

Abstract
Transcription factors (TFs) play a critical role in determining cell fate decisions by integrating developmental and environmental signals through binding to specific cis-regulatory modules and regulating spatio-temporal specificity of gene expression patterns. Precise identification of functional TF binding sites in time and space not only will revolutionize our understanding of regulatory networks governing cell fate decisions but is also instrumental to uncover how genetic variations cause morphological diversity or disease. In this review, we discuss recent advances in mapping TF binding sites and characterizing the various parameters underlying the complexity of binding site recognition by TFs.
Introduction
The production of the diverse and specialized cell types of multicellular organisms, which are encoded by the same DNA in an individual, is controlled by the precise spatial and temporal regulation of gene expression. Cis-regulatory elements (CREs), including promoters, enhancers, silencers, and insulators, modulate the spatial and temporal expression of genes via recruitment of trans-regulatory factors such as sequence-specific TFs, chromatin remodelers, and RNA polymerase II [1–9] (Figure 1). Identifying the CREs that precisely define expression activity of developmentally and physiologically important genes in time and space is a long-standing challenge in plant biology and can open new opportunities for accelerating genetic improvement of crops. Except for gene promoters that are located close to the transcription start sites (TSS), the other CREs, especially for large genomes, can be thousands or even millions of bases away from their target genes [10,11]. Moreover, although the sequence specificities and binding locations of many TFs are known, we lack adequate knowledge about the dynamics of TF-DNA interaction over time and space, nor do we understand the complexity of factors determining when and where binding sites are functional. All these make it difficult to accurately pinpoint the CREs controlling the expression pattern of a given gene. Rapid development in experimental techniques and computational methods in conjunction with intensive studies over the last two decades have advanced our knowledge on this topic, such as how TFs recognize a subset of CREs and regulate the expression of proximally located or distal target genes and how paralogous TFs recognize non-identical binding sites in vivo [12–16]. In this review, we attempt to highlight the important progress that has been made in recent years for identifying TF-DNA binding sites at genome-scale and understanding the factors that contribute to TF DNA interaction.
Figure 1.
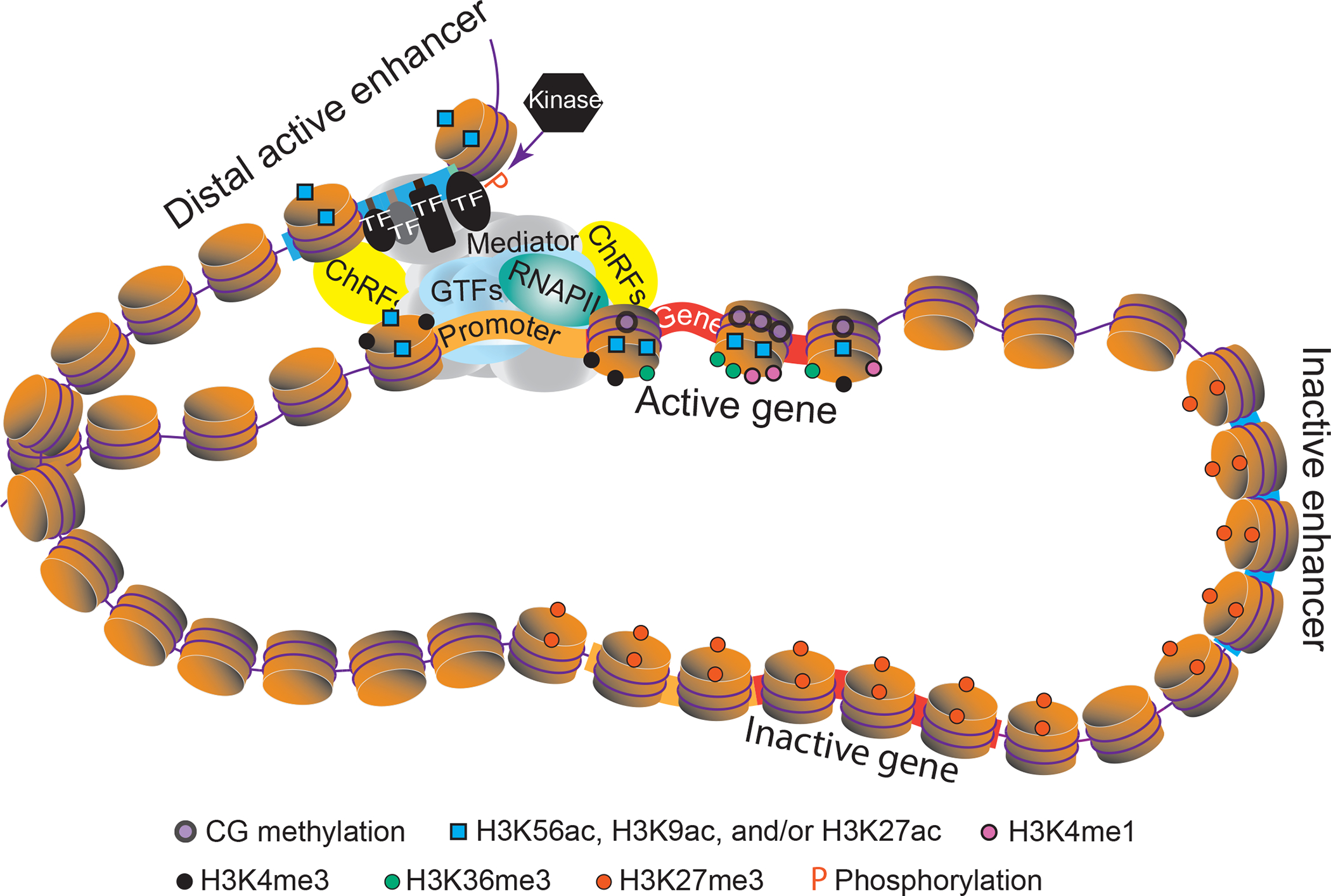
A model of tissue- or cell-type specific transcriptional regulation by distal enhancer containing multiple TF binding sites in plants. The enhancer located in an open chromatin region and harboring multiple TF motifs recruits sequence-specific TFs, which in turn leads to the recruitment of the Mediator complex, chromatin remodeling factors (ChRFs), general transcription factors (GTFs) and RNA polymerase II (RNAPII). Recent genome-wide studies in plants have suggested that active and inactive enhancers as well as genes can be defined by unique chromatin features and DNA methylation patterns. In this model, the insulator blocks unwanted interaction of the active enhancer with the depicted inactive gene. Post-translational modifications, including phosphorylation (P), may play an important role in TF-DNA binding. CG methylation represents DNA methylation in CG context. H3K56ac, histone H3 acetylation at lysine 56; H3K9ac, histone H3 acetylation at lysine 9; H3K27ac, histone H3 acetylation at lysine 27; H3K36me3, histone H3 tri-methylation at lysine 36; H3K4me1, histone H3 mono-methylation at lysine 4; H3K4me3, histone H3 tri-methylation at lysine 4; H3K27me3, histone H3 tri-methylation at lysine 27. Histone methylation and acetylation marks are represented by circle and square, respectively.
