funding from Aevi Genomics, AstraZeneca, Bioporto, Vanda, and Visterra. Mr. Pavan Khosla reports previous employment with Basepair Inc., and reports having an ownership interest in BioNano Genomics Inc., Canopy Growth Corp., GrowGeneration Corp., GSK PLC (formerly GlaxoSmithKline PLC), Pfizer Inc., Pacific Biosciences of California Inc., Nokia Oyj., and 10X Genomics Inc. R.J.F. Loos reports having consultancy agreements with Regeneron; and reports having an advisory or leadership role as Board member of the European Association of the Study of Diabetes. Dr. Simone Sanna-Cherchi reports receiving research funding from NIH/NIDDK, US Department of Defense; reports receiving honoraria from Travere Therapeutics; and reports having an advisory or leadership role with the editorial boards, with no royalties received, from Acta Biomedica, Kidney International, Journal of Nephrology, and Negative Results. Y. Shen reports having an advisory or leadership role with the Scientific Reports. All remaining authors have nothing to disclose. Funding for the CRIC study was obtained under a cooperative agreement from the NIDDK through grants U01DK060990, U01DK060984, U01DK061022, U01DK061021, U01DK061028, U01DK060980, U01DK060963, U01DK060902, and U24DK060990). The eMERGE Network was initiated and funded by National Human Genome Research Institute through grants: U01HG006828 (Cincinnati Children's Hospital Medical Center and Boston Children's Hospital), U01HG006830 (Children's Hospital of Philadelphia), U01HG006389 (Essentia Institute of Rural Health, Marshfield Clinic Research Foundation, and Pennsylvania State University), U01HG006382 (Geisinger Clinic), U01HG006375 (Group Health Cooperative and the University of Washington), U01HG006379 (Mayo Clinic), U01HG006380 (Icahn School of Medicine at Mount Sinai), U01HG006388 (Northwestern University), U01HG006378 (Vanderbilt University Medical Center), and U01HG006385 (Vanderbilt University Medical Center serving as the coordinating center). Participation of Columbia University in the eMERGE network was supported by National Human Genome Research Institute grant U01HG008680 (to C. Weng, A.G. Gharavi, and G. Hripcsak). The CKiD study was conducted by the CKiD Investigators and supported by the NIDDK, with additional funding from the National Institute of Child Health and Human Development, and the National Heart, Lung, and Blood Institute (U01-DK-66143, U01-DK-66174, U01DK-082194, U01-DK-66116). The data and biospecimens from the CKiD study reported here were supplied by the NIDDK Central Repository. The FIND study was supported by grants U01DK57292, U01DK57329, U01DK057300, U01DK057298, U01DK057249, U01DK57295, U01DK070657, U01DK057303, and U01DK57304 from the NIDDK and, in part, by the Intramural Research Program of the NIDDK; support was also received from the National Heart, Lung and Blood Institute grants U01HL065520, U01HL041654, and U01HL041652; this project has been funded in whole or in part with federal funds from the National Cancer Institute, NIH, under contract N01-CO-12400 and the Intramural Research Program of the National Cancer Institute, Center for Cancer Research; this work was also supported by the National Center for Research Resources for the General Clinical Research Center grants: Case Western Reserve University, M01-RR-000080, Wake Forest University, M01-RR-07122, Harbor-University of California, Los Angeles Medical Center, M01-RR-00425, College of Medicine, University of California, Irvine, M01-RR-00827–29, University of New Mexico, HSC M01-RR-00997, and Frederic C. Bartter, M01-RR-01346. Because A.G. Gharavi is an editor of the Journal of the American Society of Nephrology, he was not involved in the peer review process for this manuscript. A guest editor oversaw the peer review and decision-making process for this manuscript.
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases grants R01-DK080099 and U54-DK104309 (to Dr. Ali G. Gharavi) and R21-DK119802 (to Dr. Miguel Verbitsky).
