

Abstract
Background:
Multiple myeloma (MM) measurable residual disease (MRD) evaluated by flow cytometry is a surrogate for progression-free and overall survival in clinical trials. However, analysis and reporting between centers lack uniformity. We designed and evaluated a consensus protocol for MM MRD analysis to reduce inter-laboratory variation in MM MRD reporting.
Methods:
Seventeen participants from 13 countries performed blinded analysis of the same eight de-identified flow cytometry files from patients with/without MRD using their own method (Stage 1). A consensus gating protocol was then designed following survey and discussions, and the data re-analyzed for MRD and other bone marrow cells (Stage 2). Inter-laboratory variation using the consensus strategy was reassessed for another 10 cases and compared with earlier results (Stage 3).
Results:
In Stage 1, participants agreed on MRD+/MRD− status 89% and 68% of the time respectively. Inter-observer variation was high for total numbers of analyzed cells, total and normal plasma cells (PCs), limit of detection, lower limit of quantification, and enumeration of cell populations that determine sample adequacy. The identification of abnormal PCs remained relatively consistent. By consensus method, average agreement on MRD− status improved to 74%. Better consistency enumerating all parameters among operators resulted in near-unanimous agreement on sample adequacy.
Conclusion:
Uniform flow cytometry data analysis substantially reduced inter-laboratory variation in reporting multiple components of the MM MRD assay. Adoption of a harmonized approach would meet an important need for conformity in reporting MM MRD for clinical trials, and wider acceptance of MM MRD as a surrogate clinical endpoint.
INTRODUCTION:
The clinical outcome of multiple myeloma (MM) patients has significantly improved following the introduction of new and effective therapies [1]. Not all patients, however, will achieve long-term remission as measured by traditional response criteria. A growing body of evidence supports the premise that remnant myeloma plasma cells (PCs) continue to proliferate following treatment, causing relapse in some patients [2, 3]. The level of measurable residual disease (MRD), which inversely correlates with the depth of response, is directly associated with the risk of relapse and is used as a surrogate marker for progression-free and overall survival [4]. The measurement of MM MRD by various methods is also being investigated as a possible indicator of when to terminate or intensify treatment strategies to prevent relapse [5]. While an increasing complement of improved anti-myeloma therapies singly and in combination have resulted in more patients achieving deeper responses, the length of time taken for traditional outcome measures to herald meaningful results is impeding the rate of translation from discovery to regulatory approval. This is because randomized phase 3 trials take years to complete when relying on conventional endpoints of relapse/event-free and overall survival. This has prompted regulatory bodies to consider using MM MRD detection as a surrogate for biological response to reduce the time and cost of the drug approval process [6, 7].
Multi-parametric flow cytometry (MFC) is a sensitive whole cell assay platform that is well-suited for MM MRD testing and able to achieve a detection sensitivity down to 10−5 and as high as 10−6 under optimal circumstances. Multiple clinical trials have demonstrated the utility of MFC in defining an ‘immunophenotypic complete response’ (MRD negative at 10−4) in MM patients following autologous stem cell transplantation and non-transplant eligible patients treated with novel agents [8–11]. However, MM MRD level is a continuous variable, with an approximate one-year survival benefit per log reduction; and yet, no lower cut-off point for predicting outcome has been consistently demonstrated [12]. Consequently, the International Myeloma Working Group updated the recommendations for response assessment of MM in 2016, which included the introduction of a ‘flow MRD-negative’ response requiring the demonstration of MRD negativity at a further log reduction than ‘immunophenotypic complete response’ [13]. The United States Food and Drug Administration considers that MRD status by MFC, if appropriately validated and standardized, could be used as a surrogate biomarker of response replacing the usual clinical endpoints in trials evaluating next-generation novel therapies [14].
As laboratories accommodate the latest MFC-based response criterion and adopt an MM MRD assay more sensitive than previous assays, the need for inter-laboratory harmonization of the assay becomes more imperative, particularly for comparing results within and across clinical trials. The widespread implementation of MFC-based MRD testing, however, has been difficult because laboratories use different staining methods, antibody clones and conjugates, analysis strategies, and assays with different detection sensitivities [15, 16]. A survey of laboratories in the United States conducted by Flanders et al. in 2013 demonstrated substantial variability in methodologies and a 100-fold difference in assay sensitivity between laboratories [15]. The survey also found considerable disparities regarding the composition of antibody panels used for staining bone marrow samples. As a result, the definitions used to immunophenotypically define abnormal PCs differed widely among the institutions, with less than half utilizing commonly affected markers such as CD27, CD81, and CD117 when characterizing abnormal PCs [15]. After the publication of international consensus for MM MRD testing by flow cytometry, the same group revealed more laboratories were analyzing MM MRD by MFC and that the overall detection sensitivities reported had improved considerably in a 2016 follow-up survey [17]. Minimal acceptable cell acquisition numbers, implementation of universal staining methods, and a consistent data analysis strategy, however, remained lacking. Therefore, despite consensus guidance on sample staining and data acquisition, analysis and reporting, quality requirements of the assay [18–20], and a reference method [21], there remains a degree of heterogeneity between laboratories amenable to further reduction [16].
Achieving harmonization in the analysis and reporting of MM MRD by flow cytometry is an important step for the use of this biomarker within and across clinical trials, and for broader acceptance as a surrogate clinical endpoint for guiding personalized therapy in potentially all MM patients [6, 22]. An agreed reference method for the quantification of MM MRD by MFC and assessment of sample quality would also assist laboratories seeking to gain and monitor proficiency in performing the assay.
The goal of this study was to develop and demonstrate the efficacy of a consensus algorithm for MM MRD analysis in reducing inter-laboratory variation in both MRD quantification and defining sample quality, agnostic to proprietary software. Participants from seventeen laboratories around the world independently analyzed the same listmode flow cytometry standard (FCS) data files, initially using the local laboratory-established approach. The group discussed the pooled results and developed a provisional consensus analysis strategy. Participants re-analyzed the same files using the agreed approach, accommodating minor modifications to gating after further discussion. A second set of FCS files was then analyzed in the last stage using the final consensus approach. We showed that the adoption of a harmonized approach for the analysis of flow cytometry data derived from bone marrow sampled for response assessment meaningfully reduced the differences in MM MRD levels reported and in the assessment of sample adequacy by a group of international flow cytometry experts.
METHODS:
Participants
The first invitation to participate in this proficiency study was sent to sixteen laboratories with experience performing MM MRD testing by MFC. Twelve invitees responded and confirmed their desire to contribute; the remaining 4 institutions either did not respond or declined to take part in this exercise. To increase the power of the study, 10 additional participants were recruited in January 2019. Because the previous 12 participants had already completed Stage 1 of the study when the additional participants were recruited, newcomers were asked to neither share nor discuss the content of the proficiency study with those who had already submitted their Stage 1 results. Stage 1 was officially closed in March 2019 after all the participants (n = 22) returned their results. One institution was subsequently excluded after requesting removal from the study, and another participant was removed because of duplicated lab source.
All institutions who successfully completed Stage 1 were automatically invited to continue their participation in Stage 2. One institution did not respond and was excluded from all analyses. Similarly, those who completed Stage 2 were invited to participate in Stage 3 of the study, which was officially closed in August 2020. Two of the remaining 19 participants did not complete the final stage and their results were removed from all analyses. Results from the final total of 17 participants were compiled and reported in this manuscript.
Study Design
Manual data analysis can be one of the most challenging and time-consuming components of a flow cytometric experiment and is prone to inter-observer variation even among experienced practitioners [23]. To understand the degree of variation regarding MM MRD data analysis that exists, a proficiency study was conducted; each participant was asked to analyze the same data files using their preferred methods (Stage 1) vs. a harmonized approach (Stage 2 and Stage 3). Because the data files evaluated by all participants were identical, it was assumed that any observed variabilities can be attributed to how the data set was analyzed by each laboratory.
The study was reviewed and approved by the Institutional Review Board at Mayo Clinic (study ID #12-003246) and Roswell Park Comprehensive Cancer Center (study ID #00001597). In Stage 1 and Stage 2 of the study, routine bone marrow aspirate samples and FCS files from eight patients were acquired from the Clinical Flow Laboratory at Roswell Park Comprehensive Cancer Center (Buffalo, NY). The patients had either suspected MRD or no hematological malignancy. Samples were stained using the 2 tube 8-color MM MRD panel recommended by International Clinical Cytometry Society (ICCS) and European Society for Clinical Cell Analysis (ESCCA) consensus groups [18–20]. Briefly, both tubes were stained with backbone fluorochrome-conjugated antibodies consisting of CD45 PCPCy5.5, CD19 PECy7, CD56 PE, CD38 FITC, CD138 BV421, and CD27 BV510. In Tube 1, CD117 APC and CD81 APCH7 were added and in Tube 2 cKappa APC and cLambda APC-C750 were used. In Stage 3 of the study, participants analyzed 10 different FCS files which were derived from five patients at Roswell Park, and five from the Mayo Clinic (Rochester, MN). These samples were stained similarly using the antibody panel described above; see Supplementary Table 1 for a description of the antibodies source and clone used in the study. All FCS files were de-identified and blinded to disease status.
In Stage 1, participants were asked to analyze eight FCS files using their own current laboratory-defined protocol and record on a supplied Excel spreadsheet the following data: ‘Total number of cells’, ‘Normal PCs’, ‘Abnormal PCs’, ‘MRD%’, ‘Limit of Detection (LOD)’, ‘Lower Limit of Quantification (LLOQ)’, and the number of mast cells, hematogones, myeloid and erythroid precursors. Participants were also required to record their interpretation as to whether the sample was MRD positive, negative, or undetermined; and whether the sample was considered of sufficient quality to determine MRD status based on the presence of a sufficient number of the aforementioned bone marrow-associated cell populations. Individual Excel files were returned for central compilation and analysis of agreement. Stage 1 of the study was followed by a conference call where an initial consensus gating analysis strategy was drafted, and then refined after a follow-up survey.
In Stage 2 of the study, participants were asked to re-analyze the same FCS data files examined in Stage 1 using the drafted consensus gating approach. Following central review of results from Stage 2, the draft consensus document was updated to further address gating of cell populations that define sample adequacy, and a final consensus document was created. This is described in detail in the results section.
Stage 3 of the study involved analysis of a fresh set of FCS files from an additional 10 patients with or without MM MRD to verify the results were free of observer bias, using the final approach developed after Stage 2.
Statistical Analysis
For the analysis and reporting of the provided data files, participants were given the flexibility to utilize their own preferred analysis software package throughout the study. Of the 17 participants who completed all stages of the exercise, the software distribution of participants was as follows: Four used Kaluza v1.5 and v2.1 (Beckman Coulter), one used CytoPaint v1.1 (Leukobyte), one used WinList v9.1 (Verity Software House), two used FCS Express v4, v6, and v7 (De Novo Software), and nine used Infinicyt v2.0 (Cytognos).
For evaluating the degree of discrepancy of reported values between institutions, coefficient of variation (CV) was used as the primary measure. These reported values were compared between Stage 1, Stage 2, and Stage 3 to evaluate consistency of the participants in applying the consensus approach. The percent CV was calculated by dividing the standard deviation by the mean of measure and multiplied by one hundred percent. All data points that were greater than two standard deviations from the mean were regarded as outliers and removed from the analysis.
To evaluate the suitability of mast cells, hematogones, abnormal PCs, erythroid precursors, and myeloid precursors as reflections of sample adequacy, area under receiver operating characteristic curve was computed using Prism v5.03 (GraphPad). Different combinations of true positive and false positive were constructed on a bivariate plot to calculate area under curve (AUC). An ideal cell population for informing sample quality would have an AUC close to 1.00 while an AUC of 0.5 would represent a completely uninformative cell population. An AUC of > 0.5 was used as the minimum criteria to define a cell population that showed any positive correlation with sample adequacy.
RESULTS:
Stage 1: Independent Analysis of MM MRD Data Files using Existing Laboratory Approaches
In Stage 1 of the study, a total of 16 de-identified data files collected from 8 patients with suspected MM MRD were examined. Participants were asked to determine the MRD status of each sample and the frequency of normal and abnormal PCs. Using their own analytical methods and experience, 100% of the participants agreed that Samples #1 and #2 were MRD+ (Figure 1A). Samples #4, #5, #6, and #7 were considered to be MRD+ by 88%, 65%, 94%, and 88% of the participants, respectively. Samples #3 and #8 were considered MRD− by the majority of investigators (Figure 1B); however, 29% and 35%, respectively, concluded these samples to be MRD+ (Figure 1A). Five institutions reported they were unsure about the MRD status of either Samples #3 (12%) or Sample #5 (18%) (Figure 1C). The MRD level for each sample can be found in Supplementary Table 2 and the phenotypic profiles of the myeloma cells for MRD+ samples can be found in Supplementary Table 3.
Figure 1. Comparison of participant identification of MRD status between Stages 1 and 2.
