

Abstract
Loss-of-function mutations in SLC30A10 induce hereditary manganese (Mn)-induced neuromotor disease in humans. We previously identified SLC30A10 to be a critical Mn efflux transporter that controls physiological brain Mn levels by mediating hepatic and intestinal Mn excretion in adolescence/adulthood. Our studies also revealed that in adulthood, SLC30A10 in the brain regulates brain Mn levels when Mn excretion capacity is overwhelmed (e.g. after Mn exposure). But, the functional role of brain SLC30A10 under physiological conditions is unknown. We hypothesized that, under physiological conditions, brain SLC30A10 may modulate brain Mn levels and Mn neurotoxicity in early postnatal life because body Mn excretion capacity is reduced in this developmental stage. We discovered that Mn levels of pan-neuronal/glial Slc30a10 knockout mice were elevated in specific brain regions (thalamus) during specific stages of early postnatal development (postnatal day 21), but not in adulthood. Furthermore, adolescent or adult pan-neuronal/glial Slc30a10 knockouts exhibited neuromotor deficits. The neuromotor dysfunction of adult pan-neuronal/glial Slc30a10 knockouts was associated with a profound reduction in evoked striatal dopamine release without dopaminergic neurodegeneration or changes in striatal tissue dopamine levels. Put together, our results identify a critical physiological function of brain SLC30A10—SLC30A10 in the brain regulates Mn levels in specific brain regions and periods of early postnatal life, which protects against lasting deficits in neuromotor function and dopaminergic neurotransmission. These findings further suggest that a deficit in dopamine release may be a likely cause of early-life Mn-induced motor disease.
Keywords: basal ganglia, dopamine, metal homeostasis, parkinsonism, ZnT10
Graphical Abstract
Graphical Abstract.
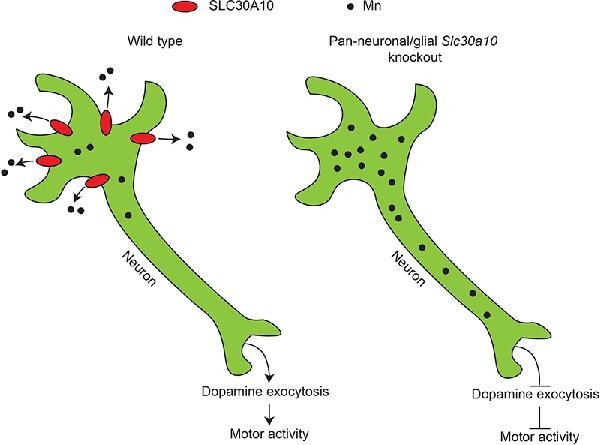
Elucidating the physiological function of SLC30A10 in the brain using mouse models, metal analyses, and neurochemistry.
Introduction
Manganese (Mn) is an essential metal, but elevated levels of Mn induce severe neurotoxicity.1–4 Excess Mn mainly accumulates in the basal ganglia, leading to motor deficits as a primary manifestation of Mn neurotoxicity.1–4 Historically, Mn neurotoxicity has been observed in the context of occupational Mn overexposure in adults, which produces a parkinsonism-like disorder.1–5 Mn-induced parkinsonism is also associated with decreased liver function (e.g. in individuals with liver disease such as cirrhosis), even in the absence of elevated Mn exposure, because Mn is primarily excreted by the liver.6 Furthermore, recent epidemiological studies have suggested that environmental Mn exposure (e.g. from sources such as drinking water or air) is an important public health problem, especially in the early life periods of infancy, childhood, and adolescence.7–19 In these developmentally sensitive life stages, environmental Mn exposure is associated with substantial impairments of motor function, such as deficits in motor coordination, skilled motor function, tremor intensity, postural stability, among others, and additionally, also induces executive function deficits.7–19 Currently there are no treatments for Mn neurotoxicity, either in adults or early life, and delineating the underlying neuronal mechanisms is essential for the development of effective interventions.
Understanding of the homeostatic control and detoxification of Mn has been transformed by the recent discoveries of two inherited disorders of Mn metabolism due to homozygous loss-of-function mutations in SLC30A10 or SLC39A14.2,20–23 Over the last few years, we characterized SLC30A10 as a specific Mn efflux transporter that reduces cellular Mn levels to protect against Mn toxicity.20,24–29 Our studies in whole-body and tissue-specific Slc30a10 knockout mice revealed that, under basal/physiological conditions (i.e. in the absence of exposure to elevated Mn) in adolescent/adult animals, SLC30A10 regulates brain Mn levels by mediating hepatic and intestinal Mn excretion, which prevents accumulation of Mn in the blood, and subsequently, in other organs including the brain.25,29 The role of SLC30A10 in Mn excretion was later confirmed by another group.30 Other studies revealed that a major physiological function of SLC39A14 is to import Mn from blood into the liver and intestines for subsequent excretion by SLC30A10.2,6,20,31 In totality, currently available evidence indicates that the excretory function of SLC30A10 and SLC39A14 in the liver and intestines plays a fundamental role in controlling blood and brain Mn levels under physiological conditions in adolescence/adulthood.2,6,20,31
In addition to the liver and intestines, we previously reported that SLC30A10 is also expressed in the basal ganglia, a primary brain region affected by Mn overexposure,1–4 and thalamus, which receives the basal ganglia output32,33 and is also targeted by Mn,34 of adult mice.29 Furthermore, basal ganglia and thalamus Mn levels of adult pan-neuronal/glial Slc30a10 knockout mice were higher than littermate controls under elevated Mn exposure, but not physiological, conditions.29 These and related other findings indicated that, in adulthood, function of SLC30A10 in the brain becomes important in reducing brain Mn levels when Mn excretion capacity is overwhelmed and excess Mn accumulates in the brain (e.g. after Mn exposure).29
A fundamental question that has not yet been answered is whether brain SLC30A10 has a function under physiological conditions. Notably, Mn excretion capacity is reduced in early postnatal life.6,35,36 In rodents, under physiological conditions, Mn excretion increases at approximately postnatal day (PND) 17–18.6,35,36 The increase in Mn excretion coincides with weaning (∼PND 21) and is likely a compensatory mechanism to adapt to the dietary change that occurs at weaning because milk has a low Mn content.20 The age-dependent change in Mn excretion raises an important, testable hypothesis—under physiological conditions, brain SLC30A10 may play a critical role in controlling brain Mn levels and modulating Mn neurotoxicity in early postnatal life when the ability to excrete Mn is lower than in later developmental stages. Here, we test this hypothesis by performing metal measurement, behavioral, imaging, and neurochemical assays in pan-neuronal/glial Slc30a10 knockout mice and their littermate controls. We report that SLC30A10 in the brain regulates Mn levels in specific brain regions and specific periods of early postnatal development, which protects against lasting deficits in neuromotor function and dopaminergic neurotransmission. These findings establish a physiological function for brain SLC30A10, and additionally, provide new insights into the mechanisms of early-life Mn-induced neuromotor disease, which are not well characterized.
Results
Pan-neuronal/glial Slc30a10 knockout mice exhibit elevated Mn levels in specific brain regions in early postnatal life
To determine whether brain SLC30A10 plays an age-dependent role in regulating brain Mn levels, we performed metal measurement assays in pan-neuronal/glial Slc30a10 knockout mice, which we have extensively described previously,29 and littermate controls in early postnatal life and adulthood. We reconfirmed tissue specificity of the pan-neuronal/glial Slc30a10 knockouts by demonstrating that SLC30A10 was depleted in the brain, but not the liver, of the knockouts (Fig. 1A). Additionally, we confirmed that SLC30A10 was expressed in the brain in early postnatal development (PND 8) (Fig. 1B). For our studies, we considered the pre-weaning period of PND 1–21 as the early postnatal period in mice. This period is (i) characterized by reduced Mn excretion capacity (as described earlier, Mn excretion in mice increases at ∼PND 17–186,36) and (ii) corresponds to human infancy and early childhood,37,38 which have high epidemiological relevance to environmental Mn exposure.7–19
Fig. 1.

SLC30A10 expression and brain metal levels of pan-neuronal/glial Slc30a10 knockouts (Slc30a10fl/fl; Nes-Cre) and littermate controls in early postnatal life without Mn exposure. (A) RT-PCR from midbrain (containing the basal ganglia and thalamus) and liver tissue of 4-month-old pan-neuronal/glial Slc30a10 knockouts or littermate controls. (B) RT-PCR from midbrain, liver, and small intestine tissue of a control mouse at PND 8. (C–D) ICP–MS from midbrain containing basal ganglia and thalamus at PND 8 (C) or PND 14 (D). For C, n = 5 controls (2 males, 3 females) and 5 knockouts (3 males, 2 females). For (D), n = 8 controls (3 males, 5 females) and 5 knockouts (all females). Mean ± SE. P > 0.05 by t-test.
