

Abstract
Early treatment with the histone deacetylase inhibitor, trichostatin A, plus nutritional support extended median survival of spinal muscular atrophy mice by 170%. Treated mice continued to gain weight, maintained stable motor function, and retained intact neuromuscular junctions long after trichostatin A was discontinued. In many cases, ultimate decline of mice appeared to result from vascular necrosis, raising the possibility that vascular dysfunction is part of the clinical spectrum of severe spinal muscular atrophy. Early spinal muscular atrophy disease detection and treatment initiation combined with aggressive ancillary care may be integral to the optimization of histone deacetylase inhibitor treatment in human patients.
Spinal muscular atrophy (SMA) is caused by mutation of the telomeric copy of the survival motor neuron gene (SMN1) and retention of the centromeric SMN2 gene in variable copy number.1 Transcripts derived from SMN2 are alternatively spliced such that the majority lacks exon 7 and codes for truncated SMN protein that is rapidly degraded; the minority codes for full-length SMN transcripts and protein.2,3 SMA is caused by deficiency of SMN protein with disease severity correlated inversely with SMN2 gene copy number and SMN protein levels.4–6
A major objective of therapeutics development for SMA is to increase SMN protein levels.7 This might be accomplished by increasing the expression of the one or more SMN2 genes present in all patients. Histone deacetylase (HDAC) inhibitors modify chromatin structure, thereby activating gene expression.8 Two HDAC inhibitors, phenylbutyrate and valproic acid, are being studied in ongoing clinical trials in SMA patients. One completed trial of phenylbutyrate failed to show efficacy in type II SMA patients.9 We have previously demonstrated that the HDAC inhibitor, trichostatin A (TSA), extends median survival of SMA mice by 19% when delivered postsymptomatically starting on postnatal day 5 (P5) until P20.10 Here, we explored whether the efficacy of TSA could be optimized.
Materials and Methods
Animals, Drug Treatment, and Nutritional Supplementation
Mice were treated with TSA and evaluated as described previously.10 Nutritional supplementation was started on P8. (See supplemental text for details of nutritional regimen and animal care.)
Pathology and Immunohistochemistry
Four treated SMA mice showing gross signs of tissue necrosis and three age-matched normal littermates were killed and fixed in formalin for complete necropsy analysis (see supplemental text). Muscles from two SMA mice and two age-matched normal littermates were processed for immunohistochemistry of neuromuscular junctions (NMJs) (see supplemental text).
Results
Early Trichostatin A Combined with Nutritional Support Dramatically Extends Survival of Spinal Muscular Atrophy Mice
At baseline, SMA mice showed abnormal motor behavior starting at birth (P1) (see Supplemental Fig 1). SMA mice receiving TSA alone starting at different early start times until P20 had significant extensions of median survival compared with vehicle-treated mice (P1 = 40%, P2 = 40%, P3 = 33%), but mice starting TSA at P6 or P7 did not (see Supplemental Fig 2).
We tested whether early TSA treatment efficacy could be further improved by combining it with aggressive nutritional support, which included infant formula by mouth and subcutaneous fluids. The formula volumes consumed by SMA pups receiving vehicle (n = 9) or TSA (n = 17) were approximately equal (Fig 1B). TSA plus nutrition extended median survival of SMA mice to 38 days (170%) (log-rank p < 0.001) compared with mice receiving vehicle plus nutrition (median survival, 14 days) (see Fig 1C). Improved survival in the TSA plus nutrition group correlated with early weight gain between P2 and P15, but not with litter size or gender (data not shown). Shorter TSA treatment intervals plus nutrition also showed some efficacy. P5 to P13 was the minimal required treatment interval (see Supplemental Fig 3). SMN protein levels were increased in SMA mice treated with TSA plus nutrition compared with untreated or nutrition alone–treated mice (see Supplemental Fig 4). The magnitude of increase was equivalent to that previously observed with TSA alone,10 suggesting that the addition of nutrition does not further increase SMN protein levels.
Fig 1.