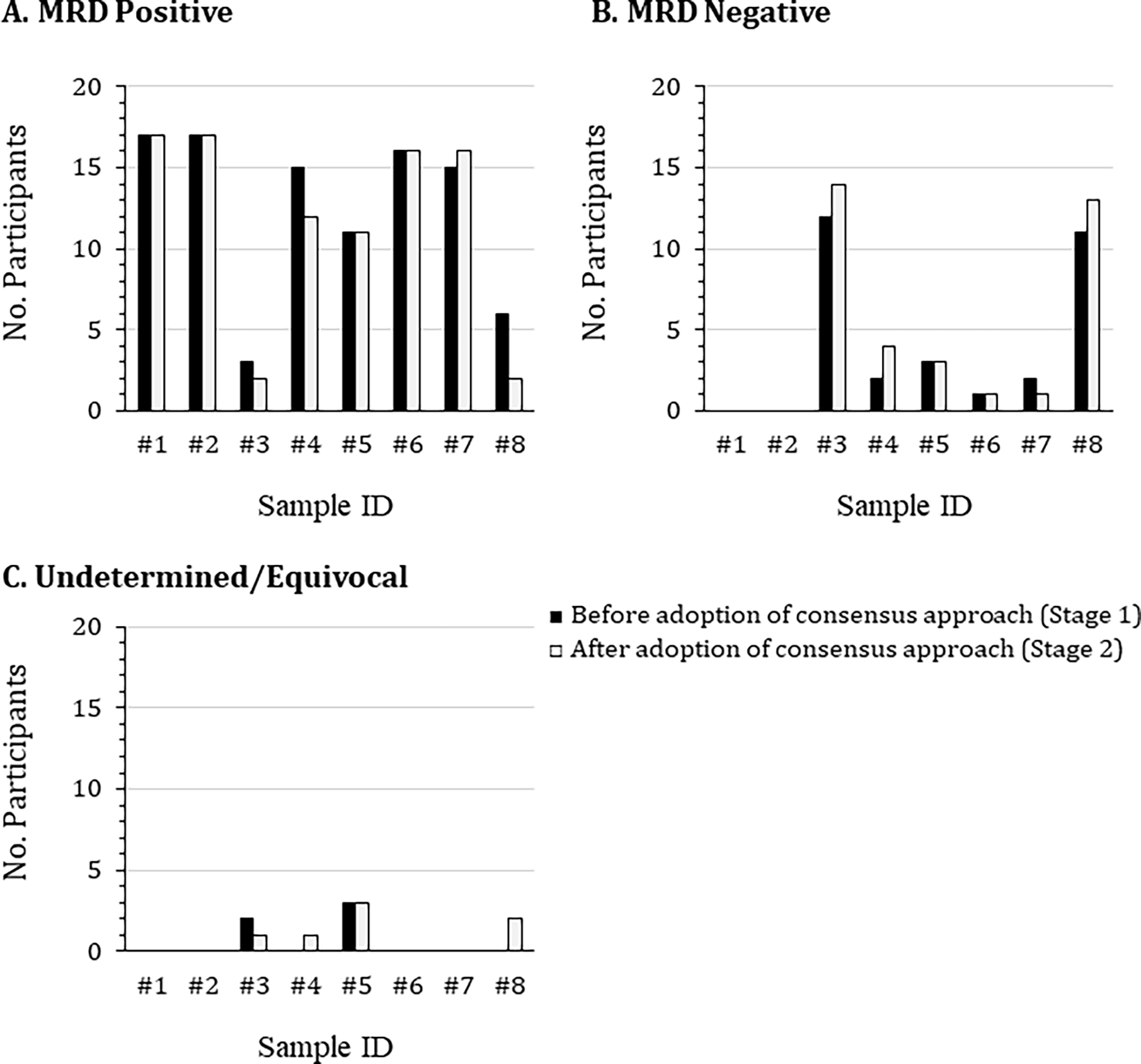
Eight de-identified flow cytometric standard (FCS) data files were distributed to 17 participants at the beginning of the study. These files related to bone marrow aspirate samples collected at Roswell Park from patients with suspected MM MRD or with no known hematological malignancy. The samples were stained using a 2 tube 8-color panel containing consensus markers agreed by a joint initiative of the International Clinical Cytometry Society and European Society for Clinical Cell Analysis. Participants first analyzed the FCS files using their own laboratory-established protocol (black bars), then repeated the analysis using the agreed upon draft consensus analysis protocol (white bars), concluding whether the samples were either (A) MRD positive, (B) MRD negative, or (C) undetermined/equivocal.
Participants were also asked to report the ‘Total Number of Analyzed Cells’, which is commonly used as the denominator to calculate percentages of each cell population in the final report. The accuracy and reproducibility of this denominator are important as it relates closely to the detection sensitivity of the assay. As shown in Table 1, there was a considerable degree of variation in the enumeration of ‘Total Number of Analyzed Cells’, ‘Total Normal PCs’, and ‘Total Abnormal PCs’ demonstrated among participants. Consequently, there was major heterogeneity in calculated LOD and LLOQ which was further compounded by participants differing in the number of cells considered sufficient for defining LOD (20 – 30 events) and LLOQ (50 – 62 events).
Table 1.
Variation in the measurement of MM MRD related parameters between participants using their own analysis method (Stage 1).
| Sample No. | #1 | #2 | #3* | #4 | #5 | #6 | #7 | #8* | Mean CV |
|---|---|---|---|---|---|---|---|---|---|
| Total Cells Analysed | |||||||||
| Mean | 6,707,285 | 6,846,778 | 2,186,043 | 6,993,562 | 7,167,311 | 7,900,975 | 7,379,873 | 4,181,342 | |
| SD | 2,526,914 | 2,188,275 | 586,730 | 2,159,761 | 2,319,217 | 2,381,478 | 2,361,154 | 1,431,501 | |
| CV, % | 37.67 | 31.96 | 26.84 | 30.88 | 32.36 | 30.14 | 31.99 | 34.24 | 32.01 |
| Total Normal PCs | |||||||||
| Mean | 2,098 | 298 | 1,014 | 302 | 3,136 | 6,696 | 423 | 332 | |
| SD | 1,031 | 118 | 435 | 148 | 1292 | 2676 | 1,382 | 165 | |
| CV, % | 49.14 | 39.60 | 42.90 | 49.01 | 41.20 | 39.96 | 326.71 | 49.70 | 79.78 |
| Total Abnormal PCs | |||||||||
| Mean | 436 | 664 | 71 | 133 | 81 | 5,355 | 8,949 | 79 | |
| SD | 234 | 311 | 187 | 96 | 60 | 2,676 | 5,612 | 150 | |
| CV, % | 53.67 | 46.84 | 263.38 | 72.18 | 74.07 | 49.97 | 62.71 | 189.87 | 101.59 |
| MRD Level, % | |||||||||
| Mean | 0.0063 | 0.0096 | 0.0034 | 0.0019 | 0.0011 | 0.0660 | 0.1117 | 0.0019 | |
| SD | 0.0026 | 0.0033 | 0.0077 | 0.0010 | 0.0007 | 0.0247 | 0.0548 | 0.0031 | |
| CV, % | 41.27 | 34.38 | 226.47 | 52.63 | 63.64 | 37.42 | 49.06 | 163.16 | 83.5 |
| Limit of Detection, % | |||||||||
| Mean | 0.0004 | 0.0004 | 0.0011 | 0.0004 | 0.0004 | 0.0003 | 0.0003 | 0.0006 | |
| SD | 0.0003 | 0.0002 | 0.0005 | 0.0002 | 0.0002 | 0.0002 | 0.0001 | 0.0003 | |
| CV, % | 75.00 | 50.00 | 45.45 | 50.00 | 50.00 | 66.67 | 33.33 | 50.00 | 52.55 |
| Lower Limit of Quantification, % | |||||||||
| Mean | 0.0009 | 0.0008 | 0.0025 | 0.0008 | 0.0008 | 0.0007 | 0.0008 | 0.0014 | |
| SD | 0.0005 | 0.0003 | 0.0008 | 0.0003 | 0.0003 | 0.0002 | 0.0003 | 0.0005 | |
| CV, % | 55.56 | 37.50 | 32.00 | 37.50 | 37.50 | 28.57 | 37.50 | 35.71 | 37.73 |
| Total Mast Cells | |||||||||
| Mean | 333 | 619 | 396 | 129 | 1,283 | 678 | 319 | 168 | |
| SD | 1,084 | 2,214 | 1,396 | 228 | 4,536 | 1,729 | 228 | 311 | |
| CV, % | 325.53 | 357.67 | 352.53 | 176.74 | 353.55 | 255.01 | 71.47 | 185.12 | 259.7 |
| Total Hematogones | |||||||||
| Mean | 50,829 | 5,429 | 18,384 | 6,883 | 64,394 | 358,146 | 7,643 | 20,138 | |
| SD | 205,308 | 18,641 | 41,067 | 9,859 | 265,795 | 489,049 | 23,486 | 24,586 | |
| CV, % | 403.92 | 343.36 | 223.38 | 143.24 | 409.33 | 136.55 | 307.29 | 122.09 | 261.15 |
Samples considered to be MRD-negative
Bold values represent maximum, minimum, and mean CVs
Post Stage 1: Creation of Initial Consensus Approach
At the completion of Stage 1, combined results were disseminated for review and discussion by teleconference. A consensus analysis algorithm and template were drafted taking into consideration discrepancies in results between participants and reasons for the elevated CVs. A major difference between the different flow cytometry software packages utilized among participants that contributed to the high CVs observed was that, in contrast to the others, Infinicyt allows merging of files such that the number of events from both Tube 1 and Tube 2 can be added into a final report. Participants using other software enumerated cells from each tube individually and reported the results from Tube 1 which had not undergone cytoplasmic processing. Consequently, the enumerated ‘Total Number of Analyzed Cells’, ‘Total PCs’, ‘Normal PCs’, ‘Abnormal PCs’, and ‘Total Number of Hematogones’ reported by Infinicyt users were twice as many as reported by users of other software. This also affected both LOD and LLOQ which were more sensitive using the Infinicyt approach versus the other software. To achieve consistency between participants and to acknowledge the overall robustness of flow-based MM MRD analysis, it was agreed in the survey following Stage 1 that the enumeration of cells from both tubes should be combined when the results are reported.
Another important observation arising out of Stage 1 was the different gating strategies employed by participants to enumerate the number of ‘Total Analyzed Cells’ (used as the denominator to calculate all percentages). Importantly, some participants used total leukocytes, whereas others used total nucleated cells. There were also differences in how Total PCs were identified, and in the discrimination between normal and abnormal PCs. The group discussed and eventually agreed on the following gating strategies and nomenclature for labelling of gating regions.
Gating Strategy: Total Number of Analyzed Cells
Four different strategies to enumerate the denominator cell population were proposed and participants agreed on the following approach to derive the total number of ‘nucleated cells’ (Figure 2). To define ‘nucleated cells’, the algorithm should first include a gate(s) on a bivariate plot of time vs. forward scatter area (FSC-A) to exclude invalid events such as air bubbles or other interruptions during event acquisition, followed by the partitioning of events on a bivariate plot of FSC-A vs. FSC-H to exclude doublets. ‘Nucleated Cells’ are then defined using a combination of FSC-A vs. side scatter area (SSC-A) and CD45 versus SSC-A to exclude debris, apoptotic, and aggregated events.
Figure 2. Consensus gating strategy for (A) ‘Total Analyzed Cells’ and (B) ‘Total PCs’.