Metal analyses early in development under physiological conditions (i.e. in the absence of elevated Mn exposure), at PND 8 or 14, performed using a section of the middle of the brain containing the developing basal ganglia and thalamus revealed that brain Mn levels of the pan-neuronal/glial knockouts were comparable to littermates (Fig. 1C)and D). Levels of other metals were also comparable between genotypes (Fig. 1C)and D). We postulated that the lack of genotype-specific effects on brain Mn in early development may reflect the milk-based diet of pups, which has a substantially lower Mn content than rodent chow.20 Notably, pups start consuming increasing amounts of Mn-enriched rodent chow in the peri-weaning period as development continues and they become large enough to access food on the top of the cage.28 Thus, there was a possibility that genotype differences in brain Mn levels may become apparent in the third week of life. To test this, we repeated the metal measurement assay at PND 21 under physiological conditions. We increased the spatial resolution of this assay by isolating basal ganglia regions (striatum, globus pallidus, and substantia nigra) and the thalamus using a brain punching technique (punching was not feasible in the PND 8 and 14 mice due to the small size of the developing brain). Importantly, knockouts exhibited a ∼40% increase in Mn levels in the thalamus (Fig. 2A; P < 0.05 for a genotype-specific difference in the thalamus using repeated measures 2-way ANOVA and Sidak's post hoc test). Mn levels in other regions were comparable between genotypes, and there were no genotype differences in levels of other metals (Fe, Cu, and Zn) in any of the regions (Fig. 2A–D). We hypothesized that the absence of genotype-specific differences in Mn levels in the basal ganglia sub-regions was likely because the increase in brain Mn levels in the pan-neuronal/glial Slc30a10 knockouts under physiological conditions was transient, modest, and approaching the sensitivity limit of the brain punch analyses by inductively coupled plasma-mass spectrometry (ICP–MS). This hypothesis predicted that more profound genotype differences in brain Mn levels, including in basal ganglia sub-regions, may be detectable when body/brain Mn levels further increase. We tested this prediction by repeating the Mn measurement assay at PND 21 after exposing mice to a low-dose oral Mn exposure regimen via water that models environmental Mn exposure in human children. After Mn treatment, Mn levels in basal ganglia sub-regions and the thalamus, combined, of the pan-neuronal/glial knockouts were significantly (∼100%) higher than littermates (Fig. 3A; P < 0.05 for main effect of genotype on mixed-effects ANOVA). Due to high within-group variation, brain region-specific genotype differences in Mn levels were not evident on post hoc analyses (Fig. 3A). The reasons for this variation are unclear, but possible explanations may relate to the brain punch technique in which metal levels cannot be normalized to tissue weight (see the “Methods” section) and animal-specific differences in Mn excretion capacity and/or chow consumption in the peri-weaning period leading to PND 21. Importantly, however, levels of other metals in the Mn-treated mice were essentially comparable across genotypes in all of the brain regions (Fig. 3B–D), implying that the main effect of genotype was specific to Mn levels (Fig. 3A; a very modest decrease in globus pallidus Zn levels was noted in the knockouts [Fig. 3D], but the biological relevance of this change is uncertain). Finally, we had previously reported that there were no differences in Mn content of basal ganglia sub-regions and thalamus of 3-month-old pan-neuronal/glial knockouts and littermates in the absence of Mn exposure when analyses were performed using brain punches or a section of the middle of the brain.29 In the current study, we reconfirmed the lack of genotype-specific differences in adult 3-month-old mice under physiological conditions using sections of the middle of the brain containing the basal ganglia and thalamus (Fig. 4; we did not repeat the brain punching in the 3-month-old mice because of the strength of our prior data29). We also confirmed that blood Mn levels of the pan-neuronal/glial knockouts were comparable to controls at all of the above time-points with or without Mn exposure (Supplementary Fig. S1A–E), implying that changes in brain Mn of the knockouts at PND 21 were not secondary to increases in blood Mn. Modest changes in blood Zn, Cu, or Fe levels were observed at some, but not all, of the time-points (Supplementary Fig. S1), but the biological relevance of these changes is unclear. Put together, the results of the metal analyses indicate that, under physiological conditions, SLC30A10 in the brain is necessary to regulate Mn levels in specific brain regions in specific periods of early postnatal life.
Fig. 2.

In the absence of Mn exposure, Mn levels in the thalamus of pan-neuronal/glial Slc30a10 knockout mice (Slc30a10fl/fl; Nes-Cre) are elevated at PND 21. (A–D) ICP–MS from brain punches at PND 21; n = 6 (2 males, 4 females) per region and genotype for each metal. Mean ± SE. *, P < 0.05 using 2-way ANOVA with repeated measures and Sidak's post hoc analyses.
Fig. 3.

After daily oral Mn exposure, Mn levels in the basal ganglia and thalamus of pan-neuronal/glial Slc30a10 knockout mice (Slc30a10fl/fl; Nes-Cre) are elevated at PND 21. (A–D) ICP–MS from brain punches obtained at PND 21 from mice treated with daily oral Mn; n = 9–15 animals (7–8 males, 2–7 females) per region and genotype for each metal. Mean ± SE. *, P < 0.05 for a main effect of genotype using mixed-effects ANOVA analysis for Mn and with Sidak's post hoc test for indicated comparison for Zn. Note that there was a main effect of Mn treatment (P < 0.05) when data from Figs. 2A)and 3A)were compared using mixed-effects ANOVA.
Fig. 4.

Mn levels in the brain of pan-neuronal/glial Slc30a10 knockouts (Slc30a10fl/fl; Nes-Cre) normalize by 3 months of age in the absence of Mn exposure. ICP–MS analyses were performed using midbrain samples. For each metal, n = 15 controls (7 males, 8 females) and 8 knockouts (5 males, 3 females). Mean ± SE. P > 0.05 by t-test.
Pan-neuronal/glial Slc30a10 knockout mice exhibit neuromotor deficits in adolescence and adulthood
To determine whether the brain Mn level elevations in the pan-neuronal/glial Slc30a10 knockouts in early postnatal life under physiological conditions, albeit brain region and developmental stage specific, are associated with motor function, we performed a set of neuromotor tests—beam balance and pole tests in early adolescence (PND 28), and the open-field test in late adolescence (7–8 wk) and young adulthood (3 months; mice reach adulthood at PND 60, and the period between weaning at PND 21 to PND 60 corresponds to adolescence37,38). The age at which each test was conducted was based on prior studies by us and others and our additional pilot assays indicating that the selected age was ideal for determining the impact of brain Mn level increases on test performance.
The beam balance test assays for the capability of a mouse to walk across a narrow beam and missteps quantify motor performance. Compared with littermate controls, pan-neuronal/glial knockouts had a significantly higher number of missteps (Fig. 5A)and B). The pole test assays for the capability of a mouse to ascend/descend a narrow pole, and time to complete the trial is a measure of motor function. We did not detect genotype differences in this test (Fig. 5C). The open-field test assesses generalized exploratory and locomotor function in rodents and has been extensively used to assay for Mn neurotoxicity.29,39–41 The first 5 min of the test reflect reactivity and exploratory behavior of an animal in a novel environment while the next 10 min is a measure of generalized locomotion. In 7–8 wk or 3-month-old mice, there were no differences between genotypes in the first 5 min of the test (Fig. 5D and E; Supplementary Fig. S2A and B). Importantly, in the next 10 min interval, the pan-neuronal/glial knockouts exhibited reduced movement (Fig. 5D and E; Supplementary S2A and B). Overall, the results of the motor tests indicate that the pan-neuronal/glial Slc30a10 knockouts develop neuromotor deficits, which are detectable in early adolescence and persist into adulthood.
Fig. 5.

Adolescent pan-neuronal/glial Slc30a10 knockout mice (Slc30a10fl/fl; Nes-Cre) exhibit Mn-dependent neuromotor deficits. (A–C) Mean or total number of missteps on the beam balance test (A and B) or mean total time for completion of the pole test (C) from three trials performed at PND 28 in mice treated daily with vehicle water orally. Mean ± SE. For A and B, n = 12 controls (4 males, 8 females) and 9 knockouts (5 males, 4 females). For C, n = 12 controls (4 males, 8 females) and 8 knockouts (4 males, 4 females). *, P < 0.05 by t-test. (D–E) Open-field test to monitor horizontal movement (D) or resting time (E) of vehicle water-exposed 7–8-wk-old mice; n = 20 controls (9 males, 11 females) and 8 knockouts (5 males, 3 females). Mean ± SE. *, P < 0.05 using repeated measures 2-way ANOVA and Sidak's post hoc analyses. (F) Open-field analyses of Mn-treated 7–8-wk-old mice; n = 18 controls (10 males, 8 females) and 11 knockouts (5 males, 6 females). Mean ± SE. *, P < 0.05 using repeated measures 2-way ANOVA and Sidak's post hoc analyses. (G) Comparison of the horizontal movement of vehicle or Mn-treated pan-neuronal/glial Slc30a10 knockouts from D and F. Mean ± SE. *, P < 0.05 by t-test. Note that there was a main effect of Mn treatment (P < 0.05) when normalized horizontal movement of vehicle or Mn-treated control or pan-neuronal/glial Slc30a10 knockout mice in the 6–15 min interval was compared using 2-way ANOVA.
