

Abstract
Routine small-molecule analysis is challenging owing to the need for high selectivity and/or low limits of quantification. This work reports a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method to quantify 14 antiepileptic drugs (AEDs) in human serum. For the optimized LC-MS/MS method described herein, we applied the guidelines outlined in the Clinical and Laboratory Standards Institute (CLSI) LC-MS C62-A document and the U.S. Food and Drug Administration (FDA) Bioanalytical Method Validation Guidance for Industry to evaluate the quality of the assay. In these studies, AED linearity, analyte recovery, matrix effects, precision, and accuracy were assessed. Using liquid chromatography-drift tube ion mobility-mass spectrometry (LC-DTIM-MS), a qualitative method was also used to increase confidence in AED identification using accurate mass and collision cross section (CCS) measurements. The LC-DTIM-MS method was also used to assess the ability of drift tube CCS measurements to aid in the separation and identification of AED structural isomers and other AEDs. These data show that another dimension of information, namely CCS measurements, provides an orthogonal dimension of structural information needed for AED analysis. Multiplexed AED measurements using LC-MS/MS and LC-DTIM-MS have the potential to enable better optimization of dosing owing to the high precision capabilities available in these types of analytical studies. Taken together, these data also show the ability to increase confidence in small-molecule identification and quantification using these analytical technologies.
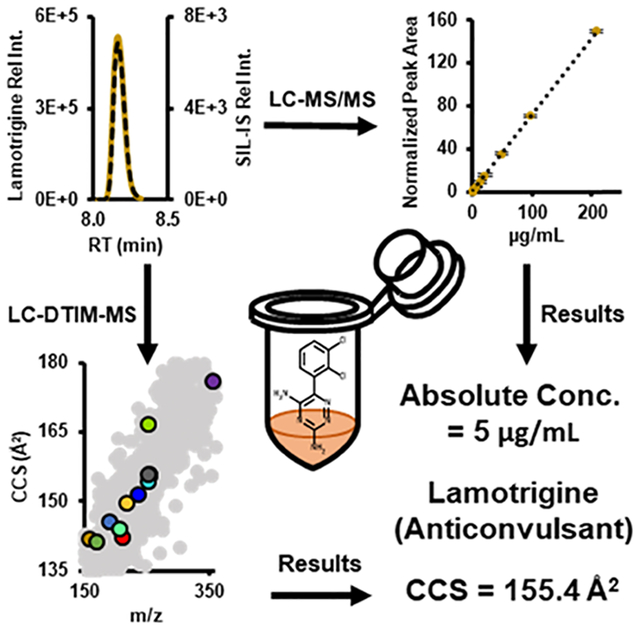
Graphical Abstract
Epilepsy is a common neurological disorder characterized by a long-term risk of recurrent seizures. Antiepileptic drugs (AEDs) are mood stabilizers not only effective at controlling seizures but also treating schizophrenia, depression, and bipolar disorder.1–4 However, many patients experience adverse side effects associated with AED usage, such as Stevens–Johnson syndrome, toxic epidermal necrolysis, liver toxicity, tremors, dizziness, nausea, and fatigue.5–7 AEDs have a narrow therapeutic range; therefore, the serum concentration of AEDs must be optimized to ensure effectiveness (control/minimize seizures) and minimize negative side effects.8–10 AED effectiveness is partially dependent upon patient-specific pharmacokinetics (PK), which include: absorption, distribution, metabolism, and excretion (ADME).11 Patient-specific PK can influence a clinician’s decision on dosage and dosing frequency of AEDs.8 Therapeutic drug management (TDM) requires iterative measurements of AEDs in serum to ensure that a patient’s AED concentration is within the therapeutic range.10,12 Developing a workflow with high precision, accuracy, sensitivity, and selectivity is essential for optimizing AED treatment for individual patients, a primary tenet of personalized medicine.13
Routine clinical TDM is often performed using quantitative immunoassays, where analyte concentration is determined as a function of antibody binding, and not through direct molecular measurements.5,6 Immunoassays are generally limited to single-drug detection and are susceptible to false positives as a result of cross-reactivity between the drug target (e.g., carbamazepine or CBZ) and related metabolites (e.g., carbamazepine-10,11 epoxide or CBZ epoxy).14–17 Supplement consumption in the United States and across the globe has also increased dramatically in recent years, which may convolute testing by introducing additional interferences.13,18 In addition, new AEDs are routinely developed, which may complicate the quantification of AEDs using less selective methodologies like immunoassays.5,19,20 Finally, patients are often prescribed more than one AED, which necessitates testing a single sample using multiple different immunoassays. However, the need for performing multiple tests on a single sample impedes cost effectiveness.21–24 Thus, an analytical workflow offering highly specific detection of multiple related drugs and drug metabolites with high sensitivity and selectivity is ideal.25
Separation of drugs by liquid chromatography and detection via absorbance at a particular wavelength (LC-UV) or by tandem mass spectrometry (LC-MS/MS) have been utilized, as alternatives to immunoassays, to quantify AEDs.25–27 However, LC-UV analyses are also susceptible to interferences from chemically similar species. Further differentiation and more specific detection techniques (such as triple-quadrupole MS/MS and liquid chromatography-drift tube ion mobility-mass spectrometry (LC-DTIM-MS)) may be necessary for AED quantification studies, particularly if structural isomers will be analyzed simultaneously.12 In addition to LC-MS/MS, LC-DTIM-MS has become an important analytical technique for characterizing drugs and drug metabolites simultaneously by molecular structure and weight.28–34 Clinical and analytical labs have utilized LC-MS/MS methods for the quantification of AEDs in serum for numerous reasons, including ruggedness, ease of use, and cost.35 LC-MS/MS methods have been developed for quantification of AEDs from different biological matrices such as dried plasma spots, plasma, and serum.25–27,36
In these studies, a complementary approach of AED analysis utilizing LC-DTIM-MS was investigated to determine if additional structural information for individual AEDs increases confidence in selectivity. Briefly, DTIM experiments provide a dimension of separation in addition to LC that can be coupled to mass analysis to potentially distinguish isobaric interferences and provide further confidence in AED identification in human serum. Furthermore, LC-DTIM-MS experiments have the potential to identify AEDs not reported in clinically relevant samples, through a combination of targeted quantitation by LC-MS/MS and semitargeted (restricted to coverage over AED ranges of interest) LC-DTIM-MS. The LC-triple-quadrupole MS/MS and LC-DTIM-MS workflow described herein offers both quantitative and qualitative analytical methods for AEDs in human serum.
Here, we present an LC-MS/MS and LC-DTIM-MS workflow that can be used in any analytical setting, including clinical, for the quantification and qualification of 14 AEDs in patient serum (Figure 1). In accordance with the Clinical and Laboratory Standards Institute (CLSI) LC-MS guidelines (C62-A)37 and US Food and Drug Administration (FDA) guidelines (Bioanalytical Method Guidance for Industry Validation),38 the quantitative LC-MS/MS method was assessed for method verification and validation. A second qualitative method for AED identification was performed using LC-DTIM-MS to assess selectivity. Collision cross section (CCS) values, which are derived from DTIM, show utility in improving the accuracy of small-molecule AED identifications. The qualitative method for AEDs was performed using an LC-DTIM-MS assay, building upon an approach detailed elsewhere.39 Taken together, these methods deliver robust quantification and qualification results for 14 AEDs and should prove beneficial for laboratories interested in increasing efficiency, sensitivity, and selectivity in AED testing.