TF recognition of DNA requires direct and indirect readout
Cocrystal structures of protein-DNA complexes contributed substantially to resolve how TFs physically bind to specific DNA sequences. These studies suggest that recognition of a short DNA sequence by a TF is achieved primarily through direct interactions between amino acid residues and the DNA base edges [17,18]. The physical contact of protein side-chains with the major or minor groove of the DNA helix is mainly established by hydrogen bonds, water-mediated hydrogen bonds, hydrophobic interactions, and/or π-interactions [17,18]. Although the direct interaction between TF amino acids and DNA bases, the so-called base readout, is critical for the formation of TF-DNA complexes, most TFs require a combination of base and shape readout (indirect readout), which is mainly driven by van der Waals interactions and electrostatic potentials, to achieve DNA-binding specificity [18,19]. In other words, most TFs need to recognize local or global structural changes within the DNA as well as direct physical or water-mediated binding with DNA bases to accurately pinpoint their specific target sites [20,21] (Figure 2). Accordingly, models incorporating DNA structure information predict TF-DNA binding sites at higher accuracy than models that use sequence information alone [19,22]. For example, using a collection of genome-wide binding sites for 216 A. thaliana TFs created by an in vitro TF binding site assay called DNA affinity purification sequencing (DAP-seq), binding site models were generated for each TF by a random forest machine learning approach that combined DNA shape features with syntax sequences in a shape-based regressor [23]. The models improved the prediction of target sites for all the TFs tested, and the features defined by the shape-based regressor could reliably pinpoint most of the distinct target sites for different TFs within the same structural family [23].
Figure 2.
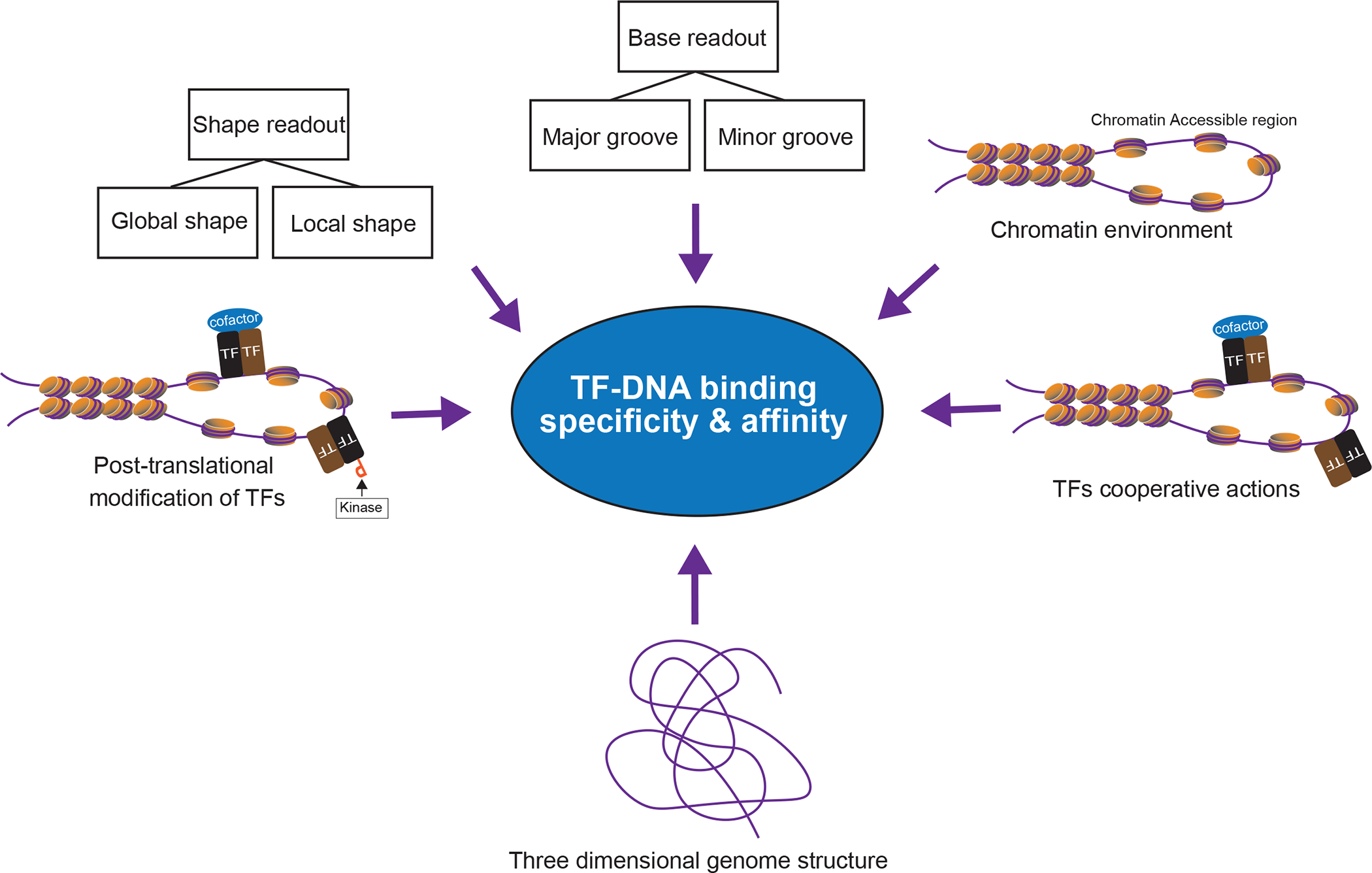
Schematic overview of mechanisms contributing to TF-DNA binding specificity and affinity.