We had performed the neuromotor tests in the absence of Mn exposure because our goal was to characterize the physiological function of brain SLC30A10. However, it was important to validate that the motor deficits of the pan-neuronal/glial Slc30a10 knockouts were induced by increased brain Mn, and were not simply a consequence of the knockout of SLC30A10. We did not attempt a rescue experiment (i.e. use low Mn chow to normalize brain Mn levels of the pan-neuronal/glial knockouts followed by behavioral analyses) because, based on our prior experience with such assays,28,29 the modest, region specific, and transient increase in brain Mn levels in the pan-neuronal/glial Slc30a10 knockouts in early postnatal life was unlikely to be normalized by reducing the Mn content of chow. Instead, we repeated the open-field assay in mice exposed to elevated oral Mn. Our expectation was that Mn-treated pan-neuronal/glial knockouts would exhibit more profound locomotor deficits than knockouts that had not been exposed to Mn. We first confirmed that, within the Mn-treated groups, pan-neuronal glial/knockouts exhibited reduced movement compared with littermates in the 6–15 min interval (Fig. 5F). Subsequent analyses of the movement of pan-neuronal/glial knockouts treated with or without Mn in the 6–15 min interval revealed that, as expected, Mn-treatment reduced the movement of the knockouts (Fig. 5G). Thus, the neuromotor phenotype of the pan-neuronal/glial Slc30a10 knockouts is induced by elevated brain Mn.
To complete the behavioral analyses, we also performed the Montoya staircase test (in the absence of Mn exposure), which assesses skilled forelimb motor function in rodents by quantitative measurements of reaching, grasping, and retrieving movements for food pellets located on descending steps of a left- and right-sided staircase.42–44 Performance in the staircase test has been reported to be impacted by early-life Mn exposure in rats.16 However, performance of 8–9-wk-old pan-neuronal/glial knockouts on this test was comparable to littermates (Supplementary Fig. S3A–E). The lack of genotype-specific differences in the staircase test may be a consequence of its lower sensitivity, particularly in mice. Despite the outcome of the staircase and pole tests, the totality of the results in Figs.1–5 indicate that, in the pan-neuronal/glial Slc30a10 knockouts, an increase in Mn levels in specific brain regions in early postnatal development leads to a lasting deficit in neuromotor function.
Pan-neuronal/glial Slc30a10 knockouts exhibit a deficit in evoked dopamine release without dopaminergic neurodegeneration
Neuronal mechanisms of early-life Mn-induced neuromotor deficits are not well understood and of adult Mn-induced parkinsonism are controversial (see the “Discussion” section). To elucidate the mechanisms leading to neuromotor deficits in the pan-neuronal/glial Slc30a10 knockouts, we focused on dopaminergic neurotransmission in the basal ganglia because (i) dopamine plays a central role in regulating motor function32,33; and (ii) some studies have implicated degeneration or dysfunction without degeneration of substantia nigra dopaminergic neurons to be the cause of adult Mn-induced parkinsonism.2,3,5 Microscopy assays revealed that the total number of dopaminergic neurons in the substantia nigra and ventral tegmental area were comparable between adult pan-neuronal/glial Slc30a10 knockout mice and their littermate controls (Fig. 6A)and B). There were also no genotype-specific changes in the total levels of striatal tissue dopamine, the dopamine metabolite 3,4-dihydroxyphenylacetic acid (DOPAC), or the DOPAC to dopamine ratio (Fig. 6C–E; we used the striatum for these assays because the substantia nigra pars compacta dopaminergic neurons project to the striatum32,33). Thus, the motor phenotype of the pan-neuronal/glial Slc30a10 knockouts could not be explained by dopaminergic neurodegeneration or reduced tissue dopamine levels.
Fig. 6.

Pan-neuronal/glial Slc30a10 knockout mice (Slc30a10fl/fl; Nes-Cre) exhibit reduced evoked dopamine release without dopaminergic neurodegeneration under physiological conditions. (A–B) Immunofluorescence to detect tyrosine hydroxylase (TH)-positive neurons in the substantia nigra pars compacta and ventral tegmental area of pan-neuronal/glial knockout mice or littermate controls. Representative images of a control and knockout animal are shown in A and quantification from n = 3 male mice per genotype in B. Mean ± SE. P > 0.05 by t-test. Bar, 500 µm. (C–E) HPLC with electrochemical detection to measure dopamine (C), DOPAC (D), and the DOPAC to dopamine ratio (E) in striatal tissue of 3-month-old pan-neuronal/glial knockouts or littermate controls; n = 7 controls (4 males, 3 females) and 8 knockouts (5 males, 3 females). Mean ± SE. P > 0.05 by t-test. (F) In vivo microdialysis to measure dopamine levels in the dorsal striatum of 3–4-month-old pan-neuronal/glial knockouts or littermate controls; n = 3 controls (1 male, 2 females) and 4 knockouts (2 males, 2 females). Mean ± SE. *, P < 0.05 using repeated measures 2-way ANOVA and Sidak's post hoc test for the difference between genotypes at the 80 min time-point. (G) The average of the first three baseline dopamine measures from F. Mean ± SE. P > 0.05 by t-test. H. The percentage increase in potassium-evoked dopamine release from F. Mean ± SE. *, P < 0.05 by t-test.
These negative results led us to contemplate in vivo microdialysis experiments to assay for extracellular dopamine levels and functional changes in dopamine release. As microdialysis is technically challenging and time intensive, before beginning these experiments, we assayed for a few other parameters also reported to be impacted by Mn exposure or altered in other models of Parkinson's disease—striatal tissue γ-aminobutyric acid (GABA) levels, glial activation/accumulation in the basal ganglia, and degeneration of noradrenergic neurons in the locus coeruleus.2,3,5,41,45,46 We did not detect obvious genotype-specific differences in these parameters (Supplementary Fig. S4A–D). We then proceeded with the in vivo microdialysis assays. Baseline dopamine levels measured in the dorsal striatum were comparable between the genotypes (Fig. 6F)and G). Importantly, however, potassium-evoked release of dopamine was markedly reduced in the pan-neuronal/glial knockouts (Fig. 6F)and H). In totality, the neurochemical and microscopy results suggest that a deficit in potassium-evoked striatal dopamine release, but not dopaminergic neurodegeneration or changes in bulk tissue dopamine content, likely leads to the neuromotor phenotype of the pan-neuronal/glial Slc30a10 knockout mice.
Discussion
We previously reported that under physiological conditions in adolescence/adulthood blood and brain Mn levels are controlled by the excretory function of SLC30A10 in the liver and intestines.25,29 We additionally reported that the role of brain SLC30A10 in adulthood is to lower brain Mn when the body burden of Mn increases and Mn excretion capacity is overwhelmed (e.g. during elevated Mn exposure) and provide additional neuroprotection from Mn neurotoxicity.29 The current work now establishes a physiological function of brain SLC30A10 in the absence of elevated Mn exposure—brain SLC30A10 is necessary to regulate Mn levels in defined brain regions and stages of early postnatal development, which protects against lasting Mn-induced deficits in neuromotor function. Overall, our current and prior results imply that brain Mn levels are controlled by the excretory function of SLC30A10 in the liver and intestines and by brain SLC30A10 in an age- and Mn exposure-dependent manner.
Compared with littermates, brain Mn levels were elevated by ∼10–15-fold in adolescent tissue-specific Slc30a10 knockout mice lacking SLC30A10 expression in the liver and intestines.29 In contrast, the brain Mn level elevations we observed in the pan-neuronal/glial knockouts in early life were restricted to the thalamus, detected only at one time-point (PND 21), and ≤1-fold of control. The modest effect of brain SLC30A10 on brain Mn levels in early postnatal life may be reflective of the unique nutritional and developmental features of this life stage during which Mn intake is low because of the naturally low Mn content of milk,20 but Mn is still critical for cellular function as highlighted by severe neurodevelopmental deficits in children harboring loss-of-function mutations in the Mn importer SLC39A8.47,48
We observed that Mn levels were elevated in the thalamus, but not the basal ganglia, of the pan-neuronal/glial Slc30a10 knockouts in early life under physiological conditions. Our inability to detect genotype differences in Mn levels in regions outside the thalamus under physiological conditions may be reflective of technical/experimental limitations (sensitivity limit of brain punch analyses by ICP–MS combined with the transient/modest Mn levels increases in the knockouts). After low-dose Mn exposure, Mn levels in the basal ganglia and the thalamus, combined, of the pan-neuronal/glial knockouts were significantly higher than littermates. This result is consistent with our prior findings, which demonstrated that Mn levels in the globus pallidus and thalamus of Mn-treated adult pan-neuronal/glial Slc30a10 knockout mice were higher than littermates.29 Moreover, while ICP–MS analysis of brain punch tissue is a sophisticated technique that improves the spatial resolution of brain tissue metal analyses, it cannot distinguish between metal accumulation within the cell bodies of neurons or axons of projecting neurons. As basal ganglia neurons project to the thalamus, the observed increase in thalamic Mn levels of the pan-neuronal/glial Slc30a10 knockouts under physiological conditions may be reflective in part of Mn level changes in the basal ganglia axonal projections. Overall, it is possible that SLC30A10 has a direct role in controlling basal ganglia Mn levels under physiological conditions, and our results do not imply that the deficits in neuromotor function and dopaminergic neurotransmission observed in the pan-neuronal/glial Slc30a10 knockouts are solely driven by increases in thalamic Mn levels.