Author Contributions
H.I. Feldman, A.G. Gharavi, I. Ionita-Laza, K. Kiryluk, S. Sanna-Cherchi, and M. Verbitsky conceptualized the study; D. Hughes, A.G. Gharavi, J.T. Glessner, E.E. Kenny, R.P. Igo, S.K. Iyengar, A. Khan, P. Khosla, P. Krithivasan, T.Y. Lim, M. Marasa, S. Sanna-Cherchi, N. Shang, N. Vena, M. Verbitsky, and J. Zhang were responsible for the data curation; D. Hughes, R.P. Igo, S.K. Iyengar, A. Khan, P. Khosla, S. Krishnamurthy, P. Krithivasan, T.Y. Lim, S. Sanna-Cherchi, N. Vena, and M. Verbitsky were responsible for the formal analysis; A.G. Gharavi and M. Verbitsky were responsible for the funding acquisition; H.I. Feldman, A.G. Gharavi, S.K. Iyengar, S. Sanna-Cherchi, M. Verbitsky, and C.S. Wong were responsible for the investigation; I. Ionita-Laza, K. Kiryluk, S. Sanna-Cherchi, and M. Verbitsky were responsible for the methodology; A.G. Gharavi and M. Verbitsky were responsible for the project administration; D.R. Crosslin, H.I. Feldman, S. Furth, J.T. Glessner, A.G. Gharavi, H. Hakonarson, G. Hripcsak, R.P. Igo, S.K. Iyengar, E.E. Kenny, K. Kiryluk, B. Levy, R.J.F. Loos, A. Parsa, S. Sanna-Cherchi, N. Shang, Y. Shen, B.A. Warady, C. Weng, and C.S. Wong were responsible for the resources; A.G. Gharavi and M. Verbitsky provided supervision; B. Levy was responsible for the validation; P. Khosla and M. Verbitsky were responsible for the visualization; A.G. Gharavi, K. Kiryluk, and M. Verbitsky wrote the original draft; D.R. Crosslin, H.I. Feldman, H. Hakonarson, G. Hripcsak, I. Ionita-Laza, S.K. Iyengar, E.E. Kenny, K. Kiryluk, A. Khan, P. Khosla, S. Krishnamurthy, P. Krithivasan, T.Y. Lim, R.J.F. Loos, M. Marasa, A. Parsa, S. Sanna-Cherchi, N. Vena, B.A. Warady, C. Weng, C.S. Wong, and J. Zhang reviewed and edited the manuscript.
Data Sharing Statement
Raw data that support the findings of these study are available from the corresponding authors upon reasonable request. The data are also available on dbGaP (https://www.ncbi.nlm.nih.gov/gap) under the following accession numbers: phs000524.v1.p1, phs000333.v1.p1, phs000360.v3.p1, phs000650.v3.p1, phs000304.v1.p1, phs000169.v1.p1, phs000092.v1.p1, phs000199.v1.p1, phs000431.v1.p1, phs000356.v2.p1, phs001584.v1.p1 (and other, pending). Some restrictions may apply according to participants’ consent and privacy protection.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/D760.
Supplemental Table 1. Known GD in cohort 2 of the CKiD study.
Supplemental Table 2. GD phenotypic associations in the CRIC cohort.
Supplemental Table 3. Multivariable proportional hazards model for death in the CRIC cohort.
Supplemental Table 4. Known GD in the eMERGE cohort.
Supplemental Table 5. GD PheWAS in the eMERGE cohort.
Supplemental Figure 1. Overall study design.