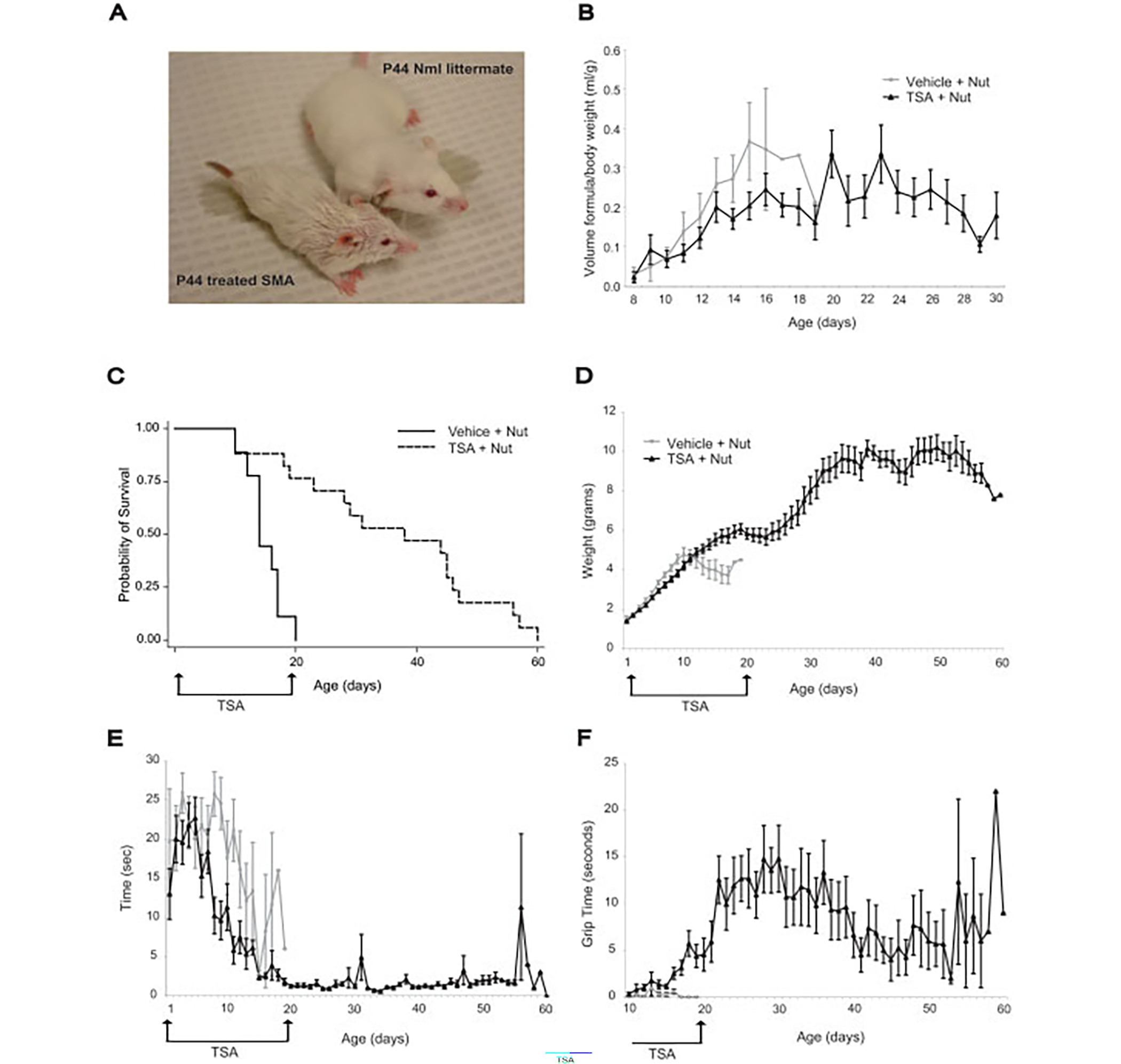
Early trichostatin A (TSA) treatment combined with nutritional support substantially improves survival and results in sustained improvement in motor function. (A) TSA-treated spinal muscular atrophy (SMA) mouse and normal littermate at postnatal day 44 (P44). The SMA mouse is smaller than the normal littermate but is upright and ambulates well. The rough coat is secondary to antibiotic ointment. (B) SMA mice treated with vehicle (n = 9; gray lines) and TSA (n = 17; black lines) consumed approximately equal volumes of formula (ml/gram of body weight). TSA-treated SMA mice continued to receive formula feedings until the fifth week of life. (C) SMA mice treated with TSA (P2-P20) plus nutrition (dashed line) had a substantial increase in median survival of 170% (38 days) compared with mice treated with vehicle plus nutrition (14 days; solid line) (log rank < 0.001). (D) SMA mice receiving TSA plus nutrition (black lines) had weight gain long after TSA was discontinued. Gray lines represent vehicle plus nutrition. (E) SMA mice receiving TSA plus nutrition (black lines) showed improved righting time during the first 2 weeks and maintained this ability thereafter. Gray lines represent vehicle plus nutrition. This difference is significant at P8 (p < 0.001). (F) SMA mice receiving TSA plus nutrition (black lines) showed improved forelimb grip time. Gray lines represent vehicle plus nutrition. This difference is significant at P13 (p = 0.04).
