

Abstract
Decreasing neurotrophic support and impaired mitochondrial bioenergetics are key mechanisms for long-term neurodegeneration and cognitive decline after traumatic brain injury (TBI). We hypothesize that preconditioning with lower and higher volumes of physical exercise upregulates the CREB-BDNF axis and bioenergetic capability, which might serve as neural reserves against cognitive impairment after severe TBI. Using a running wheel mounted in the home cage, mice were engaged in lower (LV, 48h free access, and 48h locked) and higher (HV, daily free access) exercise volumes for thirty days. Subsequently, LV and HV mice remained for additional thirty days in the home cage with the running wheel locked and were euthanized. The sedentary group had the running wheel always locked. For the same type of exercise stimulus in a given time, daily workout presents higher volume than alternate days workout. The total distance ran in the wheel was the reference parameter to confirm distinct exercise volumes. On average, LV exercise ran 27.522 m and HV exercise ran 52.076 m. Primarily, we investigate whether LV and HV protocols increase neurotrophic and bioenergetic support in the hippocampus thirty days after exercise ceased. Regardless of volume, exercise increased hippocampal pCREBSer133-CREB-proBDNF-BDNF signaling and mitochondrial coupling efficiency, excess capacity, and leak control, that may compose the neurobiological basis for neural reserves. Further, we challenge these neural reserves against secondary memory deficits triggered by a severe TBI. After thirty days of exercise LV and HV, and sedentary (SED) mice were submitted to the CCI model. Mice remained for additional thirty days in the home cage with the running wheel locked. The mortality after severe TBI was approximately 20% in LV and HV, while in the SED was 40%. Also, LV and HV exercise sustained hippocampal pCREBSer133-CREB-proBDNF-BDNF signaling, mitochondrial coupling efficiency, excess capacity, and leak control for thirty days after severe TBI. Corroborating these benefits, the mitochondrial H2O2 production linked to complexes I and II was attenuated by exercise regardless of the volume. These adaptations attenuated spatial learning and memory deficits caused by TBI. In summary, preconditioning with LV and HV exercise builds up long-lasting CREB-BDNF and bioenergetic neural reserves that preserve memory fitness after severe TBI.
Keywords: Volume of exercise, preconditioning, exercise-induced brain reserves, BDNF, mitochondrial bioenergetics, traumatic brain injury, memory deficits
Table of contents title:
Prior exercise training promotes brain resilience after TBI.
1. Introduction
Traumatic brain injury (TBI) is responsible for a high incidence of death worldwide. The forces associated with TBI inflict mechanical damage to brain tissue leading to secondary biochemical cascades that ultimately cause death, and long-term neurodegeneration associated with depression, suicide ideation, and persisting cognitive deficits (Blennow et al., 2012; Brown et al., 2011; Jorge et al., 2004; Mckee and Daneshvar, 2015; Meaney et al., 2014; Meaney and Smith, 2011). Although the mechanisms underlying secondary injury remain not fully understood, excitotoxicity, mitochondrial dysfunction, oxidative stress, and hypometabolism appear to play a collective role in worsening outcomes (Carteri et al., 2019; McGinn and Povlishock, 2015; Shiga et al., 2006; Stefani et al., 2017; Yasmin et al., 2022).
Remarkably, many of those secondary injury mechanisms may reflect a loss of interactions with regulatory molecules in the brain such as the brain-derived neurotrophic factor (BDNF). This neurotrophin is synthesized as proBDNF, which is then cleaved to active BDNF (Je et al., 2012). The extracellular cleavage of proBDNF is an important step for inducing long-term potentiation; an electrophysiological correlate of memory function (Pang et al., 2004; Patterson et al., 1996). After being secreted, BDNF binds to membrane TRkB receptors and mediates the activation of cAMP-response element-binding protein (CREB) by phosphorylation at Serine 133. This results in the upregulation of BDNF levels that provide enhanced protection of neuronal signaling through a range of interactions in the synapse (Finkbeiner et al., 1997). For example, maintenance of synaptic firing, ionic homeostasis, neurotransmitter levels, and reactive oxygen species (ROS) balance rely on appropriated mitochondrial bioenergetic function, which is under the modulation of the CREB-proBDNF-BDNF pathway. Since this neurotrophic-bioenergetic interplay is impaired by TBI it serves as a potential target candidate for therapies (Cheng et al., 2010; Correia et al., 2010b, 2010a; Zhao et al., 2021).
Nonetheless, despite the therapeutic potential of other neurotrophins, exogenous administration of BDNF after TBI does not translate into prolonged neuronal survival and cognitive benefits (Blaha et al., 2000; Conte et al., 2008). While no current pharmacological treatment modifies long-term TBI-related outcomes, studies suggest that endogenous upregulation in the CREB-BDNF pathway through lifestyle interventions like physical exercise could provide disease-modifying mechanisms to counteract secondary injury cascades after TBI (Fogelman and Zafonte, 2012; Griesbach et al., 2007; Vaynman et al., 2004). Regular physical exercise increases the expression of BDNF in the brain in a time-dependent manner, which provides neuroprotection that could benefit individuals later in life (Berchtold et al., 2005). Indeed, emerging data highlight that exercise preconditioning can protect neurons against the dysfunction and degeneration encountered in TBI (Raefsky and Mattson, 2017). The mechanisms by which prior physical exercise promotes neuroprotection after TBI are poorly understood, but many concepts that arose from longitudinal studies with Alzheimer’s disease patients seem to be suitable to TBI settings. Accordingly, it is proposed that early active lifestyle engagement leads to the formation of “neural reserves”, a term that defines the gain of plastic properties allowing to sustain cognitive function in the face of brain pathology (Cheng, 2016; McQuail et al., 2021; Stern, 2002). Evidences suggest the increase in neurotrophic factors, cerebral perfusion and metabolism, gains in white matter and gray matter volume, and decreased oxidative stress, as proxy biomarkers of exercise-induced neural reserves (Cheng, 2016). Further, the overlapping pathophysiological molecular signatures of TBI and Alzheimer’s disease such as tau hyperphosphorylation, Aβ protein accumulation, axonal pathology, and hypometabolism can be antagonized by the aforementioned factors mediated by physical exercise engagement (Gozt et al., 2021; Hermanides et al., 2021; Johnson et al., 2013, 2012, 2010; Lien et al., 2022; Yasmin et al., 2022). However, it is currently unclear how much physical activity or exercise volume is needed for neuroprotection. In the field of sports training, volume refers to the total amount of exercise performed in a given period. For example, for the same type of exercise stimulus, different volumes can be achieved by submitting individuals to daily workout or alternate days workout (Weineck, 2019).
In highly trained athletes, an increased volume of physical activity over time is essential for better performance, and thus it could be expected that more volume of activity leads to the formation of higher neurotrophic and neuroenergetic reserves in the brain. However, it is unknown whether subjects that are engaged in a lower volume of physical exercise might achieve comparable brain adaptations over time. Hence, volume is an important component of exercise prescription and may affect adherence to an active lifestyle; thus, it is critical to determine whether the manipulation of this variable before TBI might hamper secondary neurobiochemical mechanisms associated with unfavorable neurological outcomes (Garber et al., 2011; Weineck, 2019). In this context, it is challenging to understand from the neurotrophic, neuroenergetic, and cognitive point of view, if a higher volume of physical exercise promotes more effective brain reserves for neuroprotection than a lower volume of physical exercise. From a translational perspective, the outstanding question is whether an individual who exercises daily and suffers severe TBI would have better neurofunctional outcomes than an individual who exercises eventually (i.e. three times a week).
Therefore, here, we investigate the effects of lower- and higher volumes of physical exercise in building up neurotrophic and neuroenergetic reserves in relation to cognitive dysfunction after severe TBI in the mouse.
2. Material and methods
2.1. Animals and Treatment Protocols
Two-month-old CF1 male mice (n=84) were obtained from Centro de Reprodução e Experimentação de Animais de Laboratório (CREAL, UFRGS, Porto Alegre, Brazil). Mice were housed in large plexiglass cages (38 cm × 32 cm × 16 cm) in which a transparent acrylic running wheel (16 cm diameter, 8 cm wide) was attached to the stainless-steel coverage. To avoid detrimental changes associated with social isolation, we maintained n=4 mice/per cage (Muller et al., 2011). The housing temperature was kept at 22 °C under a 12-h light/12-h dark cycle (lights on at 7 a.m.) with water and food provided ad libitum. The daily and total distance running was measured through an automatic counter system, that measures each complete revolution of the running wheel using a sensor and calculates the distance using MS excel®. The information was stored in an internal micro SD card and then extracted in a computer for quantitative analysis. The running wheel and electronic system were developed by our laboratory and described elsewhere (de Carvalho et al., 2017; Dietrich et al., 2005; Muller et al., 2011; Portela et al., 2017).
Our study design comprised two settings of experiments; the first one was carried out with healthy mice exposed or not to the voluntary running wheel. The second was performed with healthy mice exposed or not to the voluntary running wheel, and subsequently submitted to TBI. To submit mice to different volumes of exercise, the rotation of the voluntary running wheel was controlled using metal clamps, that locked the wheel in position. It should be reinforced that for the same type of exercise stimulus daily workout presents comparatively higher volume than alternate days workout (Weineck, 2019). Hence, mice were randomly assigned to the following groups: sedentary (SED; running wheel constantly locked); lower-volume exercise (LV; alternating 48 h with the voluntary running wheel locked and 48 h with free access to the running wheel); and higher-volume exercise (HV, free running wheel access daily).
In the first experimental setting, we sought for specific signatures in locomotor activity, neurotrophic signaling, and mitochondrial neuroenergetic induced by preconditioning with different volumes of physical exercise (LV and HV). The protocol consisted of 15 days of free access to the exercise running wheel (habituation) followed by intermittent (48 h locked/unlocked cycle) or free unrestricted voluntary access to the running wheel for 15 additional days (manipulation of physical exercise volume; LV, and HV, respectively). This exercise regimen was adopted according to reports by Berchtold et al. which indicate that BDNF protein levels increase progressively with increasing days of running; however daily exercised mice reached difference from sedentary on day 14th meanwhile alternating days exercised mice reach difference from sedentary on day 21st (Berchtold et al., 2005). Given this, the fifteen-day habituation phase serves as an adaptation to a novel environment and provides similar baseline hippocampal BDNF levels before starting the protocol of LV and HV exercise manipulation. After thirty days, the running wheels of LV and HV exercise groups were locked, and mice spent the next thirty days without physical exercise. The sum of distance in meters (habituation plus training) ran by mice was the reference parameter to confirm LV and HV exercise. Within this time frame, mice were challenged in the open field arena and Morris water maze task and then were euthanized. The brain was dissected for analysis of neurotrophic and mitochondrial bioenergetic endpoints in the hippocampus. See experimental timeline Figure 1A.
In the second experimental setting, we interrogated whether behavioral (locomotor activity and spatial memory) and neurochemical signatures (bioenergetic and neurotrophic) associated with TBI are modulated by LV and HV exercise and if these modulations differ according to exercise volume. Initially, mice were submitted to LV, or HV exercise preconditioning protocols as above mentioned (habituation plus training). Sedentary mice have the running wheel locked during the entire protocol. After thirty days of exercise, sedentary and exercised mice were submitted to TBI, as follows: SEDTBI, LVTBI, and HVTBI (all groups submitted to craniotomy and TBI) followed by thirty-days detraining. See the experimental timeline in Figure 4A.
Thirty-days detraining period was an exploratory approach to sought for sustained BDNF increments beyond the already reported after 15 days of exercise (Fiorin et al., 2016). Also, within thirty days after TBI, rodents, and patients already present detectable neurological deficits. Hence, interactions between early neurobiochemical adaptations and long-term neurofunctional abnormalities are, in our experience, more predictable within thirty days post-TBI.
All experiments were conducted according to the NIH Guide for Care and Use of Laboratory Animals, the National Research Council’s Guide for the Care and Use of Laboratory Animals, the Brazilian Society for Neuroscience and Behavior (SBNeC), and the official governmental guidelines in compliance with the Federation of Brazilian Societies for Experimental Biology. The experiments were approved by the Ethical Committee of the Federal University of Rio Grande do Sul, Brazil (#22436).
2.2. Controlled Cortical Impact Protocol (CCI)
A severe TBI was performed by placing mice in a heated stereotaxic bed (Kopf Instruments, Tujunga, CA, USA) (37 ± 1°C). Inhalation anesthesia was provided (2.5% isoflurane) in a mixture of N2 and O2 (2:1). The anesthesia maintenance during surgical procedure was confirmed through testing withdrawal reflex of feet. An eye lubricant was applied to protect corneal membranes during the surgical procedure. Initially, a 4-mm diameter craniotomy was performed in the central part of the left parietal bone, without damaging the dura matter. Mice were subsequently submitted to a controlled cortical impact (CCI injury) induced by the Benchmark stereotaxic impactor (myNeuroLab®, Leica, St. Louis, MO, USA). The head injury was performed with a 3.0-mm diameter piston adjusted to an impact velocity of 5.7 m/s; impact duration was 100 ms and 2 mm of depth penetration on the exposed surface of the dura mater (Smith et al., 1995). After the injury, the craniotomy area was isolated using a concave lamella bonded with dental cement and the scalp was sutured (Smith et al., 1995). Lidocaine was applied locally to alleviate pain and discomfort. All surgeries were performed by the same model expert with a duration ranging from 4–7 min to avoid experimental variation, in a random order of control and treated animals.
After being removed from the stereotaxic frame, mice were left to recover from surgery in a heated chamber to maintain a body core temperature of 37°C, and were monitored up to 4 h post-injury, in which they usually recover spontaneous locomotion. Mice were then returned to the home cage and inspections were performed daily by the researches and animal care staff to check for signals of distress, discomfort and pain including bleeding and inflammation at the surgical site, piloerection, lack of spontaneous activity, changes in body temperature, decreased drinking water or food pellet consumption, and hunched position. Mice were removed from the study and humanely euthanized if they presented at least one of these phenotypes (Andersen et al., 2016; National Research Council, 2008).
