

Abstract
Genomic instability is an important driver of ageing. The accumulation of DNA damage is believed to contribute to ageing by inducing cell death, senescence and tissue dysfunction. However, emerging evidence shows that inflammation is another major consequence of DNA damage. Inflammation is a hallmark of ageing and the driver of multiple age-related diseases. Here we review the evidence linking DNA damage, inflammation and ageing, highlighting how premature ageing syndromes are associated with inflammation. We discuss the mechanisms by which DNA damage induces inflammation, such as through activation of the cGAS–STING axis and NF-κB activation by ATM. The triggers for activation of these signalling cascades are the age-related accumulation of DNA damage, activation of transposons, cellular senescence, and the accumulation of persistent R-loops. We also discuss how epigenetic changes triggered by DNA damage can lead to inflammation and ageing via redistribution of heterochromatin factors. Finally, we discuss potential interventions against age-related inflammation.
Introduction
Genomic instability is a hallmark of ageing1. DNA in the cell is constantly subjected to damaging agents from the environment as well as damage resulting from intrinsic biological processes. DNA damage, left unrepaired or repaired incorrectly, may cause deleterious mutations. DNA damage was traditionally believed to only contribute to genomic instability. Recently, it was shown that DNA damage is an inducer of inflammation both in vitro and in vivo2–5. In cell culture, DNA damage induces the expression of type I interferons (IFN) and other inflammatory factors2–4. Mechanistic studies revealed that the connection between DNA damage and inflammation is through the cytoplasmic DNA sensing pathway6–8.
Two major events are involved in this process: the presence of DNA in the cytoplasm, and the sensing of cytoplasmic DNA to induce inflammation. DNA damage results in the accumulation of cytoplasmic DNA via two pathways: first, via the formation of micronuclei and second, through the direct leakage of DNA into the cytoplasm9,10. Micronuclei emerge from chromosome mis-segregation during mitosis and in response to DNA damage9,11,12. When the double-stranded DNA breaks (DSBs) are left unrepaired or mis-repaired, this can lead to formation of acentric chromosomal fragments9. These fragments recruit the nuclear envelope to form micronuclei in the subsequent cell division9. Micronuclei are nuclei-like structures, consisting of nuclear envelope surrounding chromosome fragments13. Roughly 60% of micronuclei undergo a nuclear envelope rupture during interphase14. Nuclear envelope rupture is associated with loss of lamina, but the exact mechanism is not clear. Normally, the nuclear envelope is maintained and repaired by the endosomal sorting complexes required for transport III (ESCRT-III) complex15. It is possible that this repair is deficient in micronuclei6. This results in the leakage of the chromosomal DNA fragments from micronuclei into the cytoplasm. Cyclic GMP-AMP synthase (cGAS) is a DNA sensor that recognizes and binds cytoplasmic DNA in a sequence-independent manner16. Upon DNA binding, the cGAS protein changes the conformation of its catalytic centre and mediates the synthesis of cyclic GMP–AMP (cGAMP). cGAMP binds to and activates the adaptor protein stimulator of IFN genes (STING)16. Activated STING recruits TANK-binding kinase 1 (TBK1) to phosphorylate IFN regulatory factor 3 (IRF3), which then induces production of type I IFN17. STING also activates NF-κB through tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6)18. NF-κB cooperates with IRF3 to activate type I IFN and other inflammatory factors6,19 (Figure 1).
Figure 1: DNA damage as an inducer of inflammation.
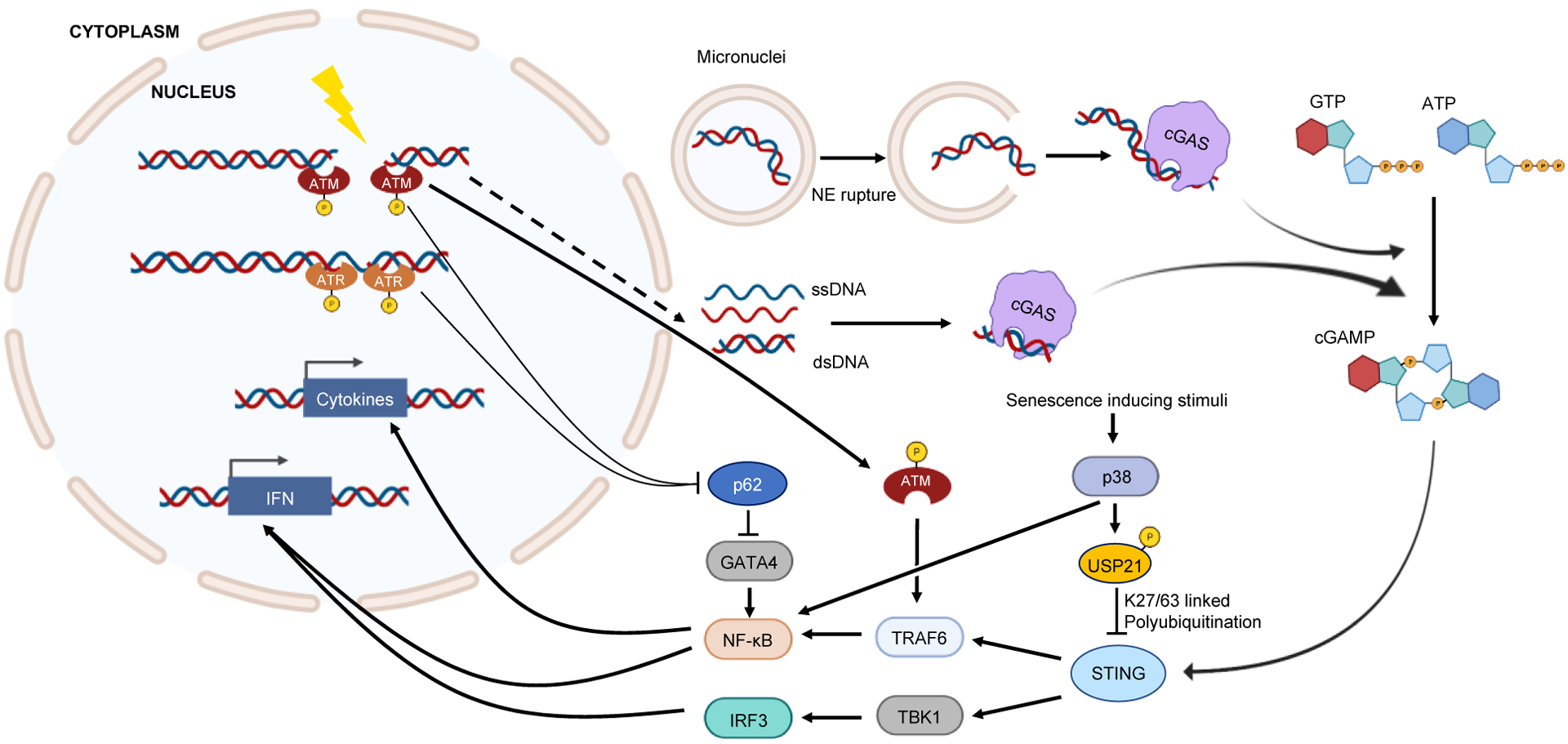
Multiple mechanisms connect DNA damage to inflammation. DNA damage may result in chromosome fragments that do not segregate properly during subsequent cell division. These chromosome fragments become surrounded by nuclear envelope forming. When the micronuclei undergo nuclear envelope rupture, the chromosome DNA is exposed to the DNA sensor cGAS. Alternatively, during DNA damage or subsequent resection and repair, DNA fragments may directly leak into the cytoplasm through a less understood mechanism (dashed line). ssDNA fragments may form double-stranded secondary structures, which can also be recognized by cGAS. DNA-bound cGAS converts GTP and ATP into cGAMP, which activates STING. Activated STING through TBK1-IRF3 and TRAF6-NF-κB, induces transcription of IFN genes and other cytokines. Additionally, proteins involved in DNA damage response can directly trigger inflammation. Depending on the type of DNA damage, ATM or ATR are activated and recruited to the DNA break sites. Activated ATM or ATR can activate NF-κB by stabilizing GATA4 protein. ATM can also stimulate NF-κB by activating TRAF6. Senescence inducing stimuli, including sustained DNA damage and telomere defects, may activate p38 pathway. p38 induces inflammation through NF-κB; it also inhibits STING-dependent IFN response through USP21-mediated K27/63 linked polyubiquitination. NE: nuclear envelope. Image was generated using biorender.com.
In addition to micronuclei, single-stranded DNA (ssDNA) or double-stranded DNA (dsDNA) can be observed in the cytoplasm following DNA damage, possibly occurring due to DNA end resection10. The leakage of DNA from the nucleus into the cytoplasm depends on the endonuclease MUS8120. MUS81 also promotes type I IFN production in prostate cancer cells20. cGAS has little to no affinity for ssDNA but can be activated by the double-stranded secondary structures of ssDNA21. This process can also contribute to the inflammation induced by DNA damage. The cGAS-STING pathway recognizes foreign nucleic acids, including DNA from viruses, bacteria, and tumours, as well as self-DNA originating from the nucleus and mitochondria22. By detecting foreign DNA and triggering inflammation, cGAS-STING pathway plays a critical role for host defense22.
Recently, studies have demonstrated direct associations between the DNA damage response and NF-κB-mediated inflammation23–25. In response to DNA damage, DSBs and single-stranded DNA breaks recruit ataxia-telangiectasia mutated (ATM) and ATM and RAD3-related (ATR), respectively, to the DNA lesion26. The transcription factor GATA4 is normally degraded by autophagy mediated by p62. Upon DNA damage, ATM or ATR inhibit p62 and thus, stabilize GATA4, which activates NF-κB to induce inflammation23. ATM was also shown to translocate to the cytosol to activate TRAF6, which eventually activates NF-κB24. Remarkably, STING can be activated by ATM independently of cGAS, leading to NF-κB activation27. Activated ATM can also activate NF-κB by degrading IκBα and phosphorylating RELA (p65) in the cytoplasm25. In summary, DNA damage has emerged as an important inducer of inflammation (Figure 1). In this Review, we will discuss various events associated with DNA damage, including DNA repair deficiencies, activation of transposons, cellular senescence, R-loop formation, and change in chromatin structure, that link inflammation and ageing.
DNA damage and premature ageing
Defective DNA repair, inflammation and premature ageing syndromes.
Mutations in genes involved in DNA repair and genome maintenance are often associated with premature ageing syndromes. Traditionally, it was believed that the pathologies seen in DNA repair disorders arise from cell death and accumulation of mutations. However, emerging evidence points to inflammation as a contributing cause to the pathogenesis of these syndromes. The connection between DNA repair defects and inflammation appears to be mediated, in many cases, by damaged DNA driving activation of the cGAS-STING signalling pathway and interferon response. Below, we review several cases where defects in DNA repair have been linked to both premature ageing and the induction of an inflammatory response.
Nucleotide excision repair syndromes.
Inherited mutations in more than a dozen of nucleotide excision repair (NER) [G] genes contribute to several human syndromes, including xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy (TTD), as well as some related syndromes with combined phenotypes28. Each syndrome has several complementation groups, with different combinations of mutant NER genes29. Xeroderma pigmentosum is characterized by high risk of skin cancer30,31. Other NER deficiency syndromes, however, display premature ageing without cancer predisposition. The XPF–ERCC1 heterodimer is a 3’-flap endonuclease that generates the primary DNA nick crucial for NER progression and is also required for DNA inter-strand crosslink (ICL) repair32–34. The R153P mutation of XPF gene in XFE progeroid syndrome results in severe progeroid symptoms without elevated cancer risk35.
Several studies showed that mouse models carrying mutations in ERCC1–XPF demonstrate increased inflammation. Ercc1−/− mice display persistent DNA damage that triggers a chronic autoinflammatory response in the adipose tissue36. The cytoplasmic phosphorylated ATM is believed to induce transcriptional de-repression of inflammatory factors. In cultured human ERCC1-deficient fibroblasts or cultured fibroblasts from the skin of Ercc1−/Δ mice (which carry a null and a hypomorphic Ercc1 alleles), there is elevated cellular senescence and production of senescence-associated secretory phenotype (SASP) [G] factors37. TNF secreted by the senescent cells induced apoptosis of the neighbouring stem cells, contributing to the premature ageing phenotypes37. Fibroblasts and tissues of ERCC1-deficient mice also display ATM-dependent activation of NF-κB, which induces SASP and senescence38.
