


Keywords: Dent disease, proximal tubule, endocytic traffic, endosome maturation, endosome acidification, megalin, proteinuria
Abstract
Significance Statement
Loss of function of the 2Cl−/H+ antiporter ClC-5 in Dent disease causes an unknown impairment in endocytic traffic, leading to tubular proteinuria. The authors integrated data from biochemical and quantitative imaging studies in proximal tubule cells into a mathematical model to determine that loss of ClC-5 impairs endosome acidification and delays early endosome maturation in proximal tubule cells, resulting in reduced megalin recycling, surface expression, and half-life. Studies in a Dent mouse model also revealed subsegment-specific differences in the effects of ClC-5 knockout on proximal tubule subsegments. The approach provides a template to dissect the effects of mutations or perturbations that alter tubular recovery of filtered proteins from the level of individual cells to the entire proximal tubule axis.
Background
Loss of function of the 2Cl−/H+ antiporter ClC-5 in Dent disease impairs the uptake of filtered proteins by the kidney proximal tubule, resulting in tubular proteinuria. Reduced posttranslational stability of megalin and cubilin, the receptors that bind to and recover filtered proteins, is believed to underlie the tubular defect. How loss of ClC-5 leads to reduced receptor expression remains unknown.
Methods
We used biochemical and quantitative imaging data to adapt a mathematical model of megalin traffic in ClC-5 knockout and control cells. Studies in ClC-5 knockout mice were performed to describe the effect of ClC-5 knockout on megalin traffic in the S1 segment and along the proximal tubule axis.
Results
The model predicts that ClC-5 knockout cells have reduced rates of exit from early endosomes, resulting in decreased megalin recycling, surface expression, and half-life. Early endosomes had lower [Cl−] and higher pH. We observed more profound effects in ClC-5 knockout cells expressing the pathogenic ClC-5E211G mutant. Alterations in the cellular distribution of megalin in ClC-5 knockout mice were consistent with delayed endosome maturation and reduced recycling. Greater reductions in megalin expression were observed in the proximal tubule S2 cells compared with S1, with consequences to the profile of protein retrieval along the proximal tubule axis.
Conclusions
Delayed early endosome maturation due to impaired acidification and reduced [Cl−] accumulation is the primary mediator of reduced proximal tubule receptor expression and tubular proteinuria in Dent disease. Rapid endosome maturation in proximal tubule cells is critical for the efficient recovery of filtered proteins.
Introduction
Patients with Dent disease, a rare, X-linked disorder caused by loss of function of the 2Cl−/H+ antiporter ClC-5, present with tubular proteinuria that typically progresses to CKD and end-stage kidney disease.1,2 There is considerable variability in disease severity, but no correlation of pathogenic mutations with disease severity has been uncovered.3–5 Studies of ClC-5 KO mice suggest that proteinuria is attributed to reduced posttranslational stability of megalin and cubilin, the proximal tubule (PT) receptors that recover filtered proteins.6–9 The specific trafficking steps affected by the loss of ClC-5 function are unknown.
Microsomes isolated from the kidney cortex of mice lacking ClC-5 have reduced rates of acidification.10,11 However, some studies of disease-causing uncoupled variants with mutations that render ClC-5 a pure Cl− channel have reported impaired endocytic uptake despite apparently normal acidification.10,12 These studies have raised questions about the relative importance of endosomal pH versus [Cl−] in modulating endocytic traffic. Moreover, it remains unclear how disruptions in endosomal ion homeostasis may affect receptor trafficking.
We used CRISPR/Cas9 to generate KO and rescued cells expressing wild-type (WT) or uncoupled ClC-5 in a highly differentiated culture model that transcriptionally resembles the S1 segment of the PT.13–18 These cell lines were used to identify delayed early endosome maturation driven by impaired acidification as the primary trafficking defect that impairs endocytic uptake in Dent disease. Studies in a newly created ClC-5 KO mouse model were consistent with this conclusion and also revealed axial differences in megalin stability that have implications for the uptake of filtered proteins as the disease progresses.
Materials and Methods
Generation of CRISPR/Cas9 ClC-5 KO Clones
Using transcriptomic information for the OK cells13 and the available Monodelphis domestica genomic sequence (MomDom5; NC_008809.1), two single-guide RNAs (sgRNA) were designed targeting the region of Clcn5, which contains the sequence for the gating glutamate (E211 in human and mouse; predicted E225 in OK cells) (Supplemental Table S1). The two sgRNAs were selected such that the cut site was within 10 nucleotides of the gating glutamate codon. The sgRNA sequences with added PAM were inserted into the pHRS surrogate reporter plasmid (PNA Bio Inc.) as previously described (Supplemental Table S1).19 CRISPR RNA (crRNA):transactivating crRNA (tracrRNA) duplexes were generated by annealing 2 nmol of target-specific crRNA (TrueGuide, Thermo Fisher Scientific) and 5 nmol tracrRNA (TrueGuide, Thermo Fisher Scientific) and then mixed in a 1:2 molar ratio with recombinant Cas9 (TrueCut Cas9 Protein v2, A36499, Thermo Fisher Scientific) to make the ribonucleoprotein (RNP) complex (75 pmol Cas9:150 pmol sgRNA).
OK cells (2×106 cells per electroporation; OK-P subclone originally obtained from Moshe Levi, Georgetown University) in 100 µl of electroporation solution (MIR 50112, Ingenio Mirus) were combined with the RNP complex and 2.5 µg of pHRS and electroporated in an Amaxa electroporator (Lonza Nucleofector II) using program D-023. Approximately 48 hours after electroporation, cells were treated with 1 mg/ml hygromycin B (Invitrogen 10687010) for 48 hours, and clones were isolated by single-cell dilution. Additional clones were isolated based on red fluorescent protein and green fluorescent protein expression by fluorescence activated cell sorting as previously described.19
DNA was isolated from individual clones using QuickExtract DNA extraction solution (Lucigen.com) following the manufacturer's protocol and amplified using primers listed in Supplemental Table S2. PCR products were purified using the Wizard SV gel and PCR clean-up system (A9281, Promega), ligated into the pGEM-T Easy Vector System (A1360, Promega) and then transformed. DNA was isolated from single colonies and submitted to the University of Pittsburgh Genomics Research Core for Sanger sequencing. The sequences were analyzed in CLC Genomics Workbench. The sequences from ten bacterial colonies were used to confirm allelic mutations in individual clones. Six ClC-5 KO clonal cell populations were recovered from the two guides, each with two distinct allelic frameshift mutations predicted to result in early termination of the protein were isolated, consistent with the female origin of OK cells.
Generation of Rescue and Uncoupled Stably Transfected Cell Lines
ClC-5 KO cl.1 cells were transfected with expression plasmids encoding hemagglutinin (HA)-tagged human WT ClC-5 (rescue) or the disease-causing uncoupled ClC-5E211G mutant (uncoupled; both provided by Dr. Stéphane Lourdel, Sorbonne Université12). After selection in 400 µg/ml geneticin, individual colonies were isolated using cloning cylinders, and expression of HA-tagged constructs was verified by Western blot.
OK Cell Culture under Continuous Orbital Shear Stress
Cells were cultured in DMEM-F12 (Sigma; D6421), 5% FBS, and 2.5× GlutaMax (Gibco; 35050-061) at 37°C and 5% CO2. Cells were seeded onto 12-mm Transwell permeable supports (Costar; 3401) in 12-well dishes at 4×105 cells in 0.5 ml medium on the apical side of the filter. The basolateral side of the filter received 1.5 ml of medium. After overnight incubation, the filters were transferred to an orbital platform shaker in the incubator and rotated at 146 rpm for 72 hours to enhance differentiation as previously described.17 Media were changed daily.
Albumin Uptake Assay
Cells cultured on filters under orbital shear stress for 72 hours as described above were incubated at 37°C under continuous orbital shear stress for 15–60 minutes with 40 µg/ml Alexa Fluor 647 albumin (A34785, Invitrogen) added apically in DMEM-F12, 2.5× Glutamax, and 25 mM HEPES (Gibco, 15630-080; DF+H media). Filters were washed three times with cold phosphate-buffered saline containing MgCl2 and CaCl2 (PBS; Sigma, D8662), filters were excised with a razor blade, and cells were solubilized in 300 µl detergent lysis buffer (50 mM Tris, pH 8.0, 62.5 mM EDTA, 1% IGEPAL, 4 mg/ml deoxycholate) for 30 minutes on a rotating platform rocker at 4°C. When comparing data from individual clones, cell-associated Alexa Fluor 647 fluorescence intensity was quantified by spectrofluorimetry normalized to the protein concentration of the lysate measured using a DC Protein Assay Kit (Bio-Rad; 5000111).
Albumin Uptake Concentration Curve and Deconvolution
ClC-5 KO and rescue clones were incubated in DF+H media for 15 minutes with Alexa Fluor 647 albumin at concentrations ranging from 10 µg/ml to 10 mg/ml. Albumin uptake was quantified by spectrofluorimetry as described above. Albumin uptake as a function of concentration was fit to a three-component model, and high-affinity and low-affinity uptake component capacities were calculated as previously described.20
Albumin Surface Binding
Cells cultured on filters under orbital shear stress for 72 hours as described above were incubated for 30 minutes on ice with 100-µg/ml Alexa Fluor 647 albumin added apically in DF+H media. Filters were washed 3× rapidly in ice-cold PBS, excised with a razor blade, and cells solubilized in 300-µl detergent lysis buffer for 30 minutes on a rotating platform rocker at 4°C. Surface-bound Alexa Fluor 647 albumin was quantified by spectrofluorimetry.
Western Blotting of Cell Lysates
Filters were washed with cold PBS, excised, and solubilized in 0.3-ml detergent lysis buffer plus 5 µg/ml leupeptin, 7 µg/ml pepstatin A, 1 mM PMSF, and Complete Protease Inhibitor EDTA-Free (Roche, 04693159001; 1 tablet/10 ml of buffer) for 20 minutes on a rotator at 4°C. The protein concentration of the lysate was measured using a DC Protein Assay Kit (Bio-Rad; 5000111). Equivalent amounts of total protein were separated by SDS-PAGE on 4% to 15% Criterion TGX (Bio-Rad; 5671083) gels. Megalin and cubilin were detected with anti-megalin antibody generously provided by Dr. Daniel Biemesderfer and Dr. Peter Aronson (Yale University, MC-220, 1:20,000,21) and polyclonal anticubilin antibody (No. 27445, validated in20,22, 1:5000), respectively.
Quantitative PCR Analysis
Cells were cultured on filter supports as described. Cells were collected using Accutase (Sigma), and RNA was extracted using the Ambion PureLink RNA mini kit (Thermo Fisher) according to the manufacturer's protocol. cDNA was synthesized from 2 µg of total RNA using the Reverse Transcriptase, High-Capacity cDNA Kit (Life Technologies). Gene expression was measured by quantitative PCR (qPCR) using the iTaq Universal SYBR Green Supermix 500 (Bio-Rad) on a CFX Connect Real-Time PCR Detection System (Bio-Rad). Gene primers used in the experiments are listed in Supplemental Table S3. The expression of each gene was normalized to Gapdh expression, and relative fold change was calculated using the equation 2δδCq where δδCq = ClC-5 KO (or uncoupled) δCq − Rescue δCq, and δCq = target gene Cq − Gapdh Cq.
Surface Biotinylation–Based Assays
Endocytosis of Megalin
After washing with cold PBS, the apical surface of cells cultured on permeable supports under shear stress was biotinylated with 1 mg/ml EZ-Link Sulfo-NHS-SS-biotin (Thermo Scientific, 21331) in 0.5-ml TEA-buffered saline (TBS; 10 mM triethanolamine-HCl, pH 7.6, 137 mM NaCl, 1 mM CaCl2) for 2 X 15 minutes on ice. The biotinylation reaction was quenched by washing with DMEM-F12 plus 5% FBS for 10 minutes on ice. Samples were rinsed once with ice-cold DF+H media, then quickly warmed to 37°C by the addition of prewarmed DF+H media and placed on an orbital shaker in the incubator (146 rpm) for 0–5 minutes. Endocytosis was stopped by washing with prechilled PBS on ice. Biotin at the cell surface was stripped by washing cells with prechilled 100 mM MESNA in Stripping Buffer (50 mM Tris-HCl pH 8.6, 100 mM NaCl, 1 mM EDTA, 0.2% BSA) for 2 × 20 minutes. A duplicate 0 minutes time point was left unstripped to estimate the fraction of total megalin at the apical surface at steady state. Residual MESNA was quenched by washing cells with ice-cold DF+H media for 10 minutes on ice. Filters were washed with ice-cold PBS, excised with a clean razor blade, and solubilized in 0.6-ml detergent lysis buffer plus 5 µg/ml leupeptin, 7 µg/ml pepstatin A, 1 mM PMSF, and Complete Protease Inhibitor EDTA-Free (Roche, 04693159001; 1 tablet/10 ml of buffer) for 20 minutes at 37°C. To determine total megalin levels, 5% of the lysate volume was reserved. From the remaining lysate, biotinylated proteins were recovered after precipitation with streptavidin agarose resin (Thermo Scientific, #20353) overnight at 4°C. Samples were analyzed by Western blot after SDS-PAGE on 4%–15% Criterion TGX gels using anti-megalin antibody.
