

Significance
The rapid emergence of multidrug-resistant bacteria is one of the greatest threats to global public health, demanding rapid delivery of new therapies to save patient lives. However, the development and clinical rollout of novel drug classes is a slow process that is unlikely to deliver a timely solution to this crisis. The reengineering of clinically approved antibiotics to evade resistance mechanisms offers a potential near- to short-term solution that takes advantage of established supply chains and clinical success. In this vein, we have designed and synthesized a unique class of shapeshifting vancomycin dimers that evade resistance acquisition, paving the way for future studies into shapeshifting antibiotic drugs to aid in the fight against resistant pathogens — including deadly VRE and MRSA.
Keywords: click chemistry, shapeshifting antibiotics, resistant bacterial infections, vancomycin, flippase
Abstract
The alarming rise in superbugs that are resistant to drugs of last resort, including vancomycin-resistant enterococci and staphylococci, has become a significant global health hazard. Here, we report the click chemistry synthesis of an unprecedented class of shapeshifting vancomycin dimers (SVDs) that display potent activity against bacteria that are resistant to the parent drug, including the ESKAPE pathogens, vancomycin-resistant Enterococcus (VRE), methicillin-resistant Staphylococcus aureus (MRSA), as well as vancomycin-resistant S. aureus (VRSA). The shapeshifting modality of the dimers is powered by a triazole-linked bullvalene core, exploiting the dynamic covalent rearrangements of the fluxional carbon cage and creating ligands with the capacity to inhibit bacterial cell wall biosynthesis. The new shapeshifting antibiotics are not disadvantaged by the common mechanism of vancomycin resistance resulting from the alteration of the C-terminal dipeptide with the corresponding d-Ala-d-Lac depsipeptide. Further, evidence suggests that the shapeshifting ligands destabilize the complex formed between the flippase MurJ and lipid II, implying the potential for a new mode of action for polyvalent glycopeptides. The SVDs show little propensity for acquired resistance by enterococci, suggesting that this new class of shapeshifting antibiotic will display durable antimicrobial activity not prone to rapidly acquired clinical resistance.
Antibiotics are powerful drugs used for fighting life-threatening infections and have transformed both human and animal health (1). However, the threat of intractable antimicrobial resistance — partly a consequence of poor antibiotic stewardship — on health care and global economies has potentially catastrophic implications (2, 3); this situation, in recent years, has been compounded by bacterial resistance emerging faster than new treatment options (4, 5).
Ancient mechanisms of resistance are increasingly accumulating in pathogenic bacteria (6–8); increased drug efflux, protein target mutations, direct deactivation of drug molecules, and even morphological changes to the bacterial cell all drive antibiotic resistance (8–10). Consequently, there is an acute need for renewed vigor and innovative strategies that will lead to new therapies for the treatment of multidrug-resistant (MDR) gram-positive and gram-negative bacterial infections (11, 12). The sustained development and flow of new (13–15) or reengineered (16) antibiotics that overcome the forces of evolution and selection pressures responsible for bacterial resistance are necessary. Through innovation, creative design, and precision synthetic chemistry, antibiotics that are impervious to, or at least less prone to the development of, resistance are within reach (17–24).
In this vein, the glycopeptide antibiotics, including vancomycin — forming a last line of defense in the fight against serious infections — stand out as a class of antibiotic with much scope for structural modification that provides them with additional and multiple synergistic mechanisms of action, as a means to reinvigorate activity against MDR pathogens (25, 26).
Clinical resistance to vancomycin first emerged in enterococci (VanA and VanB, VRE) in 1987 (27), and in Staphylococcus aureus (VRSA) in 2002 (28), caused by the late-stage remodeling of the N-terminus of peptidoglycan precursors from d-Ala-d-Ala to d-Ala-d-Lac (Fig. 1A) (29). This single-point modification to the cell wall precursors reduces vancomycin binding and reduced antimicrobial activity by 1,000-fold (30). The chemical evolution of the privileged core structures of glycopeptides, achieved by means of complex total syntheses or semi-synthetic modification, has proven a profitable strategy for overcoming the molecular basis of resistance (26), with several recently approved drugs reaching the clinic (31).
Fig. 1.

Shapeshifting vancomycin dimers (SVDs). (A) Vancomycin binding to lipid II. (B) Bullvalene, the archetypal shapeshifting molecule. (C) Bullvalene as a polyol sensing platform. (D) Shapeshifting core-tethered vancomycin dimer.
The covalent tethering of glycopeptides, such as vancomycin, to create polyvalent assemblies is also known to achieve improved activity against vancomycin-resistant bacteria (32–40). While the reasons underpinning this phenomenon are not fully understood, for “head-to-tail” (37, 38) and “back-to-back” (41, 42) dimers, the hydrogen bonds at the dimer interface are governed by the same amide units responsible for binding to the terminal d-Ala-d-Ala binding site of vancomycin, albeit through a different network of hydrogen bonds (43). This cooperative interaction results in the dimer having a greater affinity for the d-Ala-d-Ala ligand than the monomer, and the ligand-bound monomer has a higher propensity to dimerize than the free monomer (43).
Nicolaou and coworkers harnessed the power of template-accelerated synthesis to discover highly potent vancomycin dimers (44). Such dynamic combinatorial libraries rely on the high degree of reversibility of the chosen ligation reaction under a given set of conditions, but typically, these systems suffer from long equilibration times and the need for additional reagents both to mediate the reversible reactions and to freeze the equilibrium once adaptation has occurred (45). Dynamic covalent unimolecular chemical systems offer the potential to overcome these limitations yet are a far less common strategy (45, 46). Bullvalene is the archetypal fluxional molecule which, through endless Cope rearrangements, achieves a state of total degeneracy whereby there are no permanent carbon–carbon bonds (Fig. 1B) (47–49). Substituted derivatives represent unimolecular shapeshifting dynamic combinatorial libraries (50). Bode et al. have developed this concept and demonstrated a self-sorting adaptive binding polyol sensor array (Fig. 1C) (51).
