

Abstract
The major goals of the Kidney Precision Medicine Project (KPMPP) are to establish a molecular atlas of the kidney in health and disease and improve our understanding of the molecular drivers of CKD and AKI. In this clinical-pathologic-molecular correlation, we describe the case of a 38-year-old woman without any history of CKD who underwent a research kidney biopsy in the setting of AKI suspected to be due to nonsteroidal anti-inflammatory use after cesarean section delivery. The participant's histopathology was consistent with mild acute tubular injury, without significant interstitial fibrosis or tubular atrophy. This diagnosis was supported by analysis of the glomerular and tubulointerstitial proteomes. The proteomic interrogation revealed a molecular landscape that demonstrated differences in kidney prostaglandin synthesis that may be in response to nonsteroidal anti-inflammatory drugs and signs of intrarenal inflammation and fibrosis that were not evident by histopathology alone.
Keywords: acute kidney injury, proteomics, kidney biopsy, NSAID
Clinical Case
A 38-year-old White woman with no significant medical or kidney disease history (baseline serum creatinine 0.7–0.8 mg/dl) presented at 36-week gestation to a local hospital with premature rupture of membranes. She underwent successful cesarean section (C-section) delivery the following day without any reported perioperative complications. On postoperative day 0, she received three doses of the nonsteroidal anti-inflammatory drug (NSAID) ketorolac (15 mg each) intravenously. On postoperative day 1, she was transitioned to oral ibuprofen 800 mg every 6 hours. She was discharged on postoperative day 4 with instructions to continue ibuprofen. On the morning of postoperative day 5, she developed fatigue and shortness of breath. She returned to a local emergency room with a serum creatinine elevated to 2.3 mg/dl, from a baseline of 0.7 mg/dl on postoperative day 1, which was the last time it was checked after C-section. NSAIDs were stopped that morning (day 5), but her serum creatinine continued to increase and was up to 3.7 mg/dl on postoperative day 7, for which she was diagnosed with Kidney Disease: Improving Global Outcomes stage 3 AKI. She was transferred to the Johns Hopkins Hospital for further management.
On the night of transfer, the patient's serum creatinine peaked at 4.2 mg/dl and fluctuated between 4.1 and 4.2 mg/dl over the next 72 hours, beginning a downtrend to 3.8 mg/dl on the day of biopsy (postoperative day 11). Her evaluation included a kidney ultrasound that was unremarkable (left kidney 12.1 cm, right kidney 12.4 cm) and a urinalysis that showed moderate leukocyte esterase but no nitrite on dipstick with 6–10 white blood cells, 0–2 red blood cells, and 6–10 squamous epithelial cells per high-power field. A random spot urine protein-creatinine ratio was measured at 215 mg/g. Her creatinine kinase level was normal, and serological workup was unremarkable. There was no clinical evidence suggestive of thrombotic microangiopathy. She underwent kidney biopsy 10 days after C-section.
Pathology
Figure 1 highlights the major pathologic findings. Up to nine glomeruli per section were seen, none of which were obsolescent. Glomeruli were histologically unremarkable. Crescents, necrosis, and thrombi were not appreciated. Mild acute tubular injury with apical simplification and blebbing was appreciated and associated mild interstitial edema. Minimal tubular atrophy and interstitial fibrosis involving <5% of the sample were noted. Direct immunofluorescence for IgG, IgA, IgM, C3, C1q, kappa and lambda light chains, albumin, and fibrinogen were nonspecific/noncontributory. Ultrastructural evaluation by electron microscopy showed features of acute tubular injury supporting the light microscopic findings.
Figure 1.

Pathologic assessment of the kidney biopsy specimen. (A) Focal apical blebbing (*) of proximal tubular epithelium with luminal prominence (**) and (B) evidence of epithelial loss (>). (C) Scattered bland PAS-positive casts consistent with Tamm–Horsfall protein (>) and (D) interstitial extrusion of Tamm–Horsfall. (E) Interstitial edematous expansion (*) with sparse inflammation consisting of lymphocytes (>). (F) Histologically unremarkable glomeruli. (G) Focal moderate arteriolar hyaline (>).
Molecular Interrogation Methods
To complement the nephropathology evaluation, agnostic mass spectrometric proteomic analysis was performed on the glomeruli and tubulointerstitium, as previously described.1–3 Briefly, laser capture microdissection was performed on 10 µm sections from frozen tissue blocks obtained at optimal cutting temperature to isolate the glomerular and tubulointerstitial compartments from the participant and five tumor nephrectomies that served as controls. Tissues from the tumor nephrectomies were obtained from targeted benign kidney parenchyma without histologic evidence of kidney disease. Protein was extracted from the tissue isolated from each compartment. Overall, 1.7 × 106 µm2 tissue was extracted from the glomerular compartment, and 4.8 × 106 µm2 was collected from the tubulointerstitial compartment. The kidney tissue from the participant yielded 2730 glomerular and 4777 tubulointerstitial proteins on the ThermoFisher Orbitrap Eclipse mass spectrometer. A detailed description of tissue processing and mass spectrometry analysis can be found in the Supplemental Methods.
Protein abundance from the participant was compared with the protein abundance quantified from nephrectomy controls (n=5) obtained from The Ohio State University kidney biopsy repository. Figure 2 is a principal component analysis showing that the nephrectomy controls clustered together and did not overlap with the participant biopsy in both compartments. Proteins that differed significantly (by at least two-fold) are reported. Groups of proteins relevant to specific biologic processes (i.e., pathways) were considered enriched if more proteins within a given pathway were identified than what would be expected by chance. EnrichR was used to identify enriched gene ontology biologic processes in the glomeruli and tubulointerstitium of our case compared with controls and to characterize the molecular processes that may be contributing to kidney injury in this participant. Considering this was a case of AKI and histology consistent with mild acute tubular injury, we expected to see some changes at the molecular level, mostly confined to the tubulointerstitium. Similarly, because NSAIDs were considered to be the primary driver of the participant's AKI, at the molecular level, we suspected that prostaglandin expression may be altered in the participant's kidney.
Figure 2.
