

Abstract
Background
CAG‐repeat expansions in Ataxin 2 (ATXN2) are known to cause spinocerebellar ataxia type 2 (SCA2), but CAA interrupted expansions may also result in autosomal dominant Parkinson's disease (AD PD). However, because of technical limitations, such expansions are not explored in whole exome sequencing (WES) data.
Objectives
To identify ATXN2 expansions using WES data from PD cases.
Methods
We explored WES data from a cohort of 477 index cases with PD using ExpansionHunter (Illumina DRAGEN Bio‐IT Platform, San Diego, CA). Putative expansions were confirmed by combining polymerase chain reaction and fragment length analysis followed by sub‐cloning and sequencing methods.
Results
Using ExpansionHunter, we identified three patients from two families with AD PD carrying either ATXN2 22/39 or 22/37 repeats, both interrupted by four CAA repeats.
Conclusion
These findings demonstrate the usefulness of WES to detect pathogenic CAG repeat expansions, which were found in 1.7% of AD PD in the ATXN2 gene in our exome dataset.
Keywords: Parkinson's disease, repeat expansions, ATXN2 gene, whole exome sequencing, CAA interruption
Parkinson's disease (PD) is a neurodegenerative disorder characterized by a triad of symptoms: akinesia, rigidity, and rest tremor, and is associated with a good response to levodopa therapy. These symptoms are because of the degeneration of dopaminergic neurons of the substantia nigra secondary to the accumulation of aggregated α‐synuclein. 1 , 2 , 3
Although the cause of PD is commonly sporadic, Mendelian forms account for 5% to 10% of cases. 4 Disease causing variants, mostly in SNCA, LRRK2, and VPS35 have been identified in patients with autosomal dominant (AD) PD. In addition, we and others have previously described families with heterozygous expansions in Ataxin 2 (ATXN2) presenting with predominant parkinsonian symptoms and in some cases with typical AD PD. 5 , 6 , 7 , 8 , 9 , 10 , 11 As in ataxic forms of spinocerebellar ataxia type 2 (SCA2), the expanded allele contained >33 repeats, but in PD, they were often interrupted by one or more CAA codons. 12
Next‐generation sequencing (NGS) has proven to be of great diagnostic value in clinical practice, 13 but until recently, was thought to have a limited ability to assess for loci containing repeat expansions. Over the last few years, several bioinformatic tools for genome‐wide genotyping of short tandem repeats (STRs) in short read sequencing data, mainly from whole genome sequencing (WGS) data have been developed. 14 , 15 , 16 , 17 However, despite the extensive application of whole exome sequencing (WES) in routine diagnostic genetic testing and the publication of several STR detection studies, STRs have not been systematically looked for in these data. Furthermore, large‐scale assessments of the diagnostic potential of STR detection from WES have not been undertaken yet in the context of PD. In this study, we assessed the clinical use of detecting the most common PD‐related ATXN2 expansions using the ExpansionHunter software, 15 , 18 which can estimate the repeat size in the target loci in WES data from a large undiagnosed cohort enriched in familial and early‐onset PD patients.
Patients and Methods
Patients
We selected a total of 477 PD index cases (192 females, 285 males), recruited between 1990 and 2021 for whom WES data were available. In addition, WES data were also available for 98 affected relatives for co‐segregation analyses.
Clinical assessment was performed by specialists in movement disorders, mainly through the French Parkinson Disease Genetics Network 19 , 20 (the PDG group). Diagnosis of parkinsonism was established according to the United Kingdom Parkinson's Disease Society Brain Bank (PDSBB) criteria. 21 We excluded all patients with mutations in known genes causing or related to PD/parkinsonism. 19 , 20 Written informed consent was obtained from all participants, and genetic studies were approved by local ethics committees.