Patients with Cockayne syndrome display accelerated ageing in multiple systems, characterised by hearing loss, compromised vision and cognitive dysfunction, among other symptoms39,40. There are two complementation groups for Cockayne syndrome, namely Cockayne syndrome A (caused by mutation of CSA (also known as ERCC8)) and Cockayne syndrome B (caused by mutation of CSB (also known as ERCC6)), with Cockayne syndrome B accounting for the majority of Cockayne syndrome cases41. In a mouse model of Cockayne syndrome, Csbm/m mice carry mutations affecting CSB and develop fibrosis and inflammation in response to ultraviolet (UV) radiation42. In another mouse model with complete loss of NER, namely the Csa−/−Xpa−/− mouse, animals display upregulation of inflammatory markers in the brain, including ICAM1, TNF and phosphorylated NF-κB p6543. Although the molecular mechanisms responsible for induction of inflammation in Cockayne syndrome are poorly understood, it is possible that mitochondrial stress is playing a role. CSB interacts with mitochondrial transcription factor A (TFAM) and plays an important role in maintaining mitochondrial integrity44. Mitochondrial DNA stress due to loss of TFAM has been shown to trigger cGAS–STING–IFN signalling via the release of mitochondrial DNA into the cytoplasm45,46. Going forward, it will be important to test the contribution of mitochondrial DNA stress to the inflammation observed in patients with Cockayne syndrome.
DNA double-strand break repair syndromes.
Defects in DNA double-strand break (DSB) repair [G] are implicated in several premature ageing syndromes. Mutations in RecQ family of helicases result in Werner syndrome and Bloom syndrome. Werner syndrome is a segmental progeroid syndrome caused by a null mutation on the gene encoding Werner syndrome ATP-dependent helicase (WRN)47. Cells isolated from patients with Werner syndrome display genomic instability, including aberrant recombination, increased spontaneous mutations, and telomere defects48. Werner syndrome is associated with strong induction of inflammatory response and was therefore proposed as an example of inflammageing [G]49–52. Fibroblasts isolated from patients with Werner syndrome express high levels of IFNβ53. Likewise, short-term siRNA-mediated knock down of WRN in fibroblasts from healthy young individuals triggered an inflammatory gene-expression profile resembling that seen in older patients, with activated inflammatory pathways including IL-6, NF-κB, and genes associated with the IFN response48. The inflammation was believed to be activated through the p38 pathway due to telomere defects and genomic instability-induced intracellular stress49. p38 activated inflammation is believed to be largely mediated by increasing the transcriptional activity of NF-κB54. However, the induction of an interferon response suggests that the cGAS–STING pathway may also be involved in Werner syndrome, although further studies are required to provide direct evidence. Bloom syndrome is another segmental progeroid syndrome and it results from mutations in BLM helicase, which plays a critical role in genome integrity maintenance55. Cells from patients with Bloom syndrome display increased activation of the phosphorylated histone 2A (γH2AX)–ATM– serine/threonine protein kinase CHK2 (CHK2) DNA double-strand break checkpoint response pathway56. Importantly, Bloom syndrome cells accumulate micronuclei, which trigger the cytoplasmic DNA sensing cGAS–STING–IFN pathway57. While mutation of BLM results in Bloom syndrome, high levels of BLM correlate with poor prognosis in breast cancer10. This is possibly because BLM also plays a role in generating ssDNA during DSB repair by mediating end resection10. Depletion of BLM decreased cytosolic ssDNA sensing through cGAS-STING pathway10.
The Ku70–Ku80 heterodimer is required for the non-homologous end joining (NHEJ) pathway of DNA repair. Knockdown of Ku70 or Ku80 in mice results in severe premature ageing. These mice also display airway inflammation and subepithelial fibrosis, which are potentially induced by mitochondrial stress or ER stress58. Missense mutations of DNA-PK catalytic subunit (DNA-PKcs) in mice or patients derepress cGAS-mediated innate immunity59. Interestingly, DNA-PK itself has been reported to be a cytoplasmic DNA sensor, which triggers IRF3-dependend innate immunity60, providing a direct link between DNA repair and inflammation.
ATM is a serine/threonine protein kinase activated by DNA double-strand breaks. ATM initiates DSB repair responses by phosphorylating multiple downstream targets involved in cell cycle arrest, apoptosis and repair61. Inherited mutations of ATM cause Ataxia-telangiectasia, a premature ageing disorder characterized by immunodeficiency, progressive neurodegeneration, and increased risk of cancer62–64. Studies demonstrate that Ataxia-telangiectasia syndrome is associated with strong pathologic inflammation, which is responsible for many of its symptoms65. Serum levels of IL-6 and IL-8 are significantly higher in patients with Ataxia-telangiectasia than in control groups66,67. ATM-deficiency also causes elevated and prolonged inflammation in response to lung injury68. Type I and type III IFN signatures are elevated in the plasma and peripheral blood cells of patients with Ataxia-telangiectasia resulting from the accumulation of DNA in the cytoplasm69. In microglia from patients with Ataxia-telangiectasia, cytoplasmic DNA accumulates and activates the cGAS–STING pathway70 and the absent in melanoma 2 (AIM2) inflammasome70. These studies underlie the contribution of inflammation to the symptoms of Ataxia-telangiectasia syndrome through accumulation of cytoplasmic DNA and activation of the cGAS–STING pathway.
Defects in genome maintenance.
Hutchinson-Gilford progeria syndrome (HGPS) is caused by mutations on the lamin A (LMNA) gene71. The nuclear lamins are responsible for maintaining nuclear architecture by providing a scaffold for chromatin and all nuclear contents. Lamins play important roles in maintaining nuclear membrane integrity, nuclear envelope assembly, DNA replication, transcription, and maintenance of heterochromatin72. The mutant lamin A in patients with HGPS results in a mis-shaped nucleus, genome instability, dysregulated epigenome and gene expression, and premature ageing73,74. One of the hallmarks of HGPS is inflammation. Higher basal expression of components of the NLRP3 inflammasome pathway were observed in skin fibroblasts and lymphoblasts from patients with HGPS75. In an LMNAG609G/G609G mouse model, inhibition of NLRP3 inflammasome significantly extended lifespan, suggesting that inflammasome activation is a major contributor to the progeria phenotype75. In another HGPS model, cells from mice with a Zmpste24−/− mutation (affecting the Zmpste24 protease responsible for processing pre-lamin A), exhibit nuclear blebbing, micronuclei formation, and activation of the cGAS–STING pathway76. Both mouse models of HGPS are characterized by elevated expression of inflammatory cytokines and activation of ATM and NF-κB signalling pathways77.
In summary, deficiency in different genes involved in DNA repair and genome maintenance is associated with a strong induction of inflammation (Table 1). Considering the central role of inflammation in the etiology of age-related disease it is possible the induction of inflammation, rather than accumulation of mutations, plays a key role in pathogenesis of premature ageing syndromes.
Table 1.
Syndromes of DNA repair deficiency and their association with inflammation
| DNA repair pathway | Genes mutated | Syndrome | Cancer risk | Premature ageing | Inflammation | Mechanisms of inflammation |
|---|---|---|---|---|---|---|
| NER | various NER genes | Xeroderma pigmentosum (XP) | Yes | Yes | No | NA |
| NER | XPF (severe mutation) | XPF-ERCC1 progeroid (XFE) | No | Yes | Yes | Cytoplasmic p-ATM; SASP |
| NER | csa, csb | Cockayne syndrome (CS) | No | Yes | Yes | Mitochondrial stress |
| DSB | WRN | Werner syndrome (WS) | Yes | Yes (segmental) | Yes | telomere defect; possibly cGAS-STING |
| DSB | BLM | Bloom syndrome (BS) | Yes | Yes (segmental) | Yes | Micronuclei |
| Indirect | LMNA | Hutchinson-Gilford Progeria Syndrome (HGPS) | No | Yes | Yes | DNA leakage; micronuclei |
| Indirect | ATM | Ataxia-telangiectasia (AT) | Yes | Yes | Yes | Cytoplasmic DNA |
Transposons are inducers of inflammation
A major part of the mammalian genome consists of transposable elements (TEs). Mammalian TEs include retrotransposons (RTEs) [G], such as long-interspersed nuclear elements (LINEs) and short-interspersed nuclear elements (SINES), and endogenous retroviruses (ERVs)78,79. LINE elements are 6 kb-long, fully autonomous retroelements that encode two proteins necessary for retrotransposition into the genome; ORF1p, which forms homo-trimers that assemble on the LINE RNA as chaperones and ORF2p, which encodes an endonuclease and reverse transcriptase80–82. SINE elements are considerably shorter than LINEs and unlike their larger brethren, they do not encode proteins83. Rather, SINEs rely upon the protein machinery encoded and expressed by LINE elements in order to copy themselves back into the genome83,84. In humans, LINE-1 retrotransposons (L1s) and associated SINEs are the predominantly active elements, with only minor contribution from human ERVs (HERVs)85. While the vast majority of LINEs belong to evolutionarily inactive families, there are still hundreds of sequences that remain fully active and capable of autonomous self-replication86. The mechanisms by which these elements retro-transpose into the genome has been extensively covered elsewhere and will only be briefly covered here87. The protein subunits encoded by L1s preferentially associate in cis with L1 mRNA, forming a ribonuclear protein (RNP). The RNP translocates to the nucleus, where it initiates reverse-transcription of the RNA after generating a single-strand break (SSB) in the genomic DNA to prime off of. Integration of the new DNA and the subsequent generation of the second strand complete the process. SINEs retro-transpose in a similar manner, as they also associate with L1-encoded proteins to form their own RNPs. Retrotransposons are potentially mutagenic, generating insertion mutations and DNA breaks. Retrotransposons are considered genomic parasites involved in a perpetual arms race with the host cell.
Retrotransposons and disease.
Cells have evolved multiple overlapping mechanisms to suppress TE activity88,89. Failure of these mechanisms can lead to inappropriate transposon activation, which has been documented in several diseases. Historically, the contributions of RTEs to disease were primarily linked to their mutagenic activity90. Elevated activity of RTEs, especially L1s, is a hallmark of many cancers91–93. In fact, over 40% of neoplasms demonstrate elevated L1 activity94,95. However large-scale sequencing efforts showed that the majority of RTE insertions found in tumours are benign passenger events96. While several cases of insertions into tumour suppressor genes had been documented, these events are rare96. Thus, elevated RTE activity in tumours is likely a consequence rather than a cause of tumorigenesis.
In recent years, the deleterious impact of RTE activity outside of mutagenesis has become center-stage. Primarily, it appears that disruption of mechanisms of RTE suppression and the subsequent unregulated retrotransposition result in sterile inflammation [G]. Dysregulation of L1s has been demonstrated to drive inflammation in Trex1-dependent autoimmune disorders and initiate pro-inflammatory interferon expression in senescent cells97–99. Loss of tumour suppressors and disruption of genes associated with DNA repair, such as p53 and SIRT6, also elicit increased L1 activity and inflammation100,101. This pro-inflammatory activity driven by L1 is also observed in cancers with elevated L1 expression102,103. In addition to L1-induced autoimmune and pro-inflammatory diseases, Alu SINE elements in humans induce inflammation resulting in macular degeneration and Aicardi-Goutières syndrome97,104–109.
Mechanistically, RTE-induced inflammation results from RTE cDNA triggering anti-viral surveillance mechanisms in the cytoplasm, though there is also evidence that RTE-induced genomic instability may also contribute to pro-inflammatory activity through the formation of micronuclei110,111 (Figure 2). The cGAS–STING axis plays a pivotal role in the sensing of RTE cDNAs and triggering a sterile inflammation response22,111. This pathway can detect a host of extrinsic (viral, retroviral, bacterial, tumoural) and intrinsic (nuclear, mitochondrial and cytoplasmic) DNA sources. In any event, the activation of an immune response mediated by NF-κB and IRF3 produces pro-inflammatory cytokines such as IFN-α, IFN-β, IL-6, and TNF112. How RTE-derived cDNA arrives in the cytoplasm is still not fully understood. While it has been suggested that failed integration events accumulate in the nucleus and ‘leak’ out, there is recent evidence that demonstrates cytoplasmic synthesis can occur in the case of Alu elements associated with macular degeneration109.
Figure 2: RTE mobilization triggers inflammation via cytoplasmic DNA.