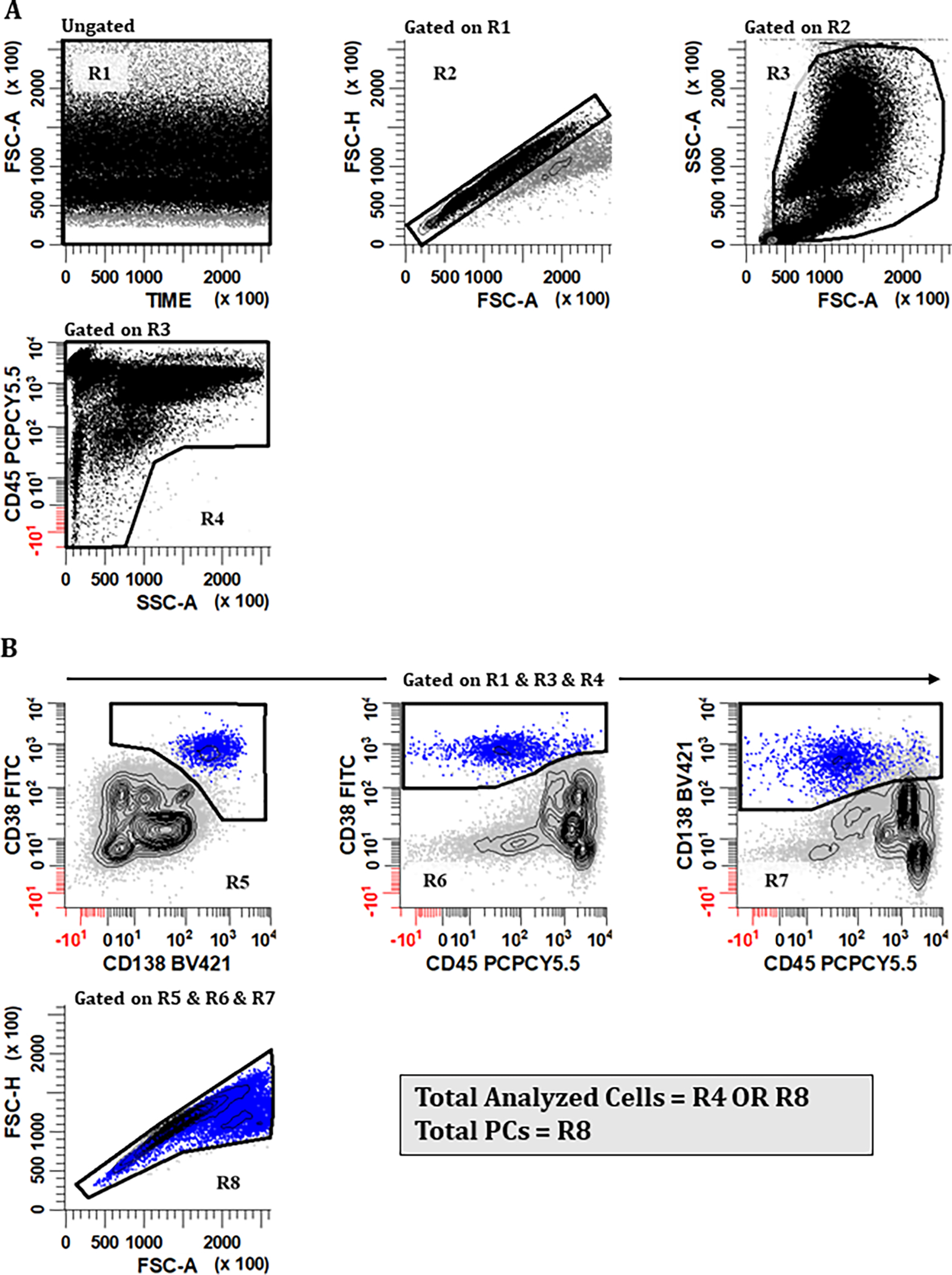
A harmonized gating approach that can be used for calculating the ‘Total Number of Analyzed Cells’ is important because it is the denominator used in all calculations of population frequencies. (A) Valid events that are singlets and free of large cell aggregates/debris are first identified using a combination of regions. To accomplish this, a region (R1) is created on a bivariate plot of Time vs FSC-A. A rectangular region (R2) is drawn on a bivariate plot of FSC-A vs. FSC-H gated on (R1) to identify singlet events. An irregular region (R3) is then placed on a bivariate plot of FSC-A vs. SSC-A gated on (R1) & (R2) to circumscribe cellular events that are not debris, apoptotic, or large aggregates. A region (R4) is drawn on a plot of SSC-A vs CD45 gated on (R1) & (R2) & (R3) to include CD45+ leukocytes, CD45− erythroid precursors, and CD45-/dim aberrant PCs. (B) For the identification of ‘Total PCs’, a combination of (R1) & (R3) & (R4) is initially used (R2 is intentionally omitted from this Boolean product). On bivariate plots of CD138 vs. CD38, CD45 vs. CD38, and CD45 vs. CD138 all gated on (R1) & (R3) & (R4), regions (R5), (R6), and (R7) are drawn, respectively, to circumscribe PCs expressing CD38br, CD138+, and CD45+/−. Events that satisfy the Boolean product (R5) & (R6) & (R7) are then displayed on a bivariate plot of FSC-A vs. FSC-H. and an irregular region (R8) is drawn to identify ‘Total PCs’, taking precaution to exclude events that are off scale based on FSC-A and SSC-A, which are considered to be true doublets. The final denominator value ‘Total Analyzed Cells’ is therefore represented by the formula (R1) & (R2) & (R3) & (R4) OR (R1) & (R3) & (R4) & (R5) & (R6) & (R7), summarized as (R4) OR (R8), which allows for the inclusion of unusually large and hyperdiploid PCs that fall outside of (R2). All plots were generated using WinList v9.1 (Verity Software House).
As shown in Figure 2A, valid events that were singlets and free of large cell aggregates/debris were first identified using a combination of regions. A region (R1) is created on a bivariate plot of Time vs FSC-A to identify events that are valid.
A rectangular region (R2) gated on (R1) is drawn on a bivariate plot of FSC-A vs. FSC-H to identify singlet events. Note: The combinations of FSC-A, FSC-H, or FSC-W used for the elimination of doublets are instrument dependent. For example, on a FACS Canto instrument (BD Biosciences), the combination of FSC-A vs. FSC-H is considered best for eliminating doublets, whereas the Navios instrument (Beckman Coulter) can additionally utilize ‘time of flight’ for this purpose.
Gated on (R1) & (R2), an irregular region (R3) is then placed on a bivariate plot of FSC-A vs. SSC-A to circumscribe cellular events that are not debris or large aggregates.
Gated on (R1) & (R2) & (R3), a large polygonal region (R4) is drawn to include CD45+ leukocytes, CD45− erythroid precursors, and CD45− aberrant PCs. Events that satisfy the Boolean logic of (R1) & (R2) & (R3) & (R4) are then considered to be the ‘Total Number of Analyzed Cells’ for calculating the percentages in MM MRD reports.
Gating Strategy: Total Plasma Cells
As described above, doublet exclusion is necessary to eliminate events consisting of two or more distinct cells. Abnormal PCs, however, can be large and hyperploid, causing them to fall within the region (based on FSC-A versus FSC-H) which most flow cytometrists would consider as ‘doublets’ and consequently eliminate them from the analysis. The consensus group agreed a modified strategy was needed to include all PCs including those with a higher area-to-height ratio. In this approach, two singlet gating strategies are combined using Boolean logic, one excluding leukocyte doublets from the ‘Total Nucleated Cells’ analysis (Figure 2A) and another identifying all PCs (Figure 2B) including those with high FSC-A to FSC-H ratio.
Bivariate plots of CD138 vs. CD38, CD45 vs. CD38, and CD45 vs. CD138 are created and gated on (R1) & (R3), intentionally omitting (R2). To these dot plots, regions (R5) & (R6) & (R7) are drawn to define PCs expressing CD38br, CD138+, and CD45+/−, respectively.
A second FSC-A versus FSC-H dot plot this time gated on (R1) & (R3) & (R5) & (R6) & (R7) is created and an irregular region (R8) is drawn to include all PCs including those not aligned in the diagonal line typically considered to be doublet events. Those events off scale based on FSC-A are considered as true doublets.
The combined events described by [(R1) & (R2) & (R3) & (R4)] OR [(R1) & (R3) & (R5) & (R6) & (R7) & (R8)] are defined as the true value representing ‘Total Number of Analyzed Cells’.
Gating Strategy: Discriminating Normal vs. Abnormal PCs
Distinguishing myeloma cells reliably and consistently from normal PCs based on their antigen expression profiles is perhaps the most challenging aspect of the analysis as there is no single typical ‘myeloma immunophenotype’. Moreover, small subpopulations of normal PCs may show antigen expression combinations mimicking multiple myeloma that are not readily apparent at lower acquisition numbers and may only be discernable as normal by cytoplasmic light chain restriction. As a discrete analysis approach that can be used by all qualified analysts to generate identical and/or reproducible results is currently unavailable, the participants considered several approaches to reliably discriminate myeloma cells from normal PCs.
In the selected strategy, the participants agreed that after total PCs were identified, CD19 and CD56 should be used to initially screen for potential abnormal PCs. Four populations of PCs are defined by quadrants (a) through (d) on a bivariate plot depicting CD56 vs. CD19 gated on Total PCs derived from Tube 1, repeating the steps for Tube 2 (Supplementary Figure 1). Then, different combinations of bivariate plots are gated on each quadrant and reviewed to gain an appreciation of the likely patient-specific myeloma phenotype for more standardized gating and enumeration (as outlined later in Figure 3). Specifically for Tube 1, bivariate plots of CD38 vs. CD117, CD45 vs. CD38, CD45 vs. CD27, and CD81 vs. CD27 are gated on quadrants (a) through (d) and the steps are repeated for Tube 2, using plots CD45 vs. CD38, CD45 vs. CD27, and cLambda vs. cKappa. Normal PCs typically express CD19+, CD56−, CD45+, CD38br, CD117−, CD81+, CD27+, and show polyclonal light chain expression. Any expression patterns that deviate from that of normal PCs would be considered suspicious and further investigation warranted.
Figure 3. Consensus gating strategy for defining ‘Abnormal PCs’ using (A) Tube 1 and (B) Tube 2.
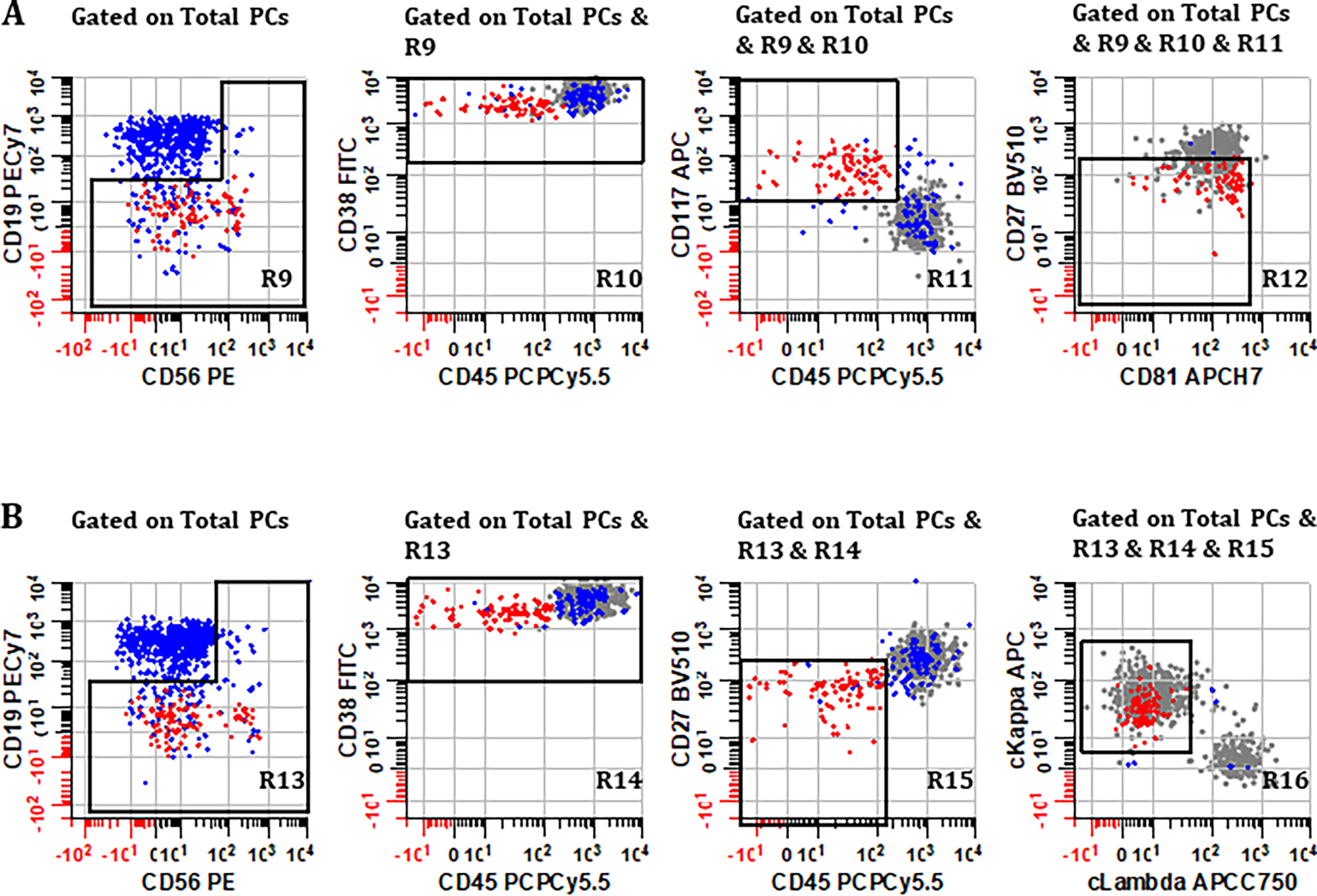
Normal PCs (blue events) typically express CD19+, CD27+, CD38br, CD45+, CD56−, CD81+, CD117−, and polytypic cytoplasmic light chains, whereas abnormal PCs (red events) usually show aberrant expression of two or more markers with demonstrable monotypic light chain expression. Gray events represent population of normal PCs that will be discarded during the sequential gating procedure; the gray events are only displayed here for illustration purposes. Using one of the cases as an example, (A) plots derived from Tube 1 gated on ‘Total PCs’ (R8) show how abnormal PCs are defined by sequentially gating on regions (R9), (R10), (R11), and (R12) created on bivariate plots of CD56 vs. CD19, CD45 vs. CD38, CD45 vs. CD117, and CD81 vs. CD27, respectively. These regions are drawn generously enough to target known deviations in normal antigen expression on PCs without inadvertently excluding abnormal events, particularly if no demarcation between normal and abnormal PC is evident on a specific bivariate plot. (B) Using plots derived from Tube 2, the sequential gating algorithm is repeated, this time focusing additionally on light chain expression of events that satisfy the Boolean product of regions (R13) & (R14) & (R15) & (R16) drawn on bivariate plots CD56 vs. CD19, CD45 vs. CD38, CD45 vs. CD27, and cLambda vs. cKappa, respectively. All plots were generated using WinList v9.1 (Verity Software House).