References
- 1.Arpegård J, Viktorin A, Chang Z, de Faire U, Magnusson PK, Svensson P: Comparison of heritability of Cystatin C- and creatinine-based estimates of kidney function and their relation to heritability of cardiovascular disease. J Am Heart Assoc 4: e001467, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fox CS, Yang Q, Cupples LA, Guo CY, Larson MG, Leip EP, et al. : Genomewide linkage analysis to serum creatinine, GFR, and creatinine clearance in a community-based population: The Framingham Heart Study. J Am Soc Nephrol 15: 2457–2461, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Connaughton DM, Bukhari S, Conlon P, Cassidy E, O’Toole M, Mohamad M, et al. : The Irish Kidney Gene Project--Prevalence of family history in patients with kidney disease in Ireland. Nephron 130: 293–301, 2015 [DOI] [PubMed] [Google Scholar]
- 4.McClellan WM, Satko SG, Gladstone E, Krisher JO, Narva AS, Freedman BI: Individuals with a family history of ESRD are a high-risk population for CKD: Implications for targeted surveillance and intervention activities. Am J Kidney Dis 53[Suppl 3]: S100–S106, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Devuyst O Knoers NV Remuzzi G Schaefer F; Board of the Working Group for Inherited Kidney Diseases of the European Renal Association and European Dialysis and Transplant Association : Rare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 383: 1844–1859, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. : Chronic kidney disease: Global dimension and perspectives. Lancet 382: 260–272, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Wühl E, van Stralen KJ, Wanner C, Ariceta G, Heaf JG, Bjerre AK, et al. : Renal replacement therapy for rare diseases affecting the kidney: An analysis of the ERA-EDTA Registry. Nephrol Dial Transplant 29[Suppl 4]: iv1–iv8, 2014 [DOI] [PubMed] [Google Scholar]
- 8.Ingelfinger JR Kalantar-Zadeh K Schaefer F World Kidney Day Steering C; World Kidney Day Steering Committee : World Kidney Day 2016: Averting the legacy of kidney disease-focus on childhood. Pediatr Nephrol 31: 343–348, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Vivante A, Hildebrandt F: Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12: 133–146, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguirre M, Rivas MA, Priest J: Phenome-wide burden of copy-number variation in the UK Biobank. Am J Hum Genet 105: 373–383, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. : Global variation in copy number in the human genome. Nature 444: 444–454, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sudmant PH Rausch T Gardner EJ Handsaker RE Abyzov A Huddleston J et al. ; 1000 Genomes Project Consortium : An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lupski JR: Genomic disorders ten years on. Genome Med 1: 42, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. : DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66: 219–232, 1991 [DOI] [PubMed] [Google Scholar]
- 15.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. : A copy number variation morbidity map of developmental delay. Nat Genet 43: 838–846, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Rivera E, Liu YP, Verbitsky M, Anderson BR, Capone VP, Otto EA, et al. : Genetic drivers of kidney defects in the DiGeorge Syndrome. N Engl J Med 376: 742–754, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, et al. : Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 91: 987–997, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, et al. : Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet 8: e1002843, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Duyvenvoorde HA, Lui JC, Kant SG, Oostdijk W, Gijsbers AC, Hoffer MJ, et al. : Copy number variants in patients with short stature. Eur J Hum Genet 22: 602–609: 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westland R, Bodria M, Carrea A, Lata S, Scolari F, Fremeaux-Bacchi V, et al. : Phenotypic expansion of DGKE-associated diseases. J Am Soc Nephrol 25: 1408–1414, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westland R, Verbitsky M, Vukojevic K, Perry BJ, Fasel DA, Zwijnenburg PJ, et al. : Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int 88: 1402–1410, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verbitsky M, Sanna-Cherchi S, Fasel DA, Levy B, Kiryluk K, Wuttke M, et al. : Genomic imbalances in pediatric patients with chronic kidney disease. J Clin Invest 125: 2171–2178, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verbitsky