The pan-neuronal/glial Slc30a10 knockouts exhibited significant neuromotor deficits in adolescence/adulthood under physiological conditions that were exacerbated by Mn exposure. The extent of the neuromotor deficits we observed in the knockouts is in the same range as those reported in prior studies of Mn neurotoxicity.39,41,45 Since brain (thalamus) Mn levels of the pan-neuronal/glial knockouts were detectably higher than littermates at PND 21, but not adulthood (this study and Taylor et al.29), our interpretation is that the lasting neuromotor dysfunction observed in the knockouts can be attributed to the modest, region-specific increase in brain Mn in early postnatal development. Important implications of these findings are that (i) function of SLC30A10 in the brain in early postnatal life is indispensable to protect against lasting Mn-induced neurotoxicity that would otherwise develop from physiological/dietary Mn intake; and (ii) mild and transient increases in brain Mn in early postnatal life are sufficient to induce lasting neuromotor deficits, which is consistent with prior studies.16,49,50 These results do not rule out the possibility of changes in brain Mn levels that were not detectable by the brain punch and ICP–MS methods used here occurring in later life stages (adolescence/adulthood) of pan-neuronal/glial Slc30a10 knockouts and contributing to neuromotor deficits.
Evoked dopamine release in the striatum of the pan-neuronal/glial Slc30a10 knockouts was significantly inhibited while several other parameters tested (e.g. tissue dopamine levels, dopaminergic neurodegeneration, glial activation, etc.) were unaffected. Viewed in conjunction with the metal analyses and behavior data, these results indicate that a modest increase in brain Mn levels in early development is sufficient to induce lasting changes in dopaminergic neurotransmission. These findings also suggest that a deficit in evoked dopamine release may likely be a primary cause of the neuromotor phenotype of the pan-neuronal/glial Slc30a10 knockouts, and by extension, of early-life Mn-induced motor disease. This is notable because the neuronal targets and mechanisms of Mn-induced motor disease are controversial. Mechanistic studies, largely in the context of adult exposures, have implicated degeneration or dysfunction of basal ganglia GABAergic or dopaminergic neurons as the cause.2,3,5 Other mechanisms, such as glutamatergic excitotoxicity, cholinergic dysfunction or neuroinflammation, which would be expected to impact basal ganglia dopaminergic or GABAergic neurons, have also been implicated.2,3,5 In contrast with the considerable, albeit conflicting, literature about Mn-induced parkinsonism in adults, understanding of the effects of early-life Mn exposure that induce motor deficits has thus far been limited. This is primarily because the focus of the few mechanistic studies on environmental Mn neurotoxicity has been on the executive function deficits that usually co-occur with motor dysfunction.15 Importantly, however, results from two recent studies support the idea that deficits in evoked dopamine release may underlie early-life Mn-induced motor disease. Analyses of another genetic mouse model of Mn neurotoxicity, full-body Slc39a14 knockout mice with ∼10-fold increases in brain Mn,25,31,51 revealed that the knockouts had a strong inhibition of evoked striatal dopamine release without dopaminergic neurodegeneration.51 An inhibition of evoked striatal dopamine release was further observed in weanling and aged adult rats exposed to Mn in early-life.50 Overall, there is an emerging body of evidence implicating deficits in stimulated striatal dopamine release, but not dopaminergic neurodegeneration, as the cause of neuromotor deficits induced by early-life increases in brain Mn.
The neurochemical findings raise the important question of what are the molecular mechanisms that lead to the inhibition of striatal dopamine release after early-life increases in brain Mn. Close examination of our microdialysis data offers some clues. We did not observe any difference between the genotypes in baseline dialysate dopamine concentrations. These samples are collected when mice are resting and not actively engaged in specific behaviors, so the dialysate dopamine mainly reflects tonic dopamine release.52 This suggests that early-life brain Mn elevations do not alter the function of the basic calcium-dependent exocytotic release mechanisms, although this needs to be confirmed using quantitative microdialysis techniques.53 On the other hand, the use of potassium perfusion into the microdialysis probe produced a dramatic 15-fold increase in dialysate dopamine in control mice, which was significantly attenuated in the knockouts. This result suggests that physiological mechanisms that require activation of dopamine circuits above resting levels may be more sensitive to the neurotoxic effects of Mn overexposure. Clearly, detailed assays are necessary in the future to delineate the molecular mechanisms of the Mn-induced inhibition of dopamine release, and to elucidate the apparent differential sensitivity of aspects of dopamine release during resting or activated states to Mn toxicity. These future studies will need to be performed with cognizance of the fact that increased dopamine retention may induce oxidative stress and produce reactive dopamine quinones,54 which may further contribute to the disease pathology. Another fundamental issue for future investigation is determining whether the observed inhibition of striatal dopamine release is due to a primary Mn-induced injury in dopaminergic neurons or a consequence of Mn-dependent changes in other neurons in the motor circuit. Finally, studies in rats have associated executive function deficits induced by early-life Mn exposure with deficits in the evoked release of dopamine, norepinephrine, and serotonin in the prefrontal cortex.50 Determining whether the mechanisms by which early-life Mn exposure inhibits release of different neurotransmitters in different brain regions are similar or divergent will be critical in the future.
In conclusion, our studies establish the physiological function of SLC30A10 in the brain and suggest that deficits in dopamine release, but not dopaminergic neurodegeneration, likely underlie early-life Mn-induced neuromotor disease.
Methods
Generation of knockout strains, age of animals, and husbandry
All mouse experiments were approved by the Institutional Animal Care and Use Committee of the University of Texas at Austin (Austin, TX, USA). We previously described the development and characterization of homozygous floxed Slc30a10 mice (Slc30a10fl/fl), in which exon 1 of Slc30a10 is flanked by loxP sites.25,28,29 Pan-neuronal/glial Slc30a10 knockouts were generated as described by us previously by crossing floxed Slc30a10 mice with the Nestin-Cre strain.29 Breeding, genotyping, and animal housing were as described previously.25,28,29,40 Chow used was Prolab RMH 1800; LabDiet #5LL2 with 160 µg Mn/g chow.
Epidemiological studies indicate that the early life stages of infancy, childhood and adolescence in humans are most relevant for environmental Mn exposure.7–19 In mice, these stages correspond to PND 1–60, with PND 1–21 representing the early postnatal period, and PND 21–60 representing the prepubescent-adolescent period.37,38 We refer to this PND 1–60 age range as “early-life.” Animals at ages PND 60 onward are considered adults.37,38
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative RT-PCR
Oral Mn treatment
Mn-treated mice received 50 mg/kg MnCl2.4H2O per day (∼10 mg absolute Mn/kg per day) beginning at birth and continuing until euthanasia. For pre-weaning exposure (birth—PND 21), Mn was administered by hand via pipette in a total volume of 5 μL (birth—PND 6) or 10 μL (PND 7—20). A 250 mg/mL MnCl2.4H2O stock was diluted in MilliQ water containing 2.5% of the natural sweetener stevia, and the required dose, based on the pup’s weight, was delivered. Vehicle-treated animals received water containing 2.5% stevia. Pre-weaning Mn delivery did not impact nursing by the dam or milk intake by the pup. Post-weaning Mn exposure was administered via drinking water containing 0.2 mg Mn/mL, based on the fact that the animals’ total water intake was ∼25% of body weight, and this Mn level provided the required 10 mg Mn/kg daily dose. Animal body weights and water intake were measured regularly, and Mn concentration was adjusted if necessary to ensure target dose delivery. Vehicle-treated animals received only water in the post-weaning period.
We used the oral Mn exposure regimen because drinking water-based oral exposure is a major source of environmental Mn exposure in human infants, children, and adolescents.20 The regimen used by us, and related similar regimens, (i) approximate the increase in Mn exposure in infants and children consuming well water contaminated with 1.5 mg Mn/L, which is the median well water concentration associated with neurological deficits in children20; (ii) induce modest increases in brain Mn that are comparable to humans20; and (iii) produce measurable behavioral deficits without overt toxicity.20
Beam balance and pole tests
These were performed as described previously with some modifications.55,56 Mice were tested individually. Equipment was from Maze Engineers (Skokie, IL, USA) and cleaned with 5% ethanol between animals. For the beam balance, a round beam of 12 mm diameter and 100 cm length with a safe box at one end was used. The subject animal was allowed to acclimatize for 30 s in the safe box. Subsequently, the ability to walk across the beam to reach the safe box was tested in three successive trials. The first trial was initiated from the midpoint of the beam. The last two trials were initiated from the end of the beam opposing the safe box. Missteps and time taken to complete the test were calculated for each trial. For the pole test, a 50 cm long round pole with an 8 mm diameter was used. This pole was placed in a large cage with sufficient bedding. Mice were placed ∼1 inch from the top of the pole facing upward. Ability of the mouse to ascend to the top of the pole, then turn to face downward, and finally descend to the bottom of the pole was assayed in three trials. Both tests were performed during the light period of the animals’ light–dark cycle.
Open-field test
The open-field test was performed exactly as described by us previously.29,40 Animals were placed individually in the open-field chamber (Opto-Varimex 4 Activity Meter, Columbus Instruments, Columbus, OH, USA) for 15 min. Data were binned into two intervals: 0–5 and 6–15 min. The first 5 min provides information about the animal's exploratory behavior, while the second 10 min provides information about general locomotion. The test was performed during the light period of the animals’ light–dark cycle. The experimenter was not in the room during testing. The open-field chamber was cleaned with 5% ethanol between animals.