Treated, Long-Lived Spinal Muscular Atrophy Mice Gain Weight, Show Stable Motor Behavior, and Retain Structurally Intact Motor Units after Discontinuation of Trichostatin A
The SMA mice treated with TSA P2–20 plus nutrition continued to grow and gain weight long after TSA was discontinued at P20 (see Figs 1A, D). These mice showed an early improvement in motor function including decreased righting time and improved forelimb grip time (see Figs 1E, F), which persisted after discontinuation of TSA. Many TSA-treated SMA mice were able to ambulate quickly and run, skills never achieved by untreated SMA mice (see Fig 1A and supplemental video). Rotorod testing in five SMA mice also showed relatively stable motor function (see Supplemental Fig 5).
Necropsy of four long-lived SMA mice (P29, P44, P57, and P60), and three age-matched normal littermates showed anterior horn cells in the ventral horn of the spinal cord with normal morphology in SMA and normal littermate mice (Fig 2D). Examination of limb muscles in SMA mice showed myofibers that were diffusely hypotrophic, but otherwise unremarkable (see Fig 2A). Examination of NMJs in the Tibialis Anterior (TA), paraspinal, and intercostal muscles in two long-lived treated SMA mice (P39 and P46) and two age-matched normal littermates (see Figs 2B, C) showed reduced size of NMJs in SMA mice, but little to no denervation (see Fig 2C).