Methods
Exons were captured using different enrichment kits: the Roche Seqcap Ez MedExome (Roche Diagnostics Corporation, Indianapolis, IN) (n = 139) or the Human Twist exome refseq 40 Mb (Twist Biosciences, San Francisco, CA) (n = 338) kits followed by 150‐bp paired‐end sequencing performed on Ilumina NovaSeq 6000 instrument (Illumina Inc, San Diego, CA). Mean coverage was 104.6× (range, 51.1–216.4×) and 25, 30, 50‐fold average sequencing depth was achieved across 73.1% (range, 55.6–102.8%), 93% (range, 51.1–205.1%) and 111.1% (range, 56.8–216.4%) of targeted regions, respectively. Read alignment and variant calling were made using an in‐house pipeline. Briefly, FastQC was used to check the quality of the reads and low‐quality reads were removed using Trimmomatic. Sequencing data were then aligned to the human reference genome hg19 using the bwa 22 , 23 suite and variant calling was performed using the HaplotypeCaller function of GATK suite or using Dragen (Illumina). Mutations in known PD and related parkinsonism genes (Table S1) were excluded using VarAFT software 24 (version 2.17–1) and Illumina DRAGEN BioIT Platform v3.9. Exomes alignment files were then analyzed with ExpansionHunter v.5.0.0 Illumina DRAGEN Bio‐IT Platform (Illumina, San Diego, CA) for targeted screening of CAG‐repeat in ATXN2.
Molecular Analysis
All likely expanded ATXN2 alleles (>33 repeats) were confirmed using polymerase chain reaction (PCR) and fragment length analysis. To further verify the number of CAG repeats and assess the presence of CAA interruptions, the amplified fragments were cloned into a pJET1,2/blunt x‐plasmid (ThermoFisher Scientific, Waltham, MA) and sequenced bi‐directionally, using primer pairs: forward (5′‐CGTGCGAGCCGGTGTATGGG‐3′) and reverse (5′‐GGCGACGCTAGAAGGCCGCT‐3′).
Violin plots representing the distribution of CAG‐repeat size according to the different exome enrichment kits were plotted in R v.4.1.1 using ggplot2. 25 , 26
Results
Patients’ Characteristics
The cohort consisted of 203 familial PD index cases 121 with at least two cases in two generations compatible with AD inheritance, 82 with at least two affected siblings compatible with autosomal recessive (AR) inheritance and 274 isolated cases. Of them, 223 were from Europe, 31 from North Africa and 223 of unknown origin. Mean age at onset of motor signs was 41 ± 12 years (range, 3–83 years) (Table S2).
Exonic Expansion Detected in WES Data
Using different exome enrichment kits for library preparation, we found that the mean coverage of the ATXN2 region depended on the enrichment kit used. As a result, the mean coverage for the Roche Seqcap Ez MedExome was 60.67 ± 27.86× (range, 28–181×) and of 85.13 ± 48.75× (range, 5–367×) for the Human Twist exome refseq 40 Mb.
CAG‐repeat lengths in index cases were estimated using ExpansionHunter. The mean estimated CAG‐repeat allele length in WES data obtained with the Roche Seqcap Ez MedExome kit was 22.27 ± 2.22 (range, 15–52), and of 22.2 ± 1.24 (range, 17–39) with the Human Twist exome refseq 40 Mb enrichment capture kit (Fig. 1). Of the 477 PD index cases, expanded CAG‐repeat alleles were detected in two, both from AD PD families for whom the ATXN2 repeat size was within the pathogenic range (≥33 CAG‐repeat) (Fig. 1). No instances of ATXN2 expansions were detected among the 274 isolated cases or the 82 AR families. Estimated confidence intervals of the ATXN2 CAG‐repeat allele sizes were 22‐22/39‐39 for index case A‐1 and 22‐22/41‐61 for index case B‐2, accounting for 1.7% of our series of 121 AD PD families. Co‐segregation analysis of additional affected relatives in family B for whom WES data was available showed an expanded allele length of 22–22/40–52 in the affected proband's uncle (individual 1), but normal CAG‐repeat alleles (22/22) in the proband's aunt (individual 4) (Fig. 2A).
FIG. 1.