Importantly, our early experience indicates that these CCI parameters translate some of the brain tissue alterations and neurological impairment found in severe TBI patients (Carteri et al., 2019; Smith et al., 1995). Particularly, severe TBI affects many young individuals worldwide, and in the region of this study there is a high incidence in young adult males relative to females (3:1). Furthermore, mortality rates are high, and secondary sequelae are very frequent in this specific population of TBI patients (Böhmer et al., 2011; de Almeida et al., 2016; Stefani et al., 2017; Strogulski et al., 2022), reinforcing the requirement of modeling severe TBI outcomes to address unsolved pertinent clinical questions.
2.3. Open Field Test
Open field (OF) was used to access spontaneous locomotion and exploratory activity 72 h after exercise as well as exercise + CCI protocols (see Figure 1A and Figure 4A, respectively). The experiments were conducted in a sound-attenuated room under low-intensity light (12 lx) using a homemade apparatus that consisted of a black-painted box measuring 50 cm × 50 cm surrounded by 50 cm-high walls. The mouse was placed in the right corner of the arena, and locomotor activity was recorded with a video camera for 10 min (Rodolphi et al., 2021). The analysis was performed using a computer-operated tracking system (Any-maze, Stoelting, Woods Dale, IL). The apparatus was cleaned with 70% alcohol between each test. The total distance traveled, distance traveled per minute, maximum speed, mean speed, total mobile time, total immobile time, and time spent in peripheral and central zones were recorded and used as indicators of locomotor activity and exploratory behavior.
2.4. Preparation of Ipsilateral Hippocampal Homogenates
Whole ipsilateral hippocampal tissue was collected thirty days after the sedentary, exercise, and exercise + CCI protocols. The hippocampus was homogenized in a buffer (320 mM sucrose, 1 mM EDTA, pH 7.4) using a pre-cooled glass potter with 10 to 15 strokes (Gnaiger, 2014). After centrifugation (1350xg for 3 min at 4 °C), we obtained mitochondria-enriched homogenates that were aliquoted for high-resolution respirometry linked to complexes, membrane potential, and hydrogen peroxide analysis. These analyses were performed simultaneously by blinded researchers within 10 min of euthanasia to the start of the analysis. Aliquots were also used for protein quantification (Pierce™ BCA Protein Assay Kit Catalog number: 23225) and immunoblotting.
2.5. Western Blotting
The ipsilateral hippocampi samples (20 μg of protein) were used to perform electrophoresis using polyacrylamide gel and PVDF membranes (Amersham, GE Healthcare, Little Chalfont, UK) (Muller et al., 2011). The following primary antibodies were used against CREB (Cell Signaling, USA ref. 9104; 1:1000), pCREBSer133(Cell Signaling, USA, ref. 9191; 1:1000), pro-BDNF (Millipore, USA, ref. ab5613; 1:1000), and BDNF (Millipore, USA, ref ab1513; 1:1000). Antibodies were incubated overnight followed by 2 h incubation with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:5000) (GE, Little Chalfont, UK) secondary antibodies. Membranes were incubated with Amersham ECL Western Blotting Detection Reagent (GE Healthcare Life Sciences, Little Chalfont, UK) for 5 min, and immunodetection used chemiluminescence via Image Quant LAS 4000 (GE, Little Chalfont, UK). The protein expression was quantified using Image J® software (Rockville, USA). The values for the optical density were expressed as a percentage of the control (Rodolphi et al., 2021).
2.6. Mitochondrial Respiratory Protocol
Oxygen consumption rates (OCR) were obtained at 37°C using a high-resolution 17Oxygraphy-2k system and recorded in real-time using DatLab software (Oroboros, Innsbruck, Austria) in respiration buffer (100 mM KCI, 75 mM mannitol, 25 mM sucrose, 5 mM phosphate, 0.05 mM EDTA, and 10 mM Tris-HC1, pH 7.4). After five minutes to establish routine OCR, a multi-substrate titration protocol was initiated with pyruvate, malate, glutamate, and succinate (PMGS; 10, 10, 20, and 10 mM, respectively) to obtain a functional system that stimulated the formation of an electrochemical gradient. Adenosine diphosphate (ADP, 2.5 mM) was then titrated to obtain maximal OxPHOS capacity (P). The oligomycin was then titrated to obtain Lomy OCR, and the maximal electron transport capacity (ETS) was obtained following titration of the proton ionophore carbonyl cyanide 4-trifluoromethoxy-phenylhydrazone (FCCP 1 μM). Non-mitochondrial respiration (ROX) was calculated after the inhibition of complex I, III, and IV with rotenone, antimycin-A, and cyanide (ROT, AA and KCN, 2, 2.5 and 5 mM, respectively) (Gnaiger, 2014). The following parameters were calculated based on the OCR: biochemical coupling efficiency ((P-Lomy)/P), OxPHOS phosphorylation capacity ((E-P)/E), ETS (E-ROX), and leak control ratio (Lomy/E) (Carteri et al., 2019; Gnaiger, 2014).
2.7. Mitochondrial Membrane Potential (ΔѰm)
The ΔѰm was obtained via the fluorescence signal (excitation wavelength of 495 nm and emission wavelength of 586 nm) of the ipsilateral hippocampal homogenates incubated in respiration buffer supplemented with 10 μM safranin-O (Spectra Max M5, Molecular Devices). The increased fluorescence units mirror the decreased ΔѰm whereas decreased fluorescence units indicate increased ΔѰm.
Incubation without substrates represents the baseline ΔѰm. Substrates for complex I, II (PMGS), and V (ADP) as well as uncoupling with FCCP (1 μM) was used to modulate the ΔѰm dynamic responses. Moreover, the fluorescence units of each coupling state were used to calculate the relative percentage of change in ΔѰm. Data are expressed as arbitrary fluorescence units (AFUs) and were normalized to protein content (Portela et al., 2017).
2.8. Mitochondrial H2O2 Production
Mitochondrial hydrogen peroxide (H2O2) production in the ipsilateral hippocampi was assessed in different mitochondrial coupling states, using samples incubated in the respiration buffer supplemented with 10 μM Amplex Red and 2 units/mL horseradish peroxidase. Baseline state H2O2 levels were measured without the presence of exogenous substrates in the incubation medium. The L (PMGS) P, and Lomy coupling states were induced to assess H2O2 production using the same substrates and inhibitors indicated in respirometry. Fluorescence was monitored at excitation (563 nm) and emission (587 nm) wavelengths with a Spectra Max M5 microplate reader (Molecular Devices, USA) (Muller et al., 2013; Portela et al., 2017).
2.9. Morris Water Maze Task
The spatial learning and memory after CCI were assessed by the Morris Water Maze adapted from Smith et. Al., 1995, (see experimental timeline Figure 4A). The animals (n = 10 per group) were submitted to a flag test (experimental day 54), and 24 h after they were trained daily in a four-trial MWM to find a hidden platform (experimental days 55 to 58). This was followed by a one-day probe test on day 59 without the platform (Figure 4A). The time spent in the target quadrant was measured as an indicator of memory retention. The analysis was performed using Any-maze software (Stoelting, Woods Dale, IL) (Muller et al., 2011).
2.10. Variable Interaction Mapping
We performed pairwise Pearson correlations for each continuous variable studied for each experimental group. We then estimated the association between paired variables using the Pearson’s product moment correlation coefficient (rho) testing a null hypothesis of rho being zero. The test statistics follow a t distribution and confidence intervals are given based on Fisher’s Z transform. These analyses and associated visualizations were obtained using the corrplot (version 0.9) and the stats (version 4.1.2) packages in R statistical environment by applying the cor and cor.mtest functions.
2.11. Statistical Analysis
To calculate survival proportions, we used the product limit (Kaplan-Meier) method (Machin et al., 2006). The results were calculated and expressed as the mean ± S.E.M. To analyze the differences between groups, we used a two-way analysis of variance (ANOVA) followed by a post-hoc Tukey test. Further, a two-way repeated measures ANOVA was used to assess statistical significance relative to minute-to-minute differences in distance travelled during the open field test. All procedures were performed using GraphPad Prism 7.0 software. The differences were considered statistically significant at p < 0.05.
3. Results
3.1. Experimental design timeline and volume of physical exercise
The experimental design is shown in Figure 1A. Sedentary mice (SED) had access to a locked running wheel assembled in the home cage. Mice in the exercise groups were submitted to 15 days of habituation in the running wheel followed by full (HV) or intermittent (LV) availability for additional 15 days. Day-by-day distance ran in the running wheel by LV and HV groups during thirty-days protocol (Figure 1B). Particularly, in the habituation phase, both groups ran a similar distance (LV, 35.416 ± 1105.89 m vs HV, 35.982 ± 652.94 m; p < 0.9687; Figure 1C). In the exercise training phase, mice allocated to HV ran significantly longer distances than LV (52.076 ± 823.31 m vs 27.522 ± 248.82 m, p < 0.0001; Figure 1D). The distance ran in the exercise habituation plus exercise training phases was greater for HV relative to LV (88.058 ± 284.47 m vs 62.938 ± 756.38 m; p < 0.0001; Figure 1E) (Results represents mean ± S.D., 4 mice per cage; n=12 per group). The cumulative distance run by mice confirmed two different volumes of training (Figures 1B and 1E).
Figure 1. Physical exercise volumes.
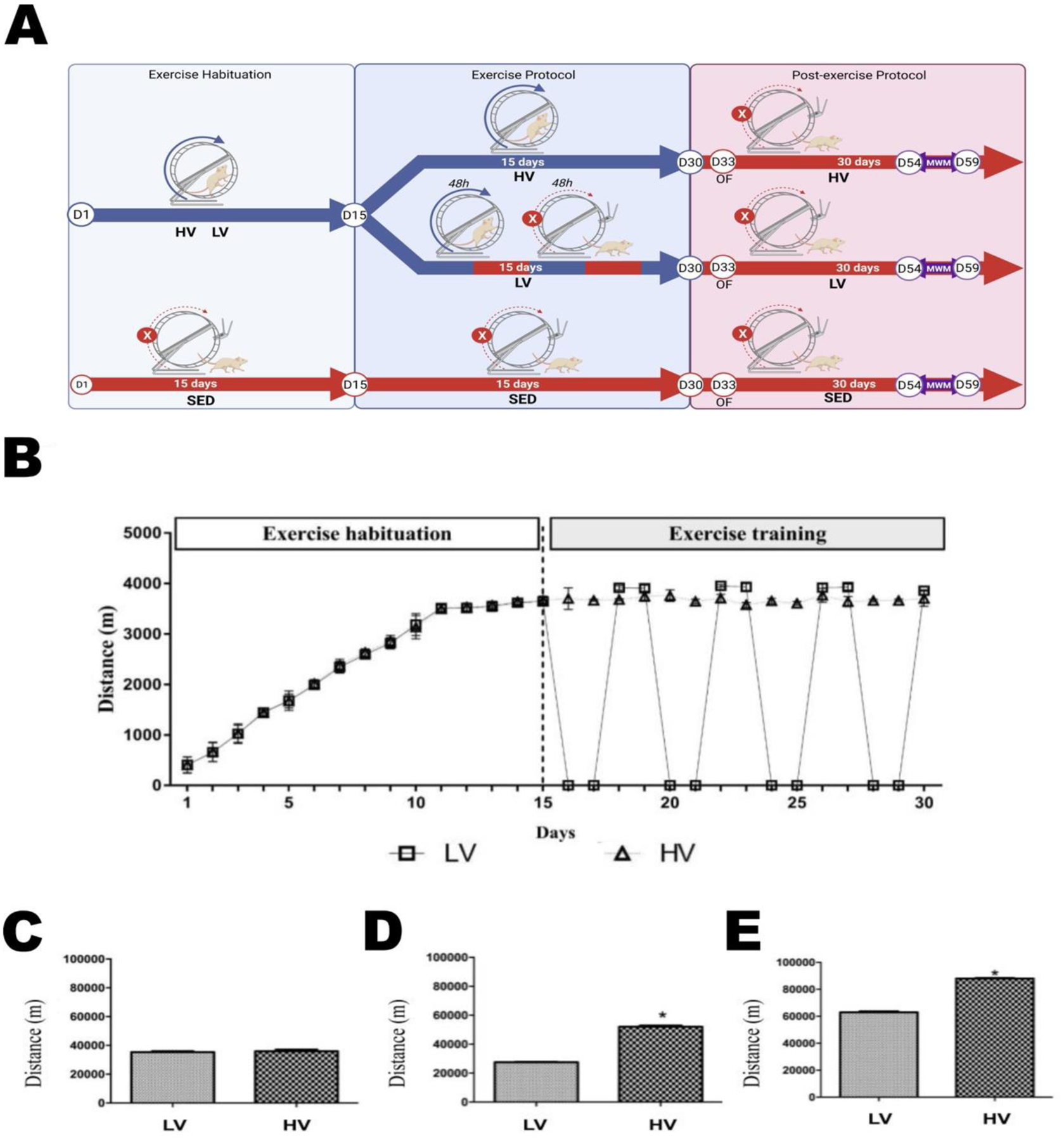
Experimental design (A). Initial 15 days of free voluntary exercise habituation in the running wheel assembled in the home cage. Additional 15 days of free voluntary exercise in the running wheel (HV, higher volume; running wheel unlocked) or intermittent access to the exercise running wheel (LV, lower volume; alternating two days with wheel locked or unlocked). Sedentary mice had the running wheel always locked. Profile of the distance ran by mice during thirty-days habituation and training exercise (B). Distance ran by mice only in the habituation phase (C). Distance ran by mice from LV- and HV groups only in the training phase (D); and the cumulative distance ran (habituation plus training) by LV and HV groups after thirty-days exercise (E). The values represent mean ± S.D. (4 mouse/per cage; n=12 per group). For the same type of exercise stimulus, more frequent engagement presents comparatively higher volume of training compared to less frequent engagement * Indicates statistical differences between HV and LV groups (p < 0.05).