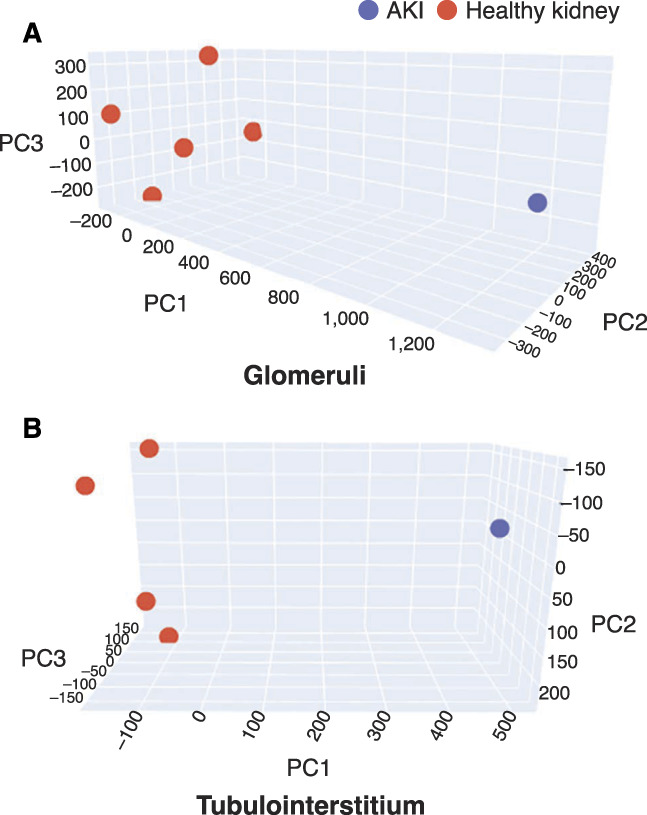
Principal component analysis (PCA) of the kidney proteome. (A) Glomerular and (B) tubulointerstitial proteome comparing participant kidney and nephrectomy control kidneys (n=5). 3D PCA plot showing nonoverlapping clustering of healthy kidney samples (red) in both glomerular and tubulointerstitial compartments compared to the participant kidney (blue).
Results
Molecular Response to AKI
Compared to control tissues, 2162 glomerular and 1418 tubulointerstitial proteins were found to be differentially regulated in the participant's biopsy. Among dysregulated tubulointerstitial proteins, 657 demonstrated higher levels and 761 demonstrated lower levels compared to controls. Dysregulated pathways identified in both compartments included suppression of energy pathways, such as mitochondrial (GO:0032543, GO:0016311, GO:0033108, GO:0042775, GO:1903578), fatty acid metabolism (GO:0019395, GO:0009062), glutathione metabolism (GO:1901687, GO:1901685), and cellular response to stress pathways (GO:0062197, GO:00801350) and increased abundance of proinflammatory pathways, including type 1 IFN (MX1, IF16, IFIT2, IRF9, IFITM3, IFIT5) and monocytes (CD163, CD14, CD58, MRC1) in the tubulointerstitial (Supplemental Figure 1) and complement activation products (C4A, C4B, C1QB, C1S, C3, CFH, CFHR1, C9, SERPING1, F2) in the glomeruli. Finally, higher levels of proteins associated with fibrosis and profibrotic pathways were found in the glomeruli and tubulointerstitium of the participant compared to controls.
Evidence of the NSAID Effects on the Kidney Parenchyma
Many proteins relevant to eicosanoid synthesis were found to be dysregulated in the participant's kidney compared to controls (Figure 3). Prostaglandin-endoperoxide synthase 1 (PTGS1), also known as cyclooxygenase-1 (COX-1), a primary target of NSAIDs, was not detected in the participant's glomeruli but was detected at low abundance in control glomeruli. PTGS1 was not detected in the tubulointerstitium of the participant's or in control kidneys. Downstream enzymes responsible for metabolizing arachidonic acid to prostaglandins, however, were present at lower levels in both compartments compared to controls. Glomerular prostaglandin synthases (PTGES2, PTGES3, PRXL2B), H2 isomerase (PTGDS), and reductases (PTGR1, PTGR3) all had three- to five-fold lower levels compared to controls. In the tubulointerstitium, PTGES2 (1.9-fold), PTGDS (1.9-fold), PRXL2B (4-fold), PTGR1 (3-fold), and prostaglandin E2 receptor (PTGER3, 2-fold) were less abundant relative to controls. By contrast, prostacyclin synthase (PTGIS) was 2.8-fold higher in the participant tubulointerstitium compared to controls but was not different than controls in the glomeruli. Thromboxane synthase (TBXSA1) was not detected, but thromboxane A2 receptor expression was similar to controls in both compartments.
Figure 3.

Differences in glomerular and tubulointerstitial proteins of prostaglandin metabolism in the participant kidney compared to controls. (A) Glomerular prostaglandin enzymes including prostamide/prostaglandin F synthase (PRXL2B), prostaglandin-H2 D-isomerase (PTGDS), prostaglandin E synthase 2 (PTGES2), prostaglandin E synthase 3 (PTGES3), prostaglandin F2 receptor negative regulator (PTGFRN), prostaglandin reductase 1 (PTGR1), prostaglandin G/H synthase 1 (PTGS1), and prostaglandin reductase 3 (ZADH2) are downregulated in the participant glomeruli compared to the controls. (B) Tubulointerstitial prostaglandin enzymes including prostamide/prostaglandin F synthase (PRXL2B), prostaglandin-H2 D-isomerase (PTGDS), prostaglandin E synthase 2 (PTGES2), prostaglandin reductase 1 (PTGR1), and prostaglandin reductase 3 (ZADH2) are downregulated in the participant TI compared to controls. Fold change of the participant proteins compared to controls is shown.
Compared to controls, glomerular and tubulointerstitial expression of vascular and endothelial cell proteins relevant to maintaining vascular tone had lower abundance as follows: glomerular vascular endothelial growth factor (VEGF, 2.5-fold), vascular cell adhesion molecule-1 (2.2-fold), angiomoitin (4-fold), and angiopoietin-related protein 6 (16-fold). Pathway analysis revealed less VEGF (P00056), angiotensin-II (P05911), and endothelin signaling (P00019) in the glomeruli of the participant. In the tubulointerstitium, VEGF was 3.3-fold lower, while the angiotensin II receptor (AGTRAP), platelet endothelial cell adhesion molecule (PECAM1), and vascular endothelial cadherin (CDH5) were all 1.7-fold lower compared to controls. Meanwhile, tubulointerstitial expression of von Willebrand factor and prothrombin were 3- and 2.5-fold higher, respectively, perhaps suggesting a hypercoagulability related to NSAIDs.4
In the setting of COX-1 inhibition by NSAIDs, arachidonic acid metabolism would be expected to shift toward leukotriene and thromboxane-2 generation. Indeed, leukotriene C4 was 2.5-fold higher in the participant's glomeruli compared to controls. However, enzymes needed to initiate arachidonic acid conversion to leukotrienes were less abundant. For example, leukotriene hydroxylases (CYP4F2, CYP4F3) were the least abundant tubulointerstitial proteins at 20- and 16-fold lower than controls, respectively, while glutathione-S transferase enzymes (GSTA1, GSTA2) were 11- and 5-fold less, respectively, in the glomeruli.