Figure 1.
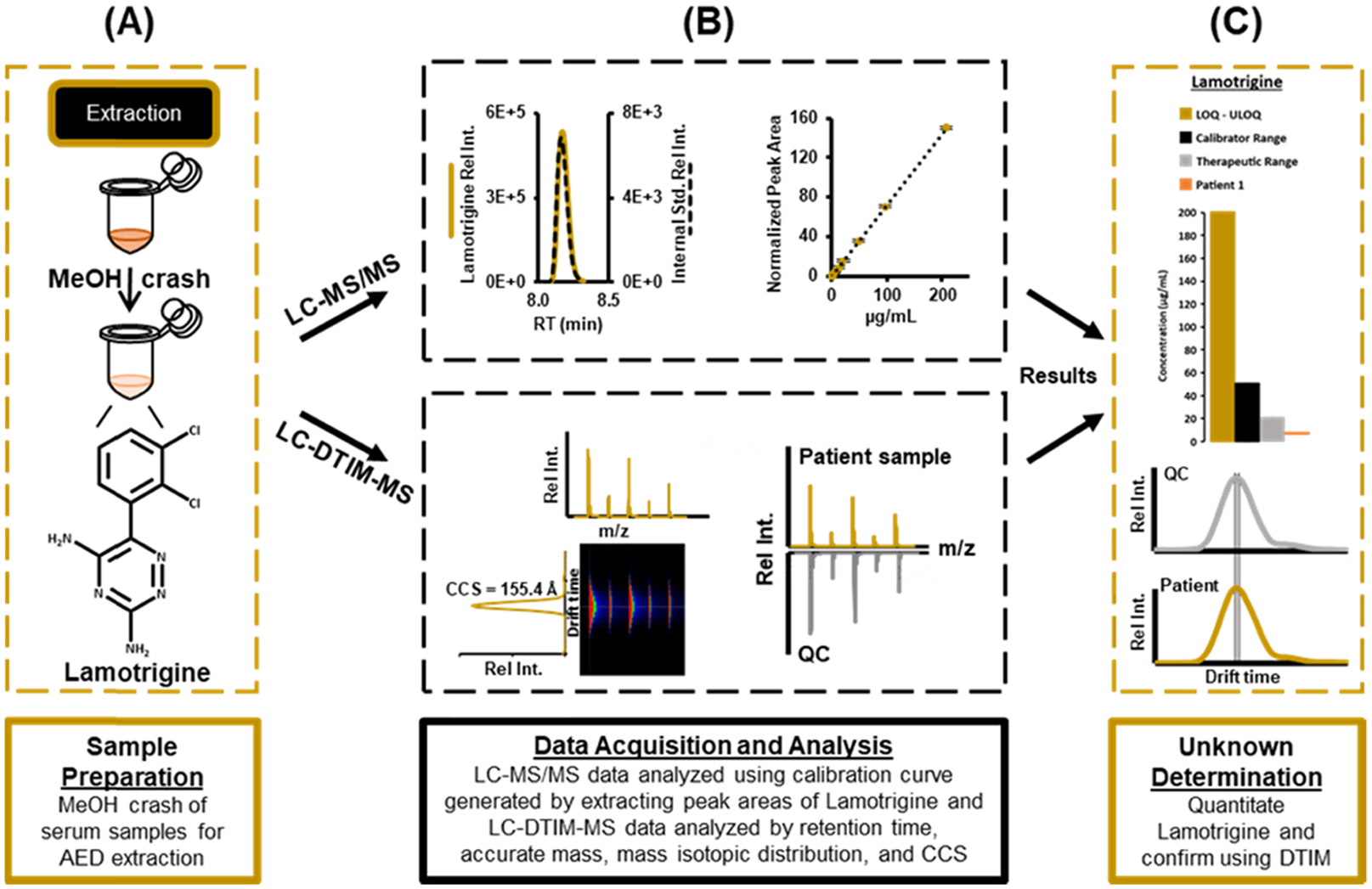
Workflow for LC-MS/MS and LC-DTIM-MS analysis beginning with (A) sample preparation, (B) data acquisition and analysis, and (C) unknown determination of AEDs in epileptic patient serum samples. In this study, the validated LC-MS/MS method provides quantitative AED concentrations and the LC-DTIM-MS method allowed for structural analysis and isomer discrimination.
EXPERIMENTAL METHODS
Standards and Chemicals.
All AEDs and stable isotopically labeled internal standards (SIL-ISs) were purchased from Sigma-Aldrich (St. Louis, MO). Optima LC/MS grade water, isopropyl alcohol, and methanol were obtained from Fisher Scientific (Hampton, NH). Optima LC/MS grade formic acid and ammonium acetate were obtained from Fisher Scientific (Hampton, NH).
Serum Samples.
Human drug-free serum and matrix-matched third party verified quality control (QC) material at low, medium, and high AED concentrations were obtained from UTAK Laboratories Incorporated (hereafter referred to as third party QC samples, Valencia, CA). QC and calibrator concentrations are outlined in Tables S1 and S3, respectively. Calibrators are samples used for linearity and/or the calibration curve. Deidentified, residual serum specimens were obtained from VUMC’s clinical toxicology laboratory in accordance with Institutional Review Board approval (IRB #172021). These samples were subjected to the extraction protocol described in Figure 1.
Human Serum Extraction and Preparation.
For all samples analyzed in these studies, a protein precipitation was performed in a 1.5 mL polypropylene microcentrifuge tube (Eppendorf, Hauppauge, NY).
Chromatographic Conditions.
For the LC-MS/MS method, AEDs were analyzed using a 3.0 × 50 mm2 reverse phase column, ZORBAX Eclipse Plus C18 Rapid Resolution HD 1.8 μm (Agilent Technologies, Santa Clara, CA) with a 2.1 × 5 mm2 1.8 μm ZORBAX Eclipse Plus C18 guard column, maintained at 40 °C for separation by ultra-high-pressure liquid chromatography (UHPLC, Agilent 1290 Infinity II system, Agilent Technologies, Santa Clara, CA). Mobile phase A consisted of water with 0.1% formic acid and 10 mM ammonium acetate. Mobile Phase B consisted of methanol with 0.1% formic acid and 10 mM ammonium acetate. The UHPLC was directly coupled online to a commercial triple-quadrupole mass spectrometer (6470, Agilent Technologies, Santa Clara, CA).
For the LC-DTIM-MS experiments, a UHPLC (Agilent 1290 Infinity I LC system, Agilent Technologies, Santa Clara, CA) was directly coupled online to a commercial DTIM-MS (6560, Agilent Technologies, Santa Clara, CA) using the same column and mobile phases as described above.
For both LC-MS/MS and LC-DTIM-MS methods, 1 μL of sample was injected at a flow rate of 800 μL/min and was analyzed using the following chromatographic conditions (17.5 min runtime including purge and equilibration times): mobile phase B was maintained at 5% for the first 3 min for an initial isocratic hold, linearly increased from 5 to 32.5% over 5.5 min, linearly increased again from 32.5 to 35% over 0.01 min, linearly increased a final time from 35 to 100% over 2.48 min, and held at 100% for 1.51 min. Mobile phase B returned to 5% by 12.5 min and was held at 5% for 5 min to re-equilibrate the column. In this method, the initial isocratic hold, final purge, and re-equilibration times were performed to ensure efficient cleaning, minimize carryover, and preserve column integrity. Additionally, the injection needle was washed with 60:40 (v:v) isopropanol: methanol followed by mobile phase starting conditions between every run to minimize carryover. A representative chromatogram of the 14 AEDs and mobile phase B gradient is shown in Figure 2A.