The evaluation of the locomotor profile post-exercise protocol using an open field (OF) test showed that neither SED, LV, or HV groups showed significant differences in distance traveled per minute (A), total distance traveled (B), total mobile time (C), total immobile time (D), and clockwise and anticlockwise rotations (CW and ACW, respectively). Also, exploratory profiles in the center and periphery zones of the OF arena were not significantly different between SED, LV, and HV groups (E) (Supplementary Figure 1) (One-way ANOVA, or two-way ANOVA with repeated measures, p > 0.05).
3.2. Lower and higher volumes of physical exercise increased hippocampal neurotrophic signaling
We next analyzed the protein content in the hippocampal tissue to characterize the concept of exercise-induced neurotrophic brain reserves. LV and HV exercise significantly increased the hippocampal phosphorylation of CREB at Ser133 (pCREBSer133/CREB) and pro-BDNF/BDNF levels relative to sedentary mice (p < 0.001; Figure 2A and B, respectively). It could be expected that a positive modulation on pCREBSer133/CREB and pro-BDNF/BDNF axis mediated by physical exercise may also benefit mitochondrial bioenergetic function.
Figure 2. Preconditioning with lower and higher volumes of physical exercise increases neurotrophic signaling.
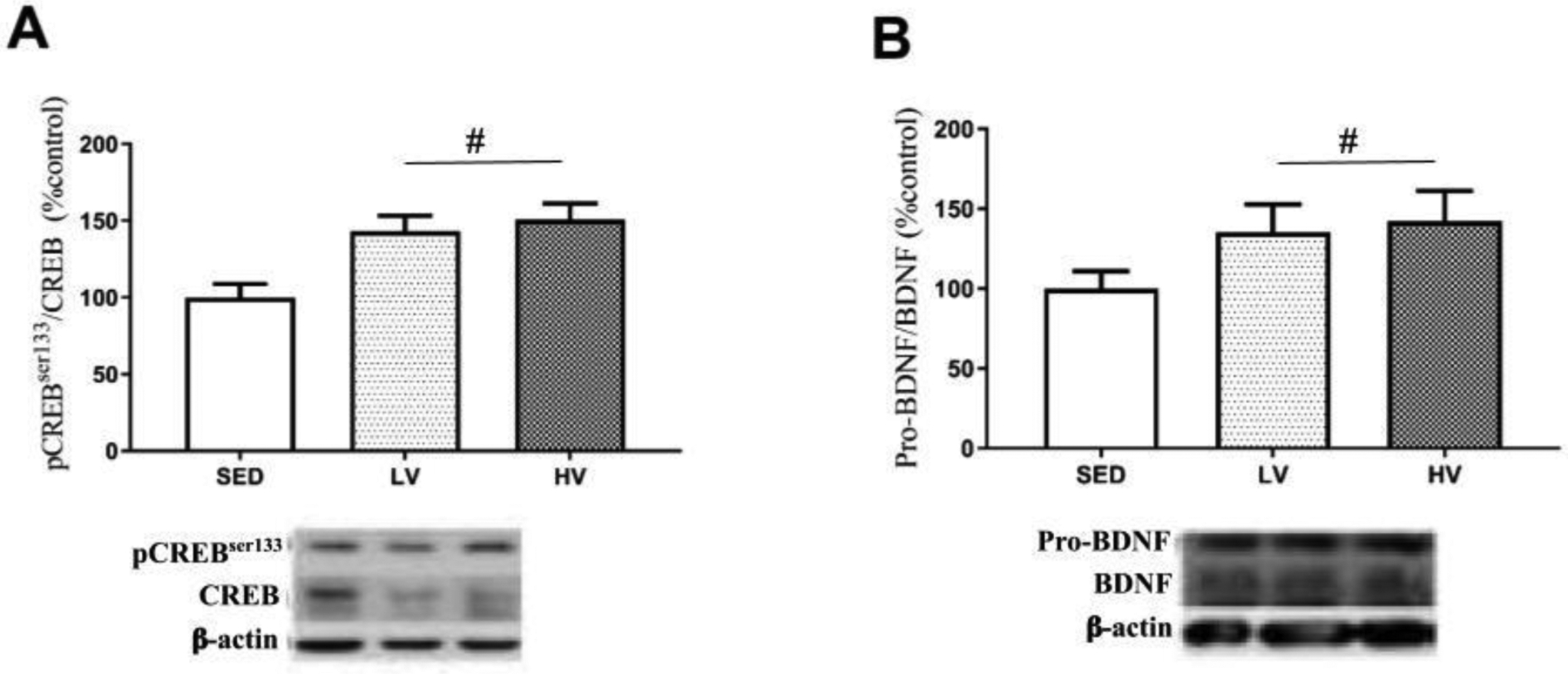
Thirty days after finishing the exercise protocol (LV and HV) the hippocampal levels of trophic proteins belonging to pCREBSer133/CREB and pro-BDNF/BDNF axis were increased relative to sedentary mice (n=8 per group). # Denotes significant statistical difference from sedentary group (SED) (p < 0.05). Abbreviations: LV, lower exercise volume; HV, higher volume.
3.3. Mitochondrial bioenergetic responses to lower and higher volumes of physical exercise
The OCR by mitochondria from the ipsilateral hippocampus is shown in Figure 3A. The maximal electron transport capacity (ETS) was significantly increased in LV and HV groups, thus suggesting that physical exercise—regardless of volume—favors an effective flux of electrons through the mitochondrial oxidative complexes (SED vs LV p < 0.013 and SED vs HV p < 0.001; Figure 3B). Leak respiration was characterized by the addition of metabolic substrates (PMGS) but not ADP. There was decreased O2 consumption in exercised mice (SED vs LV, p < 0.0175; SED vs HV, p < 0.00788; HV vs LV, p < 0.0365; Figure 3C). The OxPhos capacity increased in the exercised groups (SED vs LV, and SED vs HV, p < 0.01; Figure 3D). The biochemical coupling efficiency was significantly increased in LV and HV groups versus SED (SED vs LV p < 0.0269, SED vs HV p < 0.001). It was also higher in HV vs LV (p < 0.0112) (Figure 3E). There were no significant differences between groups in the capacity of mitochondrial membranes to generate and dissipate the electrochemical potential (Figure 3F). The mitochondrial H2O2 production was stimulated by metabolic substrates and ADP; complex V inhibitor (oligomycin) similarly decreases in the exercised groups regardless of preconditioning volume (p < 0.01; Figure 3G).
Figure 3. Prior engagement in lower- and higher volumes of physical exercise increase the mitochondrial bioenergetic capacity in hippocampus.
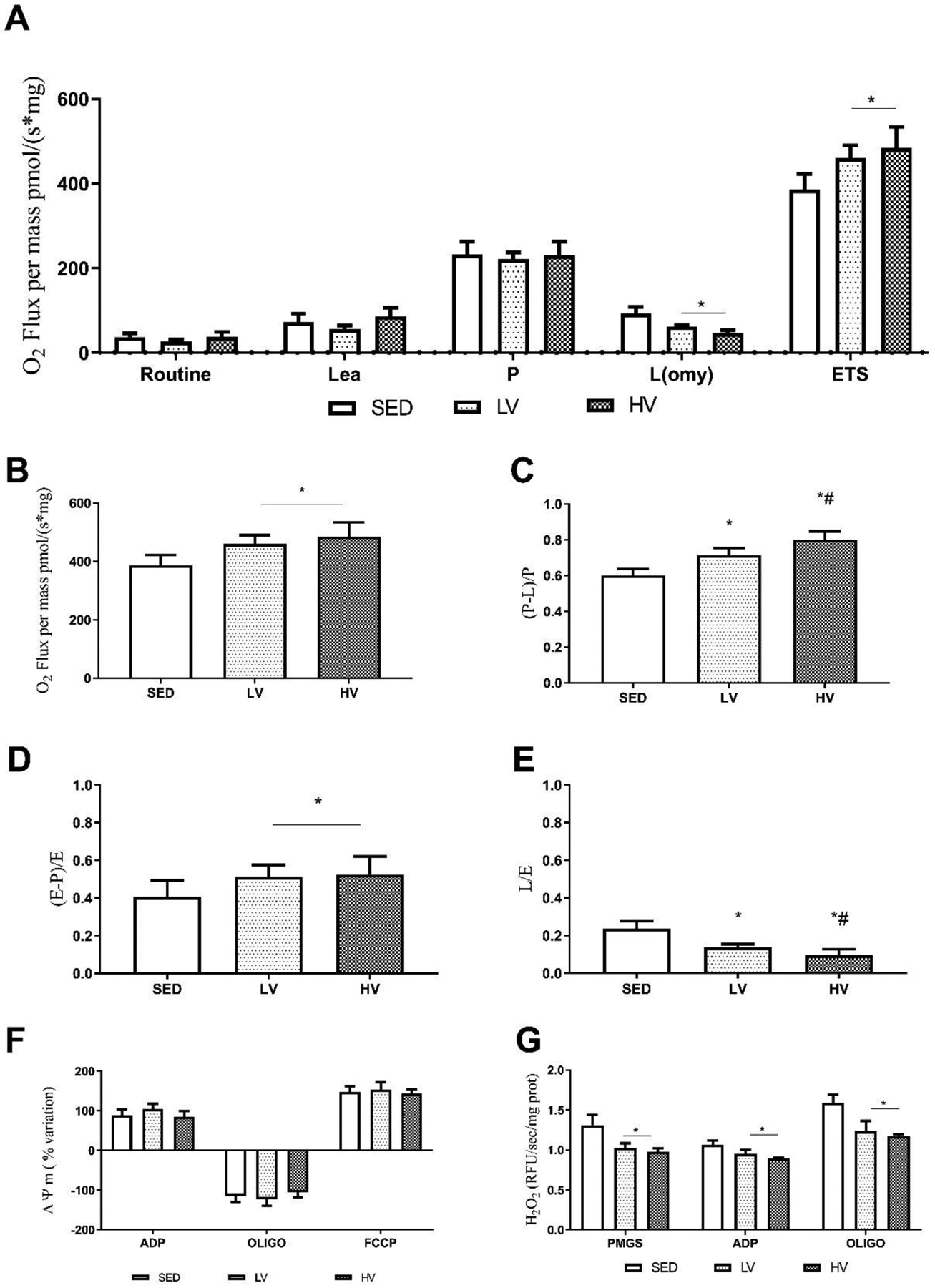
The oxygen consumption rate in hippocampal homogenates during the real-time respirometry protocol (A). Higher and lower exercise volumes (HV and LV, respectively) increased oxygen consumption rate in the maximal electron transport system (ETS) (B). Mitochondrial OxPhos capacity (P), and biochemical coupling efficiency (C, and D respectively) increased in the exercised groups (HV and LV). The sequential addition of metabolic substrates (pyruvate + malate + glutamate + succinate) decreased leak respiration (L) in exercised groups (HV and LV) (E). The formation and dissipation of mitochondrial membrane potential (ΔΨ) in energized states was similar between groups (F); H2O2 production decreased in HV and LV groups (G) (n=8). * Denotes significant difference from sedentary group (SED); # indicates significant difference compared to SED and LV groups (p < 0.05). Abbreviations: LV, lower exercise volume; HV, higher volume; SED, sedentary; OxPhos, oxidative phosphorylation; H2O2, hydrogen peroxide.
3.4. Lower and higher volumes of exercise decrease mortality after severe traumatic brain injury and does not affect the locomotor and exploratory activity
Mice were submitted to TBI after thirty days of preconditioning with lower and higher volumes of exercise (see experimental timeline Figure 4A). We increased the sample size to 17 animals per group because the head injury CCI model has 40 to 50% mortality in our experience. We found a high mortality rate in the SEDTBI group (41.18%) compared to LVTBI (16.67%) and HVTBI (23.08%) groups (Figure 4B). The number of deaths between LVTBI and HVTBI groups was not different (p < 0.486). Based on this mortality rate, the sample size remained as follows: SEDTBI (n=10), LV TBI (n=14), and HVTBI (n=13).
Figure 4. Lower and higher volumes of physical exercise preconditioning decrease mortality and do not affect locomotor activity after severe traumatic brain injury.