The Tubulointerstitial Proteome in Acute Tubular Injury
Molecular support for tubular injury is suggested by higher levels of apoptotic markers, caspase 8 (3.5-fold), toll-like receptor 3 (2.8-fold), and pathways that may reflect the loss of cell membrane integrity seen in acute tubular injury, including cholesterol (GO:0010901, GO:0034371, GO:0034370), phospholipid (GO:0033700, GO:0015914), and nuclear DNA replication (GO:0033260), in the tubulointerstitium of the participant compared to controls (Table 1).
Table 1.
Top tubulointerstitial enriched pathways
| Term | Adjusted P Value | Genes |
|---|---|---|
| Upregulated pathways | ||
| ECM organization Homo sapiens R-HSA-1474244 | 0.014 | FBN2; COL15A1; LAMA2; FN1; TNC; PLOD3; FBLN1; PSEN1; FBLN2; THBS1; FBLN5; VCAN; CAPNS1; COL5A1; P4HA2; COL7A1; ITGA11; SERPINH1; SPP1; ITGA8; COL4A5; ITGAV; ITGB6; COLGALT1 |
| Class I MHC–mediated antigen processing and presentation Homo sapiens R-HSA-983169 | 0.038 | CUL7; UBE2H; ERAP2; FBXO2; TAP2; UBE2E2; CYBB; UBE3A; HLA-A; UBE2A; PSMA7; FBXL8; SEC61A1; RNF114; RNF213; PSMD5; SEC61G; CDC27; MRC1; ITGAV; RBCK1; PSMF1; SEC24D |
| RNA polymerase II transcription Homo sapiens R-HSA-73857 | 0.038 | FYTTD1; CHTOP; TBP; RNMT; CSTF3; SUPT4H1; CDC40; ZC3H11A; ERCC2; POLR2E; POLR2H; TAF5; SNRPB |
| S phase Homo sapiens R-HSA-69242 | 0.038 | RB1; FEN1; RFC3; MCM7; MAX; RFC2; PSMA7; STAG1; PSMD5; RAD21; PSMF1; MCM6; MCM2 |
| Antigen processing-cross presentation Homo sapiens R-HSA-1236975 | 0.038 | SEC61A1; PSMD5; SEC61G; MRC1; TAP2; CYBB; ITGAV; HLA-A; PSMF1; PSMA7 |
| Elastic fiber formation Homo sapiens R-HSA-1566948 | 0.038 | FBN2; ITGA8; FBLN1; ITGAV; ITGB6; FBLN2; FBLN5 |
| IFN-α/β signaling Homo sapiens R-HSA-909733 | 0.038 | IFITM3; OAS2; STAT1; MX1; PTPN6; HLA-A; SAMHD1; IRF9; IFIT2 |
| Molecules associated with elastic fibers Homo sapiens R-HSA-2129379 | 0.038 | ITGA8; FBLN1; ITGAV; ITGB6; FBLN2; FBLN5 |
| DNA strand elongation Homo sapiens R-HSA-69190 | 0.049 | FEN1; RFC3; MCM7; RFC2; MCM6; MCM2 |
| Downregulated pathways | ||
| Metabolism Homo sapiens R-HSA-1430728 | 8.90E-09 | PECR; ECI1; CPNE6; SLC4A1; ACSM5; NUDT5; NUBP2; NUDT4; AS3MT; PPP2R1B; UROD; NAMPT; NUDT19; RPL35; ENPP1; NUDT16; PRSS3; PTGDS; PDK1; EPM2A; PHYH; RPL21; GPT2; NOSIP; PRKCA; OCRL; UPB1; FOLH1; PSME3; HPRT1; CES1; ISYNA1; COX15; ARL2; GSTT1; PIK3R2; ACMSD; SLC9A1; MTM1; HSD11B2; CYP27B1; PTDSS2; AFMID; PSAP; ESD; SLC19A3; COX11; NUP88; CHST13; PCK1; ST3GAL1; ARSB; DECR1; AGXT2; CYP4F2; CAV1; CYP4F3; MTHFS; PYCR2; ACSF3; GGCT; AIMP1; QDPR; BHMT; DAO; NAT1; RPS29; NDUFAF7; MAN2B2; PI4KA; NDUFAF1; MOCS2; SLC44A1; SLC2A1; CROT; ECSIT; PFAS; KHK; DHTKD1; HK3; CA1; ACADL; KYNU; NAPRT; ACP5; HMGCS2; CHST9; ARG2; PLA2G4F; GSTO1; ELOVL7; PTGR1; COASY; NT5C3A; ACOX2; ACOX1; ASPG; FBP1; RAPGEF3; PRPS2; AGPAT5; PDXK; OAT; NDUFB7; GSTP1; HAAO; LMF2; AGPAT3; CYP17A1; CKMT2; NT5C; NUP43; NDUFA8; AOC1; DUT; NDUFA7; MDH1; MMAB; GLYCTK; GSR; NDUFA1; FABP1; GSTZ1; RPIA; MARCKS; GALE; GSTA2; CARM1; NUP35; LPIN2 |
| Mitochondrial translation initiation Homo sapiens R-HSA-5368286 | 2.41E-04 | MRPS17; PTCD3; MRPS16; MRPL19; MRPS34; MRPL17; MRPL28; MTIF3; MRPS30; MRPL23; MRPS6; MRPL13; MRPL43; MRPL20; MRPL40 |
| Mitochondrial translation termination Homo sapiens R-HSA-5419276 | 5.47E-04 | MRPS17; PTCD3; MRPS16; MRPL19; MRPS34; MRPL17; MRPL28; MRPS30; MRPL23; MRPS6; MRPL13; MRPL43; MRPL20; MRPL40 |
| Mitochondrial translation elongation Homo sapiens R-HSA-5389840 | 5.47E-04 | MRPS17; PTCD3; MRPS16; MRPL19; MRPS34; MRPL17; MRPL28; MRPS30; MRPL23; MRPS6; MRPL13; MRPL43; MRPL20; MRPL40 |