Experimental advances in identifying transcription factor binding sites (TFBS)
Chromatin immunoprecipitation coupled with next-generation sequencing (ChIP-seq) is widely used for the identification of binding sites of a given TF in vivo [11,24] (Table 1). However, ChIP-seq data are limited by several intrinsic factors. Chromatin shearing by sonication is an irreproducible process that creates DNA fragments with variable sizes leading to generation of broad regions of read enrichment (“peaks”) where the resolution is often insufficient for precise mapping of binding sites [25]. Crosslinking is another intrinsic limiting step in ChIP-seq experiments, leading to generation of low signal-to-noise ratio peaks, false-positive binding sites, and masking of epitopes by the surrounding crosslinked proteins [26]. Moreover, systematic and broad enrichment of non-targeted TFs across ChIP-seq datasets may confound the proper interpretation of ChIP-seq data [27]. Several new approaches have been developed to tackle the limitations of ChIP-seq (Table 1). For example, ChIP-exo and ChIP-nexus improved the resolution of binding site maps by applying exonucleases to trim excess sequences [28]. CUT&RUN, CUT&Tag, and DamID use nucleases (micrococcal nuclease, Tn5 transposase, or DpnI) for DNA fragmentation and thus do not require crosslinking [29–31]. DamID further allows determination of transient TF-DNA interaction by introducing into cells the TF of interest fused to a bacterial DNA adenine methyltransferase followed by identifying the methylated adenines resulting from the TF binding events [29]. However, these methods also have specific drawbacks. For example, in DamID the target regions are broadly methylated and often do not have sufficient resolution to precisely localize the binding sites [32], while the high cost and technical complexity of ChIP-exo and ChIP-nexus limit their broad application [33,34].
Table 1.
In vivo and in vitro methods to identify TF-DNA binding sites.
| Method | Input | TF source | Time | Relative cost | Advantages | Disadvantages | References |
|---|---|---|---|---|---|---|---|
| ChIP-seq | Cross-linked chromatin | Endogenous or in vivo expressed recombinant protein | 4–5 days | medium | Can be applied for a wide range of organisms | Low resolution of binding site maps; prone to false positive and false negative errors; low signal-to-noise ratio peaks | [25–27] |
| ChIP-exo | Cross-linked chromatin | Endogenous or in vivo expressed recombinant protein | 4–5 days | high | High resolution of binding site maps | High technical complexity | [28, 34] |
| ChIP-nexus | Cross-linked chromatin | Endogenous or in vivo expressed recombinant protein | 4–5 days | high | High resolution of binding site maps | High technical complexity | [28, 33] |
| CUT&RUN | Native chromatin | Endogenous or in vivo expressed recombinant protein | 2 days | medium | High resolution of binding site maps; high signal-to-noise ratio peaks | No published report for plant TFs | [30] |
| CUT&Tag | Native chromatin | Endogenous or in vivo expressed recombinant protein | 2 days | medium | High resolution of binding site maps; high signal-to-noise ratio peaks | No published report for plant TFs | [31] |
| DamID | Native chromatin | In vivo expressed recombinant protein | 4–5 days | high | Identification of transient TF-DNA interactions | Low resolution of binding site maps | [29, 32] |
| scCC | RNA | In vivo expressed recombinant protein | 2 days | medium | Simultaneous measure of transcriptome and TF binding profiles at the single-cell level | Low resolution of binding site maps; possible modification or silencing of target gene expression due to transposon integration | [43] |
| nextPBM | Randomized synthetic DNA | Endogenous or in vivo expressed recombinant protein | 2-3 days | medium | Captures the effect of TF-protein interactions and post-translational modifications on DNA binding specificity and affinity | Lack of endogenous genome sequence and chromatin context | [41] |
| PBM | Randomized synthetic DNA | In vitro or nonnative cell expressed recombinant protein | 2 days | low | Identifies binding sequence motifs in a high-throughput manner | Lack of endogenous genome sequence and chromatin context | [15, 38] |
| SELEX-seq | Randomized synthetic DNA | In vitro or nonnative cell expressed recombinant protein | 2 days | low | Identifies binding sequence motifs in a high-throughput manner | Lack of endogenous genome sequence and chromatin context | [15, 35] |
| DAP-seq | Genomic DNA | In vitro or nonnative cell expressed recombinant protein | 2 days | low | High resolution of binding site maps in endogenous genome context; high signal-to-noise ratio peaks; can be easily performed in a high-throughput manner; can be used to dissect the direct and indirect binding sites and disentangle the cooperative action of TFs | Lack of chromatin context | [15, 36, 39] |
| Multi-DAP-seq | Genomic DNA | In vitro or nonnative cell expressed recombinant protein | 2 days | low | Can be applied to non-model organisms; high resolution of binding site maps in endogenous genome context; high signal-to-noise ratio peaks; can be easily performed in a high-throughput manner | Lack of chromatin context; potential modification of DNA binding specificity and/or affinity due to incorporation of biotinylated lysine into the TF protein sequence | [37] |
In parallel to in vivo methods, several in vitro approaches have been widely used to identify TF-DNA sequence specificity and binding locations [11,15,35,36] (Table 1). In contrast to in vivo methods, in vitro methods such as protein binding microarrays (PBM), systematic evolution of ligands by exponential enrichment-sequencing (SELEX-seq), and DNA affinity purification sequencing (DAP-seq) are relatively fast, cost effective, and can be easily applied in a high-throughput manner. In PBM and SELEX-seq, TFs are exposed to synthetic DNA oligonucleotides, while DAP-seq employs fragmented genomic DNA and captures genomic features such as DNA methylation pattern and the flanking regions of core motifs [15,35–39]. Compared to ChIP-seq, DAP-seq identifies binding sites on genomic DNA that are directly bound by the TFs and can potentially disentangle the cooperative action of a given TF with other TFs or with other cofactors from its individual activity [40]. However, it is important to consider that most in vitro methods lack cellular chromatin context, which is critical for binding site availability and TF-DNA binding in vivo. Moreover, given that in vitro methods mostly use TFs expressed in vitro or by non-native cell systems, they usually cannot capture the effect of post-translational modifications (PTMs) of TFs on DNA binding affinity [9,41]. But the effect of PTM such as phosphorylation can be achieved by phosphomimetic (asparagine/glutamine) or phospho-negative substitutions (alanine/phenylalanine) [42]. To tackle these constraints of traditional in vitro methods, some modified methods have recently been developed (Table 1). For example, Hook et al. developed the nuclear extract protein-binding array (nextPBM) to address the lack of PTMs and interaction partner/partners of TFs in genome-wide binding assays [41]. In this method, the nuclear extract is directly incubated on the microarray, an antibody specific to the TF of interest is applied, and the DNA targets of the TF are detected by measuring the fluorescence signal from a fluorophore-conjugated secondary antibody. Besides introducing chromatin context experimentally, DNA binding information from in vitro methods can be combined with data from ATAC-seq, DNase-seq or MNase-seq that identifies tissue- or cell-type specific accessible chromatin regions (ACRs) to provide reliable predictions for TF-DNA binding sites in vivo [15].