Half-Life of Surface-Biotinylated Megalin
The apical surface of filter-grown cells was biotinylated as above. Cells were rinsed once with ice-cold DF+H media then quickly warmed to 37°C by the addition of prewarmed DF+H media and placed on a rotating shaker in the incubator. At each time point starting from 1 to 8 hours, filters were rinsed in cold PBS, cells were lysed, biotinylated proteins were recovered, and samples were immunoblotted for megalin as described above.
Indirect Immunofluorescence in Cell Lines
Filters were washed in warm PBS and fixed in warm 4% paraformaldehyde (PFA) and 100-mM sodium cacodylate at ambient temperature. After two washes in PBS, the filters were quenched (PBS, 20 mM glycine, and 75 mM ammonium chloride) for 5 minutes and permeabilized for 7 minutes in quench solution containing 0.1% Triton X-100. After being washed with PBS, the filters were blocked with PBS, 1% BSA, and 0.1% saponin and incubated for 1 hour with primary antibody diluted in PBS, 0.5% BSA, and 0.025% saponin (wash buffer). The filters were washed three times, incubated for 30 minutes with secondary antibody diluted in wash buffer, and washed three times. After excising, filters were mounted onto glass slides with ProLong Gold Antifade Mountant (Molecular Probes, P36935) or ProLong Glass Antifade Mountant (Invitrogen, P36980), depending on the immersion fluid of the objective, with a No. 1.5 cover glass (Fisher Scientific, 12541A). Antibodies, sources, and dilutions used for indirect immunofluorescence in OK cells are listed in Supplemental Table S4. For labeling lysosomes, cells were incubated with 100 nM LysoTracker Red DND-99 (Invitrogen, L7528) in DF+H media for 30 minutes on a rotating shaker in the incubator before fixation.
For costaining with primary antibodies from different host species, filters were incubated with both primary antibodies simultaneously followed by both secondary antibodies.23 When costaining with primary antibodies from the same host species (e.g., Rab11 and Megalin or EEA1 and Rab7), labeling was performed sequentially. Filters were incubated with the first primary antibody for 1 h, followed by a fluorophore-conjugated F(ab) fragment for 30 minutes. Filters were then incubated with unconjugated F(ab) fragment diluted in wash buffer for 30 minutes to block any remaining unbound sites on the prior to sequential incubation the second primary and secondary antibodies.
Filters were imaged on a Leica TCA SP5 or Stellaris 8 confocal microscope using a 63× glycerol or oil (respectively) immersion objective (NA 1.4). Images were acquired with a voxel size of 45 × 45 × 130 nm (x, y, z). All images were deconvolved with Huygens Essential version 17.04 using the CMLE algorithm, with signal-to-noise ratio (SNR): 20 and 40 iterations (Scientific Volume Imaging, The Netherlands, http://svi.nl). Colocalization of two channels over the whole z-stack was determined by Manders coefficients using the JACoP plugin for ImageJ without thresholding.24,25 The Manders coefficient represents the fraction or percentage of all the positive pixels in one channel that overlap with positive pixels from another channel.26 In Supplemental Tables S7 and S8, we refer to these measurements with the following nomenclature, using EEA1 and Rab7 as an example: The percentage of total EEA1 pixels that overlap with Rab7 pixels is and the percentage of total Rab7 pixels that overlap with EEA1 pixels is . Megalin is typically abbreviated to when its colocalization is described this way.
The fractional distribution of markers over the z axis was determined by dividing the sum of pixel intensities in each plane by the total pixel intensity in the whole z-stack for each marker. The z position was normalized by dividing by the total number of z-steps in the stack. The fractional distribution of each marker was averaged across all images after interpolation. This analysis was performed in MATLAB R2021a (The MathWorks Inc., Natick, MA).
Degradation Kinetics of 125I-Albumin
Radioiodination of Albumin
Desalting columns (Thermo Scientific #89889) were prepped by washing 4× with 1% BSA (Sigma #A7030) in PBS followed by washing 2× in PBS. To begin the iodination reaction, 20 µl of 670 ng/ml iodine monochloride (ICl; Sigma #208221) in 2 mM NaCl was added to a prechilled reaction tube with 100 µl of 1 M Tris, pH 8.0 on ice. 130–160 µCi of Iodine-125 radionucleotide (125I; Perkin Elmer #NEZ033002MC) was added to the reaction tube and incubated on ice for 1 minute. The equivalent of 20 µl of 50 mg/ml of fatty acid–free bovine serum albumin (Sigma #A7030) in PBS was added to the reaction for iodination and incubated on ice for 10 minutes. To stop the reaction, 100 µl of 5 mg/ml sodium iodide (NaI; Sigma #S9538) in PBS was added. The entire reaction was loaded onto the previously prepped desalting column and washed with 300 µl of PBS, and the flow-thru was discarded. The iodinated albumin was eluted with 600 µl of PBS. To determine the efficiency of iodination, 10 µl of the flow-thru was counted in a gamma counter (Perkin Elmer Wizard2).
Quantitation of 125I-Albumin Degradation Kinetics
Cells were cultured on filter supports as described. Filters were washed 1× in DF+H media incubated with 125I-albumin in DF+H media for 7.5 minutes at 37°C under continuous orbital shear stress. Filters were washed 1× and chased in DF+H media for 10, 20, 30, 40, or 60 minutes. At each time point, the apical and basal media were collected, and the filters were excised and lysed in 500 µl of detergent lysis buffer for 20 minutes on ice. Apical media, basal media, and cell lysates were precipitated by adding ice-cold 100% trichloroacetic Acid (TCA) to a final concentration of 20% and incubated on ice for 10 minutes. Solutions were pelleted at 13,200 rpm for 10 minutes at 4°C, and 500 µl of each supernatant was transferred to 1.2 ml tubes (USA Scientific #1412-0000). Any remaining supernatant was carefully discarded, and the pellet was allowed to dry on the benchtop. Dry pellets were resuspended with 500 µl of either DF+H media (from apical or basal media) or detergent lysis buffer (from cell lysate) and transferred to 1.2 ml tubes. The amount of 125I-albumin in each sample was quantified in a gamma counter.
To calculate the fraction of internalized 125I-albumin degraded at each chase time point, all the counts in the supernatant and pellet from the apical media, basal media, and cell lysate were summed to determine the total internalized.27 The counts in just the supernatant from apical media, basal media, and cell lysate were summed to determine the total degraded. The fraction of internalized 125I-albumin degraded at each time was determined by dividing the total degraded by the total internalized. For both rescue and ClC-5 KO cells, these data were plotted versus time and fit to a logistic growth curve with the form: where is the fraction degraded, is the maximum value, is the starting value, and is the rate constant (min−1). We set and as shared between the rescue and ClC-5 KO cells, given that no albumin is degraded to start, and all the albumin is eventually degraded. We tested whether is shared between the two cell lines with the extra sum-of-squares F test. All analysis was performed with GraphPad Prism 9.3.1 (GraphPad Software, San Diego, CA).
Quantification of Endosomal Chloride Concentration
Cells were cultured on filter supports as described. After washing once with DF+H media, filters were excised and incubated with 2 mg/ml Alexa Fluor 647 dextran (AF 647; Invitrogen, D22914) in DF+H media for 15 minutes at ambient temperature. Filters were washed in PBS once and incubated in mixture of 20 mg/ml 10,10′-Bis[3-carboxypropyl]-9,9′-acridiniumdinitrate di-NHS ester- (BAC; EMP Biotech, AF-0406-D005.0-001;28)–conjugated dextran (Thermo Fisher Scientific, D1860) and 1 mg/ml tetramethylrhodamine (TMR) (Thermo Fisher Scientific, 46406)–conjugated dextran in DF+H media for 3 minutes. Filters were rapidly washed 4× in PBS and once in Opti-MEM (Gibco, 51985-034) and then imaged immediately on a Leica SP8 confocal microscope using a 40× water objective (NA 1.1). Uptake of AF 647 dextran was used to determine cell viability and the ideal focal planes for imaging so that three sequential stacks were selected blindly. Imaging parameters were optimized to minimize bleaching and allow for subsequent deconvolution: 7.5 × zoom, 0.17 µm step size, 1400 hz, bidirectional scanning. Sequential scanning was used to avoid bleed through with BAC excited with 405 nm laser (em 419–550 nm) and TMR with 552 nm laser (em 582–620 nm).
All images were deconvolved with Huygens Essential version 17.04 using the CMLE algorithm, with SNR: 10 and 40 iterations. The deconvolved images were used to select regions of interest (ROIs) containing intracellular punctae (endosomes) to omit background fluorescence signal in the extracellular space. Each ROI was a sum projection of three planes of region cropped from the deconvolved and raw images in ImageJ. The deconvolved images for each ROI were used to generate a mask only of endosomes containing both BAC and TMR signal. The ratio of TMR to BAC intensities was quantified from the raw images within each compartment identified by the mask. Generation of the mask and calculation of intensity ratio were performed using MATLAB R2021a.
A calibration curve of TMR to BAC intensity ratios over a range of [Cl−] was generated. After initial imaging, cells preloaded with BAC dextran and TMR dextran were incubated in buffers with chloride concentrations of 0, 25, and 50 mM (120 mM KCl/KNO3, 20 mM NaCl/NaNO3, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES pH 7.4; with NO3- replacing Cl−) with 10 µM each of nigericin, valinomycin, and monensin (Sigma, M5273) and 5 µM CCCP (Sigma, C2759) for 6 minutes. Images were collected, and the ratios of TMR to BAC intensities in intracellular spots were quantified as described for the experimental conditions. The ratios of TMR to BAC intensity were plotted versus chloride concentration and fit by linear regression in GraphPad Prism 9.3.1 (Supplemental Figure S7, A and B). This line was used to convert the TMR to BAC intensity ratios measured in early endosomes of live cells to [Cl−] (mM).
Quantification of Endosomal pH
Cells were cultured on filter supports as described. After washing with DF+H media, filters were excised and incubated with 2 mg/ml Alexa Fluor 568 dextran (AF 568; Invitrogen, D22912) in DF+H media for 15 minutes at ambient temperature. Filters were washed in PBS once and incubated in mixture of 6 mg/ml FITC dextran (Invitrogen, D1821) and 1.125 mg/ml AF 647 dextran (Invitrogen, D22914) in DF+H media for 3 minutes. Filters were rapidly washed 4× in PBS and once in Opti-MEM and then imaged immediately on a Leica SP8 confocal microscope using a 40× water objective (NA 1.1). Environmental chamber was set at 25°C to slow endocytic process to facilitate imaging of early endosomes. Uptake of AF 568 dextran was used to determine cell viability and the ideal focal planes for imaging so that three sequential stacks were selected blindly. Imaging parameters were optimized to minimize bleaching and allow for subsequent deconvolution: 7.5 × zoom, 0.17 µm step size, 1800 hz, bidirectional scanning with FITC excited with 488 nm laser (em 494–551 nm) and AF 647 with 633 nm laser (em 639–689 nm).
All images were deconvolved with Huygens Essential version 17.04 using the CMLE algorithm, with SNR: 10 and 40 iterations. The deconvolved images were used to select ROIs containing spots (endosomes) to avoid quantifying extracellular regions. Each ROI was a sum projection of three planes of region cropped from the deconvolved and raw images in ImageJ. The deconvolved images for each ROI were used to generate a mask only of endosome spots containing both FITC and 647 signal. The ratio of FITC to 647 intensities was quantified from the raw images within each spot identified by the mask. Generation of the mask and calculation of intensity ratio were performed using MATLAB R2021a.
A calibration curve of FITC to 647 intensity ratios over a range of known pH values was generated using the intracellular pH calibration buffer kit (Invitrogen P35379) according to manufacturer's instructions. After initial imaging, cells preloaded with FITC dextran and AF 647 dextran were incubated with each pH buffer (4.5, 5.5, 6.5, and 7.5) with 10 µM each of nigericin and valinomycin for 6 minutes. Images were collected, and the ratios of FITC to AF 647 intensities in intracellular spots were quantified as described for the experimental conditions. The ratios of FITC to AF 647 intensities within endosomes were plotted versus pH values and fit with an exponential growth curve by nonlinear regression in GraphPad Prism 9.3.1 (Supplemental Figure S7, C and D). This curve was used to convert FITC to AF 647 ratios measured in early endosomes to pH values.
Mathematical Model of Megalin Trafficking in Cell Culture Models of Dent Disease
We adapted our previously described comprehensive model of megalin traffic in parental OK cells29 to predict kinetic trafficking rates for rescue, ClC-5 KO, and uncoupled cells. The model describes the traffic of megalin from the apical surface through intracellular compartments of PT cells. Kinetic data from biotinylation-based assays and colocalization data collected from rescue, ClC-5 KO, and uncoupled cells were used in the estimation of kinetic parameters as previously described29 with minor modifications described below.