| SLC-mediated transmembrane transport Homo sapiens R-HSA-425407 | 0.004795 | SLC22A6; SLC44A1; SLC2A1; SLC4A1; SLC8A1; SLC9A1; HK3; SLC17A5; SLC16A7; NUP88; NUP43; SLC17A1; SLC34A3; SLC30A7; SLC12A3; SLC13A3; SLC6A13; SLC16A10; SLC30A1; SLC7A7; NUP35; SLC29A1; RHCG; SLC26A6; SLC27A4 |
| Organelle biogenesis and maintenance Homo sapiens R-HSA-1852241 | 0.006238 | MRPS17; UNC119B; MRPS16; ARL6; CETN2; MRPL19; MRPS34; MRPL17; MRPS30; MTIF3; TFB1M; MRPL13; MRPL20; IFT74; MRPL40; PCNT; PTCD3; MRPL28; CSNK1D; MRPL23; POLRMT; WDR35; MRPS6; MRPL43; CARM1; EXOC3; TRIP11; SSBP1 |
| Metabolism of water-soluble vitamins and cofactors Homo sapiens R-HSA-196849 | 0.009506 | MOCS2; PDXK; GSTO1; MMAB; SLC2A1; MTHFS; COASY; NAPRT; NAMPT; SLC19A3; ENPP1; ACP5; PRSS3 |
| Detoxification of reactive oxygen species Homo sapiens R-HSA-3299685 | 0.021077 | CCS; GSTP1; GSR; NUDT2; ATP7A; SOD3; PRDX6 |
| Transmembrane transport of small molecules Homo sapiens R-HSA-382551 | 0.027555 | SLC22A6; ABCB6; SLC44A1; SLC2A1; SLC4A1; ATP2C1; SLC8A1; SLC9A1; HK3; SLC17A5; ANO10; SLC16A7; NUP88; NUP43; SLC17A1; ATP7A; CLIC2; ATP6V1G3; ATP6V1C2; SLC34A3; SLC30A7; SLC12A3; SLC13A3; ABCC6; SLC6A13; ATP11C; ATP2B3; SLC16A10; SLC30A1; ABCA8; ATP2B1; CLCN7; SLC7A7; TRPV4; NUP35; SLC29A1; RHCG; SLC26A6; SLC27A4; ATP6V1B1 |
| Peroxisomal lipid metabolism Homo sapiens R-HSA-390918 | 0.038403 | PECR; PHYH; ACOX2; ACOX1; NUDT19; CROT |
| Glutathione conjugation Homo sapiens R-HSA-156590 | 0.039693 | GSTZ1; GGCT; GSTO1; GSTP1; GSTA2; ESD; GSTT1 |
ECM, extracellular matrix.
In addition to decreases in mitochondrial and metabolism pathways, we also observed lower levels of 29 transmembrane transporters, including the Na/Cl exchanger (SLC12A3, 7-fold), Na/PO4 exchanger (SLC34A3, 6.2-fold), Na/H exchanger (SLC9A1, 4-fold), long-chain fatty acid transporter (SLC27A4, 5-fold), and multiple zinc transporters. Conversely, the Na-K-2Cl cotransporter (SLC12A1) was 2.5-fold higher in the tubulointerstitium. This finding is expected because NSAIDs increase urinary concentration by blocking PGE2-mediated decrease in sodium reabsorption via the Na-K-2Cl cotransporter. The net effect of NSAIDs is higher Na-K-2Cl abundance in the thick ascending limb.
The tissue expression of previously established urine biomarkers of AKI, such as neutrophil gelatinase–associated lipocalin, liver fatty acid binding protein-1 (LFABP), kidney injury molecule-1 (KIM-1), IL-18, and insulin-like growth factor 7 (IGFBP7),5–10 was unexpected. Tubulointerstitial neutrophil gelatinase–associated lipocalin (LCN2) abundance was similar between the participant and controls, but LFABP, IL-18, and IGFBP7 levels were 2.7-, 2-, and 1.9-fold lower, respectively.
Molecular Evidence of Fibrosis
Histologic evaluation of the kidney revealed acute tubular injury, but no evidence to suggest interstitial fibrosis or tubular atrophy. However, tissue proteomic analysis of the tubulointerstitium identified higher levels of collagen and extracellular matrix (ECM) proteins. Extracellular matrix (GO:0030198) and collagen fibril (GO:0030199) organization were two of the most enriched biologic processes detected in the tubulointerstitium of the participant (Table 2). Collagen proteins, such as α-1 chain of collagen VII (COL7A1, 100-fold), α-1 chain of collagen V (COL5A1, 2.8-fold), α-5 chain of collagen 4 (COL4A5, 2.7-fold), and α-1 chain of collagen XV (COL15A1, 2.1-fold), were higher in the participant compared to controls. In addition, ECM proteins, versican (VCN, 14-fold), periostin (POSTN, 10-fold), tenascin C (TNC, 10-fold), thrombospondin-2 (THBS2, 6.5-fold), fibulin-2 (FBN2, 2.8-fold), and fibulin-5 (FBN5, 3.4-fold) were higher in the participant's tubulointerstitium compared to controls.
Table 2.