However, the chemistry of bullvalene has been limited by difficult and/or lengthy synthetic access. Recently, one of our laboratories has introduced a range of methods that provide easy and modular access to substituted bullvalenes (52). Such derivatives have the potential to act as highly specific sensing molecules that can differentiate structurally similar biomolecules from one another. Furthermore, the vast number of permutations accessed by shapeshifting molecules can be considered self-contained adaptive systems (52, 53) that respond to host–guest interactions — a feature, we posit, that could offer potential in countering the evolutionary forces of drug resistance, particularly MDR bacteria.
Herein, we describe the click chemistry synthesis of a focused library of “back-to-back” vancomycin-fused dimers connected through a fluxional bullvalene core that are circumspect to the resistance mechanisms typically associated with vancomycin-resistant bacterial strains (Fig. 1D).
Results and Discussion
Synthesis.
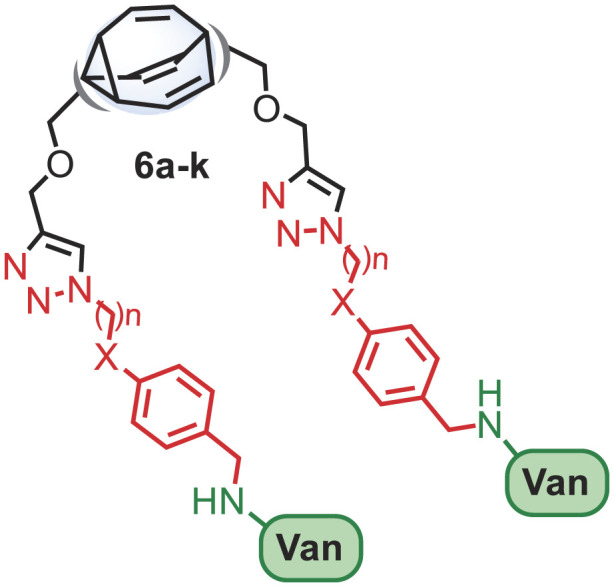
To explore the potential of shapeshifting antibiotics, we elected to focus on symmetrically disubstituted bullvalene derivatives. The design of the SVDs centered on three distinct modules that could be connected using a click chemistry approach (54): i) a shapeshifting bullvalene core, ii) a flexible linker, and iii) a vancomycin warhead connected through the vancosamine unit (Fig. 2A).
Fig. 2.

Synthesis of SVDs and controls. (A) SVD design. (B) Synthesis. (C) Control ligands.
Assembly of the complex SVDs would be achieved using sequential CuAAC (55–57) coupling of the bisacetylene functionalized bullvalene core 3 with the aromatic azides (4a–k), followed by reductive amination of the aldehyde (5a–k) with vancomycin (16). Our synthesis began from bis (methylenehydroxy) bicyclooctatetraene 1 (Fig. 2B), by treatment with propargyl bromide and sodium hydride to afford the bis-propargyl ether 2, which itself was subjected to a photochemical-induced di-π-methane rearrangement to give the bis-propargyl ether bullvalene 3. Next, double CuAAC reaction of 3 with the azides 4a–k gave a selection of aldehyde-containing linkers of varying length in good yields. Finally, reductive amination of the aldehydes 5a–k with vancomycin using sodium cyanoborohydride resulted in complete consumption of starting material (5) as indicated by thin-layer chromatography to give the target SVDs 6a–k as crude mixtures. Purification of the SVDs was achieved using preparative HPLC. Although numerous columns were required in most instances and the pure products were isolated in relatively low yields (8 to 30%), sufficient quantities (>2 mg) of each SVD were obtained to perform biological studies. Furthermore, the synthetic route was amenable to scale-up (for example, 6d was prepared on a 0.23 mmol scale to afford 244 mg of product).
The characterization of the complex SVDs (6a–k) is a significant challenge, not least due to the fluxional nature of their design (refer to SI Appendix for more information). Attempts to characterize the SVDs by NMR spectroscopy proved difficult (refer to SI Appendix, Figs S2–S4). Nevertheless, our structural assignment is supported by several characteristics of our synthesis design, including the resilience of vancomycin to the reductive amination conditions (16), the reliability and predictable outcome of click reactions, and the identification of the expected molecular mass by high-resolution mass spectrometry.
We also probed the dynamics of the shapeshifting ligands using a series of VT-NMR experiments. For bullvalenes with this substitution pattern, there are 15 possible constitutional isomers (52, 58). For bis-propargyl ether 3, the populated isomers A–F were identified, with isomers A and B predominating (a network diagram of isomer interconnections is shown in Fig. 3A, with nodes representing isomers and edges representing transition structures; also refer to SI Appendix). The isomers B, C, and F are chiral and will interconvert between enantiomers. Isomers with 1,2-adjacent substitution will be destabilized and are highlighted in red. The relatively flat isomer distribution suggests that 3 and its derivatives should explore a relatively wide range of dynamic shape characteristics with varying distance and angle constraints. This notion is supported by NMR analysis of the aldehydes 5a–k, revealing spectral features consistent with the isomer distribution of 3. As an exemplar, the room temperature and −60 °C proton spectra of 5f are shown in Fig. 3B and SI Appendix, Fig. S8. The bullvalene core alkenic (He) and aliphatic signals (Hj), as well as the signals proximal to the bullvalene (methylenes Hi, Hf, and triazole proton Hc), all show pronounced broadening at room temperature, which is resolved at −60 °C into complex signal sets. Diagnostic signals in the aliphatic region affirm the major isomers A and B, as well as unidentified minor isomers. Given the consistent isomer distribution within the series, it is possible that the SVDs 6a–k may too reflect this trend, although due to the complexity of the fluxional system and limited stability of the SVDs, a detailed analysis was precluded, and further investigation will be required.
Fig. 3.

Bullvalene isomer distribution. (A) Network analysis of dipropargyl bullvalene 3. (B) Room temperature and −60 °C 1H NMR spectra of linker 5f.