The consensus gating strategy for definitive identification and enumeration of abnormal PCs is outlined by the following sequential steps and shown in Figure 3, using one of the cases as an example. It should be noted that in practice the immunophenotypic definition of aPCs will differ from patient-to-patient. In this regard, analysts can refer to Supplementary Figure 1 as a guideline to screen for potential phenotypic aberrancy of aPCs, so that appropriate permutation of markers can be selecting during gating process.
Starting with ‘Total PCs’ derived from Tube 1 (R8), a bivariate plot of CD56 vs. CD19 is first demarcated by a broad reverse L-shaped region R9 to encompass all PCs except CD19+, CD56− (unless these are suspected as abnormal during initial screen, which is rare).
Gating on R9, a rectangular region R10 is drawn on a bivariate plot of CD45 vs. CD38 to encompass abnormal PCs, ensuring some overlap with normal PCs at this stage.
Gating on R10, a rectangular region R11 is drawn on a bivariate plot of CD45 vs. CD117, again ensuring that all abnormal PC are included with some overlap with normal PC.
Gating on R11, a final rectangular region R12 is drawn on a bivariate plot of CD81 vs. CD27 to include all remaining PC considered abnormal. Highlighting events in R12 a different color assists with fine tuning of regions R9 through R12 to arrive at a final enumeration of abnormal PC (MRD) for Tube 1.
Repeating the steps for Tube 2, starting with ‘Total PCs’ (regions R1 through R8 are identical for each tube up to this point) a reverse L-shaped region R13 is drawn on a bivariate plot of CD56 vs. CD19.
Gating on R13, a rectangular region R14 is drawn on a bivariate plot of CD45 vs. CD38, separating abnormal PC if possible.
Gating on R14, a rectangular region R15 is drawn on a bivariate plot of CD45 vs. CD27, again separating abnormal PC if possible.
Gating on R15, a final region R16 is drawn on a bivariate plot of cLambda vs. cKappa, encompassing any PC population showing abnormally deviated cytoplasmic light chain expression, and representing a final enumeration of abnormal PC (MRD) for Tube 2.
Stage 2: Adoption of Draft Harmonized Approach for Data Analysis
In Stage 2, after the draft consensus protocol was adopted, participants were asked to reanalyze the eight FCS data files they had evaluated in Stage 1. The decision as to whether the samples were MRD−, MRD+, or undetermined were compared between Stages 1 and 2 (Figure 1). Overall, there was no changes in the number of individuals calling Samples #1, #2, #5, and #6 as MRD+. The number of individuals calling Samples #3 and #8 as MRD− increased by two. Additionally, two participants who called Sample #4 as MRD+ in Stage 1 changed their interpretation to MRD− and one changed their decision on Sample #7 from MRD− to MRD+. The coefficient of variations of the analyzed parameters can be found in Supplementary Table 4.
Stage 3: Adoption of Final Harmonized Approach for Data Analysis
As prior exposure to the FCS data during Stage 1 may have influenced the results from Stage 2, in order to further evaluate how successfully the consensus approach improved consistency among participants, the study was repeated using 10 new samples from MM patients with suspected MRD (i.e., Stage 3 of the study). Like Stage 2, the participants were asked to perform flow cytometric analysis on these MM MRD data files using the draft consensus protocol and report their findings.
The overall agreement about MRD status achieved in Stage 3 was 81.8% ± 11.6% (Table 2), which was comparable to Stage 1 (83.8% ± 15.0%) and Stage 2 (85.3% ± 13.7%). When the analysis was stratified based on MRD positivity and MRD negativity, it was found that the agreement on the determination of MRD status was higher among MRD+ sample (89.4% ± 8.6%) than MRD− samples (74.1% ± 7.0%).
Table 2:
Determination of MM MRD status using consensus analysis approach (Stage 3).
| Sample (n = 10) |
Participant (n = 17) | MRD Level, % (Mean ± SD) | Agreeability, % | ||
|---|---|---|---|---|---|
| Positive | Negative | Undetermined | |||
|
| |||||
| All Samples | |||||
| Average | 81.77 | ||||
| (SD) | (11.57) | ||||
|
| |||||
| MRD Positive | |||||
| #1 | 14 | 2 | 1 | 0.00130 ± 0.00072 | 82.4 |
| #3 | 17 | 0 | 0 | 0.00179 ± 0.00091 | 100 |
| #6 | 16 | 1 | 0 | 0.00203 ± 0.00070 | 94.1 |
| #7 | 16 | 0 | 1 | 0.00251 ± 0.00058 | 94.1 |
| #10 | 13 | 3 | 1 | 0.00086 ± 0.00045 | 76.5 |
|
| |||||
| Mean | 89.42 | ||||
| (SD) | (8.62) | ||||
|
| |||||
| MRD Negative | |||||
| #2 | 1 | 15 | 1 | 0.00002 ± 0.00006 | 88.2 |
| #4 | 5 | 12 | 0 | 0.00485 ± 0.01268 | 70.6 |
| #5 | 5 | 12 | 0 | 0.00023 ± 0.00048 | 70.6 |
| #8 | 2 | 12 | 3 | 0.00005 ± 0.00008 | 70.6 |
| #9 | 3 | 12 | 2 | 0.00024 ± 0.00082 | 70.6 |
|
| |||||
| Mean | 74.12 | ||||
| (SD) | (7.04) | ||||
Cell Enumeration is more Consistent with the Adoption of Consensus Analysis Approach
To empirically test if the adoption of the consensus analysis method would improve the consistency of reported values across multiple institutes, results for each stage were compared (Figure 4, see Supplementary Table 5 for the coefficient of variations of analyzed parameters). Significant improvement in the enumeration of ‘Total Number of Analyzed Cells’ was observed when a harmonized approach was adopted (Figure 4A). The variations were significantly reduced in Stage 2 and Stage 3 when compared to Stage 1 (Stage 1 vs. Stage 2: p < 0.001; Stage 1 vs. Stage 3 (p < 0.001). This improvement remained significant when the analysis was stratified into MRD− and MRD+.
Figure 4. Comparison of participants’ agreement on MRD status before and after adoption of a consensus analysis protocol.
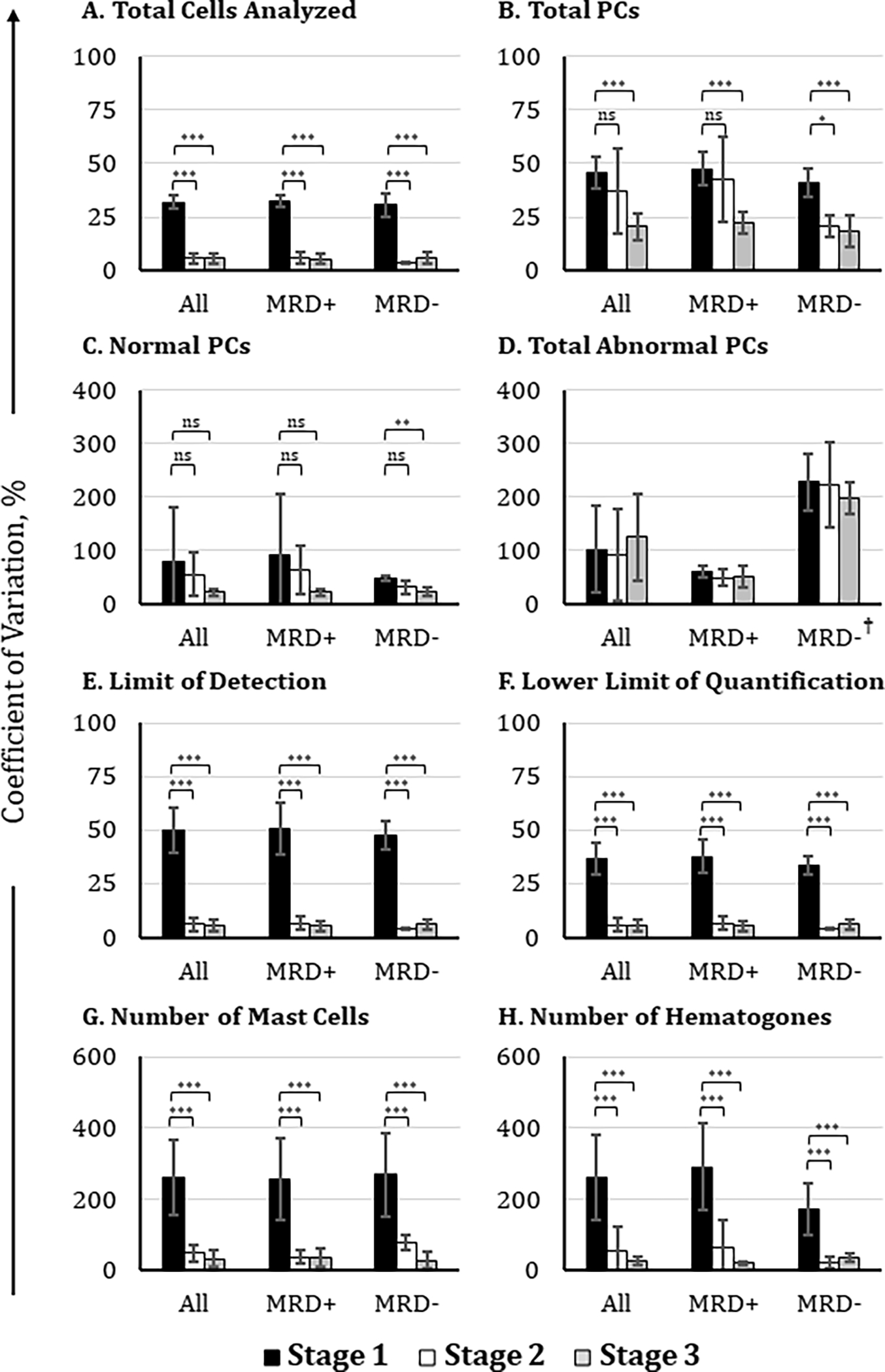
Participating institutions (n = 17) analyzed FCS data files of suspected MM MRD samples using their own laboratory-established protocol (black histograms), draft consensus analysis protocol (white histograms), and final consensus analysis protocol (gray histograms). Coefficients of variation were compared for the reported values of (A) total cells analyzed, (B) total PCs, (C) total normal PCs, (D) total abnormal PCs, (E) the limit of detection, (F) the lower limit of quantification, (G) mast cells, and (H) hematogones. The final coefficient of variation values shown here excluded outliers from the data set, which were defined as values greater than 2 standard deviations from the mean. *: p-value < 0.05; **: p-value < 0.01; ***: p-value < 0.001. †: high CV% observed because of the extremely low number of aPCs detected in MRD− samples
In Figure 4B, a significant decrease in variation was found when the consensus approach was applied to enumerate ‘Total PCs’ in Stage 3 (p < 0.001). When the CVs were stratified based on MRD status, the association between the measured CVs of MRD+ samples comparing Stage 1 vs. Stage 2 was not significant but was significantly improved between Stage 1 vs. Stage 3 (p < 0.001). In patients deemed to be MRD−, there was a significant improvement in the CV comparing both Stage 1 vs. Stage 2 (p < 0.05) and Stage 1 versus Stage 3 (p < 0.001).
There were no significant changes in the CVs of enumerated ‘Normal PCs’ between any of the stages (Figure 4C) but when stratified based on MRD status there was a significant reduction in CVs between Stage 1 and Stage 3 in samples deemed to be MRD− (p <0.01). There was no significant change in the ‘Number of Abnormal PCs’ between any of the Stages even when the analysis was stratified based on the MRD status (Figure 4D). By employing a consensus standard, significant improvements in both the ‘Limit of Detection’ and ‘Lower Limit of Quantification’ calculations were achieved with the harmonized approach (p < 0.001 in all comparisons; Figures 4E and 4F).
Sample Adequacy
The ability to detect rare abnormal PCs in the bone marrow is highly dependent on the quality of the sample collected. This observation was previously confirmed by Rawstron et al. who established that the concentration of abnormal PCs was highest in the first bone marrow aspiration and decreased significantly with each subsequent aspirate [9]. The presence of mast cells and hematogones have served as the cell populations of choice to evaluate the quality of bone marrow aspirates; these cell populations should theoretically be found only in the bone marrow of healthy individuals, although the presence of normal PCs, myeloid and erythroid precursors are also considered as evidence suggesting true bone marrow sampling [9, 19, 21]. To test if participants preferred to use mast cells or hematogones for determining sample adequacy, a regression analysis was performed to correlate the percentage of detected mast cells and hematogones with perceived sample adequacy. Our results demonstrated that the participants who identified samples to be adequate did not report a percentage of mast cells that was any different from those who claimed the samples were inadequate (Supplementary Figure 2A). This observation was also true for hematogones (Supplementary Figure 2B).