Top glomerular enriched pathways
| Term | Adjusted P Value | Genes |
|---|---|---|
| Upregulated pathways | ||
| Negative regulation of endopeptidase activity (GO:0010951) | 1.43E-08 | SERPINA3; SERPINB4; PCID2; SERPINB12; SERPINA1; SERPIND1; SERPINF1; SERPING1; SERPINA6; SERPINA7; KNG1; AGT |
| Negative regulation of peptidase activity (GO:0010466) | 1.43E-08 | SERPINA3; SERPINB4; CSTA; SERPINB12; SERPINA1; SERPIND1; SERPINF1; SERPING1; SERPINA6; SERPINA7; KNG1; AGT |
| Regulation of endopeptidase activity (GO:0052548) | 4.18E-08 | SERPINA3; SERPINB4; SERPINB12; SERPINA1; SERPIND1; HDAC1; SERPINF1; SERPING1; SERPINA6; SERPINA7; KNG1; AGT |
| Regulation of complement activation (GO:0030449) | 2.17E-07 | C4B; C1QB; C3; C4A; C1S; CFH; CFHR1; C9; SERPING1; F2 |
| Negative regulation of blood coagulation (GO:0030195) | 3.03E-07 | FGB; FGA; FGG; KRT1; SERPING1; A2M; F2; KLKB1; KNG1 |
| Neutrophil degranulation (GO:0043312) | 4.16E-07 | SERPINA3; SERPINB12; SERPINA1; PSMD14; TMEM179B; HP; HBB; C3; PRDX4; CD58; DSP; JUP; DYNLT1; ARG1; GAA; KRT1; CYBB; MIF; HRNR; RAB10; FLG2; FABP5; PKP1; DSG1; DSC1 |
| ECM organization (GO:0030198) | 0.004 | FBN2; FGB; FGA; POSTN; COL15A1; FGG; SCUBE3; VCAN; ADAMTSL4; GFOD2; COL21A1; A2M; KLKB1 |
| Downregulated pathways | ||
| PDGF signaling pathway Homo sapiens P00047 | 0.009 | MAP2K1; GSK3A; MAP2K2; RASA4B; SHC1; PDPK1; ITPR2; ITPR3; ARHGAP5; ETS1; FLI1; ARHGAP12; VAV2; RPS6KA4; RPS6KA3; ELF2; RASA1; RPS6KA1; MAPKAPK2; PIK3C3; PLCG1; GABPA; NCK1 |
| VEGF signaling pathway Homo sapiens P00056 | 0.009 | PRKCI; MAP2K1; MAP2K2; NOS3; PIK3C2A; ETS1; PTK2; MAPKAPK3; MAPKAPK2; KDR; PIK3C3; RAC1; PRKD1; PLCG1 |
| Integrin signaling pathway Homo sapiens P00034 | 0.04 | COL16A1; SHC1; LAMC3; ITGA2B; PIK3C2A; CRKL; PTK2B; FYN; RAC1; MAP2K1; MAP2K2; CAV1; PTPN12; PTK2; RAP2C; RAP2A; RAP2B; COL4A1; COL5A3; ITGA8; PIK3C3; ITGA5; CRK; BCAR1; LIMS2 |
| Angiotensin II–stimulated signaling through G proteins and β-arrestin Homo sapiens P05911 | 0.06 | MAP2K1; PLCB3; MAP2K2; GNG7; ITPR2; ITPR3; ARRB1; GNG11 |
ECM, extracellular matrix; VEGF, vascular endothelial growth factor.
Glomerular Response to AKI
Alterations to a number of glomerular pathways were identified in the participant's kidney. Enriched pathways included negative regulation of blood coagulation (GO:0030195), endopeptidase activity (GO:0010951), complement activation (GO:0030449), neutrophil degranulation (GO:0043312), and ECM organization (GO:0030198) (Table 2). Markers of mesangial and glomerular basement membrane remodeling and expansion were higher in the participant's glomeruli compared to controls. Similar to the tubulointerstitium, versican was 11-fold higher while collagens COL21A and COL15A1, fibrinogen proteins (FGA, FGB, FGG), and fibrillin 2 (FBN2) were all three- to five-fold higher, consistent with early mesangial matrix expansion. Podocyte proteins were generally unperturbed in the participant glomeruli except for podocin (NPHS2), which was 2.7-fold higher compared to controls. Further evidence for glomerular basement membrane dysregulation is demonstrated by lower levels of 55 Rho-Rac pathway proteins and their activating partners (Supplemental Figure 2).
Discussion
This recently postpartum 38-year-old woman developed AKI while receiving NSAIDs. Kidney pathology demonstrated findings consistent with very mild acute tubular injury and no evidence of chronic damage, well-preserved vasculature, and relatively normal-appearing glomeruli. The working clinical diagnosis was NSAID-induced AKI. This diagnosis was supported by analysis of the glomerular and tubulointerstitial proteomes, but the proteomic interrogation revealed a molecular landscape in the kidney that could not be detected by routine kidney histopathology.
NSAIDs block prostaglandin synthesis through the inhibition of cyclooxygenase 1 and 2.11 Kidney prostaglandins regulate vascular tone, and salt and water homeostasis. Inhibition of this homeostatic pathway causes afferent arteriolar vasoconstriction and decreased renal blood flow and GFR.12,13 NSAIDs also suppress renin secretion and angiotensin II activation, and inhibit angiogenesis.14–16 Although the half-life of ketorolac and ibuprofen are short (<4 hours), the kidney prostaglandin changes associated with NSAID-induced injury may last longer. Analysis of the glomerular and tubulointerstitial proteome generally revealed suppression of enzymes critical to arachidonic acid metabolism and prostanoid production. For example, the enzymes responsible for generating prostaglandin E2 (PGE2), the major prostaglandin responsible for regulating vascular tone in kidney,17 were expressed at lower levels in the glomeruli and tubulointerstitium of this participant. Notably, in this case, the kidney biopsy was performed 10 days after the development of clinical AKI and 5 days after NSAIDs were stopped. Thus, the observed changes to prostaglandin metabolism may have been waning. Nonetheless, these changes in the kidney parenchyma that may reflect an NSAID effect could only be identified through molecular techniques and not histology.
Pregnancy influences renal hemodynamics, and the fact this patient recently delivered could have influenced the kidney proteome and, in particular, vascular proteins. Certainly, a more ideal comparison would have been tissue obtained from a recently postpartum healthy control, but this tissue was not available. Interestingly, during pregnancy, endothelin B and the renin-angiotensin-aldosterone system are known to be upregulated,18 but this was not evident in the participant's biopsy. Instead, there were notable decreases in proteins of the renin-angiotensin-aldosterone system and markers of angiogenesis in both tissue compartments, suggesting the changes seen in this case were more likely related to NSAIDs and the AKI as opposed to pregnancy.
Proteomic changes of acute tubular injury were detected in the tubulointerstitium and included findings that suggest apoptosis, loss of cell membrane integrity, and lower levels of common transport proteins. Interestingly, early AKI biomarkers such as LFABP, IGFBP7, and IL18 were not abundantly expressed but were in fact lower in the tubulointerstitium compared to controls. These markers have previously been shown to increase in the urine during AKI, but less is known regarding tissue abundance during injury.10 In addition, how these markers change in response to NSAID-mediated injury has not been studied, nor has the (potential) contribution of the preceding pregnancy. Notably, these proteins are believed to be early biomarkers of AKI and, in many cases, precede changes in serum creatinine. Thus, it is conceivable that the tissue expression of these biomarkers was higher earlier in the course of this patient's AKI and not captured here due to timing of the kidney biopsy.