Fig 2.
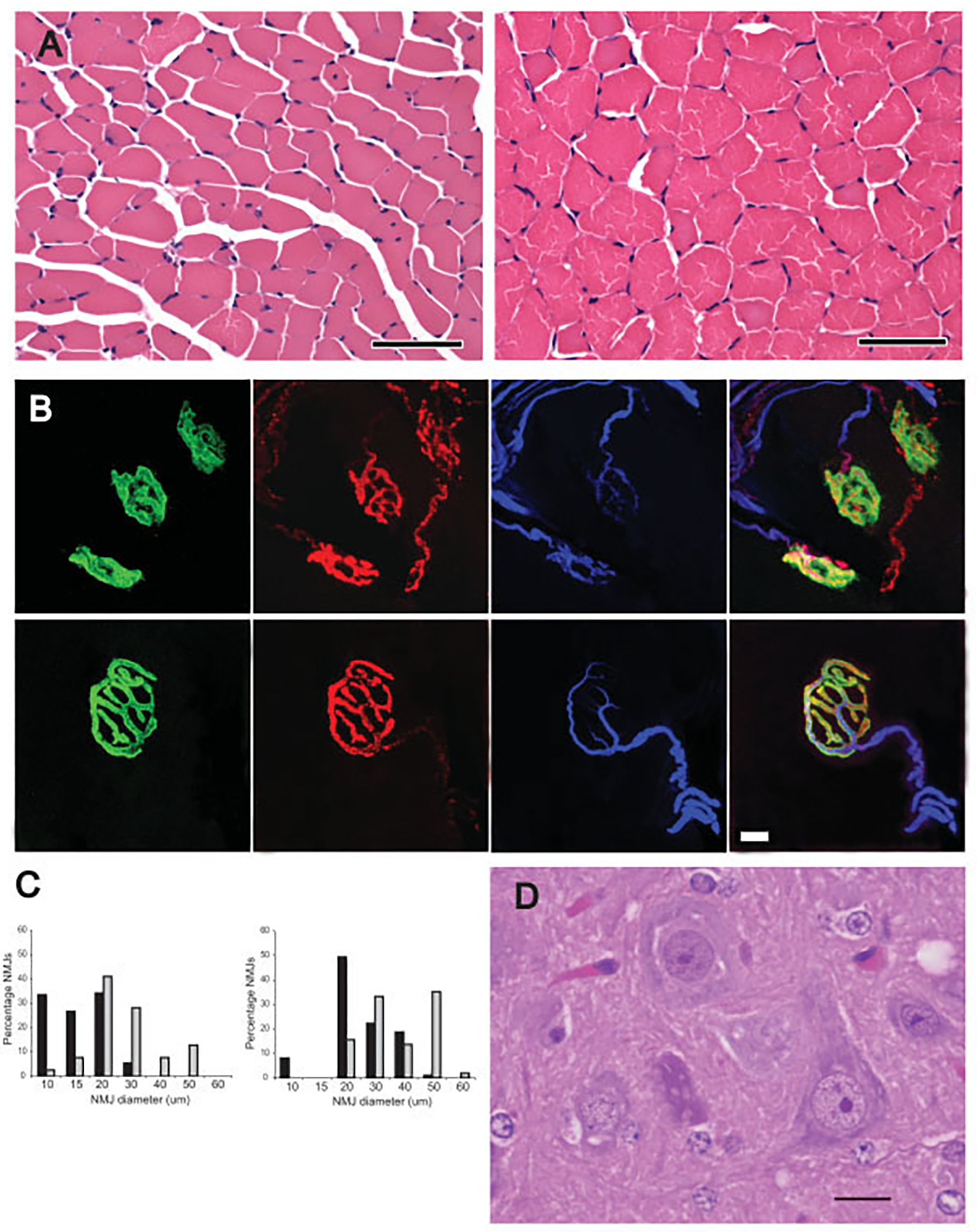
Long-lived trichostatin A (TSA)–treated spinal muscular atrophy (SMA) mice have morphologically immature but connected motor units. (A) Myofibers of the TA muscle were hypotrophic, but otherwise morphologically normal in a treated postnatal day 44 (P44) SMA mouse (left) compared with an age-matched normal littermate (right). (B) Triple staining of neuromuscular junctions (NMJs) with α-bungarotoxin (green), synaptophysin (red), and neurofilament (blue). NMJs in the paraspinal muscle of a treated P46 SMA mouse (top) were small and simplified compared with a P46 normal littermate. (C) As indicated in the Table, there was minimal evidence of denervated NMJs in the TA, intercostal, or paraspinal muscles of P39 and P46 SMA mice compared with normal littermates (NL; gray bars). Only the SMA P39 (black bars) TA muscle showed a statistically significant increase in denervation (*p = 0.005). As indicated in histograms, NMJ diameter was reduced in the paraspinal muscles of treated SMA mice compared with age-matched normal littermates. (D) Many morphologically normal anterior horn cells were present in ventral horn of lumbar spine of a treated P29 SMA mouse. Scale bars 50μm (A); 10μm (B); 30μm (C).