In vitro methods typically expressed TF proteins using expression vectors carrying coding sequence of each TF fused to an affinity tag. This is a major obstacle for the application of these methods to non-model organisms where vector collections harboring TFs are not readily available. To circumvent this bottleneck, Baumgart et al. (2021) generated a clone-free DAP-esq method called multi-DAP-seq [37]. In multi-DAP-seq, the CDS, cDNA, or genomic DNA from prokaryotic cells are used directly for PCR amplification with primers harboring all the required sequences for the in vitro transcription and translation. During translation, biotinylated lysines are incorporated into the protein sequence and biotin-tagged TF proteins are purified using streptavidin-coated beads along with the bound DNA sequences. However, it is important to note that incorporating biotinylated lysines in the protein sequences may lead to changes in protein conformation and potentially alter the DNA binding specificity and/or affinity.
The rapid development of single cell RNA-seq (scRNA-seq) methods has enabled in-depth exploration of gene expression profiles of cell types and developmental trajectories in many tissues or organs. However, the datasets themselves do not directly address how various cell types arise. Moudgil et al. recently developed the single-cell calling cards (scCC) approach that simultaneously provides transcriptome and TF binding profiles at single-cell resolution [43]. In this method, a TF fused to the hyperactive piggyback (HyPBase) transposase integrates the self-reporting transposons (SRTs) near the TF binding sites. The genomic location of SRTs were found using the transcriptome profiles, leading to cell-type-specific mapping of SRTs in combination with the transcript expression profiles in the same cell [43]. Such approach allows discovery of key factors involved in developmental dynamics and transitions between cell types. The most concerning drawback is that the integration of transposon into the target gene may lead to alteration of target gene expression including silencing (Table 1).
Given that the accessibility of binding sites for most TFs, including cell-type specific TFs, is supposed to be a prerequisite for precise gene targeting (Figure 1 and 2), ACRs are expected to vary in a cell-type specific manner over time and space. This property can be used to infer TF-DNA interaction dynamics. Single cell chromatin accessibility datasets are especially informative for this purpose: because the accessibility profiles that are specific to cell types or cell states covering a wide range of developmental trajectories could be found without the generation of transgenic lines, it is possible to observe chromatin dynamics and predict TF-DNA interaction underlying the developmental trajectories of a wide range of tissues in many plant species [6,44,45]. Marand et al. (2021) profiled 72,090 nuclei across six maize organs to explore chromatin accessibility and nuclear gene expression using scATAC-seq and scNucRNA-seq, respectively. Integrated analysis of ACRs and gene expression from single nuclei showed a similar pattern of ACRs and gene expressions across thousands of genes, suggesting that ACRs are overlapping with regions containing CREs for the genes and could be a good proxy for active transcription in maize organs [6]. Approximately 31% of ACRs showed a cell-type specific pattern, where they were notably hypomethylated, highly associated with active enhancers, and greatly enriched with TF motifs compared to non-cell-type specific ACRs or controls. In contrast to studies that used bulk tissues or organs and reported a scarcity of dynamic chromatin in plants, this and other single cell studies showed a substantial level of cell-type specific pattern of chromatin dynamics and provide an important basis for identifying cell-type specific CREs [44,45].
Mechanisms contributing to TF-DNA binding specificity beyond the core motifs
TFs bind preferentially at genomic regions harboring sequences that match the short in vitro binding motifs usually 5–11 bp long [46]. However, genome-wide analysis demonstrated that among the numerous motif-containing sequences present in the genome only a small fraction (~1%) are bound by TFs [46,47]. This suggests that the motif sequences alone do not provide sufficient information for directing TFs to their target sites [48]. Over the past few decades, many studies have been designed to uncover how TFs precisely distinguish motifs containing their genuine binding sites from other regions containing similar sequences. These studies identified multiple factors underlying target recognition, including chromatin environment, sequence and structural features of regions flanking the core recognition sequences, combinatorial action of TFs and cofactors, nuclear compartmentalization of regulatory DNA sequences (three dimensional genome architecture), PTMs of TFs, and DNA base modifications such as 5’-methylcytosine [9,12,15,48–53] (Figure 2).
In the nucleus of eukaryotic cells, long strands of genomic DNA are organized in a higher order structure called chromatin. DNA wraps around histone proteins to create nucleosomes, the fundamental unit of chromatin, and nucleosomes are found in a continuum of compactness between the densely packed heterochromatin and lightly packed euchromatin (Figure 1 and 2). The stable and compact structure of chromatin in eukaryotes constructs an inherent barrier that is not only critical for maintaining genome stability by suppressing transposon activation but is also required for inhibiting improper cell fate and developmental transitions [1,54]. Reducing the physical compaction of chromatin to make chromatinized DNA accessible for regulatory factors is a prerequisite for many DNA-based processes such as DNA replication, DNA repair, recombination, and transcription [1,55]. Thus in eukaryotes, the evolution of the intrinsically repressive chromatin structure occurred in parallel to the evolution of mechanisms such as epigenetic marks, chromatin remodelers, histone variants, and pioneer TFs to regulate accessibility of chromatin regions [1,54,56,57].