The proteomic analysis detected evidence of tissue inflammation in the glomeruli and tubulointerstitium, but there were no histologic correlates, at least by routine microscopy of intrarenal inflammation. The consequences of activating inflammation at the molecular level in a kidney with resolving AKI are not known, but it is interesting to speculate that this could be an early step in the transition of AKI to CKD, especially in conjunction with finding upregulation of several protein markers and pathways of renal fibrosis in the tubulointerstitium. Markers of fibrosis not typically present or present at low levels in normal kidneys were higher in the tubulointerstitium of the injured kidney. All these proteins have previously been linked to ECM production and fibrosis in kidney disease.19,20 For example, VCN, a ubiquitous hyaluronan proteoglycan and large ECM protein, is minimally expressed in healthy kidneys but was 14-fold higher in the tubulointerstitium of our participant compared to controls. VCN is involved in cell proliferation, adhesion, migration, and ECM assembly.21 It has previously been shown to be activated by TGF-β in fibroblasts and has been previously associated with a progressive decline in kidney function in CKD.19,21 These findings will be better understood if they can be placed in the context of a time course as opposed to this cross-sectional picture.
On the basis of the histologic injury being mainly confined to the tubulointerstitium and being relatively mild, we expected that the majority of molecular changes would be in the tubulointerstitium and also relatively mild. By contrast, significant protein perturbations were seen in the glomerular compartment despite normal-appearing histology. Furthermore, tubular energy and mitochondrial metabolism pathways were altered at the molecular level. These findings illustrate that what is seen by routine nephropathologic techniques does not necessarily represent the extent or severity of kidney injury.
The aim of the Kidney Precision Medicine Project (KPMP) is to redefine common chronic and acute kidney diseases by applying molecular techniques to enhance our clinical and histopathologic diagnoses,22 with the hope that these additions will lead to improved clinical outcomes in the future. This study is a clinicopathologic molecular correlation (CPMC) with a deeper exploration into pathophysiologic insights of a clinical scenario, similar to other CPMCs recently published through the KPMP.23 Molecular interrogation has the potential to reveal more about what is happening in the kidney than can be appreciated by histopathology alone. Notably, the molecular observations found here and in other studies within the KPMP would not be possible without participants such as the participant in this case agreeing to a research kidney biopsy. As this participant's kidney function had already begun to improve, it is unlikely a biopsy would have been performed on clinical grounds alone. Thus, the altruistic contribution of this participant and others within the KPMP to the advancement in our understanding of kidney disease cannot be overstated.
Specific to this case, mass spectrometry proteomics of the glomeruli and tubulointerstitium revealed changes in kidney prostaglandins in response to NSAIDs and identified signs of intrarenal inflammation and fibrosis that were not evident by histopathology alone. Going forward, applying -omics technologies to kidney biopsy evaluation will provide greater insights into disease pathophysiology, redefine diseases according to mechanisms, and, ultimately, improve outcomes for patients affected by AKI and CKD.
Clinical Follow-Up
After kidney biopsy, the participant's serum creatinine improved, albeit slowly. The serum creatinine was 3.8 mg/dl on the day of biopsy and fell to 3.3 mg/dl 24 hours later, at which time the participant was discharged. Testing obtained 2 days after discharge demonstrated a serum creatinine of 2.06 mg/dl. Two weeks after discharge, the participant's serum creatinine had improved to 1.22 mg/dl, still well above her healthy baseline. After 2 months, the participant's serum creatinine was 0.95 mg/dl, and not until 6 months postdischarge did the serum creatinine return to her baseline of 0.81 mg/dl. This slow resolution of clinical kidney function is less consistent with the mild histopathologic changes seen on biopsy and perhaps more consistent with the inflammatory, metabolic, and fibrotic changes seen on molecular interrogation of the biopsy.
Patient Perspective and Conclusion
Patient engagement is an essential aspect of nephrology research. Patient engagement often increases the relevance of research. Author Richard Knight is one of the patients who was involved in the general introduction of the KPMP:
The concept of precision medicine was theoretical at the onset of the KPMP. As an active participant in this CPMC study, I have the opportunity to see precision medicine in action. In reading the study note, the clinical diagnosis was NSAID-induced AKI. This final clinical diagnosis was based primarily on analyzing the tissue histology. However, the proteomic interrogation, especially in the glomerular and tubulointerstitial proteomes, suggests a more active molecular setting in the kidney undetected by routine histopathology.
The practical application of precision medicine concepts in this detailed CPMC study provides an example of the efficacy of work undertaken by KPMP researchers. Patients yearn for proper diagnosis that leads to appropriate treatment. Many patients, including myself, feel that the opportunity to help researchers, via a kidney biopsy, discover answers and insights that will enhance the quality of life for future kidney patients is such that the benefits derived outweigh the risks of a biopsy.
This CMPC study highlights this notion because the results from standard nephropathologic practices did not show the full extent of the kidney injury. As a patient, I am grateful to be part of this study highlighting the practical application of precision medicine that will soon be available to more patients with kidney diseases.