With sufficient quantities (>2 mg) of each of SVDs (6a–k) in hand, we next performed screens against drug-sensitive and -resistant bacterial strains of S. aureus and enterococci.
Antibacterial Activity.
The antibacterial properties of the SVDs (6a–k) and the covalently tethered dimeric controls, 7a–c, were evaluated against drug-sensitive and -resistant strains of S. aureus and Enterococcus, as reported in Table 1A. While all the dimers exhibited some degree of antibacterial activity, of note was the enhanced activity of the dimers against vancomycin-intermediate (VIE) and vancomycin-resistant (VRE) strains, with minimum inhibitory concentration (MIC) values up to 64-fold lower than that of vancomycin. Against VRSA, SVD 6k was the most active with an MIC of 10 µg/mL. To assess whether this improved activity was maintained across various methicillin-resistant S. aureus (MRSA) and VRE strains, SVD 6d (the most active SVD against VSE) was screened against extended panels of MRSA (ATCC® MP-3™) and VRE (ATCC® MP-1™) (Table 1B). While a similar potency against MRSA was observed relative to vancomycin, there was also a consistent increase in potency of SVD 6d, relative to vancomycin, against VRE.
Table 1.
A) Antibacterial activity of vancomycin and the SVDs 6a–k against drug-sensitive and -resistant bacterial strains (N = 3). B) Extended panels against multiple strains of MRSA, VIE, and VRE. MSSA: methicillin-sensitive S. aureus (ATCC® 9144™); MRSA: methicillin-resistant S. aureus (ATCC® BAA-1720™); VSE: vancomycin-sensitive E. faecium (ATCC® BAA-2127™); VIE: vancomycin-intermediate E. gallinarum (ATCC® 49608™); VRE: vancomycin-resistant E. faecium (ATCC® 700221™); VSSA: SH1000; and VRSA: vancomycin-resistant S. aureus (VRS1/HIP11714)
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| A | MIC (µg/mL) | ||||||||
| Compound | X | n | MSSA | MRSA | VSE | VIE | VRE | VSSA | VRSA |
| Vancomycin | – | – | 1–2 | 2 | 1 | 8–16 | 256 | 1.25 | >200 |
| 6a | CH2 | 1 | 4 | 2–4 | 1–2 | 2–4 | 32 | – | – |
| 6b | CH2 | 2 | 8 | 4–8 | 2–4 | 0.5 | 16 | – | – |
| 6c | O | 2 | 8 | 8–16 | 2–4 | 0.25–0.5 | 16 | – | – |
| 6d | O | 3 | 8–16 | 8 | 2 | 1–2 | 16 | – | >20 |
| 6e | O | 4 | 8–16 | 4 | 2–4 | 2 | 32 | 10 | >20 |
| 6f | O | 5 | 16 | 8 | 4 | 2–4 | 16 | 5 | >20 |
| 6g | O | 6 | 8 | 8 | 2–4 | 2 | 32 | – | >20 |
| 6h | O | 7 | 8 | 16–32 | 2–4 | 1 | 4 | – | – |
| 6i | O | 8 | 16 | 16 | 4 | 4 | 16–32 | 20 | >20 |
| 6j | O | 9 | 4–8 | 8 | 2 | 2–4 | 16 | – | – |
| 6k | O | 10 | 4–8 | 8 | 2–4 | 4 | >32 | 5 | 10 |
| B | MIC (µg/mL) | ||||||||
| Compound | X | n | MRSA (10 strains) | VIE (5 strains) | VRE (10 strains) | ||||
| Vancomycin | – | – | 1–4 | 8–16 | >256 | ||||
| 6d | O | 3 | 8–16 | 1–4 | 16–32 | ||||
In Vitro Toxicity.
Vancomycin and SVD 6d were assessed for their cytotoxicity in vitro using the MTT viability assay with the cytotoxic peptide NaD1 employed as a positive control (Fig. 4). At the highest dose tested of vancomycin (twofold greater than the MIC), a significant decrease in the viability of the human embryonic kidney cell line HEK293 was observed, consistent with the reports of nephrotoxicity (59, 60). Vancomycin has previously been associated with acute kidney injury, a result of the larger doses and longer duration of treatment needed to curb the increasing incidence of vancomycin-resistant strains of S. aureus and Enterococcus. Therefore, it was necessary to evaluate whether the modifications and dimerization of vancomycin reduced the toxic effects. At the same concentration as vancomycin, the SVD 6d resulted in no significant decrease in viability for either cell line, indicating that the analog is not cytotoxic to HEK293 and HepG2 (human hepatoma) cell lines, relative to the vehicle control. Further, given the difference in potency between vancomycin and SVD 6d, the highest dose of the dimer tested equates to 10-fold higher than the effective antibacterial dose, and consequently, SVD 6d may provide a wider therapeutic window than vancomycin.
Fig. 4.

In vitro toxicity profile assessed by an MTT viability assay. Viability of HEK293 cells and HepG2 cells treated with 500 μg/mL SVD 6d (blue) relative to 500 μg/mL vancomycin (orange), and 50 μM of the cytotoxic peptide NaD1 (purple), normalized to a 1% (v/v) DMSO vehicle control. Error bars: S.D. (n = 3); ns P > 0.05, *P < 0.05, ***P < 0.001.
In Vivo Infection Model.
To further explore the potential of the SVDs as unique antibiotics, we assessed their efficacy in an in vivo infection model. Specifically, the larvae of Galleria mellonella were used, which is a well-established model to study bacterial virulence and for assessing antibiotic efficacy (Fig. 5) (61, 62). When challenged with vancomycin at 20 mg/kg, a standard clinical dose, G. mellonella larval survival rate was not significantly reduced, with 75% survival at day 7. Similarly, the larvae tolerated treatment with SVD 6d with 70% survival, indicating minimum toxicity in the G. mellonella model. When challenged with VRE without treatment, larval survival was reduced to 10% by day 7. The treatment with vancomycin increased survival rates to only 40%, whereas treatment with SVD 6d retained larval survival at 70%, the same as the SVD treatment alone. This indicates that SVD 6d can successfully treat a VRE infection in the G. mellonella model.