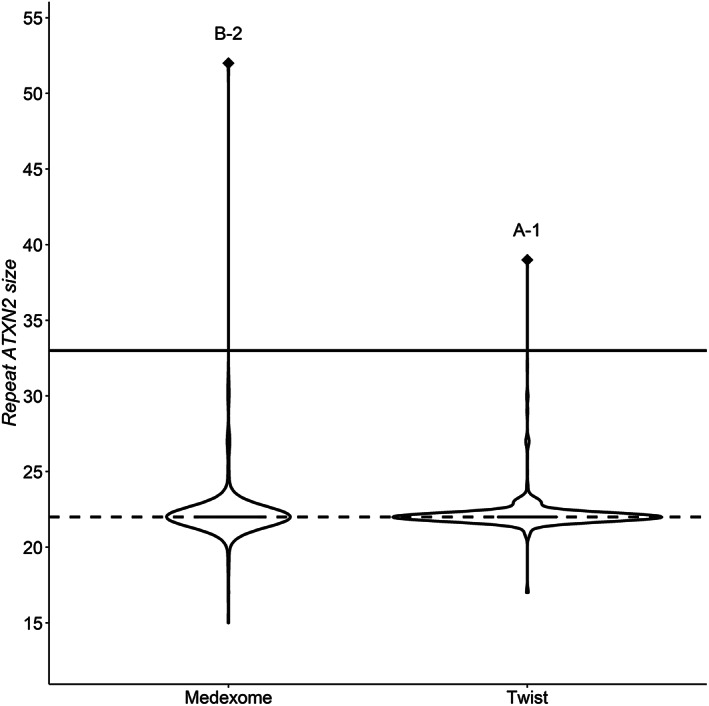
Estimated ATXN2 repeat size using ExpansionHunter according to the enrichment kits used. The solid line delineates the mean ATXN2 CAG‐repeat size to 22, estimated from Caucasian populations; the dashed line indicates the pathological threshold up to 32.
FIG. 2.
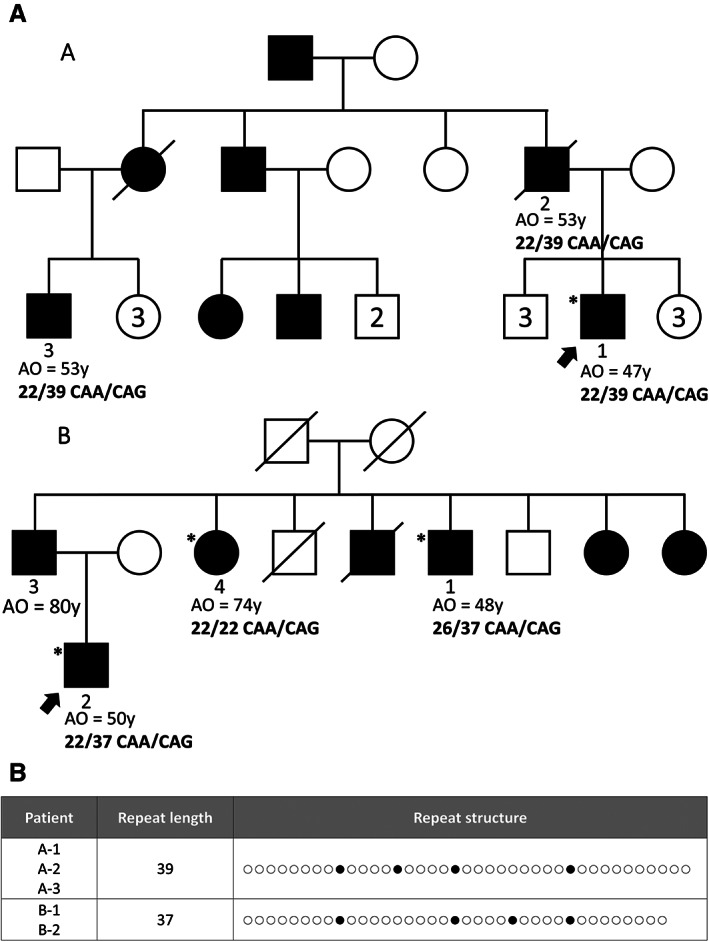
Pedigrees of families A and B with autosomal dominant Parkinson's disease (AD PD) and expanded CAG‐repeat alleles in ATXN2. A: Pedigrees of families A and B. Square = male; circles = female; black symbol = affected; white symbol = unaffected; slashed symbol = deceased. Asterisks indicate individuals with genetic testing and arrow, the proband. AO, age at onset. B: Length and structure of the expanded CAG/CAA repeats in ATXN2 for the three carriers. Open circles = CAG; filled circles = CAA interruptions.