M, Westland R, Perez A, Kiryluk K, Liu Q, Krithivasan P, et al. : The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet 51: 117–127, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verbitsky M, Krithivasan P, Batourina E, Khan A, Graham SE, Marasà M, et al. : Copy number variant analysis and genome-wide association study identify loci with large effect for vesicoureteral reflux. J Am Soc Nephrol 32: 805–820, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verbitsky M, Kogon AJ, Matheson M, Hooper SR, Wong CS, Warady BA, et al. : Genomic disorders and neurocognitive impairment in pediatric CKD. J Am Soc Nephrol 28: 2303–2309, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vivante A, Kleppa MJ, Schulz J, Kohl S, Sharma A, Chen J, et al. : Mutations in TBX18 cause dominant urinary tract malformations via transcriptional dysregulation of ureter development. Am J Hum Genet 97: 291–301, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crawford K, Bracher-Smith M, Owen D, Kendall KM, Rees E, Pardiñas AF, et al. : Medical consequences of pathogenic CNVs in adults: Analysis of the UK Biobank. J Med Genet 56: 131–138, 2019 [DOI] [PubMed] [Google Scholar]
- 28.Furth SL, Cole SR, Moxey-Mims M, Kaskel F, Mak R, Schwartz G, et al. : Design and methods of the Chronic Kidney Disease in Children (CKiD) prospective cohort study. Clin J Am Soc Nephrol 1: 1006–1015, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atkinson MA, Ng DK, Warady BA, Furth SL, Flynn JT: The CKiD study: Overview and summary of findings related to kidney disease progression. Pediatr Nephrol 36: 527–538, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parsa A Kanetsky PA Xiao R Gupta J Mitra N Limou S et al. ; FIND Consortium; and the Chronic Renal Insufficiency Cohort (CRIC) Study Investigators : Genome-wide association of CKD progression: The Chronic Renal Insufficiency Cohort Study. J Am Soc Nephrol 28: 923–934, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, et al. : Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux. Sci Rep 7: 14595, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, et al. : High-resolution mapping and analysis of copy number variations in the human genome: A data resource for clinical and research applications. Genome Res 19: 1682–1690, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Cheng R, Verbitsky M, Kisselev S, Browne A, Mejia-Sanatana H, et al. : Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med Genet 12: 104, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Padmanabhan S Melander O Johnson T Di Blasio AM Lee WK Gentilini D et al. ; Global BPgen Consortium : Genome-wide association study of blood pressure extremes identifies variant near UMOD associated with hypertension. PLoS Genet 6: e1001177, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. : Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet 43: 321–327, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer RHC, Brick L, Nugent NR, Bidwell LC, McGeary JE, Knopik VS, et al. : Examining the role of common genetic variants on alcohol, tobacco, cannabis and illicit drug dependence: Genetics of vulnerability to drug dependence. Addiction 110: 530–537, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salvi E, Kutalik Z, Glorioso N, Benaglio P, Frau F, Kuznetsova T, et al. : Genomewide association study using a high-density single nucleotide polymorphism array and case-control design identifies a novel essential hypertension susceptibility locus in the promoter region of endothelial NO synthase. Hypertension 59: 248–255, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. : Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 42: 781–785, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, Liu Z, Wang LE, Gershenwald JE, Lee JE, Prieto VG, et al. : Haplotype and genotypes of the VDR gene and cutaneous melanoma risk in non-Hispanic whites in Texas: A case-control study. Int J Cancer 122: 2077–2084, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desch KC, Ozel AB, Siemieniak D, Kalish Y, Shavit JA, Thornburg CD, et al. : Linkage analysis identifies a locus for plasma von Willebrand factor undetected by genome-wide association. Proc Natl Acad Sci U S A 110: 588–593, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matise TC Ambite JL Buyske S Carlson CS Cole SA Crawford DC et al. ; PAGE Study : The Next PAGE in understanding complex traits: Design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) Study. Am J Epidemiol 174: 849–859, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iyengar SK Sedor JR Freedman BI Kao WH Kretzler M Keller BJ et al. ; Family Investigation of Nephropathy and Diabetes (FIND) : Genome-wide association and trans-ethnic meta-analysis for advanced diabetic kidney disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet 11: e1005352, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, et al. : Diagnostic utility of exome sequencing for kidney disease. N Engl J Med 380: 142–151, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cameron-Christie S, Wolock CJ, Groopman E, Petrovski S, Kamalakaran S, Povysil G, et al. : Exome-based rare-variant analyses in CKD. J Am Soc Nephrol 30: 1109–1122, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarty CA Chisholm RL Chute CG Kullo IJ Jarvik GP Larson EB et al. ; eMERGE Team : The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics 4: 13, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crosslin DR McDavid A Weston N Nelson SC Zheng X Hart E et al. ; Electronic Medical Records and Genomics (eMERGE) Network : Genetic variants associated with the white blood cell count in 13,923 subjects in the eMERGE Network. Hum Genet 131: 639–652, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glessner JT, Li J, Desai A, Palmer M, Kim D, Lucas AM, et al. : CNV association of diverse clinical phenotypes from eMERGE reveals novel disease biology underlying cardiovascular disease. Int J Cardiol 298: 107–113, 2020 [DOI] [PubMed] [Google Scholar]
- 48.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. : PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al. : PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17: 1665–1674, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. : Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 91: 597–607, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fasel D, Verbitsky M, Sanna-Cherchi S: CNVkit – Software tools for analyzing genomic structural variants. J Am Soc Nephrol Vol. 26, Abstract Edition, 2015 [Google Scholar]
- 52.Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. : DECIPHER: Database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet 84: 524–533, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reddy UM Page GP Saade GR Silver RM Thorsten VR Parker CB et al. ; NICHD Stillbirth Collaborative Research Network : Karyotype versus microarray testing for genetic abnormalities after stillbirth. N Engl J Med 367: 2185–2193, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. : Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 367: 2175–2184, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kearney HM Thorland EC Brown KK Quintero-Rivera F South ST; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee : American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13: 680–685, 2011 [DOI] [PubMed] [Google Scholar]
- 56.South ST Lee C Lamb AN Higgins AW Kearney HM; Working Group for the American College of Medical Genetics and Genomics Laboratory Quality Assurance Committee : ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet Med 15: 901–909, 2013 [DOI] [PubMed] [Google Scholar]
- 57.Yang W Xie D Anderson AH Joffe MM Greene T Teal V et al. ; CRIC Study Investigators : Association of kidney disease outcomes with risk factors for CKD: Findings from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis 63: 236–243, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carroll RJ, Bastarache L, Denny JC: R PheWAS: Data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30: 2375–2376, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.United States Centers for Medicare and Medicaid Services : ICD-9-CM Diagnosis and Procedure Codes: Abbreviated and Full Code Titles, 2014. Available at: https://www.cms.gov/Medicare/Coding/ICD9ProviderDiagnosticCodes/codes. Accessed May 18, 2020
- 60.Feldman HI Appel LJ Chertow GM Cifelli D Cizman B Daugirdas J et al. ; Chronic Renal Insufficiency Cohort (CRIC) Study Investigators : The Chronic Renal Insufficiency Cohort (CRIC) Study: Design and methods. J Am Soc Nephrol 14[Suppl 2]: S148–S153, 2003 [DOI] [PubMed] [Google Scholar]