Prior to Stage 2, a consensus was reached among participants that a minimum of two bone marrow-derived cell populations should be evaluated to determine sample adequacy (Table 3). It was agreed mast cells should always be detectable, along with at least one other population. These were, in order of preference, hematogones, myeloid precursors, and erythroid precursors. The presence of abnormal PCs alone was not considered by the group to be a sufficient indicator of sample adequacy.
Table 3:
Analysis of participant survey following Stage 1 to determination sample adequacy
|
Option |
Sufficiency of bone marrow cell populations in defining an adequate sample as considered by each participant, n (%) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Very Sufficient | Somewhat sufficient | Somewhat insufficient | Not sufficient at all | No Response | ||
|
| ||||||
| Unweighted | ||||||
| Abnormal PCs only | 3 (18) | 4 (24) | 2 (12) | 5 (29) | 3 (18) | |
| Mast cells only | 1 (6) | 9 (53) | 4 (24) | 0 (0) | 3 (18) | |
| Hematogones only | 2 (12) | 5 (29) | 6 (35) | 1 (6) | 3 (18) | |
| Erythroid Precursors only | 0 (0) | 6 (35) | 3 (18) | 5 (29) | 3 (18) | |
| Myeloid Precursors only | 0 (0) | 6 (35) | 5 (29) | 2 (12) | 4 (24) | |
| 2 or more cell populationsa | 13 (76) | 4 (24) | 0 (0) | 0 (0) | 0 (0) | |
| All cell populations | 10 (59) | 0 (0) | 0 (0) | 1 (6) | 6 (35) | |
|
| ||||||
| Weightedb | Scores | |||||
|
|
||||||
| 4 | 3 | 2 | 1 | Weighted Total | ||
|
|
||||||
| Abnormal PCs only | 12 | 12 | 4 | 5 | 33 | |
| Mast cells only | 4 | 27 | 8 | 0 | 39 | |
| Hematogones only | 8 | 15 | 12 | 1 | 36 | |
| Erythroid Precursors only | 0 | 18 | 6 | 5 | 29 | |
| Myeloid Precursors only | 0 | 18 | 10 | 2 | 30 | |
| 2 or more cell populationsa | 52 | 12 | 0 | 0 | 64 | |
| All cell populations | 40 | 0 | 0 | 1 | 41 | |
Provided one of the cell populations is mast cells
Each response was given a weight and the results retabulated: very sufficient: 4; somewhat sufficient: 3; somewhat not sufficient: 2; not sufficient at all: 1; no response: 0 (omitted from table)
Bold type: Consensus criteria adopted for sample adequacy
To address the considerable heterogeneity seen with the enumeration of mast cells and hematogones in Stage 1 (Table 1; data for myeloid and erythroid precursors were not collected during Stage 1), explicit gating definitions for mast cells, hematogones, myeloid and erythroid precursors were developed during the creation of the draft consensus analysis strategy and applied to Stage 2. These harmonized gating strategies are described below:
Gating Strategy: Mast Cells
Only Tube 1 can be used for the identification of mast cells due to the lack of CD117 staining in Tube 2. To identify mast cells two steps are outlined below and in Figure 5A.
Figure 5. Gating strategies for identifying bone marrow derived cell populations important for defining sample quality: (A) mast cells, (B) hematogones, (C) myeloid precursors, and (D) erythroid precursors.
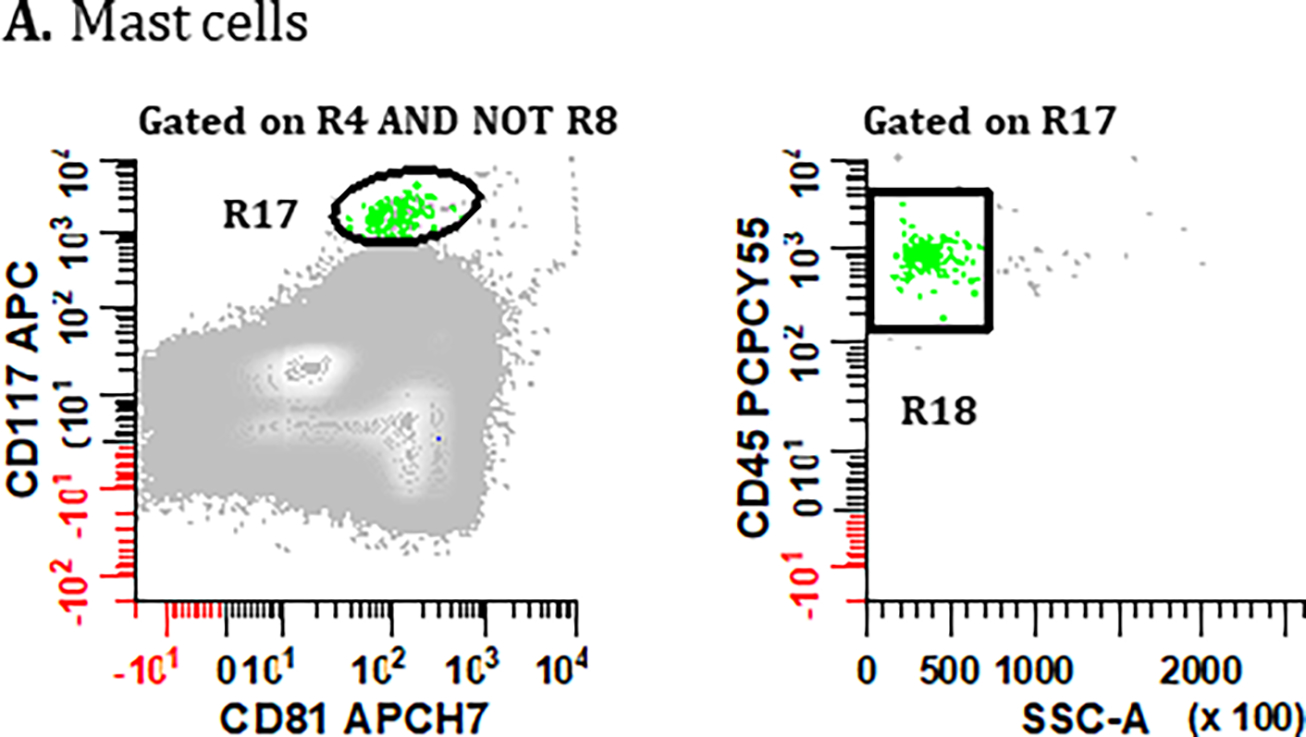
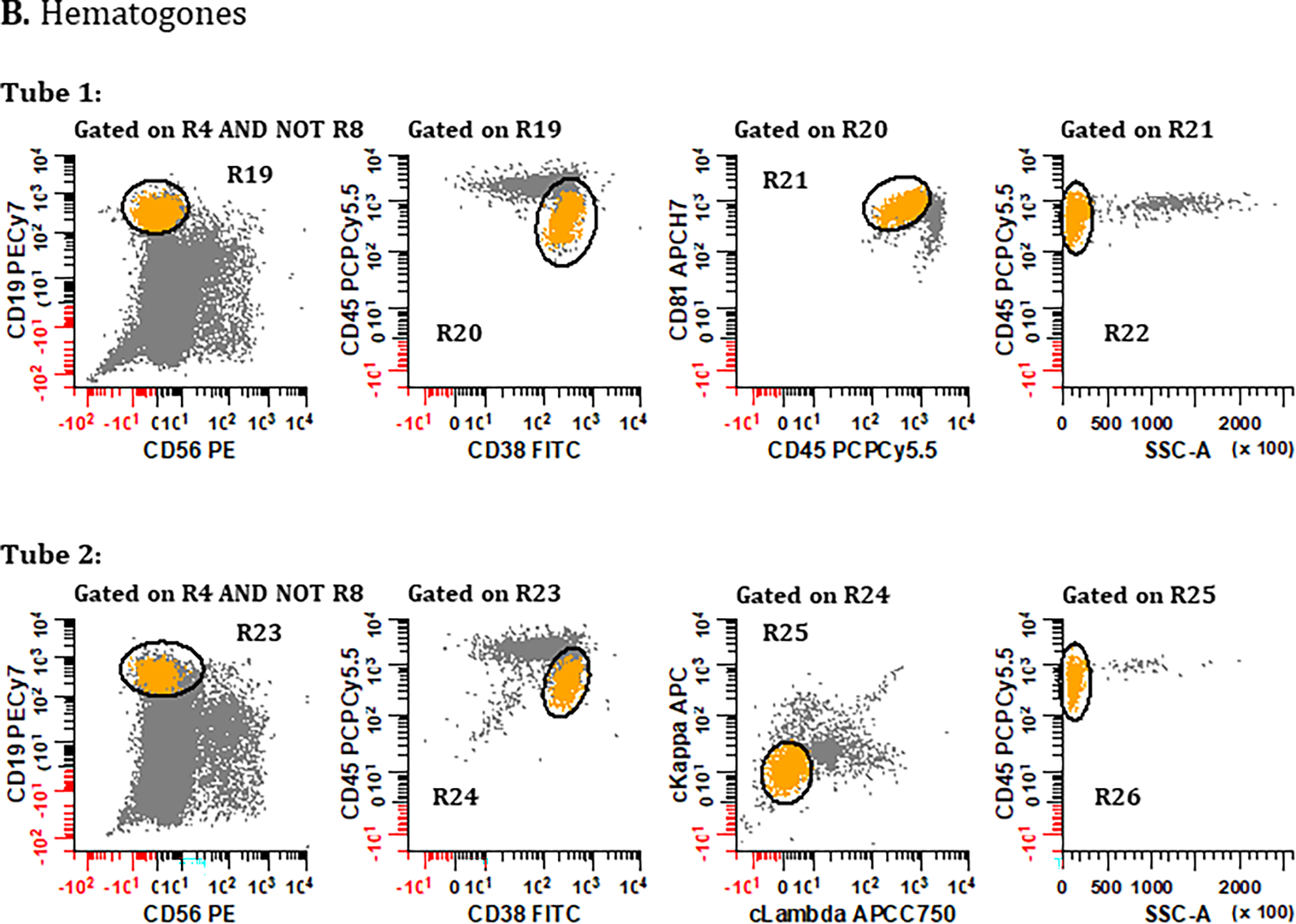
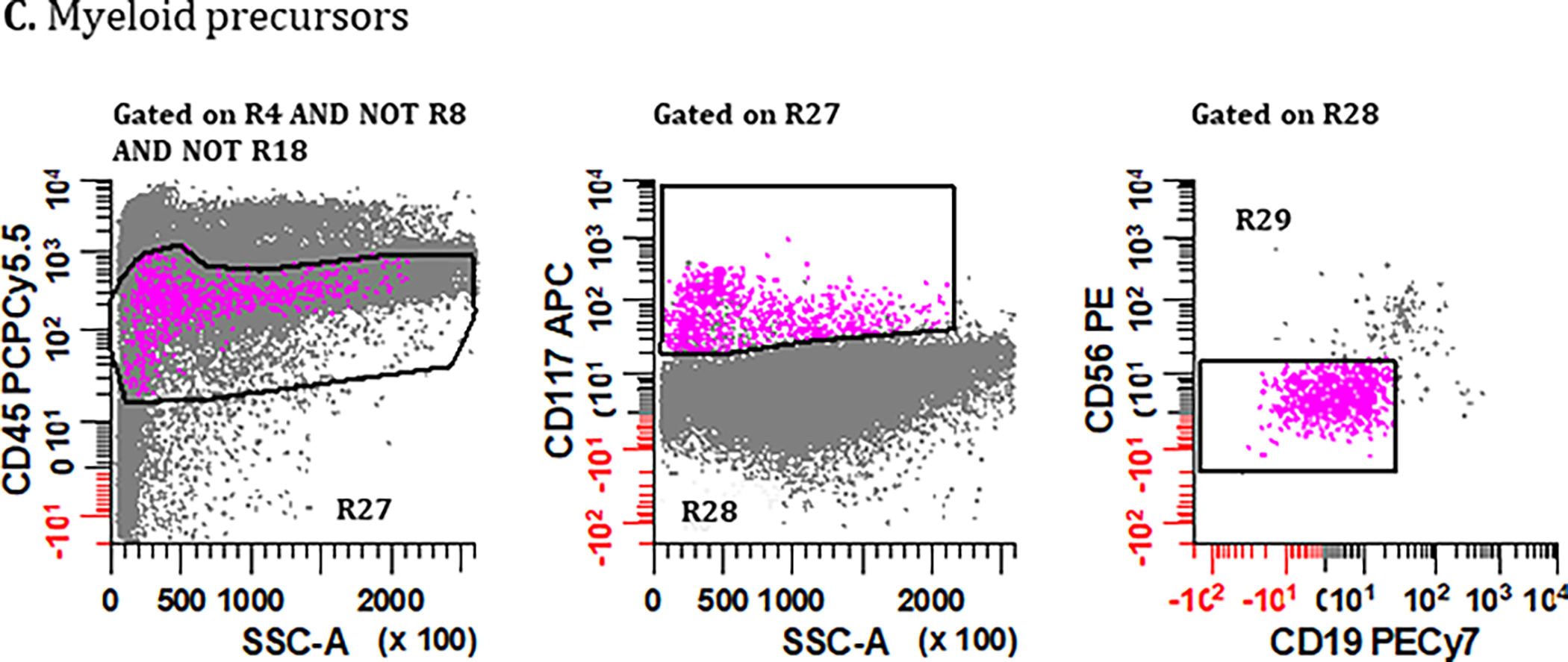
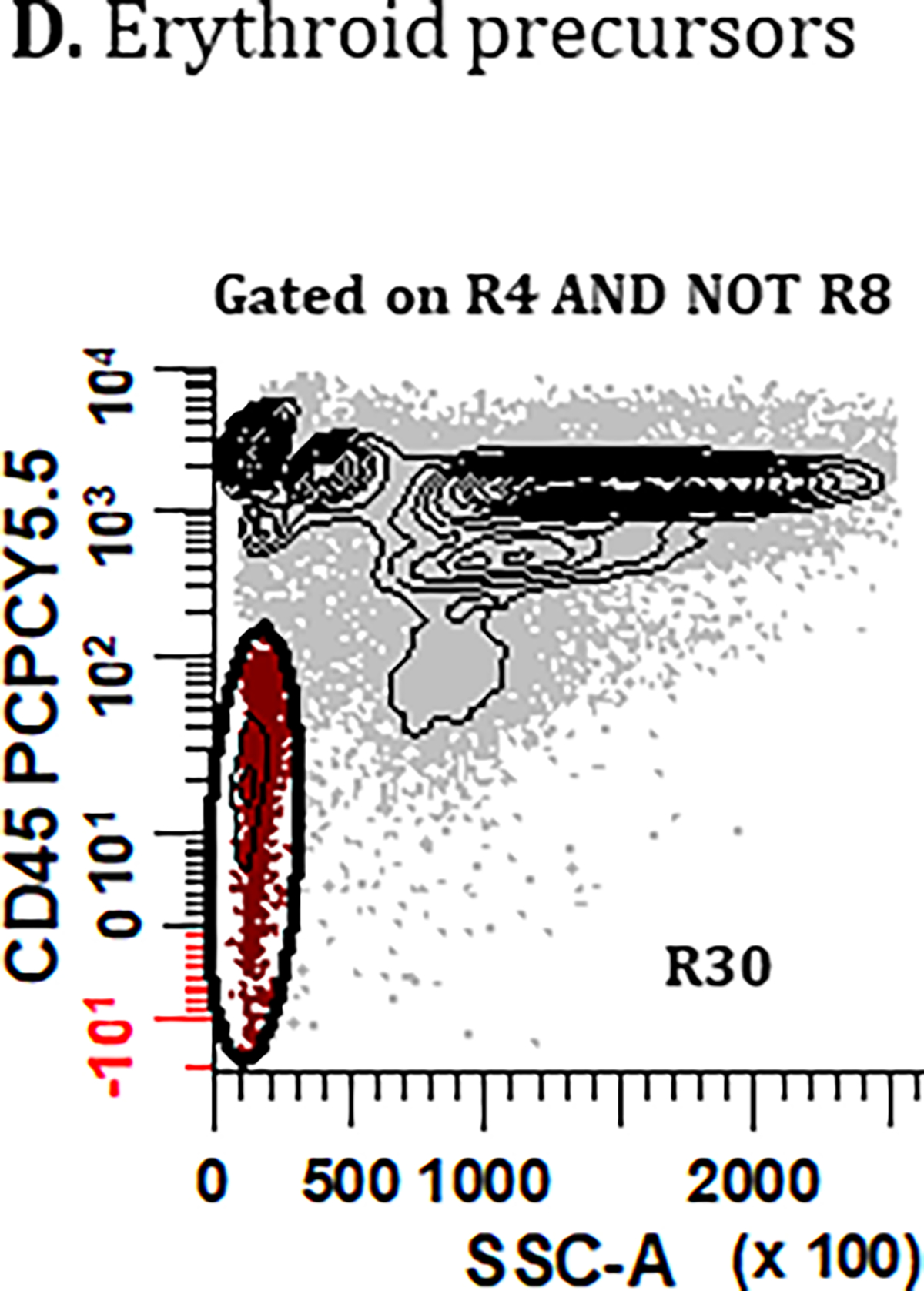
PCs are first excluded using the gating strategies displayed in Figure 2 but using the Boolean definition [(R1 & R2 & R3 & R4) AND NOT (R5 & R6 & R7)] summarized as R4 AND NOT R8. (A) To identify mast cells, a bivariate plot of CD81 vs. CD117 is then created and a region (R17) drawn to circumscribe events that are CD117br, CD81dim. A bivariate plot of SSC-A vs. CD45 gated on (R17) is then created and a region (R18) drawn to identify CD45br, SSClo events, defining the final number of ‘Mast Cells’. (B) For the identification of hematogones in Tube 1, a bivariate plot of CD56 vs. CD19 is created and a region (R19) is drawn to identify the CD56−, CD19+ events. On a bivariate plot of CD38 vs. CD45 gated on (R19), a region (R20) is drawn to identify cells that are CD38dim, CD45dim. Next, using a bivariate plot of CD45 vs. CD81 gated on (R20), a region (R21) is created to circumscribe events that are CD45dim, CD81br. Finally, on a bivariate plot of SSC-A vs. CD45 gated on (R21), a region (R22) is created to circumscribe the SSC-A low and CD45dim cells, defining the final number of ‘Hematogones’ enumerated from Tube 1. For the identification of hematogones in Tube 2, the first two bivariate plots and gating strategy used in Tube 1 are repeated, numbering the regions (R23) and (R24). The CD45 vs. CD81 plot is replaced with a bivariate plot of cLambda versus cKappa light chains gated on (R24) with a region (R25) that encompasses immunoglobulin light chain negative events. On a bivariate plot of SSC-A vs. CD45 gated on R25 an elliptical region (R26) is created to circumscribe a SSClo cluster, defining the final number of ‘Hematogones’ enumerated from Tube 2. (C) For the identification of ‘Myeloid Precursors’, mast cells are first excluded from the analysis by the Boolean formula R4 AND NOT R8 AND NOT R18. A bivariate plot of SSC-A vs. CD45 is then created and a broad irregular region (R27) is drawn to identify events that are SSC-A low, CD45dim. Using a bivariate plot of SSC-A vs. CD117 gated on (R27), a polygonal region (R28) is drawn to identify cells that are SSC-A low to medium, CD117+. Finally, using a bivariate plot of CD19 vs. CD56 gated on (R28), a region (R29) is placed to identify events that are CD19−, CD56−, which defines the final number of ‘Myeloid Precursors’. (D) Lastly, the final number of ‘Erythroid Precursors’ derived from Tube 1 is defined by an elliptical region (R30) drawn on a bivariate plot of SSC-A vs. CD45, capturing the cluster of SSC-A low/CD45− events. All plots were generated using WinList v9.1 (Verity Software House).
A bivariate plot of CD81 vs. CD117 gated on singlet cells devoid of PC is created (i.e., R4 AND NOT R8). A region (R17) is used to preliminarily define mast cells that express CD117br, CD81dim, the latter excluding any non-specific antibody binding events.