Fig. 5.

In vivo infection model using Galleria mellonella. Tolerance of the G. mellonella larvae to 20 mg/kg SVD 6d (blue dash), 20 mg/kg vancomycin (orange dash), and 1% (v/v) DMSO vehicle control (purple dash), compared to the survival of the larvae following infection with VRE (ATCC® 51575™) (purple solid) and treatment with either 20 mg/kg SVD 6d (blue solid) or 20 mg/kg vancomycin (orange solid) (N = 20).
Resistance Studies.
We envisioned that the incorporation of the fluxional bullvalene core would minimize the development of acquired resistance compared to dimers that lack the intrinsic shapeshifting characteristic. Our hypothesis is that such a dynamic core would favor the most preferred binding interactions on the cell wall, which we posit may confer reduced propensity for resistance relative to the parent drug. To validate this hypothesis, we replaced the bullvalene core of SVD 6d with aryl linkers, ortho 7a, meta 7b, and para 7c substituted (Fig. 2C; see SI Appendix), and assessed the propensity of a drug-sensitive strain of E. faecium (VSE) (ATCC® BAA-2127™) to develop resistance to the three control analogs (7a–c), SVD 6d, and vancomycin (Fig. 6).
Fig. 6.

Propensity of Enterococcus faecium to develop resistance. E. faecium (ATCC® BAA-2127™) was exposed to increasing concentrations of vancomycin, SVD 6d, control 7a, control 7b, and control 7c, up to 4 × MIC. Data are plotted as the fold-change in MIC value relative to a DMSO vehicle control (N = 2).
As anticipated, the serial passaging of VSE in the presence of vancomycin resulted in bacteria with a >32-fold increase in MIC, indicative of the emergence of a vancomycin-resistant strain of Enterococcus. Passaging with the control analogs resulted in bacteria with a 4- to 16-fold increase in MICs, signifying the emergence of early-stage resistance. Contrariwise, passaging with SVD 6d did not invoke significant resistance development in the bacterial strain, with MICs only increasing by twofold. The evidence suggests that the incorporation of a dynamic linker seems to minimize the propensity for bacteria to develop resistance. Furthermore, previous studies have predominantly focused on monomeric vancomycin analogs with modifications at the C-terminus and within the target binding pocket (18, 63, 64). While these monomers demonstrated a significantly reduced propensity for resistance development, our results constitute evidence for time-limited resistance development to tethered vancomycin dimers.
Lipid II Model Binding Assays.
The mechanisms behind the enhanced activity of vancomycin dimers are not fully understood. Ellman and coworkers, using covalent tail-to-tail dimers of vancomycin and the corresponding dimers of damaged vancomycin (the latter unable to bind to Lys-d-Ala-d-Ala or Lys-d-Ala-d-Lac), demonstrated that Lys-d-Ala-d-Lac binding is not required for the high activity of their vancomycin dimers against VRE, suggesting an alternate mode of action involving disruption of the function of proteins critical for VRE cell wall biosynthesis (33, 65, 66).
To explore the mode of binding of the back-to-back SVDs, we employed microscale thermophoresis (MST) binding studies with vancomycin and SVD 6d using the fluorescently labeled tripeptides acetyl-Lys-d-Ala-d-Ala and acetyl-Lys-d-Ala-d-Lac to determine dissociation constants (KD) (Table 2). Vancomycin was found to bind to the acetyl-Lys-d-Ala-d-Ala tripeptide with a KD of 1.0 ± 0.3 μM, which is in agreement with reported KD values for vancomycin binding to model peptides (65). On the contrary, no binding was observed with the acetyl-Lys-d-Ala-d-Lac tripeptide (33), correlating with its poor activity against VRE. The SVD 6d bound to acetyl-Lys-d-Ala-d-Ala tripeptide with a KD of 11 ± 1.9 μM, which is of the order of magnitude reported for other dimeric vancomycin species (33). However, we also observed binding of 6d to the acetyl-Lys-d-Ala-d-Lac, albeit with relatively lower affinity and a KD of 25 ± 4.1 μM. The similar affinity in binding supports the hypothesis that the bullvalene core allows for multiple binding conformations, resulting in antibacterial activity against not only vancomycin-sensitive but also vancomycin-resistant enterococci.
Table 2.
Binding affinities of vancomycin and SVD 6d for acetyl-Lys-d-Ala-d-Ala and acetyl-Lys-d-Ala-d-Lac, defined as the dissociation constant (KD) (N = 3, ±S.D.); n.b.: no binding
| Compound | KD (µM) | |
|---|---|---|
| acetyl-Lys-d-Ala-d-Ala | acetyl-Lys-d-Ala-d-Lac | |
| Vancomycin | 1.0 ± 0.3 | n.b. |
| 6d | 11 ± 1.9 | 25 ± 4.1 |
To explore the effects of the dynamic nature of bullvalene ring in our SVD 6d, we employed a recently developed native mass spectrometry-based assay where vancomycin has been shown to form a ternary complex through binding lipid II within a lipid II:MurJ complex (67). MurJ is the key flippase protein that transports lipid II across the cytoplasmic membrane. To determine the differences between vancomycin and SVD 6d actions, we first considered detecting whether there are any possible interactions with MurJ directly. For this, we added 3 µM of each of these compounds to MurJ, and no binding was observed in the respective mass spectra (bottom spectra, Fig. 7 A and B). Next, we made MurJ:lipid II complex by incubating 5 µM MurJ with 3 µM lipid II, and as expected, the spectra indicated lipid II binding to MurJ (middle spectra, Fig. 7 A and B). We then attempted to form the ternary complexes by incubating this MurJ:lipid II complex with 7 µM vancomycin and SVD 6d. While the expected ternary complex was formed with vancomycin, complete loss of lipid II binding from MurJ was observed with SVD 6d, suggesting that ligand disruption of the complex had occurred (top spectra, Fig. 7 A and B). A plausible explanation for the action of 6d may include: i) the SVD 6d interacts with other regions of lipid II such as the MurNAc, GlcNAc, or pyrophosphate group, with high affinity; (ii) the SVD 6d:lipid II complex is too large to accommodate lipid II binding to MurJ; and/or iii) the lipophilic linker of SVD 6d participates in binding to lipid II, in which case the interaction with C55PP tail may cause dissociation. Collectively, our data indicate that there may exist a different mode of action for the SVD 6d compared to vancomycin, and further advanced studies are required to unravel the complexity of this system.