PCR and fragment analysis confirmed the pathogenic CAG‐repeat allele size in all three patients, although it was overestimated by ExpansionHunter in family B (Fig. 2A). Sub‐cloning and sequencing analysis revealed the presence of four CAA interruptions within the pathogenic CAG‐repeat expanded fragment for all three patients: 39 repeats, (CAG)8‐CAA‐(CAG)4‐CAA‐(CAG)4‐CAA‐(CAG)9‐CAA‐(CAG)10 for patient A‐1 and 37 repeats, (CAG)8‐CAA‐(CAG)9‐CAA‐(CAG)4‐CAA‐(CAG)4‐CAA‐(CAG)8 for patients B‐1 and B‐2 (Fig. 2B).
Clinical Features
Patient A‐1 had a diagnosis of PD, but with an atypical initial presentation with a slowly progressive feeling of discomfort of the lower limbs, predominantly on the right side, at age 47. At age 57, he had asymmetric brisk reflexes of the four limbs and discrete bradykinesia predominantly in the right lower limb. No rest tremor was observed. The dopamine transporter (DAT) scan performed at age 56 showed asymmetrical decreased uptake in the caudate nucleus and putamen. The magnetic resonance imaging (MRI) was normal, with no cerebellar atrophy reported. l‐dopa treatment only partially relieved his symptoms. He had spasticity with preserved strength, other than just mild weakness in the right lower limb, and became wheelchair bound at the age of 58. At age 58, there was an increase in upper limb rigidity and the development of mild dysarthria, which was not l‐dopa‐responsive. He had increased body mass index (BMI) and presented an obstructive sleep apnea syndrome that required positive airway pressure therapy. His father (A‐2) had a l‐dopa‐responsive akineto‐rigid syndrome with a marked asymmetric resting tremor and was diagnosed with typical PD. His cousin (A‐3) developed an asymmetric akineto‐rigid syndrome at age 53, which responded well to l‐dopa therapy and was diagnosed with PD. The presence of an ATXN2 expanded allele was confirmed by genetic testing in the clinical setting in his father and his cousin.
The two patients from family B (patients 1 and 2) with ATXN2 expanded alleles presented with typical l‐dopa‐responsive parkinsonism, diagnosed at ages 48 and 50, respectively. A DAT scan performed in patient B‐2 confirmed dopaminergic denervation. In patient B‐1, the positive response to treatment was quantified with an Unified Parkinson's Disease Rating Scale III score of 36 on “off” state, which decreased to 14 on medication. Interestingly, the third affected relative was also diagnosed with PD at an older age of onset (74 years), but with normal ATXN2 repeat alleles, suggesting that she had idiopathic PD unrelated to the familial ATXN2‐related PD. No signs of ataxia or oculomotor abnormality were reported in any of these three patients.
Discussion
WES has been used in multiple settings as a first‐line diagnostic test for several neurological disorders, but was previously thought to have a low ability to detect repeat expansions. Several tools have been recently developed to detect repeat expansions from whole exome/genome sequencing in the research setting. We used ExpansionHunter here, which was previously reported as the most sensitive and specific bioinformatics tool, 27 to genotype ATXN2 CAG‐repeats in our WES data from a large cohort enriched of undiagnosed familial and early‐onset PD patients. Using the same setting, Méreaux et al, 28 compared the results of ExpansionHunter with the sizing of the ATXN2 repeats by PCR followed by capillary electrophoresis in 247 ataxic patients and found that both specificity and sensitivity reached 100%. Our data showed that WES combined with ExpansionHunter can reliably distinguish between non‐expanded and expanded alleles at the ATXN2 locus. However, a good coverage (>10×) is required, which might depend on the enrichment kit used.