Gating on R17, a bivariate plot of SSC-A vs. CD45 is created, and a region (R18) drawn around a plausible cluster of CD45br, SSClo events to enumerate a final pure population of ‘Mast Cells’.
Gating Strategy: Hematogones
Despite difference in markers between Tube 1 and Tube 2 of the MM MRD panel, hematogones can be determined from each using slightly different strategies. For comparison, the coefficient of determination (R2) between reported percentages of hematogones in Tube 1 and Tube 2 in this study was 82.4%. The consensus identification of hematogones is outlined below and in Figure 5B:
Starting with singlet cells devoid of PC (i.e., R4 AND NOT R8) derived from Tube 1, a region R19 is first drawn on a bivariate plot of CD56 vs. CD19 to identify all CD19+ B cells excluding CD56+ non-specific coincident events.
Gating on R19, a region R20 is drawn on a bivariate plot of CD38 vs. CD45 placed to separate CD38+, CD45dim hematogones from mature B cells.
Gating on R20, this crudely defined population of hematogones is further refined by placing a region (R21) on a bivariate plot of CD45 vs. CD81 to identify events that are CD45dim, CD81br.
Gating on R21, a bivariate plot of SSC-A vs. CD45 is created, and a region (R22) drawn to exclude contaminating events with high SSC, encompassing a plausible cluster of CD45+, SSClo events that represent the final enumeration of hematogones derived from Tube 1.
Steps 1 and 2 are repeated using Tube 2, creating R23 on a bivariate plot of CD56 vs. CD19; and gating on R23, creating region R24 on a bivariate plot of CD38 vs. CD45.
Gating on R24, a bivariate plot of cLambda vs. cKappa is created to demarcate dual light chain negative B cell precursors using a region R25.
Lastly, gating on R25, a bivariate plot of SSC-A vs. CD45 is created, and a region (R26) drawn to exclude contaminating events with high SSC, encompassing a plausible cluster of CD45+, SSClo events that represent the final enumeration of hematogones derived from Tube 2.
Gating Strategy: Myeloid Precursors
Myeloid precursors can only be identified in Tube 1, relying on CD117 expression, and the following steps outline the consensus identification as shown in Figure 5C.
Starting with singlet cells devoid of both PCs and mast cells (i.e., R4 AND NOT R8 AND NOT R18) a bivariate plot of SSC-A vs. CD45 is created, and an irregular region R27 is drawn to encompass events that are SSC low, CD45dim.
Gating on R27, a region R28 is drawn on a bivariate plot of SSC-A vs. CD117 to broadly capture CD117+ events containing myeloid precursors including SSC-A heterogeneous promyelocytes.
Lastly, gating on R28, a bivariate plot of CD56 vs. CD19 is created and a region R29 drawn to exclude contaminating CD19+ and/or CD56+ coincident events, encompassing a plausible cluster of true CD45+, CD117+, and SSC-A variable events that represent the final enumeration of myeloid precursors and promyelocytes derived from Tube 1.
Gating Strategy: Erythroid Precursors
The identification of erythroid precursors could technically be achievable in both Tube 1 and Tube 2 of the MM MRD panel. The consensus from the group was, however, that only Tube 1 should be utilized (Figure 5D) because the additional fixation/permeabilization, and washing steps employed for Tube 2 will reduce the number of nucleated erythroid cells. Conversely, it should be noted that any degree of incomplete lysis in Tube 1 will potentially lead to an erroneously higher count of erythroid precursors and therefore this bone marrow-derived cell population was given the lowest priority for assessing sample adequacy. Consensus gating of erythroid precursors begins with a bivariate plot of SSC-A vs. CD45 gated on singlet cells devoid of PCs (i.e., R4 AND NOT R8). A region (R30) is used to encompass a cluster of SSClo, CD45-/dim events that represent the final enumeration of erythroid precursors derived from Tube 1.
Increased Reproducibility of Mast Cells and Hematogones Enumeration
Adoption of consensus gating significantly reduced variability in the enumeration of ‘Mast Cells’ and ‘Hematogones’ observed between Stage 1 and Stage 2 or Stage 3 (p < 0.001) (Figures 4G and 4H). This improvement remained when the analysis was stratified into MRD− and MRD+ subcategories.
Determination of Sample Adequacy Cut-off Values for Marrow Constituents
Receiver-operating characteristic (ROC) analysis was performed to evaluate the ability of mast cells, hematogones, myeloid precursors, and erythroid precursors to serve as surrogates of bone marrow adequacy (Figure 6). The presence of more than 0.0015% mast cells was determined to be the most reliable indicator of sample adequacy (Figure 6A; p value < 0.001). According to the consensus achieved in this study, if mast cells were present at ≥ 0.0015% of total nucleated cells, at least one other marrow population should be assessed; otherwise, the sample would automatically be considered inadequate for MM MRD assessment. While mast cells indicate that bone marrow has been sampled, their presence alone provide no further assurance that the sample is not hemodilute or hypoplastic. The presence of hematogones at a minimum concentration of 0.025% emerged as the second-best indicator of sample adequacy (Figure 6B; p value < 0.001). The presence of myeloid precursors at a minimum concentration of 0.146% can also be used for determining sample adequacy (Figure 6C; p value < 0.002). Finally, the presence of erythroid precursors at a minimum frequency of 0.754% can be used, although it was considered the least favorable parameter based on lowest AUC and vulnerability to variation in erythrocyte lysis during sample preparation (Figure 6D; p value < 0.002).
Figure 6. Determination of sample adequacy based on the number of mast cells, hematogones, myeloid and erythroid precursors.