Endocytic Rate
The biotinylated megalin remaining at the surface during the brief endocytosis period can be represented as:
| (1) |
where is the biotinylated megalin remaining at the surface at time and . The data obtained from the endocytosis biotinylation assay represent the percent of surface megalin that is internalized over time and is equivalent to . To estimate the endocytic rate of megalin, the data from 0 to 5 minutes were log-transformed and fit with a simple linear regression model such that:
| (2) |
The slopes of the lines of best fit for rescue and ClC-5 KO cells were compared with the extra sum-of-squares F-test to see whether they were significantly different. With a P-value=0.3860, it was determined that the slopes were the same for both datasets. A line was fit to the dataset combining data from both ClC-5 KO and rescue cells, and the shared fractional endocytic rate is equivalent to the slope. Then, we tested whether the slope of the line of best for data from uncoupled cells was significantly different from the shared value determined from rescue and ClC-5 KO cells with the extra sum-of-squares F-test. With a P-value=0.0045, it was determined that the slope was significantly different, and therefore, the fractional endocytic rate in uncoupled cells is significantly different from that of the rescue and ClC-5 KO cells. Statistical analysis was performed in GraphPad Prism 9.3.1. The fit endocytic rates are shown in Supplemental Table S8.
Intracellular Trafficking Rates
The steady state distribution of megalin among endocytic compartments was calculated using the megalin colocalization values in Supplemental Table S7 and the colocalization values for the overlap between different combinations of EEA1, Rab7, and Rab11a in Supplemental Table S8 as described previously. Standard error was propagated accordingly. For overlap values with LysoTracker, we used the same values we found in the OK parental cells. This steady state distribution was used to estimate intracellular kinetic rates with the assumption that 51% of recycling megalin traffics through apical vacuoles (AVs) () for all cell lines.29 The predicted fractional rates for all three cell lines are shown in Supplemental Table S8. Supplemental Figure S7, C–E shows how the values of , , change with the value of for rescue, ClC-5 KO, and uncoupled cells. At every value of , the estimated value of and is smaller in ClC-5 KO and uncoupled cells compared with rescue cells.
CRISPR/Cas9 ClC-5 KO Mice
All animal protocols conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. To generate a CRISPR/Cas9 ClC-5 KO mouse line, 129-Elite Mouse 129S2/SvPasCrl (Charles River) zygotes freshly produced by in vitro fertilization were injected with a mixture of 0.33-µM EnGen Cas9 protein (New England Biolabs, Cat.No. M0646T), the Clcn5-E211-guide3 sgRNA (21 ng/µl) and the Clcn5-E211X-HDR ssODN (0.5 µM). The next day, 2-cell embryos were transferred to the oviducts of pseudopregnant CD1 female recipients. After gestation, tail genomic DNA for the CRISPR edited pups were tested by PCR and sequencing. We identified a founder male carrying a 189 bp deletion that includes half of exon 6 and the intron junction. This mutation prevents splicing of intron 6 results in KO of ClC-5. The founder mouse was backcrossed to WT C57BL/6J mice for three generations to produce offspring carrying the mutation. All oligos used in the generation and characterization of this mouse line are listed in Supplemental Table S5.
Male WT and ClC-5 KO littermates from the N3 generation (approximately 10–12 weeks old; five of each genotype) were anesthetized using isoflurane. Urine was collected directly from the bladder after opening of the body cavity. We were unable to collect urine from one ClC-5 KO mouse. The mice were perfused intracardially with cold PBS, the right kidneys were removed, and blood vessels were clamped. The right kidneys were rinsed in cold PBS and stored at 4°C. Then, left kidneys were fixed initially by perfusion with cold 4% PFA in cacodylate buffer (pH 7.4). The upper and lower poles of the left kidneys were removed and fixed further for 2 hours in 4% PFA on ice, for immunofluorescence. After fixation, tissue was dehydrated in 30% sucrose and embedded in O.C.T. compound (Scigen 4583) at −80°C for sectioning. The centers of each were further fixed in 10% formalin in PBS overnight at ambient temperature and then dehydrated in 70% ethanol. These were paraffin embedded, sectioned, and stained with hematoxylin and eosin (H&E) by the University of Pittsburgh Biospecimen Core.
Western Blotting
With 30 minutes of harvest, the capsule and then cortex of the right kidneys were removed. The cortex was pressed through a 150-µm sieve and collected in cold PBS. Approximately one quarter of the collected tissue was spun down at 2000g for 2 minutes at 4°C. The supernatant was carefully removed, and the pelleted tissue was lysed in 1 ml detergent lysis buffer plus 5 µg/ml leupeptin, 7 µg/ml pepstatin A, 1 mM PMSF, and Complete Protease Inhibitor EDTA-Free (Roche, 04693159001; 1 tablet/10 ml of buffer) for 30 minutes on a rotator at 4°C. The protein concentration of the lysate was measured using a DC Protein Assay Kit (Bio-Rad; 5000111). Equivalent amounts of total protein were separated by SDS-PAGE on 4% to 15% Criterion TGX gels. Megalin, cubilin, NHE3, and SGLT2 were detected with anti-megalin antibody generously provided by Dr. Daniel Biemesderfer and Dr. Peter Aronson (Yale University, MC-220, 1:20,000,21), anticubilin antibody (No. 27445, validated in20,22, 1:5000), and anti-SGLT2 antibody (Abcam, ab85626, 1:500), respectively.
Proteinuria Analysis
Equal volumes of urine were separated by SDS-PAGE on 4%–15% Criterion TGX Stain-Free gels (Bio-Rad, 5678084) to visualized total protein in urine. Albumin and beta-2 microglobulin (β2m) were detected with antialbumin antibody (Abcam, ab207327, 1:1000) and anti-β2m antibody (Cell Signaling, 59035, 1:1000), respectively. Creatinine concentration was determined using the Creatinine Enzymatic kit (Pointe Scientific, C7548-480) after a 1:20 dilution according to manufacturer's instructions.
Indirect Immunofluorescence in Mouse Kidney Sections
Cryostat sections (10 µm) from three mice of each genotype were placed on slides, rehydrated in PBS for 30 minutes, permeabilized with 0.1% Triton X-100 for 10 minutes, and blocked with 1% BSA, 5% normal goat serum, and 0.1% saponin for 15 minutes. Sections were stained sequentially and blocked to prevent cross-species binding of goat anti-mouse secondary to rat primary. Sections were blocked again with 100 µg/ml goat anti-mouse unconjugated F(ab) fragments (Jackson Immuno, 115-007-003) in 0.5% BSA and 0.1% saponin for 15 minutes. Sections were incubated for 1 hour with mouse anti-megalin antibody (Santa Cruz, sc-515750, 1:100) washed three times for 5 minutes each in wash buffer (1× PBS, 0.5% BSA, and 0.025% saponin) and incubated for 30 minutes with goat anti-mouse F(ab) fragment Alexa Fluor 647 secondary (Jackson Immuno, 115-547-003, 1:500). After being washed three times in wash buffer, sections were blocked with 100 µg/ml goat anti-mouse in 0.5% BSA and 0.1% saponin for 15 minutes. After rinsing with PBS, sections were incubated for 1 hour with rabbit anti-SGLT2 antibody (Abcam, ab85626, 1:400) and rat anti-LAMP1 antibody (DSHB, 1D4B, 1:100), washed three times for 5 minutes each in wash buffer and incubated for 30 minutes with goat anti-rabbit Alexa Fluor 568 secondary (Invitrogen, A11036, 1:500) and goat anti-rat Alexa Fluor 488 secondary (Invitrogen, A11077, 1:500). After washing two times in wash buffer and once in PBS, sections were mounted using with ProLong Glass Antifade Mountant with a No 1.5 cover glass (Thermo Scientific, 3422).
Sections were imaged on the Leica SP8 confocal microscope with a 63× oil objective (NA 1.4). Images were acquired with a voxel size of 40 × 40 × 130 nm (x, y, z). Images were deconvolved with Huygens Essential version 17.04 using the CMLE algorithm, with SNR: 20 and 40 iterations. Masks were drawn by hand in ImageJ to only include the region within the end-on tubule of interest and to exclude background binding of antimouse secondary to the outside of tubules (see Supplemental Figure S6F). The colocalization of two channels over the entire z-stack only within the mask region was determined by Manders coefficients, which were calculated using MATLAB R2021a.
To determine the average apical to basal fractional distribution of SGLT2, megalin, and LAMP1, line scans were drawn on single planes of merged images in ImageJ. Lines were drawn beginning in the lumen above SGLT2 staining through cells perpendicular to the megalin staining ending past the LAMP1 staining. The fractional distribution of each channel was calculated by dividing the intensity profiles along the length of the lines by the sum intensity of each line. Lines were aligned by the location of the SGLT2 maximum and averaged after interpolation. The alignment and averaging of lines were performed using MATLAB R2021a.
Axial Megalin Distribution
Tile scans of WT and ClC-5 KO mouse kidney sections stained for megalin and SGLT2 were imaged on the Leica SP8 confocal microscope with a 40× oil objective (NA 1.3). Images were acquired with a voxel size of 446 × 446 × 1000 nm (x, y, z). The individual tiles were merged into a single stack. Sum projections were generated for each stack. In ImageJ, line scans were drawn across longitudinally oriented proximal tubules perpendicular to the outside edges in the sum projections. The intensity information at each point along the length of the line was stored. For each line, the intensity along the line was summed between the local maxima that correspond to the outside edges of the tubule using MATLAB R2021a. Bright staining on the outside of the tubule is due to background binding of the secondary and served as a marker for the outside edge. SGLT2 staining was used to delineate between tubules corresponding to the S1 and S2 segments. SGLT2 positive tubules were defined as S1. SGLT2 negative tubules in the cortical region, if possible, within medullary rays, were defined as S2.
Axial Model of Protein Uptake along the PT
Model predictions of albumin and β2m fluxes along the PT were obtained using a published model of protein uptake.30 The model distinguishes between receptor-mediated endocytosis using cubilin, megalin, as well as fluid-phase uptake. All parameters for WT mice were kept unchanged except for the affinity of cubilin to β2m (set to 223 mM herein), which we adjusted to better match measurements of β2m fractional urinary excretion under normal conditions. In the simulations of protein uptake in CLC-5 KO mice, we (1) increased the GFR by 31%, relative to WT mice, based on the averaged measured increase in creatinine excretion; (2) modified the relative distribution of cubilin and megalin in S1, S2, and S3, based on measured differences in axial expression; and (3) lowered the maximum uptake capacity () of cubilin and megalin by 60% each, to match the measured change in the fractional urinary excretion of albumin.
To determine the distribution of megalin (and cubilin) in CLC-5 KO mice, we multiplied the expression in S1, S2, and S3 in WT mice by the KO-to-WT intensity ratio in that segment (e.g., by 7863/14,786 in S1, by 5555/15,837 in S2, and by the average 0.438 in S3). We then recalculated the fractional receptor expression in each segment. The WT and ClC-5 KO receptor distributions are listed in Supplemental Table S6.
Results
Generation of ClC-5 KO and Rescue PT Cell Models
We used CRISPR/Cas9 technology to generate six ClC-5 KO clones in OK cells (Figure 1, A and B and Supplemental Figure S1A). All clones had markedly reduced levels of albumin uptake (>50%) compared with parental OK cells (Supplemental Figure S1B). Uptake of albumin was fully rescued by the expression of HA-tagged human WT ClC-5 in the two rescue clones we isolated (Figure 1, C and D and Supplemental Figure S1B). Our remaining studies primarily compared ClC-5 KO and rescue cell lines to minimize effects of clonal variability.
Figure 1.