- 61.Lash JP Go AS Appel LJ He J Ojo A Rahman M et al. ; Chronic Renal Insufficiency Cohort (CRIC) Study Group : Chronic Renal Insufficiency Cohort (CRIC) Study: Baseline characteristics and associations with kidney function. Clin J Am Soc Nephrol 4: 1302–1311, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanna-Cherchi S, Sampogna RV, Papeta N, Burgess KE, Nees SN, Perry BJ, et al. : Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med 369: 621–629, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, et al. : Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet 81: 1057–1069, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, et al. : Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet 92: 221–237, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, et al. : Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry 66: 947–956, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, et al. : Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70: 886–897, 2011 [DOI] [PubMed] [Google Scholar]
- 67.Collins RL Glessner JT Porcu E Lepamets M Brandon R Lauricella C et al. ; Epi25 Consortium; Estonian Biobank Research Team : A cross-disorder dosage sensitivity map of the human genome. Cell 185: 3041–3055.e25, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lata S, Marasa M, Li Y, Fasel DA, Groopman E, Jobanputra V, et al. : Whole-exome sequencing in adults with chronic kidney disease: A pilot study. Ann Intern Med 168: 100–109, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanna-Cherchi S, Khan K, Westland R, Krithivasan P, Fievet L, Rasouly HM, et al. : Exome-wide association study identifies GREB1L mutations in congenital kidney malformations. Am J Hum Genet 101: 1034, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sampson MG, Coughlin CR, 2nd, Kaplan P, Conlin LK, Meyers KE, Zackai EH, et al. : Evidence for a recurrent microdeletion at chromosome 16p11.2 associated with congenital anomalies of the kidney and urinary tract (CAKUT) and Hirschsprung disease. Am J Med Genet A 152A: 2618–2622, 2010 [DOI] [PubMed] [Google Scholar]
- 71.Bellanné-Chantelot C, Clauin S, Chauveau D, Collin P, Daumont M, Douillard C, et al. : Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54: 3126–3132, 2005 [DOI] [PubMed] [Google Scholar]
- 72.Borlot F, Regan BM, Bassett AS, Stavropoulos DJ, Andrade DM: Prevalence of pathogenic copy number variation in adults with pediatric-onset epilepsy and intellectual disability. JAMA Neurol 74: 1301–1311, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liabeuf S, Pepin M, Franssen CFM, Viggiano D, Carriazo S, Gansevoort RT, et al. : Chronic kidney disease and neurological disorders: Are uraemic toxins the missing piece of the puzzle? Eur. Ren. Assoc. 37: ii33–ii44, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hooper SR, Laney N, Radcliffe J, Moodalbail D, Hartung EA, Ruebner RL, et al. : Executive functioning in children, adolescents, and young adults with chronic kidney disease. J Dev Behav Pediatr 36: 734–742, 2015 [DOI] [PubMed] [Google Scholar]
- 75.Kogon AJ, Kim JY, Laney N, Radcliffe J, Hooper SR, Furth SL, et al. : Depression and neurocognitive dysfunction in pediatric and young adult chronic kidney disease. Pediatr Nephrol 34: 1575–1582, 2019 [DOI] [PubMed] [Google Scholar]
- 76.Kurella M, Chertow GM, Fried LF, Cummings SR, Harris T, Simonsick E, et al. : Chronic kidney disease and cognitive impairment in the elderly: The health, aging, and body composition study. J Am Soc Nephrol 16: 2127–2133, 2005 [DOI] [PubMed] [Google Scholar]
- 77.Yaffe K Ackerson L Kurella Tamura M Le Blanc P Kusek JW Sehgal AR et al. ; Chronic Renal Insufficiency Cohort Investigators : Chronic kidney disease and cognitive function in older adults: Findings from the Chronic Renal Insufficiency Cohort cognitive study. J Am Geriatr Soc 58: 338–345, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Männik K, Mägi R, Macé A, Cole B, Guyatt AL, Shihab HA, et al. : Copy number variations and cognitive phenotypes in unselected populations. JAMA 313: 2044–2054, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. : CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505: 361–366, 2014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data that support the findings of these study are available from the corresponding authors upon reasonable request. The data are also available on dbGaP (https://www.ncbi.nlm.nih.gov/gap) under the following accession numbers: phs000524.v1.p1, phs000333.v1.p1, phs000360.v3.p1, phs000650.v3.p1, phs000304.v1.p1, phs000169.v1.p1, phs000092.v1.p1, phs000199.v1.p1, phs000431.v1.p1, phs000356.v2.p1, phs001584.v1.p1 (and other, pending). Some restrictions may apply according to participants’ consent and privacy protection.