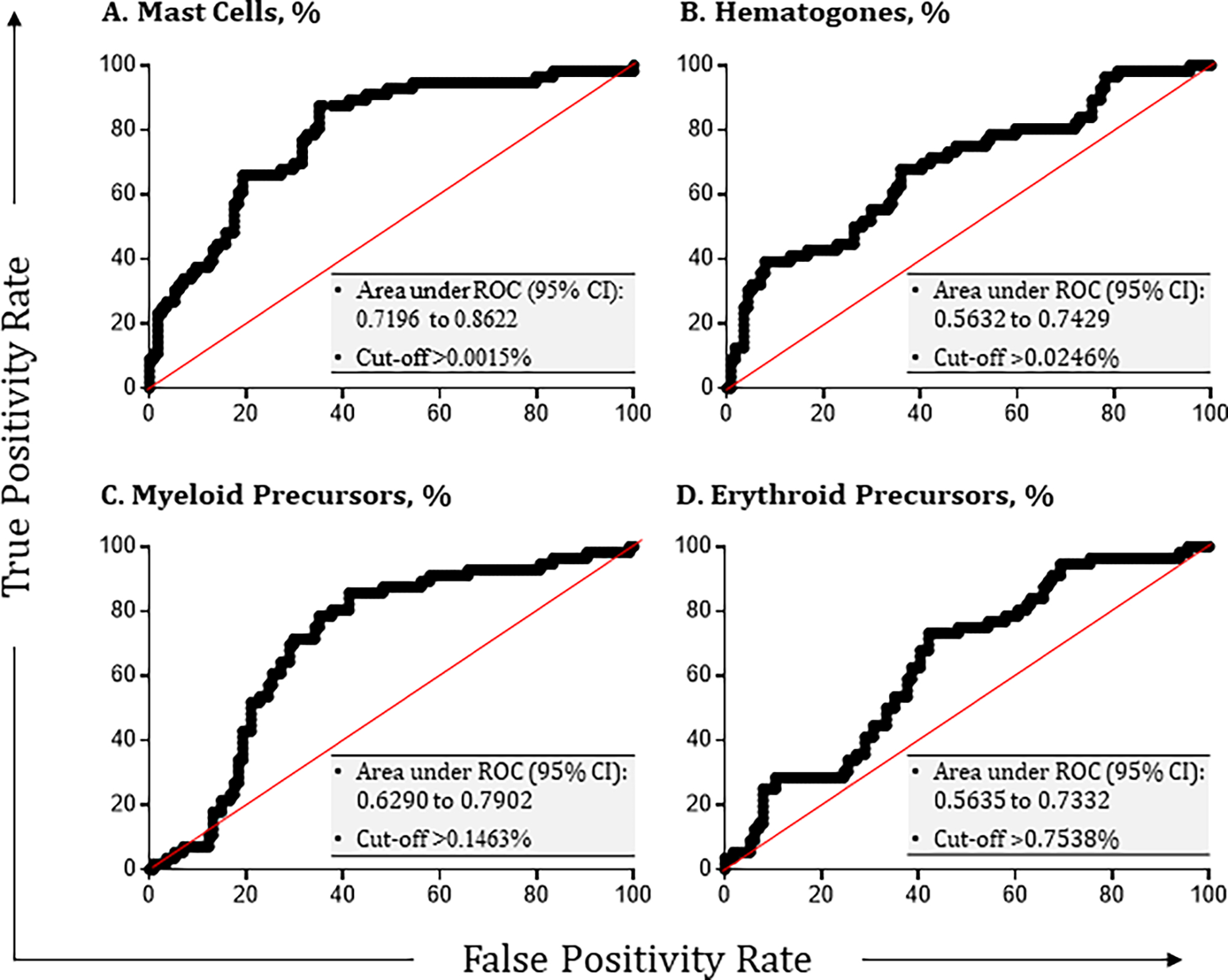
Optimal receiver-operating characteristic (ROC) curves are depicted for the concentrations of (A) mast cells, (B) hematogones, (C) myeloid precursors, and (D) erythroid precursors in any given sample that resulted in the greatest area under curve (AUC) and predictive values of each individual parameter for MM MRD assessment (p < 0.05). Different combinations of true positive and false positive MRD results were constructed based on concentrations of these cell populations, which were dependent upon the total number of bone marrow cells counted. The area under curve (AUC) was calculated using Prism (GraphPad). Each optimal cut-off was then established by the group as the minimum requirement used in the consensus protocol for determining sample adequacy.
To assess whether adopting these cut-offs would improve consistency in the determination of sample adequacy, the percentages of participants considering MM MRD samples as adequate, inadequate, or equivocal were compared before and after applying the consensus hierarchy. As indicated in Table 4, when participants reported the status of sample adequacy without employing harmonized cut-offs, the agreeability was only 72.4% ± 16.9%. Agreement improved to 97.1% ± 5.7% after harmonized cut-offs were applied (noting unanimity for 7 out of the ten samples).
Table 4:
Agreement between participants on sample adequacy before and after adopting consensus cut-offsa.
| Sample Number | Agreeability of Participants on the Outcome, % | |
|---|---|---|
|
|
||
| Beforeb | Afterc | |
|
| ||
| #1 | 88.2 | 100.0 |
| #2 | 64.7 | 94.1 |
| #3 | 64.7 | 100.0 |
| #4 | 88.2 | 100.0 |
| #5 | 94.1 | 100.0 |
| #6 | 88.2 | 100.0 |
| #7 | 52.9 | 100.0 |
| #8 | 76.5 | 82.4 |
| #9 | 58.8 | 100.0 |
| #10 | 47.1 | 94.1 |
|
| ||
| Mean | 72.4 | 97.1 |
| SD | 16.9 | 5.7 |
All samples were truly adequate except for Sample #8, which most of the participants agreed was hemodiluted.
No standardized cut-offs applied
Standardized cut-offs applied; cut-offs were determined based on ROC analysis (Figure 6). Sample was defined as adequate if mast cells were ≥0.002% and one other cell population was present above cut-off. I.e., Hematogones ≥0.025%, myeloid precursors ≥0.146%, or erythroid precursors ≥0.754%. If mast cells were present <0.002%, the sample was considered inadequate regardless of the presence of other BM cells or residual disease.
DISCUSSION:
Monitoring of MM MRD by flow cytometry is now recommended by the International Myeloma Working Group to categorize responses to therapy that are deeper than those conventionally defined by electrophoresis, light chain ratio, and morphology [13]. Several clinical trials have collectively demonstrated MM MRD evaluated by MFC to be a powerful predictor of both disease-free and overall survival [8, 10, 24]. Flow cytometry is applicable in over 95% of patients, has a low-cost barrier, uses instrumentation available in many institutions, has a relatively short turnaround, is semi-quantitative, and can discriminate between normal and abnormal PCs [5, 7, 25]. An additional advantage of MFC is the inherent ability to check for sample quality within the assay by measuring other bone marrow-associated cell populations. In the clinical flow setting, data analysis is an important part of the workflow, and good practices are expected to produce high-quality patient report in a reasonable turnaround time. In order to meet the increasing demand of flow cytometric interpretation, there is an interest to implement automated analysis for the detection of MM MRD and EuroFlow has published articles describing its utility while comparing expert-based vs. automated data analysis [21]. Automated analysis can speed up data analysis because it classifies a very high percentage of the different cell types contained in the sample, but it currently does not discern normal from abnormal PCs. It is the analyst’s responsibility to correctly classify the abnormal events. Thus, even when automated approach is employed, the analyst needs to manually classify and verify abnormal events to determine if anything was missed by the software. Moreover, an analyst can only fully appreciate the spectrum and biology of the disease by performing manual analysis.
Manual data analysis by MFC, however, is subjective and can represent a major source of variation between sites. To ensure high-quality data and consistent results from one facility to another, a standardized approach for the analysis of flow cytometric MM MRD data is essential. A series of consensus documents have been published addressing sample processing and panel design, data collection and analysis, and assay validation and quality control [18–20]. However, these guidelines lack detailed instructions specifying exactly how flow cytometric data should be analyzed to produce consistent results, both for the quantification of MRD and defining sample adequacy.
Indeed, the need for further harmonization was highlighted in a recent United Kingdom National External Quality Assessment Scheme international inter-laboratory assessment of MM MRD in a ‘wet’ survey of the ability of eight laboratories to assess for MM MRD in four serially diluted spiked samples of a normal harvested human stem cell product [16]. While this group of participants showed relatively good concordance despite heterogeneity of instruments, panels, sample preparation and analysis, they concluded that there was potential for additional reduction in inter-laboratory variation if MM MRD assays were further standardized. The study did not address other cell populations relating to sample quality. A recent consensus document of the Blood and Marrow Transplant Clinical Trials Network for international harmonization in performing and reporting minimal residual disease assessment in multiple myeloma trials further highlighted the increasing need for harmonization of the MM MRD assay for the reliable and efficient translation of therapeutic advances in multiple myeloma [22]. Our goal was to address this area of need by bringing together experienced active participants from the ICCS and ESCCA who are currently performing MM MRD analysis in a clinical context to develop a consensus document with detailed instructions on the identification and enumeration of all cell populations important for MM MRD assessment. This was to include a consensus on defining sample adequacy, using result-driven, dry-run exercises where all investigators analyzed identical data sets.
A total of 17 institutions from 13 countries participated in and completed this MM MRD proficiency study. The study was designed to comprise three stages during which the participants analyzed MM MRD FCS data files first using their own in-house strategy and then using a consensus protocol. All participants examined the same data files to confine observed variation to how individuals analyzed and reported the data. In Stage 1, key areas of discrepancy were identified and used to focus our discussions, out of which arose a software independent consensus manual data analysis protocol. The merit of this protocol was tested in Stage 2 of the study, where individuals were asked to reanalyze the same data set they evaluated in Stage 1. At the conclusion of Stage 2, everybody’s results were reviewed by all participants. A new set of FCS data files were analyzed in Stage 3 during which participants were asked to submit additional details about sample adequacy.
As expected, the adoption of a harmonized approach to data analysis and reporting improved the overall consistency between participants. Improvements were most notable in the enumeration of ‘Total Analyzed Cells’, calculation of LOD and LLOQ, and in defining sample adequacy. The agreed gating regions and strategies described in the final protocol are compatible with all commercially available flow cytometry software packages, and operators performing manual analysis should achieve similar values when the procedures are strictly followed.
A significant reduction in the variation of the reported number of ‘Total Number of Analyzed Cells’ was observed between Stages 1 and 2. This improvement was in part because a common gating strategy to define total cells was developed. However, to achieve additional consistency, following Stage 1 the group agreed to combine ‘Total Number of Analyzed Cells’, ‘Total PCs’, ‘Abnormal PCs’, and ‘Hematogones’ from both tubes in their final calculations as it was apparent that software packages handled the data differently. Notably, Infinicyt™ automatically combined FCS data from both tubes in the final report, whereas the other software packages used in this study did not. The enumeration of mast cells, myeloid precursors, and erythroid precursors was based only on Tube 1 because of the lack of CD117 and the two lysis steps in Tube 2. However, the advantage of combining other common data from both tubes for MRD assessment is the increased calculated total number of abnormal PCs detected which when properly validated amplifies the detection limits (i.e., LOD and LLOQ) of the flow assay.
While the implementation of a consensus approach improved the overall detection and quantification of total and normal PCs, there was no significant change in the enumeration of abnormal PCs, likely because all participants were expert at detecting myeloma cells. While consistency in defining the total number of ‘Abnormal PCs’ did not change, the percentage of ‘Abnormal PCs’ did significantly improve due to better conformity with the enumeration of the denominator cell population. This observation confirmed the reliability of the consensus method, demonstrating that it facilitated the output of consistent results obtained by manual analysis of MM MRD FCS data. The greatest heterogeneity in the interpretation of MRD status was seen in the MRD negative cases and the MRD+ samples with the lowest frequency of positive cells. For example, in Sample #10 the average number of reported abnormal events was 68 when Tube 1 and Tube 2 were combined, which was closed to the LLOQ. This observation highlights the need for additional studies at the MM MRD cut-off point. This is a necessary and critical development if the flow community is to provide reproducible results between laboratories given the important role of MM MRD as an indicator of depth of response, and its increasing use as a surrogate biological end point in clinical trials informing drug development and ultimately therapeutic decisions.
While the enumeration of all cell populations except ‘Abnormal PCs’ became substantially more homogenous with the implementation of the consensus analysis approach, there were, however, some individuals who reported values that were at least two standard deviations beyond the mean. These participants were asked for further details to understand the differences in data interpretation and given the opportunity to submit a revised analysis. Most of these instances were the result of reporting errors. On rare occasion, there was a misunderstanding on how to implement the consensus approach.
