


Keywords: GRK4, cell signaling, cystic kidney, genetics and development, kidney development, cilia, polymorphisms, TOR serine-threonine kinases
Abstract
Significance Statement
G protein–coupled receptor kinase 4 (GRK4) regulates renal sodium and water reabsorption. Although GRK4 variants with elevated kinase activity have been associated with salt-sensitive or essential hypertension, this association has been inconsistent among different study populations. In addition, studies elucidating how GRK4 may modulate cellular signaling are sparse. In an analysis of how GRK4 affects the developing kidney, the authors found that GRK4 modulates mammalian target of rapamycin (mTOR) signaling. Loss of GRK4 in embryonic zebrafish causes kidney dysfunction and glomerular cysts. Moreover, GRK4 depletion in zebrafish and cellular mammalian models results in elongated cilia. Rescue experiments suggest that hypertension in carriers of GRK4 variants may not be explained solely by kinase hyperactivity; instead, elevated mTOR signaling may be the underlying cause.
Background
G protein–coupled receptor kinase 4 (GRK4) is considered a central regulator of blood pressure through phosphorylation of renal dopaminergic receptors and subsequent modulation of sodium excretion. Several nonsynonymous genetic variants of GRK4 have been only partially linked to hypertension, although these variants demonstrate elevated kinase activity. However, some evidence suggests that function of GRK4 variants may involve more than regulation of dopaminergic receptors alone. Little is known about the effects of GRK4 on cellular signaling, and it is also unclear whether or how altered GRK4 function might affect kidney development.
Methods
To better understand the effect of GRK4 variants on the functionality of GRK4 and GRK4's actions in cellular signaling during kidney development, we studied zebrafish, human cells, and a murine kidney spheroid model.
Results
Zebrafish depleted of Grk4 develop impaired glomerular filtration, generalized edema, glomerular cysts, pronephric dilatation, and expansion of kidney cilia. In human fibroblasts and in a kidney spheroid model, GRK4 knockdown produced elongated primary cilia. Reconstitution with human wild-type GRK4 partially rescues these phenotypes. We found that kinase activity is dispensable because kinase-dead GRK4 (altered GRK4 that cannot result in phosphorylation of the targeted protein) prevented cyst formation and restored normal ciliogenesis in all tested models. Hypertension-associated genetic variants of GRK4 fail to rescue any of the observed phenotypes, suggesting a receptor-independent mechanism. Instead, we discovered unrestrained mammalian target of rapamycin signaling as an underlying cause.
Conclusions
These findings identify GRK4 as novel regulator of cilia and of kidney development independent of GRK4's kinase function and provide evidence that the GRK4 variants believed to act as hyperactive kinases are dysfunctional for normal ciliogenesis.
Introduction
The zebrafish embryo is an elegant model for kidney research because of the simplicity of its developing kidney consisting of only two nephrons. As in humans, the zebrafish kidney comprises a glomerulus followed by an unlooped pronephric tubule with a proximal and a distal compartment.1 Filtration occurs in the glomerulus and if defective, is apparent as proteinuria that can be detected by analysis of the water in which the embryos were housed. Kidney dysfunction may become apparent by the formation of edema, for example, in the pericardiac cavity or brain ventricles. Owing to transparency of zebrafish embryos, morphological analyses are highly feasible and reporter lines allow for easy scoring of glomerular cyst formation. Pronephric cilia develop and function similarly to cilia in mammalian kidney, making zebrafish also an excellent model for renal cilia research.2 Indeed, zebrafish are often used to assess ciliopathies such as cystic kidney disease.
G protein–coupled receptor kinase 4 (GRK4) is one member of a family of seven protein kinases that phosphorylate and desensitize activated G protein–coupled receptors (GPCRs). Loss of GRKs results in overactive GPCRs and pathological conditions. However, permanent uncoupling of GPCRs due to GRK-mediated hyperphosphorylation may be detrimental, too. GRK4 phosphorylates dopaminergic receptors in the proximal tubules of adult kidneys. This induces higher expression and more incorporation of the sodium hydrogen exchanger 3 (NHE3) into the tubule cell membrane causing increased sodium reabsorption from the tubule's lumen.3–5 As a result, blood volume and blood pressure rise, while GRK4 loss counteracts these effects.4,6 The extent of GRK4's regulatory actions on kidney function has become evident by the identification of three genetic variants (p.R65L, rs2960306; p.A142V, rs1024323; p.A486V, rs1801058) in different hypertensive cohorts. Experimental validation of these variants revealed elevated kinase activity toward dopaminergic receptors, suggesting constitutive receptor uncoupling as a cause of hypertension.7-9 p.R65L and p.A142V were most reliably associated with either essential or salt-sensitive hypertension.10–12 The third variant p.A486V seems to inversely correlate with the risk of developing hypertension.12,13 When modeled in mice, p.A142V expression triggered spontaneous high blood pressure (HBP), while in p.R65L transgenic mice, HBP could be provoked on high salt intake.14 Thus, certain genetic variants seem to predispose to hypertension, although additional factors likely modulate the variants' effects.10,15,16 Moreover, in untreated individuals, p.A142V had no effect on sodium excretion17 suggesting that GRK4 variants may function more broadly than only by regulation of dopaminergic receptors. Little is known, however, about the impact of GRK4 on cellular signaling and whether this involved the regulation of GPCRs. It is also unclear whether alterations to GRK4 function might affect kidney development and if so, by what mechanism.
Therefore, we investigated the actions of GRK4 in cellular signaling during kidney development. Using a series of different readouts and methods, we establish that GRK4 limits cilium elongation. We demonstrate that the observed phenotypes are independent of GRK4 kinase activity, but likely due to unleashed mammalian target of rapamycin (mTOR) signaling. We also provide, for the first time, evidence that genetic variants of GRK4, which had previously been designated kinase-hyperactive, lack functionality regarding renal cilia.
Materials and Methods
Materials, Cells, and Zebrafish Lines
Rotundine and fenoldopam were purchased from Sigma, SCH-39166 hydrobromide from Biomol, and recombinant PDGF-AA from BioLegend. FITC-dextran was obtained from MedChemExpress (catalog no. HY-128868) and rapamycin from LC Laboratories. HEK293T cells were from American Type Culture Collection; hTert immortalized 1BR3 human fibroblasts were from the collection of the Genome Damage and Stability Center at the University of Sussex, United Kingdom. Murine inner medullary collecting duct (IMCD3) cells were from Soeren Lienkamp. RPE-1 cells were kindly provided by Travis Stracker, National Institutes of Health. The Tg(wt1b:eGFP) zebrafish line was a kind gift of Christoph Englert.18
Cloning
To generate a template for riboprobe synthesis, a 637 bp fragment of the zebrafish grk4 coding sequence was inserted into pCRII using TOPO TA cloning (Invitrogen). The zebrafish grk4 open reading frame was PCR amplified from cDNA made from RNA of 24 hours postfertilization (hpf) embryos and cloned into pCS2+ through ClaI and StuI restriction sites. A human GRK4 clone encoding GRK4α was obtained from Dharmacon (clone MHS6278-211687744) and cloned into pCS2+ through ClaI and StuI. All variants of zebrafish and human GRK4 used in this study were generated using site-directed mutagenesis starting with the expression plasmids described above. To generate a construct deficient in the RGS homology (RH) domain, amino acids (aa) 39–180 of human GRK4 or the respective aa of zebrafish Grk4 were deleted using site-directed mutagenesis. To verify knockdown efficiency in zebrafish, the open reading frame of zebrafish grk4 along with parts of it 5′-UTR was cloned into pCS2+ through BamHI and EcoRI.
Generation of Capped RNA
Capped RNA required for microinjection into fertilized zebrafish eggs was generated from template plasmids as given above after linearization using NotI and subsequent in vitro transcription using either Ambion's SP6 mMessage mMachine kit or the AmpliCap SP6 High Yield Message Maker Kit (Cellscript).
Zebrafish Husbandry and Manipulation
Adult zebrafish were kept under standardized conditions in a water recycling tank system, which automatically monitors and adjusts water temperature, pH, and conductivity (Tecniplast), in a 14-hour light and 10-hour dark cycle. Husbandry and manipulations of zebrafish have been approved by local authorities (Veterinary Care Unit at Ulm University and University of Tübingen, respectively, and the animal welfare commissioner of the regional board for scientific animal experiments in Tübingen, Germany). Experiments were performed according to the European Union Directive 86/609/EEC for the protection of animals used for experimental and other scientific purposes. For experiments, either standard wild-type (wt) lines such as AB and EK were used or the wt1b-GFP transgenic kidney reporter line expressing GFP in the glomerulus and the proximal part of the pronephros. Fertilized eggs were generated by natural mating, and the resulting eggs were randomly distributed into the different treatment groups. Knockdown of Grk4 in zebrafish was achieved by antisense morpholino oligonucleotide (MO) microinjection into the yolk of 1–2 cell stage embryos using a Femtojet microcompressor and a Narishige micromanipulator.19 The MOs (Gene-Tools) used in this study were a translation-blocking MO against the UTR of zebrafish Grk4 with the following sequence: 5′-GTCAGAAGAAAGATCCCATGCAGTC and a control MO containing a five-base mismatch: 5′-GTGACAACAAACATCCCATCCAGTC. Embryos were raised at 28.5°C until the desired stages. No blinding of the experimentor was performed because the Grk4 loss-of-function phenotypes were too obvious to ensure blinding during analyses. A zebrafish mutant of Grk4 was generated using CRISPR/Cas9-mediated gene editing with the guide RNA: 5-CAUUACAGGGUAUUGGGCAAG (IDT) resulting in a 6 bp in frame deletion from position 580–585 in the ORF of grk4. For experiments, embryos of this mutant line were generated by heterozygous crosses. Drug treatments always started at tail-bud stage until 48 hpf, when the embryos were analyzed.
Generation and Genotyping of Grk4 Mutant Zebrafish
Genomic DNA was isolated by boiling zebrafish tissue in 50 mM NaOH for 10 minutes followed by rapid cooling to room temperature and buffering using 1/10 volume 1 M Tris pH8. PCR was performed using Taq polymerase with Thermopol buffer (NEB) and a common reverse primer with allele-specific forward primers: WT Fw: 5′-TGGGATTTACGGTGCATTGTTATTAGTGAA, MutFw: 5′-GACATTACAGGGTATTGGGCGGAT, Rev: 5′-ACCACTGATAACGCCCATTCAATCAAATG. The resulting bands (WT: 493 bp, mutant: 487 and 385 bp) were resolved on a 2% agarose gel.