Figure 2.
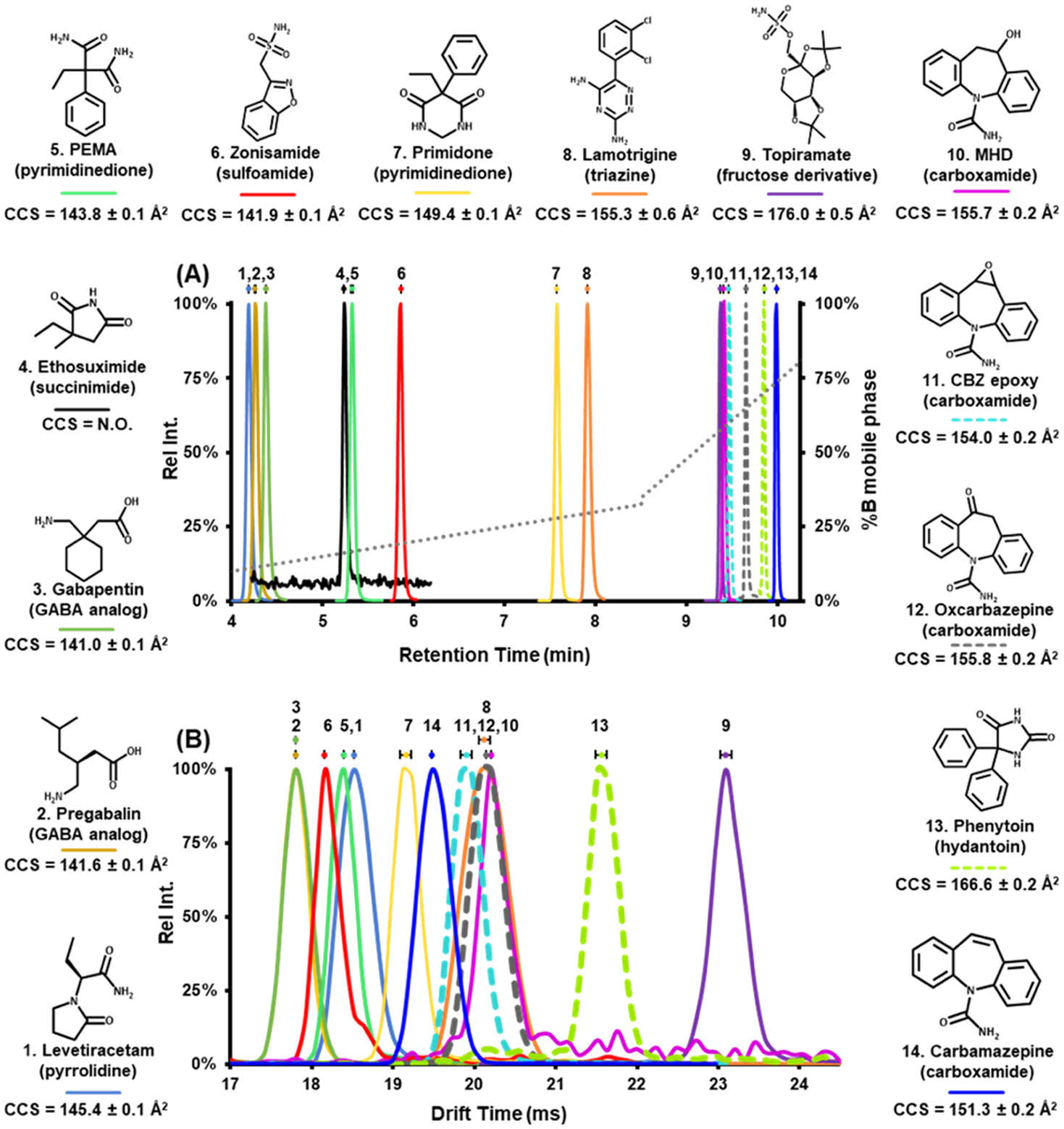
Structures of the 14 AEDs annotated with experimental CCS measurements. (A) LC-MS/MS analysis showing an LC chromatogram (min) of 14 AEDs in pooled human serum (quality control low). Dashed chromatograms are the three constitutional isomeric AEDs (C15H12N2O2, 253.0977 Da). The dotted gray line represents %B mobile phase gradient. Standard error bars (n = 3 process replicates) represent overlapping of retention times, and in this LC method, all AED retention times were shown to be statistically significant and distinct. (B) Flow injection analysis (FIA)-ion mobility-MS showing experimental DTIM spectra (ms) of 13 AED neat standards in the gas phase. Standard error bars (n = 3 technical replicates) were used to demonstrate overlapping drift times. Pregabalin/gabapentin and lamotrigine/MHD/oxcarbazepine exhibit nonstatistically significant different drift times. Dashed drift times represent the three constitutional isomeric AEDs (C15H12N2O2, 253.0977 Da), and all three have statistically significant distinct drift times, thus can be separated from each other in drift time space. It is important to note that AEDs that have similar retention times (e.g., topiramate and MHD) have different baseline resolved drift times. These data illustrate the utility in using both drift time and retention time measurements.
MS/MS and DTIM-MS Conditions.
For LC-MS/MS experiments, AEDs were analyzed in positive ionization mode using the Jet Stream ESI source (Agilent Technologies, Santa Clara, CA) coupled to a triple-quadrupole mass spectrometer (6470, Agilent Technologies). Nitrogen was used as both the nebulizing gas and the collision gas. AED transitions were collected using scheduled multiple reaction monitoring (MRM) with a Δ 2 min (±1 min) retention time window for each chromatographic peak. MRM transitions are listed in Table 1. Mass spectrometry conditions were optimized on a per-molecule basis, including compound-dependent parameters (e.g., fragmentor voltage, collision energy voltage, and cell accelerator voltage) by flow injection analysis (FIA) to maximize sensitivity. Table 1 also denotes the stable isotopically labeled internal standards (SIL-IS) used for normalization, transitions, limits of quantification (LOQs), therapeutic ranges, recoveries, and matrix effects. MHD-13C6 was used for CBZ, CBZ epoxy, phenytoin, and oxcarbazepine normalization owing to its similar RT and structural properties. Quantifier transitions (Quant m/z) are characteristic fragments or product ions of AED precursor ions used to quantitate AED concentrations. Representative chromatograms of AEDs and SIL-ISs at individual LOQs are available in Figure S1. Data were acquired using Agilent’s MassHunter Workstation Data Acquisition software and analyzed using Skyline (MacCoss Lab),40,41 Agilent’s MassHunter Quantitative Analysis, MassHunter Qualitative Analysis, Microsoft PowerPoint, and Microsoft Excel.
Table 1.