In this study, we identified two AD PD families with ATXN2 expanded alleles, accounting for 1.7% (2/121) of AD PD in our cohort, consistent with our previous study 5 (2%) and that of another Caucasian population 10 (1.5%). In these studies, PD patients with ATXN2 expansions were only observed in AD PD cases. Indeed, ATXN2 expansion frequency varies depending on ethnicity and family history of parkinsonism: up to 2.5% in Caucasians and up to 8.7% in Asians in familial PD, but is much lower in sporadic cases (ranging from 0% to 2.2% in all populations). 29 SCA2‐parkinsonism is associated with relatively short ATXN2 CAG‐repeat expansion ranging from 33 to 43 in exon 1 of ATXN2, but with one to four CAA interruptions. In comparison, patients presenting with pure SCA2‐ataxia harbor a wider range of expansion sizes from 32 to over 200, usually without CAA interruptions. 5 Interestingly, in the last years, several studies have shown that intermediate repeat size (29–32) in ATXN2 could act as genetic risk factors or modifiers in other neurodegenerative diseases, such as amyotrophic lateral sclerosis, frontotemporal dementia, progressive supranuclear palsy, Alzheimer's disease, and multisystem atrophy. 30 , 31 , 32 , 33 , 34 , 35
In conclusion, our findings provide guidance for the implementation of ATXN2 CAG‐repeat analysis in routine diagnostic clinical exome sequencing. Moreover, our results show that systematic STR evaluation may increase diagnostic yield of exome sequencing by identifying SCA2 Parkinsonism cases. This could contribute to a significant percentage of AD PD families, who might have been previously undiagnosed because of the lack of systematic testing.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the first draft, B. Review and Critique; (4) Clinical investigation.
F.C.: 1A, 1C, 3A, 3B
T.C.: 1A
C.T.:1A, 1C, 3B
M.F.: 1C
S.N.: 1C
A.L.F.A.: 1C
T.G.: 1A
J.G.: 1A
M.A.: 4
L.L.M.: 3B, 4.
N.L.F.: 4
C.T.: 4
J.C.C.: 1B, 3B, 4
S.L.: 1A, 1B, 3B
A.B.: 1A, 1B, 3B, 4
Study Group: The French clinicians’ network for Parkinson's disease genetics (the PDG group) members: 4. All authors have read and agreed to the published version of the manuscript.
Disclosures
Ethical Compliance Statement: Informed consent was obtained from all participants, and genetic studies were approved by local ethics committees (INSERM, CCPPRB du Groupe Hospitalier Pitié‐Salpêtrière, Paris, France, No. 44814). We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: This work was supported by the Fondation pour la Recherche Médicale (FRM) (MND202004011718), the Fondation de France, France‐Parkinson Association, la Fédération pour la Recherche sur le Cerveau (FRC) and the French program “Investissements d'avenir” (ANR‐10‐IAIHU‐06). J.C.C. has served in advisory boards for Biogen, Denali, Idorsia, Prevail Therapeutic, Servier, Theranexus, and UCB. L.L.M. has served in advisory marketing board for Sanofi. Other authors report no conflicts of interest related to the paper.
Financial Disclosures for the Previous 12 Months: F.C. and C.T. have been employed by Paris Brain Institute (ICM) during this work. M.A. has received grants from AbbVie, Merz, Orkyn, Reata, Ipam, Ena Pharma, and Asdia outside of this work. L.L.M. has received grants from French Parkinson's Disease Association, Fondation de France, French Ministry National PHRC, Paris Brain Institute ICM Big Brain Theory and Paris Brain Institute ICM Neurocatalyst outside of this work. J.C.C. has received grants from Sanofi and The Michael J. Fox Foundation outside of this work. S.L. has received grants from FRM. A.B. has received grants from Fondation Roger de Spoelberch and Greater Paris University Hospitals (APHP).