In Situ Hybridization
The standard protocol by Thisse and Thisse20 was followed to perform whole mount in situ hybridization on fixed zebrafish embryos. A digoxigenin (DIG)-labeled anti–sense-riboprobe against grk4 was generated by linearization of a template plasmid containing a 637 bp fragment of grk4 (OTTDART00000036285) and in vitro transcription using SP6 polymerase (NEB) and the DIG RNA labeling mix (Sigma).
Immunofluorescence
Forty eight hpf zebrafish embryos were immunostained essentially as described in Burkhalter et al.,21 and tails of stained embryos were mounted between two coverslips in a small drop of Vectashield mounting medium (Vectorlabs). Antibody staining in fibroblasts was performed with cells grown on coverslips. In brief, after fixation with either 4% PFA or ice-cold methanol, cells were permeabilized with 0.1% Triton X-100 and subsequently blocked with 10% normal goat serum (Vectorlabs) in PBS. Primary antibodies diluted in blocking buffer were incubated over night at 4°C. After three washes in PBS, cells were incubated with fluorescently labeled secondary antibodies diluted 1:1000 in blocking buffer. After three more washes, coverslips were mounted onto slides using Vectastain containing DAPI (Vectorlabs) and the coverslips were sealed with nail polish. IMCD3 spheroids were processed for immunofluorescence essentially as described in the study by Giles et al.22
The following antibodies were used in this study: mouse anti-acetylated tubulin (1:500, SCBT, clone 6-11B-1 and 1:500, Sigma, cat. no. T6793), rabbit anti-γTubulin (1:500, Sigma-Aldrich, cat. no. T5192), rabbit anti-Flag (1:200, Cell Signaling cat. no. 2368S), and rabbit anti-ZO1 (1:1,000, Proteintech, cat. no. 21773-1-AP). Primary antibodies were detected using Alexa Fluor–labeled secondary antibodies (1:1000, Molecular Probes).
Imaging
Z-stacks of immunostained samples were imaged on a Leica TCS SP5II confocal microscope with a section distance of 0.3 µm or a Leica Stellaris 5 with a system-optimized section distance. The neurite tracer tool in FIJI was used to measure cilium length. GFP fluorescence to assess knockdown efficiency in zebrafish was imaged using a Leica M205 FCA microscope equipped with a Leica DFC9000GT sCMOS camera. A Leica M125 upright microscope equipped with a Leica IC80 HD camera was used to visualize live zebrafish or whole mount in situ hybridization in fixed zebrafish.
Fluid Excretion Assay
Pronephric function was assessed by measuring the relative fluorescence of FITC-dextran injected into the pericardiac cavity according to a previous protocol.23 In brief, fertilized eggs were injected with either CTRL or Grk4 MO and treated with N-Phenylthiourea from tail-bud stage on to prevent pigmentation. At 48 hpf, embryos were anesthetized using tricaine and injected with 5 mg/ml FITC-dextran (MW 10,000) into the pericardiac cavity and incubated in the dark. At 0.5, 3, 5, and 24 hours after dye injection (T1, T2, T3, and T4, respectively), all embryos were imaged with the same exposure settings. Fluorescence intensities were measured using Image J.
Cell Culture of HEK Cells
HEK293 cells were cultured in DMEM containing 10% heat-inactivated FCS and 1% penicillin/streptomycin (all from Gibco) at 37°C in an incubator with 5% CO2. siRNA SMART Pools (Dharmacon) were transfected using Lipofectamine RNAiMax, while plasmids alone or siRNAs along with plasmids were transfected using Lipofectamine 3000 (both Life Technologies). Gene editing using CRISPR-Cas9 technology was achieved using two guide RNAs (IDT) and recombinant Cas9 nuclease (IDT) targeting the following sequences in human GRK4: 5′-CGATAGGAAGACGTCTCTTC and 5′-ATGATGAGGACCGAAGTGAT. Individual clones were picked and screened by PCR and Sanger sequencing. mTOR inhibition was achieved by 48 hours treatment with 30 nM rapamycin dissolved in DMSO. For the assessment of mTOR pathway activity cells were serum and amino acid starved for 90 minutes and then restimulated for 30 minutes using aa (Sigma).
Cell Culture of Human Fibroblasts and RPE-1 Cells
hTert-immortalized 1BR3 human fibroblasts were cultured as previously described24 in MEMα supplemented with 10% FCS and 1% penicillin/streptomycin (all from Gibco) at 37°C in an incubator with 5% CO2. Knockdown was achieved by transfection of SMART Pool siRNAs (Dharmacon) using Lipofectamine RNAiMax (Life Technologies). Nucleofection of siRNA SMART Pool along with plasmid DNA was performed essentially as described with an Amaxa nucleofector II, the Amaxa Cell Line Nucleofector Kit R (both Lonza), and program U-023.25 To induce cilium formation, cells were changed 2 days after transfection or nucleofection, respectively, from culture medium into ciliogenesis medium containing only 0.1% FCS and maintained for 3 days under starvation conditions. To induce PDGF receptor α signaling, cells were essentially treated as described before26: After 3 days of starvation, cells were stimulated for 3 minutes with 50 ng/ml PDGF-AA or DMSO as control and immediately lysed for Western blotting. RPE-1 cells were cultured in a DMEM:F12 medium (Sigma) containing 10% FCS and 1% penicillin/streptomycin under the same conditions as human fibroblasts. Cilia were induced by serum starvation for 72 hours. All cell lines are regularly monitored for mycoplasma contamination.
3D Culture of mIMCD3 Cells
Murine IMCD3 (mIMCD3) cells were cultured in DMEM/F12 containing 10% FCS and 1% penicillin/streptomycin (all from Gibco) at 37°C in an incubator with 5% CO2. siRNA SMART Pools (Dharmacon) and plasmids were transfected using Lipofectamine 3000 (Thermo Fisher) before transfer into 3D matrix. Spheroid culture and subsequent immunostaining was exactly performed as described before22 on chambered coverslips.
β-Arrestin Recruitment Assay Using Intermolecular Bioluminescence Resonance Energy Transfer Sensors
β-arrestin recruitment was measured in a CRISPR/Cas 9-generated GRK2, 3, 5, and 6 quadruple knockout HEK293 cell line (ΔQ-GRK;27) and a CRISPR control cell line (Control), transfected with empty lentiCRISPR v2 plasmid, in a NanoBRET-based assay. 1.6×106 cells were seeded in 6 cm dishes and transfected the next day with 1.4 μg of parathyroid hormone 1 receptor (PTH1R) C-terminally fused to a Halo-Tag, 0.35 μg of β-arrestin2 (βArr2) C-terminally coupled to NanoLuciferase (NanoLuc), and 0.25 μg of human GRK5, zebrafish GRK4, zebrafish GRK4-c.580-585del, zebrafish GRK4-K216,217M, or empty vector. Transfection was performed according to the Effectene transfection reagent manual by Qiagen (catalog no. 301427). The following day, 40,000 cells per well were seeded into 96-well plates (Brand, catalog no. 781965), coated with poly-D-lysine. Halo-ligand (618) (Promega, catalog no. G980A) was added in a ratio of 1:2000 to the cell suspension. In addition, a mock labeling condition lacking the Halo-ligand (618) was seeded for each transfection. After 24 hours, the cells were washed twice with measuring buffer (140 mM NaCl, 10 mM HEPES, 5.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2; pH 7.3) and measuring buffer containing the NanoLuc-substrate furimazine (Promega, catalog no. N157B) was added in a ratio of 1:35,000. Donor luminescence and acceptor emission were collected simultaneously using a custom filter and a dual PMT setup in a Synergy Neo2 plate reader (Biotek). The donor luminescence was separated from acceptor emission using a custom filter cube fitted with a 555-nm dichroic mirror and a 620/15 bandpass filter. The measurement was performed for 3 min before and 5 min after stimulation with the indicated concentration of PTH(1–34) (Bachem, catalog no. 4011474) in measuring buffer. The initial bioluminescence resonance energy transfer (BRET) ratio was corrected for labeling efficiency by subtracting the values measured for mock labeling conditions. Halo-corrected BRET changes were calculated by the division of the corrected and averaged values measured subsequent to ligand addition by the respective, corrected and averaged baseline values. For the final dynamic Δ net BRET change, this corrected BRET change was divided by the vehicle control.
Co-Immunoprecipitation (CoIP)
HEK293 cells were seeded in 10 cm dishes and transfected using Lipofectamine 3000 (Life technologies). Two days after transfection cells were lysed, protein interactions cross-linked using formaldehyde and immunoprecipitated using anti-Flag M2 beads (Sigma) essentially as described before.21 One percent of the whole cell lysate and the whole eluate of the beads were used for Western blotting (see below). Please, note: GRK4 is endogenously not detectable with commercially available antibodies and requires also under overexpression conditions very high expression levels to be detected by Western blotting.
Western Blotting
HEK cells were seeded into a 12-well plate and transiently transfected. Two days after transfection, cells were washed once with PBS and each well was lysed with 100 µL lysis buffer (50 mM HEPES, 250 mM NaCl, 2 mM EDTA, 10% glycerol, and 0.5% NP-40) containing proteinase inhibitors (Complete Mini, Roche) and phosphatase inhibitors. The lysates were cleared by 1 min centrifugation, and 20 µl lysate was mixed with 5 µl 5× Laemmli loading buffer. After 10-minute denaturation at 60°C, proteins were separated on a 4%–12% Bolt Bis-Tris-Gel (Novex) using 1× MES buffer. Proteins were transferred onto nitrocellulose membranes, and blots were blocked in 3% milk in TBS containing 0.1% Tween-20. HEK cells for mTOR pathway activity and fibroblast cells for PDGF receptor activity were lysed using a SDS-based buffer containing 1% SDS, 0.1 M Tris pH 6.8, plus phosphatase and proteinase inhibitors. Protein content was then measured using a BCA kit (Sigma), and equal amounts of protein were used for Western blotting. 24 hpf zebrafish embryos were deyolked as described previously21 before lysis. The following antibodies were used: rabbit anti-GRK4 antibody (1:200, order no. HPA028737, Prestige Antibodies, Sigma), mouse anti-GAPDH (1:500, Acris Antibodies, clone 6C5), rabbit anti-Flag (1:1,000, Cell Signaling, cat. no. 2368), rabbit anti-Flag M2 (1:1000, Sigma, cat. no. F7425), rabbit anti-AKT (1:1000, Cell Signaling, clone C67E7, lot no. 17), mouse anti-phosphoAKT (1:1000, Rockland cat. no. 200-301268S, lot no. 27843), rabbit anti-mTOR (1:1,000, Cell Signaling, cat. no. 2983S), mouse anti-S6 (1:500, Cell Signaling, cat. no. 2317), rabbit anti-phospho-S6 (1:500, Cell Signaling, cat. no. 5364), and mouse anti-β actin (1:10,000, Sigma, cat. no. A1978). Dopamine receptors 1 and 5 were detected using antibodies from Proteintech (1:1,000, cat. no. 17934-1-AP and 20310-1-AP). Alexa Fluor–labeled secondary antibodies were used (1:5000) to visualize bands on an iBright FL1500 imaging system (Life Technologies).