Long-Lived, Trichostatin A–Treated Spinal Muscular Atrophy Mice Develop Progressive Vascular Necrosis
Long-lived, TSA-treated SMA mice that lived to 27 days or longer developed tissue ischemia (see Supplemental Table). Blue then black discoloration started at the tip of the tail (Fig 3A), and proximal extension caused complete loss of the tail (see Fig 3C) and anus and pelvic necrosis in some mice. Ischemia in the hind limbs was first evident in one or two digits and evolved to complete loss of the hind feet and distal leg in some mice (see Fig 3C and Table). Four mice showed tissue necrosis in the pinnae of the ears (see Fig 3B).
Fig 3.
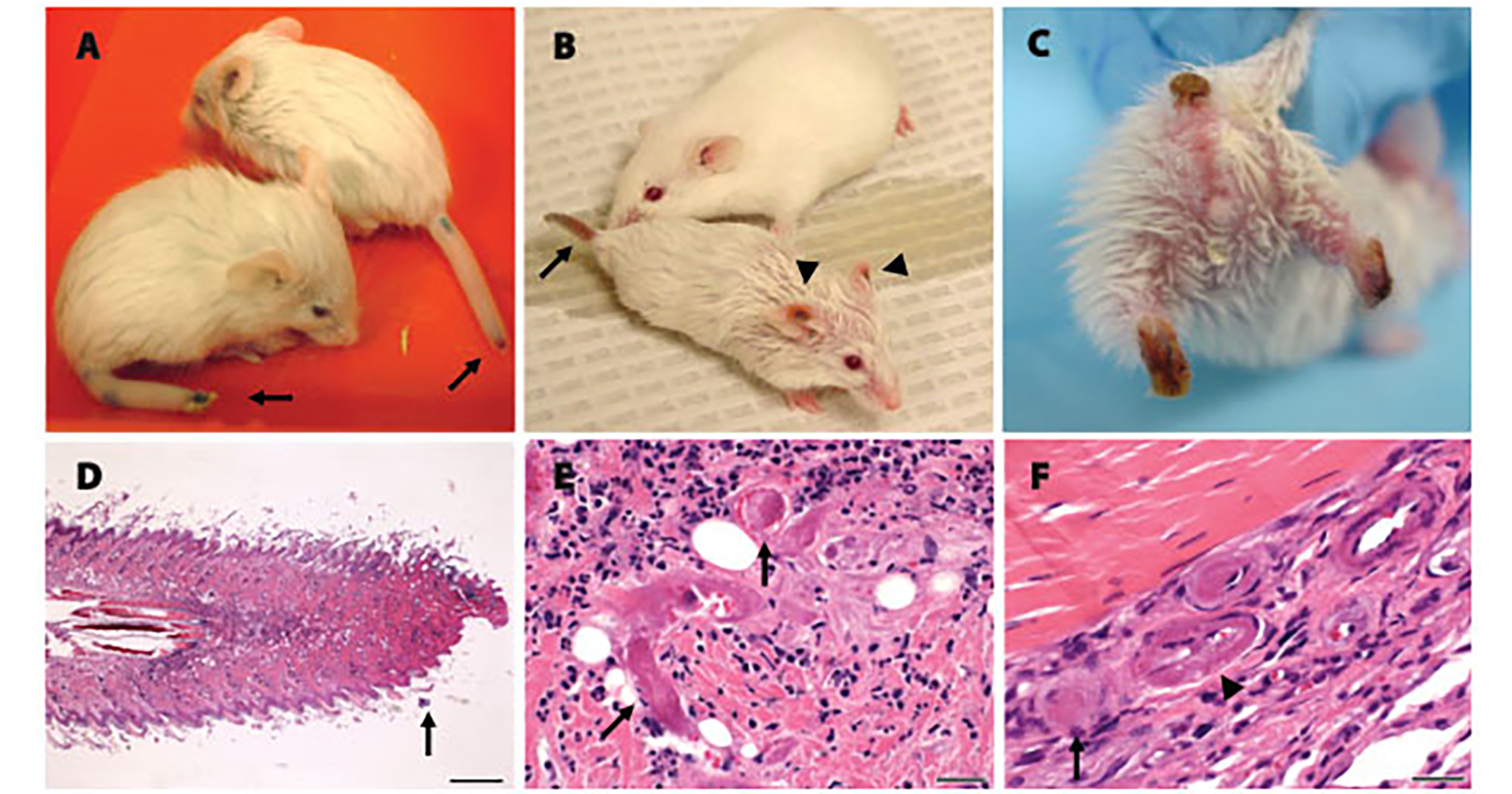
Long-lived trichostatin A (TSA)–treated spinal muscular atrophy (SMA) mice show progressive distal necrosis and small-vessel thrombosis. (A) Two postnatal day 25 (P25) SMA mice showing early necrosis at the tips of the tails. (B) A P44 SMA mouse and age-matched normal littermate. The SMA mouse shows necrosis of the tail (arrow) and ears (arrowheads). (C) A P47 SMA mouse showing severe tail and bilateral hind-limb necrosis resulting in significant tissue loss. (D) Hematoxylin-and-eosin–stained longitudinal section of the tail from a P29 SMA mouse demonstrated necrosis at the tail tip. Scale bar = 400μm. (E) High-power view of the tail just proximal to the junction of viable and necrotic tissue. Arrows highlight two thrombosed small vessels. Scale bar = 25μm. (F) High-power view of foot from a P57 SMA mouse showing four vessels. One vessel indicated by the arrow is thrombosed and one vessel indicated by the arrowhead shows fibrinoid necrosis in the vessel wall.
Table.
Percentage of denervated NMJ (mean ± SEM)
| Tibialis anterior | Intercostal | Paraspinal | |
|---|---|---|---|
|
| |||
| P39 SMA | *3.8 ± 1.4 | 2.2 ± 1.1 | 7.5 ± 2.9 |
|
| |||
| P39 NL | 0 | 2.9 ± 2.0 | 3.6 ± 1.6 |
|
| |||
| P46 SMA | 0 | 1.0 ± 1.0 | 2.0 ± 1.0 |
|
| |||
| P46 NL | 0 | 0 | 2.3 ± 1.3 |