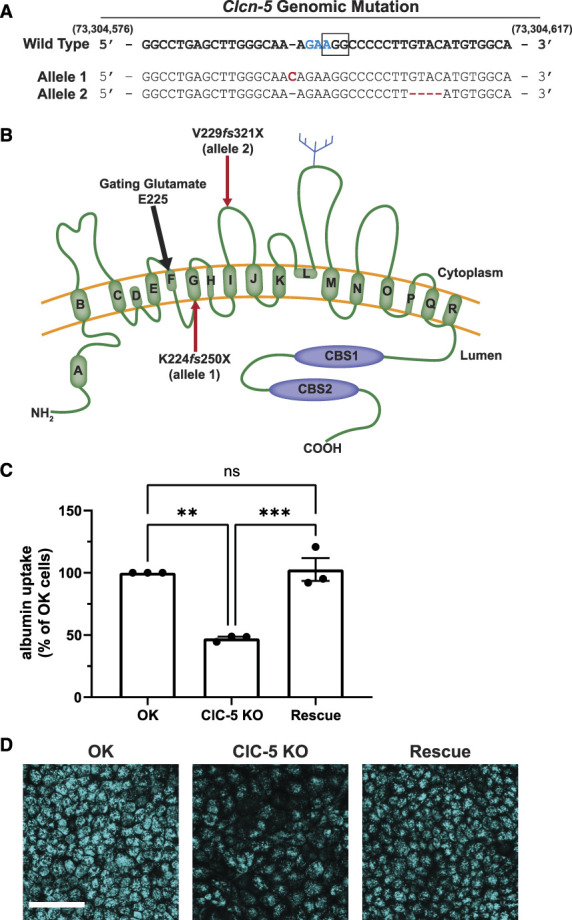
CRISPR/Cas9 KO of ClC-5 impairs albumin uptake in OK cells and is rescued by the expression of human wild-type (WT) ClC-5. Two sgRNAs were designed to target the region containing the gating glutamate of ClC-5 in OK cells. (A) Genomic sequence showing allelic insertion and deletion mutations (red) predicted to result in early termination of protein translation in ClC-5 knockout clone 1 (KO cl.1). The WT genomic DNA sequence correlates to nucleotides 73,304,582-73,304,617 in the MonDom5 genome. The codon encoding the gating glutamate in ClC-5 is highlighted in blue, and the PAM sequence for CRISPR/Cas9 guide 2 used to generate clones is denoted by the black box. Cells from KO cl.1 were transfected with constructs encoding human wild-type HA-tagged ClC-5 (rescue). (B) Schematic representation of the ClC-5 monomer protein structure. The gating glutamate (approximate location indicated by the black arrow) is predicted to correspond to amino acid 225 in the opossum sequence (211 in human and mouse) within the CLC Voltage Gated Domain. Red arrows indicate approximate location of early termination sites resulting in the two mutant alleles in KO cl.1. (C) Cells from the OK parental population, KO cl.1, and rescue cl.1 were incubated for 30 minutes with 40 µg/ml Alexa Fluor 647 albumin and then solubilized. Uptake of fluorescent albumin was quantified by spectrofluorimetry, normalized to the protein concentration of the cell lysate, and then normalized to uptake in the OK parental population. **P=0.001 and ***P=0.0008 (n=3 experiments) using the one-way ANOVA Tukey multiple comparison test. (D) Representative maximum projection images of fluorescent albumin uptake after 30 minutes incubation with 40 µg/ml Alexa Fluor 647 albumin in parental OK, KO cl.1, and rescue cl.1 cells. Scale bar: 50 µm.
Consistent with the overall reduction in albumin uptake, ClC-5 KO cells had 38% fewer surface binding sites for albumin compared with rescue cells (Supplemental Figure S2A). Moreover, deconvolution of albumin uptake concentration curves revealed comparable reductions in the uptake of albumin using high-affinity binding to cubilin versus lower-affinity uptake mediated by megalin (Supplemental Figure S2, B and C).20 We conclude that KO of ClC-5 results in coordinate effects on the trafficking and stability of both megalin and cubilin receptors.
Megalin Expression and Trafficking Kinetics in ClC-5 KO and Rescue Cells
Western blotting revealed that ClC-5 KO cells have 16% and 20% reductions in total megalin and cubilin, respectively, compared with rescue cells (Figure 2A, Supplemental Figure S2D), with no change in mRNA (Figure 2B). In addition, at steady state, the fraction of total megalin present at the apical surface of ClC-5 KO cells was numerically lower but not statistically significant when compared with rescue cells (5.1% versus 6.2%, P-value=0.1463, Figure 2C, equating to approximately 31% fewer apical receptors). We did not measure a difference in the fractional endocytic rate of surface megalin (Figure 2D). Thus, we estimate that 31% fewer megalin receptors are internalized per unit time in ClC-5 KO compared with rescue cells. Quantitation of the loss of apically biotinylated megalin over time revealed a significantly faster rate of decay and a shorter half-life in ClC-5 KO compared with rescue cells when fit with a simple exponential decay model (P-value=0.0228; t1/2=3.49 hours versus 4.58 hours, respectively) (Figure 2E). The increased degradation kinetics without any change in the fractional endocytic rate suggests that loss of ClC-5 impairs the trafficking of megalin through intracellular compartments.
Figure 2.
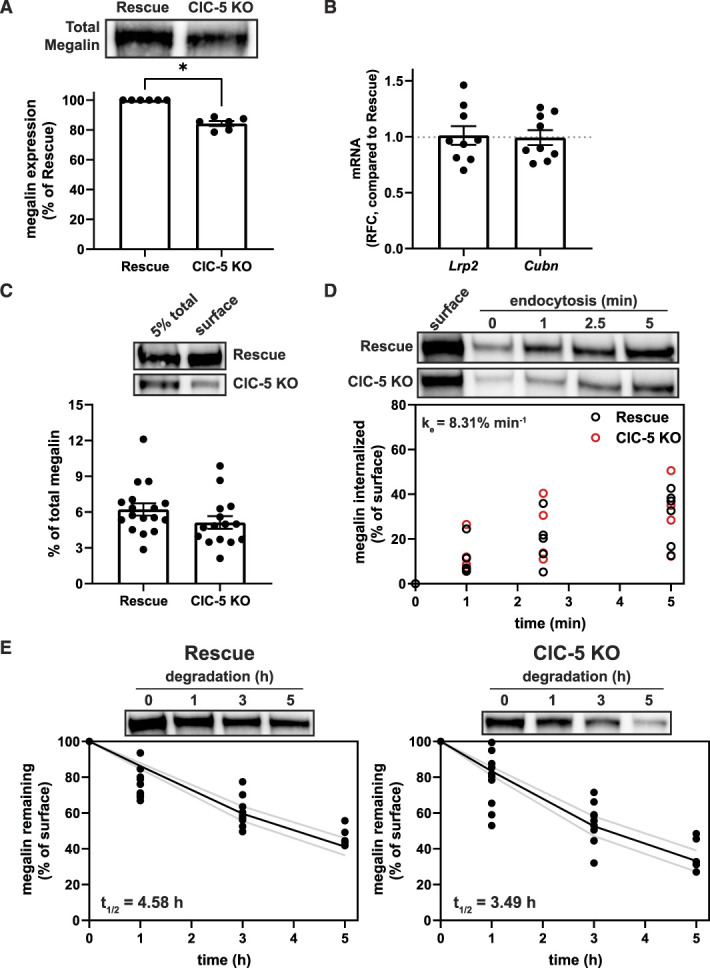
Expression and trafficking of megalin is impaired in ClC-5 KO cells. (A) Equivalent amounts of protein from rescue and ClC-5 KO cell lysates were blotted for megalin. The quantified band intensity for each independent experiment was normalized to rescue cells. A representative blot is shown above the graph. *P=0.0169 (n=6 experiments) using one-way ANOVA Tukey multiple comparison test. (B) megalin (Lrp2) and cubilin (Cubn) transcripts in rescue and ClC-5 KO cells were quantified by quantitative PCR and normalized to Gapdh transcript levels. Relative fold change (RFC) of ClC-5 KO cells compared with rescue cells (normalized to 1; dotted line) is plotted. (C) The apical surfaces of rescue and ClC-5 KO cells were biotinylated, and total (5%) and surface megalin were quantified by blotting. Representative blots are shown above the graph. (D) Endocytosis kinetics of rescue and ClC-5 KO cells were quantified using the biotinylation stripping approach described in Methods, and internalized megalin was quantified as a percentage of surface megalin at t=0. Data from five ClC-5 KO and six rescue experiments are plotted and were used to calculate the fractional endocytic rate for rescue and ClC-5 KO cells as described in Methods, shown in upper left corner. Representative blots are shown above the graph. (E) Data from 11 independent experiments were used to fit (black line) the degradation rate of apically biotinylated megalin in rescue and ClC-5 KO cells using estimates of fractional endocytic rate (Figure 2D and Supplemental Table S8) and the fraction of megalin at the apical surface (gray lines: 95% CI). The estimated half-life of surface megalin is shown in the bottom left corner of each graph. Representative blots are shown above each graph.
Distribution of Intracellular Megalin in Endocytic Compartments
To explore which trafficking step(s) are affected by ClC-5 KO, we determined the intracellular distribution of megalin. Rescue and ClC-5 KO cells were costained to label megalin with markers of apical early endosomes (AEEs; EEA1), AVs (Rab7), dense apical tubules (DATs; Rab11a), and lysosomes (LysoTracker), and colocalization was quantified (Figure 3, Supplemental Table S7). The fraction of megalin colocalizing with EEA1 was significantly higher in ClC-5 KO versus rescue cells (15.1% versus 8.31%, P-value=0.0437).
Figure 3.
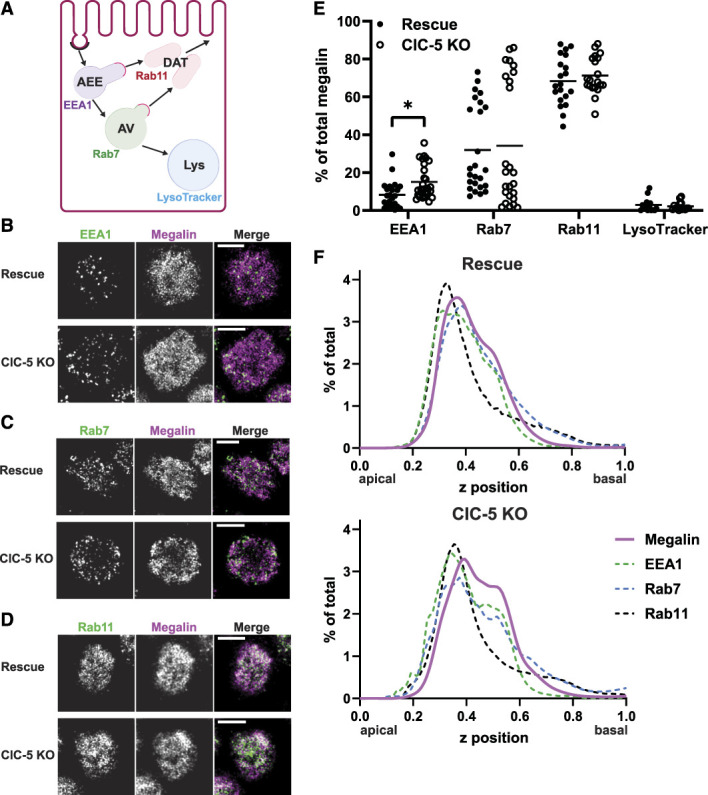
Steady state distribution of intracellular megalin is altered in ClC-5 KO cells. (A) Schematic of markers used to label endocytic compartments in cells: apical early endosomes (AEEs, labeled with anti-EEA1), apical vacuoles (AVs, labeled with anti-Rab7), dense apical tubules (DATs, labeled with anti-Rab11a), and lysosomes (labeled with LysoTracker Red). Adapted from Shipman et al., 2022, Function.29 (B–F) Rescue and ClC-5 KO cells on permeable supports were fixed and processed to detect megalin colocalization with the markers shown in panel A. Representative sum projection images of six planes were cropped to show a region of high colocalization of megalin with (B) EEA1, (C) Rab7, and (D) Rab11a within a single cell. Scale bars: 5 µm. Note the doming of the apical surface in these cells where subapical endocytic compartments are concentrated. (E) Megalin colocalization with each marker was quantified by Manders coefficient over the entire z-stack and plotted as the percent of total megalin. Each point represents a single z-stack image. *P=0.0437 (n=23–27) using the one-way ANOVA Tukey multiple comparison test. (F) The average fractional distribution of EEA1 (n=51–52), Rab7 (n=55), Rab11a (n=50), and megalin (n=82) over the z axis of a cell, from 0 (apical) to 1 (basal), is plotted for rescue (top) and ClC-5 KO cells (bottom). Note the basolateral shift in megalin distribution in ClC-5 KO compared with rescue cells.
Apical endocytic compartments mature rapidly in PT cells, resulting in overlap between endocytic markers. Manders coefficients were quantified in rescue and ClC-5 KO cells costained to label pairs of endocytic markers (Supplemental Figure S3 and Supplemental Table S8). Compared with rescue cells, ClC-5 KO cells had significantly reduced overlap between EEA1 and Rab11a, consistent with a reduction in DATs exiting from AEEs (Supplemental Figure S3B).
We also quantified the average fractional distribution over the z axis of megalin, EEA1, Rab7, and Rab11 in ClC-5 KO and rescue cells (Figure 3F). There was a pronounced shift of megalin, EEA1, and Rab7 toward the basolateral aspect of ClC-5 KO cells compared with rescue cells. This profile is consistent with slower conversion of AEEs to AVs and increased retention of megalin in sorting rather than recycling compartments.
Mathematical Model of Megalin Traffic in ClC-5 KO Versus Rescue Cells
We integrated our kinetic and colocalization data to adapt a mathematical model that predicts intracellular trafficking rates of megalin in ClC-5 KO and rescue cells29 (Figure 4A). We used colocalization data from pairs of markers (Supplemental Figure S3) to estimate the true steady state distribution of megalin among endocytic compartment as described in Methods (Figure 4B). In both ClC-5 KO and rescue cells, more than half of total megalin is localized to DATs at steady state. However, ClC-5 KO cells have a reduced fraction of megalin at the surface and an increased fraction localized to AEEs.
Figure 4.