Assessment of Proteinuria
At 48 hpf, 60 embryos of each condition were transferred to a fresh petri dish and incubated in 4 ml embryo medium for another 24 hours. The medium was then completely collected and stored frozen at −20°C until use. Proteins were precipitated by adding one volume of 100% trichloroacetic acid to four volumes of embryo medium in a 2 ml tube. The precipitate was centrifuged at full speed in a microcentrifuge at 4°C for 5 minutes. The resulting pellet was washed twice using 200 µl cold acetone and afterward dried at 95°C for 5 minutes. Proteins were resolved on a 4%–12% NuPage Bis-tris Gel (Novex) after addition of 2× Laemmli sample buffer and heating to 80°C for 10 minutes. Proteins were visualized by Coomassie staining (Quick Coomassie solution, Generon).
Cell Size Analysis
HEK cells were seeded at the same density into six-well plates. After two days, cells were trypsinized and spun down. The pellets were resuspended in 50 µl PBS, and images of two replicates were taken using a Countess II cell counter (Life Technologies). The cell diameter was measured using FIJI. Countess test beads (Life Technologies) were used for normalization.
qPCR Analysis
Total RNA was extracted using Zymo Research's RNA Microprep Kit, and equal amounts of RNA were transcribed into cDNA with the Protoscript II Kit (NEB). qPCR was performed with the Luna probe master mix (NEB), the Universal Probe system, and a Lightcycler 480 II instrument (both Roche). Information regarding qPCR assays (primers and probes) is available on request.
Mouse Breeding
Mice lacking Grk4 and Grk5 were kept according to standard procedures in accordance with a Duke University–approved animal use Institutional Animal Care and Use Committee protocol. Individual knockout generation has been described previously.28,29 Double knockouts were bred from double heterozygous breeding pairs.
Statistics
Data were analyzed using Prism 9 (GraphPad). Data were first assessed for normal distribution using a Shapiro-Wilk normality test before the respective parametric or nonparametric test was applied. Datasets containing more than two conditions were analyzed with an ANOVA-based test. For all tests, the α level was set to 0.05.
Results
Zebrafish Grk4 Regulates Kidney Function during Development
To advance the understanding of GRK4 function in the kidney, we used zebrafish because of their many advantages.30 Using in situ hybridization on embryos, we detected Grk4 in the eye, nervous system, lateral line primordium, and developing kidney. Interestingly, all these tissues present ciliated structures (Figure 1A). In adult zebrafish, Grk4 transcripts were detected in testis, brain, heart, kidney, skin, and eye, similar to mouse tissues, in which Grk4 seems even more widely expressed (Supplemental Figure 1). We generated a Grk4 loss-of-function model by injection of a translation blocking MO against the 5′-UTR and start codon of zebrafish grk4. The knockdown efficiency was tested with a construct consisting of parts of the 5′-UTR fused to the open reading frame of Grk4 tagged by a GFP. Coinjection of capped RNA of this construct along with a control MO (CTRL MO) produced green fluorescent embryos, while coinjection with the Grk4 MO efficiently prevented fluorescence, suggesting that the MO depletes zebrafish embryos of Grk4 expression (Figure 1B).
Figure 1.

Depletion of GRK4 results in kidney dysfunction in zebrafish embryos. (A) Left: At 24 hpf, grk4 RNA can be detected in the eye, brain, sc, cvp, and the pronephric tubule (higher magnification). Right: At 48 hpf, grk4 is also expressed in the ba, fin buds, notochord, and llp. Higher magnification images below show expression along the pronephric tubules. Scale bars: 100 µm. (B) Strategy and verification of Grk4 knockdown. Capped RNA containing parts of the 5′-UTR and encoding for a GFP-tagged zebrafish Grk4 was coinjected along with a CTRL MO or a MO complementary to Grk4. The Grk4 MO efficiently prevents green fluorescence from Grk4-GFP, while injection of the CTRL MO does not. Scale bar: 500 µm. (C) Live images of 48 hpf zebrafish embryos left noninjected or injected with either a CTRL MO or a MO targeted against Grk4. Arrow: hydrocephalus. Scale bar: 500 µm. (D) Grk4-depleted embryos develop hydrocephalus, which can be partially rescued by coinjection of RNA-encoding zebrafish Grk4. Mean±SEM. Circles show individual experiments. n=4 experiments with 136–182 embryos in total. One-way ANOVA with Sidak multiple comparison test. ****P <0.0001 (CTRL MO versus Grk4 MO), *P =0.0293 (Grk4 MO versus Grk4 MO+grk4 RNA). (E) Hydrocephalus on Grk4 depletion can be partially rescued using RNA-encoding human GRK4 (GRK4α). Mean±SEM. Circles show individual experiments. n=73–238 embryos in total. One-way ANOVA with Sidak multiple comparison test. ****P <0.0001. (F) Grk4 morphants suffer from proteinuria because embryo medium, in which Grk4 MO embryos were raised, contains protein. SDS protein gel stained with Coomassie blue. Dashed line indicates gel splice to remove irrelevant lanes. (G) The developing zebrafish kidney consists of two pronephric tubules arising from a gl. Analysis of 48 hpf Tg (wt1b:eGFP) embryos reveals that loss of Grk4 is associated with glomerulus cyst formation, which can be prevented by supplementation with human GRK4 RNA. Scale bar: 200 µm. Graph displays quantification of cyst formation as mean±SEM. Circles show individual experiments. n=4 experiments with 95–176 embryos in total. One-way ANOVA with Sidak multiple comparison test. ****P <0.0001. ba, branchial arches; cvp, caudal vein plexus; gl, glomerulus; llp, lateral line progenitor; NI, noninjected zebrafish; sc, spinal cord.
Grk4 MO injection resulted in zebrafish with a distinct body curvature, smaller eyes, and hydrocephalus reminiscent of ciliopathy phenotypes, while injection of the five-base mismatch CTRL MO did not (Figure 1C). Reconstitution with zebrafish Grk4 rescued hydrocephalus formation partially, yet significantly (Figure 1D). Similarly, coinjection of RNA encoding full-length human GRK4 (GRK4α, Supplemental Figure 2A) ameliorated the hydrocephalus (Figure 1E).
In zebrafish, only one Grk4 protein with high conservation to human and mouse full-length GRK4 is present (Supplemental Figure 3). In humans, in-frame skipping of exons 2, 15, or both generates three additional transcripts, GRK4β, GRK4γ, and GRK4δ, additionally to full-length GRK4 (GRK4α) (Supplemental Figure 2A).31 We tested the ability of these three shorter transcript variants to rescue the phenotype. All four transcript variants could be expressed in zebrafish embryos (Supplemental Figure 2B) and also rescued hydrocephalus formation (Supplemental Figure 2C). Based on RT-PCR experiments on cDNA of human fibroblasts or HEK cells, respectively, which produced only a single band for GRK4 (data not shown), we concluded that GRK4α is the predominantly expressed transcript variant. Hence, we performed all subsequent rescue experiments with GRK4α.
Furthermore, we concluded that edema formation is likely due to impaired kidney function upon Grk4 knockdown because we did not observe substantial impairment of heart function or morphogenesis (data not shown).
As readout for kidney dysfunction, we assessed glomerular filtration. Only embryos injected with Grk4 MO developed proteinuria, suggesting a glomerular filtration defect in the absence of Grk4 (Figure 1F). Glomeruli, however, can exhibit dysfunctions other than poor filtration and may develop cysts due to cilia defects.32 Consistent with the ciliopathy-like phenotype, we observed a higher occurrence of glomerular cyst formation after Grk4 knockdown, which was rescued by reconstitution with human GRK4 (Figure 1G, Supplemental Figure 2D).
Depletion of Grk4 Affects Kidney Tubule Morphology and Renal Cilia
Next, we examined the pronephric tubules by immunostaining cilia and apical cell borders. Grk4 knockdown caused dilated pronephric tubules, which was rescued by coinjection of RNA encoding zebrafish Grk4 (Figure 2A). The proximal tubules further showed massive expansion of pronephric cilia (Figure 2A). Cilia measured in the distal part of the pronephros, where individual cilia can be reliably distinguished, were elongated (Figure 2B). In addition, a fluid excretion assay demonstrated ciliary dysfunction upon Grk4 depletion (Figure 2C), which was confirmed by qPCR for target genes of the Hedgehog pathway (Supplemental Figure 4), which relies on functional cilia.33
Figure 2.