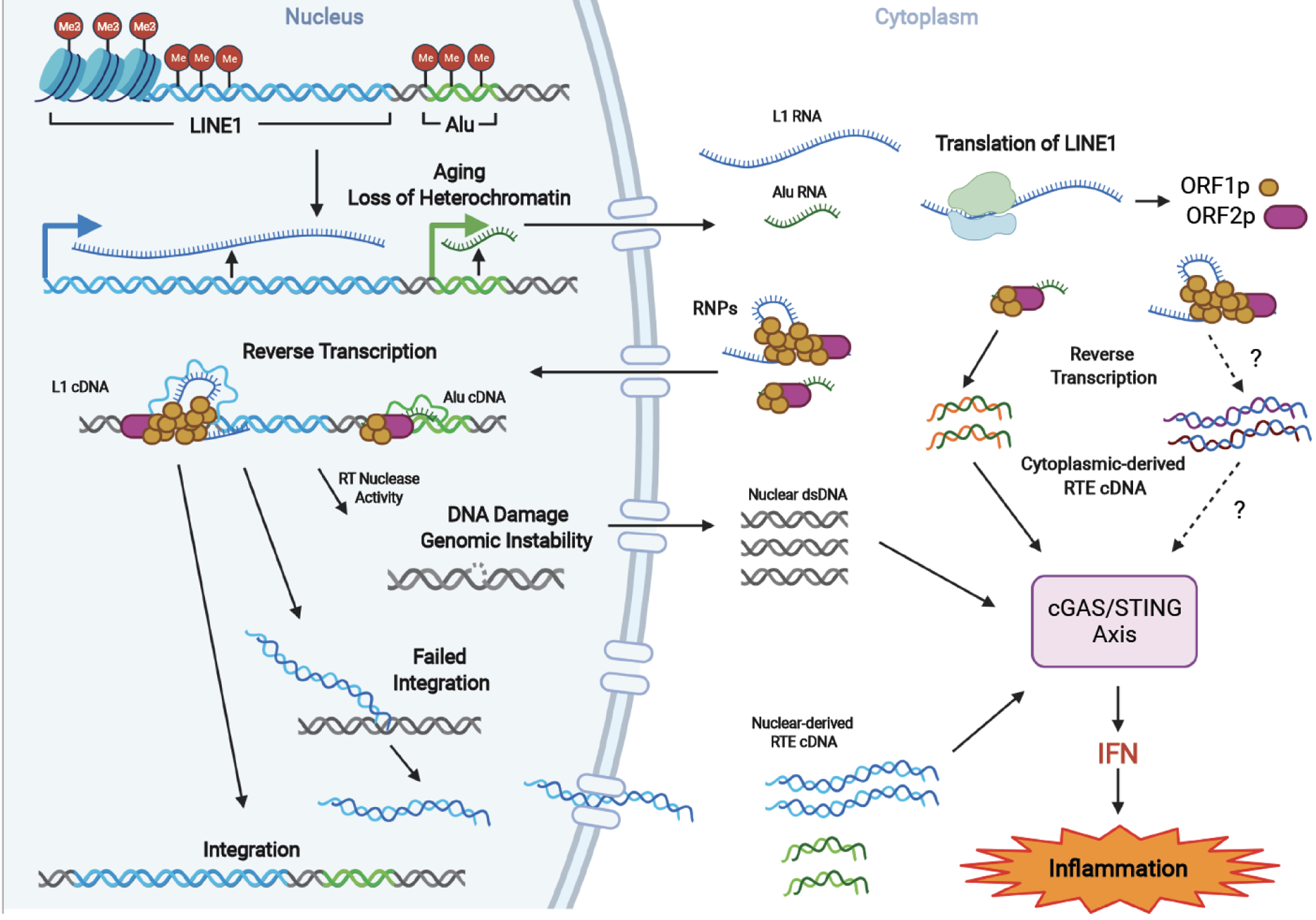
Ageing-related loss of heterochromatin marks on RTEs results in their transcriptional activation and retrotransposition. In the cytoplasm, Alu elements produce ssDNA via self-priming utilizing LINE1 RT machinery. Simar process may take place for L1s. In the nucleus, RNPs initiate RTE integration by generating DNA nicks and reverse transcription, which induces DNA damage. Additionally, unsuccessful integration produces DNA flaps, which are processed to RNA-DNA hybrid fragments. The RTE RNA-DNA hybrids resulting from reverse transcription either in the nucleus or cytoplasm and DNA fragments resulting from genomic damage are recognized by cytoplasmic cGAS, triggering the cGAS-STING signaling, leading to IFN production and sterile inflammation. Dashed lines indicate steps that require further experimental evidence.
Retrotransposons promote ageing via induction of inflammation.
While the immediate impact of genetic and stress-induced RTE activation and inflammation are clearly demonstrated in the pathologies they induce, there lies a more insidious threat within the RTE-induced inflammation pathway. Indeed, loss of heterochromatin, sterile inflammation and the mobilization of RTEs are also hallmarks of ageing. Aside from the RTE-induced inflammatory signature observed in senescent cells (which accumulate with age), progeroid SIRT6-knockout mice and aged wild-type mice also demonstrate elevated interferon production triggered by cGAS-sensing of LINE cDNAs in the cytoplasm98,100,113. In cell-free DNA detected in blood, L1 methylation changes and circulating Alu DNAs are also observed to correlate with advanced age114–116. SINE elements also contribute to ageing pathologies, as is the case with age-related macular degeneration104,109. Additionally, SINEs carry over one third of total CpG methylations within the genome, which become hypomethylated with age117–119. The ubiquitous nature of age-related sterile inflammation gave rise to the concept of inflammageing, where sterile inflammation serves as a driving force of multiple age-related pathologies and the ageing process itself120. The sources of sterile inflammation are not fully understood and are believed to result from cell debris, accumulation of senescent cells among other factors. RTE-induced inflammation points to a novel source of age-related inflammation where ageing cells gradually lose control over endogenous genomic parasites triggering a response similar to that of a persistent viral infection (Figure 2).
Tumour suppression by retrotransposons: the bright side.
Besides the deleterious effects triggered by RTEs in the context of ageing, emerging evidence indicate beneficial effects of transposons as tumour suppressors. This role of RTEs may be the reason these parasites have not been eliminated from the mammalian genomes. In human cancer cells, DNA-demethylating agents activate ERVs, which form double-stranded RNA (dsRNA) and trigger IFN response to kill the cancer cells121,122. Likewise, ablation of lysine-specific histone demethylase 1A (LSD1) derepresses ERVs and triggers cancer cell death through cytoplasmic dsRNA sensing pathway, where the dsRNA sensors TLR3 and MDA5 trigger IFN response123. Forced reactivation of Alu-derived dsRNA inhibited human cancer cell growth, which was further synergized by loss of ADAR1, a dsRNA-editing enzyme124,125. These studies suggest that transposons may have a potential tumour suppressive function through viral mimicry. Recently, it was demonstrated that L1s trigger genomic instability in myeloid leukemia, showing a tumour-suppressive effect126.
A recent study using a naturally long-lived and cancer-resistant rodent, the blind mole rat [G] (Spalax), revealed that RTEs can serve as a tumour-suppressor through a natural process127. Blind mole rats are extremely resistant to both spontaneous and induced tumorigenesis. Their fibroblasts display a unique phenotype termed concerted cell death upon over-proliferation128. It was demonstrated that, due to their naturally weak DNA methyltransferase 1 (DNMT1) activity, rapidly proliferating cells in blind mole rats lose global DNA methylation and derepress RTEs. Activated RTEs form cytoplasmic RNA–DNA hybrids and activate the cGAS–STING–IFN pathway to kill the over-proliferating cells and prevent cancer127. Remarkably, pro-inflammatory genes are repressed in the normal tissue of middle-aged blind mole rats, indicating that concerted cell death is activated specifically in over-proliferating cells to avoid widespread inflammation. In summary, these results suggest that ‘domesticated’ RTEs were naturally co-opted to serve as a tumour suppressor. Hence, RTEs can be seen as a double-edged sword, acting as a tumour-suppressor in young age, but later contributing to inflammageing.
Cellular senescence and inflammation
Cellular senescence is a status of permanent cell-cycle arrest in response to stress or telomere shortening. Senescence has evolved as a tumor-suppressor mechanism to limit cell proliferation129. However, over time accumulation of senescent cells compromises tissue function and is one of the hallmarks of ageing1. First observed in replicative exhaustion of human primary fibroblasts (replicative senescence)130, cellular senescence was later identified to also result from activation of oncogenes (oncogene-induced senescence, OIS) and sustained DNA damage (stress-induced premature senescence, SIPS)26.
Senescent cells exhibit inflammatory secretory phenotypes.
Senescent cells were traditionally viewed as zombie cells, contributing to ageing by altering tissue function and integrity131. Later studies, however, revealed more complicated effects beyond the cell-autonomous level. Senescent cells were shown to secrete a spectrum of inflammatory factors, which were termed the senescence-associated secretory phenotype (SASP)132. SASP has deleterious effects by promoting inflammation and, potentially, tumour progression in neighbouring cells133. Both the overexpression of the oncogene RAS and the loss of the tumour suppressor p53 can enhance the development of SASP, suggesting that SASP may be a consequence of a persistent DNA-damage response134.
SASP is triggered by cytoplasmic DNA sensing.
Recent studies have shown that SASP is dependent on cytoplasmic nucleic acid-sensing pathways in senescent cells111,135. One characteristic of senescent cells is the micronuclei-like structures observed in the cytoplasm, termed cytoplasmic chromatin fragments (CCFs)111,136. The CCFs contain fragments of chromatin blebbing from the nucleus to cytoplasm. The mechanism of CCF formation in senescent cells is incompletely understood and is associated with the loss of nuclear membrane integrity136. This is associated with the loss of lamin B1, a biomarker of senescence136,137. It has been shown that in RAS-induced senescence, the degradation of lamin B1 is mediated by the autophagy-related protein LC3/ATG8, which directly interacts with lamin B1 and the lamin-associated domains on the chromatin138 targeting them to the lysosome for degradation.
The CCFs in senescent cells are positive for the DNA damage response marker γH2AX and negative for DNA repair protein 53BP1136, suggesting that the CCF formation is associated with DNA damage. 53BP1 is a DNA repair protein that inhibits resections of DSBs139. Interestingly, 53BP1 is also shown to suppress the formation of CCFs140,141. It was shown that irradiation-induced senescence was associated with mitochondrial dysfunction and production of reactive oxygen species (ROS), which activated JUN N-terminal kinase (JNK). JNK interacts with 53BP1 to regulate the formation of CCFs140. However, the exact mechanism of JNK–53BP1 interaction and its regulation of CCF formation remain unclear.
Cytoplasmic DNA, the DNA part of CCFs, is sensed by the cytoplasmic DNA sensor cGAS, which triggers STING and innate immune response16 (Figure 3). This model is supported by the observation that DNA damage-induced cytoplasmic DNA fragments contain both γH2AX and cGAS142. Importantly, while cGAS senses cytoplasmic DNA presented in senescence, cGAS is also essential for maintenance of senescence142. Downstream of STING are two signalling pathways, namely the IFN pathway mediated by IRF3 and the SASP pathway induced by NF-κB143. The IFN promoter contains elements responsive to both IRF3 and NF-κB; therefore, mechanistically, both pathways can activate IFN responses, and the combination of IRF3 and NF-κB results in a synergetic effect19,144. The induction of senescence in mouse embryonic fibroblasts (MEFs) by different methods activated both IFN and SASP responses, which promoted senescence in a paracrine manner145. However, in a similar study using primary human lung fibroblasts from the IMR90 cell line, senescence induction using HRASV12 [G] transduction or treatment with the chemotherapeutic drug etoposide only activated the SASP, but not an IFN response135. The conditioned media from senescent cells, which contains SASP factors and especially IL-1α, suppressed IFN induction by double-stranded DNA transfection135. It was speculated that the failure to induce a type I IFN response resulted from the inhibition of STING by p38 MAP kinase (MAPK) in senescent cells54,135. Indeed, it was shown that activated p38 phosphorylates USP21, stimulating deubiquitination of STING leading to in STING inhibition146. Interestingly, exogenous IL-1β was recently reported to activate an IFN response through the release of mtDNA, which was recognized by cGAS147. These different observations may have arisen from the use of different cell types and/or methods to induce senescence. Recently it was reported that topoisomerase 1 (TOP1)–DNA cleavage complex (TOP1cc) is necessary and sufficient for cGAS to recognize cytoplasmic chromatin to trigger SASP during senescence148. This process is stabilized by the chromatin architecture protein HMGB2, suggesting chromatin-associated proteins are involved in cGAS activation in senescence148. Studying the mechanisms responsible for the choice of the downstream pathways will provide a better understanding for the role of inflammation in senescence. Interestingly, recent evidence showed that cGAS-STING pathway also activates autophagy through LC3 lipidation independent of the TBK1 and interferon induction149. More strikingly, the ancient origin of STING homologue (NvSTING) identified from the sea anemone Nematostella vectensis lacks the C-terminal domain required for TBK1 activation150. NvSTING still possesses the ability to induce autophagy, suggesting that the cGAS-STING pathway has originally evolved as part of autophagic response but was later co-opted to serve as a DNA sensor for immune response149,150. Thus, providing a link between autophagy, another established hallmark of aging, and inflammation.