AED MRM Transition List, Limit of Quantification (LOQ), Therapeutic Range, Recovery, and Matrix Effects
| no. | AED | Q1 (m/z) | Q3 (m/z) | LOQ (μg/mL) | therapeutic range (μg/mL) | recovery ± %CV | matrix effect ± %CV |
|---|---|---|---|---|---|---|---|
| 1 | levetiracetam | 171 | 154 | 0.2 | 5–40 | 112 ± 7% at 25 μg/mL | 92 ± 12% at 25 μg/mL |
| leveti.racetam.-D6 | 177 | 160 | |||||
| 2 | pregabalin | 160 | 142 | 0.2 | 4–20 | 116 ± 7% at 13 μg/mL | 103 ± 9% at 13 μg/mL |
| pregabalin-13C3 | 163 | 145 | |||||
| 3 | gabapentin | 172 | 154 | 0.2 | 2–20 | 120 ± 7% at 13 μg/mL | 104 ± 10% at 13 μg/mL |
| gabapentin-13C3 | 175 | 157 | |||||
| 4 | ethosuximide | 142 | 114 | 6.3 | 40–100 | 80 ± 18% at 50 μg/mL | 76 ± 11% at 50 μg/mL |
| zonisamide-13C6 | 219 | 138 | |||||
| 5 | PEMA | 207 | 162 | 0.2 | 95 ± 7% at 25 μg/mL | 50 ± 3% at 25 μg/mL | |
| zonisamide-13C6 | 219 | 138 | |||||
| 6 | zonisamide | 213 | 132 | 0.2 | 10 – 40 | 114 ± 6% at 25 μg/mL | 100 ± 10% at 25 μg/mL |
| zonisamide-13C6 | 219 | 138 | |||||
| 7 | primidone | 219 | 162 | 0.2 | 5–12 | 87 ± 8% at 12 μg/mL | 63 ± 4% at 12 μg/mL |
| lamotrigine-13C, 15N4 | 262 | 215 | |||||
| 8 | lamotrigine | 256 | 211 | 0.2 | 1–20 | 111 ± 7% at 13 μg/mL | 101 ± 10% at 13 μg/mL |
| lamotrigine-13C, 15N4 | 262 | 215 | |||||
| 9 | topiramate | 362 | 265 | 0.4 | 10–20 | 52 ± 2% at 13 μg/mL | 47 ± 4% at 13 μg/mL |
| topiramate-D12 | 374 | 276 | |||||
| 10 | MHD | 255 | 237 | 0.2 | 10 – 40 | 104 ± 8% at 25 μg/mL | 98 ± 11% at 25 μg/mL |
| MHD-13C6 | 261 | 200 | |||||
| 11 | CBZ epoxy | 253 | 236 | 0.1 | 4–12 | 62 ± 6% at 13 μg/mL | 86 ± 19% at 13 μg/mL |
| MHD-13C6 | 261 | 200 | |||||
| 12 | oxcarbazepine | 253 | 180 | 0.7 | 10 – 40 | 88 ± 15% at 25 μg/mL | 80 ± 10% at 25 μg/mL |
| MHD-13C6 | 261 | 200 | |||||
| 13 | phenytoin | 253 | 104 | 0.8 | 1–2.5 | 59 ± 6% at 25 μg/mL | 62 ± 7% at 25 μg/mL |
| MHD-13C6 | 261 | 200 | |||||
| 14 | carbamazepine | 237 | 194 | 0.1 | 4–12 | 60 ± 6% at 13 μg/mL | 91 ± 3% at 13 μg/mL |
| MHD-13C6 | 261 | 200 |
For FIA-DTIM-MS and LC-DTIM-MS analysis, AEDs were analyzed in positive ionization mode using the Jet Stream ESI source (Agilent Technologies, Santa Clara, CA) coupled to a DTIM mass spectrometer (6560, Agilent Technologies) using previously described instrumental settings and methods.39,42–45 Briefly, the LC-DTIM-MS method consisted of a single-field drift time analysis using nitrogen drift gas with the drift tube at a temperature of 30 °C, a pressure of 4.0 Torr, and an electric field of 17.3 V/cm for 30 s. For LC-DTIM-MS analyses, a calibrated single-field CCS method was used to calculate CCS values via the Mason–Schamp equation.39 Data was analyzed using MassHunter Qualitative Analysis, MassHunter IM-MS Browser, and Microsoft Excel. A representative plot of the 13 AED ion mobility profiles is shown in Figure 2B. The primary measurement dimension of drift time is used rather than collision cross section to overlay the different mass species without alterations in expected ion mobility resolving power (i.e., changes in peak width arising from nonlinear conversion from drift time to cross section).
Method Validation.
Guidelines established by the FDA Guidance for Industry Bioanalytical Method Validation were used to validate the LC-MS/MS AED assay.37,38 Analytical figures of merit including LOQ, accuracy, precision, carryover, stability, selectivity, recovery, and matrix effect were assessed. We did not perform dilution effects and partial volume validation studies because none of the clinical samples were diluted as received. The full volume of samples (100 μL) was used for analysis.
RESULTS AND DISCUSSION
LC-MS/MS and FIA-DTIM-MS.
The range of retention times (min) from LC-MS/MS and ion mobility drift times (ms) from FIA-DTIM-MS are shown in Figure 2A,B, respectively. Collectively, these data demonstrate the structural diversity among the AEDs/anticonvulsants outlined in these studies. Under these conditions, ethosuximide was not observed using the FIA-DTIM-MS method, where we observed an LOQ for this AED approximately 1 order of magnitude higher in concentration than all other AEDs in this study (as shown in Figure S1). Chromatographically, all AEDs were shown to display statistically significant separation with minimal overlapping retention times (Figure 2B). For those AEDs that did not exhibit baseline separation chromatographically, the data show that there were statistically significant distinct mobility profiles with baseline separation for two AEDs, topiramate and MHD (Figure 2A,B). Structural isomers (CBZ epoxy, oxcarbazepine, and phenytoin) demonstrated statistically significant separation in both chromatographic and ion mobility profiles (Figure 2). Separation of structurally isomeric AEDs, such as CBZ epoxy, oxcarbazepine, and phenytoin (identical chemical formulas), can also be observed in Figure 3 in which these three AEDs have the same isotopic m/z but different CCS values. The structural diversity of AEDs is also shown in Figure 3; these data exemplify the presence of unique best fit trendlines with ±10% deviation from the best fit line for 13 AEDs. The deviation demonstrates the structural properties of AEDs and lays the groundwork for using CCS AED measurements as an identifier in untargeted and/or targeted studies.34 Taken together, these data strengthen the confidence in identifying which AEDs a particular patient is taking through internal validation of workflows and also affords the ability to characterize potentially unknown or known structurally similar drugs or druglike compounds that may be in a patient’s serum/plasma sample as chromatographically coeluting interferences. Such situations can complicate accurate quantification of the clinical panel,33 which ultimately may lead to treatment decisions for a particular patient that might be different with improved accuracy.
Figure 3.