Chromatin environment and genome organization govern TF-DNA binding
Comparison of genome-wide TF binding datasets with chromatin accessibility profiles revealed relatively high overlap between ACRs with TF binding motifs [7,58] (Figure 1). Chromatin accessibility is commonly measured in genome-wide scale using DNase-seq, FAIRE-seq, ATAC-seq, and MNase-seq. ACRs in plants with small genome size are mostly located within 2 kb upstream of the gene bodies, and higher percentage of distal ACRs (dACRs) are found for increased genome size. However, increased level of dACRs is not directly proportional to genome size [59]. For example, around 6% of ACRs of A. thaliana (135 Mbp genome) are dACRS located more than 2 kb away from the nearest gene, whereas the percentage of dACRs in Z. mays (~2365 Mbp genome) is around 32.5% [5]. Lu et al. analyzed the genome and epigenomes of 13 plant species and found that genic and proximal ACRs of active genes, which are within 2kb from the nearest gene, were marked by H3K4me3, H3K56ac, and/or H3K36me3. They also reported that genes flanking H3K56ac dACRs are usually highly expressed, suggesting that the H3K56ac marked dACRs might be predictive of active enhancers with functional TF binding sites [5] (Figure 1).
Beyond the regulatory functions of the genome modulated by nucleotide sequences in linear space, the three-dimensional (3D) genome organization also contributes to the fine tuning of genome functions (Figure 2). The 3D genome is highly dynamic in response to environmental signals and developmental cues and regulates gene expression predominantly through long-range chromatin interactions [60]. In maize, 3D genome organization contributes to transcriptional regulation of two agronomically important genes, teosinte branched1 (tb1) and UNBRANCHED3 (UB3), via chromatin loops that bring distal enhancers to the close proximity of tb1 and UB3 promoters [61–63]. High resolution and accurate identification of chromatin loops connecting ACRs to genes at the single cell level can provide a 3D view of functional CREs and TFs responsible for cell fate specification [6,53,64,65].
Cooperative actions between TFs
Cooperative action of TFs is a widespread mechanism that leads to diversification of DNA binding affinity and specificity and subsequently functional complexity of TFs in eukaryotes [12,24,40,66,67] (Figure 2). For example, it is long known that the cooperative action of MADS-box TFs, which is critical for floral organ specification, mediates their unique DNA binding specificity and affinity. However, our knowledge about how the combinatorial action of MADS-box TFs determines floral organ identity at the systems level is still limited [40,68]. Lai et al. recently used a combination of sequential DAP-seq with ChIP-seq and RNA-seq to explore genome-wide binding sites of the heterodimeric and heterotetrameric complexes of SEPALLATA3 (SEP3) and AGAMOUS (AG), as well as the SEP3 homooligomer [40]. They showed that SEP3 and SEP3-AG targeted distinct binding sites but also had many overlapping binding targets. Furthermore, the tetrameric SEP3-AG complex exhibited increased DNA binding affinity throughout the genome compared to the dimeric SEP3Δtet-AG complex while placing a greater restriction on the spacing between the DNA-binding motifs, resulting in more efficient binding of the tetrameric complex to some regions that were weakly accessible to the dimeric SEP3Δtet-AG complex. Another example of cooperative TF action comes from the AUXIN RESPONSE FACTOR (ARF) family of TFs, through which the phytohormone auxin controls almost all aspects of plant growth and development. Recent studies suggest that spacing, direction, and order of the DNA binding motifs by the ARF homo- and heterodimers play a key role in differential binding affinity and specificity of the ARF subfamilies [66,69].
TF-DNA binding and post-translational modifications
PTMs of TFs are critical for targeting the TFs to the desired subcellular compartments and for regulating their transcriptional activity, especially for many TFs involved in hormone signaling responses [70]. PTMs may also alter DNA binding affinity of the TF (Figure 1 and 2). For example, the TF WRKY33, involved in disease resistance by regulating camalexin biosynthesis, is phosphorylated by the mitogen-activated protein kinases (MAPKs) and CALCIUM-DEPENDENT PROTEIN KINASES (CPKs). Whereas MAPKs phosphorylate the C-terminus of WRKY33 and promote its transactivation activity, CPKs phosphorylate the N-terminus of WRKY33 and enhance its DNA-binding affinity, which is required for the full activity of WRKY33 in camalexin biosynthesis [9].
Intrinsically disordered regions of TFs contribute to binding specificity of orthologous TFs
Recent studies have shown that low-affinity TF binding sites, which can evolve rapidly, are vital in fine tuning binding specificity of TFs and developmental robustness in plants and animals [11,71]. Crocker et al. [72] suggest an inverse correlation between sequence affinity and specificity: whereas high-affinity binding sites are targeted by multiple TFs from the same family, clusters of low-affinity binding sites provide higher specificity for a unique TF within a family and thus leading to recognition of non-identical binding sites by paralogous TFs [14]. Intrinsically disordered regions (IDRs) of TFs, which exhibit low similarity between distant orthologs, also play an important role in guiding the TFs to broad target regions in which DNA binding domains recognize their sequence motifs [73]. Importantly, the whole IDR but not a specialized domain within it contributes to the binding specificity of the TFs. Therefore, IDRs likely provide another mechanism besides clusters of low-affinity binding sites that contribute to the binding specificity of related TFs.
Conclusion and future perspectives
Recent studies have demonstrated that many features beyond the core sequence motifs are critical for TF binding site recognition, and incorporating these features has improved models for binding site prediction. However, the current measurements of TF-DNA interactions are mostly qualitative, so going forward it is important to develop techniques and models that provide quantitative information that can be linked to quantitative measurements of gene expression. Although technological and methodological advances have substantially reduced the required time and cost of large-scale experiments for identifying TF binding sites, the catalog of TF binding sites remains incomplete even in the model plant A. thaliana and very limited in many plants including important crop species. Moreover, different isoforms of TFs may show diverse binding specificities [74], so systematic assessment of TFs splicing variants require special attention.
Wide applications of single cell genomics approach in many plant species have started to uncover factors that drive developmental dynamics, cell type transitions and evolution. Integrating single cell or nuclei transcriptomes and chromatin dynamics with TF binding site assays will revolutionize our understanding of gene regulatory networks underlying plant development and response to the environment.
Acknowledgements
This work was supported by the grants from the National Science Foundation Plant Genome Research Program (IOS-1916804) and National Institutes of Health (5R35GM138143) to S.C.H.