Supplementary Material
Footnotes
*Kidney Precision Medicine Project: Richard Knight, Stewart H. Lecker, Isaac Stillman, Steve Bogen, Laurence H. Beck, Sushrut Waikar, Gearoid M. McMahon, Astrid Weins, Mia R. Colona, Nir Hacohen, Paul J. Hoover, Mark Aulisio, William S. Bush, Dana C. Crawford, John O'Toole, Emilio Poggio, John Sedor, Leslie Cooperman, Stacey Jolly, Leal Herlitz, Jane Nguyen, Agustin Gonzalez-Vicente, Ellen Palmer, Dianna Sendrey, Carissa Vinovskis, Petter M. Bjornstad, Paul Appelbaum, Jonathan M. Barasch, Andrew S. Bomback, Vivette D. D'Agati, Krzysztof Kiryluk, Karla Mehl, Pietro A. Canetta, Ning Shang, Olivia Balderes, Satoru Kudose, Shweta Bansal, Theodore Alexandrov, Helmut Rennke, Tarek M. El-Achkar, Yinghua Cheng, Pierre C. Dagher, Michael T. Eadon, Kenneth W. Dunn, Katherine J. Kelly, Timothy A. Sutton, Daria Barwinska, Michael J. Ferkowicz, Seth Winfree, Sharon Bledsoe, Marcelino Rivera, James C. Williams Jr., Ricardo Melo Ferreira, Chirag R. Parikh, Celia P. Corona Villalobos, Steven Menez, Avi Rosenberg, Sylvia E. Rosas, Neil Roy, Mark Williams, Evren U. Azeloglu, Cijang He, Ravi Iyengar, Jens Hansen, Yuguang Xiong, Brad Rovin, Samir Parikh, John P. Shapiro, Christopher R. Anderton, Ljiljana PasaTolic, Dusan Velickovic, Jessica Lukowski, George (Holt) Oliver, Joseph Ardayfio, Jack Bebiak, Keith Brown, Catherine E. Campbell, John Saul, Anna Shpigel, Christy Stutzke, Robert Koewler, Taneisha Campbell, Lynda Hayashi, Nichole Jefferson, Glenda V. Roberts, Roy Pinkeney, Olga Troyanskaya, Rachel Sealfon, Katherine R. Tuttle, Yury Goltsev, Kun Zhang, Blue B. Lake, Zoltan G. Laszik, Garry Nolan, Patrick Boada, Minnie Sarwal, Tara Sigdel, Paul J. Lee, Rita R. Alloway, Steve Woodle, Heather Ascani, Ulysses G.J. Balis, Jeffrey B. Hodgin, Matthias Kretzler, Chrysta Lienczewski, Laura H. Mariani, Rajasree Menon, Becky Steck, Yougqun He, Edgar Otto, Jennifer Schaub, Victoria M. Blanc, Sean Eddy, Ninive C. Conser, Jinghui Luo, Paul M. Palevsky, Matthew Rosengart, John A. Kellum, Daniel E. Hall, Parmjeet Randhawa, Mitchell Tublin, Raghavan Murugan, Michele M. Elder, James Winters, Charles E. Alpers, Kristina N. Blank, Jonas Carson, Ian H. De Boer, Ashveena L. Dighe, Jonathan Himmelfarb, Sean D. Mooney, Stuart Shankland, Kayleen Williams, Christopher Park, Frederick Dowd, Robyn L. McClelland, Stephen Daniel, Andrew N. Hoofnagle, Adam Wilcox, Stephanie M. Grewenow, Kumar Sharma, Manjeri Venkatachalam, Guanshi Zhang, Annapurna Pamreddy, Hongping Ye, Richard Montellano, Robert D. Toto, Miguel Vazquez, Simon C. Lee, R. Tyler Miller, Orson W. Moe, Jose Torrealba, Nancy Wang, Asra Kermani, Kamalanathan Sambandam, Harold Park, S. Susan Hedayati, Christopher Y. Lu, Sanjay Jain, Anitha Vijayan, Joseph P. Gaut, Dennis Moledina, Francis P. Wilson, Ugochukwu Ugwuowo, and Tanima Arora.
Published online ahead of print. Publication date available at www.cjasn.org.
Disclosures
R. Knight reports employment with American Association of Kidney Patients; honoraria from American Kidney Fund, Johns Hopkins Center for Health Equity, Labcorp, Northwestern University, Novartis, Otsuka, and Personalized Medicine Coalition; advisory or leadership roles for NIDDK Advisory Council, Scientific Advisory Board for the “Rescuing Kidneys at Risk of Discard” project, and SRTR Review Committee; serving as AAKP President and a member of Quality Insights Patient Advisory Committee; and other interests or relationships with Bowie State University Board of Advisors and SRTR Review Committee. S. Menez reports consulting fees from the Dedham Group and research funding from RenalytixAI. C.R. Parikh reports consultancy agreements with Genfit Biopharmaceutical Company; ownership interest in RenalytixAI; research funding from National Heart, Lung and Blood Institute (NHLBI) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK); advisory or leadership roles for Genfit Biopharmaceutical Company and RenalytixAI; and is a coinventor on a pending patent, “Methods and Systems for Diagnosis of Acute Interstitial Nephritis,” that is subject to an option for a license agreement with RenalytixAI, Inc. S.V. Parikh reports consultancy agreements with Alexion, Aurinia Pharmaceuticals, Bristol Myers Squibb, GlaxoSmithKline, and Kezar Life Sciences; research funding from Aurinia Pharmaceuticals, NIH-NIDDK, and Serono-EMD; grant support from Aurinia Pharmaceuticals and Biogen Idec; and royalties from UpToDate.com. A.Z. Rosenberg reports research funding from NIH and NKF; honoraria from Georgetown University, Ichilov Hospital (Tel Aviv, Israel), and Stony Brook University; and an advisory or leadership role for Escala. B. Rovin reports consultancy agreements with Alexion, AstraZeneca, Aurinia, Biocryst, Biogen, BMS, Bristol Myers Squibb, Calliditas, Chemocentryx, Corrona, EMD-Serono/Merck, Exagen, Galapagos, Genentech, Horizon, Human Genome Sciences (GSK), Idorsia, Janssen, Lupus Foundation of America, MedImmune, Morphosys, Novartis, Omeros, Otsuka, Resonance, Retrophin, RILITE Foundation, Roche, and Vistera; research funding from Biogen; honoraria from Alexion, AstraZeneca, Aurinia, Biocryst, Biogen, BMS, Calliditas, Chemocentryx, Corrona, EMD-Serono/Merck, Exagen, Galapagos, Genentech, Horizon, Human Genome Sciences (GSK), Idorsia, Janssen, MedImmune, Morphosys, Novartis, Omeros, Otsuka, Resonance, Retrophin, RILITE Foundation, Roche, and Vistera; advisory or leadership roles for ASN Kidney Week, CureGN, KDIGO, Kidney International, Kidney International Reports, Lupus Foundation of America, Nephrology Dialysis and Transplantation, and UpToDate; and a lot of work with the ASN—mostly educational courses, work with the ISN and NKF, and work with the LFA. The remaining authors have nothing to disclose.
Funding
S. Menez receives grant support from K23DK128538 and research support from RenalytixAI. Research reported in this manuscript was supported by the National Institute of Diabetes and Digestive and the Kidney Diseases (NIDDK) KPMP (www.kpmp.org). The KPMP is funded by the following grants from the NIDDK: U2C DK114886, UH3DK114861, UH3DK114866, UH3DK114870, UH3DK114908, UH3DK114915, UH3DK114926, UH3DK114907, UH3DK114920, UH3DK114923, UH3DK114933, and UH3DK114937. S. Madhavan received grant support from K08DK123411.