Montoya staircase test
In the Montoya staircase test, animals are confined to a staircase apparatus and trained to grasp food pellets from different step levels in staircases on the left and right side of the animal. Procedures were performed similarly to those described previously.16 Animals were placed individually into the staircase apparatus for 15 min. In this apparatus (Campden Instruments Limited, Model #80301 mouse), each staircase had eight descending steps with one well per step. Steps 1 through 6 (with step 1 being closest to the animal) were baited with three 5 mg food pellets (Lab Supply, AIN-76-A Purified Rodent Tablet). Thus, there were a total of six pellets (three on the left staircase and three on the right staircase) available per step. To enhance the sensitivity of this assay, pellets were colored with food dye (McCormick & Co., Baltimore, MD, USA) such that each color corresponded to a specific step as previously described.16,44 To increase motivation to grasp the pellets, animals were placed on food restriction throughout training and testing. After completing the test, each cage was provided ∼2–2.5 g of rodent chow per animal each day (our animals normally consume ∼3 g of rodent chow per animal per day). Body weights were measured daily during training and testing, and the amount of rodent chow provided was adjusted as needed to ensure animals remained within 90–95% of their original body weight. There were two stages of this experiment: training (days 1–11) and testing (days 12–16). Training was broken up into three segments: days 1–5, days 6–10, and day 11. Between each training session, animals were allowed 2 days of free feeding to ensure each mouse maintained proper body weight. All testing occurred during the light period of the animals’ light–dark cycle. Once testing was complete, animals were removed from the staircase apparatus and returned to their home cage. The staircase apparatus was cleaned with 70% ethanol between animals.
Skilled forelimb performance was assayed using the following parameters: total number of pellets eaten, the average number of pellets taken per step during testing, the average number of pellets eaten per step during testing, success rate per step, and the percentage of pellets misplaced per step. For each step, the number of pellets taken was calculated as the number of initial pellets−the number of pellets remaining in the assigned well. The number of pellets eaten was calculated as the number of initial pellets−(the number of pellets remaining in the assigned well + the number of pellets misplaced + the number of pellets lost). The success rate was calculated as (pellets eaten/pellets taken) ×100. The percentage of pellets misplaced was calculated as (pellets misplaced/pellets taken) ×100. For these calculations, pellets misplaced described any pellets that were on any step (steps 1–7) except the assigned step. Pellets lost described any pellets that were on step 8 or lost to the floor of the apparatus as animals were unable to reach pellets in these locations.
Immunofluorescence assays
Whole brain was collected and post-fixed in 4% paraformaldehyde for at least 48 h. Following fixation, tissue was placed in a 30% sucrose solution in phosphate-buffered saline (PBS) until the brains sunk (∼48 h) and then placed in PBS. For sectioning, whole brains were embedded in optimal cutting temperature (OCT) compound and frozen in cold isopentane (Thermo Fischer Scientific, Waltham, MA, USA). Tissue was sliced on a cryostat (set to ∼−18°C) in 20 μm sections. Sections were placed free-floating in PBS and stored at 4°C until staining. For staining, sections were transferred to a 24-well plate with one section per well and processed in the following order: three 10 min washes in PBS at room temperature, 1 h incubation in 1% Triton X-100 (in PBS) at room temperature, 1 h incubation in blocking solution (50 mM glycine; 0.5% Triton X-100; 5% fetal bovine serum in PBS) at room temperature, overnight incubation in the primary antibody at 4°C, five 10 min PBS washes at room temperature, 2 h incubation in the secondary antibody at room temperature, 10 min incubation in 4′,6-diamidino-2-phenylindole (DAPI), and five 10 min PBS washes at room temperature. Sections were placed on a shaker for each step. Primary antibodies (anti-tyrosine hydroxylase, Immunostar, Cat# 22941; anti-GFAP, Abcam, Cat#Ab7260; and anti-Iba-1, Wako Chemicals, Cat# 019-19741), secondary antibodies (Alexa Fluor; Thermo Fisher Scientific), and 4′,6-diamidino-2-phenylindole (DAPI) were diluted 1:1000 in blocking solution. After staining, sections were placed on glass slides. Coverslips were placed over the sections using mounting media (90% glycerol in 100 mM Tris-HCl; pH 8.0) and sealed with clear nail polish. Images were captured using a Nikon swept-field confocal with an inverted Nikon TiE inverted microscope and an iXon Ultra EMCCD camera (Andor Technology, UK). The objective was a Nikon 20×/0.75 numerical aperture objective. Images were captured as a large image with stitching. Automated image analysis was performed using the NIS Elements software.
Surgical procedures
All surgical procedures were performed using aseptic techniques. Surgeries were performed similarly to procedures previously described.57,58 Briefly, mice were anesthetized with isoflurane (2.5–3% for induction, 1–2% for maintenance) and secured on a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). A guide cannula (8 mm long, 20 gauge; Ziggy's Tubes and Wires, Inc., Sparta, TN, USA) was implanted to target the dorsal striatum using the following coordinates: +2.3 mm lateral to Bregma, +0.7 mm anterior to Bregma, 1.5 mm ventral to the skull surface. The guide cannula was secured using one stainless steel screw (Antrin Minature Specialties, Inc., Fallbrook, CA, USA) and dental cement. A wire loop for tethering during in vivo microdialysis was also secured to the skull with dental cement. An obturator was inserted into the guide cannula to prevent the cannula from clogging. Mice were given a local anesthetic (bupivacaine, 5 mg/kg; Covetrus North America) and a general analgesic (Rimadyl, 5 mg/kg; Zoetis, Parsippany, NJ, USA) subcutaneously during surgery. Following surgery, mice were allowed to recover on heat until fully awake and sternal. All mice were singly housed and provided food and water ad libitum. Mice were given the general analgesic Rimdayl every 12 h for 2 days post-surgery and as needed until microdialysis. General health and body weight were monitored daily.
In vivo microdialysis
Microdialysis probes (2 mm active area) were constructed similar to Job et al. (2006).59 Probes were inserted under light anesthesia (1–2% isoflurane) at least 4 days post-surgery. Following probe insertion, animals were placed in individual microdialysis chambers, and normal artificial cerebrospinal fluid (ACSF; 149 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2.2H20, 1.2 mM MgCl2.6H20, 5.4 mM D-glucose, and 0.05 mM ascorbic acid) was perfused at 0.2 μL/min overnight. The following morning, the flow rate was increased to 1 μL/min at least 2 h before sample collection. Sample collection occurred at least 12 h after probe insertion. Three baseline samples were collected at 20 min intervals. For the high KCl sample, animals were perfused with ACSF containing 50 mM KCl (NaCl concentration was adjusted to maintain isotonicity) for 10 min, then returned to normal ACSF for the remaining 10 min. At least three additional baseline samples were collected. After the final baseline sample collection, calcium-free ACSF was perfused for at least 2 h, and samples were collected to determine the calcium dependency of dopamine release. All samples were frozen on dry ice immediately after collection and stored at –80°C until analysis by high-performance liquid chromatography (HPLC). Animals were perfused with PBS and 4% paraformaldehyde, and whole brain was collected and post-fixed in 4% paraformaldehyde. Tissue was sliced on a vibratome in 100 μm sections to validate probe locations (Supplementary Fig. S5).
Liquid chromatography
Concentrations of dopamine and DOPAC were determined using HPLC with electrochemical detection. For dopamine and DOPAC analysis of striatal tissue, tissue preparation was performed similarly to previous work.60 Briefly, the striatum was dissected by hand, flash-frozen in liquid nitrogen, and stored at –80°C. To prepare tissue for HPLC analyses, 300 μL of 0.2 M perchloric acid per 10 mg of wet tissue was added. Samples were sonicated and 20 μL were removed to measure protein content (Bio-Rad Protein assay dye, #5000006). The remainder was centrifuged at 14 000 ×g for 10 min, syringe filtered (0.22 μm), and kept on ice until injection. Samples not used for immediate injection were stored at −80°C. The mobile phase consisted of 0.027 g 1-octanesulfonic acid sodium salt monohydrate (OSA), 3.011 g citric acid monohydrate, 0.02 g ethylenediaminetetraacetic acid disodium salt (EDTA), and 1.017 g sodium dihydrogen phosphate dihydrate dissolved in 1 L MilliQ water, pH 4.6, and 7% (v/v) methanol. A Luna C18(2) LC Column (3 μm particle size, 100 Å, 150 × 2 mm; Phenomenex, Torrence, CA, USA), guard column (BDS-Hypersil-C18, 5 μm particle size, 20 × 2.1 mm), and VT03 (2 mm glassy carbon electrode) were used. The flow rate was set to 0.2 mL/min, the oxidizing potential was +0.65 V, and the injection volume was full loop.
For GABA analyses using striatal samples, tissue preparation was exactly as described earlier. GABA derivatization was performed as described by us.61 Briefly, the working solution was made by dissolving 11 mg of o-phthalaldehyde in 250 μL of 95% ethanol, 250 μL 1 M sodium sulfite, and 4.5 mL sodium tetraborate decahydrate (pH adjusted to 10.4 using 4 M sodium hydroxide; borate). The working solution was stored at 4°C. For the derivatization, 9 μL of sample or standard was combined with 2 μL borate and 0.5 μL working solution, then mixed and incubated in the dark at room temperature for 10 min. The mobile phase consisted of 15.6008 g sodium dihydrogen phosphate dihydrate and 0.1863 g EDTA dissolved in 1 L MilliQ water, pH 4.5, and 27% (v/v) methanol. The column and guard column were the same as for dopamine and DOPAC analysis, and a SenCell (2 mm glassy carbon electrode, salt-bridge reference; Antec Scientific, The Netherlands) was used. The flow rate was set to 0.05 mL/min, the oxidizing potential was +0.70 V, and the injection volume was full loop.