Loss of Grk4 impacts on cilia formation and function in the zebrafish pronephros. (A) Grk4 morphant pronephric tubules are dilated compared with tubules of control-injected zebrafish, but coinjection of grk4 RNA reduces dilatation efficiently. Confocal z-stacks of 48 hpf zebrafish pronephric tubules. Green, anti-PKCζ staining apical cell borders of the tubule; red, anti-acTub. to visualize cilia. Scale bars: 10 µm. Graph shows n=23 (CTRL MO), 21 (Grk4 MO), and 34 (Grk4 MO+grk4) embryos. Kruskal-Wallis test with Dunn multiple comparison test. ****P< 0.0001. (B) Individual cilia from distal ends of pronephric tubules of injected zebrafish are longer after depletion of Grk4. Scale bar: 5 µm. Graph summarizes n=151 (CTRL MO) and 179 (Grk4 MO) cilia. Two-tailed Mann-Whitney test, ****P< 0.0001. (C) Grk4-depleted embryos clear fluids more slowly than control embryos. Representative images of embryos injected with FITC-dextrane into the pericardiac cavity shortly after injection (T1) and 24 hours later. Graphs depict disappearance of fluorescence in pericardiac cavity 0.5 (T1), 3 (T2), 5 (T3), and 24 hours postinjection (T4) in individual fish. P =0.0387, Mann-Whitney test. (D) Western blot of HEK cells transiently transfected with Grk4 wt or Grk4 mutated to match the zebrafish Cas9-edited allele, demonstrating lower expression of mutant Grk4, compared with wt. GAPDH as loading control. (E) pronephric tubules stained as in (A) of 48 hpf wt and grk4-mutant embryos showing a similar dilatation as in knockdown embryos. Scale bars: 10 µm. Left graph shows proximal pronephros diameter in wt and Grk4 mutant embryos at 48 hpf. n=14 (grk4+/+) and 15 (grk4mut/mut) embryos. Two-tailed Welch test. **P=0.0030. Graph on right displays cilia measurements in the distal pronephros of Grk4 mutants. n=124 (grk4+/+) and 123 (grk4mut/mut) cilia from 4 to 5 embryos, respectively. Two-tailed Welch test. ****P< 0.0001. All graphs: Red line indicates median. Circles represent individual experiments. acTub., acetylated tubulin; PKC, protein kinase C.
To further confirm that these phenotypes were due to depletion of Grk4 and not nonspecific off-target effects, we generated a mutant. CRISPR/Cas9-mediated gene editing resulted in in-frame deletion of 2 aa at positions 194 and 195 (c.580-585del) (Supplemental Figure 3). To test whether this deletion affects Grk4 function, we expressed the mutant cDNA in HEK293 cells and compared it with wt Grk4-transfected cells using Western blot. Deletion of aa194-195 strongly reduced the expression of Grk4 (Figure 2D, Supplemental Figures 3 and 5), suggesting this mutant mimics depletion of Grk4 using MO-mediated knockdown. Pronephric tubule dilatation was also apparent in homozygous Grk4 mutants, and, as in Grk4 knockdown embryos, cilia in the pronephros were longer (Figure 2E). Finally, we injected Grk4 MO along with Grk4 RNA containing this mutation. The deletion mutant was incapable of rescuing either hydrocephalus or cyst formation (Supplemental Figure 6). Thus, decreased expression of Grk4 results in kidney and cilia defects.
GRK4 Regulates Cilia in Human Fibroblasts
To test whether GRK4 controls ciliogenesis per se, we turned to human fibroblasts, in which overexpressed GRK4 localizes to punctate structures, microtubules, and cilia (Figure 3A). Transfection of an siRNA SMART Pool along with a plasmid encoding zebrafish Grk4 substantially lowered endogenous fibroblast GRK4 expression and produced robust expression of zebrafish Grk4 (Figure 3B). Such GRK4 knockdown significantly increased cilium length. Reconstitution with zebrafish Grk4 normalized cilium length (Figure 3, C and D). To assess cilium function in fibroblasts, we performed a PDGF-AA assay, which stimulates ciliary signaling using the PDGF receptor and results in AKT phosphorylation.34 This revealed that primary cilia are functionally impaired in the absence of GRK4 (Figure 3E, Supplemental Figure 7). Taken together, we identified GRK4 as novel regulator of ciliogenesis.
Figure 3.
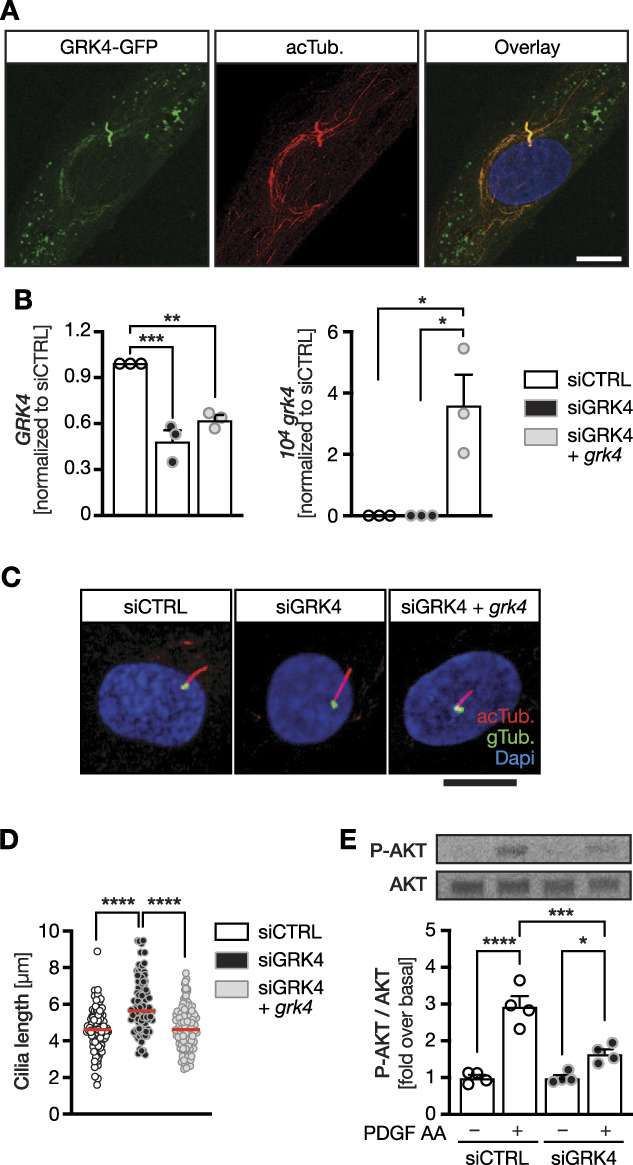
Loss of GRK4 causes cilia defects in human fibroblasts. (A) Confocal z-stack of human fibroblasts transiently transfected with a plasmid encoding GFP-tagged GRK4 and stained for acTub. (red). Scale bar: 10 µm. B, GRK4 levels can be efficiently depleted from human fibroblasts using a pool of four siRNAs. Human immortalized fibroblasts were nucleofected with control siRNAs, siRNAs against GRK4 and EV, or a plasmid encoding zebrafish Grk4 for rescue. Left panel shows qPCR for GRK4, and right panel shows qPCR for zebrafish Grk4 by nucleofection, both normalized to SDHA as housekeeping gene. Mean±SEM. n=3 nucleofections. One-way ANOVA with Sidak multiple comparison test. ***P=0.0003, **P=0.0019, *P=0.013. (C) Representative confocal z-stacks of nucleofected human fibroblasts stained for acTub. (red, cilia) and gTub. (green, basal body). Scale bar: 10 µm. (D) Primary cilia of fibroblasts are significantly longer upon knockdown of GRK4, which can be rescued by conucleofection of zebrafish Grk4. Red line indicates median. Circles represent individual cells. n=105 (siCTRL), 97 (siGRK4), and 117 (siGRK4+grk4) cilia. Kruskal-Wallis test with Dunn multiple comparison test, ****P< 0.0001. (E) Knockdown of GRK4 renders cilia dysfunctional as shown for AKT phosphorylation upon stimulation with PDGF AA for 3 minutes. Representative blots. Graph depicts mean±SEM with individual experiments shown as circles. acTub., acetylated tubulin; EV, empty vector; gTub., γTubulin.
Kinase Function is Dispensable for GRK4 Action in Ciliogenesis
The main regulatory action of GRK4 on kidney function has been assumed to be its kinase activity toward dopaminergic receptors. We tested whether this is also true during embryonic development using kinase-dead mutant GRK4. We mutated zebrafish Grk4 similarly as it has previously been done to render human GRK4 kinase-dead (Grk4 K216,217M) (Supplemental Figure 3). To control for Grk4's inability to phosphorylate and desensitize GPCRs, we measured recruitment of βArr2 to the PTH1R (Figure 4A). GRK-mediated phosphorylation of ligand-activated GPCRs increases the affinity of βArr2 for the receptor and βArr2 translocates from the cytosol to the activated receptor.35 While overexpression of wt Grk4 efficiently produced recruitment of βArr2 (similarly to human GRK5), Grk4 K216,217M and the newly generated zebrafish mutant (Grk4 c.580-585del) failed to do so. We concluded that Grk4 K216,217M is likely kinase-dead.
Figure 4.

Grk4 does not require kinase activity during cilium formation. (A) Verification that Grk4 K216,217M resembles a kinase-dead version of Grk4 and that Grk4 c.580-585del is unable to modify GPCRs. Graph shows concentration-dependent recruitment of βArrestin 2 (βArr2) to the PTH1R on stimulation with PTH peptide. Recruitment was assessed using BRET. GRK5 served as positive control. n=4 (EV, Grk4, GRK5) and n=3 (Grk4 K216,217M, Grk4 c.580-585del). (B) Immunofluorescence of fibroblasts after nucleofection with siRNAs and plasmids. Reconstitution of siGRK4 cells successfully rescues cilium length. Red: cilia (acTub.), green: centrioles (gTub.), DAPI: nucleus. Scale bar: 10 µm. (C) Kinase-dead Grk4 rescues cilium length in human fibroblasts. n=98 (siCTRL), 95 (siGRK4), and 104 (siGRK4+grk4 K216,217M) cilia. Kruskal-Wallis test with Dunn multiple comparison test, ****P< 0.0001, **P=0.0026 (siCTRL versus siGRK4+grk4 K216,217M). Red line: median. (D) Cyst formation can be partially rescued using kinase-dead GRK4 (GRK4 K216,217M). Mean±SEM. Circles show individual experiments. n=8 experiments with 197–253 embryos in total. RM one-way ANOVA with Holm-Šídák multiple comparison test. ****P< 0.0001. (E) Kinase-dead GRK4 ameliorates edema formation upon Grk4 knockdown. Mean±SEM. Circles show individual experiments. n=4 experiments with 110–121 embryos in total. Brown-Forsythe and Welch ANOVA test. ***P=0.0004 (CTRL MO versus Grk4 MO), **P=0.0061 (Grk4 MO versus Grk4 MO+GRK4 K216,217M). (F) Cilia projecting into the lumen of mouse IMCD3 spheroids are longer after Grk4 depletion and can be rescued by wt zebrafish Grk4 and a kinase-dead variant. Representative z-stacks of spheroids, apical cell borders stained for ZO1 (green), and cilia in magenta. n=266 (siCTRL), 360 (siGRK4), 284 (siGrk4+grk4), and 292 (siGRK4+grk4 K216,217M) cilia. Kruskal-Wallis test with Dunn multiple comparison test, ****P< 0.0001. Red line: median. EV, empty vector; gTub., γTubulin.