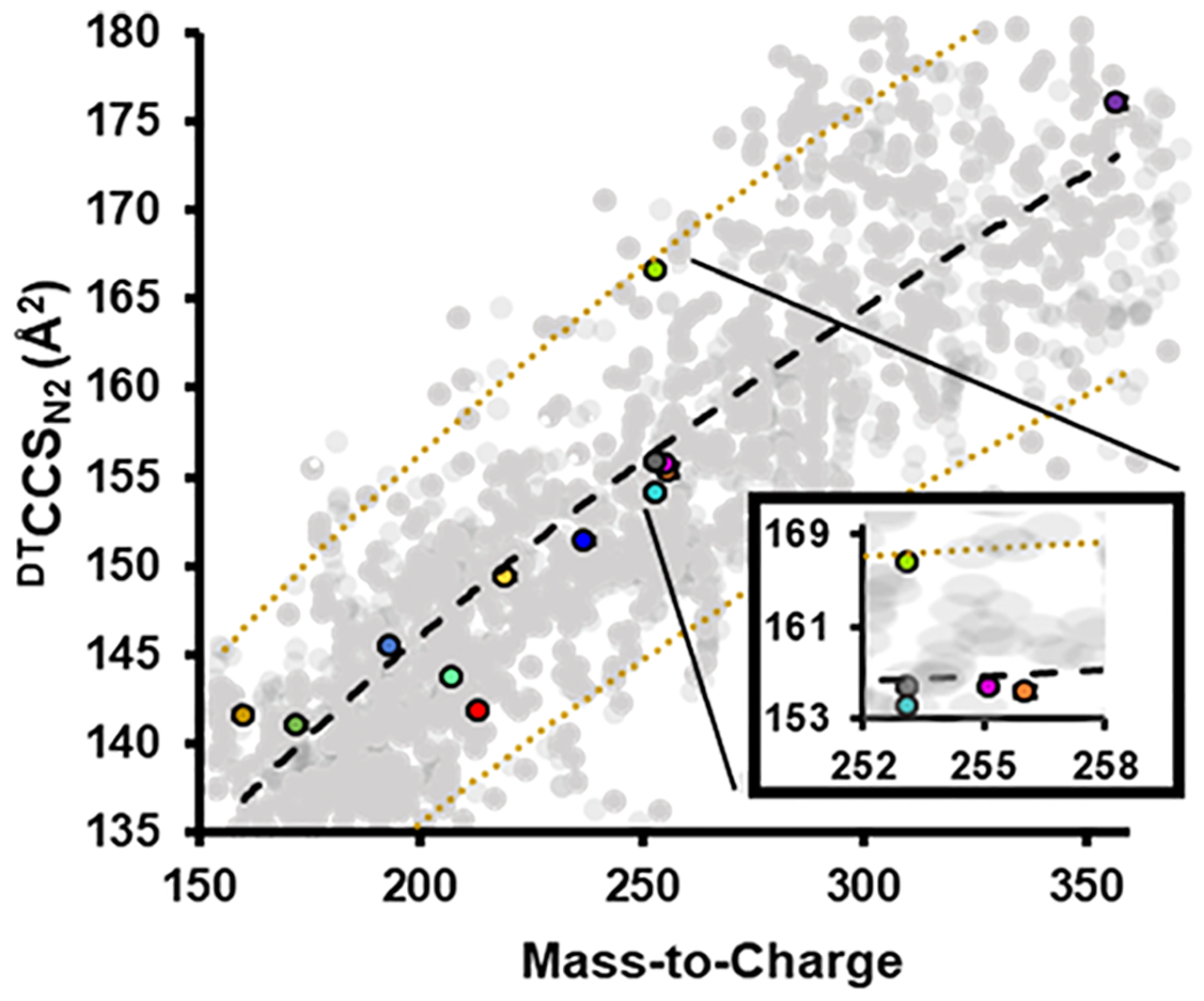
Conformational space analysis showing DTCCSN2 values for the AEDs investigated using FIA-DTIM-MS with neat standards. Included is a black dashed trendline representing the best fit line of the data fit to a power function. Also shown (in gold) are dashed lines representing ±10% deviation from the best fit line. Measured AEDs were within ±10% of the best fit line. Error bars represent standard errors and are for most values within the scale of the marker (n = 3 technical replicates). The gray data points represent ~1700 entries available in the CCS compendium over this CCS and m/z range. These data span multiple classes of compounds.34
Method Validation, Comparison, and Application.
For individual AEDs, the recovery and matrix effect studies (Table 1 and Figure S6) showed high reproducibility (<20% CV). Recovery studies for topiramate, phenytoin, CBZ, and CBZ epoxy were <80%. However, the QC samples for these molecules did meet the acceptability criteria for precision and accuracy (<15% CV and <15% bias, respectively); therefore, the extraction did not negatively affect reproducibility (Figure S2). We did not observe any significant matrix effects for 9 out of the 14 AEDs (i.e., ionization suppression or ionization enhancement matrix effect >80%). Fortunately, the SIL-IS of these AEDs successfully compensated for recovery and/or matrix effects; therefore, the matrix effect values were >80% (e.g., levetiracetam, pregabalin, gabapentin, zonisamide, lamotrigine, MHD, and oxcarbazepine).46 Five of the AEDs exhibited a matrix effect <80% (topiramate, ethosuximide, PEMA, primidone, and phenytoin). For these AEDs, we were unable to use the exact SIL-IS and therefore their recoveries and/or matrix effects were <80%.46 The SIL-IS used for oxcarbazepine was a physiochemical mimic (MHD-13C6); the calculated recovery and matrix effect studies were >80%. On the other hand, the SIL-IS used for topiramate has 12 deuterons causing it to display an isotopic effect and elute earlier (<5 s) than topiramate. Because of these shifts in elution time, we observed <80% recovery and matrix effect for topirimate.47–49 We anticipate that using either carbon-13 or nitrogen-15 SIL-ISs for AEDs exhibiting recoveries and/or matrix effects <80% could resolve these findings that were observed in complex human serum samples.37 In these studies, a verified third party vendor provided the quality control samples; these samples were used to assess and validate precision, accuracy, recovery, and matrix effects. These results demonstrate that the LC-MS/MS quantitative method described herein is fit for purpose. Taken together, these results suggest that the analytical method reported here minimizes the impacts of endogenous interferences from human serum and/or from concomitant medications and allows for both reliable and reproducible quantification of the 14 AEDs.
The developed LC-MS/MS method meets the criteria for routine clinical TDM owing to the results of the linearity, accuracy, precision, carryover, recovery, matrix effects, selectivity, and stability studies. The LOQ was established and adequately brackets the therapeutic range via linearity studies (Tables 1 and S2). Precision and accuracy of all AEDs in third party QCs were <15% CV and 15% bias (Figure S2). There was no significant carryover (Figure S5), instability (Figure S4), recovery (Table 1), and matrix effects (Table 1) that negatively impacted the quantification of all 14 AEDs.