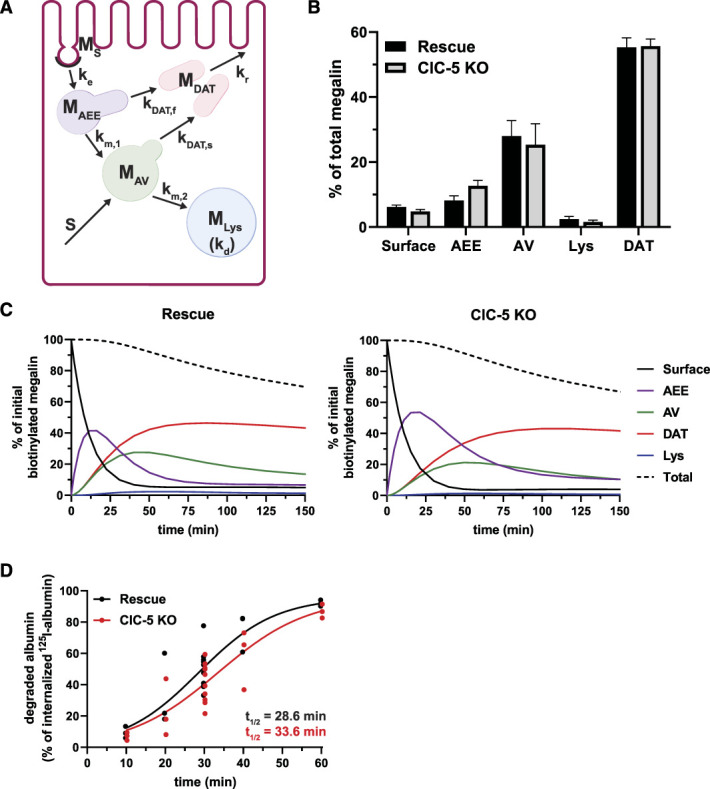
Kinetic model of megalin trafficking reveals impaired endosome maturation in ClC-5 KO cells. (A) Graphical representation of the model of megalin traffic displaying endosomal compartments with coefficients denoting the kinetic rates of megalin traffic between these compartments. Megalin is internalized from the apical surface into AEEs at rate . Megalin within AEEs enters DATs through the fast recycling route at rate or matures to AVs at rate . Megalin within AVs enters DATs through the slow recycling route at rate or is transferred to lysosomes at rate . Megalin within lysosomes is degraded at rate , and newly synthesized megalin () enters the system through AVs. Adapted from Shipman et al., 2022, Function.29 (B) Megalin steady state distribution among the endocytic compartments, surface, AEEs, AVs, DATs, and lysosomes (Lys), in rescue and ClC-5 KO cells (mean±SEM), which was calculated based on the average fractional colocalization of megalin with each marker in Figure 3 and the overlaps between the markers in Supplemental Figure S3. (C) The predicted temporal route of megalin biotinylated at the apical surface through each compartment in rescue (left) and ClC-5 KO (right) cells is plotted. In this simulation, the synthesis rate is set to zero because no newly biotinylated megalin is created after initial labeling. The total remaining biotinylated megalin over time is plotted as the black dashed line. Note the greater accumulation of biotinylated megalin in AEEs and the delayed accumulation in DATs in ClC-5 KO cells compared with rescue. (D) Rescue and ClC-5 KO cells were incubated with 125I-albumin and chased. At each time point the apical and basal media was collected cells were lysed. The fraction of degraded 125I-albumin at each time point was determined after TCA precipitation and fit to a logistic growth curve. The rate constant is significantly smaller in ClC-5 KO cells compared with rescue cells: 0.0873 versus 0.1025 min−1. P-value= 0.0195 using the extra sum-of-squares F test.
Using our calculated steady state distributions and experimentally measured rates in Figure 2, we estimated the intracellular trafficking kinetic rates in ClC-5 KO and rescue cells (Supplemental Table S9). The predicted kinetic rates indicate that endosome maturation is considerably delayed in ClC-5 KO cells, with 47% reductions in the fractional rates of megalin traffic to AVs and DATS from AEEs ( and ) compared with rescue cells. Figure 4C tracks the predicted temporal distribution of megalin molecules biotinylated at the apical plasma membrane at time 0 minute. In ClC-5 KO cells, there is an increased accumulation and longer residence time of megalin in AEEs. This results in a delay in the accumulation of megalin in DATs, corresponding to a decrease in the overall recycling of megalin per unit time.
Consistent with the delayed endosome maturation in ClC-5 KO cells predicted by our model, the measured rate of degradation of internalized 125I-albumin was significantly slower in ClC-5 KO cells compared with rescue cells, although the fate of degraded products was similar (Figure 1D). In both cell lines, albumin was almost entirely degraded by 1 hour after internalization, and primarily only degraded albumin was released into the apical and basal media (Supplemental Figure S4).
An Uncoupled ClC-5 Mutant Disrupts Endocytic Traffic to a Greater Extent than ClC-5 KO
Alterations in endosomal Cl− homeostasis have been suggested to underpin the pathologic defect in Dent disease.10 To explore this, we generated clones of ClC-5 KO cells stably expressing the disease-causing mutant ClC-5E211G12 (Figure 3A). Surprisingly, expression of this uncoupled mutant in ClC-5 KO cells resulted in a further reduction in both albumin uptake (nearly 70% reduction versus rescue cells) and megalin expression (32% reduction), with no change in cell morphology or megalin mRNA levels (Figure 3, B and C, Supplemental Figure S5, A–C). The fraction of total megalin localized to the apical surface in uncoupled cells was similar to that in rescue cells (6.7% versus 6.2%, respectively; Supplemental Figure S5D; compare with Figure 2C), translating to an estimated 30% reduction in apical megalin receptors. Consistent with this, surface binding sites for albumin were comparably reduced in ClC-5 KO and uncoupled cell lines (Supplemental Figure S5E).
The fractional endocytic rate of megalin was reduced by 25% in uncoupled cells compared with rescue and ClC-5 KO cells (P-value=0.0045; Figure 3D; compare with Figure 2D). This results in 43% fewer megalin receptors internalized per unit time compared with rescue cells. Consistent with its reduced endocytic rate, the half-life of apically biotinylated megalin in uncoupled cells is longer than in ClC-5 KO and comparable with rescue cells (4.51 hours, Supplemental Figure S5F). Quantitative imaging revealed a significantly greater fraction of megalin colocalized with EEA1 (17.5%) and significantly smaller fraction of megalin colocalized with Rab11a (54.1%) (Supplemental Figure S5E and Supplemental Table S7) with no difference in the overlap of endocytic markers themselves compared with rescue cells (Supplemental Figure S6, A–D and Supplemental Table S8). Along the z axis, distributions of megalin, EEA1, and Rab7 exhibited greater basolateral shifts in uncoupled cells compared with both ClC-5 KO and rescue cells (Supplemental Figure S6E). We quantified the number and total area of EEA1-positive compartments per cell (Supplemental Figure S6, F and G). Although not statistically significant, the average EEA1-positive areas were higher in both the ClC-5 KO and uncoupled cell lines. Together, these data point to a more pronounced impairment in megalin traffic in uncoupled versus ClC-5 KO cells because of a reduced rate of endocytosis in addition to prolonged retention within the endolysosomal system.
Supplemental Table S9 shows the intracellular trafficking rates predicted by our kinetic model of megalin traffic in rescue, ClC-5 KO, and uncoupled cells as a percentage of megalin trafficked from the originating compartment per minute. Figure 5E graphically depicts the steady state distribution and trafficking rates of megalin receptors in rescue, ClC-5 KO, and uncoupled cells for every 10,000 megalin molecules in a rescue cell. Despite fewer total molecules, more megalin is present in AEEs of ClC-5 KO and uncoupled cells, and fewer molecules are trafficked from this compartment to DATs or AVs. Moreover, although fewer megalin molecules are internalized/min in uncoupled cells, they have an even greater accumulation of megalin in AEEs and even slower AEE exit rates than ClC-5 KO cells.
Figure 5.
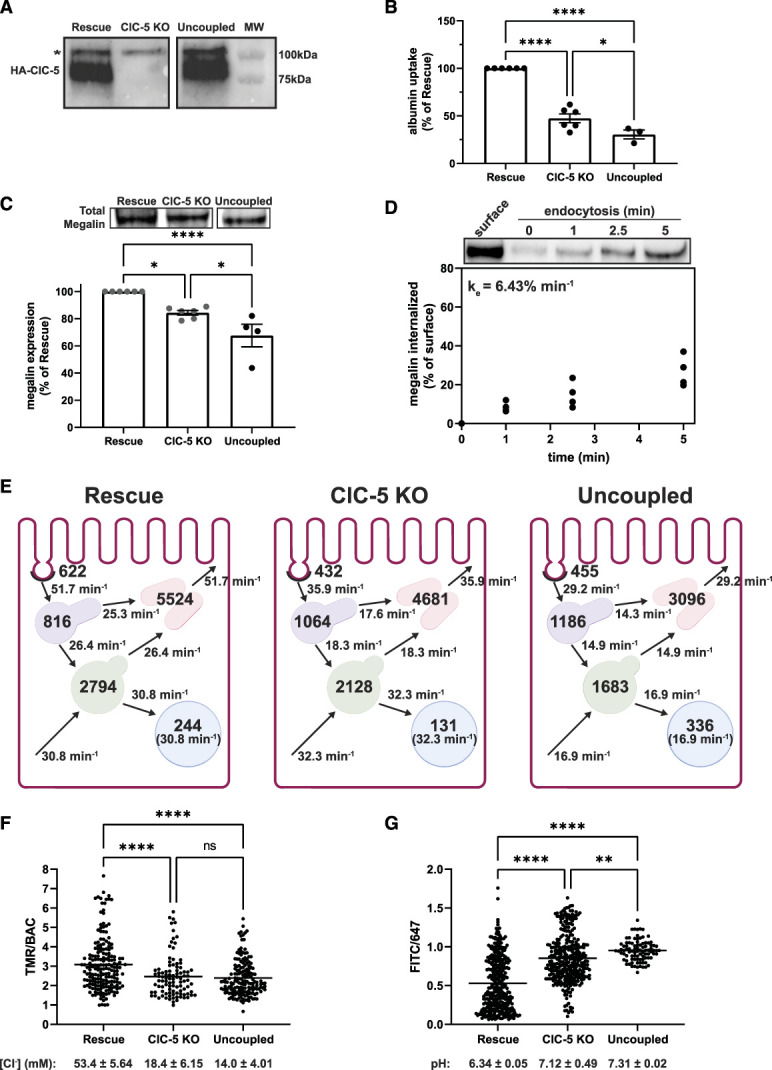
The uncoupled mutant ClC-5E211G impairs endocytic function more than knockout. Equivalent amounts of protein from rescue, ClC-5 KO, and uncoupled lysates were blotted using anti-HA antibody to detect heterologously expressed wild-type (rescue) and ClC-5E211G (uncoupled). A representative blot is shown confirming comparable expression of HA-tagged protein in rescue and uncoupled clones (predicted molecular weight of ClC-5–80 kDa). Blot was cropped to remove middle lane. *Indicates a background band appearing in all three cell lines. (B) Uptake of fluorescent albumin in rescue, ClC-5 KO, and uncoupled cells was quantified by spectrofluorimetry as described in Methods and then normalized to uptake in rescue cells. **P=0.0283 and ****P<0.0001 (n=3–6 experiments) using the one-way ANOVA Tukey multiple comparison test. (C) Equivalent amounts of protein from rescue, ClC-5 KO, and uncoupled cells were blotted for megalin, and the quantified band intensities in each experiment were normalized to rescue cells. *P=0.0213 and ***P=0.0001 (n=4–6) using the one-way ANOVA Tukey multiple comparison test. A representative blot is shown above the graph. Blot was cropped so that data could be presented in the same order as the rest of the figure. The data for rescue and ClC-5 KO are the same as shown in Figure 2A and are indicated in dark gray. (D) Endocytosis kinetics of uncoupled cells was quantified using the biotinylation stripping approach described in Methods. Data from four independent experiments are plotted, and a representative blot is shown above the graph. The fractional endocytic rate was calculated as described in Methods and is shown in the upper left corner. (E) Graphical representation of the model of megalin traffic in rescue, ClC-5 KO, and uncoupled cells. Values with the compartments denote steady state distribution of 10,000 molecules of megalin in rescue, 8437 molecules in ClC-5 KO, and 6756 molecules in uncoupled based on the calculated differences in total megalin expression in panel C. Kinetic rate values are shown as the number of megalin molecules trafficked per minute from the originating compartment. (F) Rescue, ClC-5 KO, and uncoupled cells were incubated for 3 minutes with a mixture of BAC- and TMR-dextrans to load early endosomes and then rapidly imaged by confocal microscopy. The ratio of TMR to BAC intensities within endosomes was quantified as described in Methods. Each point represents a single endosome. ****P<0.0001 (n=97–185) using the one-way ANOVA Tukey multiple comparison test. The intensity ratios were converted to [Cl−] (mM) based on the calibration curve shown in Supplemental Figure S6A. The average [Cl−] (±SEM) for each cell line is shown below the graph. (G) Rescue, ClC-5 KO, and uncoupled cells were incubated for 3 minutes with a mixture of FITC- and Alexa Fluor 647-dextrans to load early endosomes and rapidly imaged by confocal microscopy. The ratio of FITC to 647 intensities within endosomes was quantified as described in Methods. Each point represents a single endosome. ****P<0.0001 and *P=0.0363 (n=91–348) using the one-way ANOVA Tukey multiple comparison test. The intensity ratios were converted to pH based on the calibration curve shown in Supplemental Figure S6B. The average pH (±SEM) for each cell line is shown below the graph.