Human fibroblasts were nucleofected with siRNAs plus Grk4 K216,217M and serum-starved to induce ciliogenesis. As before, GRK4 knockdown led to cilia elongation. Interestingly, Grk4 K216,217M fully rescued cilium length. In fact, cilia were even a bit shorter than in siCTRL-nucleofected fibroblasts (Figure 4, B and C), suggesting that GRK4's kinase function is dispensable for normal ciliogenesis. Similarly, cysts occurred less frequently when kinase-dead GRK4 RNA was coinjected into Grk4-depleted embryos (Figure 4D). Consistent with improvement in kidney function, we further observed less hydrocephalus formation (Figure 4E). Finally, in an organoid model derived from murine IMCD3 kidney cells, cilia were longer on Grk4 depletion (Figure 4F, Supplemental Figure 8). Consistent with the data in zebrafish and human cells, both wt and kinase-dead zebrafish Grk4 rescued cilium length (Figure 4F). Thus, GRK4 seems to control cilia biology not only in zebrafish but also in mammalian cells.
GRK4 Likely Controls Cilia Independent of Its Action on Dopaminergic Receptors
GRK4 has been reported to regulate kidney function by desensitizing dopaminergic receptors. Furthermore, activation of dopaminergic receptors seems to elongate primary cilia.36 If GRK4 action at dopaminergic receptors was important for the phenotypes, cilium elongation could be due to loss of desensitization and constitutive activity of dopamine receptors. Hence, antagonism at dopaminergic receptors would reduce cilium length. Intriguingly, however, the dopaminergic receptor antagonist rotundine increased cilium length in siCTRL- and in siGRK4-transfected fibroblasts (Supplemental Figure 9A). Similarly, SCH-39166, a distinct dopamine receptor blocker, neither reduced cilium length in GRK4-depleted fibroblasts (Supplemental Figure 9B), suggesting that GRK4 conveys regular ciliogenesis and kidney development in a kinase-independent and potentially dopamine receptor-independent fashion. Moreover, dopamine receptor stimulation did not cause a significant change in cilium length (Supplemental Figure 9C), although both dopamine D1 and D5 receptors were present in the treated fibroblasts (Supplemental Figure 9, D and E). We corroborated this finding in RPE-1 cells (Supplemental Figure 9F), which were reported to express dopaminergic receptors and are routinely used for cilia analyses.37 We also did not detect cilia dysfunction phenotypes upon dopamine treatment of zebrafish embryos (Supplemental Figure 9, G–I).
GRK4 Variants with Increased Kinase Activity are Dysfunctional Toward Cilia
The two hypertension-associated GRK4 variants p.R65L and p.A142V (Supplemental Figures 3 and 10) display elevated kinase activity and have been classified as factors for enhanced sodium reabsorption and increased blood pressure.5 However, the linkage between hypertension and these variants has been equivocal because p.A142V does not modulate sodium excretion in all individuals.17 Interestingly, neither variant rescued hydrocephalus formation to a similar extent as wt GRK4 (Figure 5, A and B). There was also no positive effect on cyst formation or cilia length on coinjection of either variant (Figure 5, C and D). In summary, these data demonstrate that neither GRK4 variant retains full functionality and emphasize further the irrelevance of kinase activity of GRK4 regarding kidney development and function. Instead, the data indicate the importance of the RH domain of GRK4 for cilium biology. Indeed, deletion of the RH domain of human and zebrafish GRK4, respectively, rendered GRK4 unable to rescue either phenotype in zebrafish or human fibroblasts (Supplemental Figure 11).
Figure 5.
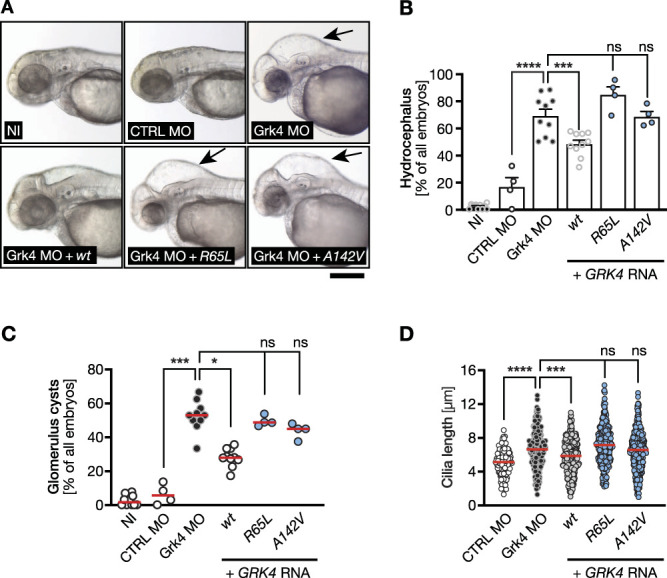
The GRK4 variants p.R65L and p.A142V are unable to correct the Grk4 depletion phenotypes. (A) Live images of 48 hpf zebrafish embryos. Hydrocephalus is still present on coinjection of RNA encoding human GRK4 R65L and A142V, respectively, while wt RNA can decrease hydrocephalus development. Scale bar: 200 µm. (B) The GRK4 variants R65L and A142V display reduced ability to prevent hydrocephalus formation upon Grk4 depletion. Mean±SEM. Circles show individual experiments. n=4–10 experiments with 98–386 embryos in total. One-way ANOVA with Sidak multiple comparison test. ****P< 0.0001 (CTRL MO versus Grk4 MO), ***P=0.0005 (Grk4 MO versus Grk4 MO+GRK4 wt), ns=0.0880 (Grk4 MO versus Grk4 MO+GRK4 R65L), and >0.9999 (Grk4 MO versus Grk4 MO+GRK4 A142V). (C) None of the two GRK4 variants is able to significantly prevent glomerular cyst formation in Grk4 LOF zebrafish. Red line indicates median. n=4–10 experiments with 93–305 embryos in total. Kruskal-Wallis test with Dunn multiple comparison test. ***P=0.0008 (CTRL MO versus Grk4 MO), *P=0.0200 (Grk4 MO versus Grk4 MO+GRK4 wt), ns, P>0.9999 (Grk4 MO versus Grk4 MO+GRK4 R65L), and >0.9999 (Grk4 MO versus Grk4 MO+GRK4 A142V). (D) Neither of the analyzed patient variants can rescue cilium length after Grk4 knockdown in the zebrafish pronephros. Red line indicates median. n=133–379 cilia from 7 to 10 embryos. Kruskal-Wallis test with Dunn multiple comparison test. ****P< 0.0001 (CTRL MO versus Grk4 MO), ***P=0.0004 (Grk4 MO versus Grk4 MO+GRK4 wt), ns, P=0.0616 (Grk4 MO versus Grk4 MO+GRK4 R65L), and >0.9999 (Grk4 MO versus Grk4 MO+GRK4 A142V).
GRK4 Limits mTOR Signaling Activity
The mTOR is a master regulator of cellular protein synthesis, growth, and proliferation.38 Previously, we and others reported that elevated mTOR pathway activity can also elongate cilia.21,39 We further demonstrated that GRK5, another member of the GRK4 subfamily, dampens mTOR signaling activity.21 Indeed, co‐immunoprecipitation experiments using Flag-tagged GRK4 revealed that GRK4 interacts with mTOR (Figure 6A and Supplemental Figure 12). We hence tested whether the mTOR inhibitor rapamycin affects cilium elongation in GRK4 knockdown fibroblasts. Cilium length was significantly ameliorated by rapamycin administration (Figure 6B). Furthermore, GRK4-depleted HEK cells were larger, which was reversed by rapamycin or expression of zebrafish Grk4 (Figure 6C, Supplemental Figure 13). A similar effect of mTOR pathway activation was apparent in GRK4 knockout HEK cells: Absence of GRK4 increased cell size, while treatment with rapamycin reversed this effect (Figure 6D). Western blots of these cells demonstrated that basal and stimulated mTOR pathway activity were elevated in the absence of GRK4 (Figure 6E, Supplemental Figure 12). Moreover, zebrafish embryos depleted of Grk4 also displayed higher mTOR signaling than control embryos (Figure 6F, Supplemental Figure 12). Rapamycin also reduced cilium elongation in Grk4-depleted embryos and rescued to a low, but significant extent cyst formation (Figure 6, G and H). In summary, loss of GRK4 function in human cells or zebrafish embryos results in elevated mTOR signaling.
Figure 6.