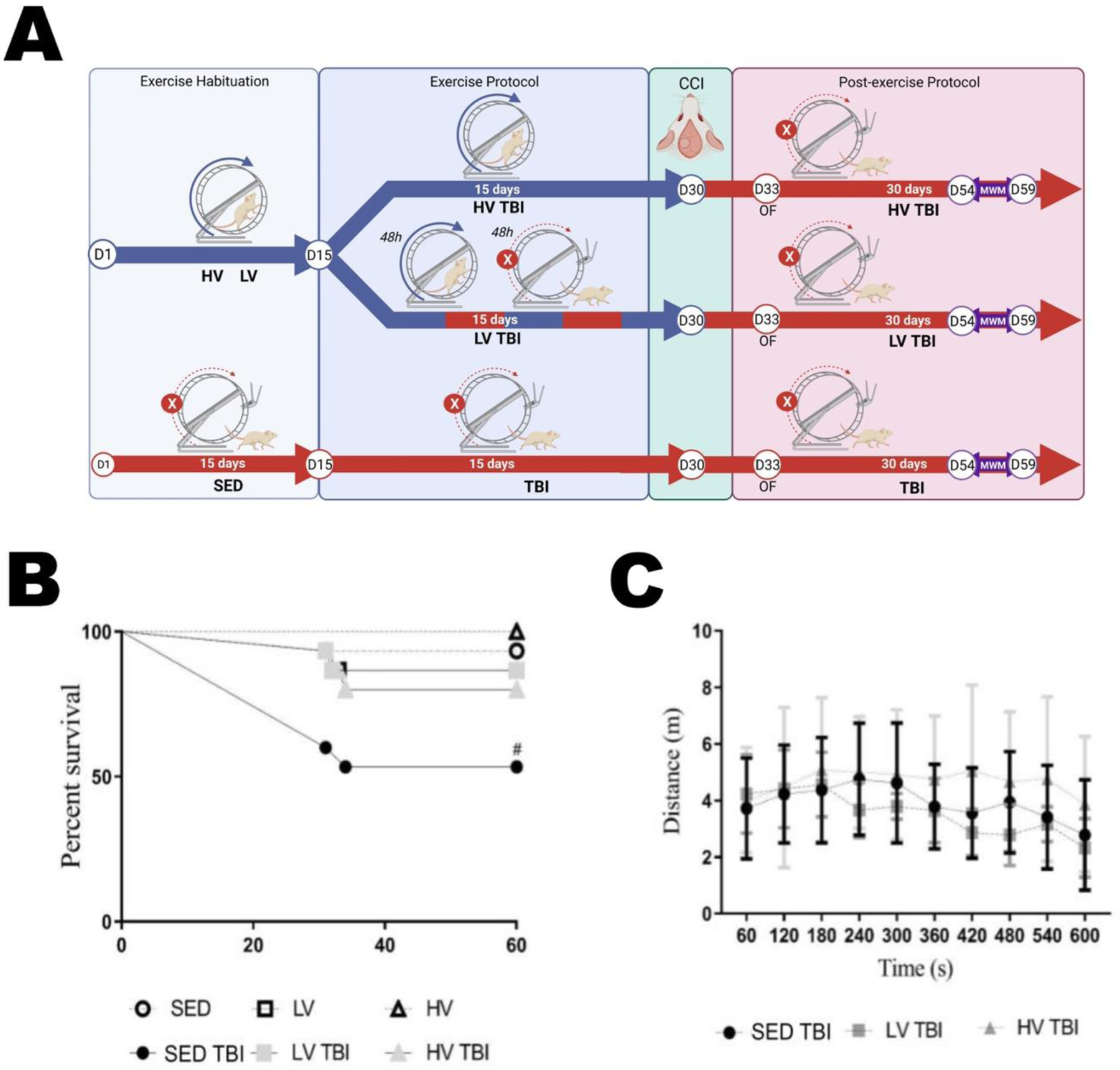
Mice were submitted to thirty days LV and HV exercise protocols plus TBI shortly after resulting in LVTBI, HVTBI and SEDTBI groups (A). The percent mortality after a head injury was 41.18% for SEDTBI, 16.67% LVTBI, and 23.08% HVTBI (B). After TBI the sample size was: SEDTBI (n=10), LVTBI (n=14), and HVTBI (n=13). The locomotor activity assessed by the distance traveled per minute in the open field arena was not different between groups (C). # Denotes statistically significant difference compared to other groups. Abbreviations: LV, lower exercise volume; HV, higher volume; SED, sedentary; TBI, traumatic brain injury.
Considering that after thirty days voluntary running wheel exercise did not alter the locomotor and exploratory activity of LV and HV groups, in this setting of experiment we next sought to investigate whether a severe TBI inflicted soon after the thirty-day exercise protocol (Figure 4A) might change this behavioral phenotype. Three days after TBI there were no significant differences in the distance traveled (m) between groups across 10 min evaluation in the open field test (OF) (Figure 4C) implying no detectable locomotor deficits. Additional locomotor parameters like total distance traveled (A), total mobile time (B), total immobile time (C), clockwise and anticlockwise rotations (E), and the exploratory profile in the center and periphery of OF arena (D) did not show significant differences between SEDTBI, LVTBI and HVTBI groups (Supplementary Figure 2).
3.5. Preconditioning with lower- and higher volumes of physical exercise preserves neurotrophic and neuroenergetic support up to thirty days after severe TBI
After TBI, the mice remained for thirty days without access to the running wheel. The brain was then dissected, and hippocampal tissue was collected for analysis. We found a significant increase in hippocampal levels of pCREBSer133/CREB and pro-BDNF/BDNF in exercised TBI groups (LVTBI and HVTBI) relative to SEDTBI (n=7 per group, p < 0.01 and p < 0.001; Figure 5A and B, respectively).
Figure 5. Preconditioning with different volumes of exercise sustain neurotrophic and neuroenergetic brain reserves thirty days after severe traumatic brain injury.
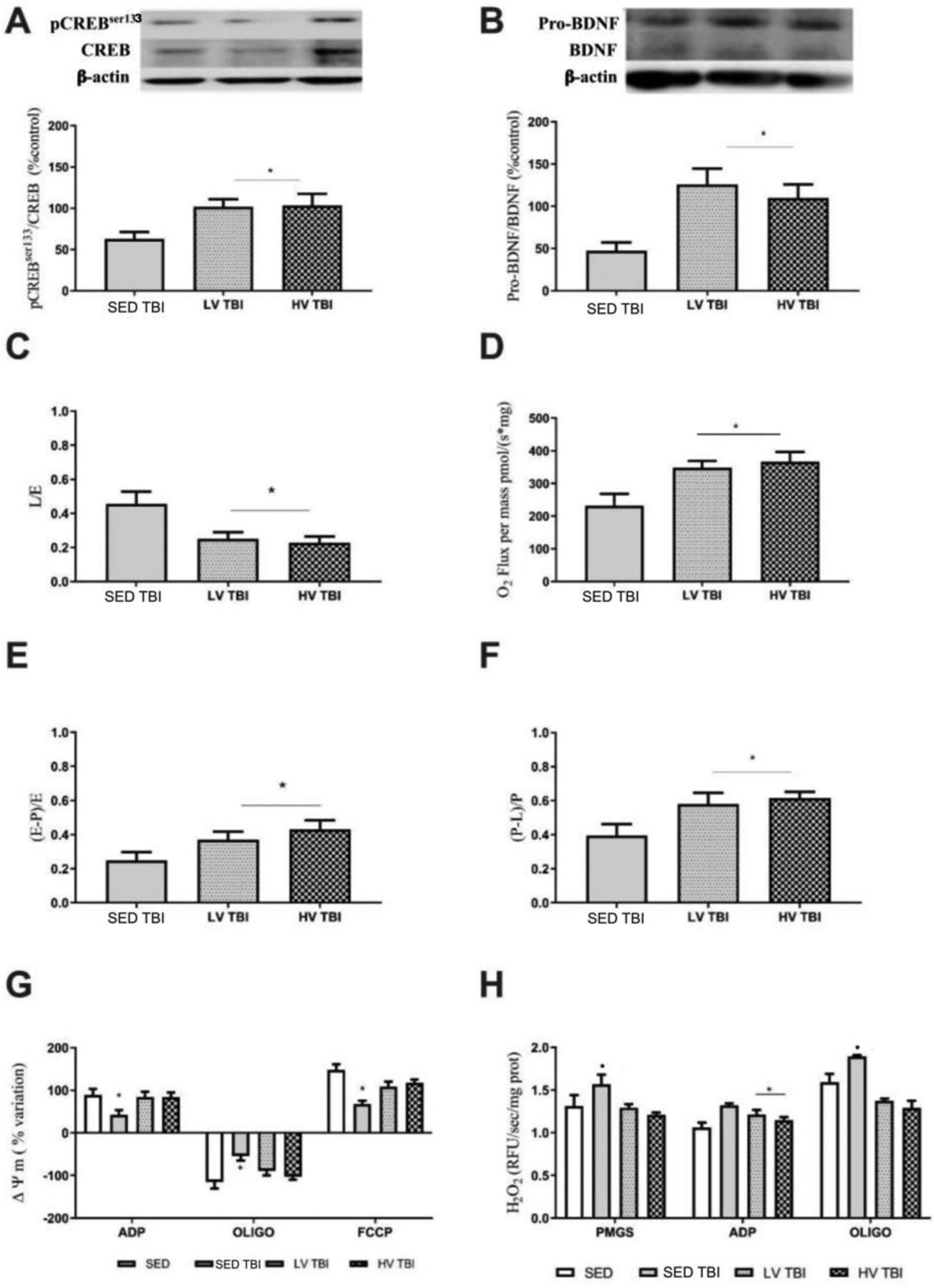
Engagement in lower and higher exercise volumes (LV and HV, respectively) prior to TBI sustained the hippocampal levels of pCREBSer133/CREB (A) and pro-BDNF/BDNF (B). The mitochondrial bioenergetic endpoints including leak respiration (C), maximal ETS capacity (D), biochemical coupling efficiency (E) and the OxPhos capacity (F) was significantly improved in LVTBI and HVTBI relative to SEDTBI group. Mitochondrial membrane potential (ΔΨm) and mitochondrial H2O2 production was improved in LVTBI and HVTBI groups (G and H, respectively). Pointed lines in A, B, C, D, and F represent the values of SED group (n=8 per group). * Denotes difference from SEDTBI group when p < 0.05). Abbreviations: LV, lower exercise volume; HV, higher volume; SED, sedentary; OxPhos, oxidative phosphorylation.
The oxygen consumption linked to maximal ETS capacity was increased in LVTBI and HVTBI groups relative to SEDTBI (p < 0.001, Figure 5C). In the leak respiration, both exercised TBI groups had decreased oxygen consumption versus the SEDTBI group (p < 0.01; Figure 5D). The OxPhos capacity was significantly increased in exercised TBI groups compared to SEDTBI (SEDTBI vs LVTBI p < 0.0251 and SEDTBI vs HVTBI p < 0.0133; Figure 5E). Similarly, the biochemical coupling efficiency was significantly different in exercised TBI groups relative to SED TBI (SEDTBI vs LVTBI and SEDTBI vs HVTBI; both comparisons p < 0.01; Figure 5F).
While the SEDTBI group showed altered formation and dissipation of mitochondrial membrane potential, LVTBI and HVTBI groups sustained this membrane-linked function at the level of the uninjured SED group. There were no significant differences in the capacity of the mitochondrial membrane in generating and dissipating the electrochemical potential (ADP state p < 0.0149, oligomycin state p < 0.0274, and FCCP state p < 0.0312, n=7 per group; Figure 5G).
The mitochondrial H2O2 production stimulated by metabolic substrates (PMGS), ADP, and by complex V inhibitor (oligomycin) increased in the SED TBI group relative to LVTBI and HVTBI groups (PMGS state p < 0.01, ADP state p < 0.0475, and oligomycin state p < 0.001; Figure 5H). The LVTBI and HVTBI groups had H2O2 production similar to SED uninjured mice (n=7 per group; Figure 5H).
3.6. Lower- and higher exercise volumes strengthen the functional associations between neurotrophic and neuroenergetic variables
The correlogram shows the magnitude of associations between molecular and metabolic endpoints modulated by the effect of exercise and TBI (Figure 6). Mice submitted to lower- and higher volumes of exercise increased the magnitude of positive (+1) and negative (−1) associations between variables compared with sedentary mice (Figure 6A, B, and C).
Figure 6. Pairwise correlation of neurotrophic and mitochondrial metabolic variables.
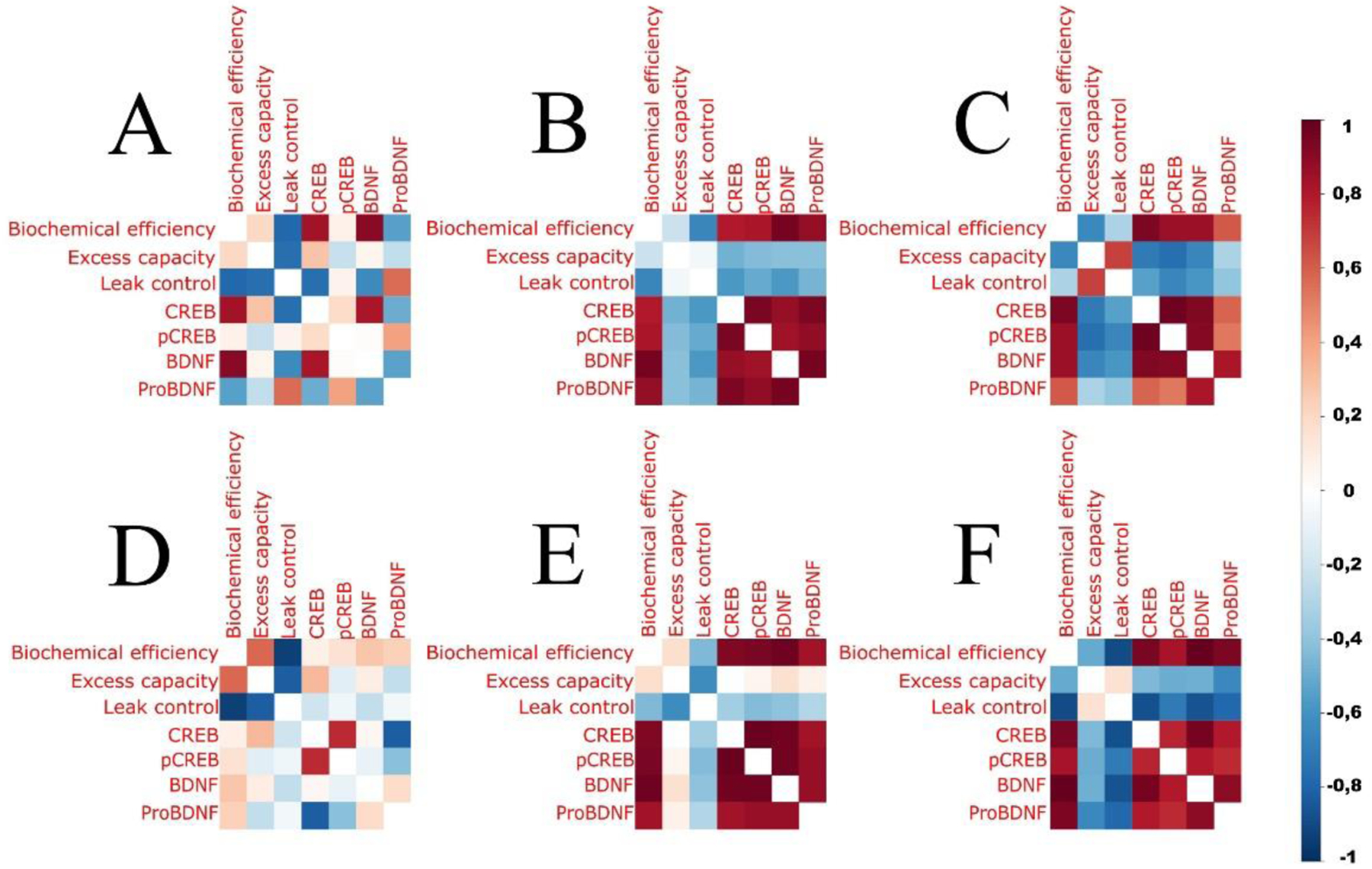
The correlation profiles between neurotrophic and neuroenergetic variables in sedentary mice (A) and in mice submitted to preconditioning with lower (B) and higher (C) volumes of exercise. Traumatic brain injury induced changes in the associations between variables in sedentary mice (D), and both lower (E) and higher (F) exercise volumes preconditioning sustained the associations between neurotrophic and neuroenergetic variables in mice submitted to head impact at level similar to exercise alone (B and C) (n=8 per group).