Figure 3: Mechanisms of senescence induced inflammation.
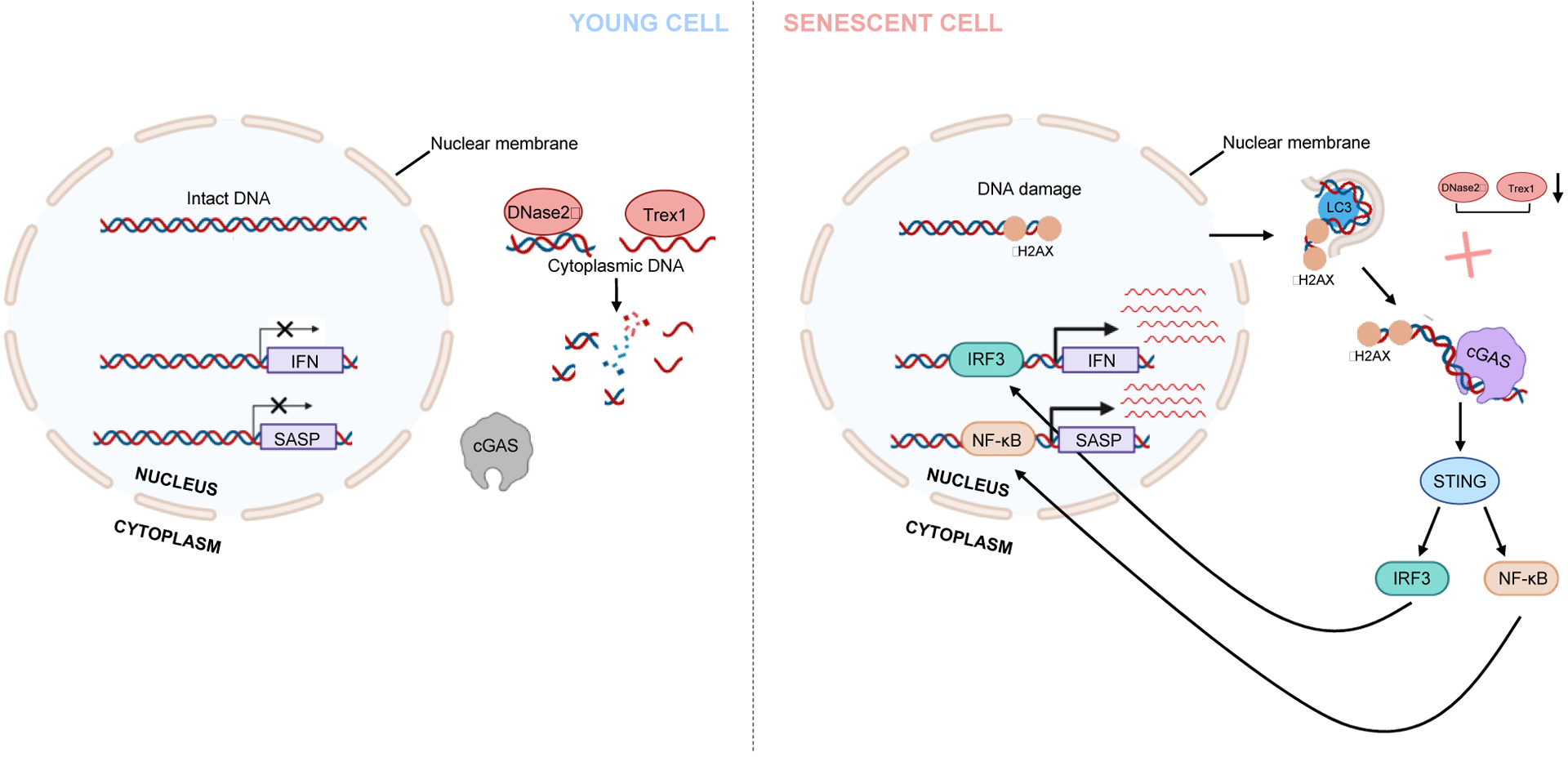
Cellular senescence induces inflammation via cytoplasmic nucleic acid sensing pathway. Three major events participate in the generation of cytoplasmic DNA. One of the hallmarks of senescence is the loss of lamin B1. The autophagy-related protein LC3 interacts with lamina and lamina-associated domains (LADs) of the chromatin and directs lamin B1 to autophagy. This process results in the loss of nuclear membrane integrity, contributing to the leaking of chromatin to the cytoplasm. Sustained DNA damage response (DDR) is another hallmark of senescence. The cytoplasmic DNA is positive for γH2AX and negative of 53BP1, suggesting that the formation of cytoplasmic DNA is associated with DDR. Two cytoplasmic DNases, DNase 2α and Trex1, are responsible for degrading excessive cytoplasmic DNA. Both enzymes are down-regulated in senescence through a negative transcriptional regulation. Therefore, in senescence, loss of nuclear membrane integrity promotes the accumulation of cytoplasmic DNA, which is further stabilized by the down-regulation of DNases. Cytoplasmic DNA is sensed by the DNA the sensor cGAS, which triggers STING and its downstream pathways, including IRF3-IFN and NF-κB-SASP pathways.
Negative regulation of cytoplasmic DNA.
To prevent unintended activation of inflammation, deoxyribonucleases (DNases) in the cytoplasm digest excessive DNA, serving as a negative regulator of cytoplasmic DNA. There are two major DNases in the cytoplasm: the DNase 2α (encoded by DNaseII) and Trex1 (originally designated DNaseIII). DNase 2α is located in the lysosome and plays an important role in engulfment-mediated DNA degradation151. Deficiency of DNase 2α results in type I IFN-mediated autoinflammation and promotes an ageing phenotype in mice152,153. The ageing phenotype can be rescued in Dnase2α−/− STING−/− mice, suggesting its dependence on cGAS–STING pathway153. Similarly, deficiency of Trex1, a DNase located in the cytosol, also induces accumulation of cytoplasmic DNA that triggers innate immune responses154,155. Intriguingly, both DNases are down-regulated in senescent cells, contributing to aberrant cytoplasmic DNA sensing and inflammation156,157. The down-regulation of DNase 2α and Trex1 in senescent cells is believed to occur on the mRNA level, through the repression of the transcriptional factor E2F, which is repressed by the p16-pRb pathway during senescence156. It is tempting to propose that the down-regulation of the cytoplasmic DNases synergizes with the loss of nuclear membrane integrity, to contribute to inflammation in senescence through cytoplasmic DNA sensing (Figure 3).
R-loops in age-related inflammation
R-loops as indicators of genomic integrity.
Both DNA synthesis and the transcription of RNA involve accessing chromatin-bound DNA and unwinding of the double helix to form DNA–RNA hybrids. These three-stranded structures, known as R-loops, are frequently found at sites of high transcriptional activity and repetitive sequences158. Additionally, R-loops are formed during immunoglobulin class-switching providing access to ssDNA necessary for activation-induced cytidine deaminase to initiate this process159. R-loops also play a prominent role at telomeres, where they are formed by association of the TERRA long noncoding RNA (lncRNA) with telomeric DNA160. Thus, R-loops play critical roles in normal physiology and constitute ~5% of the human genome at any given time161.
R-loops are thought to be transitory and are removed once their function is completed. RNaseHs are employed by the cell to remove the RNA from these structures162,163. Several DNA-RNA unwinding factors, such as SENATAXIN, BLM and WRN also contribute to the breakdown and resolution of R-loops164–166. This tight regulation of R-loop number, location and persistence is necessary to maintain genomic integrity. Indeed, perturbation of several chromatin maintenance pathways, including BRCA1 and BRCA2, the NER pathway via XPG and XPF, and Fanconi anemia pathway increase R-loop abundance167–171.
Inappropriate R-loop formation and accumulation portends imminent disaster for a cell. Several recent studies have demonstrated R-loops to be drivers of genomic instability and abundant sources for DNA damage. A large portion of the genomic instability attributed to aberrant R-loop activity has been ascribed to stalled replication fork progression, though more recent reports indicate that specific subsets of R-loops and those associated with DNA damage sites may be the main drivers172–174. The exposure of ssDNA in R-loops allows endonucleases, such as XPG, XPF, and FEN1, to cleave the exposed DNA, producing single-strand and double-strand breaks166,174–176. Loss of tumour suppressors BRCA1 and BRCA2 coincide with genomic instability and over-abundance of R-loops, which are believed to mediate the loss of stability and an increase in DSBs169.
R-loops drive sterile inflammation and intersect several ageing pathways.
Given the mounting body of evidence that links DSBs with genomic instability and the onset of ageing pathologies, it appears highly probable that aberrant R-loop formation and persistence may contribute to ageing177,178. Indeed, R-loops are already associated with cancer and neurodegenerative diseases, as well as autoimmunity169,170,179–183. Defects in genes involved in R-loop biology, such as WRN, ERCC1, EPF and XPG are also associated with progeria in human and/or model organisms35,166,184,185. In C. elegans, loss of H3K9 histone methylation results in shortened lifespan, infertility and R-loop accumulation186,187. Loss of histone methylation is also correlated with aging and the erroneous expression of transposons and satellite repeat elements in C. elegans186–188. In fact, it appears that increased R-loop formation and the expression of TE and satellite DNA are closely linked, both in their occurrence and their correlation with aging, and future work elucidating the contributions from each to inflammation will be of high interest. Additionally, a recent study in zebra fish has indicated that R-loops are capable of triggering inflammatory cascades via the cGAS–STING pathway110,189. In Ercc1−/− knockout mice, R-loop accumulation results in the release of ssDNA to the cytoplasm, where it is recognized by the canonical cGAS–STING pathway, triggering the innate immune response190. Curiously, cGAS has recently been shown to function as a suppressor of genomic instability by slowing replication forks, which suggests that cGAS may be directly inhibiting R-loop formation191. Given their ability to be recognized by cGAS and activate an innate immune response, R-loops have entered the stage as potential drivers of inflammageing (Figure 4) though much work has yet to be done to determine their contribution.
Figure 4: R-loops accumulate with age and drive inflammation.
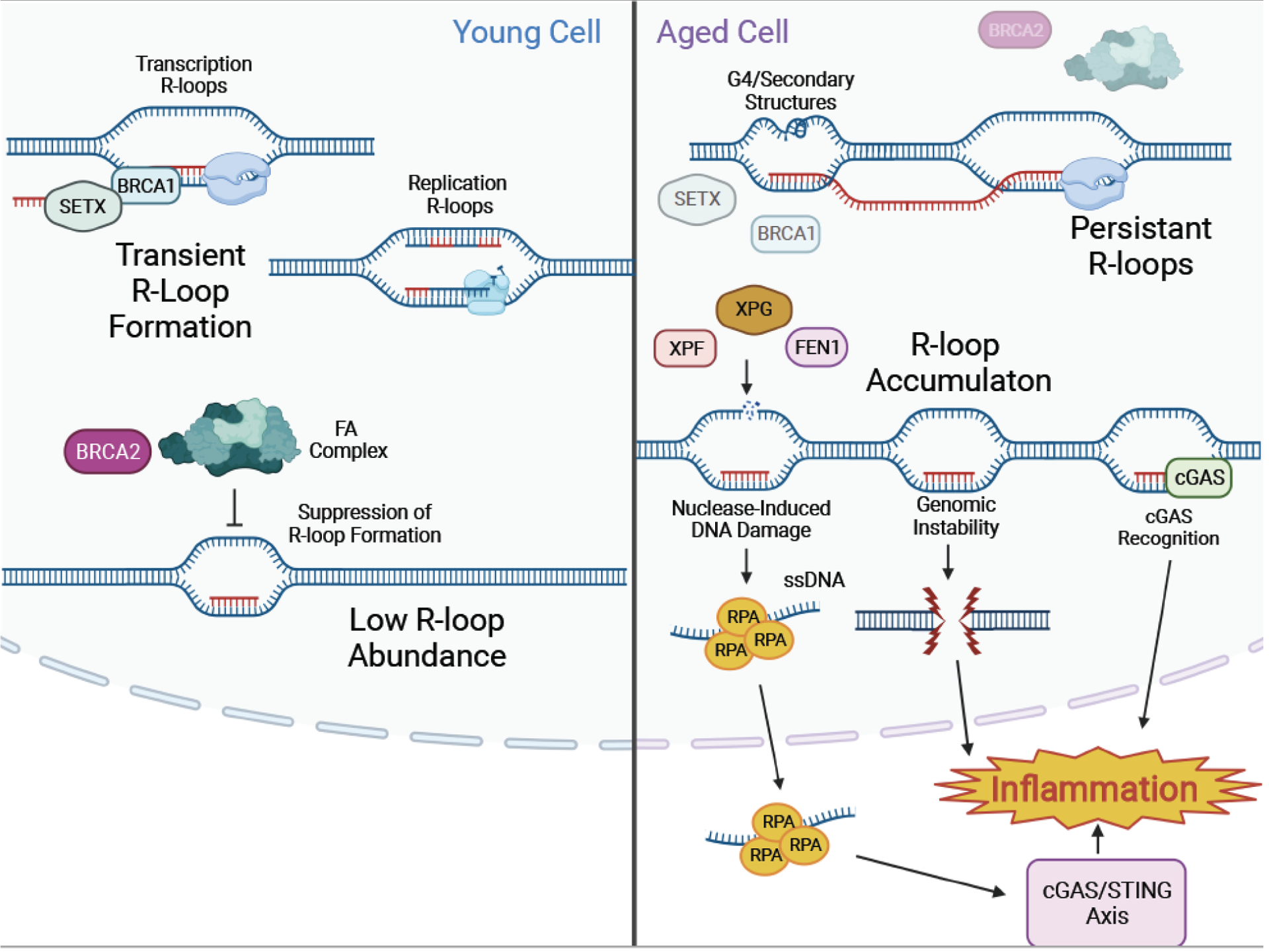
R-loops form in healthy young cells during regular cellular processes, including transcription and DNA replication. Proteins such as BRCA1 and SETX ensure RNA displacement and R-loops resolution. FA complex proteins and BRCA2 also ensure that R-loops remain transitory. In older cells, R-loops accumulate and persist within the genome, due to failure of processes responsible for their resolution. Secondary DNA structures, such as G-quadraplexes, contribute to unresolved R-loop accumulation. Persistent R-loops leave displaced ssDNA strands exposed to endonucleases, such as XPF, XPG and FEN1, leading to genomic instability and inflammation. Persistent R-loops can also be recognized by cGAS-STING and trigger sterile inflammation.