For dopamine analysis in dialysates, the mobile phase (0.50 g OSA, 0.101 g sodium 1-decanesulfonate, 0.128 g EDTA, and 11.081 g sodium dihydrogen phosphate dihydrate) was dissolved in 1 L MilliQ water, pH 5.6, and 12% methanol. A Luna C18(2) LC Column (3 μm particle size, 100 Å, 50 × 1 mm; Phenomenex) and SenCell (2 mm glassy carbon electrode, salt-bridge reference; Antec Scientific) were used. The flow rate was set to 0.1 mL/min, the oxidizing potential was +0.45 V, and the injection volume was 5 μL. Samples were thawed immediately before injection. Samples that did not exhibit calcium dependency (>50% of dopamine concentration of preceding baseline measure) and statistical outliers determined using the GraphPad Outlier Calculator (https://www.graphpad.com/quickcalcs/grubbs1/) were excluded from analyses.
Chromeleon 6.8 Data System Software (Thermo Fisher Scientific) was used for all data acquisition. For all analyses, the signal-to-noise ratio for each sample was greater than 2.
ICP–MS metal analyses
ICP–MS metal analyses from bulk tissue samples and blood were performed as described in our previous papers.25,28,29 To summarize here, brain samples were either a section of the midbrain or brain punches.25,28,29 ICP–MS data were normalized to tissue dry weight for the bulk midbrain sections.25,28,29 Brain punch samples were collected essentially as described by us previously.29 Briefly, the whole brain was collected, flash-frozen in liquid nitrogen, and stored at −80°C until ready for use. The tissue was then embedded in OCT in an embedding mold. The tissue and OCT were frozen on cold isopentane. Brains were sliced in 500 µm sections on a cryostat and, for each animal, one punch (1.25 mm in diameter) was taken from the same region of each hemisphere. The two punches were placed together in a 1.5 mL tube and processed for ICP–MS. Weights of fresh frozen brain punch samples were too low for accurate measurement, and therefore, similar to our previous work,29 ICP–MS data for these samples are expressed as “per brain punch.”
Chemicals
Unless otherwise noted, all chemicals were from Thermo Fisher Scientific or Sigma-Aldrich.
Statistical analyses
All statistical analyses were performed using the PRISM 9 software (Graphpad, Inc., La Jolla, CA, USA). Comparisons between multiple groups were performed using 1- or 2-way ANOVA with appropriate post hoc analyses. Comparisons between two groups were performed using Student's t-test. Comparisons that involved multiple groups and time points, brain regions, or steps were performed using 2-way ANOVA with repeated measures or mixed-effects analysis and appropriate post hoc analyses. Sexes were combined for analyses because of the lack of clear sex-specific differences. For all testing, P < 0.05 was considered statistically significant. “Asterisks” in figures indicate a statistically significant difference.
Supplementary Material
Contributor Information
Cherish A Taylor, Division of Pharmacology & Toxicology, College of Pharmacy; and Institute for Neuroscience, The University of Texas at Austin, Austin, TX 78712, USA.
Stephanie M Grant, Division of Pharmacology & Toxicology, College of Pharmacy; and Institute for Neuroscience, The University of Texas at Austin, Austin, TX 78712, USA.
Thomas Jursa, Department of Microbiology and Environmental Toxicology, University of California at Santa Cruz, Santa Cruz, CA 95064, USA.
Ashvini Melkote, Division of Pharmacology & Toxicology, College of Pharmacy; and Institute for Neuroscience, The University of Texas at Austin, Austin, TX 78712, USA.
Rebecca Fulthorpe, Division of Pharmacology & Toxicology, College of Pharmacy; and Institute for Neuroscience, The University of Texas at Austin, Austin, TX 78712, USA.
Michael Aschner, Department of Molecular Pharmacology, Albert Einstein College of Medicine, Bronx NY 10461, USA.
Donald R Smith, Department of Microbiology and Environmental Toxicology, University of California at Santa Cruz, Santa Cruz, CA 95064, USA.
Rueben A Gonzales, Division of Pharmacology & Toxicology, College of Pharmacy; and Institute for Neuroscience, The University of Texas at Austin, Austin, TX 78712, USA.
Somshuvra Mukhopadhyay, Division of Pharmacology & Toxicology, College of Pharmacy; and Institute for Neuroscience, The University of Texas at Austin, Austin, TX 78712, USA.
Funding
Supported by National Institutes of Health/National Institute of Environmental Health Sciences (NIH/NIEHS) R01 ES024812, R01 ES031574 and R01 ES031574-S1 (all to S.M.); and National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) F99 NA124142 (C.A.T.).
Authors contributions
C.A.T., S.M.G., T.J., A.M., and R.F.: Performed experiments. C.A.T., D.R.S., R.A.G., and S.M.: Designed experiments and interpreted the data. C.A.T., D.R.S., R.A.G., and S.M.: Analyzed data and wrote the paper.
Conflicts of interest
The authors declare no financial conflicts of interest with this study.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
References
- 1. Aschner M., Erikson K. M., Herrero Hernandez E., Tjalkens R., Manganese and its Role in Parkinson's Disease: from Transport to Neuropathology, NeuroMol. Med., 2009, 11(4), 252–266. 10.1007/s12017-009-8083-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balachandran R. C., Mukhopadhyay S., McBride D., Veevers J., Harrison F. E., Aschner M., Haynes E. N., Bowman A. B., Brain Manganese and the Balance between Essential Roles and Neurotoxicity, J. Biol. Chem., 2020, 295(19), 6312–6329. 10.1074/jbc.REV119.009453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kwakye G. F., Paoliello M. M., Mukhopadhyay S., Bowman A. B., Aschner M., Manganese-Induced Parkinsonism and Parkinson's Disease: Shared and Distinguishable Features, Int. J. Environ. Res. Public Health, 2015, 12(7), 7519–7540. 10.3390/ijerph120707519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bowman A. B., Aschner M., Considerations on Manganese (Mn) Treatments for In Vitro Studies, Neurotoxicology, 2014, 41, 141–142. 10.1016/j.neuro.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guilarte T. R., Gonzales K. K., Manganese-Induced Parkinsonism Is Not Idiopathic Parkinson's Disease: Environmental and Genetic Evidence, Toxicol. Sci., 2015, 146(2), 204–212. 10.1093/toxsci/kfv099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gurol K. C., Aschner M., Smith D. R., Mukhopadhyay S., Role of Excretion in Manganese Homeostasis and Neurotoxicity: a Historical Perspective, Am. J. Physiol. Gastrointest. Liver Physiol., 2022, 322(1), G79–G92. 10.1152/ajpgi.00299.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lucchini R. G., Guazzetti S., Zoni S., Donna F., Peter S., Zacco A., Salmistraro M., Bontempi E., Zimmerman N. J., Smith D. R, Tremor, Olfactory and Motor Changes in Italian Adolescents Exposed to Historical Ferro-Manganese Emission, Neurotoxicology, 2012, 33(4), 687–696. 10.1016/j.neuro.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rugless F., Bhattacharya A., Succop P., Dietrich K. N., Cox C., Alden J., Kuhnell P., Barnas M., Wright R., Parsons P. J., Praamsma M. L., Palmer C. D., Beidler C., Wittberg R., Haynes E. N, Childhood Exposure to Manganese and Postural Instability in Children Living Near a Ferromanganese Refinery in Southeastern Ohio, Neurotoxicol. Teratol., 2014, 41, 71–79. 10.1016/j.ntt.2013.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takser L., Mergler D., Hellier G., Sahuquillo J., Huel G., Manganese, Monoamine Metabolite Levels at Birth, and Child Psychomotor Development, Neurotoxicology, 2003, 24(4-5), 667–674. 10.1016/S0161-813X(03)00058-5 [DOI] [PubMed] [Google Scholar]
- 10. Rodriguez-Agudelo Y., Riojas-Rodriguez H., Rios C., Rosas I., Sabido Pedraza E., Miranda J., Siebe C., Texcalac J. L., Santos-Burgoa C., Motor Alterations Associated with Exposure to Manganese in the Environment in Mexico, Sci. Total Environ., 2006, 368(2-3), 542–556. 10.1016/j.scitotenv.2006.03.025 [DOI] [PubMed] [Google Scholar]
- 11. Standridge J. S., Bhattacharya A., Succop P., Cox C., Haynes E., Effect of Chronic Low Level Manganese Exposure on Postural Balance: a Pilot Study of Residents in Southern Ohio, J. Occup. Environ. Med., 2008, 50(12), 1421–1429. 10.1097/JOM.0b013e3181896936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim Y., Bowler R. M., Abdelouahab N., Harris M., Gocheva V., Roels H. A., Motor Function in Adults of an Ohio Community with Environmental Manganese Exposure, Neurotoxicology, 2011, 32(5), 606–614. 10.1016/j.neuro.2011.07.011 [DOI] [PubMed] [Google Scholar]