Footnotes
Conflict of interest statement
The authors declare that they have no competing interests.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
References
- 1.Hajheidari M, Koncz C, Bucher M: Chromatin evolution-key innovations underpinning morphological complexity. Frontiers in plant science 2019, 10:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jayavelu ND, Jajodia A, Mishra A, Hawkins RD: Candidate silencer elements for the human and mouse genomes. Nature communications 2020, 11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurbidaeva A, Purugganan M: Insulators in plants: progress and open questions. Genes 2021, 12:1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu B, Zhang W, Zhang T, Liu B, Jiang J: Genome-wide prediction and validation of intergenic enhancers in Arabidopsis using open chromatin signatures. The Plant Cell 2015, 27:2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu Z, Marand AP, Ricci WA, Ethridge CL, Zhang X, Schmitz RJ: The prevalence, evolution and chromatin signatures of plant regulatory elements. Nature Plants 2019, 5:1250–1259. [DOI] [PubMed] [Google Scholar]
- 6. Marand AP, Chen Z, Gallavotti A, Schmitz RJ: A cis-regulatory atlas in maize at single-cell resolution. Cell 2021, 184:3041–3055. e3021. ••The authors profiled chromatin accessibility and nuclear gene expression using scATAC-seq and scNucRNA-seq, respectively. They identified 31,660 unique ACRs using scATAC-seq. After scanning the genomic location of the identified unique ACRs, they found 165,913 CREs, which covered around 4% of the maize genome. Approximately 31% of ACRs showed a cell-type specific pattern. The authors discovered that the cell-type-specific ACRs were less diverged compared to non-cell-type specific ACRs and sequence polymorphisms within regions of cell-type specific ACRs showed more frequent association with phenotypic variation predicted by genome-wide association studies (GWAS), suggesting that the cell-type specific ACRs could be a good target for genome editing and extending the phenotypic range of traits critical for plant biomass.
- 7.Ricci WA, Lu Z, Ji L, Marand AP, Ethridge CL, Murphy NG, Noshay JM, Galli M, Mejía-Guerra MK, Colomé-Tatché M: Widespread long-range cis-regulatory elements in the maize genome. Nature plants 2019, 5:1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ritter EJ, Niederhuth CE: Intertwined evolution of plant epigenomes and genomes. Current Opinion in Plant Biology 2021, 61:101990. [DOI] [PubMed] [Google Scholar]
- 9. Zhou J, Wang X, He Y, Sang T, Wang P, Dai S, Zhang S, Meng X: Differential phosphorylation of the transcription factor WRKY33 by the protein kinases CPK5/CPK6 and MPK3/MPK6 cooperatively regulates camalexin biosynthesis in Arabidopsis. Plant Cell 2020, 32:2621–2638. •This study highlights the importance of post-translational modifications in TF-DNA binding affinity.
- 10.Deschamps S, Crow JA, Chaidir N, Peterson-Burch B, Kumar S, Lin H, Zastrow-Hayes G, May GD: Chromatin loop anchors contain core structural components of the gene expression machinery in maize. BMC genomics 2021, 22:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajheidari M, Wang Y, Bhatia N, Vuolo F, Franco-Zorrilla JM, Karady M, Mentink RA, Wu A, Oluwatobi BR, Müller B: Autoregulation of RCO by low-affinity binding modulates cytokinin action and shapes leaf diversity. Current Biology 2019, 29:4183–4192. e4186. [DOI] [PubMed] [Google Scholar]
- 12.Ibarra IL, Hollmann NM, Klaus B, Augsten S, Velten B, Hennig J, Zaugg JB: Mechanistic insights into transcription factor cooperativity and its impact on protein-phenotype interactions. Nature communications 2020, 11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inukai S, Kock KH, Bulyk ML: Transcription factor–DNA binding: beyond binding site motifs. Current opinion in genetics & development 2017, 43:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kribelbauer JF, Rastogi C, Bussemaker HJ, Mann RS: Low-affinity binding sites and the transcription factor specificity paradox in eukaryotes. Annual review of cell and developmental biology 2019, 35:357–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Malley RC, Huang S-sC, Song L, Lewsey MG, Bartlett A, Nery JR, Galli M, Gallavotti A, Ecker JR: Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 2016, 165:1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen N, Zhao J, Schipper JL, Zhang Y, Bepler T, Leehr D, Bradley J, Horton J, Lapp H, Gordan R: Divergence in DNA specificity among paralogous transcription factors contributes to their differential in vivo binding. Cell systems 2018, 6:470–483. e478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seeman NC, Rosenberg JM, Rich A: Sequence-specific recognition of double helical nucleic acids by proteins. Proceedings of the National Academy of Sciences 1976, 73:804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson KA, Wetmore SD: Combining crystallographic and quantum chemical data to understand DNA-protein π-interactions in nature. Structural Chemistry 2017, 28:1487–1500. [Google Scholar]
- 19. Schnepf M, von Reutern M, Ludwig C, Jung C, Gaul U: Transcription factor binding affinities and DNA shape readout. Iscience 2020, 23:101694. •This study reports the genome-wide contribution of thirteen DNA shape features and electrostatic potential to TF-DNA binding specificities for thirteen Drosophila TFs from eight different binding domain families. The high-performance fluorescence anisotropy (HiP-FA) technique was used for high sensitivity measurement of TF affinity for any given DNA sequence at large scale. Given that more than 90% of the variance in the shape features can be captured by considering only dinucleotide dependencies, the authors used the dinucleotide position weight matrices determined from binding affinities to capture the shape readout contribution at each position in the binding site. They found that TFs widely used shape features for binding site recognition independently from the type of their DNA binding domain.
- 20.Lara-Gonzalez S, Dantas Machado AC, Rao S, Napoli AA, Birktoft J, Di Felice R, Rohs R, Lawson CL: The RNA polymerase α subunit recognizes the DNA shape of the upstream promoter element. Biochemistry 2020, 59:4523–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh RK, Mukherjee A: Molecular Mechanism of the Intercalation of the SOX-4 Protein into DNA Inducing Bends and Kinks. The Journal of Physical Chemistry B 2021, 125:3752–3762. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Sagendorf JM, Chiu T-P, Pasi M, Perez A, Rohs R: Expanding the repertoire of DNA shape features for genome-scale studies of transcription factor binding. Nucleic acids research 2017, 45:12877–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sielemann J, Wulf D, Schmidt R, Bräutigam A: Local DNA shape is a general principle of transcription factor binding specificity in Arabidopsis thaliana. Nature communications 2021, 12:1–8. •This study combined DNA structural features with syntax sequences to predict TF-DNA binding specificities and demonstrated that models incorporating DNA shape features predicted TF-DNA binding specificities with higher accuracy.