Age-related epigenetic changes
Organismal ageing and cellular senescence are associated with changes in chromatin structure. Recently, it was shown that DNA damage-induced epigenetic drift and loss of cellular identity play causal roles in promoting ageing192,193. Mice in which mild levels of DNA breaks were induced by the I-PpoI homing endonuclease displayed an altered epigenetic landscape and premature ageing phenotype192,193. It has to be noted, however, that in a similar mouse model, the I-PpoI induced DSB may result in significant DNA fusions194 which may also contribute to the aging phenotype. Many of the changes occurring in the epigenome are stochastic in nature; however, there is a well-documented trend for the loss of heterochromatin on RTEs and other repetitive sequences. One of the theoretical mechanisms proposed to underlie ageing-associated loss of heterochromatin is referred to as ‘redistribution of heterochromatin modifiers’. This theory suggests a link between DNA damage and ageing involving the Sirtuin family members SIRT1 and SIRT6195,196. SIRT1 and SIRT6 are epigenetic enzymes responsible for the formation and maintenance of heterochromatin197,198. Both SIRT1199 and SIRT6200 have the second job, where they are recruited to DSBs to promote DNA repair in response to DNA damage. Aged and senescent cells accumulate higher levels of DNA damage which cause re-localization of SIRT1 and SIRT6 to DSBs, limiting Sirtuins’ availability for binding to heterochromatin, resulting in loss of heterochromatin199. SIRT6 is specifically implicated in repressing RTEs100,113. Upon DNA damage, SIRT6 relocalizes to DSBs leaving RTEs unattended, which leads to RTE activation113 and induction of IFN responses98,100.
In summary, DNA damage promotes epigenetic changes that lead to the redistribution of epigenetic modifies such as SIRT1 and SIRT6, loss of heterochromatin and RTE silencing. RTE activation results in formation of cytoplasmic dsRNA or DNA, which are recognized by dsRNA- and DNA-sensing mechanisms leading to activation of IFN responses and fueling age-related pathologies (Figure 5).
Figure 5: Epigenetic changes triggered by life-long DNA damage lead to induction of RTEs and inflammaging.
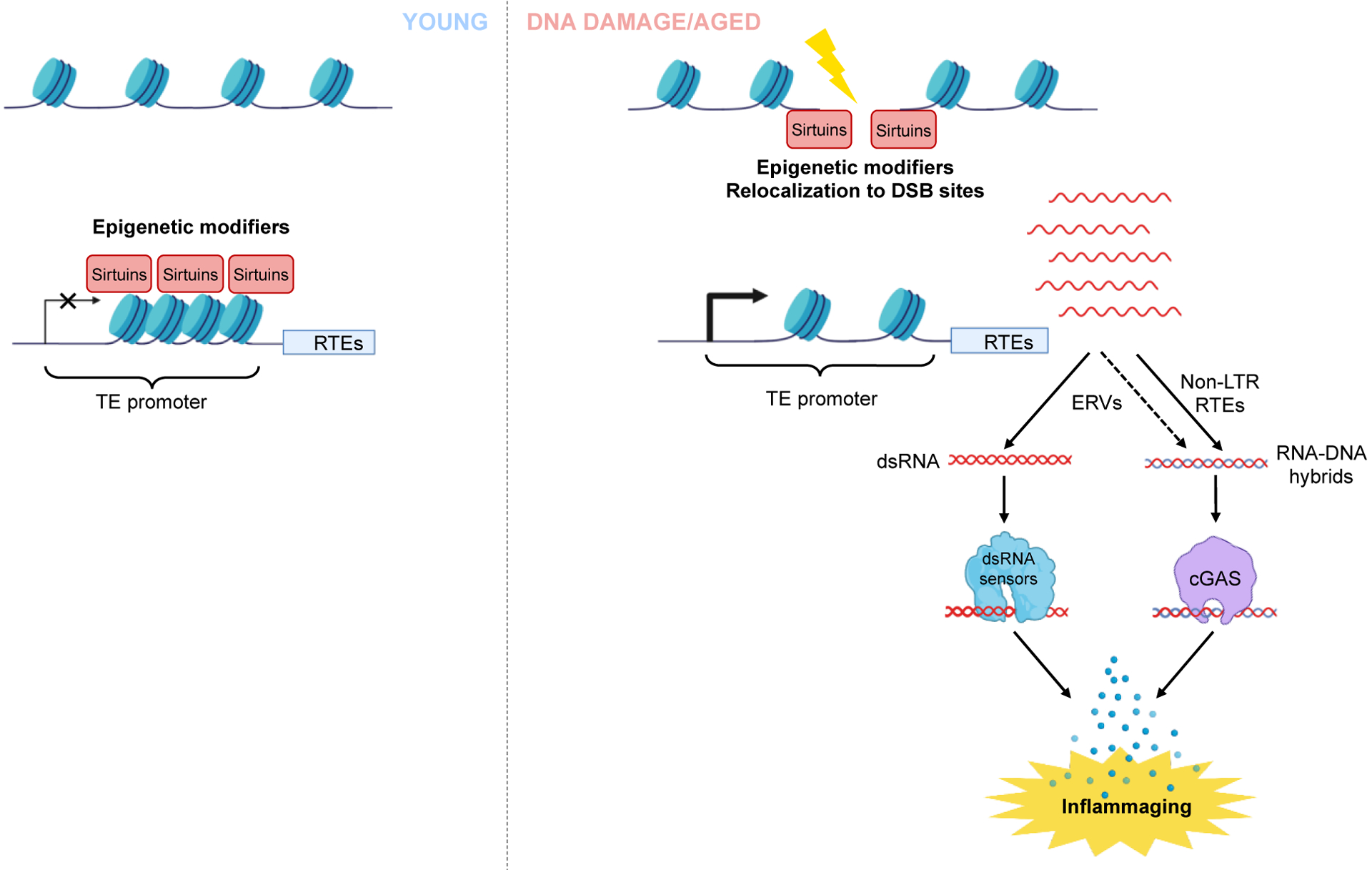
DNA damage can induce redistribution of heterochromatin modifiers such as proteins of the Sirtuin family, SIRT1 and SIRT6. In young cells, these Sirtuins silence RTEs and other repetitive elements by maintaining heterochromatin. Upon DNA damage, Sirtuins re-localize to DSB sites, leading to the activation of RTEs. Activated RTEs result in the formation of cytoplasmic RNA or DNA, which trigger the RNA or DNA sensing pathway to induce inflammation. This mechanism provides a link between DNA damage and inflammation through chromatin change.
Translation to the clinic
What can be done to suppress age-related inflammation? Multiple anti-inflammatory drugs are available and new interventions are being developed. Remarkably, several well-known drugs such as aspirin and quinacrine attenuate the cGAS–STING signallingaxis, and new inhibitors are being actively developed201–204. Recent insights into the role of cytoplasmic nucleic acid-sensing have also provided new targets for attenuating inflammation. Members of the PYHIN family of nucleic acid sensors — which includes AIM2 and IFI16 — play a pivotal role in clearing exposed cytosolic DNA and maturing pro-inflammatory cytokines such as IL-1β and IL-18205. This pathway has gained attention in recent years and due to its role in DNA sensing, its components are attractive candidates for inhibiting inflammatory responses causes by cytoplasmic DNA species. Additionally, inhibitors of the NLRP3 inflammasome, which lies downstream of nucleic acid sensing pathways, are currently in development and have demonstrated promising results in suppressing pro-inflammatory disorders such as autoimmune encephalomyelitis206. More recently, it was demonstrated that extracellular vesicles (EVs) can be utilized to deliver nucleases to cells with cytoplasmic DNA-induced inflammation, resulting in suppression of chronic inflammation and NF-κB nuclear localization190. What’s more, the use of EV-delivered nucleases was demonstrated to work in live animals lacking Ercc1, where they successfully suppressed pro-inflammatory factors such as IFNα and matrix metalloproteinase 9 (MMP9)190.
The repurposing of anti-HIV drugs, such as nucleotide reverse transcriptase inhibitors (NRTIs), has shown promise in treating RTE-related inflammation97,98,100,207. NRTI treatment has been shown to reduce Alu-induced macular degeneration and SASP factors in aged animals98,100,106. However, the chronic use of NRTIs can elicit several side effects, including hepatotoxicity and may even contribute to neuronal inflammation and chronic pain208. It is worth noting that current NRTI use in patients targets high retroviral load pathologies associated with HIV. As such, lower doses may be sufficient for suppressing endogenous RTEs and carry a lower risk of side effects. In additional, other anti-retroviral drugs, such as non-nucleoside reverse transcriptase inhibitors (NNRTIs) have fewer side effects compared to NRTIs and have also been demonstrated to suppress RTE activity209. NNRTIs specific to the L1 reverse transcriptase could be developed to provide effective protection from RTE-induced inflammation with minimal off-target effects.
Outside of targeting the initiators of inflammation, strategies that support regulators of the genome and epigenome have shown promise. The most safe and accessible strategies involve lifestyle modification, such as exercise and caloric restriction. Indeed, it has been shown that patients that introduce exercise into their routines show reduced inflammation signals, and caloric restriction studies also show a reduction in TNF and C-reactive protein (CRP) levels210–212. Alternatively, caloric restriction mimetics, such as supplemental NAD+, metformin, and rapamycin, have become attractive candidates for eliciting the beneficial results of caloric restriction, including reduced inflammation213. As many of these substances are already in use in the clinic, developing safe regimens of these drugs to treat age-related inflammation could be the most immediate approach.
Future perspectives
Genomic instability and inflammation are two major hallmarks of ageing. Only recently, however, has the link between DNA damage and inflammation been revealed. This link helps explain the age-related changes in these pathways and provides yet another axis to the underlying causes of ageing. While anti-inflammatory drugs are an obvious solution to suppress age-related inflammation, they overwhelmingly target the downstream effectors of inflammation. The most common class, NSAIDs, are known to illicit many negative side effects, including kidney, gastrointestinal, and cardiovascular issues214. The more desirable approach would be to target the root causes of inflammageing.
Developing strategies to improve DNA repair and ameliorate age-related genomic instability may be the most attractive line of research. While all animal species experience DNA damage, not all species are equal in their DNA repair capacity, indicating that there exists room for improvement177. Remarkably, the ability to repair DNA double strand breaks correlates positively with species longevity177 with SIRT6 being the key factor responsible for more efficient DNA repair in longer-lived species.