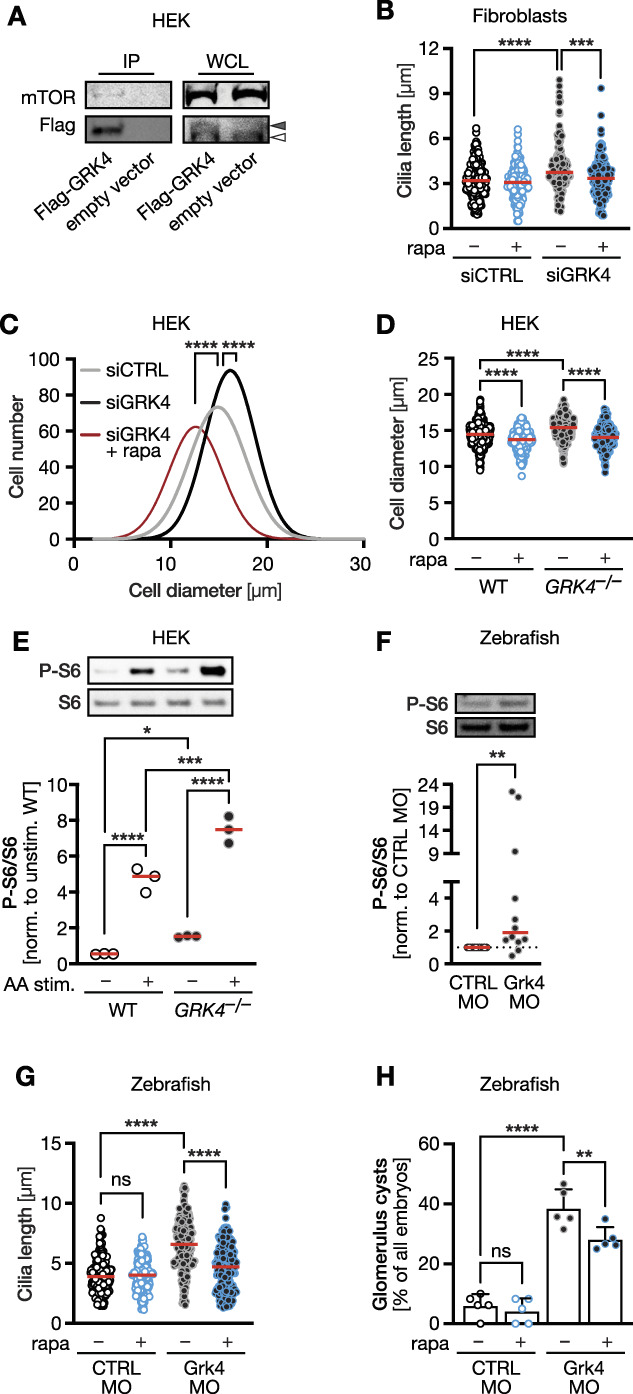
GRK4 negatively regulates mTOR signaling. (A) Co-IP experiments in control- or Flag-tag GRK4-transfected HEK293 cells demonstrating an interaction with endogenously expressed mTOR. Representative blots of one of three Co-IPs. Open arrowhead indicates nonspecific band in Flag-blot, filled arrowhead GRK4 band. (B) Cilium length in human fibroblasts is normalized by the mTOR pathway inhibitor rapamycin. n=190 (siCTRL), 173 (siCTRL+rapa), 152 (siGRK4), and 156 (siGRK4+rapa) cilia. Kruskal-Wallis test with Dunn multiple comparison test, ****P< 0.0001, ***P< 0.001. (C) HEK cells depleted of GRK4 are larger. Rapamycin treatment rescues cell size. n=398 (siCTRL), 473 (siGRK4), and 328 (siGRK4+rapa) cells. Kruskal-Wallis test with Dunn multiple comparison test, ****P< 0.0001. (D) GRK4 knockout HEK cells have a larger diameter, which is rescued by rapamycin. n=518 (WT), 465 (WT+rapa), 483 (GRK4−/−), and 326 (GRK4−/−+rapa) cells. Kruskal-Wallis test with Dunn multiple comparison test, ****P<0.0001. (E) mTOR pathway activity as measured by S6 phosphorylation is elevated in the absence of GRK4. n=4 experiments. One-way ANOVA with Holm-Sidak multiple comparison test. ****P<0.0001, ***P=0.0003, *P=0.0438. (F) Twenty four hpf zebrafish embryos depleted of Grk4 display higher mTOR pathway activity. n=4 experiments. Two-tailed Wilcoxon rank test. **P=0.0068. (G) Rapamycin reduces the cilium length in the pt of zebrafish depleted of Grk4. n=165 (CTRL MO), 167 (CTRL MO=rapa), 215 (siGRK4), and 234 (siGRK4+rapa) cells. Kruskal-Wallis test with Dunn multiple comparison test, ****P<0.0001. (H) Rapamycin partially rescues cyst formation in Grk4 morphant embryos. n=5 experiments with 69 (CTRL MO), 78 (CTRL MO=rapa), 81 (siGRK4), and 84 (siGRK4+rapa) embryos analyzed. One-way ANOVA test with Sidak multiple comparison test, ****P<0.0001, **P=0.0093. Red line indicates median in (B) and (D)–(H). WCL, whole cell lysate.
GRK4's Inhibitory Function on mTOR Signaling Does Not Require Kinase Activity
Finally, we transfected either wt or kinase-dead GRK4 or patient variants into GRK4 knockout HEK cells and assessed cell size. Kinase-dead GRK4 rescued cell size as efficiently as wt GRK4. The patient variants p.R65L and p.A142V, however, were unable to rescue (Figure 7, A and B, Supplemental Figure 14). We also measured protein content as further readout for mTOR activity. HEK cells deficient in functional GRK4 contained more protein than control cells. Treatment with rapamycin significantly reduced the protein amount per cell (Figure 7C). This effect was also rescued by kinase-dead GRK4 with similar efficacy as the wt protein. However, none of the tested patient variants was able to do so (Figure 7D). We conclude that kinase activity is irrelevant for GRK4 limiting mTOR signaling.
Figure 7.
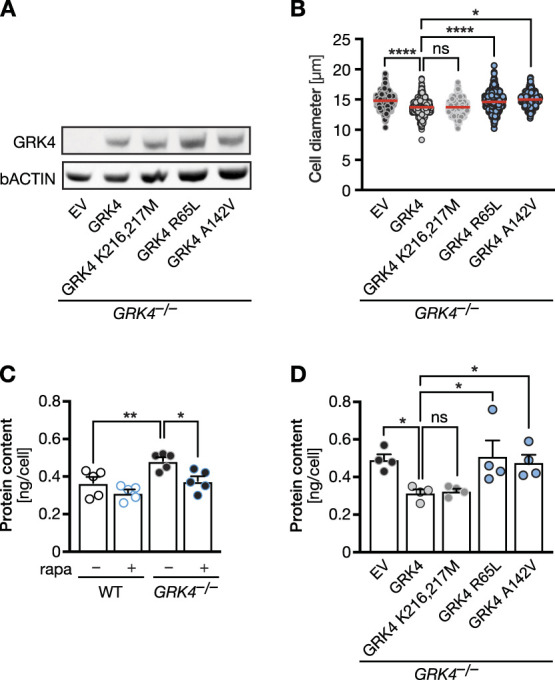
Kinase activity is dispensable for mTOR pathway activity. (A) Western blot showing that human GRK4 variants are expressed in GRK4 knockout cells after transient transfection. Empty vector (EV) or plasmids encoding human GRK4 variants were transiently transfected. n=2. (B) Expression of human kinase-dead GRK4 (K216,217M) significantly reduces cell size similar to WT GRK4, while patient-derived variants do not. n=4 experiments with 641 (EV), 820 (WT), 776 (K215,216M), 739 (GRK4 R65L), and 675 (GRK4 A142V) cells. Kruskal-Wallis test with Dunn multiple comparisons test. ns: P=0.1299, *P=0.0140, and ****P< 0.0001. (C) Protein content in GRK4 KO cells is increased but can be normalized by rapamycin treatment. n=4 experiments. One-way ANOVA with Sidak multiple comparisons test. **P=0.0070 and *P=0.0130. (D) Protein content in GRK4 KO cells can be rescued by kinase-dead GRK4, but not by patient variants. n=4 experiments. One-way ANOVA with Holm-Sidak multiple comparison test. ns: P=0.8687, *P=0.0241 (EV versus GRK4 WT), 0.0169 (EV versus GRK4 R65L), and (0.0276 EV versus GRK4 A142V). EV, empty vector.
Discussion
We here report an unanticipated function of GRK4 during kidney development. Reduction of Grk4 expression in zebrafish embryos results in glomerular filtration defects and reduces kidney function. In addition, glomerular cysts develop and pronephric cilia are abnormally long, revealing Grk4 as a novel regulator of cilium biology. This conclusion is fortified by additional phenotypes on Grk4 loss of function such as pinheads, smaller eyes, otolith defects, and a distinct curvature, which were not the focus of this study, but are all consistent with a cilium defect.40 We identified upregulation of mTOR signaling on GRK4 depletion. Pharmacological blockade of mTOR under such conditions restored normal cilium length.
GRK4 is the least studied GRK. While the roles of GRK5 in cardiomyopathy41 and the function of GRK6 in addiction and inflammation28,42 have been well established, much less attention has been paid to the third member of this GRK subfamily, GRK4. The identification of genetic variants in hypertensive patients has focused research on GRK4 in kidney and hypertension, yet a detailed analysis of GRK4 loss of function has never been reported. This is partially due to GRK4 being originally detected only in very few tissues such as testes31 and knockout mice being viable and fertile with no obvious defects. Any embryonic study was hence considered unnecessary. Double knockouts of GRK4 and 5, however, seem to be partially lethal (Table 1) suggesting that GRK5 possibly compensates for the loss of GRK4. In fact, previously we discovered that GRK5 also affects on cilia, with cilia growing longer when the close homolog of GRK5 was knocked down in zebrafish embryos.21 This is highly similar to the cilia phenotypes in this study, although the massive pronephric dilatation we observed on Grk4 knockdown did not occur in Grk5-depleted embryos. It remains unclear, however, whether the embryonic phenotypes observed in this study can be similarly detected in mouse embryos.
Table 1.
GRK4/GRK5 double knockouts are not born at Mendelian ratios
| Number of Mice | GRK4+/− GRK5+/− × GRK4+/− GRK5+/− | |
|---|---|---|
| Total | 136 | |
| Expected number/percentage | 8.5 | 6.25% |
| Actual number/percentage | 3 | 2.21% |
Mice lacking both GRK4 and GRK5 are not born at the expected Mendelian ratios. Offspring of double heterozygous matings should have yielded 6.25% double homozygotes, which is 8–9 mice of 136 in total. However, only three double homozygotes were born.
So far, all damaging effects mediated by GRK4 had been attributed to its kinase activity. The genetic variants investigated in here possess elevated kinase activity,8 which leads to constitutive phosphorylation of dopamine receptors in the proximal tubule and uncoupling from their G protein. Consecutively, NHE3 is upregulated and inserted into the plasma membrane to facilitate increased sodium reabsorption and hence diffusion of water into the blood causing HBP.5 Whether such a sodium reuptake mechanism was also relevant for the phenotypes observed in here, remains a subject of further studies. Adult fish apparently regulate sodium levels differently than humans,43 although zebrafish embryos express a NHE3 homolog in their proximal pt (data not shown). Moreover, incubation in rising sodium concentrations did not trigger comparable phenotypes with Grk4 depletion (data not shown). The cilia phenotypes in our hands could also not be explained by altered dopamine receptor activity. This finding together with the growing evidence that GRKs can act independently of their kinase activity44 suggest a novel molecular mechanism for GRK4's actions. This is further emphasized by our observation that the N-terminal part of the RH-domain is important for the functionality of GRK4. GRK5, for instance, propagates cardiac hypertrophy independent of its kinase activity through scaffolding of the transcription factor NFAT and genomic DNA.45 GRK5 further limits excessive NFκB activity through direct binding and nuclear translocation of IκB.46 Importantly, kinase-dead GRK5 functioned just the same as kinase-proficient GRK5 indicating that GRKs can function as adapter proteins and may even shuttle proteins into certain subcellular compartments. Our results similarly suggest that GRK4 has kinase-independent functions. The fact that none of the genetic variants are able to rescue any of the phenotypes, while wt and kinase-dead GRK4 can, suggests an unanticipated protein-protein interaction function of GRK4. Of note, both variants tested are located within the RH domain of GRK4. In GRK5, it is also the RH domain that interacts with IκB.46 Interestingly, the RH domain of GRK5 has even become a lead compound for the development of peptides to alter the effects of damaging NFκB signaling.47 The RH domain is also the binding site for GPCRs,48,49 and mutations in this region may increase interaction with GPCRs,50 indicating that the RH domain serves a dual role or at least forms the basis for diverse GRK-mediated signal transduction. In fact, deletion of the RH-domain rendered GRK4 unable to rescue any of the phenotypes observed here.