Fig. 7.

Mass spectrometry. Native mass spectrometry suggests additional modes of action for SVD 6d. (A) mass spectra of vancomycin with MurJ (Bottom), lipid II with MurJ (Middle), and vancomycin with MurJ:lipid II complex (Top). Ternary complex of MurJ:lipid II:vancomycin is observed (top spectra). (B) mass spectra of SVD 6d with MurJ (Bottom), lipid II with MurJ (Middle), and SVD 6d with MurJ:lipid II complex (Top). No ternary complex and complete loss of lipid II binding from MurJ (top spectra). Charge state 6+ is shown in both cases.
Conclusion
The development of vancomycin derivatives that act by multiple mechanisms of action and decrease resistance susceptibility has been previously disclosed (18, 68–81). Three semi-synthetic vancomycin analogs (82), telavancin (2009), dalbavancin (2014), and oritavancin (2015), have made it to the clinic for the treatment of MRSA infection (26). By taking advantage of the fluxional shapeshifting properties of substituted bullvalene, we have developed an unconventional style of covalently linked vancomycin dimers as unique antibiotics. This effort has provided prototype antibiotics with independent mechanisms of action targeting VRE (and VRSA), for which vancomycin is ineffective. Given the extreme threat of drug-resistant bacteria, the CDC has placed VRE on its serious threat list (83), and the WHO (84) placed it fourth on its list of drug-resistant bacteria that pose the greatest threat to human health. While glycopeptide and other antibiotics have been endowed with features that avoid many mechanisms of resistance (85), the potential benefits of molecular shapeshifting have, until now, been overlooked. The resilience of shapeshifting vancomycin dimers (SVDs) to the onset of antibiotic resistance and strong binding to acetyl-Lys-d-Ala-d-Lac render this molecular class of ligand attractive for further development. We posit that the observed enhancements arise through dynamic adaptive binding interactions imparted by the shapeshifting bullvalene core, and through destabilization of the complex formed between the flippase MurJ and lipid II. The effect of the linker chain length also appears to play a major part in the activity against bacterial species as demonstrated, for example, by the potency of 6d and 6k against VRS and VRSA species, respectively. We believe this work showcases the potential of shapeshifting hydrocarbons in drug discovery and sets the stage for further studies.
Materials and Methods
Chemical Synthesis.
Vancomycin was purchased from SavMart pharmacy and used without further purification. All other reagents were purchased from Sigma Aldrich and used without further purification. Yields refer to chromatographically pure and/or HPLC pure isolated compounds. Due to the fluxional nature of 5a–k and 6a–k, high-resolution mass spectrometry was utilized as a primary means of structural confirmation. For general procedures and characterization data, refer to SI Appendix.
Bacterial Growth Conditions.
The methicillin-sensitive Staphylococcus aureus strain (ATCC® 9144™), methicillin-resistant S. aureus Panel (ATCC® MP-3™), and the vancomycin-resistant Enterococci Panel (ATCC® MP-1™) were sourced from the American Type Culture Collection (ATCC). All bacterial strains were cultured in tryptic soy broth (TSB) or on tryptic soy agar (TSA) plates at 37 °C. Vancomycin-sensitive S. aureus strain SH1000 was obtained from S. Foster and vancomycin-resistant S. aureus (VRSA) strain VRS1 was from BEI Resources.
Cell Lines.
The human embryonic kidney (HEK293) and human liver (HepG2) cells were maintained through tri-weekly passaging. The cells were cultured in Dulbecco’s Modified Eagle Media (DMEM) and supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin/streptomycin, and glucose at a final concentration of 4.5 g/L for the HEK293 cells and 1 g/L for the HepG2 cells. The cell lines were incubated at 37 °C and 5% CO2.
Evaluation of Antibacterial Activity.
Minimum inhibitory concentration (MIC) values were determined using a broth microdilution method according to guidelines defined by the Clinical Laboratory Standards Institute (refer to SI Appendix for details). Plates were incubated at 37 °C for 20 h and growth was assessed by measuring the absorbance at 600 nm. The MIC value was defined as the lowest concentration of compound where no bacterial growth was observed. Experiments were repeated with 3 biological replicates.
Cytotoxicity Screening.
The toxicity assays using human cell lines HEK293 and HepG2 were assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) viability assay. Experiments were performed with three technical replicates with the cytotoxic peptide NaD1, at 50 μM, employed as a positive control and 1% (v/v) DMSO as the vehicle control. Unpaired two-tailed Student’s t tests were performed using GraphPad Prism software (version 8.0.2).
Galleria mellonella Infection Model.
Bacterial suspensions prepared from an overnight culture of VRE (ATCC® 51575™) were injected into Galleria mellonella larvae (20 larvae per treatment) via the hemolymph. Control larvae were injected with 20 μL 1 × PBS. After a 30 min recovery, larvae were injected with their respective treatment, vancomycin at 20 mg/kg, SVD 6d at 20 mg/kg, or 5% (v/v) DMSO. Larvae were then incubated statically at 37 °C and observed daily for 7 d post-injection.
Resistance Studies.
VSE (ATCC® BAA-2127™) was continuously exposed to the analogs using the extended gradient MIC method from 0.25 × MIC up to 4 × MIC. After a 48 h incubation with the compounds at 4 × MIC, aliquots of the cultures were streaked onto drug-free TSA plates. The MIC values were then determined using the broth microdilution method (refer to SI Appendix). Vehicle used was 1% (v/v) DMSO, and experiments were carried out in biological duplicate.
Microscale Thermophoresis.