To test the robustness of the model conclusions, we determined the sensitivity of the predicted intracellular trafficking rates to possible error in our biochemical measurements (Supplemental Figure S7). Assuming there is no discordant error in the measurements of the fraction of megalin at the surface or in the endocytic rate, the maturation of AEEs ( and ) is always slower in ClC-5 KO and uncoupled cells, compared with rescue (Supplemental Figure S7, A and B).
We performed sensitivity analysis on the number of megalin molecules internalized per minute in ClC-5 KO and uncoupled cells to determine whether any perturbations could be made to recover the endocytic capacity (Supplemental Figure S8). Increasing the rates describing entry into recycling DATs from AEEs and AVs ( and ) have the greatest positive effect on the number of receptors internalized per minute, while the rate of exit from DATS to the apical surface () has no effect on this parameter. This suggests that increasing receptor sorting efficiency or the formation of DATs could improve protein recovery in Dent disease, but that stimulation of exocytosis would be ineffectual.
We used ratiometric imaging to quantify [Cl−] in early endosomes of rescue, ClC-5 KO, and uncoupled cells (Figure 5F). The average [Cl−] in rescue cells was 53.4 mM (±5.65 mM), consistent with expected values.28 By contrast, endosomal [Cl−] in both ClC-5 KO and uncoupled cells was considerably lower (18.4±6.15 mM and 14.0±4.01 mM, respectively; P-values <0.0001) and was comparable with reported cytosolic [Cl−] in cells.31,32 We used a similar approach to quantify the pH of early endosomes (Figure 5G). Whereas the measured pH in rescue cells (pH=6.34) was consistent with previous reports of early endosomes,33 the endosomal pH in ClC-5 KO cells was significantly increased (pH=7.11; P-value<0.0001). In addition, we observed a greater impairment in acidification of early endosomes in uncoupled cells (pH=7.31; P-value=0.0363 versus ClC-5 KO). Thus, proper acidification requires the antiporter activity of ClC-5.
Megalin Distribution and Traffic in WT and ClC-5 KO Mouse S1 Cells
We compared the distribution of megalin in the S1 segment of WT mice and a newly generated ClC-5 KO mouse model that recapitulates phenotypic characteristics of Dent disease (Supplemental Figure S10, Figure 6A). Fixed kidney sections were costained to label megalin, the S1 segment apical marker SGLT2, and LAMP1.34 Similar to previous studies, we were unable to resolve distributions of Rab11a, EEA1, and Rab7 within the subapical region of mouse PT cells.29,35 Megalin colocalization with SGLT2 and LAMP1 in images of end-on tubules was quantified by Manders coefficient (Figure 6, B and C). Less megalin colocalized with SGLT2 in S1 segments of ClC-5 KO mice cells compared with WT (10.8% versus 19.3%, P-value=0.0805), with no difference in LAMP1 colocalization. Similar to OK ClC-5 KO cells, megalin was shifted basolaterally in S1 segments of ClC-5 KO mice (Figure 6D). These data are consistent with the increased intracellular retention and reduced recycling predicted by our model of megalin trafficking in OK ClC-5 KO cells.
Figure 6.

ClC-5 KO alters the distribution of megalin in mouse S1 PT cells. (A) Equivalent amounts of protein from WT and ClC-5 KO mouse kidney cortex lysates were blotted for megalin, cubilin, and SGLT2. The quantified band intensity was normalized to the average of the WT mice. Each symbol corresponds to a different mouse. **P=0.0061 (n=5) using the Welch t-test and ****P<0.0001 (n=5) using an unpaired t-test. (B–D) Cortical kidney sections from 10- to 12-week-old male WT and ClC-5 KO mice were stained to label SGLT2, megalin, and LAMP1. (B) Representative sum projection images (10 planes) of megalin colocalization with SGLT2 and LAMP1 in an end-on tubule are shown. Scale bar: 10 µm. The weak staining on the basal aspect of the tubule in the megalin channel is due to background binding of the secondary antibody. (C) Colocalization of megalin with SGLT2 (top) and LAMP1 (bottom) in WT and ClC-5 KO mice, quantified by Manders coefficient over the entire z-stack, is plotted as a percent of total megalin. Each point represents a single end-on tubule with different symbols indicating different mice as in panel A. ****P<0.0001 (n=24) using an unpaired t-test. (D) The average fractional distribution of SGLT2, megalin, and LAMP1 in cells from S1 segment of WT (top; n=17 from two mice) and ClC-5 KO (bottom; n=22 from two mice) mice is plotted over distance (arbitrary units) from lumen. Note the basolateral shift of the megalin distribution of ClC-5 KO compared with WT mice. (E) Graphical representation of the abridged model of megalin traffic in S1 segment of mouse WT and ClC-5 KO PT. Megalin is divided into a surface pool (); an intracellular pool within endosomes () that comprises AEEs, AVs, and DATs; and a lysosomal pool (). Values within the pools denote the steady state distribution of as a percent of total megalin in WT (left) and ClC-5 KO (middle) mice. Kinetic rate values are the percentage of megalin in the originating compartment trafficked per minute. The synthesis () rate and degradation () rates represent the percent of total megalin synthesized per minute and megalin in that is degraded per minute, respectively. The table (right) shows the steady state distribution of megalin in WT and ClC-5 KO mice in megalin molecules for every 10,000 molecules in WT and 1512 molecules in ClC-5 KO, based on the calculated differences in total megalin expression in panel A. Adapted from Shipman et al., 2022, Function.29
We used colocalization data to inform an abridged model of megalin traffic in mouse S1 segments in WT and ClC-5 KO mice.29 We estimated the steady state distribution of megalin using colocalization with SGLT2 and LAMP1. In the absence of direct kinetic measurements in vivo, we assumed the same fractional endocytic rate () and the same fractional degradation rate in lysosomes () as we measured in rescue cells. Figure 6E compares megalin distribution and kinetic trafficking rates in the S1 segments of WT and ClC-5 KO mice. The model predicts a 42% decrease in the fractional recycling rate of megalin () in ClC-5 KO compared with the WT mice. Figure 6E also includes a table comparing the estimated steady state distribution of every 10,000 molecules of megalin in WT S1 segments and corresponding 1512 molecules in ClC-5 KO S1 segments. Although we cannot estimate the maturation rate of AEEs into AVs, the primary defect in megalin traffic in ClC5 KO mice is consistent with impaired endosome maturation as predicted by our cell culture model.
ClC-5 KO in Mice Differentially Affects Megalin Expression along the Length of the PT
We quantified the relative expression of megalin in S1 versus S2 segments in tile scans of fixed kidney sections (Figure 7, A–D). WT mice have higher megalin expression in S2 compared with S1 segments (15,837 [±271.4] versus 14,786 [±428.9], P-value=0.0379), consistent with previously published transcriptomic data (Figure 7, A and C).16 Megalin in both the S1 and S2 segments of ClC-5 KO mice is significantly reduced compared with WT (P-value<0.0001), but there is a much greater reduction in the S2 segment compared with S1 (5555 [±185.8] versus 7863 [±218.8], P-value<0.0001; Figure 7, B and D).
Figure 7.
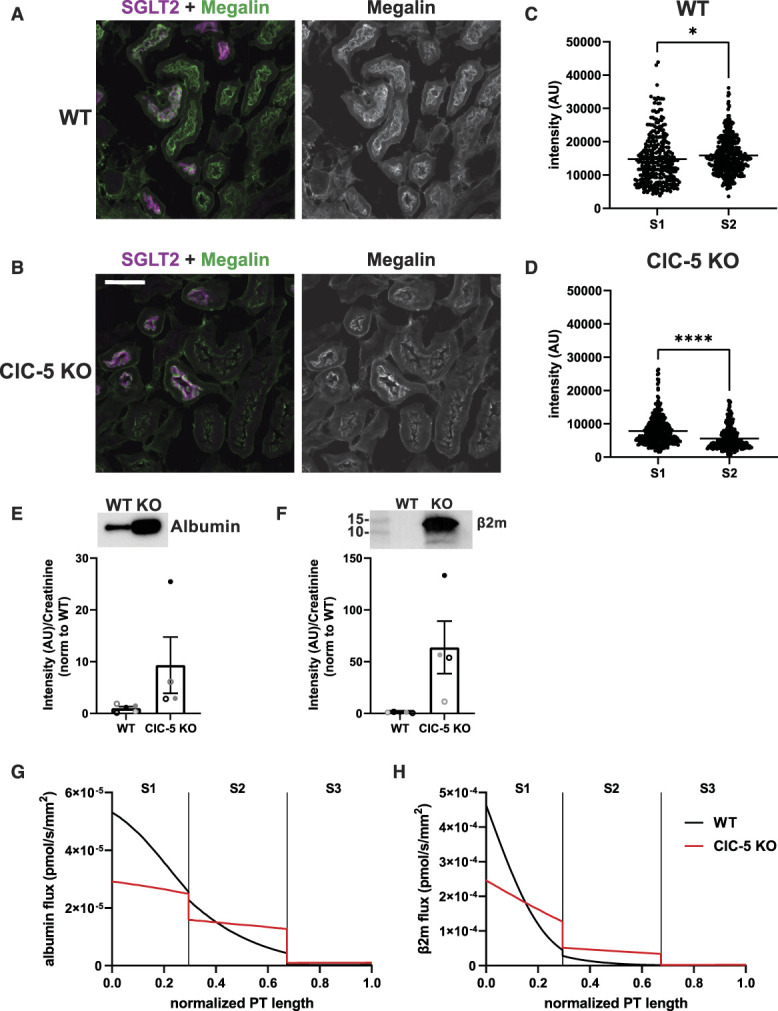
Axial megalin distribution and model. (A–B) Representative sum projection images from tile scans of WT and ClC-5 KO mice showing megalin expression in PTs with and without SGLT2 expression. Scale bar: 50 µm. (C–D) Quantification of megalin intensity in regions with (S1) and without (S2) SGLT2 expression in WT and ClC-5 KO mice. Each point represents the summed megalin intensity along a single line scan. S1 v S2: *P=0.0379 (n=307–406, from 1 mouse) and ****P<0.0001 (n=380–331, from 1 mouse) using the Welch t-test. (E–F) Equal volumes of urine collected from WT and CLC-5 KO mice were separated by SDS-PAGE and were blotted for (E) albumin and (F) β2m. Band intensity was normalized to creatinine concentration and then to the average of WT mice. Each symbol represents a different mouse and corresponds to the same mice in Figure 6, A and C. Representative blots are shown above the graphs. (G–H) Predicted fluxes of albumin (G) and β2m (H) along the normalized length of the PT per nephron in WT and ClC-5 KO mice. Vertical lines denote the boundaries between S1, S2, and S3 subsegments. Of note, our model predicts that, despite the greater loss of megalin in S2, the uptake of both albumin and β2m is shifted toward S2 in ClC-5 KO mice because the endocytic capacity of S1 is significantly reduced while GFR or flow rate through the tubule is increased. The predicted albumin reabsorption in the S2 segment in WT and ClC-5 KO, respectively, is 20.8% versus 33.7% of total reabsorption. The predicted β2m reabsorption in the S2 segment in WT and ClC-5 KO, respectively, is 17.9% versus 3.6% of total reabsorption. The fractional excretion in WT and ClC-5 KO mice, respectively, is 3.03% versus 34.7% for albumin and 0.111% versus 14.06% for β2m.
Using this altered axial distribution of megalin and measures of the relative increase in urinary excretion of albumin and β2m in ClC-5 KO mice (Figure 7, E and F), we modified our previously published model of axial protein uptake along the length of the PT30 as described in detail in Methods. We assumed a 60% reduction in Vmax, which incorporates changes in both the overall expression and the cellular distribution of megalin and cubilin in ClC-5 KO mice to match the measured increase in fractional albumin excretion. The model predicts that ClC-5 KO in mice results in a shift in the axial profile of both albumin and β2m uptake to the S2 segment (Figure 7, G and H).