In the search of a signaling module regulated by GRK4 in kidney and cilia biology, we discovered the mTOR pathway, an important physiological regulator of cell growth and pathological contributor to cystic kidney disease.32 GRK4, like GRK5,21 limits mTOR signaling so that loss of either GRK results in elongated, dysfunctional cilia. It is highly likely that GRK4 does so by protein-protein interaction with the mTOR complex because we detected an interaction between GRK4 and mTOR. Moreover, mTOR signaling occurs at the lysosome,51 and GRK4 was found in a punctate pattern in fibroblasts. Further experiments are required to investigate whether these punctae are lysosomes.
How important are these findings for the clinics and management of hypertension? In mammals, GRK4 is expressed in the proximal tubule, in the ascending limb of Henle loop, and in the glomerulus,4 similar to expression in zebrafish embryos. Altered GRK4 expression during embryonic development influences kidney function later in life,52 which may suggest that any effects we observed in zebrafish embryos may lead to changes to renal function in adult. It is not known whether GRK4 variants predispose to glomerular cysts, one major factor contributing to hypertension.32 However, a correlation between GRK4 and glomerular filtration has been made. In a cohort of Chinese hypertensive patients, the occurrence of an intronic single nucleotide polymorphism in GRK4 (rs2488815) correlated with filtration rate.53 Whether this SNP alters expression of GRK4 remains untested.
Taken together, we have characterized the outcome of GRK4 depletion during vertebrate kidney development and identified a novel, kinase-independent function of GRK4 in cilium biology that affects on pronephric cilia and cyst formation (Figure 8). The fact that the cilium defect observed in Grk4-depleted zebrafish could be well-reproduced in human fibroblasts and a murine model of kidney spheroids suggests that our findings do not apply to zebrafish only, but may apply to human tissues, including the kidney. The full extent of GRK4's function in mammalian cilia biology, however, remains to be investigated.
Figure 8.
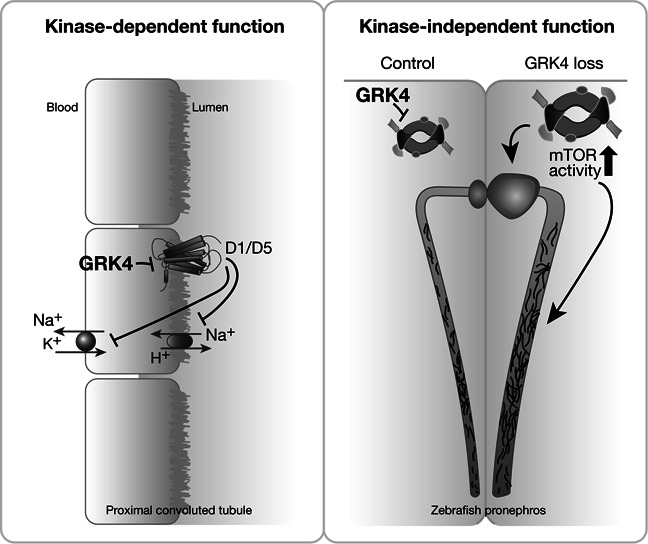
Summary figure. GRK4 has previously been shown to regulate renal sodium reabsorption by phosphorylating dopaminergic receptors. In this study, we provide evidence that GRK4 has also kinase-independent functions, at least in the developing kidney.
Acknowledgments
We thank the Cell Culture Collection at the University of Sussex for providing the 1BR3 fibroblast line, Soeren Lienkamp for the IMCD3 cells, Travis Stracker for RPE-1 cells, Christoph Englert for the transgenic fish line, and the animal care takers at University Hospital Tübingen for excellent fish care. We thank Oliver Wenzke for initial experiments assessing cell size and mTOR signaling. We are further grateful for additional grant support by the Fortüne program of the University Hospital Tübingen (grant no. F1534033) and the Fundació La Marató de TV3 (grant no. 202019-32).
Footnotes
J.G. and L.D.M. contributed equally to this work.
Published online ahead of print. Publication date available at www.jasn.org.
Present address: Richard T. Premont, Harrington Discovery Institute, University Hospitals Cleveland Medical Center, Cleveland, Ohio; and Institute for Transformative Molecular Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio.
Disclosures
R.T. Premont reports ownership interest: Bluebird Bioscience, CRISPR Therapeutics, Editas, Gilead, Intellia, Lexicon Pharmaceuticals, Nektar Therapeutics, Regenex Bio, Sangamo Therapeutics, and Trevena. The remaining authors have nothing to disclose,
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (German Research Foundation, grant number PH144/6-1) and the IZKF program of the University Hospital Tübingen (grant no. 2020-2-09) to M. Philipp. L.D. Maerz was a fellow of the International Graduate School in Molecular Medicine at Ulm University (funded by DFG).
Author Contributions
M. Philipp conceptualized the study; J. Gerhards, L.D. Maerz, and E.S.F. Matthees were responsible for data curation; M.D. Burkhalter, C. Donow, J. Gerhards, C. Hoffmann, L.D. Maerz, E.S.F. Matthees, B. Moepps, M. Philipp, and R.T. Premont were responsible for investigation; M.D. Burkhalter, L.D. Maerz, E.S.F. Matthees, and M. Philipp were responsible for methodology; M.D. Burkhalter and M. Philipp provided supervision; C. Hoffmann and M. Philipp were responsible for resources; M. Philipp was responsible for funding acquisition, project administration, and visualization; M. Philipp wrote the original draft; and M.D. Burkhalter, C. Hoffmann, M. Philipp, and R.T. Premont reviewed and edited the manuscript.
Data Sharing Statement
All data used in this study are available in this article.
Supplement Material
This article contains the following supplemental material online at http://links.lww.com/JSN/D730.
Supplemental Figure 1. Widespread GRK4 expression in adult zebrafish and mice.
Supplemental Figure 2. All four human GRK4 splicing isoforms rescue the Grk4 LOF phenotype.
Supplemental Figure 3. Protein alignment of human, mouse, and zebrafish GRK4.
Supplemental Figure 4. Reduced ciliary signaling in Grk4-depleted zebrafish embryos.
Supplemental Figure 5. Uncropped Western blot.
Supplemental Figure 6. The Grk4 mutation generated by gene editing results in loss of function.
Supplemental Figure 7. Uncropped Western blot.
Supplemental Figure 8. Verification of knockdown efficiency and expression of cotransfected zebrafish Grk4 variants.
Supplemental Figure 9. Dopamine receptor modulation does not alter cilium length.
Supplemental Figure 10. Expression of human GRK4α variants p.R65L and p.A142V does not provoke a phenotype.
Supplemental Figure 11. The RH domain of GRK4 is required for full function.
Supplemental Figure 12. Uncropped Western blots.
Supplemental Figure 13. Rescue of increased cell size by transfection of zebrafish Grk4.
Supplemental Figure 14. Uncropped Western blot.