Histological examination showed tissue necrosis of the tail involving the skin and deeper tissues including bone (see Fig 3D). Three of four mice showed necrosis of the hind limbs, one mouse showed necrosis of the pelvis extending into the colon, and one mouse showed necrosis of the ear pinnae. At the junctions of viable and necrotic tissue, several thrombosed small blood vessels were evident (see Figs 3E, F). Rare vessels showed fibrinoid necrosis within the vessel wall. No vessel abnormalities or necrosis were evident elsewhere. Progressive necrosis was the presumed cause of death in these mice.
Discussion
Although HDAC inhibitors are a promising class of drugs for the treatment of SMA, questions remain regarding which compounds are most efficacious, when drugs should be delivered during the disease course, and what ancillary care should be administered during drug therapy. We have studied TSA, a potent and specific HDAC inhibitor, in SMA mice and have shown that early TSA combined with nutritional support results in a remarkable increase in median survival (170%) compared with early TSA alone (40%).
Adequate nutrition has long been a concern in severe SMA patients who have profound muscle wasting and are at high risk for entering a fasting state with resulting hypoglycemia and muscle breakdown.11,12 Recently, Oskoui and colleagues13 showed that the improved survival of type I SMA patients observed in recent years can be attributed, in part, to aggressive nutritional support with gastrostomy feedings. Here, nutritional supplementation dramatically improved the efficacy of TSA. Nutrition may unmask the true therapeutic effects of HDAC inhibitors by preventing starvation and catabolism, improving the overall health of the mouse, and minimizing drug toxicity. Alternatively, TSA and nutrition may directly interact in some way to improve the function of SMA motor units.
Long-lived mice treated with TSA plus nutrition showed stable motor function long after TSA was discontinued. At late stages, myofibers were hypotrophic, suggesting incomplete postnatal growth, as we have previously observed in younger SMA mice,10 but NMJs were structurally intact with little evidence of ongoing denervation. This suggests that an early, time-limited course of TSA not only improved function in the short-term but also produced long-lasting stabilization of the SMA motor unit.