The associations between variables present in sedentary mice were disrupted in SED TBI (SED vs SEDTBI; Figure 6A vs Figure 6D). Mice submitted to LVTBI and HVTBI sustained the magnitude of correlations in a profile similar to exercise alone (LVTBI vs LV; Figure 6E vs Figure 6B) and (HVTBI vs HV; Figure 6F vs Figure 6C). Also, the correlation profile in SEDTBI is different from LVTBI and HVTBI (Figure 6D vs 6E and 6F, respectively). In general, these findings demonstrate that exercise improves neurotrophic and bioenergetic functional connections, as well as exercise, attenuates the loss of these connections after TBI.
3.7. Exercise preconditioning attenuates long-term spatial memory deficits after TBI
All groups equally performed the flag test in the Morris water maze indicating no apparent visual deficits induced by a TBI (Figure 7A). In the training days of the Morris water maze, as an indicator of memory acquisition, the TBI groups spent more time searching the hidden platform, particularly throughout training days 1 and 4. In contrast, mice preconditioned with LV and HV exercise showed no significant learning deficits (Figure 7B).
Figure 7. Effects of lower- and higher physical exercise volumes and severe traumatic brain injury on spatial memory.
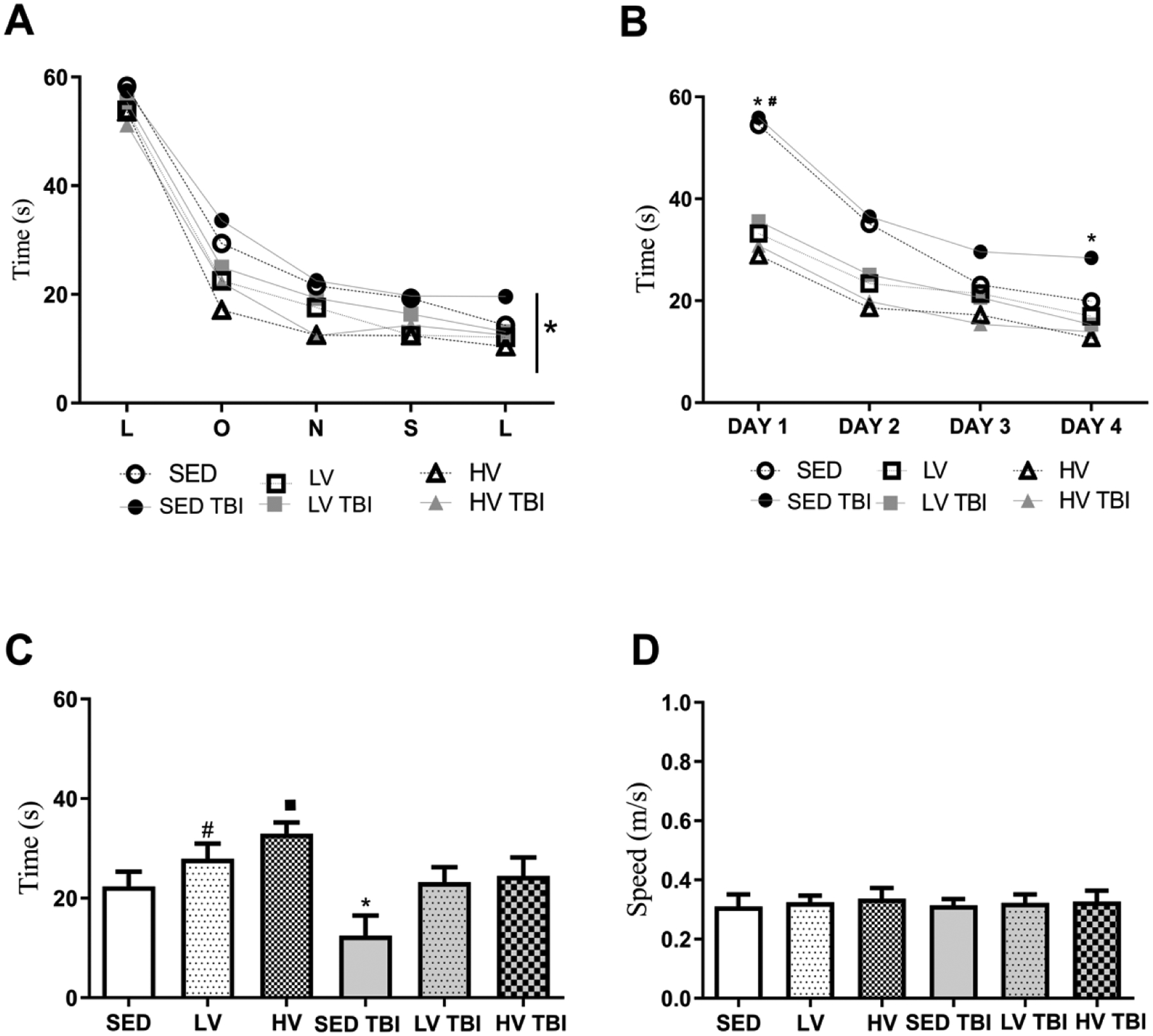
All groups learned to find the platform in the flag test (A). In the acquisition phase of the Morris water maze task (MWM), the SEDTBI group spent more time searching for the hidden platform than other groups (B). In the test phase without the platform, both exercised groups (LV and HV) spent more time in the quadrant zone than SED groups but the memory performance of HV group was significantly more pronounced than LV. SEDTBI displayed decrease in the time spent in the quadrant zone but mice submitted to exercise preconditioning (LVTBI and HVTBI) showed attenuation of this memory deficit (C). The swimming speed was similar between groups (D) (n=8 per group). # Denotes statistical difference from SED. # Denotes statistical difference from SED and LV groups. * Denotes significant difference from other groups. Abbreviations: LV, lower exercise volume; HV, higher volume; SED, sedentary; OxPhos, oxidative phosphorylation; TBI, traumatic brain injury.
In the probe trial, as an indicator of memory retention, mice engaged in HV of exercise remained in the target zone longer to SED and LV groups (SED, p < 0.0001; LV, p = 0.0139). LV also showed increased time in the target zone relative to SED (SED vs LV; p < 0.005) (Figure 7C).
We also showed that the SEDTBI group spent less time in the target quadrant relative to LVTBI and HVTBI groups (p < 0.0001 for both). LVTBI and HVTBI groups showed no significant differences relative to SED, LV, and HV groups (Figure 7C). No differences in average speed were observed between groups (Figure 7D). Taken together, these results confirm the beneficial effects of exercise on the spatial learning and memory fitness and highlight its prophylactic importance against neurofunctional deficits after severe TBI.
4. Discussion
Here, we found that preconditioning with lower and higher volumes of voluntary physical exercise increases neurotrophic and bioenergetic brain reserves, which were associated with lower mortality acutely after injury, and attenuation of long-term memory deficits after severe TBI.
The pathophysiological mechanisms of TBI involve the upregulation of neurodegenerative pathways and the downregulation of neuroprotective and adaptive responses. This balance can be influenced by both innate, and acquired processes mediated by factors like active lifestyle (Raefsky and Mattson, 2017). Indeed, exercise intervention promotes cellular and molecular adaptive responses that are often proposed as therapy after TBI (Archer et al., 2012; Cunnane et al., 2020; Fogelman and Zafonte, 2012; Itoh et al., 2011a, 2011b). The present results further demonstrate the prophylactic potential of exercise-induced brain adaptations for increased neuronal resiliency to acute damage (Fiorin et al., 2016; Zhao et al., 2015). This resiliency likely contributes to reducing mortality in the first hour after severe TBI, in which the primary damage mechanisms like the rupture of cell membranes, axonal damage, and glutamate-induced excitotoxicity are more prominent (Corrigan et al., 2017). Actually, in a clinical study, we demonstrated that higher cerebrospinal fluid glutamate levels at ICU admission predicted brain death within 3 days after severe TBI, whereas patients with lower glutamate levels survived (Stefani et al., 2017). It is known that exercise-mediated synaptic adaptations, and increased astrocyte proliferation and glutamate-uptake, which could remove extracellular glutamate excess and counteracts the primary excitotoxic mechanisms, ultimately decreasing acute mortality (Dietrich et al., 2005; Li et al., 2021, 2005). However, this assumption was not addressed in this study and is currently speculative.
Although countless numbers of pre-clinical TBI models have allowed to recognize pathophysiological mechanisms and identify potential therapeutic targets, no therapies demonstrating high efficacy in pre-clinical models have advanced toward clinical treatments that improve the survival rates and long-term neurological deficits due to TBI (Smith et al., 2021). Accordingly, a clinical review about the protocols for management of severe TBI and its supporting body of evidence over the past 20 years, pointed out for only three positive recommendations (classes 1 and 2), which actually indicates a limited therapeutic arsenal to treat patients underwent this life-threatening pathology (Volovici et al., 2019). The intrinsic limitations of preclinical studies and poor translatable evidences provided by randomized clinical trials performed with small sample size and single-center contribute to the persisting high rates of mortality and lifelong unfavorable outcome after severe TBI (Bragge et al., 2016) but also highlights the importance of building bridges between preclinical and clinical studies to avoid conceptual mismatches, and thus translate findings into benefits for patients (Smith et al., 2021). In the context of this work, we showed an elevated mortality (40%) acutely after CCI in non-exercised mice, which seems to replicate the unfavorable neurological outcome found in severe TBI patients (Glasgow coma scale ≤ 8) in the first hours of hospital admission (Strogulski et al., 2022). Further, routine assessments of righting reflex time acutely post-surgery (Kline et al., 2010), and modified neurological severity score 24h post-injury (6-point scale, assessing reflexes and motor function) (Carteri et al., 2019) confirmed the severity of neurological deficits of TBI groups (data not shown). Though most CCI models use parameters that induce relatively low mortality, the animals typically are awake and ambulating shortly after injury and display relatively minor motor dysfunction. These outcomes do not reflect the circumstances in severe TBI in humans – a disconnect of modeling that has been highlighted in a recent consensus publication on shortcomings of current TBI models (Smith et al., 2021). Accordingly, we adapted the CCI model to mimic severe TBI in humans, who display pronounced neurological dysfunction after the injury with a very high mortality. Notably, mortality rates above our study was already reported using CCI parameters of 4.5 m/s with a deformation 1.5 mm below the dura (Miao et al., 2015). The other most common rodent TBI model, fluid percussion, also presents relatively high mortality (over 20%) even at levels where the surviving animals do not display severe symptoms. While the increased mortality reported here is a consequence of approximating our preclinical model with acute clinical settings, it is important to state that mortality reduction is often not accompanied by more patients experiencing favorable functional outcomes, but rather by an increased number of survivors in a vegetative or severely disabled state (Menon and Maas, 2015; Nichol et al., 2015). Despite the high mortality, our study tries to overcome preclinical limitations and bring into the light this dichotomy, and additionally offers a platform for studying mechanisms and therapeutic targets linked to survival and favorable functional outcomes after severe TBI. Moreover, rather than treatment we highlighted how preventive physical exercise, as proposed by many vascular, cardiac and metabolic disorders, could shield against mechanisms leading to death and long-term consequences of severe TBI.
Given this, we next explored whether lower and higher exercise volumes (LV and HV) modulate the classical components behind synaptic plasticity and adaptive responses to challenges, i.e., neurotrophic and bioenergetic support. We found that the pCREBSer133-proBDNF-BDNF axis in the hippocampus was upregulated regardless of the volume of exercise. Remarkably, such adaptations were detected thirty days after the interruption of exercise, which highlights volume (total running distance in a voluntary running wheel) as a target variable that promotes and sustains neurotrophic support over the long-term.
Previous work showed that BDNF levels correlate with running distance in the voluntary running wheel and mediate the benefits of brain exercise to the brain (Cotman and Berchtold, 2002). Accordingly, the inhibition of the TRkB receptor blocked the benefits of voluntary exercise on CREB levels, neuroplasticity, and cognitive function thus confirming such functional interplay (Vaynman et al., 2004, 2003). Although exercise-induced benefits for the brain are associated with BDNF levels/signaling, there remains debate in the field regarding which exercise protocol is most effective. A previous study showed that a low-intensity exercise using a treadmill increased TRkB-BDNF-CREB and improved spatial memory, whereas high-intensity exercise suppressed neurotrophic benefits and impaired spatial memory (Wu et al., 2020). Sedentary overweight men submitted to moderate-intensity continuous training and high-intensity interval training displayed similar improvements in blood BDNF levels and cognitive performance (de Lima et al., 2022). Another study showed that high-intensity exercise increased blood BDNF levels and improved the locomotor capabilities of neurologically healthy subjects depending on the Val66Met polymorphism (Helm et al., 2017).