Supporting information
Table S1. List of Parkinson's disease‐associated genes excluded from our cohort of patients
Table S2. Characteristics of the study population according to the different exome enrichment kits used
Acknowledgments
We thank the patients and their families. Part of this work was carried out the DNA and Cell Bank of the Institut du Cerveau et de la Moëlle épinière (ICM). We gratefully acknowledge Sylvie Forlani, Ludmila Jornea, and Yassaman Ghassab for sample preparation. Part of this work was carried out through the iGenSeq core facility. We gratefully acknowledge Yannick Marie and Agnes Rastetter for the sequencing of whole exome. We would like to thank Dr Mireille Ferrari‐Henquinet for providing patient clinical data.
Relevant disclosures and conflict of interest are listed at the end of this article.
The authors report no sources of funding and no conflicts of interest.
References
- 1. de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol 2006;5(6):525–535. 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2. Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α‐Synuclein in Lewy bodies. Nature 1997;388(6645):839–840. 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24(2):197–211. 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 4. Lunati A, Lesage S, Brice A. The genetic landscape of Parkinson's disease. Rev Neurol 2018;174(9):628–643. 10.1016/j.neurol.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 5. Charles P, Camuzat A, Benammar N, et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology 2007;69(21):1970–1975. 10.1212/01.wnl.0000269323.21969.db. [DOI] [PubMed] [Google Scholar]
- 6. Kim JM, Hong S, Kim GP, et al. Importance of low‐range CAG expansion and CAA interruption in SCA2 Parkinsonism. Arch Neurol 2007;64(10):1510–1518. 10.1001/archneur.64.10.1510. [DOI] [PubMed] [Google Scholar]
- 7. Lu CS, Wu Chou YH, Kuo PC, Chang HC, Weng YH. The parkinsonian phenotype of spinocerebellar ataxia type 2. Arch Neurol 2004;61(1):35–38. 10.1001/archneur.61.1.35. [DOI] [PubMed] [Google Scholar]
- 8. Payami H, Nutt J, Gancher S, et al. SCA2 may present as levodopa‐responsive parkinsonism. Mov Disord 2003;18(4):425–429. 10.1002/mds.10375. [DOI] [PubMed] [Google Scholar]
- 9. Shan DE, Liu RS, Sun CM, Lee SJ, Liao KK, Soong BW. Presence of spinocerebellar ataxia type 2 gene mutation in a patient with apparently sporadic Parkinson's disease: clinical implications. Mov Disord 2004;19(11):1357–1360. 10.1002/mds.20212. [DOI] [PubMed] [Google Scholar]
- 10. Simon‐Sanchez J, Hanson M, Singleton A, et al. Analysis of SCA‐2 and SCA‐3 repeats in Parkinsonism: evidence of SCA‐2 expansion in a family with autosomal dominant Parkinson's disease. Neurosci Lett 2005;382(1–2):191–194. 10.1016/j.neulet.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 11. Wang JL, Xiao B, Cui XX, et al. Analysis of SCA2 and SCA3/MJD repeats in Parkinson's disease in mainland China: Genetic, clinical, and positron emission tomography findings. Mov Disord 2009;24(13):2007–2011. 10.1002/mds.22727. [DOI] [PubMed] [Google Scholar]
- 12. Coarelli G, Brice A, Durr A. Recent advances in understanding dominant spinocerebellar ataxias from clinical and genetic points of view. F1000Research 2018;7:F1000 Faculty Rev‐1781. 10.12688/f1000research.15788.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vissers LELM, van Nimwegen KJM, Schieving JH, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med 2017;19(9):1055–1063. 10.1038/gim.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dashnow H, Lek M, Phipson B, et al. STRetch: detecting and discovering pathogenic short tandem repeat expansions. Genome Biol 2018;19(1):121. 10.1186/s13059-018-1505-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dolzhenko E, van Vugt JJFA, Shaw RJ, et al. Detection of long repeat expansions from PCR‐free whole‐genome sequence data. Genome Res 2017;27(11):1895–1903. 10.1101/gr.225672.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Halman A, Oshlack A. Accuracy of short tandem repeats genotyping tools in whole exome sequencing data. F1000Research 2020;9:200. 