Author Contributions
R. Knight, S. Menez, C.R. Parikh, S.V. Parikh, and B. Rovin conceptualized the study; S. Menez, S.V. Parikh, A.Z. Rosenberg, and J. Shapiro were responsible for data curation; S. Madhavan, S.V. Parikh, A.Z. Rosenberg, and J. Shapiro were responsible for formal analysis; S. Menez and S.V. Parikh were responsible for investigation; S.V. Parikh was responsible for methodology; C.R. Parikh, S.V. Parikh, and B. Rovin provided supervision; S. Madhavan, S.V. Parikh, A.Z. Rosenberg, and J. Shapiro were responsible for visualization; C.R. Parikh and B. Rovin were responsible for funding acquisition; R. Knight, S. Menez, and S.V. Parikh wrote the original draft; and R. Knight, S. Madhavan, S. Menez, C.R. Parikh, S.V. Parikh, A.Z. Rosenberg, B. Rovin, and J. Shapiro reviewed and edited the manuscript.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/CJN/B76.
Supplemental Figure 1. Overexpression of IFN and monocyte pathways in participant tubulointerstitium compared to controls.
Supplemental Figure 2. Downregulation of RhoGTPase pathway markers in participant glomeruli compared to controls.
References
- 1.Ayoub I, Shapiro JP, Song H, et al. Establishing a case for anti-complement therapy in membranous nephropathy. Kidney Int Rep. 2021;6(2):484-492. doi: 10.1016/j.ekir.2020.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satoskar AA, Shapiro JP, Bott CN, et al. Characterization of glomerular diseases using proteomic analysis of laser capture microdissected glomeruli. Mod Pathol. 2012;25(5):709-721. doi: 10.1038/modpathol.2011.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satoskar AA, Shapiro JP, Jones M, et al. Differentiating Staphylococcus infection-associated glomerulonephritis and primary IgA nephropathy: a mass spectrometry-based exploratory study. Sci Rep. 2020;10(1):17179. doi: 10.1038/s41598-020-73847-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ungprasert P, Srivali N, Wijarnpreecha K, Charoenpong P, Knight EL. Non-steroidal anti-inflammatory drugs and risk of venous thromboembolism: a systematic review and meta-analysis. Rheumatology (Oxford). 2015;54(4):736-742. doi: 10.1093/rheumatology/keu408 [DOI] [PubMed] [Google Scholar]
- 5.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62(1):237-244. doi: 10.1046/j.1523-1755.2002.00433.x [DOI] [PubMed] [Google Scholar]
- 6.Kamijo A, Sugaya T, Hikawa A, et al. Urinary excretion of fatty acid-binding protein reflects stress overload on the proximal tubules. Am J Pathol. 2004;165(4):1243-1255. doi: 10.1016/S0002-9440(10)63384-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kashani K, Al-Khafaji A, Ardiles T, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care. 2013;17(1):R25. doi: 10.1186/cc12503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365(9466):1231-1238. doi: 10.1016/S0140-6736(05)74811-X [DOI] [PubMed] [Google Scholar]
- 9.Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. 2005;16(10):3046-3052. doi: 10.1681/ASN.2005030236 [DOI] [PubMed] [Google Scholar]
- 10.Rizvi MS, Kashani KB. Biomarkers for early detection of acute kidney injury. J Appl Lab Med. 2017;2(3):386-399. doi: 10.1373/jalm.2017.023325 [DOI] [PubMed] [Google Scholar]
- 11.Abramson SB, Weissmann G. The mechanisms of action of nonsteroidal antiinflammatory drugs. Arthritis Rheum. 1989;32(1):1-9. doi: 10.1002/anr.1780320102 [DOI] [PubMed] [Google Scholar]
- 12.Clive DM, Stoff JS. Renal syndromes associated with nonsteroidal antiinflammatory drugs. N Engl J Med. 1984;310(9):563-572. doi: 10.1056/NEJM198403013100905 [DOI] [PubMed] [Google Scholar]
- 13.Patrono C, Dunn MJ. The clinical significance of inhibition of renal prostaglandin synthesis. Kidney Int. 1987;32(1):1-12. doi: 10.1038/ki.1987.164 [DOI] [PubMed] [Google Scholar]
- 14.Hörl WH. Nonsteroidal anti-inflammatory drugs and the kidney. Pharmaceuticals (Basel). 2010;3(7):2291-2321. doi: 10.3390/ph3072291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones MK, Wang H, Peskar BM, et al. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: Insight into mechanisms and implications for cancer growth and ulcer healing. Nat Med. 1999;5(12):1418-1423. doi: 10.1038/70995 [DOI] [PubMed] [Google Scholar]
- 16.Peti-Peterdi J, Komlosi P, Fuson AL, et al. Luminal NaCl delivery regulates basolateral PGE2 release from macula densa cells. J Clin Invest. 2003;112(1):76-82. doi: 10.1172/JCI18018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Xia W, Zhao F, et al. Prostaglandins in the pathogenesis of kidney diseases. Oncotarget. 2018;9(41):26586-26602. doi: 10.18632/oncotarget.25005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung KL, Lafayette RA. Renal physiology of pregnancy. Adv Chronic Kidney Dis. 2013;20(3):209-214. doi: 10.1053/j.ackd.2013.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bülow RD, Boor P. Extracellular matrix in kidney fibrosis: more than just a scaffold. J Histochem Cytochem. 2019;67(9):643-661. doi: 10.1369/0022155419849388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genovese F, Manresa AA, Leeming DJ, Karsdal MA, Boor P. The extracellular matrix in the kidney: a source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair. 2014;7(1):4. doi: 10.1186/1755-1536-7-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudnicki M, Perco P, Neuwirt H, et al. Increased renal versican expression is associated with progression of chronic kidney disease. PLoS One. 2012;7(9):e44891. doi: 10.1371/journal.pone.0044891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Boer IH, Alpers CE, Azeloglu EU, et al. Rationale and design of the Kidney Precision Medicine Project. Kidney Int. 2021;99(3):498-510. doi: 10.1016/j.kint.2020.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel J, Torrealba JR, Poggio ED, et al. Molecular signatures of diabetic kidney disease hiding in a patient with hypertension-related kidney disease: A clinical pathologic molecular correlation. Clin J Am Soc Nephrol. 2021;17(4):594-601. doi: 10.2215/CJN.10350721 [DOI] [PMC free article] [PubMed] [Google Scholar]