Here, we additionally found that both volumes of exercise avoided the deterioration of pCREBSer133-pro-BDNF-BDNF signaling in the brain after a severe TBI. Of note, a similar 4 weeks running wheel exercise protocol led to an acute (24 h) activation of the BDNF-CREB expression in the cortex after TBI, and a concomitant attenuation of apoptotic markers linked to mitochondrial cytochrome c release and consequent caspase activation (Zhao et al., 2015). While these authors did not investigate increments in BNDF-CREB beyond 24 h, they reported long-term (between 14 to 28 days post-injury) neuroprotective effects over classic secondary alterations linked to TBI, including impaired in the spatial and recognition memory processing, cytoskeleton destabilization, and non-trophic microglial phenotypes (Zhao et al., 2015). Also, a prolonged (6 weeks) swim training period before TBI led to an increase in the hippocampal BDNF levels at 24 h and 15 days post-injury, and preserved recognition memory (Fiorin et al., 2016). On the 15th day after exercise and TBI, the BDNF level was approximately 100% above exercised sham group and approximately 150% above the sedentary TBI group. Moreover, hippocampal BDNF level in the exercise and TBI group was higher at the 15th day relative to 24 h post-injury, ultimately suggesting potentiating effect over time. The findings of the present study further indicate that our exercise preconditioning protocol may sustain hippocampal neurotrophic reserves for up to thirty days, as revealed by the increased pCREBSer133-CREB-proBDNF-BDNF level. Although, clinical and preclinical studies have demonstrated that BDNF remains upregulated throughout several weeks of exercise engagement (Berchtold et al., 2005; Russo-Neustadt et al., 1999; Szuhany and Otto, 2020) doubts remains regarding the kinetics of this neurotrophin level/expression after an exercise training protocol cease. Particularly, our study design does not allow us to further elucidate this relevant concern, which can be stated here as a limitation. Nevertheless, a study with healthy adults showed a positive correlation between increased hippocampal volume and serum BDNF level after one-year of moderate aerobic exercise training, but only hippocampal volume correlated with improved memory function. The authors suggest that morphological adaptations including cell proliferation or increased dendritic branching and vascularization might explain the benefits on memory performance parameters (de la Rosa et al., 2019; Erickson et al., 2011; Levine et al., 1995; von Bohlen und Halbach and von Bohlen und Halbach, 2018). Further, five-week moderate aerobic exercise training caused sustained elevation of serum BDNF levels at rest (basal 10.3 vs rest 16.8 pg/mL) in healthy men (Zoladz et al., 2008). These results emphasize that physical exercise actually mounts and subsequently sustains BDNF-induced adaptations through a vast array of components (Berchtold et al., 2005, 2002; Liu and Nusslock, 2018).
In fact, exercise-induced activation of the pCREBSer133-pro-BDNF-BDNF pathway mediates adaptive mitochondrial responses that may improve bioenergetic function (Mattson, 2012; Raefsky and Mattson, 2017). These adaptations likely reflect the clearance of dysfunctional mitochondria, increased metabolic enzyme activity, and protein composition of complexes (Huang et al., 2019; Kinni et al., 2011). Of note, the PGC1α levels (effector of biogenesis and mitochondrial efficiency) seen here were not modified by exercise or TBI after thirty days of intervention (data not shown) (Mattson, 2012; Raefsky and Mattson, 2017). Nonetheless, we found that improved mitochondrial bioenergetic function induced by LV and HV exercises reflect an increased capacity of electron transference, respiratory capacity coupled with ATP synthesis (see Figure 3 B, C, D, E), and decreased H2O2 production stimulated or not by metabolic substrates (Figure 3G). Our real-time respirometry approach confirms a previous report showing that exercise induced a simultaneous up-regulation of proteins involved in energy metabolism and neuroplasticity (Ding et al., 2006). In contrast to our results, prior work with treadmill training did not affect mitochondrial respiration linked to complex I, complex II, and FoF1 ATP synthase in the brain of adult mice implying no gain in bioenergetic capacity (Gusdon et al., 2017). Despite controversies regarding exercise protocols, most studies show that exercise improves brain mitochondrial complex function, increases FoF1 ATP-synthase activity, increases control of reactive oxygen species production, and provides neuroprotection and health benefits. These data suggest that exercise exerts an integral regulation on bioenergetic machinery ultimately leading to the synchronic activity of oxidative metabolism, which may counteract the mechanism of neurodegeneration (Cheng et al., 2010; Mattson, 2012; Mattson and Arumugam, 2018; Raefsky and Mattson, 2017).
It is widely recognized that TBI also exacerbates ROS production, which implies mitochondrial dysfunction at the level of ΔΨm, electron transference, and respiratory complexes (Yonutas et al., 2016). We found that both volumes of exercise preconditioning sustained the BCE score relative to sedentary TBI animals, thus implying a high efficiency of mitochondrial metabolic machinery toward ATP synthesis (Nedel et al., 2021). Both exercise volumes attenuated mitochondrial proton leak and H2O2 production after TBI. Prior work showed increased antioxidant defenses (Nrf2, SOD) induced by exercise preconditioning, thus corroborating a prophylactic role against TBI-induced oxidative damage (Fiorin et al., 2016; Hill et al., 2017; Ji et al., 2012b, 2012a). Further, voluntary exercise preconditioning attenuated the upregulation of Bid and Puma, proteolytic proteins involved in the permeabilization of the mitochondrial membrane after TBI (Zhao et al., 2015). In agreement with this published data, we found that prior exercise engagement avoided the disruption of ΔΨm (formation and dissipation) caused by TBI, thus providing a functional indication of preserved mitochondrial membrane integrity—this is also corroborated by a decrease in H2O2 production and sustained BCE. Values of mitochondrial-derived respiratory endpoints including leak, OxPhos, and electron transference capacity uphold the benefits of both volumes of exercise in sustained various components of mitochondrial bioenergetic function after TBI.
Although both volumes of exercise enhance neurotrophic and bioenergetic brain reserves, these adaptations underscore the importance of avoiding unfavorable neurological outcomes associated with TBI, i.e., memory impairment (Brown et al., 2011). A significant number of TBI patients may experience persistent neuropsychiatric symptoms. Based on a translational perspective, our protocol focuses on how preventive physical exercise engagement interplays with long-term memory deficits associated with TBI. Particularly, impaired memory processing after TBI is attributed to a combination of selective hippocampal and axonal injury associated with deleterious secondary biochemical cascades (Thompson et al., 2005). Remarkably, a TBI in previously exercised mice did not impair the daily performance in the learning phase of the Morris water maze. Specifically, exercised mice spent more time in the target quadrant looking for the removed platform, which indicates memory consolidation of the previous training. This suggests that LV and HV mediate cognitive reserves likely through the pCREBSer133-pro-BDNF-BDNF pathway and mitochondrial bioenergetic efficiency. While these findings may encompass translational relevance to TBI it should be noticed that only male animals were used, and, therefore, any conclusions in females require further experimental confirmation. Also, due to technological limitations, we were not able to phenotype the individual activity of each 4 mice in the running wheel, and this may also be stated as a limitation of this study. Nevertheless, relative to our previous experience and internal controls using the same system, we found a similar distance ran during thirty-days of voluntary exercise in the running wheel (de Carvalho et al., 2017; Dietrich et al., 2005; Muller et al., 2012, 2011; Portela et al., 2017).
5. Conclusion
In summary, preconditioning with lower and higher exercise volumes similarly promotes long-lasting BDNF and mitochondrial bioenergetic reserves that appear associated with preserved memory function after a severe TBI. These results highlight a link between lifestyle and brain reserves against TBI.
Supplementary Material
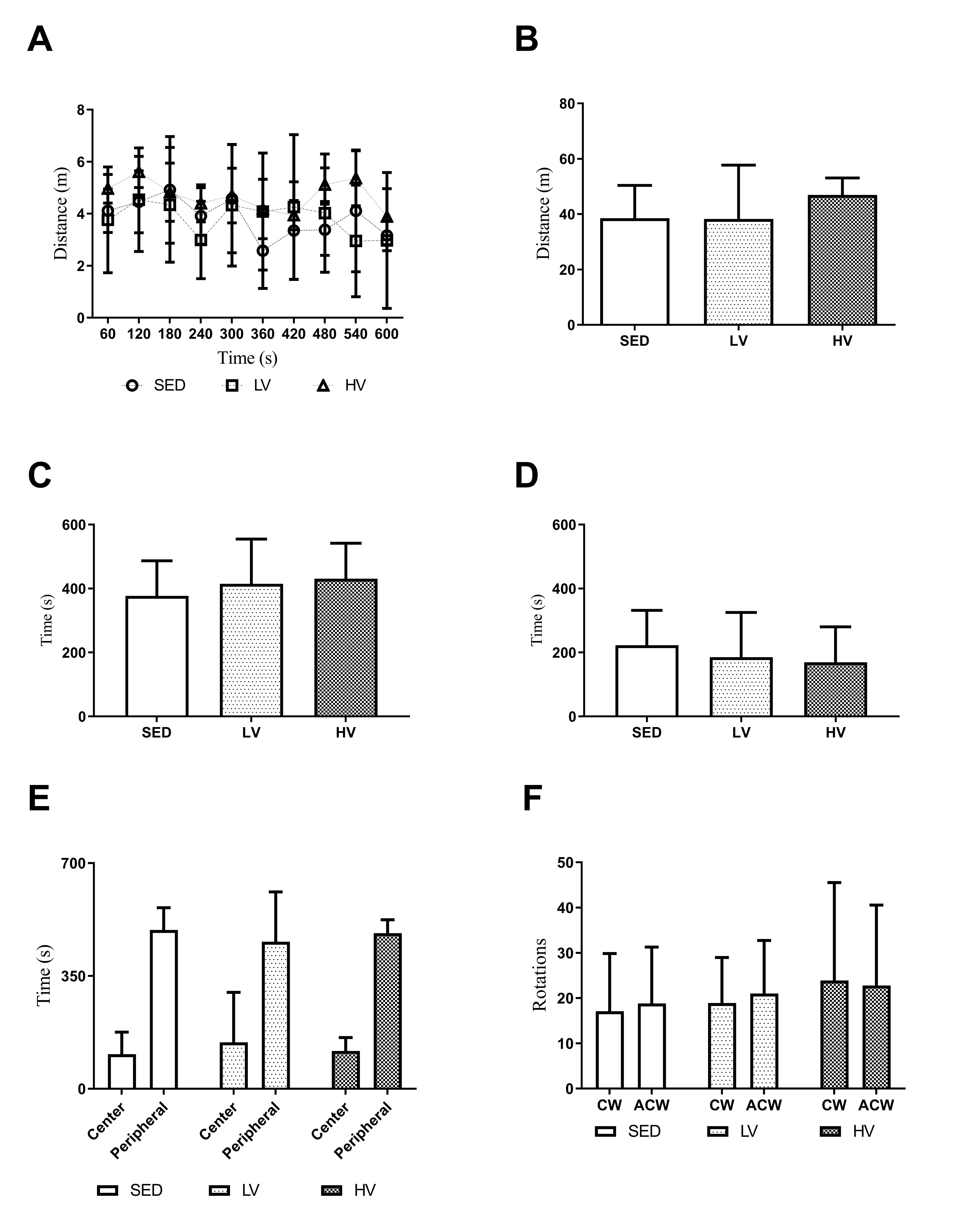
Supplementary Figure 1. Open field test parameters after thirty-days exercise. Mice were submitted to SED (sedentary; running wheel always locked), LV (lower volume; alternating two days with wheel locked or unlocked), and HV exercise (HV, higher volume; running wheel unlocked). Parameters representing the locomotor activity; Distance travelled per minute in the open field arena in 10 min evaluation (A); Total distance ran by mice (B); mobile time (C), total immobile time (D) and clockwise and anticlockwise rotations (CW and ACW, respectively) (F). Also, exploratory profile in the center and periphery zones of the open field arena (E) (n=10 per group). There were no significant statistical differences between SED, LV and HV groups in locomotor and exploratory parameters (p > 0.05).
{kind=link}
Supplementary Figure 2. Open field test parameters following thirty days of exercise preconditioning and subsequent traumatic brain injury. Mice were submitted to SEDTBI (sedentary plus TBI; running wheel always locked), LVTBI (lower volume plus TBI; alternating two days with wheel locked or unlocked), and HVTBI (HV, higher volume plus TBI; running wheel unlocked). Parameters representing locomotor activity; Total distance traveled in the open field arena in 10 min (A); Total mobile time (B); Total immobile time (C), clockwise and anticlockwise rotations (E). Exploratory profile in the center and periphery of the open field arena (D). For all evaluations, there were no significant statistical differences between groups (p > 0.05).
{kind=link}
Highlights.
Lower and higher volume of exercise built up hippocampal brain reserves
pCREBSer133-CREB-proBDNF-BDNF and bioenergetics increased regardless volume of exercise
TBI impaired neurotrophic and neuroenergetic support and induced cognitive deficits
Neurotrophic and neuroenergetic brain reserves attenuated spatial memory deficits
Acknowledgments
The authors thank to Mrs. Candace Gantt and Mr. Doug Markgraf, former TBI patients from the Hospital of the University of Pennsylvania (HUP) whose life inspired this work. The authors also would like to thank the Center for Brain Injury and Repair, University of Pennsylvania (CBIR-UPENN) for the technical and scientific support, and to Fundação de Pesquisa do Estado do Rio Grande do Sul - FAPERGS - for supporting international cooperation.
Funding
This work was supported by the Brazilian Agencies/Programs FAPERGS #1010267, FAPERGS/PPSUS#17/2551-0001, FAPERGS/PRONEX#16/2551-0000499-4, CNPq Program Science without Borders #4011645/2012-6, and CNPq INNT #5465346/2014-6, Programa de Internacionalização de Ciência FAPERGS/CAPES #19/25510000717-5. This research was made possible with support from National Institutes of Health grants R01NS092398 (DHS), R01NS038104 (DHS), R01NS094003 (DHS), R01EB021293 (DHS), U54NS115322 (WS and DHS), as well as support from Paul G. Allen Family Foundation (DHS).
Abbreviations
- TBI
Traumatic brain injury
- CCI
Controlled cortical impact
- LV
lower exercise volume
- HV
higher exercise volume
- SED
sedentary
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author disclosure statement
The authors disclose no competing financial and personal relationships with people or organizations that could inappropriately influence the results of this work.
Declaration of Competing Interest
None.
Availability of data
The data of this work will be made available to researchers who provide a methodologically sound proposal for analyses to achieve aims in the approved proposal, immediately following article publication. Please address the proposals directly to roskaportela@gmail.com.