10.12688/f1000research.22639.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ, Bahlo M. Detecting expansions of tandem repeats in cohorts sequenced with short‐read sequencing data. Am J Hum Genet 2018;103(6):858–873. 10.1016/j.ajhg.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dolzhenko E, Deshpande V, Schlesinger F, et al. ExpansionHunter: a sequence‐graph‐based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019;35(22):4754–4756. 10.1093/bioinformatics/btz431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lesage S, Houot M, Mangone G, et al. Genetic and phenotypic basis of autosomal dominant Parkinson's disease in a large multi‐center cohort. Front Neurol 2020;11:11. Accessed September 6, 2022. 10.3389/fneur.2020.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lesage S, Lunati A, Houot M, et al. Characterization of recessive Parkinson disease in a large multicenter study. Ann Neurol 2020;88(4):843–850. 10.1002/ana.25787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55(3):181–184. 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25(14):1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows–Wheeler transform. Bioinformatics 2010;26(5):589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Desvignes JP, Bartoli M, Delague V, Krahn M, Miltgen M, Béroud C, Salgado D. VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res 2018;46(W1):W545–W553. 10.1093/nar/gky471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. R Core Team . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2022. https://www.R-project.org/. [Google Scholar]
- 26. Wickham H. Ggplot2: elegant graphics for data analysis. New York: Springer‐Verlag; 2016. https://ggplot2.tidyverse.org. [Google Scholar]
- 27. van der Sanden BPGH, Corominas J, de Groot M, et al. Systematic analysis of short tandem repeats in 38,095 exomes provides an additional diagnostic yield. Genet Med 2021;23(8):1569–1573. 10.1038/s41436-021-01174-1. [DOI] [PubMed] [Google Scholar]
- 28. Méreaux JL, Davoine CS, Coutelier M, et al. Fast and reliable detection of repeat expansions in spinocerebellar ataxia using exomes. J Med Genet 2023;jmg‐2022‐108924. Online ahead of print. 10.1136/jmg-2022-108924. [DOI] [PubMed] [Google Scholar]
- 29. Park H, Kim HJ, Jeon BS. Parkinsonism in spinocerebellar ataxia. Biomed Res Int 2015;2015:125273. 10.1155/2015/125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corrado L, Carlomagno Y, Falasco L, et al. A novel peripherin gene (PRPH) mutation identified in one sporadic amyotrophic lateral sclerosis patient. Neurobiol Aging 2011;32(3):552.e1–552.e6. 10.1016/j.neurobiolaging.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 31. Lattante S, Millecamps S, Stevanin G, et al. Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology 2014;83(11):990–995. 10.1212/WNL.0000000000000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fournier C, Anquetil V, Camuzat A, et al. Interrupted CAG expansions in ATXN2 gene expand the genetic spectrum of frontotemporal dementias. Acta Neuropathol Commun 2018;6(1):41. 10.1186/s40478-018-0547-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mongelli A, Sarro L, Rizzo E, et al. Multiple system atrophy and CAG repeat length: a genetic screening of polyglutamine disease genes in Italian patients. Neurosci Lett 2018;678:37–42. 10.1016/j.neulet.2018.04.044. [DOI] [PubMed] [Google Scholar]
- 34. Rosas I, Martínez C, Clarimón J, et al. Role for ATXN1, ATXN2, and HTT intermediate repeats in frontotemporal dementia and Alzheimer's disease. Neurobiol Aging 2020;87:139.e1–139.e7. 10.1016/j.neurobiolaging.2019.10.017. [DOI] [PubMed] [Google Scholar]
- 35. Glass JD, Dewan R, Ding J, et al. ATXN2 intermediate expansions in amyotrophic lateral sclerosis. Brain 2022;145(8):2671–2676. 10.1093/brain/awac167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of Parkinson's disease‐associated genes excluded from our cohort of patients
Table S2. Characteristics of the study population according to the different exome enrichment kits used