The quantitative LC-MS/MS measurements were compared to Vanderbilt University Medical Center (VUMC) clinical toxicology laboratory’s LC-UV measurements, many samples of which contained multiple AEDs (nine contrived samples—Table S4 and 21 patient samples—Figure S3). The data obtained indicate that the method presented here is comparable to the validated LC-UV method. Of the 30 samples analyzed, ~23% of the samples contained additional AEDs that were not reported in the LC-UV method (these AEDs include levetiracetam, lamotrigine, MHD, pregabalin, and gabapentin) but were identified using the workflows described herein. Furthermore, the LC-UV method was unable to distinguish pregabalin and gabapentin from each other and required a third party LC-MS/MS laboratory for analysis. Figure S3A shows and compares the data generated from the same 21 samples but analyzed using these methods: the LC-MS/MS and LC-DTIM-MS methods developed herein, LC-UV measurements, third party LC-MS/MS analyses (pregabalin and gabapentin), and an immunoassay (CBZ). We performed LC-DTIM-MS analyses to identify AEDs that were present in individual patients but not reported (Figure S3B). To assess the utility of the DTIM dimension in identifying targeted analytes and/or selectivity, Figure 4 illustrates the theoretical isotope distribution patterns for three AEDs (generated from a single-patient sample analyzed in the same batch) as well as data obtained when extracting (1) MS only dimension, (2) LC-MS dimensions, (3) DTIM-MS dimensions, and (4) combined LC-DTIM-MS dimensions. These data collectively illustrate that orthogonal separation tools, such as DTCCSN2 measurements, provide additional selectivity and confidence in analyte identification. Specifically, the LC-DTIM-MS data separated concomitant interferences and increased confidence in AED identification for all three AEDs. These data also show the ability of the DTIM dimension to remove concomitant species otherwise not separated in the LC and MS dimensions (see zonisamide, Figure 4).
Figure 4.
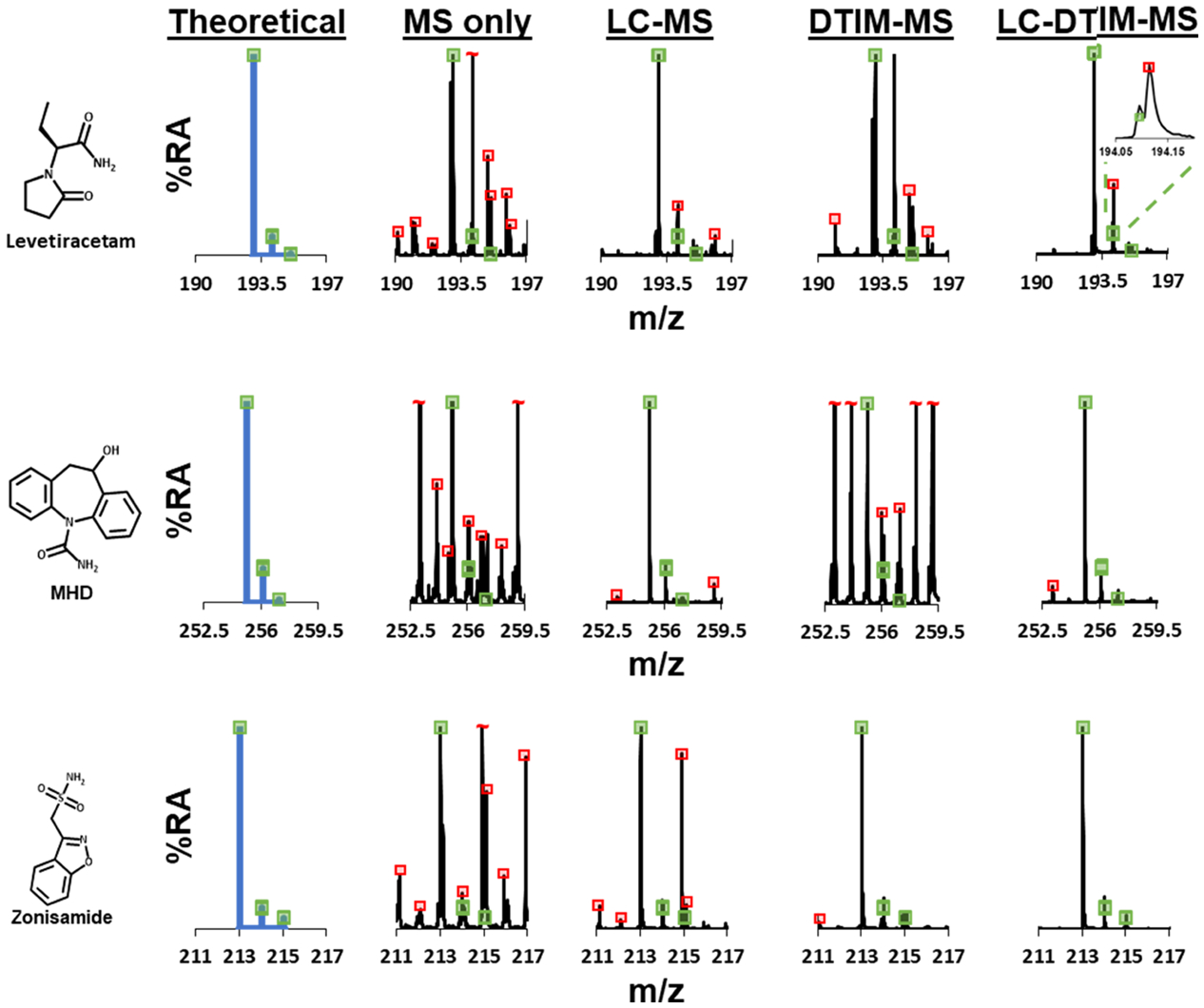
Isotope distribution patterns for three AEDs (levetiracetam, MHD, and zonisamide) from a single-patient serum sample (patient 11) analyzed by LC-DTIM-MS, where mass spectra were extracted for the listed specific dimensions. The MS only dimension extracted the entire chromatogram and ion mobility spectrum. The LC-MS dimension extracted the AED chromatograms for levetiracetam, MHD, and zonisamide, while the entire ion mobility spectrum was extracted. The DTIM-MS dimension extracted the entire chromatogram, while AED ion mobility spectra for levetiracetam, MHD, and zonisamide were extracted. The LC-DTIM-MS dimension extracted the AED chromatograms and ion mobility spectra for levetiracetam, MHD, and zonisamide. The green boxes represent theoretical mass isotopic distribution matches (±5% height deviation) and can be used to increase confidence in AED assignment. The mass spectra for MS only, LC-MS, DTIM-MS, and LC-DTIM-MS also show red boxes. These red boxes represent isotopic distribution mismatches or concomitant interferences that were observed in the complex sample. In this data, numerous interference peaks were observed in the MS only dimension. The separation power by both LC and DTIM dimensions filtered numerous interference peaks from the AED of interest, especially when these separation dimensions were combined.
By integrating the LC-MS/MS and LC-DTIM-MS work-flows, the process for monitoring AEDs is simplified and provides additional clinical information. Since TDM quantification was not ordered for levetiracetam, lamotrigine, and MHD, these were not reported in the clinical samples but monitored in the present work as they are continually included in the workflow. Furthermore, carbamazepine would be tested using an immunoassay method and through always incorporating carbamazepine in these workflows could remove the time and expense related to performing an additional antibody-based testing platform from these processes.
Importantly, the qualitative aspects of the LC-DTIM-MS analyses directly complement the quantitative LC-MS/MS results. While it is possible to potentially combine both workflows onto a single LC-DTIM-MS instrument, these instruments are typically outfitted with a time-of-flight mass analyzer rather than using a triple-quadrupole configuration for fundamental sampling reasons described elsewhere.43 To draw benchmark comparisons with conventional validated triple-quadrupole MS/MS methods, which are the gold standard in routine testing laboratories, we chose to separate the quantitative and qualitative aspects of both triple-quadrupole MS/MS and DTIM-MS platforms, respectively. Our goal is to demonstrate the unique capabilities that DTIM-MS offer for resolving interferences while having a wide breadth of molecular coverage. In this work, we demonstrate the viability of DTIM-MS for clinical and routine testing laboratories and benchmark against standardized methodologies using triple-quadrupole MS/MS methods.