The levels of many DNA repair proteins decline with age along with the DNA repair capacity, but rescue of individual factors does not necessarily result in improved repair215. Remarkably, overexpression of regulators of DNA repair and chromatin, such as SIRT1 and SIRT6, rescues DNA repair in pre-senescent cells215–217. SIRT1 and SIRT6 also play important roles in epigenome maintenance. DNA damage-induced redistribution of these factors results in RTE activation leading to inflammation. Another outstanding question remains in how regulators of RTEs change with age and the relationship between different suppression mechanisms. While there are several seemingly independent pathways involved in RTE suppression, including TREX1, APOBEC3 family members, RNAaseL, ADAR1, MOV10, SIRT1 and SIRT6 little is known about how these factors change with age in terms of their abundance and expression.
More research needs to be done to understand how epigenome maintenance can be improved and ageing chromatin rejuvenated to restore the youthful order. Exploration into differences in epigenetic regulation between animal species with diverse lifespans is likely to reveal critical components that impact the ageing process. Epigenome could be rejuvenated and RTEs silenced by strategies such as partial reprogramming218,219 or activation of SIRT6. Recently, several chemical activators of SIRT6 have been reported220–223, including natural compounds such as the seaweed polysaccharide fucoidan224,225. These new and promising interventions, combined with regiments of older drugs repurposed for ageing systems may provide a new approach to attenuating age-related inflammation.
Box: Histone modifications, DNA damage and age-related inflammation:
DNA damage induces rapid changes to histone marks surrounding the damage site.228,229 Most of these changes are reversed after repair is complete, however, some may persist and accumulate during lifetime. Histone marks undergo changes during aging that includes loss of histone H3K9me3 as well as epigenetic drifts in H3K4me3, H3K27me3, and H3K36me3 marks.230–232 Repetitive regions and TEs tend to lose repressive marks, while some gene promoters gain repressive marks230,231. It has also been observed that aging-related chromatin remodeling correlates with transcription of endogenous retroviruses and pro-inflammation pathways233. Some of these changes may be attributed to clonal selection such as repressive marks on promoters of genes that negatively regulate cell cycle, while others may represent the scars left after repair of DNA damage. While both DNA repair and aging lead to changes in histone marks, and the loss of repressive marks on TEs is likely to promote inflammation through TE activation and R-loop formation, the exact histone code connecting these events remains to be determined.
Glossary terms:
- Nucleotide excision repair (NER)
a DNA repair pathway that recognizes and removes bulky DNA lesions such as those formed by UV light. NER involves damage recognition, nicks flanking the lesion and removal of the damaged DNA strand, filling in the gap by DNS polymerase and ligation of the nicks to restore intact DNA molecule. Deficiency in NER is associated with cancer predisposition and premature aging syndromes. For review see 226
- senescence-associated secretory phenotype (SASP)
a phenotype associated with cellular senescence, which express and secrete a wide variety of inflammatory factors including cytokines, chemokines, and growth factors.
- Double-stranded DNA break (DSB) repair
DNA repair pathway that repairs lesions where both DNA strands are broken. There are two major pathways of DSB repair, homologous recombination (HR) that uses sister chromatid as a template for repair and nonhomologous DNA end joining (NHEJ) that ligates the broken ends without regard for homology. Defects in DNA repair are associated with several premature aging and cancer predisposition syndromes. For review see 227
- Inflammageing
chronic, low-grade sterile inflammation frequently observed during ageing
- HRASV12
an oncoprotein of small GTPase HRAS carrying a constitutively activated mutation on codon Val-12.
- Sterile Inflammation
inflammation arising in the absence of a pathogen or external inflammatory stimuli.
- Retrotransposons (RTEs)
genetic elements abundant in mammalian genomes that move throughout the genome by copy-paste mechanism involving reverse transcription and may accumulate in the cytoplasm triggering inflammation
- Blind mole rat
a subterranean rodent of the Muroidea superfamily, characterized by long maximum lifespan (>21 years) and resistance to cancer.
Footnotes
Conflict of Interest
VG is a consultant for Elysium, Centaura, Genflow Bio, and Do Not Age. Other authors declare no conflicting interests.
References
- 1.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217, doi: 10.1016/j.cell.2013.05.039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brzostek-Racine S, Gordon C, Van Scoy S & Reich NC The DNA damage response induces IFN. J Immunol 187, 5336–5345, doi: 10.4049/jimmunol.1100040 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondo T et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proceedings of the National Academy of Sciences of the United States of America 110, 2969–2974, doi: 10.1073/pnas.1222694110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartlova A et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 42, 332–343, doi: 10.1016/j.immuni.2015.01.012 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Sistigu A et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nature medicine 20, 1301–1309, doi: 10.1038/nm.3708 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Li T & Chen ZJ The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215, 1287–1299, doi: 10.1084/jem.20180139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Vugt M & Parkes EE When breaks get hot: inflammatory signaling in BRCA1/2-mutant cancers. Trends Cancer 8, 174–189, doi: 10.1016/j.trecan.2021.12.003 (2022). [DOI] [PubMed] [Google Scholar]
- 8.Reislander T, Groelly FJ & Tarsounas M DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell 80, 21–28, doi: 10.1016/j.molcel.2020.07.026 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Fenech M et al. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 26, 125–132, doi: 10.1093/mutage/geq052 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Erdal E, Haider S, Rehwinkel J, Harris AL & McHugh PJ A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev 31, 353–369, doi: 10.1101/gad.289769.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackenzie KJ et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465, doi: 10.1038/nature23449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding SM et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470, doi: 10.1038/nature23470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krupina K, Goginashvili A & Cleveland DW Causes and consequences of micronuclei. Curr Opin Cell Biol 70, 91–99, doi: 10.1016/j.ceb.2021.01.004 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatch EM, Fischer AH, Deerinck TJ & Hetzer MW Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60, doi: 10.1016/j.cell.2013.06.007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raab M et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362, doi: 10.1126/science.aad7611 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Sun L, Wu J, Du F, Chen X & Chen ZJ Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791, doi: 10.1126/science.1232458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka Y & Chen ZJ STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5, ra20, doi: 10.1126/scisignal.2002521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abe T & Barber GN Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol 88, 5328–5341, doi: 10.1128/JVI.00037-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwanaszko M & Kimmel M NF-kappaB and IRF pathways: cross-regulation on target genes promoter level. BMC Genomics 16, 307, doi: 10.1186/s12864-015-1511-7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho SS et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 44, 1177–1189, doi: 10.1016/j.immuni.2016.04.010 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Herzner AM et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol 16, 1025–1033, doi: 10.1038/ni.3267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Motwani M, Pesiridis S & Fitzgerald KA DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 20, 657–674, doi: 10.1038/s41576-019-0151-1 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Kang C et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612, doi: 10.1126/science.aaa5612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinz M et al. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol Cell 40, 63–74, doi: 10.1016/j.molcel.2010.09.008 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Fang L et al. ATM regulates NF-kappaB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic acids research 42, 8416–8432, doi: 10.1093/nar/gku529 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.d’Adda di Fagagna F Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 8, 512–522, doi: 10.1038/nrc2440 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Dunphy G et al. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol Cell 71, 745–760 e745, doi: 10.1016/j.molcel.2018.07.034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niedernhofer LJ, Bohr VA, Sander M & Kraemer KH Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: molecules to patients. Mech Ageing Dev 132, 340–347, doi: 10.1016/j.mad.2011.06.004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moraes MC, Neto JB & Menck CF DNA repair mechanisms protect our genome from carcinogenesis. Front Biosci (Landmark Ed) 17, 1362–1388, doi: 10.2741/3992 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Cleaver JE Defective repair replication of DNA in xeroderma pigmentosum. Nature 218, 652–656, doi: 10.1038/218652a0 (1968). [DOI] [PubMed] [Google Scholar]
- 31.Cleaver JE Xeroderma pigmentosum: a human disease in which an initial stage of DNA repair is defective. Proceedings of the National Academy of Sciences of the United States of America 63, 428–435, doi: 10.1073/pnas.63.2.428 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sijbers AM et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 86, 811–822, doi: 10.1016/s0092-8674(00)80155-5 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Niedernhofer LJ et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Molecular and cellular biology 24, 5776–5787, doi: 10.1128/MCB.24.13.5776-5787.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein Douwel D et al. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54, 460–471, doi: 10.1016/j.molcel.2014.03.015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niedernhofer LJ et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038–1043, doi: 10.1038/nature05456 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Karakasilioti I et al. DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab 18, 403–415, doi: 10.1016/j.cmet.2013.08.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim DE et al. Deficiency in the DNA repair protein ERCC1 triggers a link between senescence and apoptosis in human fibroblasts and mouse skin. Aging Cell 19, e13072, doi: 10.1111/acel.13072 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao J et al. ATM is a key driver of NF-kappaB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging (Albany NY) 12, 4688–4710, doi: 10.18632/aging.102863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cockayne EA Dwarfism with retinal atrophy and deafness. Arch Dis Child 11, 1–8, doi: 10.1136/adc.11.61.1 (1936). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karikkineth AC, Scheibye-Knudsen M, Fivenson E, Croteau DL & Bohr VA Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res Rev 33, 3–17, doi: 10.1016/j.arr.2016.08.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laugel V Cockayne syndrome: the expanding clinical and mutational spectrum. Mech Ageing Dev 134, 161–170, doi: 10.1016/j.mad.2013.02.006 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Majora M et al. HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci Transl Med 10, doi: 10.1126/scitranslmed.aam7510 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Kajitani GS et al. Neurovascular dysfunction and neuroinflammation in a Cockayne syndrome mouse model. Aging (Albany NY) 13, 22710–22731, doi: 10.18632/aging.203617 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berquist BR, Canugovi C, Sykora P, Wilson DM 3rd & Bohr VA Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic acids research 40, 8392–8405, doi: 10.1093/nar/gks565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.West AP et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557, doi: 10.1038/nature14156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chung KW et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab 30, 784–799 e785, doi: 10.1016/j.cmet.2019.08.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oshima J, Sidorova JM & Monnat RJ Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev 33, 105–114, doi: 10.1016/j.arr.2016.03.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turaga RV et al. The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell cycle 8, 2080–2092, doi: 10.4161/cc.8.13.8925 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Davis T & Kipling D Werner Syndrome as an example of inflamm-aging: possible therapeutic opportunities for a progeroid syndrome? Rejuvenation Res 9, 402–407, doi: 10.1089/rej.2006.9.402 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Goto M et al. Multiplex cytokine analysis of Werner syndrome. Intractable Rare Dis Res 4, 190–197, doi: 10.5582/irdr.2015.01035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goto M, Chiba J, Matsuura M, Iwaki-Egawa S & Watanabe Y Inflammageing assessed by MMP9 in normal Japanese individuals and the patients with Werner syndrome. Intractable Rare Dis Res 5, 103–108, doi: 10.5582/irdr.2016.01028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goto M et al. Aging-associated inflammation in healthy Japanese individuals and patients with Werner syndrome. Experimental gerontology 47, 936–939, doi: 10.1016/j.exger.2012.08.010 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Yu Q et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell reports 11, 785–797, doi: 10.1016/j.celrep.2015.03.069 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freund A, Patil CK & Campisi J p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. The EMBO journal 30, 1536–1548, doi: 10.1038/emboj.2011.69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amor-Gueret M Bloom syndrome, genomic instability and cancer: the SOS-like hypothesis. Cancer Lett 236, 1–12, doi: 10.1016/j.canlet.2005.04.023 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Rao VA et al. Endogenous gamma-H2AX-ATM-Chk2 checkpoint activation in Bloom’s syndrome helicase deficient cells is related to DNA replication arrested forks. Molecular cancer research : MCR 5, 713–724, doi: 10.1158/1541-7786.MCR-07-0028 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Gratia M et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J Exp Med 216, 1199–1213, doi: 10.1084/jem.20181329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rehman R et al. Noncanonical role for Ku70/80 in the prevention of allergic airway inflammation via maintenance of airway epithelial cell organelle homeostasis. Am J Physiol Lung Cell Mol Physiol 319, L728–L741, doi: 10.1152/ajplung.00522.2019 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Sun X et al. DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nature communications 11, 6182, doi: 10.1038/s41467-020-19941-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferguson BJ, Mansur DS, Peters NE, Ren H & Smith GL DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047, doi: 10.7554/eLife.00047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blackford AN & Jackson SP ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 66, 801–817, doi: 10.1016/j.molcel.2017.05.015 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Sirbu BM & Cortez D DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 5, a012724, doi: 10.1101/cshperspect.a012724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boder E & Sedgwick RP Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics 21, 526–554 (1958). [PubMed] [Google Scholar]