References
- 1.Gerlach GF, Wingert RA. Kidney organogenesis in the zebrafish: insights into vertebrate nephrogenesis and regeneration. Wiley Interdiscip Rev Dev Biol. 2013;2(5):559-585. doi: 10.1002/wdev.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanke N Staggs L Schroder P, et al. Zebrafishing" for novel genes relevant to the glomerular filtration barrier. Biomed Res Int. 2013;2013(3):658270. doi: 10.1155/2013/658270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fraga S, Jose PA, Soares-da-Silva P. Involvement of G protein-coupled receptor kinase 4 and 6 in rapid desensitization of dopamine D1 receptor in rat IEC-6 intestinal epithelial cells. Am J Physiol Regul Integr Comp Physiol. 2004;287(4):R772-R779. doi: 10.1152/ajpregu.00208.2004 [DOI] [PubMed] [Google Scholar]
- 4.Sanada H Yatabe J Midorikawa S, et al. Amelioration of genetic hypertension by suppression of renal G protein-coupled receptor kinase type 4 expression. Hypertension. 2006;47(6):1131-1139. doi: 10.1161/01.hyp.0000222004.74872.17 [DOI] [PubMed] [Google Scholar]
- 5.Zeng C, Sanada H, Watanabe H, Eisner GM, Felder RA, Jose PA. Functional genomics of the dopaminergic system in hypertension. Physiol Genomics. 2004;19(3):233-246. doi: 10.1152/physiolgenomics.00127.2004 [DOI] [PubMed] [Google Scholar]
- 6.Watanabe H, Xu J, Bengra C, Jose PA, Felder RA. Desensitization of human renal D1 dopamine receptors by G protein-coupled receptor kinase 4. Kidney Int. 2002;62(3):790-798. doi: 10.1046/j.1523-1755.2002.00525.x [DOI] [PubMed] [Google Scholar]
- 7.Felder RA Sanada H Xu J, et al. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci U S A. 2002;99(6):3872-3877. doi: 10.1073/pnas.062694599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gildea JJ Shah I Weiss R, et al. HK-2 human renal proximal tubule cells as a model for G protein-coupled receptor kinase type 4-mediated dopamine 1 receptor uncoupling. Hypertension. 2010;56(3):505-511. doi: 10.1161/hypertensionaha.110.152256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rankin ML, Marinec PS, Cabrera DM, Wang Z, Jose PA, Sibley DR. The D1 dopamine receptor is constitutively phosphorylated by G protein-coupled receptor kinase 4. Mol Pharmacol. 2006;69(3):759-769. doi: 10.1124/mol.105.019901 [DOI] [PubMed] [Google Scholar]
- 10.Speirs HJ, Katyk K, Kumar NN, Benjafield AV, Wang WY, Morris BJ. Association of G-protein-coupled receptor kinase 4 haplotypes, but not HSD3B1 or PTP1B polymorphisms, with essential hypertension. J Hypertens. 2004;22(5):931-936. doi: 10.1097/00004872-200405000-00014 [DOI] [PubMed] [Google Scholar]
- 11.Zhu H Lu Y Wang X, et al. The G protein-coupled receptor kinase 4 gene affects blood pressure in young normotensive twins. Am J Hypertens. 2006;19(1):61-66. doi: 10.1016/j.amjhyper.2005.07.007 [DOI] [PubMed] [Google Scholar]
- 12.Martinez Cantarin MP Ertel A Deloach S, et al. Variants in genes involved in functional pathways associated with hypertension in African Americans. Clin Transl Sci. 2010;3(6):279-286. doi: 10.1111/j.1752-8062.2010.00242.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Xi B. Pooled analyses of the associations of polymorphisms in the GRK4 and EMILIN1 genes with hypertension risk. Int J Med Sci. 2012;9(4):274-279. doi: 10.7150/ijms.4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z Armando I Asico LD, et al. The elevated blood pressure of human GRK4γA142V transgenic mice is not associated with increased ROS production. Am J Physiol Heart Circ Physiol. 2007;292(5):H2083-H2092. doi: 10.1152/ajpheart.00944.2006 [DOI] [PubMed] [Google Scholar]
- 15.Diao Z Asico LD Villar VAM, et al. Increased renal oxidative stress in salt-sensitive human GRK4γ486V transgenic mice. Free Radic Biol Med. 2017;106:80-90. doi: 10.1016/j.freeradbiomed.2017.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhatnagar V O'Connor DT Brophy VH, et al. G-protein-coupled receptor kinase 4 polymorphisms and blood pressure response to metoprolol among African Americans: sex-specificity and interactions. Am J Hypertens. 2009;22(3):332-338. doi: 10.1038/ajh.2008.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staessen JA Kuznetsova T Zhang H, et al. Blood pressure and renal sodium handling in relation to genetic variation in the DRD1 promoter and GRK4. Hypertension. 2008;51(6):1643-1650. doi: 10.1161/hypertensionaha.107.109611 [DOI] [PubMed] [Google Scholar]
- 18.Bollig F Perner B Besenbeck B, et al. A highly conserved retinoic acid responsive element controls wt1a expression in the zebrafish pronephros. Development. 2009;136(17):2883-2892. doi: 10.1242/dev.031773 [DOI] [PubMed] [Google Scholar]
- 19.Tena TC, Philipp M. Assessing smoothened-mediated Hedgehog signaling in zebrafish. Methods Cell Biol. 2016;132:147-164. doi: 10.1016/bs.mcb.2015.10.001 [DOI] [PubMed] [Google Scholar]
- 20.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3(1):59-69. doi: 10.1038/nprot.2007.514 [DOI] [PubMed] [Google Scholar]
- 21.Burkhalter MD, Fralish GB, Premont RT, Caron M, Philipp M. Grk5l controls heart development by limiting mTOR signaling during symmetry breaking. Cell Rep. 2013;4(4):625-632. doi: 10.1016/j.celrep.2013.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giles RH, Ajzenberg H, Jackson PK. 3D spheroid model of mIMCD3 cells for studying ciliopathies and renal epithelial disorders. Nat Protoc. 2014;9(12):2725-2731. doi: 10.1038/nprot.2014.181 [DOI] [PubMed] [Google Scholar]
- 23.Christou-Savina S, Beales PL, Osborn DPS. Evaluation of zebrafish kidney function using a fluorescent clearance assay. J Vis Exp. 2015;96(96):e52540. doi: 10.3791/52540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casar Tena T Maerz LD Szafranski K, et al. Resting cells rely on the DNA helicase component MCM2 to build cilia. Nucleic Acids Res. 2019;47(1):134-151. doi: 10.1093/nar/gky945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burkhalter MD Sridhar A Sampaio P, et al. Imbalanced mitochondrial function provokes heterotaxy via aberrant ciliogenesis. J Clin Invest. 2019;129(7):2841-2855. doi: 10.1172/jci98890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maerz LD, Burkhalter MD, Schilpp C, Wittekindt OH, Frick M, Philipp M. Pharmacological cholesterol depletion disturbs ciliogenesis and ciliary function in developing zebrafish. Commun Biol. 2019;2(1):31. doi: 10.1038/s42003-018-0272-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drube J Haider RS Matthees ESF, et al. GPCR kinase knockout cells reveal the impact of individual GRKs on arrestin binding and GPCR regulation. Nat Commun. 2022;13(1):540. doi: 10.1038/s41467-022-28152-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gainetdinov RR Bohn LM Sotnikova TD, et al. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron. 2003;38(2):291-303. doi: 10.1016/s0896-6273(03)00192-2 [DOI] [PubMed] [Google Scholar]
- 29.Gainetdinov RR Bohn LM Walker JK, et al. Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron. 1999;24(4):1029-1036. doi: 10.1016/s0896-6273(00)81048-x [DOI] [PubMed] [Google Scholar]
- 30.Sander V Salleh L Naylor RW, et al. Transcriptional profiling of the zebrafish proximal tubule. Am J Physiol Renal Physiol. 2019;317(2):F478-F488. doi: 10.1152/ajprenal.00174.2019 [DOI] [PubMed] [Google Scholar]
- 31.Premont RT Macrae AD Stoffel RH, et al. Characterization of the G protein-coupled receptor kinase GRK4. J Biol Chem. 1996;271(11):6403-6410. doi: 10.1074/jbc.271.11.6403 [DOI] [PubMed] [Google Scholar]
- 32.Duong Phu M, Bross S, Burkhalter MD, Philipp M. Limitations and opportunities in the pharmacotherapy of ciliopathies. Pharmacol Ther. 2021;225(2):107841. doi: 10.1016/j.pharmthera.2021.107841 [DOI] [PubMed] [Google Scholar]
- 33.Philipp M, Caron MG. Hedgehog signaling: is Smo a G protein-coupled receptor? Curr Biol. 2009;19(3):R125-R127. doi: 10.1016/j.cub.2008.12.010 [DOI] [PubMed] [Google Scholar]
- 34.Schneider L Clement CA Teilmann SC, et al. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15(20):1861-1866. doi: 10.1016/j.cub.2005.09.012 [DOI] [PubMed] [Google Scholar]
- 35.Caron MG, Barak LS. A brief history of the beta-arrestins. Methods Mol Biol. 2019;1957:3-8. doi: 10.1007/978-1-4939-9158-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Upadhyay VS, Muntean BS, Kathem SH, Hwang JJ, AbouAlaiwi WA, Nauli SM. Roles of dopamine receptor on chemosensory and mechanosensory primary cilia in renal epithelial cells. Front Physiol. 2014;5:72. doi: 10.3389/fphys.2014.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai IC, Adams KA, Tzeng JA, Shennib O, Tan PL, Katsanis N. Genome-wide suppressor screen identifies USP35/USP38 as therapeutic candidates for ciliopathies. JCI Insight. 2019;4(22):e130516. doi: 10.1172/jci.insight.130516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Casar Tena T, Burkhalter MD, Philipp M. Left-right asymmetry in the light of TOR: an update on what we know so far. Biol Cell. 2015;107(9):306-318. doi: 10.1111/boc.201400094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan S, Li J, Diener DR, Choma MA, Rosenbaum JL, Sun Z. Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proc Natl Acad Sci U S A. 2012;109(6):2021-2026. doi: 10.1073/pnas.1112834109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poprzeczko M Bicka M Farahat H, et al. Rare human diseases: model organisms in deciphering the molecular basis of primary ciliary dyskinesia. Cells. 2019;8(12):1614. doi: 10.3390/cells8121614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfleger J, Gresham K, Koch WJ. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat Rev Cardiol. 2019;16(10):612-622. doi: 10.1038/s41569-019-0220-3 [DOI] [PubMed] [Google Scholar]
- 42.Kavelaars A Vroon A Raatgever RP, et al. Increased acute inflammation, leukotriene B4-induced chemotaxis, and signaling in mice deficient for G protein-coupled receptor kinase 6. J Immunol. 2003;171(11):6128-6134. doi: 10.4049/jimmunol.171.11.6128 [DOI] [PubMed] [Google Scholar]
- 43.Hwang PP, Chou MY. Zebrafish as an animal model to study ion homeostasis. Pflugers Arch. 2013;465(9):1233-1247. doi: 10.1007/s00424-013-1269-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watari K, Nakaya M, Kurose H. Multiple functions of G protein-coupled receptor kinases. J Mol Signal. 2014;9(1):1. doi: 10.1186/1750-2187-9-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hullmann JE Grisanti LA Makarewich CA, et al. GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ Res. 2014;115(12):976-985. doi: 10.1161/circresaha.116.304475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorriento D Ciccarelli M Santulli G, et al. The G-protein-coupled receptor kinase 5 inhibits NFκB transcriptional activity by inducing nuclear accumulation of IκBα. Proc Natl Acad Sci U S A. 2008;105(46):17818-17823. doi: 10.1073/pnas.0804446105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gambardella J Ciccarelli M Del Giudice C, et al. A novel small peptide inhibitor of NFκB, RH10, blocks oxidative stress-dependent phenotypes in cancer. Oxid Med Cell Longev. 2018;2018(5):1-9. doi: 10.1155/2018/5801807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He Y Gao X Goswami D, et al. Molecular assembly of rhodopsin with G protein-coupled receptor kinases. Cell Res. 2017;27(6):728-747. doi: 10.1038/cr.2017.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Komolov KE Du Y Duc NM, et al. Structural and functional analysis of a β2-adrenergic receptor complex with GRK5. Cell. 2017;169(3):407-421.e16. doi: 10.1016/j.cell.2017.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liggett SB Cresci S Kelly RJ, et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med. 2008;14(5):510-517. doi: 10.1038/nm1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290-303. doi: 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X Luo H Chen C, et al. Prenatal lipopolysaccharide exposure results in dysfunction of the renal dopamine D1 receptor in offspring. Free Radic Biol Med. 2014;76:242-250. doi: 10.1016/j.freeradbiomed.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montasser ME Shimmin LC Gu D, et al. Variation in genes that regulate blood pressure are associated with glomerular filtration rate in Chinese. PLoS One. 2014;9(3):e92468. doi: 10.1371/journal.pone.0092468 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study are available in this article.