The results in Figures 2–4 and S3 show that the use of DTIM as a molecular descriptor in addition to accurate mass and retention time for AEDs and AED structural isomer analyses has great potential for rapidly characterizing and identifying structurally similar AEDs. Figure 3 presents a scatter plot of CCS versus m/z with 13 AEDs shown in color and the gray background points being entries over this CCS and m/z range in the Unified CCS compendium, which could be considered potential endogenous biological background.34,42 This type of 2D DTIM-MS projection as conformational space analysis is indicative of differences in gas-phase molecular packing efficiency, which is related to molecular structure.42 Previously, the community has shown that individual biomolecular classes such as oligonucleotides, carbohydrates, peptides, and lipids occupy distinct regions in conformational space.50 However, in the small-molecule/metabolite region of conformation space, these distinctions are much less pronounced. This appears to also be the case for the AEDs or anticonvulsants.33,39 Nevertheless, the results in Figure 3 illustrate that each of the AEDs and structural isomer or isobaric AEDs were separated from each other in conformational space based on their structural properties (e.g., the isobars CBZ epoxy, oxcarbazepine, and phenytoin). CBZ epoxy and oxcarbazepine are both carboxamides and phenytoin is a hydantoin (Figure 2). Both CBZ epoxy and oxcarbazepine structures correspond to constrained dibenzazepine structures resulting in more compact collision cross sections. The phenytoin structure is characterized by unconstrained phenyl moieties, which yield a higher degree of freedom and likely results in the larger observed collision cross section across the three isomers. Further support for this observation is noted in that the two phenyl functional groups in phenytoin are not coplanar. The structural differences between the two carboxamides are the epoxide group on CBZ epoxy and the ketone group on oxcarbazepine. As these three AEDs have the same chemical formula and are isobaric, it is these functional group differences that provide for structural separation via DTIM (DTCCSN2 = 154.0 Å2, DTCCSN2 = 155.8 Å2, and DTCCSN2 = 166.6 Å2, for CBZ epoxy, oxcarbazepine, and phenytoin, respectively). These structural separations of isobaric AEDs occur within the timescale of traditional liquid chromatographic and ion mobility techniques.51
To further show the utility of the ion mobility separation capabilities, we analyzed a single-patient sample that contained levetiracetam, MHD, and zonisamide (Figure 4). This data shows the mass spectra from the complex patient sample when analyzed using filtering from the four different modes of separation, specifically: MS only separation, LC-MS separation, DTIM-MS separation, and integrated LC-DTIM-MS separation. Individual mass spectra were compared to the theoretical isotopic distribution for each of these drugs. Mass spectra generated using the LC-MS dimension successfully removed numerous interferences when compared to DTIM-MS (see levetiracetam and MHD, specifically). The DTIM-MS dimension mass spectra for zonisamide removed concomitant species that the LC-MS dimension was unable to separate. The combined LC-DTIM-MS analyses yielded the least number of concomitant species and provide relatively interference free mass spectra. The qualitative LC-DTIM-MS results shown in this manuscript demonstrate that measured AED CCS values in human serum samples can be matched to pre-existing CCS values from AED standards. These data allow for an increase in confidence in annotated features.52 Specifically, Figure S3B shows data generated from AED QCs and patient samples with drift time alignments of levetiracetam, lamotrigine, zonisamide, pregabalin, and MHD, where the QCs (n = 3) confirm the identifications of the patient samples (n = 1) and thus show the ability for LC-DTIM-MS to increase confidence in identifications. These results also highlight the benefits of adding the DTIM dimension in studying complex samples. Moreover, the CCS values for the AEDs measured in this work can be readily incorporated into existing CCS libraries for inclusion into targeted and/or untargeted LC-DTIM-MS workflows. As CCS values have been previously shown to be highly reproducible (<1.0% difference in an interlab study),39 CCS values for the 13 AEDs observed in our FIA-DTIM-MS analyses can be used as reference values for other laboratories. Utilizing database CCS matching provides additional molecular confidence in annotating features in targeted and/or untargeted studies, especially when combined with other molecular descriptors such as mass defect, isotope ratio patterning, and fragmentation methods.34,52–55
CONCLUSIONS
Polypharmacy is common for epileptic patients, and many hospital laboratories use a combination of immunoassays, LC-UV, and/or LC-MS/MS assays for TDM testing of AEDs. The single, multiplexed, quantitative LC-MS/MS method outlined here would enable laboratories to simultaneously quantitate 14 AEDs when in-house mass spectrometry platforms are available. The qualitative LC-DTIM-MS analysis utilized accurate mass measurement and CCS values for improved confidence in identifications generated from the LC-MS/MS method, and additionally increased confidence in identifying AEDs previously unreported in patient samples. The tradeoff between breadth and depth in molecular characterization utilizing DTIM-MS methods is the reduction in sensitivity over targeted triple-quadrupole MS/MS (MRM) methodologies. A unique advantage of DTIM-MS methods is the ability to perform simultaneous MS/MS experiments for all species on the timescale of the chromatography and ion mobility dimensions. The quantitative and qualitative assays presented here are specific and offer quantitative separation of structural isobaric isomers (e.g., CBZ epoxy, oxcarbazepine, and phenytoin) that should enable therapeutic decisions to be made with higher confidence.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Jody C. May, Bailey S. Rose, Simona G. Codreanu, Charles M. Nichols, James N. Dodds, and Andrzej Balinski for contributions in various stages for advice and technical expertise. Financial support for aspects of this research was provided by The National Institutes of Health (National Cancer Institute R03CA222452). This work was supported in part using the resources of the Center for Innovative Technology at Vanderbilt University.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03172.
LC-MS/MS data, LC-ion mobility-MS data, and samples presented in this work (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c03172
The authors declare no competing financial interest.