- 64.Shiloh Y & Lederman HM Ataxia-telangiectasia (A-T): An emerging dimension of premature ageing. Ageing Res Rev 33, 76–88, doi: 10.1016/j.arr.2016.05.002 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Zaki-Dizaji M, Akrami SM, Azizi G, Abolhassani H & Aghamohammadi A Inflammation, a significant player of Ataxia-Telangiectasia pathogenesis? Inflamm Res 67, 559–570, doi: 10.1007/s00011-018-1142-y (2018). [DOI] [PubMed] [Google Scholar]
- 66.McGrath-Morrow SA et al. Elevated serum IL-8 levels in ataxia telangiectasia. J Pediatr 156, 682–684 e681, doi: 10.1016/j.jpeds.2009.12.007 (2010). [DOI] [PubMed] [Google Scholar]
- 67.McGrath-Morrow SA, Collaco JM, Detrick B & Lederman HM Serum Interleukin-6 Levels and Pulmonary Function in Ataxia-Telangiectasia. J Pediatr 171, 256–261 e251, doi: 10.1016/j.jpeds.2016.01.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saunders RA et al. Elevated inflammatory responses and targeted therapeutic intervention in a preclinical mouse model of ataxia-telangiectasia lung disease. Sci Rep 11, 4268, doi: 10.1038/s41598-021-83531-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gul E et al. Type I IFN-related NETosis in ataxia telangiectasia and Artemis deficiency. J Allergy Clin Immunol 142, 246–257, doi: 10.1016/j.jaci.2017.10.030 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Song X, Ma F & Herrup K Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. The Journal of neuroscience : the official journal of the Society for Neuroscience 39, 6378–6394, doi: 10.1523/JNEUROSCI.0774-19.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eriksson M et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298, doi: 10.1038/nature01629 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK & Spann TP Nuclear lamins: building blocks of nuclear architecture. Genes & development 16, 533–547, doi: 10.1101/gad.960502 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Goldman RD et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proceedings of the National Academy of Sciences of the United States of America 101, 8963–8968, doi: 10.1073/pnas.0402943101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gonzalo S & Kreienkamp R DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome. Curr Opin Cell Biol 34, 75–83, doi: 10.1016/j.ceb.2015.05.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gonzalez-Dominguez A et al. Inhibition of the NLRP3 inflammasome improves lifespan in animal murine model of Hutchinson-Gilford Progeria. EMBO Mol Med 13, e14012, doi: 10.15252/emmm.202114012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mu X et al. Cytoskeleton stiffness regulates cellular senescence and innate immune response in Hutchinson-Gilford Progeria Syndrome. Aging Cell 19, e13152, doi: 10.1111/acel.13152 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Osorio FG et al. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes & development 26, 2311–2324, doi: 10.1101/gad.197954.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921, doi: 10.1038/35057062 (2001). [DOI] [PubMed] [Google Scholar]
- 79.Mouse Genome Sequencing C et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562, doi: 10.1038/nature01262 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Dewannieux M & Heidmann T Endogenous retroviruses: acquisition, amplification and taming of genome invaders. Curr Opin Virol 3, 646–656, doi: 10.1016/j.coviro.2013.08.005 (2013). [DOI] [PubMed] [Google Scholar]
- 81.Hancks DC & Kazazian HH Jr Active human retrotransposons: variation and disease. Curr. Opin. Genet. Dev 22, 191–203, doi: 10.1016/j.gde.2012.02.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Richardson SR et al. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol Spectr 3, MDNA3-0061-2014, doi: 10.1128/microbiolspec.MDNA3-0061-2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bourque G et al. Ten things you should know about transposable elements. Genome Biol 19, 199, doi: 10.1186/s13059-018-1577-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deininger PL & Batzer MA Alu repeats and human disease. Mol Genet Metab 67, 183–193, doi: 10.1006/mgme.1999.2864 (1999). [DOI] [PubMed] [Google Scholar]
- 85.Kury P et al. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol Med 24, 379–394, doi: 10.1016/j.molmed.2018.02.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boissinot S & Sookdeo A The Evolution of LINE-1 in Vertebrates. Genome Biol Evol 8, 3485–3507, doi: 10.1093/gbe/evw247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gorbunova V et al. The role of retrotransposable elements in ageing and age-associated diseases. Nature 596, 43–53, doi: 10.1038/s41586-021-03542-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Di Giacomo M et al. Multiple epigenetic mechanisms and the piRNA pathway enforce LINE1 silencing during adult spermatogenesis. Mol Cell 50, 601–608, doi: 10.1016/j.molcel.2013.04.026 (2013). [DOI] [PubMed] [Google Scholar]
- 89.Yang F & Wang PJ Multiple LINEs of retrotransposon silencing mechanisms in the mammalian germline. Semin Cell Dev Biol 59, 118–125, doi: 10.1016/j.semcdb.2016.03.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kazazian HH Jr. & Moran JV Mobile DNA in Health and Disease. N Engl J Med 377, 361–370, doi: 10.1056/NEJMra1510092 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rodic N LINE-1 activity and regulation in cancer. Front Biosci (Landmark Ed) 23, 1680–1686, doi: 10.2741/4666 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Burns KH Transposable elements in cancer. Nat Rev Cancer 17, 415–424, doi: 10.1038/nrc.2017.35 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Belancio VP, Roy-Engel AM & Deininger PL All y’all need to know ‘bout retroelements in cancer. Semin Cancer Biol 20, 200–210, doi: 10.1016/j.semcancer.2010.06.001 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodic N et al. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am J Pathol 184, 1280–1286, doi: 10.1016/j.ajpath.2014.01.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sciamanna I, De Luca C & Spadafora C The Reverse Transcriptase Encoded by LINE-1 Retrotransposons in the Genesis, Progression, and Therapy of Cancer. Front Chem 4, 6, doi: 10.3389/fchem.2016.00006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burns KH Our Conflict with Transposable Elements and Its Implications for Human Disease. Annu Rev Pathol 15, 51–70, doi: 10.1146/annurev-pathmechdis-012419-032633 (2020). [DOI] [PubMed] [Google Scholar]
- 97.Thomas CA et al. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 21, 319–331 e318, doi: 10.1016/j.stem.2017.07.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.De Cecco M et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78, doi: 10.1038/s41586-018-0784-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stetson DB, Ko JS, Heidmann T & Medzhitov R Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598, doi: 10.1016/j.cell.2008.06.032 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Simon M et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab 29, 871–885 e875, doi: 10.1016/j.cmet.2019.02.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tiwari B et al. p53 directly represses human LINE1 transposons. Genes Dev 34, 1439–1451, doi: 10.1101/gad.343186.120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bregnard C et al. Upregulated LINE-1 Activity in the Fanconi Anemia Cancer Susceptibility Syndrome Leads to Spontaneous Pro-inflammatory Cytokine Production. EBioMedicine 8, 184–194, doi: 10.1016/j.ebiom.2016.05.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tunbak H et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat Commun 11, 5387, doi: 10.1038/s41467-020-19170-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaneko H et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471, 325–330, doi: 10.1038/nature09830 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tao SS et al. TREX1 As a Potential Therapeutic Target for Autoimmune and Inflammatory Diseases. Curr Pharm Des 25, 3239–3247, doi: 10.2174/1381612825666190902113218 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Fukuda S et al. Alu complementary DNA is enriched in atrophic macular degeneration and triggers retinal pigmented epithelium toxicity via cytosolic innate immunity. Sci Adv 7, eabj3658, doi: 10.1126/sciadv.abj3658 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morita M et al. Gene-targeted mice lacking the Trex1 (DNase III) 3’-->5’ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24, 6719–6727, doi: 10.1128/MCB.24.15.6719-6727.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ahmad S et al. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 172, 797–810 e713, doi: 10.1016/j.cell.2017.12.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fukuda S et al. Cytoplasmic synthesis of endogenous Alu complementary DNA via reverse transcription and implications in age-related macular degeneration. Proc Natl Acad Sci U S A 118, doi: 10.1073/pnas.2022751118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li T & Chen ZJ The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med, doi: 10.1084/jem.20180139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miller KN et al. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184, 5506–5526, doi: 10.1016/j.cell.2021.09.034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hopfner KP & Hornung V Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 21, 501–521, doi: 10.1038/s41580-020-0244-x (2020). [DOI] [PubMed] [Google Scholar]
- 113.Van Meter M et al. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nature communications 5, 5011, doi: 10.1038/ncomms6011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Teo YV et al. Cell-free DNA as a biomarker of aging. Aging Cell 18, e12890, doi: 10.1111/acel.12890 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mahmood W et al. Aging-associated distinctive DNA methylation changes of LINE-1 retrotransposons in pure cell-free DNA from human blood. Sci Rep 10, 22127, doi: 10.1038/s41598-020-79126-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 116.Ponomaryova AA et al. Aberrant Methylation of LINE-1 Transposable Elements: A Search for Cancer Biomarkers. Cells 9, doi: 10.3390/cells9092017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schmid CW Human Alu subfamilies and their methylation revealed by blot hybridization. Nucleic Acids Res 19, 5613–5617, doi: 10.1093/nar/19.20.5613 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gentilini D et al. Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age (Dordr) 35, 1961–1973, doi: 10.1007/s11357-012-9463-1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jintaridth P & Mutirangura A Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics 41, 194–200, doi: 10.1152/physiolgenomics.00146.2009 (2010). [DOI] [PubMed] [Google Scholar]
- 120.Franceschi C et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908, 244–254, doi: 10.1111/j.1749-6632.2000.tb06651.x (2000). [DOI] [PubMed] [Google Scholar]
- 121.Chiappinelli KB et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162, 974–986, doi: 10.1016/j.cell.2015.07.011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Roulois D et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 162, 961–973, doi: 10.1016/j.cell.2015.07.056 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sheng W et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563 e519, doi: 10.1016/j.cell.2018.05.052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu H et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med 25, 95–102, doi: 10.1038/s41591-018-0302-5 (2019). [DOI] [PubMed] [Google Scholar]
- 125.Mehdipour P et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 588, 169–173, doi: 10.1038/s41586-020-2844-1 (2020). [DOI] [PubMed] [Google Scholar]
- 126.Gu Z et al. Silencing of LINE-1 retrotransposons is a selective dependency of myeloid leukemia. Nat Genet 53, 672–682, doi: 10.1038/s41588-021-00829-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao Y et al. Transposon-triggered innate immune response confers cancer resistance to the blind mole rat. Nat Immunol 22, 1219–1230, doi: 10.1038/s41590-021-01027-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gorbunova V et al. Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc Natl Acad Sci U S A 109, 19392–19396, doi: 10.1073/pnas.1217211109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Campisi J Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522, doi: 10.1016/j.cell.2005.02.003 (2005). [DOI] [PubMed] [Google Scholar]
- 130.Hayflick L & Moorhead PS The serial cultivation of human diploid cell strains. Exp Cell Res 25, 585–621, doi: 10.1016/0014-4827(61)90192-6 (1961). [DOI] [PubMed] [Google Scholar]
- 131.Campisi J From cells to organisms: can we learn about aging from cells in culture? Experimental gerontology 36, 607–618, doi: 10.1016/s0531-5565(00)00230-8 (2001). [DOI] [PubMed] [Google Scholar]
- 132.Coppe JP et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868, doi: 10.1371/journal.pbio.0060301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Coppe JP, Desprez PY, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118, doi: 10.1146/annurev-pathol-121808-102144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rodier F et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11, 973–979, doi: 10.1038/ncb1909 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dou Z et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406, doi: 10.1038/nature24050 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ivanov A et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202, 129–143, doi: 10.1083/jcb.201212110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Freund A, Laberge RM, Demaria M & Campisi J Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23, 2066–2075, doi: 10.1091/mbc.E11-10-0884 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dou Z et al. Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109, doi: 10.1038/nature15548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bunting SF et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254, doi: 10.1016/j.cell.2010.03.012 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vizioli MG et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes & development 34, 428–445, doi: 10.1101/gad.331272.119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Miller KN, Dasgupta N, Liu T, Adams PD & Vizioli MG Cytoplasmic chromatin fragments-from mechanisms to therapeutic potential. Elife 10, doi: 10.7554/eLife.63728 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang H, Wang H, Ren J, Chen Q & Chen ZJ cGAS is essential for cellular senescence. Proceedings of the National Academy of Sciences of the United States of America 114, E4612–E4620, doi: 10.1073/pnas.1705499114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Barber GN STING: infection, inflammation and cancer. Nature reviews. Immunology 15, 760–770, doi: 10.1038/nri3921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Honda K, Takaoka A & Taniguchi T Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360, doi: 10.1016/j.immuni.2006.08.009 (2006). [DOI] [PubMed] [Google Scholar]