The binding affinities between the compounds and the tripeptides acetyl-Lys-d-Ala-d-Ala or acetyl-Lys-d-Ala-d-Lac were investigated by microscale thermophoresis (MST) employing the Monolith NT.115 instrument (NanoTemper Technologies) as described in SI Appendix. The tripeptides were labeled with an amine-reactive dye using the Monolith Protein Labeling Kit RED-NHS 2nd Generation (NanoTemper Technologies) following the manufacturer’s instructions. Aqueous solutions of the compounds (final concentration of 200 μM to 6.1 nM) were mixed 1:1 with the labeled tripeptides. Reactions were incubated at 25 °C for 5 min prior to loading into Monolith NT.115 Capillaries (NanoTemper Technologies). Measurements were performed at 25 °C using 20% LED power and 20% IR laser power (30 s ON time, 5 s OFF time). Data from three independent measurements were fitted to the single binding model (equation below) using the signal from Thermophoresis + T-jump within the NT Analysis software, version 1.5.41 (NanoTemper Technologies).
Native Mass Spectrometry.
Prior to mass spectrometry analysis, purified MurJ (see SI for preparation) was buffer exchanged into 200 mM ammonium acetate pH 8.0 and 2 × critical micelle concentration of LDAO using Biospin-6 (BioRad) column and introduced directly into the mass spectrometer using gold-coated capillary needles (prepared in-house). Data were collected on a Q-Exactive UHMR mass spectrometer (ThermoFisher). The instrument parameters were as follows: capillary voltage 1.1 kV, quadrupole selection from 1,000 to 20,000 m/z range, S-lens RF 100%, collisional activation in the HCD cell 200 V, trapping gas pressure setting 7.5, temperature 200 °C, and resolution of the instrument 12,500. The noise level was set at 3 rather than the default value of 4.64. No in-source dissociation was applied. Data were analyzed using Xcalibur 4.2 (Thermo Scientific) software package.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Cold Spring Harbor Laboratory for developmental funds from the National Cancer Institute Cancer Center Support Grant 5P30CA045508 (J.E.M.). We thank the Australian Research Council for funding (J.E.M.) (Future Fellowship; FT170100156) (T.P.S.d.C.) (DECRA Fellowship; DE190100806). We thank the National Health and Medical Research Council of Australia (T.P.S.d.C.) (APP1091976). We thank the New Zealand Marsden Fund (T.F.) (Fast Start Grant; 15-MAU-154). C.V.R’s laboratory is supported by a Medical Research Council program grant (MR/V028839/1) awarded to C.V.R. and J.R.B. J.R.B. holds a Royal Society University Research Fellowship (URF\R1\211567). P.W. acknowledges the support of a Medical Research Council U.K. Programme Grant (MR/N010477/1). J.A.W. is supported by the Defence Science Institute, an initiative of the State Government of Victoria, and is the recipient of a Research Training Program scholarship.
Author contributions
P.W., J.R.B., C.V.R., T.F., T.P.S.d.C., and J.E.M. designed research; A.O., J.A.W., O.Y., S.S., R.A.K., R.M.J., E.M., and J.R.B. performed research; A.O., J.A.W., O.Y., S.S., R.A.K., J.A.H., E.M., P.W., J.R.B., C.V.R., T.F., and T.P.S.d.C. analyzed data; and A.O., J.A.W., O.Y., P.W., R.A.K., J.A.H., J.R.B., C.V.R., T.F., T.P.S.d.C., and J.E.M. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Carol V. Robinson, Email: carol.robinson@chem.ox.ac.uk.
Thomas Fallon, Email: thomas.fallon@newcastle.edu.au.
Tatiana P. Soares da Costa, Email: tatiana.soaresdacosta@adelaide.edu.au.
John E. Moses, Email: moses@cshl.edu.
Data, Materials, and Software Availability
Experimental data for all new compounds prepared during these studies are provided in SI Appendix of this manuscript. All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Hutchings M., Truman A., Wilkinson B., Antibiotics: past, present and future. Curr. Opin. Microbiol. 51, 72–80 (2019). [DOI] [PubMed] [Google Scholar]
- 2.O’Neill J. “Antimicrobial resistance: Tackling a crisis for the health and wealth of nations” (Government of the United Kingdom, 2014). https://amr-review.org/Publications.html. [Google Scholar]
- 3.O’Neill J., “Tackling drug-resistant infections globally: Final report and recommendations” (Government of the United Kingdom, 2016) (November 22, 2021). https://amr-review.org/Publications.html. [Google Scholar]
- 4.Butler M. S., Paterson D. L., Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 73, 329–364 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandes P., Martens E., Antibiotics in late clinical development. Biochem. Pharmacol. 133, 152–163 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Martínez J. L., Baquero F., Andersson D. I., Predicting antibiotic resistance. Nat. Rev. Microbiol. 5, 958–965 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Davies J., Davies D., Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller W. R., Munita J. M., Arias C. A., Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti-infect. Ther. 12, 1221–1236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laws M., Shaaban A., Rahman K. M., Antibiotic resistance breakers: Current approaches and future directions. FEMS Microbiol. Rev. 43, 490–516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ojkic N., Serbanescu D., Banerjee S., Antibiotic resistance via bacterial cell shape-shifting. mBio 13, e00659–22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamad B., The antibiotics market. Nat. Rev. Drug. Discov. 