- 24.Tu X, Mejía-Guerra MK, Franco JAV, Tzeng D, Chu P-Y, Shen W, Wei Y, Dai X, Li P, Buckler ES: Reconstructing the maize leaf regulatory network using ChIP-seq data of 104 transcription factors. Nature communications 2020, 11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Jonge WJ, Brok M, Kemmeren P, Holstege FC: An optimized chromatin immunoprecipitation protocol for quantification of protein-DNA interactions. STAR protocols 2020, 1:100020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li R, Grimm SA, Wade PA: CUT&Tag-BS for simultaneous profiling of histone modification and DNA methylation with high efficiency and low cost. Cell Reports Methods 2021:100118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worsley Hunt R, Wasserman WW: Non-targeted transcription factors motifs are a systemic component of ChIP-seq datasets. Genome biology 2014, 15:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biswas A, Narlikar L: Resolving diverse protein–DNA footprints from exonuclease-based ChIP experiments. Bioinformatics 2021, 37:i367–i375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez JM, Schinke A-L, Brooks MD, Pasquino A, Leonelli L, Varala K, Safi A, Krouk G, Krapp A, Coruzzi GM: Transient genome-wide interactions of the master transcription factor NLP7 initiate a rapid nitrogen-response cascade. Nature communications 2020, 11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meers MP, Bryson TD, Henikoff JG, Henikoff S: Improved CUT&RUN chromatin profiling tools. Elife 2019, 8:e46314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao X, Feng S, Zhao T, Guan X: Efficient chromatin profiling of H3K4me3 modification in cotton using CUT&Tag. Plant methods 2020, 16:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szczesnik T, Ho JW, Sherwood R: Dam mutants provide improved sensitivity and spatial resolution for profiling transcription factor binding. Epigenetics & Chromatin 2019, 12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Q, Johnston J, Zeitlinger J: ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nature biotechnology 2015, 33:395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rossi MJ, Lai WK, Pugh BF: Simplified ChIP-exo assays. Nature communications 2018, 9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Käppel S, Eggeling R, Rümpler F, Groth M, Melzer R, Theißen G: DNA-binding properties of the MADS-domain transcription factor SEPALLATA3 and mutant variants characterized by SELEX-seq. Plant molecular biology 2021, 105:543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai X, Vega-Léon R, Hugouvieux V, Blanc-Mathieu R, van der Wal F, Lucas J, Silva CS, Jourdain A, Muino JM, Nanao MH: The intervening domain is required for DNA-binding and functional identity of plant MADS transcription factors. Nature Communications 2021, 12:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baumgart LA, Lee JE, Salamov A, Dilworth DJ, Na H, Mingay M, Blow MJ, Zhang Y, Yoshinaga Y, Daum CG: Persistence and plasticity in bacterial gene regulation. Nature methods 2021, 18:1499–1505. [DOI] [PubMed] [Google Scholar]
- 38.Kim JS, Chae S, Jun KM, Lee G-S, Jeon J-S, Kim KD, Kim Y-K: Rice protein-binding microarrays: a tool to detect cis-acting elements near promoter regions in rice. Planta 2021, 253:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M, Huang S-SC: DNA Affinity Purification Sequencing (DAP-Seq) for Mapping Genome-Wide Transcription Factor Binding Sites in Plants. In Accelerated Breeding of Cereal Crops. Edited by: Springer; 2022:293–303. [Google Scholar]
- 40.Lai X, Stigliani A, Lucas J, Hugouvieux V, Parcy F, Zubieta C: Genome-wide binding of SEPALLATA3 and AGAMOUS complexes determined by sequential DNA-affinity purification sequencing. Nucleic acids research 2020, 48:9637–9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hook H, Zhao RW, Bray D, Keenan JL, Siggers T: High-Throughput Analysis of the Cell and DNA Site-Specific Binding of Native NF-κB Dimers Using Nuclear Extract Protein-Binding Microarrays (NextPBMs). In NF-κB Transcription Factors. Edited by: Springer; 2021:43–66. [DOI] [PubMed] [Google Scholar]
- 42.Andrilenas KK, Ramlall V, Kurland J, Leung B, Harbaugh AG, Siggers T: DNA-binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer-specific gene regulation. Nucleic acids research 2018, 46:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moudgil A, Wilkinson MN, Chen X, He J, Cammack AJ, Vasek MJ, Lagunas T Jr, Qi Z, Lalli MA, Guo C: Self-reporting transposons enable simultaneous readout of gene expression and transcription factor binding in single cells. Cell 2020, 182:992–1008. e1021. ••The authors developed the single-cell calling cards (scCC) approach for genome scale studies of transcriptome and TF binding at single-cell resolution. This method can be used to identify key factors contributing to organ development and transition between cell types.
- 44. Dorrity MW, Alexandre CM, Hamm MO, Vigil A-L, Fields S, Queitsch C, Cuperus JT: The regulatory landscape of Arabidopsis thaliana roots at single-cell resolution. Nature communications 2021, 12:1–12. • The authors utilized a droplet-based approach to profile 5283 nuclei of the A. thaliana root samples and identify 7290 unique CREs insertions by single-cell ATAC-seq (scATAC-seq). The distribution of the examined nuclei grouped into distinct clusters based on ATAC-seq profiles and most of the clusters are annotated to distinct root cell types according to known marker genes, indicating the differential profile of ACRs between clusters of cells according to their identity. Approximately 30% of identified ACRs show a cell-type specific pattern and they also find significant motif enrichment for at least one TF family in all cell types through scanning dynamic ACRs.