- 13. Mergler D., Baldwin M., Belanger S., Larribe F., Beuter A., Bowler R., Panisset M., Edwards R., de Geoffroy A., Sassine M. P., Hudnell K., Manganese Neurotoxicity, a Continuum of Dysfunction: Results from a Community Based Study, Neurotoxicology, 1999, 20(2-3), 327–342. [PubMed] [Google Scholar]
- 14. Oulhote Y., Mergler D., Barbeau B., Bellinger D. C., Bouffard T., Brodeur M. E., Saint-Amour D., Legrand M., Sauve S., Bouchard M. F., Neurobehavioral Function in School-Age Children Exposed to Manganese in Drinking Water, Environ. Health Perspect., 2014, 122(12), 1343–1350. 10.1289/ehp.1307918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu W., Xin Y., Li Q., Shang Y., Ping Z., Min J., Cahill C. M., Rogers J. T., Wang F., Biomarkers of Environmental Manganese Exposure and Associations with Childhood Neurodevelopment: a Systematic Review and Meta-Analysis, Environ. Health, 2020, 19(1), 104. 10.1186/s12940-020-00659-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beaudin S. A., Nisam S., Smith D. R., Early Life versus Lifelong Oral Manganese Exposure Differently Impairs Skilled Forelimb Performance in Adult Rats, Neurotoxicol. Teratol., 2013, 38, 36–45. 10.1016/j.ntt.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bouchard M. F., Sauve S., Barbeau B., Legrand M., Brodeur M. E., Bouffard T., Limoges E., Bellinger D. C., Mergler D., Intellectual Impairment in School-Age Children Exposed to Manganese from Drinking Water, Environ. Health Perspect., 2011, 119(1), 138–143. 10.1289/ehp.1002321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khan K., Wasserman G. A., Liu X., Ahmed E., Parvez F., Slavkovich V., Levy D., Mey J., van Geen A., Graziano J. H.,, Factor-Litvak P. Manganese Exposure from Drinking Water and Children's Academic Achievement, Neurotoxicology, 2012, 33(1), 91–97. 10.1016/j.neuro.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wasserman G. A., Liu X., Parvez F., Ahsan H., Levy D., Factor-Litvak P., Kline J., van Geen A., Slavkovich V., LoIacono N. J., Cheng Z., Zheng Y., Graziano J. H., Water Manganese Exposure and Children's Intellectual Function in Araihazar, Bangladesh, Environ. Health Perspect., 2006, 114(1), 124–129. 10.1289/ehp.8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taylor C. A., Tuschl K., Nicolai M. M., Bornhorst J., Gubert P., Varao A. M., Aschner M., Smith D. R., Mukhopadhyay S., Maintaining Translational Relevance in Animal Models of Manganese Neurotoxicity, J. Nutr., 2020, 150(6), 1360–1369. 10.1093/jn/nxaa066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tuschl K., Clayton P. T., Gospe S. M. Jr, Gulab S., Ibrahim S., Singhi P., Aulakh R., Ribeiro R. T., Barsottini O. G., Zaki M. S., Del Rosario M. L., Dyack S., Price V., Rideout A., Gordon K., Wevers R. A., Chong W. K., Mills P. B, Syndrome of Hepatic Cirrhosis, Dystonia, Polycythemia, and Hypermanganesemia Caused by Mutations in SLC30A10, a Manganese Transporter in Man, Am. J. Hum. Genet., 2012, 90(3), 457–466. 10.1016/j.ajhg.2012.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tuschl K., Meyer E., Valdivia L. E., Zhao N., Dadswell C., Abdul-Sada A., Hung C. Y., Simpson M. A., Chong W. K., Jacques T. S., Woltjer R. L., Eaton S., Gregory A., Sanford L., Kara E., Houlden H., Cuno S. M., Prokisch H., Valletta L., Tiranti V., Younis R., Maher E. R., Spencer J., Straatman-Iwanowska A., Gissen P., Selim L. A., Pintos-Morell G., Coroleu-Lletget W., Mohammad S. S., Yoganathan S., Dale R. C., Thomas M., Rihel J., Bodamer O. A., Enns C. A., Hayflick S. J., Clayton P. T., Mills P. B., Kurian M. A., Wilson S. W., Mutations in SLC39A14 Disrupt Manganese Homeostasis and Cause Childhood-Onset Parkinsonism-Dystonia, Nat. Commun., 2016, 7(1), 11601. 10.1038/ncomms11601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quadri M., Federico A., Zhao T., Breedveld G. J., Battisti C., Delnooz C., Severijnen L. A., Di Toro Mammarella L., Mignarri A., Monti L., Sanna A., Lu P., Punzo F., Cossu G., Willemsen R., Rasi F., Oostra B. A., van de Warrenburg B. P., Bonifati V., Mutations in SLC30A10 Cause Parkinsonism and Dystonia with Hypermanganesemia, Polycythemia, and Chronic Liver Disease, Am. J. Hum. Genet., 2012, 90(3), 467–477. 10.1016/j.ajhg.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leyva-Illades D., Chen P., Zogzas C. E., Hutchens S., Mercado J. M., Swaim C. D., Morrisett R. A., Bowman A. B., Aschner M., Mukhopadhyay S., SLC30A10 Is a Cell Surface-Localized Manganese Efflux Transporter, and Parkinsonism-Causing Mutations Block Its Intracellular Trafficking and Efflux Activity, J Neurosci., 2014, 34(42), 14079–14095. 10.1523/JNEUROSCI.2329-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu C., Hutchens S., Jursa T., Shawlot W., Polishchuk E. V., Polishchuk R. S., Dray B. K., Gore A. C., Aschner M., Smith D. R., Mukhopadhyay S., Hypothyroidism Induced by Loss of the Manganese Efflux Transporter SLC30A10 May be Explained by Reduced Thyroxine Production, J. Biol. Chem., 2017, 292(40), 16605–16615. 10.1074/jbc.M117.804989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zogzas C. E., Aschner M., Mukhopadhyay S., Structural Elements in the Transmembrane and Cytoplasmic Domains of the Metal Transporter SLC30A10 Are Required for Its Manganese Efflux Activity, J. Biol. Chem., 2016, 291(31), 15940–15957. 10.1074/jbc.M116.726935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zogzas C. E., Mukhopadhyay S., Putative Metal Binding Site in the Transmembrane Domain of the Manganese Transporter SLC30A10 Is Different from that of Related Zinc Transporters, Metallomics, 2018, 10(8), 1053–1064. 10.1039/c8mt00115d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hutchens S., Liu C., Jursa T., Shawlot W., Chaffee B. K., Yin W., Gore A. C., Aschner M., Smith D. R., Mukhopadhyay S., Deficiency in the Manganese Efflux Transporter SLC30A10 Induces Severe Hypothyroidism in Mice, J. Biol. Chem., 2017, 292(23), 9760–9773. 10.1074/jbc.M117.783605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taylor C. A., Hutchens S., Liu C., Jursa T., Shawlot W., Aschner M., Smith D. R., Mukhopadhyay S., SLC30A10 Transporter in the Digestive System Regulates Brain Manganese under Basal Conditions while Brain SLC30A10 Protects against Neurotoxicity, J. Biol. Chem., 2019, 294(6), 1860–1876. 10.1074/jbc.RA118.005628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mercadante C. J., Prajapati M., Conboy H. L., Dash M. E., Herrera C., Pettiglio M. A., Cintron-Rivera L., Salesky M. A., Rao D. B., Bartnikas T. B., Manganese Transporter Slc30a10 Controls Physiological Manganese Excretion and Toxicity, J. Clin. Invest., 2019, 129(12), 5442–5461. 10.1172/JCI129710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jenkitkasemwong S., Akinyode A., Paulus E., Weiskirchen R., Hojyo S., Fukada T., Giraldo G., Schrier J., Garcia A., Janus C., Giasson B., Knutson M. D., SLC39A14 Deficiency Alters Manganese Homeostasis and Excretion Resulting in Brain Manganese Accumulation and Motor Deficits in Mice, Proc. Natl. Acad. Sci., 2018, 115(8), E1769–E1E78. 10.1073/pnas.1720739115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gerfen C. R., Surmeier D. J., Modulation of Striatal Projection Systems by Dopamine, Annu. Rev. Neurosci., 2011, 34(1), 441–466. 10.1146/annurev-neuro-061010-113641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Obeso J. A., Rodriguez-Oroz M. C., Benitez-Temino B., Blesa F. J., Guridi J., Marin C., Rodriguez M., Functional Organization of the Basal Ganglia: Therapeutic Implications for Parkinson's Disease, Mov. Disord., 2008, 23(Supp S3), S548–S559. 10.1002/mds.22062 [DOI] [PubMed] [Google Scholar]
- 34. Ma R. E., Ward E. J., Yeh C. L., Snyder S., Long Z., Gokalp Yavuz F., Zauber S. E., Dydak U., Thalamic GABA Levels and Occupational Manganese Neurotoxicity: Association with Exposure Levels and Brain MRI, Neurotoxicology, 2018, 64, 30–42. 10.1016/j.neuro.2017.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ballatori N., Miles E., Clarkson T. W., Homeostatic Control of Manganese Excretion in the Neonatal Rat, Am. J. Physiol. Regul. Integr. Comp. Physiol., 1987, 252(5), R842–R847. 10.1152/ajpregu.1987.252.5.R842 [DOI] [PubMed] [Google Scholar]