9, 675–676 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Baker S. J., Payne D. J., Rappuoli R., Gregorio E., Technologies to address antimicrobial resistance. Proc. Natl. Acad. Sci. U.S.A. 115, 12887–12895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong W., et al. , Designer broad-spectrum polyimidazolium antibiotics. Proc. Natl. Acad. Sci. U.S.A. 117, 31376–31385 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Culp E. J., et al. , Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature 578, 582–587 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Ling L. L., et al. , A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silverman S. M., Moses J. E., Sharpless K. B., Reengineering antibiotics to combat bacterial resistance: Click chemistry [1,2,3]-triazole vancomycin dimers with potent activity against MRSA and VRE. Chem. Eur. J. 23, 79–83 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Write P. M., Seiple I. B., Myers A. G., The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed. 53, 8840–8869 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okano A., Isley N. A., Boger D. L., Peripheral modifications of [Ψ[CH2NH]Tpg4]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc. Natl. Acad. Sci. U.S.A. 114, E5052–E5061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitcheltree M. J., et al. , A synthetic antibiotic class overcoming bacterial multidrug resistance. Nature 599, 507–512 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q., et al. , Synthetic group A streptogramin antibiotics that overcome Vat resistance. Nature 586, 145–150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang M., Mahasenan K. V., Hermoso J. A., Mobashery S., Unconventional antibacterials and adjuvants. Acc. Chem. Res. 54, 917–929 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konreddy A. K., Rani G. U., Lee K., Choi Y., Recent drug-repurposing-driven advances in the discovery of novel antibiotics. Curr. Med. Chem. 26, 5363–5388 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Domalaon R., Idowu T., Zhanel G. G., Schweizer F., Antibiotic hybrids: the next generation of agents and adjuvants against gram-negative pathogens? Clin. Microbiol. Rev. 31, e00077–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miethke M., et al. , Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 5, 726–749 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore M. J., et al. , Next-generation total synthesis of vancomycin. J. Am. Chem. Soc. 142, 16039–16050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaskovich M. A. T., et al. , Developments in glycopeptide antibiotics. ACS Infect. Dis. 4, 715–735 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leclercq R., Derlot E., Duval J., Courvalin P., Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus Faecium. N. Engl. J. Med. 319, 157–161 (1988). [DOI] [PubMed] [Google Scholar]
- 28.Weigel L. M., et al. , Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302, 1569–1571 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Bugg T. D. H., et al. , Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: Biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 30, 10408–10415 (1991). [DOI] [PubMed] [Google Scholar]
- 30.McComas C. C., Crowley B. M., Boger D. L., Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-Bond and repulsive lone pair contributions. J. Am. Chem. Soc. 125, 9314–9315 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Cooper R. D. G., et al. , Reductive alkylation of glycopeptide: Synthesis and antibacterial activity. J. Antibiot. 49, 575–581 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Ashford P. A., Bew S. P., Recent advances in the synthesis of new glycopeptide antibiotics. Chem. Soc. Rev. 41, 957–978 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Sundram U. N., Griffin J. H., Nicas T. I., Novel vancomycin dimers with activity against vancomycin-resistant Enterococci. J. Am. Chem. Soc. 118, 13107–13108 (1996). [Google Scholar]
- 34.Griffin J. H., et al. , Multivalent drug design. synthesis and in vitro analysis of an array of vancomycin dimers. J. Am. Chem. Soc. 125, 6517–6531 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Xing B., et al. , Multivalent antibiotics via metal complexes: Potent divalent vancomycins against vancomycin-resistant Enterococci. J. Med. Chem. 46, 4904–4909 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Jia Z., O’Mara M. L., Zuegg J., Cooper M. A., Mark A. E., Vancomycin: Ligand recognition, dimerization and super-complex formation. FEBS J. 280, 1294–1307 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Rao J., Whitesides G. M., Tight binding of a dimeric derivative of vancomycin with dimeric l-Lys-d-Ala-d-Ala. J. Am. Chem. Soc. 119, 10286–10290 (1997). [Google Scholar]
- 38.Staroske T., Williams D. H., Synthesis of covalent head-to-tail dimers of vancomycin. Tetrahedron Lett. 39, 4917–4920 (1998). [Google Scholar]
- 39.Nicolaou K. C., et al. , Solid- and solution-phase synthesis of vancomycin and vancomycin analogues with activity against vancomycin-resistant bacteria. Chem. Eur. J. 7, 3798–3823 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Nicolaou K. C., et al. , Synthesis and biological evaluation of vancomycin dimers with potent activity against vancomycin-resistant bacteria: Target-accelerated combinatorial synthesis. Chem. Eur. J. 7, 3824–3843 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Adamczyk M., Moore J. A., Rege S. D., Yu Z., Investigations into self-association of vancomycin covalent dimers using surface plasmon resonance technology. Bioorg. Med. Chem. Lett. 9, 2437–2440 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Stack D. R., Thompson R. C., “Covalently linked dimers of glycopeptide antibiotics” (1997).