Discussion
Our studies here using differentiated ClC-5 KO and rescue cells pinpoint a delay in early endosome maturation as the specific trafficking step affected by the loss of ClC-5 antiporter activity. Our model predicts how this delay affects membrane trafficking at each subsequent step along the apical endocytic pathway of PT cells. The ability to integrate data from multiple approaches into a kinetic model was instrumental in revealing how a relatively small change in a single step leads to profound reduction in endocytic uptake of filtered proteins. Our ClC-5 KO cell culture model recapitulates previous findings in ClC-5 KO mice including reduced surface expression of megalin and delayed degradation of internalized ligands.8 Delayed degradation of internalized albumin in ClC-5 KO cells indicates that loss of function of ClC-5 has a general impact on AEE maturation rather than just on the traffic of megalin in the PT. Our data also show that expression of a disease-causing ClC-5 uncoupled mutant has more deleterious effects on membrane traffic than loss of protein expression. Moreover, because megalin and cubilin levels were only marginally reduced in ClC-5 KO compared with rescued cells, our data suggest that the reduction in protein levels of these receptors is not itself the cause of tubular proteinuria in Dent disease. Finally, our data reveal axial changes in megalin expression in ClC-5 KO mice that are predicted to impair the spatial uptake of filtered proteins.
Previous studies have implicated endosomal recycling as the cause of tubular proteinuria in several genetic diseases, including Dent disease.8,32 Indeed, the observed redistribution of NHE3 to intracellular compartments observed in ClC-5 KO mice and OK cells was previously attributed to reduced exocytosis or recycling.6,36 Similarly, endocytic recycling has been implicated as the cause of tubular proteinuria in patients with mutations in EHD137 and may also underlie the reduced uptake of filtered proteins in Lowe syndrome.38,39 Instead, we find that the rate of endosome maturation profoundly affects the efficiency of apical endocytic traffic in PT cells. Specifically, integrating quantitative information on the distribution of intracellular megalin into our kinetic model of megalin traffic allowed us to determine that the trafficking steps most highly affected in Dent disease are the two rates dictating traffic out of AEEs. Delayed endosome maturation is consistent with the decreased receptor surface expression and increased colocalization of megalin with EEA1, as well as with the reduced acidification that we observed in AEEs. Studies in other cell types have shown that impaired endosome acidification results in the delayed delivery of cargo to lysosomes and impaired recycling of receptors40,41 but how acidification modulates the rate of early endosome maturation is unknown. However, precedence exists for acidification-dependent activation of GTPases bound to the cytosolic face of endosomes, as has been demonstrated for the V-ATPase–dependent activation of Arf6 by the GTP exchange factor ARNO.9,42,43 Defective endosome maturation has also been shown to underlie defective trafficking of nephrin in Drosophila nephrocytes carrying mutations in the Rab11 GTPase–activating protein TBC1D88 that result in nephrotic syndrome.44
Our studies also distinguish between pH and [Cl−] homeostasis as the primary mediator of endocytic dysfunction in Dent disease. Cells lacking ClC-5 antiporter function had significantly increased early endosomal pH, compared with rescue cells, and in fact, uncoupled cells exhibited more profound defects in endosomal pH than ClC-5 KO cells. There are conflicting reports on whether uncoupling mutants perturb endosome acidification.10,12,45 These discrepancies likely reflect the experimental systems used to quantify acidification. Some studies expressed ClC-5 mutants in HEK293T cells, which maintain expression of endogenous ClC-5.12,45 Another study monitored acidification in microsomal vesicles isolated from E211A knock-in versus WT mice and found comparable results.10 However, it is likely that the nonphysiologically high [Cl−] in the buffer used in these experiments dictated endosomal [Cl−] and drove acidification. Our studies are more representative of uncoupled ClC-5 activity in vivo because we measured endosomal [Cl−] and pH in intact cells lacking ClC-5.
Consistent with the increased effect on acidification, the uncoupled mutant also demonstrated greater impairments in megalin distribution, and trafficking, as well as in albumin uptake compared with ClC-5 KO. We found that a larger fraction of megalin colocalized with EEA1 and a smaller fraction colocalized with Rab11a than in both rescue and ClC-5 KO cells. Our mathematical model of megalin traffic predicted a 60% reduction in AEE exit rates ( and ), greater than the (47%) reduction predicted for ClC-5 KO cells. Uncoupled cells also exhibited a more pronounced basolateral shift in the distribution of megalin compared with ClC-5 KO cells.
The more profound effects of expressing the uncoupled mutant compared with ClC-5 KO are likely a function of its Cl− channel activity and suggest that nontransport activities of ClC-5, including its reported interactions with cofilin, kinesin, aspartyl aminopeptidase, and/or megalin, play a minor role in the normal function of the antiporter.46–49 Unlike ClC-5 KO cells, the fractional rate of megalin endocytosis was reduced in uncoupled cells, suggesting a direct effect on internalization. Slower acidification or uncoating of internalized endocytic vesicles may reduce the turnover of endocytic machinery required to maintain rapid endocytosis. Expression of the uncoupled mutant also led to increased accumulation of megalin in lysosomes and a slower degradation rate. These data suggest that there may be greater downstream effects on the endolysosomal system in uncoupled cells compared with simple KO. This enhanced effect of the uncoupled mutant on endosomal function and albumin uptake compared with absence of ClC-5 expression may contribute to the phenotypic variability observed in Dent disease.4,5,50 Indeed, patients expressing uncoupled mutations in ClC-5 were observed to develop kidney dysfunction very early in life.12,45 Consistent with this, a recent review of Dent disease causing variants in Europe revealed a higher prevalence of later-stage CKD (stage 2 or greater) in patients with mutations affecting the pore region of ClC-5 compared with mutations resulting in early termination.51
By contrast with the modest (approximately 20%) reduction in megalin and cubilin expression in ClC-5 KO cells, megalin and cubilin expression was reduced by 85% and 73%, respectively, in cortical kidney tissue of ClC-5 KO mice compared with WT. This large reduction in total receptor expression is congruent with previous reports in other ClC-5 KO mouse models.6,52 Strikingly, we found that the reduction in megalin is considerably greater in S2 segments of ClC-5 KO mice compared with S1. These findings may explain in part the greater reduction in megalin expression observed in mice because OK cells are transcriptionally most representative of the S1 segment.13,14,22 Consistent with our data in OK cells, we observed a slight reduction in the fraction of megalin colocalized with SGLT2 in the S1 segment and a basolateral shift in the distribution of megalin in ClC-5 KO mice, with no difference in the expression or distribution of SGLT2. Our mathematical model of megalin trafficking in the S1 segment predicts a 42% reduction in the overall fractional recycling rate, compared with 19% in KO OK cells. Our recent model comparing differentiated OK cells and WT mice concluded that the volume of endocytic flux is greater and more rapid in vivo, which could explain the more profound effect of ClC-5 KO in mice compared with our cell culture model.29 ClC-5 KO mice exhibited evident kidney disease even at early ages, and other contributing factors such as inflammatory responses may contribute to worsened PT function in vivo. With that in mind, our cell culture model provides an ideal model to investigate the direct molecular mechanisms that disrupt endocytic trafficking when endosome acidification is impaired.
Our studies also raise new questions that require future attention and suggest potential targets for therapy to treat Dent disease. At the cellular level, understanding the molecular mechanism by which acidification drives endosome maturation in the PT may lead to development of specific modulators of the PT apical endocytic pathway that may be of clinical benefit. In addition, the sensitivity analysis of our kinetic model of megalin traffic in Dent disease cells, which predicts the effects of manipulating individual steps in membrane traffic on the overall efficiency of the pathway, is of particular use. This analysis clearly demonstrates that increasing the rate of DAT formation from AEEs would have the most profound effect in restoring endocytic uptake in ClC-5 KO and uncoupled cells. By contrast, increasing the recycling rate of megalin in DATs to the surface results in no discernible change to endocytic capacity. On a larger scale, the preferential loss of megalin expression in the S2 segment that we observed has important consequences for the progression of Dent disease. Models for the uptake of filtered proteins predict that high levels of megalin expressed in the S2 segment provide a low-affinity, high-capacity uptake pathway for albumin under nephrotic conditions.30,53 The reduction in the size of this reservoir could contribute to the rapid progression of albuminuria in some patients with Dent disease. Understanding where and how the loss of ClC-5 activity leads to impaired PT function over time may inform treatment strategies to ameliorate the progression of Dent disease.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful to Stéphane Lourdel for generously providing the WT and ClC-5E211G constructs and Daniel Biemesderfer and Peter Aronson for kindly providing the anti-megalin antibody. We thank Sebastien Gingras and the University of Pittsburgh Innovative Technologies Development Core for producing ClC-5 KO mouse founder male, the University of Pittsburgh Biospecimen Core for sectioning kidneys and performing the H&E staining, the University of Pittsburgh Unified Flow Core for assistance in the isolation of ClC-5 KO single cells by fluorescence activated cell sorting, and the University of Pittsburgh Genomics Core for Sanger sequencing of plasmids and PCR products. We appreciate guidance from Megan Gliozzi and Gerry Hammond in generating the ClC-5 KO cell line.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Disclosures
A. Edwards reports employer: Sensory Cloud (spouse); consultancy: Sanofi (spouse); ownership interest: Sensory Cloud (spouse); patents or royalties: Sensory Cloud (spouse); and advisory or leadership role: Sensory Cloud (spouse). O.B. Kashlan reports ownership interest: Moderna. R.J. Tan reports advisory or leadership role: National Kidney Foundation Serving the Alleghenies Medical Advisory Board (volunteer) and Physiological Reports Journal Editorial Board and other interests or relationships: American Heart Association, National Kidney Foundation. O.A. Weisz reports consultancy: for a European company through GLG corp., Judo Biosciences; research funding: SRO from Aktis Oncology; and honoraria: Aktis Oncology paid directly to research funds; and other interests and relationships: member of the JASN editorial board. All remaining authors have nothing to disclose.
Funding
This work was funded by National Institutes of Health grants R01 DK118726 (O.A. Weisz), R01 DK125049 (O.A. Weisz), S10 OD021627 (O.A. Weisz), F31 DK121394 (K.E. Shipman), T32DK007052 (K.E. Shipman), and the Pittsburgh Center for Kidney Research (P30DK079307), and by the American Society of Nephrology Pre-Doctoral Fellowship Award (K.E. Shipman).
Author Contributions
Conceptualization: Katherine E. Shipman, Ora A. Weisz.
Formal analysis: Aurélie Edwards, Ossama B. Kashlan, Katherine E. Shipman.
Investigation: Catherine J. Baty, Isabella A. Cowan, Mona Gerges, Kimberly R. Long, Allison L. Marciszyn, Youssef Rbaibi, Katherine E. Shipman, Roderick J. Tan.
Methodology: Catherine J. Baty, Katherine E. Shipman.
Visualization: Katherine E. Shipman.
Writing – original draft: Katherine E. Shipman, Ora A. Weisz.
Writing – review & editing: Catherine J. Baty, Isabella A. Cowan, Aurélie Edwards, Mona Gerges, Ossama B. Kashlan, Kimberly R. Long, Allison L. Marciszyn, Youssef Rbaibi, Katherine E. Shipman, Roderick J. Tan, Ora A. Weisz.
Data Sharing Statement
All data used in this study are available in this article.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/D733.
Table S1. Oligonucleotides used to target Cas9 mediated KO of Clcn5.
Table S2. Primer set used to sequence targeted region of Clcn5 in OK cells.
Table S3. Primers for qPCR in OK cells.
Table S4. Antibodies for indirect immunofluorescence in OK cells.
Table S5. Oligos for generation and characterization of ClC-5 KO mice.
Table S6. The fractional distribution of cubilin and megalin for WT and ClC-5 KO mice used in the axial model of protein uptake along the PT.
Table S7. Average fractional colocalization (Manders coefficient) of megalin with endocytic markers for rescue, ClC-5 KO, and uncoupled cells.
Table S8. Average fractional overlap (Manders coefficient) between endocytic markers for rescue, ClC-5 KO, and uncoupled cells.
Table S9. Fractional trafficking kinetics of megalin in rescue, ClC-5 KO, and uncoupled cells as percentage of megalin in the originating compartment per minute (mean ± SEM).
Figure S1. Impact of ClC-5 KO and rescue on endocytic uptake of albumin confirmed in multiple clonal lines.
Figure S2. Expression of megalin and cubilin receptors is coordinately affected by ClC-5 KO.
Figure S3. Quantitation of the overlap between endocytic markers in rescue and ClC-5 KO cells.
Figure S4. Polarity of degraded 125I-albumin excretion is similar in rescue and ClC-5 KO cells.
Figure S5. Megalin expression is altered by the expression of the uncoupled ClC-5 mutant.
Figure S6. Megalin distribution is altered by the expression of uncoupled ClC-5 mutant.
Figure S7. Simulated effect of error in biochemical measurements and assumptions on intracellular trafficking kinetics.
Figure S8. Sensitivity of megalin internalization to intracellular trafficking kinetics.
Figure S9. Calibration curves for chloride concentrations and pH.
Figure S10. Characterization of ClC-5 KO mice.