- 145.Gluck S et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19, 1061–1070, doi: 10.1038/ncb3586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen Y et al. p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J Exp Med 214, 991–1010, doi: 10.1084/jem.20161387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aarreberg LD et al. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell 74, 801–815 e806, doi: 10.1016/j.molcel.2019.02.038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhao B et al. Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nature communications 11, 908, doi: 10.1038/s41467-020-14652-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gui X et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266, doi: 10.1038/s41586-019-1006-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kranzusch PJ et al. Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2’,3’ cGAMP Signaling. Mol Cell 59, 891–903, doi: 10.1016/j.molcel.2015.07.022 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Evans CJ & Aguilera RJ DNase II: genes, enzymes and function. Gene 322, 1–15, doi: 10.1016/j.gene.2003.08.022 (2003). [DOI] [PubMed] [Google Scholar]
- 152.Rodero MP et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nature communications 8, 2176, doi: 10.1038/s41467-017-01932-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lan YY et al. Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell 18, e12901, doi: 10.1111/acel.12901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jutte BB et al. Intercellular cGAMP transmission induces innate immune activation and tissue inflammation in Trex1 deficiency. iScience 24, 102833, doi: 10.1016/j.isci.2021.102833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Berndt N et al. Photosensitivity and cGAS-dependent type I IFN activation in lupus patients with TREX1 deficiency. J Invest Dermatol, doi: 10.1016/j.jid.2021.04.037 (2021). [DOI] [PubMed] [Google Scholar]
- 156.Takahashi A et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9, 1249, doi: 10.1038/s41467-018-03555-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Han X et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress-induced senescence. The Journal of biological chemistry 295, 4451–4463, doi: 10.1074/jbc.RA119.010734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Niehrs C & Luke B Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol 21, 167–178, doi: 10.1038/s41580-019-0206-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yu K, Chedin F, Hsieh CL, Wilson TE & Lieber MR R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4, 442–451, doi: 10.1038/ni919 (2003). [DOI] [PubMed] [Google Scholar]
- 160.Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E & Lingner J Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318, 798–801, doi: 10.1126/science.1147182 (2007). [DOI] [PubMed] [Google Scholar]
- 161.Sanz LA et al. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol Cell 63, 167–178, doi: 10.1016/j.molcel.2016.05.032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cheng L, Wang W, Yao Y & Sun Q Mitochondrial RNase H1 activity regulates R-loop homeostasis to maintain genome integrity and enable early embryogenesis in Arabidopsis. PLoS Biol 19, e3001357, doi: 10.1371/journal.pbio.3001357 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cerritelli SM & Crouch RJ Ribonuclease H: the enzymes in eukaryotes. FEBS J 276, 1494–1505, doi: 10.1111/j.1742-4658.2009.06908.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Skourti-Stathaki K, Proudfoot NJ & Gromak N Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42, 794–805, doi: 10.1016/j.molcel.2011.04.026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chang EY et al. RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability. J Cell Biol 216, 3991–4005, doi: 10.1083/jcb.201703168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marabitti V et al. ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells. Nucleic Acids Res 47, 3485–3502, doi: 10.1093/nar/gkz025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.D’Alessandro G et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat Commun 9, 5376, doi: 10.1038/s41467-018-07799-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kennedy RD & D’Andrea AD The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev 19, 2925–2940, doi: 10.1101/gad.1370505 (2005). [DOI] [PubMed] [Google Scholar]
- 169.Bhatia V et al. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365, doi: 10.1038/nature13374 (2014). [DOI] [PubMed] [Google Scholar]
- 170.Hatchi E et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell 57, 636–647, doi: 10.1016/j.molcel.2015.01.011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sollier J et al. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56, 777–785, doi: 10.1016/j.molcel.2014.10.020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gan W et al. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25, 2041–2056, doi: 10.1101/gad.17010011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Costantino L & Koshland D Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability. Mol Cell 71, 487–497 e483, doi: 10.1016/j.molcel.2018.06.037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Goulielmaki E et al. The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat Commun 12, 3153, doi: 10.1038/s41467-021-23505-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cristini A et al. Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep 28, 3167–3181 e3166, doi: 10.1016/j.celrep.2019.08.041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Promonet A et al. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat Commun 11, 3940, doi: 10.1038/s41467-020-17858-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tian X et al. SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 177, 622–638 e622, doi: 10.1016/j.cell.2019.03.043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Niedernhofer LJ et al. Nuclear Genomic Instability and Aging. Annu Rev Biochem 87, 295–322, doi: 10.1146/annurev-biochem-062917-012239 (2018). [DOI] [PubMed] [Google Scholar]
- 179.Grunseich C et al. Senataxin Mutation Reveals How R-Loops Promote Transcription by Blocking DNA Methylation at Gene Promoters. Mol Cell 69, 426–437 e427, doi: 10.1016/j.molcel.2017.12.030 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Crow YJ et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38, 910–916, doi: 10.1038/ng1842 (2006). [DOI] [PubMed] [Google Scholar]
- 181.Suraweera A et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol 177, 969–979, doi: 10.1083/jcb.200701042 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Becherel OJ et al. A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum Mol Genet 24, 5759–5774, doi: 10.1093/hmg/ddv296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gunther C et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125, 413–424, doi: 10.1172/JCI78001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Barnhoorn S et al. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet 10, e1004686, doi: 10.1371/journal.pgen.1004686 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Trego KS et al. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle 10, 1998–2007, doi: 10.4161/cc.10.12.15878 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zeller P et al. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat Genet 48, 1385–1395, doi: 10.1038/ng.3672 (2016). [DOI] [PubMed] [Google Scholar]
- 187.Padeken J et al. Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev 33, 436–451, doi: 10.1101/gad.322495.118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li CL et al. Region-specific H3K9me3 gain in aged somatic tissues in Caenorhabditis elegans. PLoS Genet 17, e1009432, doi: 10.1371/journal.pgen.1009432 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Weinreb JT et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev Cell 56, 627–640 e625, doi: 10.1016/j.devcel.2021.02.006 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chatzidoukaki O et al. R-loops trigger the release of cytoplasmic ssDNAs leading to chronic inflammation upon DNA damage. Sci Adv 7, eabj5769, doi: 10.1126/sciadv.abj5769 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chen H et al. cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv 6, doi: 10.1126/sciadv.abb8941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yang J-H et al. Erosion of the Epigenetic Landscape and Loss of Cellular Identity as a Cause of Aging in Mammals. Preprint at bioRxiv, doi: 10.1101/808642 (2019). [DOI] [Google Scholar]
- 193.Hayano M et al. DNA Break-Induced Epigenetic Drift as a Cause of Mammalian Aging. Preprint at bioRxiv, doi: 10.1101/808659 (2019). [DOI] [Google Scholar]
- 194.Kim J, Sturgill D, Tran AD, Sinclair DA & Oberdoerffer P Controlled DNA double-strand break induction in mice reveals post-damage transcriptome stability. Nucleic Acids Res 44, e64, doi: 10.1093/nar/gkv1482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sedivy JM, Banumathy G & Adams PD Aging by epigenetics--a consequence of chromatin damage? Exp Cell Res 314, 1909–1917, doi: 10.1016/j.yexcr.2008.02.023 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tsurumi A & Li WX Global heterochromatin loss: a unifying theory of aging? Epigenetics 7, 680–688, doi: 10.4161/epi.20540 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Vaquero A et al. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16, 93–105, doi: 10.1016/j.molcel.2004.08.031 (2004). [DOI] [PubMed] [Google Scholar]
- 198.Vaquero A et al. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 450, 440–444, doi: 10.1038/nature06268 (2007). [DOI] [PubMed] [Google Scholar]
- 199.Oberdoerffer P et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918, doi: 10.1016/j.cell.2008.10.025 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mao Z et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446, doi: 10.1126/science.1202723 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Haag SM et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273, doi: 10.1038/s41586-018-0287-8 (2018). [DOI] [PubMed] [Google Scholar]
- 202.Lama L et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun 10, 2261, doi: 10.1038/s41467-019-08620-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Dai J et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell 176, 1447–1460 e1414, doi: 10.1016/j.cell.2019.01.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Piscianz E et al. Reappraisal of Antimalarials in Interferonopathies: New Perspectives for Old Drugs. Curr Med Chem 25, 2797–2810, doi: 10.2174/0929867324666170911162331 (2018). [DOI] [PubMed] [Google Scholar]
- 205.Broz P & Dixit VM Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420, doi: 10.1038/nri.2016.58 (2016). [DOI] [PubMed] [Google Scholar]
- 206.Coll RC et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21, 248–255, doi: 10.1038/nm.3806 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fowler BJ et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 346, 1000–1003, doi: 10.1126/science.1261754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yuan S, Shi Y, Guo K & Tang SJ Nucleoside Reverse Transcriptase Inhibitors (NRTIs) Induce Pathological Pain through Wnt5a-Mediated Neuroinflammation in Aging Mice. J Neuroimmune Pharmacol 13, 230–236, doi: 10.1007/s11481-018-9777-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dai L, Huang Q & Boeke JD Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC biochemistry 12, 18, doi: 10.1186/1471-2091-12-18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Petersen AM & Pedersen BK The anti-inflammatory effect of exercise. J Appl Physiol (1985) 98, 1154–1162, doi: 10.1152/japplphysiol.00164.2004 (2005). [DOI] [PubMed] [Google Scholar]
- 211.Ott B et al. Effect of caloric restriction on gut permeability, inflammation markers, and fecal microbiota in obese women. Sci Rep 7, 11955, doi: 10.1038/s41598-017-12109-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Meydani SN et al. Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: a randomized controlled trial in non-obese humans. Aging (Albany NY) 8, 1416–1431, doi: 10.18632/aging.100994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gabande-Rodriguez E, Gomez de Las Heras MM & Mittelbrunn M Control of Inflammation by Calorie Restriction Mimetics: On the Crossroad of Autophagy and Mitochondria. Cells 9, doi: 10.3390/cells9010082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wongrakpanich S, Wongrakpanich A, Melhado K & Rangaswami J A Comprehensive Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis 9, 143–150, doi: 10.14336/AD.2017.0306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mao Z et al. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A 109, 11800–11805, doi: 10.1073/pnas.1200583109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Huang J et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS One 3, e1710, doi: 10.1371/journal.pone.0001710 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Chen Y et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res 47, 8563–8580, doi: 10.1093/nar/gkz592 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ocampo A et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 167, 1719–1733 e1712, doi: 10.1016/j.cell.2016.11.052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sarkar TJ et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nature communications 11, 1545, doi: 10.1038/s41467-020-15174-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Klein MA et al. Mechanism of activation for the sirtuin 6 protein deacylase. J Biol Chem 295, 1385–1399, doi: 10.1074/jbc.RA119.011285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Huang Z et al. Identification of a cellularly active SIRT6 allosteric activator. Nat Chem Biol 14, 1118–1126, doi: 10.1038/s41589-018-0150-0 (2018). [DOI] [PubMed] [Google Scholar]
- 222.Shang JL, Ning SB, Chen YY, Chen TX & Zhang J MDL-800, an allosteric activator of SIRT6, suppresses proliferation and enhances EGFR-TKIs therapy in non-small cell lung cancer. Acta Pharmacol Sin 42, 120–131, doi: 10.1038/s41401-020-0442-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Chen Y et al. The SIRT6 activator MDL-800 improves genomic stability and pluripotency of old murine-derived iPS cells. Aging Cell 19, e13185, doi: 10.1111/acel.13185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bonkowski MS & Sinclair DA Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17, 679–690, doi: 10.1038/nrm.2016.93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Rahnasto-Rilla MK et al. The Identification of a SIRT6 Activator from Brown Algae Fucus distichus. Mar Drugs 15, doi: 10.3390/md15060190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Marteijn JA, Lans H, Vermeulen W & Hoeijmakers JH Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15, 465–481, doi: 10.1038/nrm3822 (2014). [DOI] [PubMed] [Google Scholar]
- 227.Scully R, Panday A, Elango R & Willis NA DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20, 698–714, doi: 10.1038/s41580-019-0152-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Price BD & D’Andrea AD Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354, doi: 10.1016/j.cell.2013.02.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hauer MH & Gasser SM Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 31, 2204–2221, doi: 10.1101/gad.307702.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhang W, Qu J, Liu GH & Belmonte JCI The ageing epigenome and its rejuvenation. Nat Rev Mol Cell Biol 21, 137–150, doi: 10.1038/s41580-019-0204-5 (2020). [DOI] [PubMed] [Google Scholar]
- 231.Saul D & Kosinsky RL Epigenetics of Aging and Aging-Associated Diseases. Int J Mol Sci 22, doi: 10.3390/ijms22010401 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Benayoun BA, Pollina EA & Brunet A Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol 16, 593–610, doi: 10.1038/nrm4048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Benayoun BA et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29, 697–709, doi: 10.1101/gr.240093.118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]