- 43.Williams D. H., Maguire A. J., Tsuzuki W., Westwell M. S., An analysis of the origins of a cooperative binding energy of dimerization. Science 280, 711–714 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Nicolaou K. C., et al. , Target-accelerated combinatorial synthesis and discovery of highly potent antibiotics effective against vancomycin-resistant bacteria. Angew. Chem. Int. Ed. 39, 3823–3828 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Silver L. L., Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lippert A. R., Naganawa A., Keleshian V. L., Bode J. W., Synthesis of phototrappable shape-shifting molecules for adaptive guest binding. J. Am. Chem. Soc. 132, 15790–15799 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Doering W. von E., Roth W. R., A rapidly reversible degenerate cope rearrangement: Bicyclo[5.1.0]octa-2,5-diene. Tetrahedron 19, 715–737 (1963). [Google Scholar]
- 48.Rowan S. J., Cantrill S. J., Cousins G. R. L., Sanders J. K. M., Stoddart J. F., Dynamic covalent chemistry. Angew. Chem. Int. Ed. 41, 898–952 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Rasmussen B., Sørensen A., Beeren S. R., Pittelkow M., “Dynamic combinatorial chemistry” in Organic Synthesis and Molecular Engineering (John Wiley & Sons Ltd, 2013), pp. 393–436. [Google Scholar]
- 50.Jin Y., Yu C., Denman R. J., Zhang W., Recent advances in dynamic covalent chemistry. Chem. Soc. Rev. 42, 6634–6654 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Teichert J. F., Mazunin D., Bode J. W., Chemical sensing of polyols with shapeshifting boronic acids as a self-contained sensor array. J. Am. Chem. Soc. 135, 11314–11321 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Yahiaoui O., Pašteka L. F., Judeel B., Fallon T., Synthesis and analysis of substituted bullvalenes. Angew. Chem. Int. Ed. 57, 2570–2574 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Patel H. D., Tran T. H., Sumby C. J., Pašteka L. F., Fallon T., Boronate ester bullvalenes. J. Am. Chem. Soc. 142, 3680–3685 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Smedley C. J., et al. , Diversity Oriented Clicking (DOC): Divergent synthesis of SuFExable pharmacophores from 2-substituted-Alkynyl-1-sulfonyl fluoride (SASF) Hubs. Angew. Chem. Int. Ed. 59, 12460–12469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan T. R., Hilgraf R., Sharpless K. B., Fokin V., Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 6, 2853–2855 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Kolb H. C., Finn M. G., Sharpless K. B., Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Tornøe C. W., Christensen C., Meldal M., Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Schröder G., Oth J. F. M., Recent chemistry of bullvalene. Angew. Chem. Int. Ed. 6, 414–423 (1967). [Google Scholar]
- 59.Filippone E. J., Kraft W. K., Farber J. L., The nephrotoxicity of vancomycin. Clin. Pharm. Therap. 102, 459–469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gyamlani G., et al. , Vancomycin-associated acute kidney injury in a large veteran population. Am. J. Nephrol. 49, 133–142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cutuli M. A., et al. , Galleria mellonella as a consolidated in vivo model hosts: New developments in antibacterial strategies and novel drug testing. Virulence 10, 527–541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai C. J. Y., Loh J. M. S., Proft T., Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 7, 214–229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blaskovich M. A. T., et al. , Protein-inspired antibiotics active against vancomycin- and daptomycin-resistant bacteria. Nat. Commun. 9, 22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mishra N. M., et al. , Iterative chemical engineering of vancomycin leads to novel vancomycin analogs with a high in vitro therapeutic index. Front. Microbiol. 9, 1175 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jain R. K., Trias J., Ellman J. A., D-Ala-D-Lac binding is not required for the high activity of vancomycin dimers against vancomycin resistant enterococci. J. Am. Chem. Soc. 125, 8740–8741 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Popieniek P. H., Pratt R. F., A fluorescent ligand for binding studies with glycopeptide antibiotics of the vancomycin class. Anal. Biochem. 165, 108–113 (1987). [DOI] [PubMed] [Google Scholar]
- 67.Bolla J. R., et al. , Direct observation of the influence of cardiolipin and antibiotics on lipid II binding to MurJ. Nat. Chem. 10, 363–371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Silver L. L., Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6, 41–55 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Kerns R., et al. , The role of hydrophobic substituents in the biological activity of glycopeptide antibiotics. J. Am. Chem. Soc. 122, 12608–12609 (2000). [Google Scholar]
- 70.Gu W., Chen B., Ge M., Design and synthesis of new vancomycin derivatives. Bioorg. Med. Chem. Lett. 24, 2305–2308 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Yarlagadda V., Akkapeddi P., Manjunath G. B., Haldar J., Membrane active vancomycin analogues: A strategy to combat bacterial resistance. J. Med. Chem. 57, 4558–4568 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Yarlagadda V., Samaddar S., Paramanandham K., Shome B. R., Haldar J., Membrane disruption and enhanced inhibition of cell-wall biosynthesis: a synergistic approach to tackle vancomycin-resistant bacteria. Angew. Chem. Int. Ed. 54, 13644–13649 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Yarlagadda V., et al. , Glycopeptide antibiotic to overcome the intrinsic resistance of Gram-negative bacteria. ACS Infect. Dis. 2, 132–139 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Wu Z.-C., Isley N. A., Boger D. L., N-Terminus alkylation of vancomycin: Ligand binding affinity, antimicrobial activity, and site-specific nature of quaternary trimethylammonium salt modification. ACS Infect. Dis. 4, 1468–1474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dhanda G., Sarkar P., Samaddar S., Haldar J., Battle against vancomycin-resistant bacteria: Recent developments in chemical strategies. J. Med. Chem. 62, 3184–3205 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Antonoplis A., Zang X., Wegner T., Wender P. A., Cegelski L., Vancomycin–arginine conjugate inhibits growth of carbapenem-resistant E. coli and targets cell-wall synthesis. ACS Chem. Biol. 14, 2065–2070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marschall E., Cryle M. J., Tailhades J., Biological, chemical, and biochemical strategies for modifying glycopeptide antibiotics. J. Biol. Chem. 294, 18769–18783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu Z.-C., Isley N. A., Okano A., Weiss W. J., Boger D. L., C1-CBP-vancomycin: impact of a vancomycin C-terminus trimethylammonium cation on pharmacological properties and insights into its newly introduced mechanism of action. J. Org. Chem. 85, 1365–1375 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Umstätter F., et al. , Vancomycin resistance is overcome by conjugation of polycationic peptides. Angew. Chem. Int. Ed. 59, 8823–8827 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma C., He N., Ou Y., Feng W., Design and synthesis of new vancomycin derivatives. ChemistrySelect 5, 6670–6673 (2020). [Google Scholar]
- 81.Chen L., et al. , Vancomycin analogues active against vanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc. Natl. Acad. Sci. U.S.A. 100, 5658–5663 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Butler M. S., Hansford K. A., Blaskovich M. A. T., Halai R., Cooper M. A., Glycopeptide antibiotics: Back to the future. J. Antibiot. 67, 631–644 (2014). [DOI] [PubMed] [Google Scholar]
- 83.Centers for Disease Control and Prevention (U.S.), Antibiotic resistance threats in the United States, 2019. 10.15620/cdc:82532. (January 28, 2022). [DOI]
- 84.WHO publishes list of bacteria for which new antibiotics are urgently needed (2017) (February 16, 2022). https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed#:~:text=.
- 85.James R. C., Pierce J. G., Okano A., Xie J., Boger D. L., Redesign of glycopeptide antibiotics: back to the future. ACS Chem. Biol. 7, 797–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Experimental data for all new compounds prepared during these studies are provided in SI Appendix of this manuscript. All study data are included in the article and/or SI Appendix.