References
- Andersen ML, Lopes De Souza R, Sebben A, Augusta M, Rodrigues C, Machado R, Aurélio M, Bomfim D, Paes De Almeida J, Pinto N, Ribeiro V, Júnior R, 2016. Normativas do CONCEA para Produção, Manutenção ou Utilização de Animais em Atividades de Ensino ou Pesquisa Científica, 3rd ed. [Google Scholar]
- Archer T, Svensson K, Alricsson M, 2012. Physical exercise ameliorates deficits induced by traumatic brain injury. Acta Neurol Scand 125, 293–302. 10.1111/J.1600-0404.2011.01638.X [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Chinn G, Chou M, Kesslak JP, Cotman CW, 2005. Exercise primes a molecular memory for brain-derived neurotrophic factor protein induction in the rat hippocampus. Neuroscience 133, 853–861. 10.1016/J.NEUROSCIENCE.2005.03.026 [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Patrick Kesslak J, Cotman CW, 2002. Hippocampal brain-derived neurotrophic factor gene regulation by exercise and the medial septum. J Neurosci Res 68, 511–521. 10.1002/JNR.10256 [DOI] [PubMed] [Google Scholar]
- Blaha GR, Raghupathi R, Saatman KE, McIntosh TK, 2000. Brain-derived neurotrophic factor administration after traumatic brain injury in the rat does not protect against behavioral or histological deficits. Neuroscience 99, 483–493. 10.1016/S0306-4522(00)00214-1 [DOI] [PubMed] [Google Scholar]
- Blennow K, Hardy J, Zetterberg H, 2012. The neuropathology and neurobiology of traumatic brain injury. Neuron 76, 886–899. 10.1016/J.NEURON.2012.11.021 [DOI] [PubMed] [Google Scholar]
- Böhmer AE, Oses JP, Schmidt AP, Perón CS, Krebs CL, Oppitz PP, D’Avila TT, Souza DO, Portela LV, Stefani MA, 2011. Neuron-specific enolase, S100B, and glial fibrillary acidic protein levels as outcome predictors in patients with severe traumatic brain injury. Neurosurgery 68, 1624–1630. 10.1227/NEU.0B013E318214A81F [DOI] [PubMed] [Google Scholar]
- Bragge P, Synnot A, Maas AI, Menon DK, Cooper DJ, Rosenfeld J. v., Gruen RL, 2016. A State-of-the-Science Overview of Randomized Controlled Trials Evaluating Acute Management of Moderate-to-Severe Traumatic Brain Injury. J Neurotrauma 33, 1461–1478. 10.1089/NEU.2015.4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AW, Moessner AM, Mandrekar J, Diehl NN, Leibson CL, Malec JF, 2011. A survey of very-long-term outcomes after traumatic brain injury among members of a population-based incident cohort. J Neurotrauma 28, 167–176. 10.1089/NEU.2010.1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carteri RB, Kopczynski A, Rodolphi MS, Strogulski NR, Sartor M, Feldmann M, de Bastiani MA, Duval Wannmacher CM, de Franceschi ID, Hansel G, Smith DH, Portela LV, 2019. Testosterone Administration after Traumatic Brain Injury Reduces Mitochondrial Dysfunction and Neurodegeneration. J Neurotrauma 36, 2246–2259. 10.1089/NEU.2018.6266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Hou Y, Mattson MP, 2010. Mitochondria and neuroplasticity. ASN Neuro 2, 243–256. 10.1042/AN20100019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng ST, 2016. Cognitive Reserve and the Prevention of Dementia: the Role of Physical and Cognitive Activities. Current Psychiatry Reports 2016 18:9 18, 1–12. 10.1007/S11920-016-0721-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte V, Raghupathi R, Watson DJ, Fujimoto S, Royo NC, Marklund N, Stocchetti N, McIntosh TK, 2008. TrkB gene transfer does not alter hippocampal neuronal loss and cognitive deficits following traumatic brain injury in mice. Restor Neurol Neurosci 26, 45–56. [PMC free article] [PubMed] [Google Scholar]
- Correia SC, Carvalho C, Cardoso S, Santos RX, Santos MS, Oliveira CR, Perry G, Zhu X, Smith MA, Moreira PI, 2010a. Mitochondrial preconditioning: a potential neuroprotective strategy. Front Aging Neurosci 2. 10.3389/FNAGI.2010.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA, 2010b. Mitochondria: the missing link between preconditioning and neuroprotection. J Alzheimers Dis 20 Suppl 2. 10.3233/JAD-2010-100669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan F, Arulsamy A, Teng J, Collins-Praino LE, 2017. Pumping the Brakes: Neurotrophic Factors for the Prevention of Cognitive Impairment and Dementia after Traumatic Brain Injury. J Neurotrauma 34, 971–986. 10.1089/NEU.2016.4589 [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC, 2002. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci 25, 295–301. 10.1016/S0166-2236(02)02143-4 [DOI] [PubMed] [Google Scholar]
- Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, Beal MF, Bergersen LH, Brinton RD, de la Monte S, Eckert A, Harvey J, Jeggo R, Jhamandas JH, Kann O, la Cour CM, Martin WF, Mithieux G, Moreira PI, Murphy MP, Nave KA, Nuriel T, Oliet SHR, Saudou F, Mattson MP, Swerdlow RH, Millan MJ, 2020. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov 19, 609–633. 10.1038/S41573-020-0072-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida CER, de Sousa Filho JL, Dourado JC, Gontijo PAM, Dellaretti MA, Costa BS, 2016. Traumatic Brain Injury Epidemiology in Brazil. World Neurosurg 87, 540–547. 10.1016/J.WNEU.2015.10.020 [DOI] [PubMed] [Google Scholar]
- de Carvalho AK, da Silva S, Serafini E, de Souza DR, Farias HR, de Bem Silveira G, Silveira PCL, de Souza CT, Portela LV, Muller AP, 2017. Prior Exercise Training Prevent Hyperglycemia in STZ Mice by Increasing Hepatic Glycogen and Mitochondrial Function on Skeletal Muscle. J Cell Biochem 118, 678–685. 10.1002/JCB.25658 [DOI] [PubMed] [Google Scholar]
- de la Rosa A, Solana E, Corpas R, Bartrés-Faz D, Pallàs M, Vina J, Sanfeliu C, Gomez-Cabrera MC, 2019. Long-term exercise training improves memory in middle-aged men and modulates peripheral levels of BDNF and Cathepsin B. Sci Rep 9. 10.1038/S41598-019-40040-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima NS, de Sousa RAL, Amorim FT, Gripp F, Diniz e Magalhães CO, Henrique Pinto S, Peixoto MFD, Monteiro-Junior RS, Bourbeau K, Cassilhas RC, 2022. Moderate-intensity continuous training and high-intensity interval training improve cognition, and BDNF levels of middle-aged overweight men. Metab Brain Dis 37, 463–471. 10.1007/S11011-021-00859-5 [DOI] [PubMed] [Google Scholar]
- Dietrich MO, Mantese CE, Porciuncula LO, Ghisleni G, Vinade L, Souza DO, Portela L. v., 2005. Exercise affects glutamate receptors in postsynaptic densities from cortical mice brain. Brain Res 1065, 20–25. 10.1016/J.BRAINRES.2005.09.038 [DOI] [PubMed] [Google Scholar]
- Ding Q, Vaynman S, Souda P, Whitelegge JP, Gomez-Pinilla F, 2006. Exercise affects energy metabolism and neural plasticity-related proteins in the hippocampus as revealed by proteomic analysis. Eur J Neurosci 24, 1265–1276. 10.1111/J.1460-9568.2006.05026.X [DOI] [PubMed] [Google Scholar]
- Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, Kim JS, Heo S, Alves H, White SM, Wojcicki TR, Mailey E, Vieira VJ, Martin SA, Pence BD, Woods JA, McAuley E, Kramer AF, 2011. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A 108, 3017–3022. 10.1073/PNAS.1015950108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME, 1997. CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031–1047. 10.1016/S0896-6273(00)80395-5 [DOI] [PubMed] [Google Scholar]
- Fiorin FDS, Ferreira APDO, Ribeiro LR, Silva LFA, de Castro MRT, da Silva LRH, da Silveira MEP, Zemolin APP, Dobrachinski F, de Oliveira SM, Franco JL, Soares FA, Furian AF, Oliveira MS, Fighera MR, Royes LFF, 2016. The Impact of Previous Physical Training on Redox Signaling after Traumatic Brain Injury in Rats: A Behavioral and Neurochemical Approach. J Neurotrauma 33, 1317–1330. 10.1089/NEU.2015.4068 [DOI] [PubMed] [Google Scholar]
- Fogelman D, Zafonte R, 2012. Exercise to enhance neurocognitive function after traumatic brain injury. PM R 4, 908–913. 10.1016/J.PMRJ.2012.09.028 [DOI] [PubMed] [Google Scholar]
- Garber CE, Blissmer B, Deschenes MR, Franklin BA, Lamonte MJ, Lee IM, Nieman DC, Swain DP, 2011. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc 43, 1334–1359. 10.1249/MSS.0B013E318213FEFB [DOI] [PubMed] [Google Scholar]
- Gnaiger E, 2014. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. Bioenergetics Communications 2014, 2–2. [Google Scholar]
- Gozt AK, Hellewell SC, Thorne J, Thomas E, Buhagiar F, Markovic S, van Houselt A, Ring A, Arendts G, Smedley B, van Schalkwyk S, Brooks P, Iliff J, Celenza A, Mukherjee A, Xu D, Robinson S, Honeybul S, Cowen G, Licari M, Bynevelt M, Pestell CF, Fatovich D, Fitzgerald M, 2021. Predicting outcome following mild traumatic brain injury: protocol for the longitudinal, prospective, observational Concussion Recovery (CREST) cohort study. BMJ Open 11. 10.1136/BMJOPEN-2020-046460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Gómez-Pinilla F, Hovda DA, 2007. Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J Neurotrauma 24, 1161–1171. 10.1089/NEU.2006.0255 [DOI] [PubMed] [Google Scholar]
- Gusdon AM, Callio J, Distefano G, O’Doherty RM, Goodpaster BH, Coen PM, Chu CT, 2017. Exercise increases mitochondrial complex I activity and DRP1 expression in the brains of aged mice. Exp Gerontol 90, 1–13. 10.1016/J.EXGER.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm EE, Matt KS, Kirschner KF, Pohlig RT, Kohl D, Reisman DS, 2017. The influence of high intensity exercise and the Val66Met polymorphism on circulating BDNF and locomotor learning. Neurobiol Learn Mem 144, 77–85. 10.1016/J.NLM.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanides J, Hong YT, Trivedi M, Outtrim J, Aigbirhio F, Nestor PJ, Guilfoyle M, Winzeck S, Newcombe VFJ, Das T, Correia MM, Carpenter KLH, Hutchinson PJA, Gupta AK, Fryer TD, Pickard JD, Menon DK, Coles JP, 2021. Metabolic derangements are associated with impaired glucose delivery following traumatic brain injury. Brain 144, 3492–3504. 10.1093/BRAIN/AWAB255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RL, Singh IN, Wang JA, Hall ED, 2017. Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem Int 111, 45–56. 10.1016/J.NEUINT.2017.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wang X, Zhu Y, Li Z, Zhu YT, Wu JC, Qin ZH, Xiang M, Lin F, 2019. Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci Ther 25, 796–807. 10.1111/CNS.13114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Imano M, Nishida S, Tsubaki M, Hashimoto S, Ito A, Satou T, 2011a. Exercise increases neural stem cell proliferation surrounding the area of damage following rat traumatic brain injury. J Neural Transm (Vienna) 118, 193–202. 10.1007/S00702-010-0495-3 [DOI] [PubMed] [Google Scholar]
- Itoh T, Imano M, Nishida S, Tsubaki M, Hashimoto S, Ito A, Satou T, 2011b. Exercise inhibits neuronal apoptosis and improves cerebral function following rat traumatic brain injury. J Neural Transm (Vienna) 118, 1263–1272. 10.1007/S00702-011-0629-2 [DOI] [PubMed] [Google Scholar]
- Je HS, Yang F, Ji Y, Nagappan G, Hempstead BL, Lu B, 2012. Role of pro-brain-derived neurotrophic factor (proBDNF) to mature BDNF conversion in activity-dependent competition at developing neuromuscular synapses. Proc Natl Acad Sci U S A 109, 15924–15929. 10.1073/PNAS.1207767109/SUPPL_FILE/SM04.AVI [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Okonkwo DO, Cheng JP, Alexander H, Clark RSB, Kochanek PM, Wipf P, Kagan VE, Bayir H, 2012a. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci 15, 1407–1413. 10.1038/NN.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Tyurina YY, Tang M, Feng W, Stolz DB, Clark RSB, Meaney DF, Kochanek PM, Kagan VE, Bayír H, 2012b. Mitochondrial injury after mechanical stretch of cortical neurons in vitro: biomarkers of apoptosis and selective peroxidation of anionic phospholipids. J Neurotrauma 29, 776–788. 10.1089/NEU.2010.1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2013. Axonal pathology in traumatic brain injury. Exp Neurol 246, 35–43. 10.1016/J.EXPNEUROL.2012.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2012. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol 22, 142–149. 10.1111/J.1750-3639.2011.00513.X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2010. Traumatic brain injury and amyloid-β pathology: a link to Alzheimer’s disease? Nat Rev Neurosci 11, 361–370. 10.1038/NRN2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge RE, Robinson RG, Moser D, Tateno A, Crespo-Facorro B, Arndt S, 2004. Major depression following traumatic brain injury. Arch Gen Psychiatry 61, 42–50. 10.1001/ARCHPSYC.61.1.42 [DOI] [PubMed] [Google Scholar]
- Kinni H, Guo M, Ding JY, Konakondla S, Dornbos D, Tran R, Guthikonda M, Ding Y, 2011. Cerebral metabolism after forced or voluntary physical exercise. Brain Res 1388, 48–55. 10.1016/J.BRAINRES.2011.02.076 [DOI] [PubMed] [Google Scholar]