- 36. Miller S. T., Cotzias G. C., Evert H. A., Control of Tissue Manganese: Initial Absence and Sudden Emergence of Excretion in the Neonatal mouse, Am. J. Physiol. Legacy Content, 1975, 229(4), 1080–1084. 10.1152/ajplegacy.1975.229.4.1080 [DOI] [PubMed] [Google Scholar]
- 37. Semple B. D., Blomgren K., Gimlin K., Ferriero D. M., Noble-Haeusslein L. J., Brain Development in Rodents and Humans: Identifying Benchmarks of Maturation and Vulnerability to Injury across Species, Prog. Neurobiol., 2013, 106-107, 1–16. 10.1016/j.pneurobio.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brust V., Schindler P. M., Lewejohann L., Lifetime Development of Behavioural Phenotype in the House Mouse (Mus musculus), Front. Zool., 2015, 12(S1), S17. 10.1186/1742-9994-12-S1-S17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jursa T., Smith D. R., Ceruloplasmin Alters the Tissue Disposition and Neurotoxicity of Manganese, but Not Its Loading onto Transferrin, Toxicol. Sci., 2009, 107(1), 182–193. 10.1093/toxsci/kfn231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu C., Jursa T., Aschner M., Smith D. R., Mukhopadhyay S., Up-regulation of the Manganese Transporter SLC30A10 by Hypoxia-Inducible Factors Defines a Homeostatic Response to Manganese Toxicity, Proc. Natl. Acad. Sci., 2021, 118(35), e2107673118. 10.1073/pnas.2107673118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu X., Sullivan K. A., Madl J. E., Legare M., Tjalkens R. B., Manganese-Induced Neurotoxicity: the Role of Astroglial-Derived Nitric Oxide in Striatal Interneuron Degeneration, Toxicol. Sci., 2006, 91(2), 521–531. 10.1093/toxsci/kfj150 [DOI] [PubMed] [Google Scholar]
- 42. Montoya C. P., Campbell-Hope L. J., Pemberton K. D., Dunnett S. B., The “Staircase Test”: a Measure of Independent Forelimb Reaching and Grasping Abilities in Rats, J. Neurosci. Methods, 1991, 36(2-3), 219–228. 10.1016/0165-0270(91)90048-5 [DOI] [PubMed] [Google Scholar]
- 43. Baird A. L., Meldrum A., Dunnett S. B., The Staircase Test of Skilled Reaching in Mice, Brain Res. Bull., 2001, 54(2), 243–250. 10.1016/S0361-9230(00)00457-3 [DOI] [PubMed] [Google Scholar]
- 44. Kloth V., Klein A., Loettrich D., Nikkhah G., Colour-Coded Pellets Increase the Sensitivity of the Staircase Test to Differentiate Skilled Forelimb Performances of Control and 6-Hydroxydopamine Lesioned Rats, Brain Res. Bull., 2006, 70(1), 68–80. 10.1016/j.brainresbull.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 45. Krishna S., Dodd C. A., Hekmatyar S. K., Filipov N. M., Brain Deposition and Neurotoxicity of Manganese in Adult Mice Exposed via the Drinking Water, Arch. Toxicol., 2014, 88(1), 47–64. 10.1007/s00204-013-1088-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rommelfanger K. S., Edwards G. L., Freeman K. G., Liles L. C., Miller G. W., Weinshenker D., Norepinephrine Loss Produces More Profound Motor Deficits than MPTP Treatment in Mice, Proc. Natl. Acad. Sci., 2007, 104(34), 13804–13809. 10.1073/pnas.0702753104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boycott K. M., Beaulieu C. L., Kernohan K. D., Gebril O. H., Mhanni A., Chudley A. E., Redl D., Qin W., Hampson S., Kury S., Tetreault M., Puffenberger E. G., Scott J. N., Bezieau S., Reis A., Uebe S., Schumacher J., Hegele R. A., McLeod D. R., Galvez-Peralta M., Majewski J., Ramaekers V. T., Care4Rare Canada C., Nebert D. W., Innes A. M., Parboosingh J. S., Abou Jamra R., Autosomal-Recessive Intellectual Disability with Cerebellar Atrophy Syndrome Caused by Mutation of the Manganese and Zinc Transporter Gene SLC39A8, Am. J. Hum. Genet., 2015, 97(6), 886–893. 10.1016/j.ajhg.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Park J. H., Hogrebe M., Gruneberg M., DuChesne I., von der Heiden A. L., Reunert J., Schlingmann K. P., Boycott K. M., Beaulieu C. L., Mhanni A. A., Innes A. M., Hortnagel K., Biskup S., Gleixner E. M., Kurlemann G., Fiedler B., Omran H., Rutsch F., Wada Y., Tsiakas K., Santer R., Nebert D. W., Rust S., Marquardt T., SLC39A8 Deficiency: a Disorder of Manganese Transport and Glycosylation, Am. J. Hum. Genet., 2015, 97(6), 894–903. 10.1016/j.ajhg.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Conley T. E., Beaudin S. A., Lasley S. M., Fornal C. A., Hartman J., Uribe W., Khan T., Strupp B. J., Smith D. R., Early Postnatal Manganese Exposure Causes Arousal Dysregulation and Lasting Hypofunctioning of the Prefrontal Cortex Catecholaminergic Systems, J. Neurochem., 2020, 153,5, 631–649. 10.1111/jnc.14934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lasley S. M., Fornal C. A., Mandal S., Strupp B. J., Beaudin S. A., Smith D. R., Early Postnatal Manganese Exposure Reduces Rat Cortical and Striatal Biogenic Amine Activity in Adulthood, Toxicol. Sci., 2020, 173(1), 144–155. 10.1093/toxsci/kfz208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rodichkin A. N., Edler M. K., McGlothan J. L., Guilarte T. R., Behavioral and Neurochemical Studies of Inherited Manganese-Induced Dystonia-Parkinsonism in SLC39A14-Knockout Mice, Neurobiol. Dis., 2021, 158, 105467. 10.1016/j.nbd.2021.105467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zweifel L. S., Parker J. G., Lobb C. J., Rainwater A., Wall V. Z., Fadok J. P., Darvas M., Kim M. J., Mizumori S. J., Paladini C. A., Phillips P. E., Palmiter R. D., Disruption of NMDAR-Dependent Burst Firing by Dopamine Neurons Provides Selective Assessment of Phasic Dopamine-Dependent Behavior, Proc. Natl. Acad. Sci., 2009, 106(18), 7281–7288. 10.1073/pnas.0813415106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lonnroth P., Jansson P. A., Smith U., A Microdialysis Method Allowing Characterization of Intercellular Water Space in Humans, Am. J. Physiol. Endocrinol. Metab., 1987, 253(2), E228–E231. 10.1152/ajpendo.1987.253.2.E228 [DOI] [PubMed] [Google Scholar]
- 54. Monzani E., Nicolis S., Dell'Acqua S., Capucciati A., Bacchella C., Zucca F. A., Mosharov E. V., Sulzer D., Zecca L., Dopamine C. L.,, Oxidative Stress and Protein-Quinone Modifications in Parkinson's and Other Neurodegenerative Diseases, Angew. Chem. Int. Ed., 2019, 58(20), 6512–6527. 10.1002/anie.201811122 [DOI] [PubMed] [Google Scholar]
- 55. Tallaksen-Greene S. J., Sadagurski M., Zeng L., Mauch R., Perkins M., Banduseela V. C., Lieberman A. P., Miller R. A., Paulson H. L., Albin R. L., Differential Effects of Delayed Aging on Phenotype and Striatal Pathology in a Murine Model of Huntington Disease, J. Neurosci., 2014, 34(47), 15658–15668. 10.1523/JNEUROSCI.1830-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Balkaya M., Krober J., Gertz K., Peruzzaro S., Endres M., Characterization of Long-Term Functional Outcome in a Murine Model of Mild Brain Ischemia, J. Neurosci. Methods, 2013, 213(2), 179–187. 10.1016/j.jneumeth.2012.12.021 [DOI] [PubMed] [Google Scholar]
- 57. Tang A., George M. A., Randall J. A., Gonzales R. A., Ethanol Increases Extracellular Dopamine Concentration in the Ventral Striatum in C57BL/6 Mice, Alcohol. Clin. Exp. Res., 2003, 27(7), 1083–1089. 10.1097/01.ALC.0000075825.14331.65 [DOI] [PubMed] [Google Scholar]
- 58. Ramachandra V., Phuc S., Franco A. C., Gonzales R. A., Ethanol Preference Is Inversely Correlated with Ethanol-Induced Dopamine Release in 2 Substrains of C57BL/6 Mice, Alcohol. Clin. Exp. Res., 2007, 31(10), 1669–1676. 10.1111/j.1530-0277.2007.00463.x [DOI] [PubMed] [Google Scholar]
- 59. MO J., Ramachandra V., Anders S., Low M. J., Gonzales R. A., Reduced Basal and Ethanol Stimulation of Striatal Extracellular Dopamine Concentrations in Dopamine D2 Receptor Knockout Mice, Synapse, 2006, 60(2), 158–164. 10.1002/syn.20283 [DOI] [PubMed] [Google Scholar]
- 60. Moreno J. A., Yeomans E. C., Streifel K. M., Brattin B. L., Taylor R. J., Tjalkens R. B., Age-Dependent Susceptibility to Manganese-Induced Neurological Dysfunction, Toxicol. Sci., 2009, 112(2), 394–404. 10.1093/toxsci/kfp220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zandy S. L., Gonzales R. A., GABA Uptake Inhibition Reduces In Vivo Extraction Fraction in the Ventral Tegmental Area of Long Evans Rats Measured by Quantitative Microdialysis Under Transient Conditions, Neurochem. Res., 2018, 43(2), 306–315. 10.1007/s11064-017-2424-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.