- 45.Farmer A, Thibivilliers S, Ryu KH, Schiefelbein J, Libault M: Single-nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression in Arabidopsis roots at the single-cell level. Molecular Plant 2021, 14:372–383. [DOI] [PubMed] [Google Scholar]
- 46.Jana T, Brodsky S, Barkai N: Speed–specificity trade-offs in the transcription factors search for their genomic binding sites. Trends in Genetics 2021, 37:421–432. [DOI] [PubMed] [Google Scholar]
- 47.Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK: An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dror I, Golan T, Levy C, Rohs R, Mandel-Gutfreund Y: A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome research 2015, 25:1268–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crisp PA, Marand AP, Noshay JM, Zhou P, Lu Z, Schmitz RJ, Springer NM: Stable unmethylated DNA demarcates expressed genes and their cis-regulatory space in plant genomes. Proceedings of the National Academy of Sciences 2020, 117:23991–24000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gurdon J, Javed K, Vodnala M, Garrett N: Long-term association of a transcription factor with its chromatin binding site can stabilize gene expression and cell fate commitment. Proceedings of the National Academy of Sciences 2020, 117:15075–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang SsC, Ecker JR: Piecing together cis-regulatory networks: Insights from epigenomics studies in plants. Wiley Interdisciplinary Reviews: Systems Biology and Medicine 2018, 10:e1411. [DOI] [PubMed] [Google Scholar]
- 52.Sönmezer C, Kleinendorst R, Imanci D, Barzaghi G, Villacorta L, Schübeler D, Benes V, Molina N, Krebs AR: Molecular co-occupancy identifies transcription factor binding cooperativity in vivo. Molecular Cell 2021, 81:255–267. e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun Y, Dong L, Zhang Y, Lin D, Xu W, Ke C, Han L, Deng L, Li G, Jackson D: 3D genome architecture coordinates trans and cis regulation of differentially expressed ear and tassel genes in maize. Genome biology 2020, 21:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baile F, Gómez-Zambrano Á, Calonje M: Roles of Polycomb complexes in regulating gene expression and chromatin structure in plants. Plant Communications 2021:100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu L-Y, Shang G-D, Wang F-X, Gao J, Wan M-C, Xu Z-G, Wang J-W: Dynamic chromatin state profiling reveals regulatory roles of auxin and cytokinin in shoot regeneration. Developmental Cell 2022. [DOI] [PubMed] [Google Scholar]
- 56.Lai X, Blanc-Mathieu R, Grandvuillemin L, Huang Y, Stigliani A, Lucas J, Thévenon E, Loue-Manifel J, Turchi L, Daher H: The LEAFY floral regulator displays pioneer transcription factor properties. Molecular Plant 2021, 14:829–837. [DOI] [PubMed] [Google Scholar]
- 57.Mivelaz M, Cao A-M, Kubik S, Zencir S, Hovius R, Boichenko I, Stachowicz AM, Kurat CF, Shore D, Fierz B: Chromatin fiber invasion and nucleosome displacement by the Rap1 transcription factor. Molecular cell 2020, 77:488–500. e489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen X, Yu B, Carriero N, Silva C, Bonneau R: Mocap: large-scale inference of transcription factor binding sites from chromatin accessibility. Nucleic acids research 2017, 45:4315–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maher KA, Bajic M, Kajala K, Reynoso M, Pauluzzi G, West DA, Zumstein K, Woodhouse M, Bubb K, Dorrity MW: Profiling of accessible chromatin regions across multiple plant species and cell types reveals common gene regulatory principles and new control modules. The Plant Cell 2018, 30:15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baroux C: Three-dimensional genome organization in epigenetic regulations: cause or consequence? Current Opinion in Plant Biology 2021, 61:102031. [DOI] [PubMed] [Google Scholar]
- 61.Golicz AA, Bhalla PL, Edwards D, Singh MB: Rice 3D chromatin structure correlates with sequence variation and meiotic recombination rate. Communications biology 2020, 3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Du Y, Liu L, Peng Y, Li M, Li Y, Liu D, Li X, Zhang Z: UNBRANCHED3 expression and inflorescence development is mediated by UNBRANCHED2 and the distal enhancer, KRN4, in maize. PLoS genetics 2020, 16:e1008764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Savadel SD, Hartwig T, Turpin ZM, Vera DL, Lung P-Y, Sui X, Blank M, Frommer WB, Dennis JH, Zhang J: The native cistrome and sequence motif families of the maize ear. PLoS genetics 2021, 17:e1009689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li E, Liu H, Huang L, Zhang X, Dong X, Song W, Zhao H, Lai J: Long-range interactions between proximal and distal regulatory regions in maize. Nature communications 2019, 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng Y, Xiong D, Zhao L, Ouyang W, Wang S, Sun J, Zhang Q, Guan P, Xie L, Li W: Chromatin interaction maps reveal genetic regulation for quantitative traits in maize. Nature communications 2019, 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Freire-Rios A, Tanaka K, Crespo I, Van der Wijk E, Sizentsova Y, Levitsky V, Lindhoud S, Fontana M, Hohlbein J, Boer DR: Architecture of DNA elements mediating ARF transcription factor binding and auxin-responsive gene expression in Arabidopsis. Proceedings of the National Academy of Sciences 2020, 117:24557–24566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nie Y, Shu C, Sun X: Cooperative binding of transcription factors in the human genome. Genomics 2020, 112:3427–3434. [DOI] [PubMed] [Google Scholar]
- 68.Smaczniak C, Muiño JM, Chen D, Angenent GC, Kaufmann K: Differences in DNA binding specificity of floral homeotic protein complexes predict organ-specific target genes. The Plant Cell 2017, 29:1822–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kato H, Mutte SK, Suzuki H, Crespo I, Das S, Radoeva T, Fontana M, Yoshitake Y, Hainiwa E, van den Berg W: Design principles of a minimal auxin response system. Nature plants 2020, 6:473–482. [DOI] [PubMed] [Google Scholar]
- 70.Yu Z, Zhang F, Friml J, Ding Z: Auxin signaling: Research advances over the past 30 years. Journal of Integrative Plant Biology 2022. [DOI] [PubMed] [Google Scholar]
- 71.Crocker J, Noon EP-B, Stern DL: The soft touch: low-affinity transcription factor binding sites in development and evolution. Current topics in developmental biology 2016, 117:455–469. [DOI] [PubMed] [Google Scholar]
- 72.Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Alsawadi A, Valenti P, Plaza S, Payre F: Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 2015, 160:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brodsky S, Jana T, Mittelman K, Chapal M, Kumar DK, Carmi M, Barkai N: Intrinsically disordered regions direct transcription factor in vivo binding specificity. Molecular cell 2020, 79:459–471. e454. ••TFs contain intrinsically disordered regions (IDRs), which are known to be involved in protein-protein interactions. This study showed that long IDRs played a key role in TF-DNA binding specificity even when they were away from the DNA binding domain.
- 74.Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O’Hanlon D, Sung H-K, Alvarez M, Talukder S, Pan Q, Mazzoni EO: An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 2011, 147:132–146. [DOI] [PubMed] [Google Scholar]