As treated long-lived SMA mice aged, they developed progressive vascular necrosis. Necrosis is unlikely to be due to TSA toxicity because it was never seen in similarly treated normal littermates. Furthermore, necrosis has been previously observed in other lines of SMA mice that have not received HDAC inhibitors.14–16 Vascular dysfunction is therefore likely to be a consequence of SMN deficiency. Although this phenomena might be specific to mice, digital ischemia and necrosis has recently been observed in a few patients with severe SMA (Kathryn Swoboda and Alexandra Araujo, personal communication). Recent studies increasingly support an interdependent relation between nerves and vessels.17,18 Further work is required to understand the function of SMN in vessels and how vascular dysfunction may contribute to motor neuron dysfunction in SMA.
Here we have shown that HDAC inhibition delivered early in life and combined with aggressive nutritional support can result in a substantial and sustained amelioration of the SMA phenotype in mice. These studies indicate that, although HDAC inhibitors remain a promising therapy for SMA patients, the timing of drug delivery is likely to be critical for efficacy. The role of nutrition in SMA urgently requires further understanding as optimized nutrition combined with HDAC inhibitors may be more effective than either alone. Vascular deficits are likely to be a direct result of SMN protein deficiency, and as patients with severe SMA live longer, vascular abnormalities may become a more commonly observed clinical manifestation of SMA.
Supplementary Material
Acknowledgments
This study was supported by the National Institute of Neurological Disorders and Stroke (NINDS) Career Transition Award, K22-NS0048199-01, C.J.S.; NINDS Competitive Fellowship Award, B.G.B.) NINDS Intramural Funds and Families of Spinal Muscular Atrophy, CJS.
We thank Drs M. Sendtner and A. Burghes for generously providing the original breeding pairs for the SMA transgenic mice.
References
- 1.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–165. [DOI] [PubMed] [Google Scholar]
- 2.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A 1999;96: 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999;8:1177–1183. [DOI] [PubMed] [Google Scholar]
- 4.Feldkotter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002;70:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997;16:265–269. [DOI] [PubMed] [Google Scholar]
- 6.Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 1997;6:1205–1214. [DOI] [PubMed] [Google Scholar]
- 7.Sumner CJ. Therapeutics development for spinal muscular atrophy. NeuroRx 2006;3:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26:5541–5552. [DOI] [PubMed] [Google Scholar]
- 9.Mercuri E, Bertini E, Messina S, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 2007;68:51–55. [DOI] [PubMed] [Google Scholar]
- 10.Avila AM, Burnett BG, Taye AA, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest 2007;117:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruce AK, Jacobsen E, Dossing H, Kondrup J. Hypoglycaemia in spinal muscular atrophy. Lancet 1995;346:609–610. [DOI] [PubMed] [Google Scholar]
- 12.Orngreen MC, Zacho M, Hebert A, et al. Patients with severe muscle wasting are prone to develop hypoglycemia during fasting. Neurology 2003;61:997–1000. [DOI] [PubMed] [Google Scholar]
- 13.Oskoui M, Levy G, Garland CJ, et al. The changing natural history of spinal muscular atrophy type 1. Neurology 2007;69: 1931–1936. [DOI] [PubMed] [Google Scholar]
- 14.Hsieh-Li HM, Chang JG, Jong YJ, et al. A mouse model for spinal muscular atrophy. Nat Genet 2000;24:66–70. [DOI] [PubMed] [Google Scholar]
- 15.Tsai LK, Tsai MS, Lin TB, et al. Establishing a standardized therapeutic testing protocol for spinal muscular atrophy. Neurobiol Dis 2006;24:286–295. [DOI] [PubMed] [Google Scholar]
- 16.Gavrilina TO, McGovern VL, Workman E, et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle specific SMN expression has no phenotypic effect. Hum Mol Genet 2008;17:1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmeliet P Blood vessels and nerves: common signals, pathways and diseases. Nat Rev Genet 2003;4:710–720. [DOI] [PubMed] [Google Scholar]
- 18.Weinstein BM. Vessels and nerves: marching to the same tune. Cell 2005;120:299–302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.