As the data files were de-identified with unobtainable medical history, it was not surprising that some cases were more difficult to analyze than others. Sample #7 of the first data set was problematic in both Stage 1 and Stage 2; we concluded the patient had likely received daratumumab. Daratumumab is an anti-CD38 immunotherapy that has gained widespread use in recent years. While daratumumab is a highly effective immunotherapy for MM patients with relapsed or refractory disease, it impedes the detection of PCs by MFC for up to six months following administration [26]. Our current consensus document was not developed to evaluate samples from patients exposed to daratumumab. Relying on CD138/CD45 for detecting PCs is suboptimal and alternative gating antibody conjugates to CD38, such as CD229, CD319, and VS38c are under active investigation [27]. Nevertheless, the consensus gating strategy is adaptable to MM MRD panels that contain alternative PC gating antigens to derive Total PCs [R8] (Figure 2B); and the identification of hematogones is not completely dependent on CD38 expression (Figure 5B). Moreover, the gating strategies outlined in this document are also applicable to single tube MM MRD panels provided the consensus markers are included.
Sample #8 in Stage 3 was found to have an abnormal mature B cell population which expressed CD45, CD38, kappa light chain, and was negative for CD138. Five of seventeen participants considered this population positive for MM MRD. This highlights the dual difficulty of not having all of the ideal clinical information prior to assay and the importance of context in the use of high sensitivity flow cytometric assays [28].
The evaluation of MM MRD is confounded by sample quality, therefore the determination of sample adequacy is an important aspect of MM MRD analysis. Sample quality may be compromised during collection, transportation, and storage. MM is well recognized as a patchy disease and the entire disease or substantial numbers of PCs may be missed if an uninvolved or marginally involved site is aspirated. PCs are also well recognized to be lost during the making of single-cell suspension required for flow cytometry, most likely because of PC fragility and adherence to stromal components. The underestimation of PCs can be further exacerbated by the infiltration of blood during the collection of a bone marrow sample, and this increases with each aspirate [7, 29]. Because of these issues, the frequency of PCs from marrow aspirate samples is typically underestimated by MFC assessment when compared to morphology [30]. However, unlike peripheral blood samples where one can request a replacement, bone marrow aspirates are difficult to obtain and considered precious samples. Therefore, guidelines that promote objective and reproducible determination of sample adequacy represent a valuable adjunct for the clinical laboratory to inform the quality and degree of certainty of a MM MRD result based on the enumeration of mast cells, hematogones, myeloid, and erythroid precursors.
Currently, there is no standardized strategy for the identification or specific cut-offs for sufficient enumeration of these bone marrow-derived cell populations in MM MRD samples. Significant heterogeneity in the determination of sample adequacy was found during Stage 1 of our study. To address this issue following Stage 1, a survey was distributed where participants were asked to rank in order of preference the marrow cell types that they considered most representative of a satisfactory quality bone marrow aspirate. The presence of mast cells was considered by consensus as the most important indicator. If mast cells are absent, the quality of the sample is considered suboptimal for accurate MM MRD assessment. If mast cells are detected, most participants felt the presence of an additional hematopoietic population, either hematogones, myeloid precursors, or erythroid precursors would more ideally represent satisfactory sample quality. Potential cut-offs for these cell populations were also investigated using ROC analysis to inform sample adequacy more objectively. Retrospective analysis of data obtained during Stage 3 using these cut-offs confirmed a substantial improvement in the consistency among participants in determining sample adequacy.
To the best of our knowledge, there is only one other study that provided an overall assessment of the quality of the patient samples through identification of bone marrow specific cell populations, published by the EuroFlow group [21]. The cut-off values for informing sample quality generated from our study were lower than those published by EuroFlow which may relate to the small sample sizes used by both groups. We recommend that a larger study be performed. To this point, the EuroFlow has recently updated the reference values incorporating higher numbers of subjects into their latest database which for mast cells and hematogones are generally comparable to our results (unpublished personal communications).
In conclusion, our study revealed considerable heterogeneity in the analysis and reporting of MM MRD FCS data files by a diverse group of international experts, which was rectified by adopting a consensus approach. The significant improvement in conformity was due to the standardization of a methodical gating algorithm used for the identification and enumeration of MRD, the denominator, and other bone marrow specific cell populations, and their cut-offs provide objective information on sample quality internal to the assay. Achieving a harmonized analysis strategy for MM MRD assessment by flow cytometry represents a further important step toward the assay being adopted by regulatory agencies as a surrogate biological marker of response for both accelerated drug development in clinical trials and for informing therapy. The analysis algorithm presented in this consensus paper is also proposed as a useful reference tool for the guidance and assessment of proficiency in MM MRD FCS data analysis within and between laboratories that perform MRD testing by flow cytometry.
Supplementary Material
Gating strategy to initially screen for potential abnormal PCs. A bivariate plot of CD56 vs. CD19 gated on ‘Total PCs’ is divided into quadrants (a) CD19+, CD56−, (b) CD19+, CD56+, (c) CD19−, CD56−, and (d) CD19−, CD56+. Using data from Tube 1, bivariate plots of CD38 vs. CD117, CD45 vs. CD38, CD45 vs. CD27, and CD81 vs. CD27 are drawn and phenotypic expression profiles of PCs in each quadrant are examined for any deviation from normal. This strategy is repeated on data derived from Tube 2 using bivariate plots of CD45 vs. CD38, CD45 vs. CD27, and cLambda vs. cKappa gated on quadrants (a) through (d). All plots were generated using WinList v9.1 (Verity Software House).
Comparison of hematogone and mast cell percentages in marrow aspirate determined as adequate vs. hemodilute/equivocal. The mean percentages of (A) mast cells and (B) hematogones were counted and compared between samples determined to be adequate (black bars) vs. inadequate (white bars) for each sample in Stage 1. A paired Student’s T test was performed to determine if the differences were of any significance. There was no evidence to suggest that participants who claimed the samples as hemodilute reported percentages of hematogones and mast cells that were statistically different than those who claimed the samples as adequate.
ACKNOWLEDGEMENT:
Flow cytometry services were provided by the Flow and Image Cytometry Resource Facility at the Roswell Park Comprehensive Cancer Center, which is supported in part by the NCI Cancer Center Support Grant 5P30 CA016056. The author takes this opportunity to thank Maryalice Stetler-Stevenson, Head of Flow Cytometry Unit, Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA for the opportunity to work on this consensus document; Maryalice was very instrumental in the conception of this study.
Footnotes
CONFLICT OF INTEREST
None of the authors have any conflict of interest to declare.
BIBLIOGRAPHY:
- 1.Anderson KC, Progress and Paradigms in Multiple Myeloma. Clinical Cancer Research, 2016. 22(22): p. 5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang W-C and Lin S-F, Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. BioMed Research International, 2015. 2015: p. 341430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borrello I, Can we change the disease biology of multiple myeloma? Leukemia research, 2012. 36 Suppl 1(0 1): p. S3–S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paiva B, van Dongen JJM, and Orfao A, New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood, 2015. 125(20): p. 3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maclachlan KH, et al. , Minimal residual disease in multiple myeloma: defining the role of next generation sequencing and flow cytometry in routine diagnostic use. Pathology, 2021. 53(3): p. 385–399. [DOI] [PubMed] [Google Scholar]
- 6.Landgren O and Owen RG, Better therapy requires better response evaluation: Paving the way for minimal residual disease testing for every myeloma patient. Cytometry B Clin Cytom, 2016. 90(1): p. 14–20. [DOI] [PubMed] [Google Scholar]
- 7.Rawstron AC, Paiva B, and Stetler-Stevenson M, Assessment of minimal residual disease in myeloma and the need for a consensus approach. Cytometry B Clin Cytom, 2016. 90(1): p. 21–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paiva B, et al. , Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood, 2008. 112(10): p. 4017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawstron AC, et al. , Report of the European Myeloma Network on multiparametric flow cytometry in multiple myeloma and related disorders. Haematologica, 2008. 93(3): p. 431–8. [DOI] [PubMed] [Google Scholar]
- 10.Rawstron AC, et al. , Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol, 2013. 31(20): p. 2540–7. [DOI] [PubMed] [Google Scholar]
- 11.San Miguel JF, et al. , Immunophenotypic evaluation of the plasma cell compartment in multiple myeloma: a tool for comparing the efficacy of different treatment strategies and predicting outcome. Blood, 2002. 99(5): p. 1853–6. [DOI] [PubMed] [Google Scholar]
- 12.Rawstron AC, et al. , Minimal residual disease in myeloma by flow cytometry: independent prediction of survival benefit per log reduction. Blood, 2015. 125(12): p. 1932–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S, et al. , International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol, 2016. 17(8): p. e328–46. [DOI] [PubMed] [Google Scholar]
- 14.Landgren O, et al. , Flow cytometry detection of minimal residual disease in multiple myeloma: Lessons learned at FDA-NCI roundtable symposium. Am J Hematol, 2014. 89(12): p. 1159–60. [DOI] [PubMed] [Google Scholar]
- 15.Flanders A, Stetler-Stevenson M, and Landgren O, Minimal residual disease testing in multiple myeloma by flow cytometry: major heterogeneity. Blood, 2013. 122(6): p. 1088–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott SD, et al. , Assessment of plasma cell myeloma minimal residual disease testing by flow cytometry in an international inter-laboratory study: Is it ready for primetime use? Cytometry Part B: Clinical Cytometry, 2019. 96(3): p. 201–208. [DOI] [PubMed] [Google Scholar]
- 17.Salem D, et al. , Myeloma minimal residual disease testing in the United States: Evidence of improved standardization. American Journal of Hematology, 2016. 91(12): p. E502–E503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stetler-Stevenson M, et al. , Consensus guidelines for myeloma minimal residual disease sample staining and data acquisition. Cytometry B Clin Cytom, 2016. 90(1): p. 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arroz M, et al. , Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. Cytometry B Clin Cytom, 2016. 90(1): p. 31–9. [DOI] [PubMed] [Google Scholar]
- 20.Oldaker TA, Wallace PK, and Barnett D, Flow cytometry quality requirements for monitoring of minimal disease in plasma cell myeloma. Cytometry B Clin Cytom, 2016. 90(1): p. 40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flores-Montero J, et al. , Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa LJ, et al. , International harmonization in performing and reporting minimal residual disease assessment in multiple myeloma trials. Leukemia, 2021. 35(1): p. 18–30. [DOI] [PubMed] [Google Scholar]
- 23.Keeney M, et al. , A QA Program for MRD Testing Demonstrates That Systematic Education Can Reduce Discordance Among Experienced Interpreters. Cytometry B Clin Cytom, 2018. 94(2): p. 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roussel M, et al. , Front-line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: a phase II study by the Intergroupe Francophone du Myelome. J Clin Oncol, 2014. 32(25): p. 2712–7. [DOI] [PubMed] [Google Scholar]
- 25.Soh KT, Tario JD Jr., and Wallace PK, Diagnosis of Plasma Cell Dyscrasias and Monitoring of Minimal Residual Disease by Multiparametric Flow Cytometry. Clin Lab Med, 2017. 37(4): p. 821–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Courville EL, et al. , VS38 Identifies Myeloma Cells With Dim CD38 Expression and Plasma Cells Following Daratumumab Therapy, Which Interferes With CD38 Detection for 4 to 6 Months. Am J Clin Pathol, 2020. 153(2): p. 221–228. [DOI] [PubMed] [Google Scholar]
- 27.Soh KT, et al. , CD319 (SLAMF7) an Alternative Marker for Detecting Plasma Cells in the Presence of Daratumumab or Elotuzumab. Cytometry B Clin Cytom, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sommer U, et al. , High-sensitivity flow cytometric assays: Considerations for design control and analytical validation for identification of Rare events. Cytometry B Clin Cytom, 2021. 100(1): p. 42–51. [DOI] [PubMed] [Google Scholar]
- 29.Manasanch EE, et al. , Flow cytometric sensitivity and characteristics of plasma cells in patients with multiple myeloma or its precursor disease: influence of biopsy site and anticoagulation method. Leukemia & lymphoma, 2015. 56(5): p. 1416–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Quran SZ, et al. , Assessment of bone marrow plasma cell infiltrates in multiple myeloma: the added value of CD138 immunohistochemistry. Human pathology, 2007. 38(12): p. 1779–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gating strategy to initially screen for potential abnormal PCs. A bivariate plot of CD56 vs. CD19 gated on ‘Total PCs’ is divided into quadrants (a) CD19+, CD56−, (b) CD19+, CD56+, (c) CD19−, CD56−, and (d) CD19−, CD56+. Using data from Tube 1, bivariate plots of CD38 vs. CD117, CD45 vs. CD38, CD45 vs. CD27, and CD81 vs. CD27 are drawn and phenotypic expression profiles of PCs in each quadrant are examined for any deviation from normal. This strategy is repeated on data derived from Tube 2 using bivariate plots of CD45 vs. CD38, CD45 vs. CD27, and cLambda vs. cKappa gated on quadrants (a) through (d). All plots were generated using WinList v9.1 (Verity Software House).
Comparison of hematogone and mast cell percentages in marrow aspirate determined as adequate vs. hemodilute/equivocal. The mean percentages of (A) mast cells and (B) hematogones were counted and compared between samples determined to be adequate (black bars) vs. inadequate (white bars) for each sample in Stage 1. A paired Student’s T test was performed to determine if the differences were of any significance. There was no evidence to suggest that participants who claimed the samples as hemodilute reported percentages of hematogones and mast cells that were statistically different than those who claimed the samples as adequate.