Contributor Information
Don E. Davis, Jr., Center for Innovative Technology, Department of Chemistry, Institute of Chemical Biology, Institute for Integrative Biosystems Research and Education, Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, Tennessee 37235, United States
Stacy D. Sherrod, Center for Innovative Technology, Department of Chemistry, Institute of Chemical Biology, Institute for Integrative Biosystems Research and Education, Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, Tennessee 37235, United States
Randi L. Gant-Branum, Center for Innovative Technology, Department of Chemistry, Institute of Chemical Biology, Institute for Integrative Biosystems Research and Education, Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, Tennessee 37235, United States
Jennifer M. Colby, Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee 37235, United States
John A. McLean, Center for Innovative Technology, Department of Chemistry, Institute of Chemical Biology, Institute for Integrative Biosystems Research and Education, Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, Tennessee 37235, United States
REFERENCES
- (1).Brandt C; Fueratsch N; Boehme V; Kramme C; Pieridou M; Villagran A; Woermann F; Pohlmann-Eden B Epilepsy Behav 2007, 11, 133–139. [DOI] [PubMed] [Google Scholar]
- (2).Gupta MA; Pur DR; Vujcic B; Gupta AK Clin. Dermatol 2018, 36, 756–764. [DOI] [PubMed] [Google Scholar]
- (3).Prabhavalkar KS; Poovanpallil NB; Bhatt LK Front. Pharmacol 2015, 6, No. 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Spina E; Perugi G Epileptic Disord 2004, 6, 57–75. [PubMed] [Google Scholar]
- (5).Patsalos PN; Spencer EP; Berry DJ Ther. Drug Monit 2018, 40, 526. [DOI] [PubMed] [Google Scholar]
- (6).Patsalos PN; Berry DJ; Bourgeois BFD; Cloyd JC; Glauser TA; Johannessen SI; Leppik IE; Tomson T; Perucca E Epilepsia 2008, 49, 1239–1276. [DOI] [PubMed] [Google Scholar]
- (7).Inada A; Oyama S; Niinomi I; Wakabayashi T; Iwanaga K; Hosohata KJ Clin. Pharm. Ther 2019, 44, 775–779. [DOI] [PubMed] [Google Scholar]
- (8).Rogawski MA; Löscher W Nat. Rev. Neurosci 2004, 5, 553–564. [DOI] [PubMed] [Google Scholar]
- (9).Levy RH; Schmidt D Epilepsia 1985, 26, 199–205. [DOI] [PubMed] [Google Scholar]
- (10).St. Louis EK Curr. Neuropharmacol 2009, 7, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Balestrini S; Sisodiya SM Neurosci. Lett 2018, 667, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Krasowski MD; McMillin GA Clin. Chim. Acta 2014, 436, 224–236. [DOI] [PubMed] [Google Scholar]
- (13).Meador KJ; Loring DW Neurology 2016, 86, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shibata M; Hashi S; Nakanishi H; Masuda S; Katsura T; Yano I Biomed. Chromatogr 2012, 26, 1519–1528. [DOI] [PubMed] [Google Scholar]
- (15).Krasowski MD; Siam MG; Iyer M; Ekins S Ther. Drug Monit 2009, 31, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dasgupta A Ther. Drug Monit 2012, 34, 496–506. [DOI] [PubMed] [Google Scholar]
- (17).Krasowski MD; Siam MG; Iyer M; Pizon AF; Giannoutsos S; Ekins S Clin. Chem 2009, 55, 1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dasgupta A Ther. Drug Monit 2008, 30, 212–217. [DOI] [PubMed] [Google Scholar]
- (19).Krasowski MD Pharmaceuticals 2010, 3, 1909–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Patsalos PN; Zugman M; Lake C; James A; Ratnaraj N; Sander JW Epilepsia 2017, 58, 1234–1243. [DOI] [PubMed] [Google Scholar]
- (21).Alexander HB; Broshek DK; Quigg M Epilepsy Behav 2018, 78, 96–99. [DOI] [PubMed] [Google Scholar]
- (22).Baftiu A; Feet SA; Larsson PG; Burns ML; Henning O; Sætre E; Molden E; Granas AG; Johannessen SI; Landmark CJ Epilepsy Res 2018, 139, 35–42. [DOI] [PubMed] [Google Scholar]
- (23).Bjørke AB; Nome CG; Falk RS; Gjerstad L; Taubøll E; Heuser K Seizure 2018, 61, 63–70. [DOI] [PubMed] [Google Scholar]
- (24).Beiske GAG; Holmøy T; Beiske AG; Johannessen SI; Landmark CJ Mult. Scler. Int 2015, 2015, No. 317859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).D’Urso A; Cangemi G; Barco S; Striano P; DʼAvolio A; de Grazia U Ther. Drug Monit 2019, 41, 331–339. [DOI] [PubMed] [Google Scholar]
- (26).Kuhn J; Knabbe C Talanta 2013, 110, 71–80. [DOI] [PubMed] [Google Scholar]
- (27).Yin L; Shi M; Wang T; Zhang M; Zhao X; Zhang Y; Gu J Chromatographia 2016, 80, 137–143. [Google Scholar]
- (28).May JC; Goodwin CR; McLean JA Curr. Opin. Biotechnol 2015, 31, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chouinard CD; Wei MS; Beekman CR; Kemperman RHJ; Yost RA Clin. Chem 2016, 62, 124–133. [DOI] [PubMed] [Google Scholar]
- (30).Sherrod SD; McLean JA Clin. Chem 2016, 62, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).May JC; Gant-Branum RL; McLean JA Curr. Opin. Biotechnol 2016, 39, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dodds JN; May JC; McLean JA Anal. Chem 2017, 89, 12176–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hines KM; Ross DH; Davidson KL; Bush MF; Xu L Anal. Chem 2017, 89, 9023–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Picache JA; Rose BS; Balinski A; Leaptrot KL; Sherrod SD; May JC; McLean JA Chem. Sci 2019, 10, 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Miura M; Takahashi N Drug Metab. Pharmacokinet 2016, 31, 12–20. [DOI] [PubMed] [Google Scholar]
- (36).Farouk F; ElKady EF; Azzazy HME Biomed. Chromatogr 2017, 31, No. e3921. [DOI] [PubMed] [Google Scholar]
- (37).Clinical and Laboratory Standards Institute. Liquid Chromatography-Mass Spectrometry Methods; Approved Guidelines (C62-A), No. 1–56238-977–7; 2014; pp 1–71. [Google Scholar]
- (38).US Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation, No. FDA-2013-D-1020; 2018; pp 1–41. [Google Scholar]
- (39).Stow SM; Causon TJ; Zheng X; Kurulugama RT; Mairinger T; May JC; Rennie EE; Baker ES; Smith RD; McLean JA; et al. Anal. Chem 2017, 89, 9048–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Henderson CM; Shulman NJ; MacLean B; Hoofnagle AN; MacCoss MJ Clin. Chem 2017, 64, 408–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).May JC; Goodwin CR; Lareau NM; Leaptrot KL; Morris CB; Kurulugama RT; Mordehai A; Klein C; Barry W; Darland E; et al. Anal. Chem 2014, 86, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).May JC; McLean JA Anal. Chem 2015, 87, 1422–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).May JC; McLean JA Annu. Rev. Anal. Chem 2016, 9, 387–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dodds JN; May JC; McLean JA Anal. Chem 2017, 89, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Matuszewski BK; Constanzer ML; Chavez-Eng CM Anal. Chem 2003, 75, 3019–3030. [DOI] [PubMed] [Google Scholar]
- (47).Tanaka N; Thornton ER J. Am. Chem. Soc 1977, 99, 7300–7307. [Google Scholar]
- (48).Reed DR; Kass SR J. Am. Soc. Mass Spectrom 2001, 12, 1163–1168. [DOI] [PubMed] [Google Scholar]
- (49).Wang S; Cyronak M; Yang EJ Pharm. Biomed. Anal 2007, 43, 701–707. [DOI] [PubMed] [Google Scholar]
- (50).Fenn LS; Kliman M; Mahsut A; Zhao SR; McLean JA Anal. Bioanal. Chem 2009, 394, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Nichols CM; Dodds JN; Rose BS; Picache JA; Morris CB; Codreanu SG; May JC; Sherrod SD; McLean JA Anal. Chem 2018, 90, 14484–14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Dodds JN; Hopkins ZR; Knappe DRU; Baker ES Anal. Chem 2020, 92, 4427–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Poland JC; Schrimpe-Rutledge AC; Sherrod SD; Flynn CR; McLean JA Anal. Chem 2019, 91, 14417–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Harris RA; Leaptrot KL; May JC; McLean JA TrAC, Trends Anal. Chem 2019, 116, 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Leaptrot KL; May JC; Dodds JN; McLean JA Nat. Commun 2019, 10, No. 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.