References
- 1.Wrong OM, Norden AG, Feest TG. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87(8):473-493. [PubMed] [Google Scholar]
- 2.Lloyd SE Pearce SH Günther W, et al. Idiopathic low molecular weight proteinuria associated with hypercalciuric nephrocalcinosis in Japanese children is due to mutations of the renal chloride channel (CLCN5). J Clin Invest. 1997;99(5):967-974. doi: 10.1172/jci119262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gianesello L Ceol M Bertoldi L, et al. Genetic analyses in dent disease and characterization of CLCN5 mutations in kidney biopsies. Int J Mol Sci. 2020;21(2):516. doi: 10.3390/ijms21020516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mansour-Hendili L Blanchard A Le Pottier N, et al. Mutation update of the CLCN5 gene responsible for dent disease 1. Hum Mutat. 2015;36(8):743-752. doi: 10.1002/humu.22804 [DOI] [PubMed] [Google Scholar]
- 5.Anglani F D’Angelo A Bertizzolo LM, et al. Nephrolithiasis, kidney failure and bone disorders in Dent disease patients with and without CLCN5 mutations. Springerplus. 2015;4(1):492. doi: 10.1186/s40064-015-1294-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piwon N, Günther W, Schwake M, Bösl MR, Jentsch TJ. ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408(6810):369-373. doi: 10.1038/35042597 [DOI] [PubMed] [Google Scholar]
- 7.Wang SS Devuyst O Courtoy PJ, et al. Mice lacking renal chloride channel, CLC-5, are a model for Dent’s disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum Mol Genet. 2000;9(20):2937-2945. doi: 10.1093/hmg/9.20.2937 [DOI] [PubMed] [Google Scholar]
- 8.Christensen EI Devuyst O Dom G, et al. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci USA. 2003;100(14):8472-8477. doi: 10.1073/pnas.1432873100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shipman KE, Weisz OA. Making a dent in Dent disease. Function (Oxf). 2020;1:zqaa017. doi: 10.1093/function/zqaa017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Novarino G, Weinert S, Rickheit G, Jentsch TJ. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science. 2010;328(5984):1398-1401. doi: 10.1126/science.1188070 [DOI] [PubMed] [Google Scholar]
- 11.Günther W, Piwon N, Jentsch TJ. The ClC-5 chloride channel knock-out mouse - an animal model for Dent’s disease. Pflügers Archiv. 2003;445(4):456-462. doi: 10.1007/s00424-002-0950-6 [DOI] [PubMed] [Google Scholar]
- 12.Bignon Y Alekov A Frachon N, et al. A novel CLCN5 pathogenic mutation supports Dent disease with normal endosomal acidification. Hum Mutat. 2018;39(8):1139-1149. doi: 10.1002/humu.23556 [DOI] [PubMed] [Google Scholar]
- 13.Eshbach ML Sethi R Avula R, et al. The transcriptome of the Didelphis virginiana opossum kidney OK proximal tubule cell line. Am J Physiol Renal Physiol. 2017;313(3):F585-F595. doi: 10.1152/ajprenal.00228.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park HJ Fan Z Bai Y, et al. Transcriptional programs driving shear stress-induced differentiation of kidney proximal tubule cells in culture. Front Physiol. 2020;11:587358. doi: 10.3389/fphys.2020.587358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JW, Chou C-L, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol. 2015;26(11):2669-2677. doi: 10.1681/ASN.2014111067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L, Chou C-L, Knepper MA. A comprehensive map of mRNAs and their isoforms across all 14 renal tubule segments of mouse. J Am Soc Nephrol. 2021;32(4):897-912. doi: 10.1681/ASN.2020101406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long KR Shipman KE Rbaibi Y, et al. Proximal tubule apical endocytosis is modulated by fluid shear stress via an mTOR-dependent pathway. Mol Biol Cell. 2017;28(19):2508-2517. doi: 10.1091/mbc.e17-04-0211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren Q Gliozzi ML Rittenhouse NL, et al. Shear stress and oxygen availability drive differential changes in opossum kidney proximal tubule cell metabolism and endocytosis. Traffic. 2019;20(6):448-459. doi: 10.1111/tra.12648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gliozzi ML Espiritu EB Shipman KE, et al. Effects of proximal tubule shortening on protein excretion in a lowe syndrome model. J Am Soc Nephrol. 2020;31(1):67-83. doi: 10.1681/ASN.2019020125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren Q Weyer K Rbaibi Y, et al. Distinct functions of megalin and cubilin receptors in recovery of normal and nephrotic levels of filtered albumin. Am J Physiol Renal Physiol. 2020;318(5):F1284-F1294. doi: 10.1152/ajprenal.00030.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou Z, Chung B, Nguyen T, Mentone S, Thomson B, Biemesderfer D. Linking receptor-mediated endocytosis and cell signaling: evidence for regulated intramembrane proteolysis of megalin in proximal tubule. J Biol Chem. 2004;279(33):34302-34310. doi: 10.1074/jbc.m405608200 [DOI] [PubMed] [Google Scholar]
- 22.Long KR Rbaibi Y Bondi CD, et al. Cubilin-megalin-and Dab2-dependent transcription revealed by CRISPR/Cas9 knockout in kidney proximal tubule cells. Am J Physiol Renal Physiol. 2022;322(1):F14-F26. doi: 10.1152/ajprenal.00259.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Negoescu A Labat-Moleur F Lorimier P, et al. F(ab) secondary antibodies: a general method for double immunolabeling with primary antisera from the same species. Efficiency control by chemiluminescence. J Histochem Cytochem. 1994;42(3):433-437. doi: 10.1177/42.3.7508473 [DOI] [PubMed] [Google Scholar]
- 24.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(3):213-232. doi: 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 25.Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671-675. doi: 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-colour confocal images. J Microsc. 1993;169(3):375-382. doi: 10.1111/j.1365-2818.1993.tb03313.x [DOI] [PubMed] [Google Scholar]
- 27.Long KR, Rbaibi Y, Gliozzi ML, Ren Q, Weisz OA. Differential kidney proximal tubule cell responses to protein overload by albumin and its ligands. Am J Physiol Renal Physiol. 2020;318(3):F851-F859. doi: 10.1152/ajprenal.00490.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonawane ND, Thiagarajah JR, Verkman AS. Chloride concentration in endosomes measured using a ratioable fluorescent Cl- indicator: evidence for chloride accumulation during acidification. J Biol Chem. 2002;277(7):5506-5513. doi: 10.1074/jbc.m110818200 [DOI] [PubMed] [Google Scholar]
- 29.Shipman KE, Long KR, Cowan IA, Rbaibi Y, Baty CJ, Weisz OA. An adaptable physiological model of endocytic megalin trafficking in opossum kidney cells and mouse kidney proximal tubule. Function. 2022;3(6):zqac046. doi: 10.1093/function/zqac046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edwards A, Long KR, Baty CJ, Shipman KE, Weisz OA. Modelling normal and nephrotic axial uptake of albumin and other filtered proteins along the proximal tubule. J Physiol. 2022;600(8):1933-1952. doi: 10.1113/jp282885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stauber T, Jentsch TJ. Chloride in vesicular trafficking and function. Annu Rev Physiol. 2013;75(1):453-477. doi: 10.1146/annurev-physiol-030212-183702 [DOI] [PubMed] [Google Scholar]
- 32.Devuyst O, Luciani A. Chloride transporters and receptor-mediated endocytosis in the renal proximal tubule. J Physiol (Lond). 2015;593(18):4151-4164. doi: 10.1113/jp270087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy RF, Powers S, Cantor CR. Endosome pH measured in single cells by dual fluorescence flow cytometry: rapid acidification of insulin to pH 6. J Cell Biol. 1984;98(5):1757-1762. doi: 10.1083/jcb.98.5.1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vallon V Platt KA Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22(1):104-112. doi: 10.1681/ASN.2010030246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grieco G Janssens V Gaide Chevronnay HP, et al. Vps34/PI3KC3 deletion in kidney proximal tubules impairs apical trafficking and blocks autophagic flux, causing a Fanconi-like syndrome and renal insufficiency. Sci Rep. 2018;8(1):14133. doi: 10.1038/s41598-018-32389-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Z Jin S Duan X, et al. Chloride channel (Clc)-5 is necessary for exocytic trafficking of Na+/H+ exchanger 3 (NHE3). J Biol Chem. 2011;286(26):22833-22845. doi: 10.1074/jbc.m111.224998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Issler N Afonso S Weissman I, et al. A founder mutation in EHD1 presents with tubular proteinuria and deafness. J Am Soc Nephrol. 2022;33(4):732-745. doi: 10.1681/ASN.2021101312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vicinanza M Di Campli A Polishchuk E, et al. OCRL controls trafficking through early endosomes via PtdIns4,5P2-dependent regulation of endosomal actin. EMBO J. 2011;30(24):4970-4985. doi: 10.1038/emboj.2011.354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berquez M Gadsby JR Festa BP, et al. The phosphoinositide 3-kinase inhibitor alpelisib restores actin organization and improves proximal tubule dysfunction in vitro and in a mouse model of Lowe syndrome and Dent disease. Kidney Int. 2020;98(4):883-896. doi: 10.1016/j.kint.2020.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basu SK, Goldstein JL, Anderson RG, Brown MS. Monensin interrupts the recycling of low density lipoprotein receptors in human fibroblasts. Cell. 1981;24(2):493-502. doi: 10.1016/0092-8674(81)90340-8 [DOI] [PubMed] [Google Scholar]
- 41.Cupers P, Veithen A, Hoekstra D, Baudhuin P, Courtoy PJ. Three unrelated perturbations similarly uncouple fluid, bulk-membrane, and receptor endosomal flow in rat fetal fibroblasts. Biochem Biophys Res Commun. 1997;236(3):661-664. doi: 10.1006/bbrc.1997.7033 [DOI] [PubMed] [Google Scholar]
- 42.Maranda B Brown D Bourgoin S, et al. Intra-endosomal pH-sensitive recruitment of the Arf-nucleotide exchange factor ARNO and Arf6 from cytoplasm to proximal tubule endosomes. J Biol Chem. 2001;276(21):18540-18550. doi: 10.1074/jbc.m011577200 [DOI] [PubMed] [Google Scholar]
- 43.Hurtado-Lorenzo A Skinner M Annan JE, et al. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol. 2006;8(2):124-136. doi: 10.1038/ncb1348 [DOI] [PubMed] [Google Scholar]
- 44.Milosavljevic J Lempicki C Lang K, et al. Nephrotic syndrome gene TBC1D8B is required for endosomal maturation and nephrin endocytosis in drosophila. J Am Soc Nephrol. 20222022;33(12):2174-2193. doi: 10.1681/ASN.2022030275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satoh N Yamada H Yamazaki O, et al. A pure chloride channel mutant of CLC-5 causes Dent’s disease via insufficient V-ATPase activation. Pflügers Archiv. 2016;468(7):1183-1196. doi: 10.1007/s00424-016-1808-7 [DOI] [PubMed] [Google Scholar]
- 46.Hryciw DH, Jenkin KA, Simcocks AC, Grinfeld E, McAinch AJ, Poronnik P. The interaction between megalin and ClC-5 is scaffolded by the Na+-H+ exchanger regulatory factor 2 (NHERF2) in proximal tubule cells. Int J Biochem Cell Biol. 2012;44(5):815-823. doi: 10.1016/j.biocel.2012.02.007 [DOI] [PubMed] [Google Scholar]
- 47.Reed AAC Loh NY Terryn S, et al. CLC-5 and KIF3B interact to facilitate CLC-5 plasma membrane expression, endocytosis, and microtubular transport: relevance to pathophysiology of Dent’s disease. Am J Physiol Renal Physiol. 2010;298(2):F365-F380. doi: 10.1152/ajprenal.00038.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hryciw DH, Wang Y, Devuyst O, Pollock CA, Poronnik P, Guggino WB. Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines. J Biol Chem. 2003;278(41):40169-40176. doi: 10.1074/jbc.m307890200 [DOI] [PubMed] [Google Scholar]
- 49.Lee A Slattery C Nikolic-Paterson DJ, et al. Chloride channel ClC-5 binds to aspartyl aminopeptidase to regulate renal albumin endocytosis. Am J Physiol Renal Physiol. 2015;308(7):F784-F792. doi: 10.1152/ajprenal.00322.2014 [DOI] [PubMed] [Google Scholar]
- 50.Blanchard A Curis E Guyon-Roger T, et al. Observations of a large Dent disease cohort. Kidney Int. 2016;90(2):430-439. doi: 10.1016/j.kint.2016.04.022 [DOI] [PubMed] [Google Scholar]
- 51.Burballa C Cantero-Recasens G Prikhodina L, et al. ; DENT study group. Clinical and genetic characteristics of Dent’s Disease type 1 in Europe. Nephrol Dial Transpl. 2022:gfac310. doi: 10.1093/ndt/gfac310 [DOI] [PubMed] [Google Scholar]
- 52.Gabriel SS Belge H Gassama A, et al. Bone marrow transplantation improves proximal tubule dysfunction in a mouse model of Dent disease. Kidney Int. 2017;91(4):842-855. doi: 10.1016/j.kint.2016.11.016 [DOI] [PubMed] [Google Scholar]
- 53.Weisz OA. Endocytic adaptation to functional demand by the kidney proximal tubule. J Physiol. 2021;599(14):3437-3446. doi: 10.1113/jp281599 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study are available in this article.