- Kline AE, McAloon RL, Henderson KA, Bansal UK, Ganti BM, Ahmed RH, Gibbs RB, Sozda CN, 2010. Evaluation of a combined therapeutic regimen of 8-OH-DPAT and environmental enrichment after experimental traumatic brain injury. J Neurotrauma 27, 2021–2032. 10.1089/NEU.2010.1535/ASSET/IMAGES/LARGE/FIGURE7.JPEG [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR, 1995. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci U S A 92, 8074–8077. 10.1073/PNAS.92.17.8074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien R, Furlano J, Nagamatsu L, 2022. Resistance training improves white matter structural connectivity in older adults at-risk for cognitive decline. Alzheimers Dement 18 Suppl 9, e068795. 10.1002/ALZ.068795 [DOI] [Google Scholar]
- Li F, Geng X, Yun HJ, Haddad Y, Chen Y, Ding Y, 2021. Neuroplastic Effect of Exercise Through Astrocytes Activation and Cellular Crosstalk. Aging Dis 12, 1644–1657. 10.14336/AD.2021.0325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ding YH, Rafols JA, Lai Q, McAllister JP, Ding Y, 2005. Increased astrocyte proliferation in rats after running exercise. Neurosci Lett 386, 160–164. 10.1016/J.NEULET.2005.06.009 [DOI] [PubMed] [Google Scholar]
- Liu PZ, Nusslock R, 2018. Exercise-mediated neurogenesis in the hippocampus via BDNF. Front Neurosci 12, 52. 10.3389/FNINS.2018.00052/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machin David., Cheung Y.Bun., Parmar MKB, Parmar MKB, 2006. Survival Analysis: A Practical Approach, 2nd ed. Wiley. [Google Scholar]
- Mattson MP, 2012. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab 16, 706–722. 10.1016/J.CMET.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Arumugam T. v., 2018. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab 27, 1176. 10.1016/J.CMET.2018.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn MJ, Povlishock JT, 2015. Cellular and molecular mechanisms of injury and spontaneous recovery. Handb Clin Neurol 127, 67–87. 10.1016/B978-0-444-52892-6.00005-2 [DOI] [PubMed] [Google Scholar]
- Mckee AC, Daneshvar DH, 2015. The neuropathology of traumatic brain injury. Handb Clin Neurol 127, 45. 10.1016/B978-0-444-52892-6.00004-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuail JA, Dunn AR, Stern Y, Barnes CA, Kempermann G, Rapp PR, Kaczorowski CC, Foster TC, 2021. Cognitive Reserve in Model Systems for Mechanistic Discovery: The Importance of Longitudinal Studies. Front Aging Neurosci 12, 532. 10.3389/FNAGI.2020.607685/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney DF, Morrison B, Bass CD, 2014. The mechanics of traumatic brain injury: a review of what we know and what we need to know for reducing its societal burden. J Biomech Eng 136. 10.1115/1.4026364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney DF, Smith DH, 2011. Biomechanics of Concussion. Clin Sports Med 30, 19. 10.1016/J.CSM.2010.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon DK, Maas AIR, 2015. EPO in traumatic brain injury: Two strikes…but not out? The Lancet 386, 2452–2454. 10.1016/S0140-6736(15)00387-6 [DOI] [PubMed] [Google Scholar]
- Miao W, Bao TH, Han JH, Yin M, Yan Y, Wang WW, Zhu YH, 2015. Voluntary exercise prior to traumatic brain injury alters miRNA expression in the injured mouse cerebral cortex. Brazilian Journal of Medical and Biological Research 48, 433–439. 10.1590/1414-431X20144012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AP, Gnoatto J, Moreira JD, Zimmer ER, Haas CB, Lulhier F, Perry MLS, Souza DO, Torres-Aleman I, Portela L. v., 2011. Exercise increases insulin signaling in the hippocampus: physiological effects and pharmacological impact of intracerebroventricular insulin administration in mice. Hippocampus 21, 1082–1092. 10.1002/HIPO.20822 [DOI] [PubMed] [Google Scholar]
- Muller AP, Haas CB, Camacho-Pereira J, Brochier AW, Gnoatto J, Zimmer ER, de Souza DO, Galina A, Portela LV, 2013. Insulin prevents mitochondrial generation of H2O2 in rat brain. Exp Neurol 247, 66–72. 10.1016/J.EXPNEUROL.2013.03.007 [DOI] [PubMed] [Google Scholar]
- Muller AP, Zimmer ER, Haas CB, Oses JP, Martimbianco De Assis A, Galina A, Souza DO, Portela LV, 2012. Physical exercise exacerbates memory deficits induced by intracerebroventricular STZ but improves insulin regulation of H2O2 production in mice synaptosomes. J Alzheimers Dis 30, 889–898. 10.3233/JAD-2012-112066 [DOI] [PubMed] [Google Scholar]
- National Research Council, 2008. Recognition and Alleviation of Distress in Laboratory Animals, Recognition and Alleviation of Distress in Laboratory Animals. National Academies Press (US). 10.17226/11931 [DOI] [PubMed] [Google Scholar]
- Nedel WL, Kopczynski A, Rodolphi MS, Strogulski NR, de Bastiani M, Montes THM, Abruzzi J, Galina A, Horvath TL, Portela LV, 2021. Mortality of septic shock patients is associated with impaired mitochondrial oxidative coupling efficiency in lymphocytes: a prospective cohort study. Intensive Care Med Exp 9. 10.1186/S40635-021-00404-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol A, French C, Little L, Haddad S, Presneill J, Arabi Y, Bailey M, Cooper DJ, Duranteau J, Huet O, Mak A, McArthur C, Pettilä V, Skrifvars M, Vallance S, Varma D, Wills J, Bellomo R, 2015. Erythropoietin in traumatic brain injury (EPO-TBI): A double-blind randomised controlled trial. The Lancet 386, 2499–2506. 10.1016/S0140-6736(15)00386-4 [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B, 2004. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306, 487–491. 10.1126/SCIENCE.1100135 [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER, 1996. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16, 1137–1145. 10.1016/S0896-6273(00)80140-3 [DOI] [PubMed] [Google Scholar]
- Portela L. v., Brochier AW, Haas CB, de Carvalho AK, Gnoato JA, Zimmer ER, Kalinine E, Pellerin L, Muller AP, 2017. Hyperpalatable Diet and Physical Exercise Modulate the Expression of the Glial Monocarboxylate Transporters MCT1 and 4. Mol Neurobiol 54, 5807–5814. 10.1007/S12035-016-0119-5 [DOI] [PubMed] [Google Scholar]
- Raefsky SM, Mattson MP, 2017. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic Biol Med 102, 203–216. 10.1016/J.FREERADBIOMED.2016.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolphi MS, Kopczynski A, Carteri RB, Sartor M, Fontella FU, Feldmann M, Hansel G, Strogulski NR, Portela L. v., 2021. Glutamate transporter-1 link astrocytes with heightened aggressive behavior induced by steroid abuse in male CF1 mice. Horm Behav 127, 104872. 10.1016/J.YHBEH.2020.104872 [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, Beard RC, Cotman CW, 1999. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology 21, 679–682. 10.1016/S0893-133X(99)00059-7 [DOI] [PubMed] [Google Scholar]
- Shiga T, Ikoma K, Katoh C, Isoyama H, Matsuyama T, Kuge Y, Kageyama H, Kohno T, Terae S, Tamaki N, 2006. Loss of neuronal integrity: a cause of hypometabolism in patients with traumatic brain injury without MRI abnormality in the chronic stage. Eur J Nucl Med Mol Imaging 33, 817–822. 10.1007/S00259-005-0033-Y [DOI] [PubMed] [Google Scholar]
- Smith DH, Kochanek PM, Rosi S, Meyer R, Ferland-Beckham C, Prager EM, Ahlers ST, Crawford F, 2021. Roadmap for Advancing Pre-Clinical Science in Traumatic Brain Injury. https://home.liebertpub.com/neu 38, 3204–3221. 10.1089/NEU.2021.0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, Mcintosh TK, 1995. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 12, 169–178. 10.1089/NEU.1995.12.169 [DOI] [PubMed] [Google Scholar]
- Stefani MA, Modkovski R, Hansel G, Zimmer ER, Kopczynski A, Muller AP, Strogulski NR, Rodolphi MS, Carteri RK, Schmidt AP, Oses JP, Smith DH, Portela L. v., 2017. Elevated glutamate and lactate predict brain death after severe head trauma. Ann Clin Transl Neurol 4, 392–402. 10.1002/ACN3.416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y, 2002. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc 8, 448–60. [PubMed] [Google Scholar]
- Strogulski NR, Stefani MA, Böhmer AE, Hansel G, Rodolphi MS, Kopczynski A, de Oliveira VG, Stefani ET, Portela J. v., Schmidt AP, Oses JP, Smith DH, Portela L. v., 2022. Cerebrospinal fluid purinomics as a biomarker approach to predict outcome after severe traumatic brain injury. J Neurochem 161, 173–186. 10.1111/JNC.15590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuhany KL, Otto MW, 2020. Assessing BDNF as a mediator of the effects of exercise on depression. J Psychiatr Res 123, 114–118. 10.1016/J.JPSYCHIRES.2020.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, Mcintosh TK, 2005. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma 22, 42–75. 10.1089/NEU.2005.22.42 [DOI] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F, 2004. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci 20, 2580–2590. 10.1111/J.1460-9568.2004.03720.X [DOI] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F, 2003. Interplay between brain-derived neurotrophic factor and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience 122, 647–657. 10.1016/j.neuroscience.2003.08.001 [DOI] [PubMed] [Google Scholar]
- Volovici V, Steyerberg EW, Cnossen MC, Haitsma IK, Dirven CMF, Maas AIR, Lingsma HF, 2019. Evolution of Evidence and Guideline Recommendations for the Medical Management of Severe Traumatic Brain Injury. J Neurotrauma 36, 3183–3189. 10.1089/NEU.2019.6474 [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach O, von Bohlen und Halbach V, 2018. BDNF effects on dendritic spine morphology and hippocampal function. Cell Tissue Res 373, 729–741. 10.1007/S00441-017-2782-X [DOI] [PubMed] [Google Scholar]
- Weineck J, 2019. Optimales Training: leistungsphysiologische Trainingslehre unter besonderer Berücksichtigung des Kinder- und Jugendtrainings, 17th ed. [Google Scholar]
- Wu Y, Deng F, Wang J, Liu Y, Zhou W, Qu L, Cheng M, 2020. Intensity-dependent effects of consecutive treadmill exercise on spatial learning and memory through the p-CREB/BDNF/NMDAR signaling in hippocampus. Behavioural brain research 386. 10.1016/J.BBR.2020.112599 [DOI] [PubMed] [Google Scholar]
- Yasmin A, Jokivarsi K, Poutiainen P, Pitkänen A, Gröhn O, Immonen R, 2022. Chronic hypometabolism in striatum and hippocampal network after traumatic brain injury and their relation with memory impairment - [18F]-FDG-PET and MRI 4 months after fluid percussion injury in rat. Brain Res 1788. 10.1016/J.BRAINRES.2022.147934 [DOI] [PubMed] [Google Scholar]
- Yonutas HM, Vekaria HJ, Sullivan PG, 2016. Mitochondrial specific therapeutic targets following brain injury. Brain Res 1640, 77–93. 10.1016/J.BRAINRES.2016.02.007 [DOI] [PubMed] [Google Scholar]
- Zhao J, Qu D, Xi Z, Huan Y, Zhang K, Yu C, Yang D, Kang J, Lin W, Wu S, Wang Y, 2021. Mitochondria transplantation protects traumatic brain injury via promoting neuronal survival and astrocytic BDNF. Transl Res 235, 102–114. 10.1016/J.TRSL.2021.03.017 [DOI] [PubMed] [Google Scholar]
- Zhao Z, Sabirzhanov B, Wu J, Faden AI, Stoica BA, 2015. Voluntary Exercise Preconditioning Activates Multiple Antiapoptotic Mechanisms and Improves Neurological Recovery after Experimental Traumatic Brain Injury. J Neurotrauma 32, 1347. 10.1089/NEU.2014.3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoladz JA, Pilc A, Majerczak J, Grandys M, Zapart-Bukowska J, Duda K, 2008. Endurance training increases plasma brain-derived neurotrophic factor concentration in young healthy men. J Physiol Pharmacol 59 Suppl 7, 119–32. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Open field test parameters after thirty-days exercise. Mice were submitted to SED (sedentary; running wheel always locked), LV (lower volume; alternating two days with wheel locked or unlocked), and HV exercise (HV, higher volume; running wheel unlocked). Parameters representing the locomotor activity; Distance travelled per minute in the open field arena in 10 min evaluation (A); Total distance ran by mice (B); mobile time (C), total immobile time (D) and clockwise and anticlockwise rotations (CW and ACW, respectively) (F). Also, exploratory profile in the center and periphery zones of the open field arena (E) (n=10 per group). There were no significant statistical differences between SED, LV and HV groups in locomotor and exploratory parameters (p > 0.05).
Supplementary Figure 2. Open field test parameters following thirty days of exercise preconditioning and subsequent traumatic brain injury. Mice were submitted to SEDTBI (sedentary plus TBI; running wheel always locked), LVTBI (lower volume plus TBI; alternating two days with wheel locked or unlocked), and HVTBI (HV, higher volume plus TBI; running wheel unlocked). Parameters representing locomotor activity; Total distance traveled in the open field arena in 10 min (A); Total mobile time (B); Total immobile time (C), clockwise and anticlockwise rotations (E). Exploratory profile in the center and periphery of the open field arena (D). For all evaluations, there were no significant statistical differences between groups (p > 0.05).
Data Availability Statement
The data of this work will be made available to researchers who provide a methodologically sound proposal for analyses to achieve aims in the approved proposal, immediately following article publication. Please address the proposals directly to roskaportela@gmail.com.